
Fa 06 CSE182
CSE182-L11
Protein sequencing and Mass Spectrometry
Fa 06 CSE182
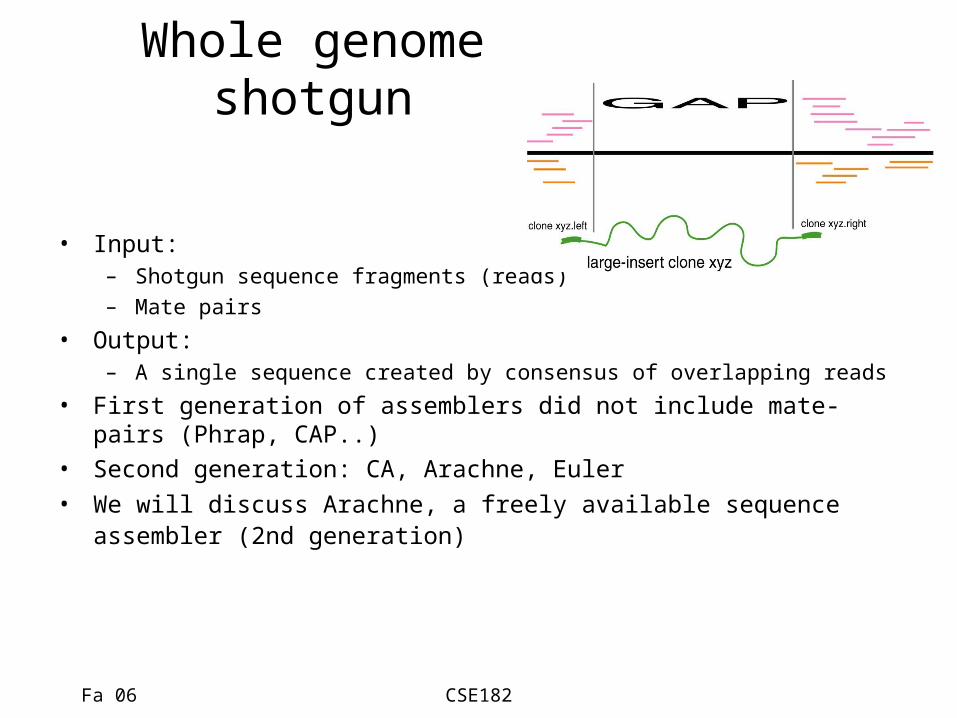
Whole genome shotgun
• Input: – Shotgun sequence fragments (reads)– Mate pairs
• Output:– A single sequence created by consensus of overlapping reads
• First generation of assemblers did not include mate-pairs (Phrap, CAP..)
• Second generation: CA, Arachne, Euler• We will discuss Arachne, a freely available sequence assembler
(2nd generation)

Fa 06 CSE182
Arachne (also celera assembler)
• Overlap– Problem 1: Large all against all computation
• Fast overlap computation using k-mer hashing.
• Layout– Problem 2: Small contigs with 10X coverage– Solution 2: Use mate-pairs to build super-contigs– Problem 3: Repetitive structure of the genome.

Fa 06 CSE182
Problem 3: Repeats

Fa 06 CSE182
Repeats & Chimerisms
• 40-50% of the human genome is made up of repetitive elements.
• Repeats can cause great problems in the assembly!
• Chimerism causes a clone to be from two different parts of the genome. Can again give a completely wrong assembly

Fa 06 CSE182
Repeats
• How can you detect if your fragment overlap is due to a repeat?

Fa 06 CSE182
Repeat detection
• Lander Waterman strikes again!• The expected number of clones in a Repeat containing island
is MUCH larger than in a non-repeat containing island (contig).• Thus, every contig can be marked as Unique, or non-unique. In
the first step, throw away the non-unique islands.
Repeat

Fa 06 CSE182
Detecting Repeat Contigs 1: Read Density
• Compute the log-odds ratio of two hypotheses:
• H1: The contig is from a unique region of the genome.
• The contig is from a region that is repeated at least twice

Fa 06 CSE182
Detecting Chimeric reads
• Chimeric reads: Reads that contain sequence from two genomic locations.
• Good overlaps: G(a,b) if a,b overlap with a high score
• Transitive overlap: T(a,c) if G(a,b), and G(b,c)
• Find a point x across which only transitive overlaps occur. X is a point of chimerism

Fa 06 CSE182
Contig assembly
• Reads are merged into contigs upto repeat boundaries.
• (a,b) & (a,c) overlap, (b,c) should overlap as well. Also, – shift(a,c)=shift(a,b)+shift(b,c)
• Most of the contigs are unique pieces of the genome, and end at some Repeat boundary.
• Some contigs might be entirely within repeats. These must be detected

Fa 06 CSE182
Creating Super Contigs

Fa 06 CSE182
Supercontig assembly
• Supercontigs are built incrementally• Initially, each contig is a supercontig.• In each round, a pair of super-contigs is merged until no
more can be performed.• Create a Priority Queue with a score for every pair of
‘mergeable supercontigs’.– Score has two terms:
• A reward for multiple mate-pair links• A penalty for distance between the links.

Fa 06 CSE182
Supercontig merging
• Remove the top scoring pair (S1,S2) from the priority queue.
• Merge (S1,S2) to form contig T.• Remove all pairs in Q containing S1 or S2
• Find all supercontigs W that share mate-pair links with T and insert (T,W) into the priority queue.
• Detect Repeated Supercontigs and remove

Fa 06 CSE182
Repeat Supercontigs
• If the distance between two super-contigs is not correct, they are marked as Repeated
• If transitivity is not maintained, then there is a Repeat
Fa 06 CSE182
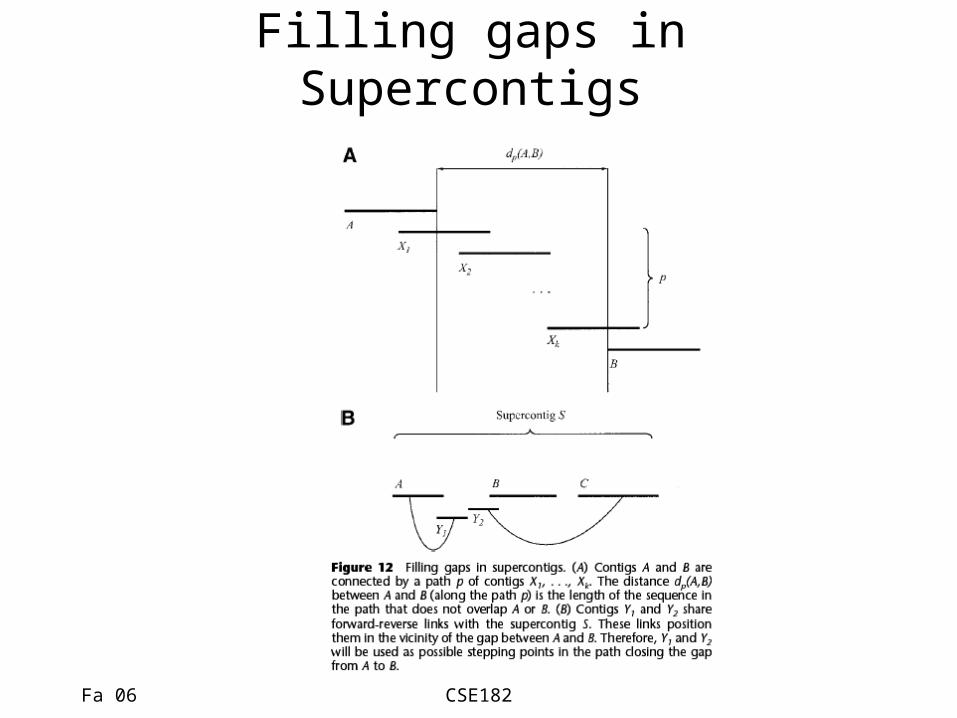
Filling gaps in Supercontigs

Fa 06 CSE182
Consensus Derivation
• Consensus sequence is created by converting pairwise read alignments into multiple-read alignments.
• The final sequence is reported as a consensus for each of the super contigs.
• The supercontigs themselves are ordered using physical markers.
• Gaps are filled in using directed sequencing efforts.

Fa 06 CSE182
Summary
• Whole genome shotgun is now routine:– Human, Mouse, Rat, Dog, Chimpanzee..– Many Prokaryotes (One can be sequenced in a day)– Plant genomes: Arabidopsis, Rice – Model organisms: Worm, Fly, Yeast
• A lot is not known about genome structure, organization and function.– Comparative genomics offers low hanging fruit
Fa 06 CSE182
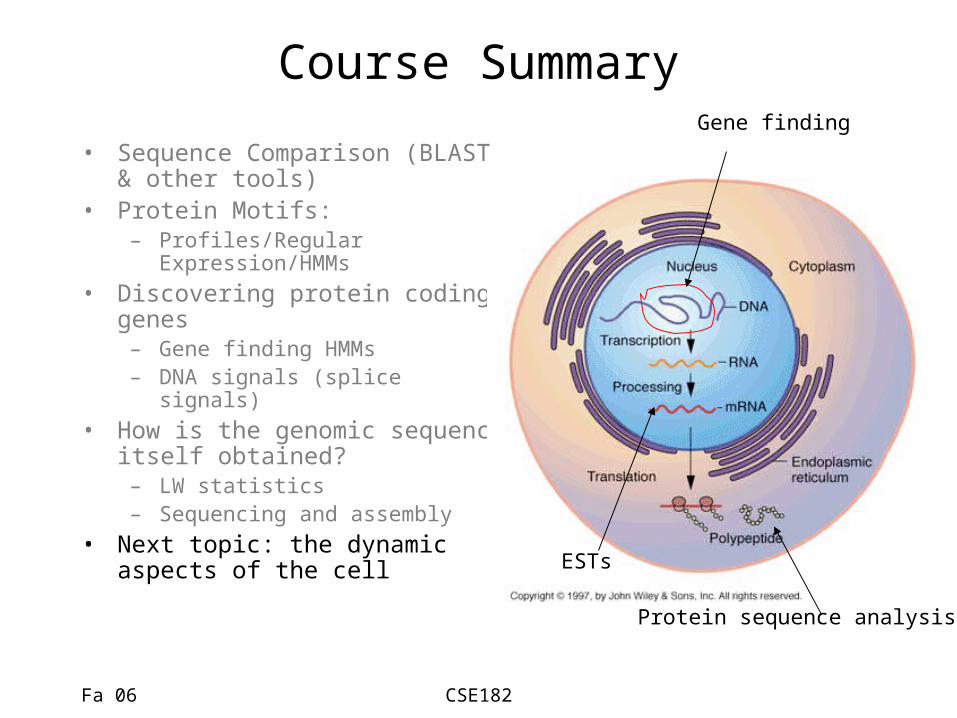
Course Summary
• Sequence Comparison (BLAST & other tools)
• Protein Motifs: – Profiles/Regular
Expression/HMMs• Discovering protein coding
genes– Gene finding HMMs– DNA signals (splice signals)
• How is the genomic sequence itself obtained?– LW statistics– Sequencing and assembly
• Next topic: the dynamic aspects of the cell
Protein sequence analysis
ESTs
Gene finding

Fa 06 CSE182
Dynamic aspects of cellular function
• Expressed transcripts– Microarrays,….
• Expressed proteins– Mass spectrometry,..
• Protein-protein interactions (protein networks)• Protein-DNA interactions• Population studies
Fa 06 CSE182
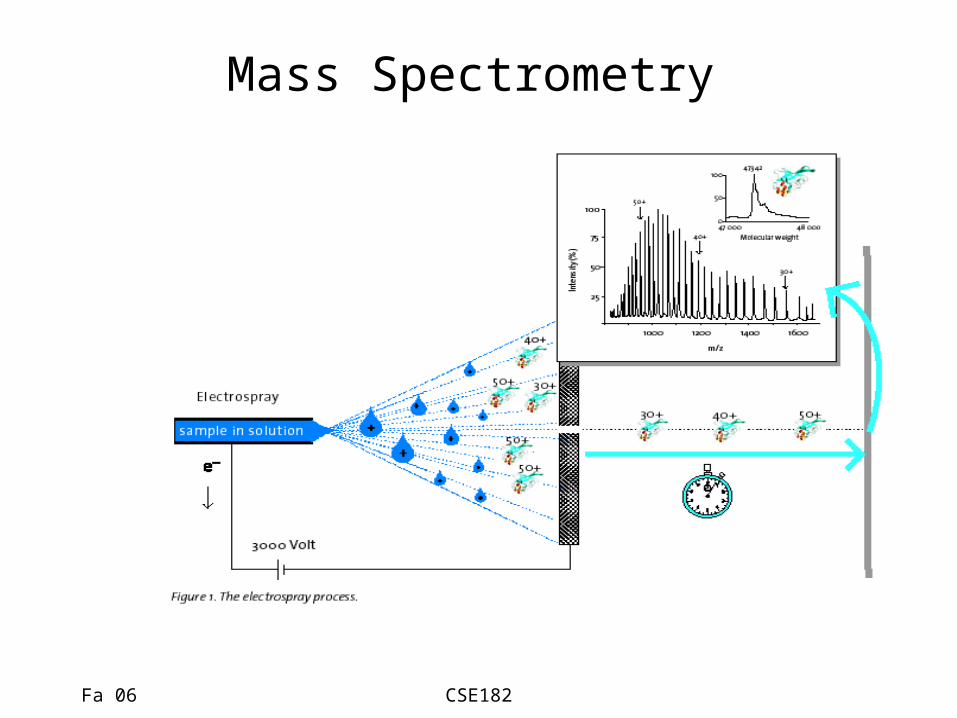
Mass Spectrometry

Fa 06 CSE182
Nobel citation ’02

Fa 06 CSE182
The promise of mass spectrometry
• Mass spectrometry is coming of age as the tool of choice for proteomics– Protein sequencing, networks, quantitation,
interactions, structure….
• Computation has a big role to play in the interpretation of MS data.
• We will discuss algorithms for – Sequencing, Modifications, Interactions..
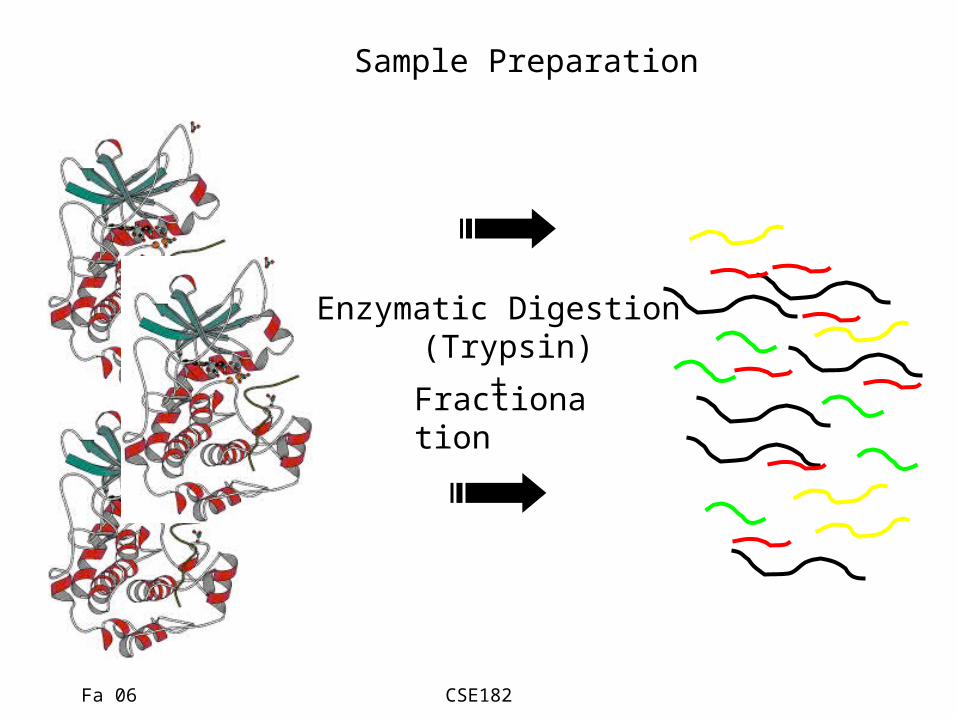
Fa 06 CSE182
Sample Preparation
Enzymatic Digestion (Trypsin)
+Fractionation
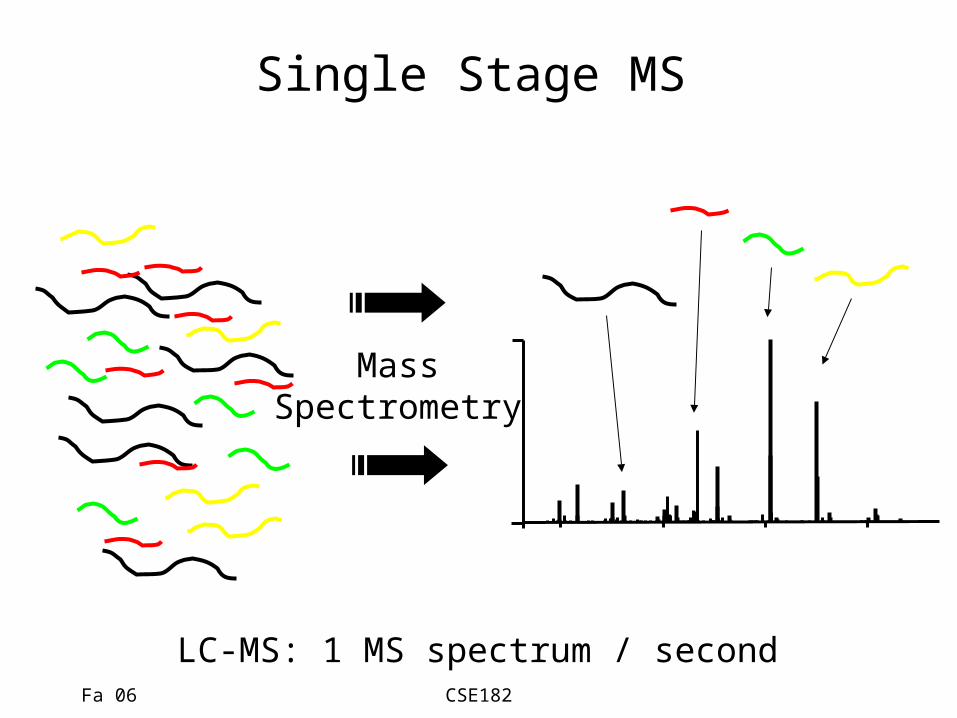
Fa 06 CSE182
Single Stage MS
MassSpectrometry
LC-MS: 1 MS spectrum / second

Fa 06 CSE182
Tandem MS
Secondary Fragmentation
Ionized parent peptide

Fa 06 CSE182
The peptide backbone
H...-HN-CH-CO-NH-CH-CO-NH-CH-CO-…OH
Ri-1 Ri Ri+1
AA residuei-1 AA residuei AA residuei+1
N-terminus C-terminus
The peptide backbone breaks to formfragments with characteristic masses.
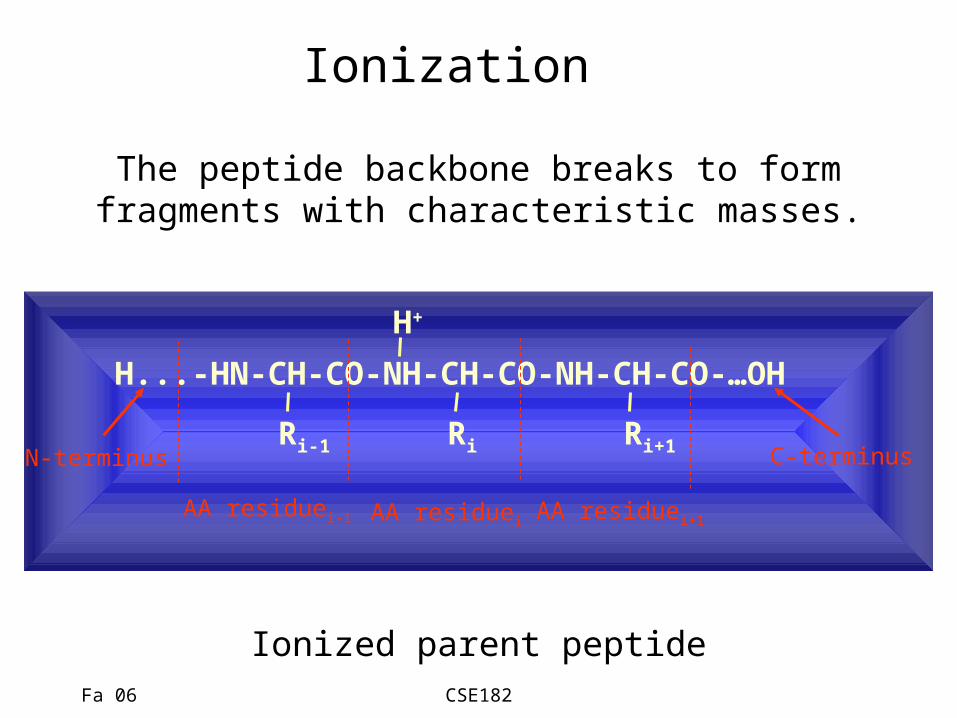
Fa 06 CSE182
Ionization
H...-HN-CH-CO-NH-CH-CO-NH-CH-CO-…OH
Ri-1 Ri Ri+1
AA residuei-1 AA residuei AA residuei+1
N-terminus C-terminus
The peptide backbone breaks to formfragments with characteristic masses.
Ionized parent peptide
H+
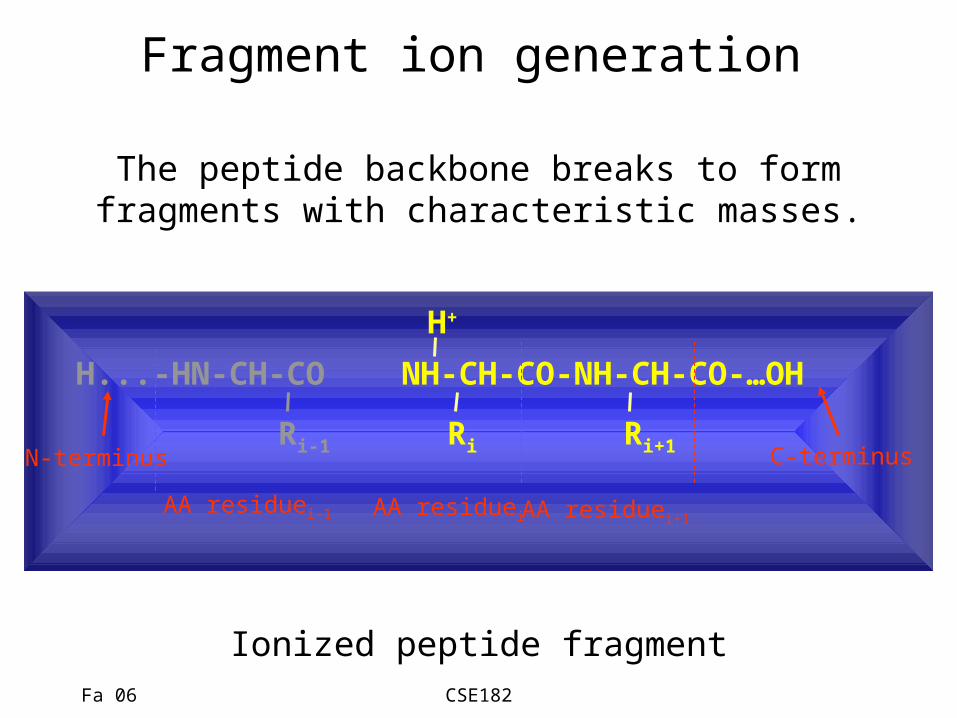
Fa 06 CSE182
Fragment ion generation
H...-HN-CH-CO NH-CH-CO-NH-CH-CO-…OH
Ri-1 Ri Ri+1
AA residuei-1 AA residuei AA residuei+1
N-terminus C-terminus
The peptide backbone breaks to formfragments with characteristic masses.
Ionized peptide fragment
H+
CSE182Fa 06
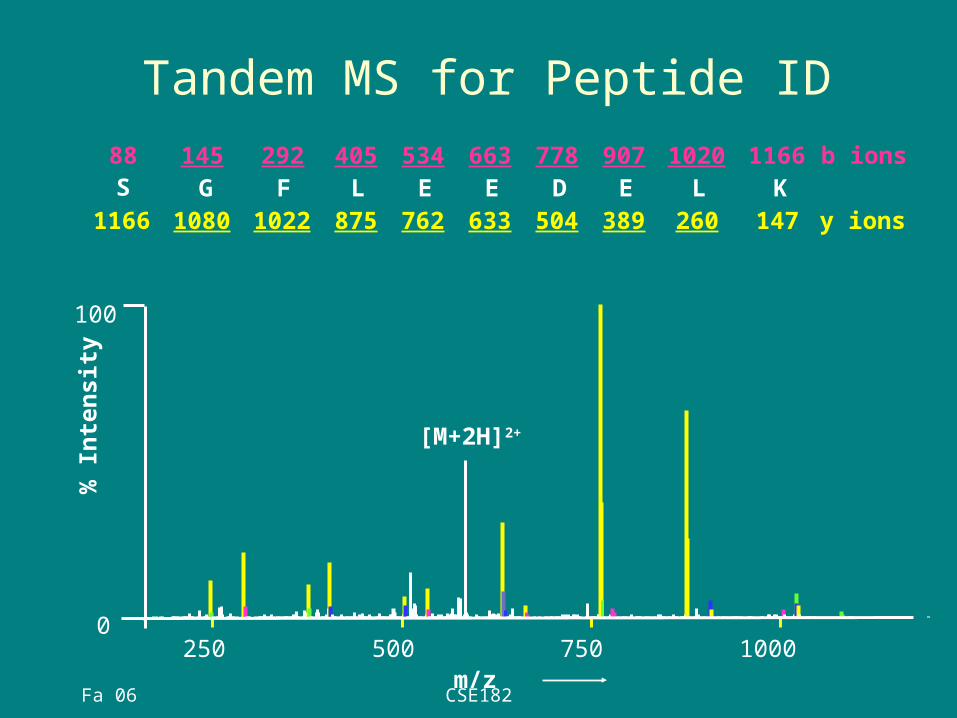
Tandem MS for Peptide ID
147K
1166L
260
1020E
389
907D
504
778E
633
663E
762
534L
875
405F
1022
292G
1080
145S
1166
88
y ions
b ions
100
0250 500 750 1000
[M+2H]2+
m/z
% I
nte
nsit
y
CSE182Fa 06
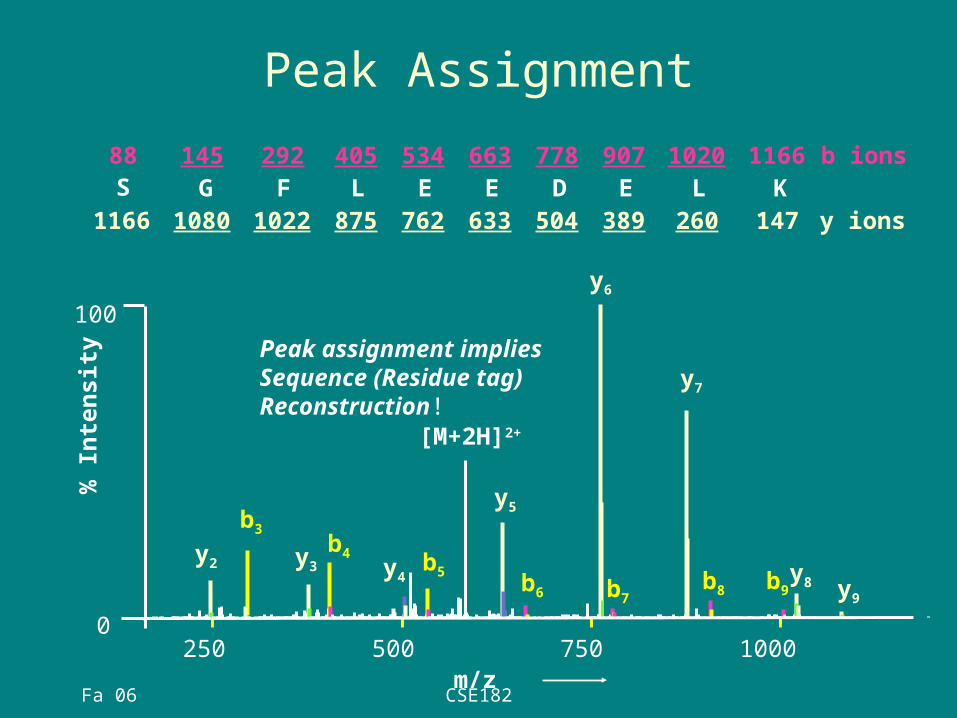
Peak Assignment
147K
1166L
260
1020E
389
907D
504
778E
633
663E
762
534L
875
405F
1022
292G
1080
145S
1166
88
y ions
b ions
100
0250 500 750 1000
y2 y3 y4
y5
y6
y7
b3b4 b5 b8 b9
[M+2H]2+
b6 b7 y9
y8
m/z
% I
nte
nsit
y Peak assignment impliesSequence (Residue tag) Reconstruction!

Fa 06 CSE182
Database Searching for peptide ID
• For every peptide from a database– Generate a hypothetical spectrum– Compute a correlation between observed and
experimental spectra– Choose the best
• Database searching is very powerful and is the de facto standard for MS.– Sequest, Mascot, and many others

Fa 06 CSE182
Spectra: the real story
• Noise Peaks• Ions, not prefixes & suffixes• Mass to charge ratio, and not mass
– Multiply charged ions
• Isotope patterns, not single peaks

CSE182Fa 06
Peptide fragmentation possibilities(ion types)
-HN-CH-CO-NH-CH-CO-NH-
RiCH-R’
ai
bici
xn-iyn-i
zn-i
yn-i-1
bi+1
R”
di+1
vn-i wn-i
i+1
i+1
low energy fragments high energy fragments

Fa 06 CSE182
Ion types, and offsets
• P = prefix residue mass• S = Suffix residue mass• b-ions = P+1• y-ions = S+19• a-ions = P-27

Fa 06 CSE182
Mass-Charge ratio
• The X-axis is not mass, but (M+Z)/Z– Z=1 implies that peak is at M+1– Z=2 implies that peak is at (M+2)/2
• M=1000, Z=2, peak position is at 501
• Quiz: Suppose you see a peak at 501. Is the mass 500, or is it 1000?
Fa 06 CSE182
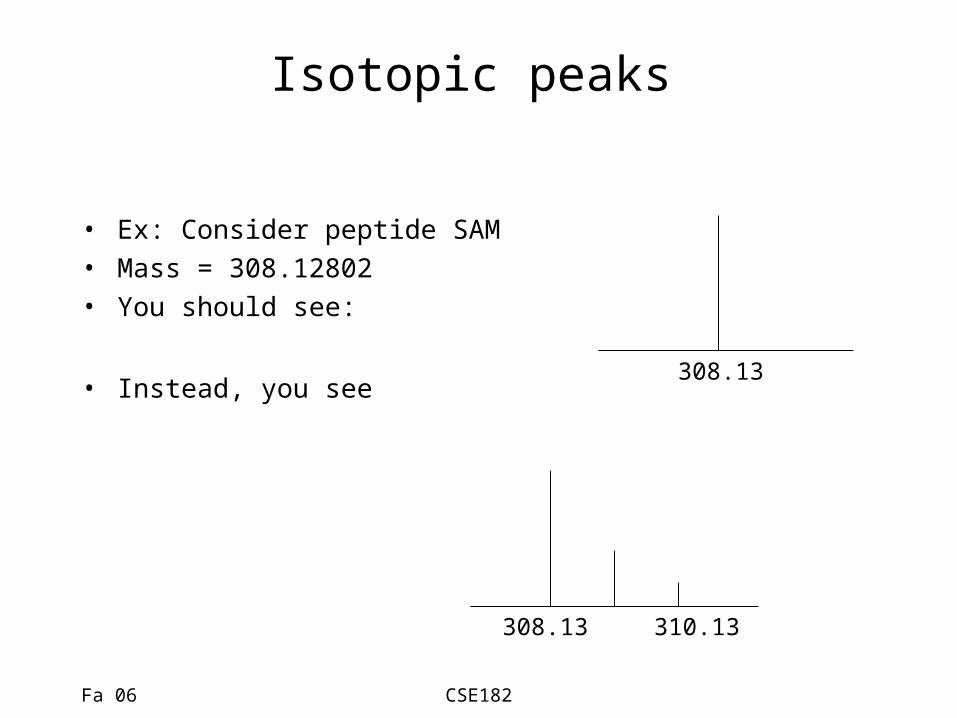
Isotopic peaks
• Ex: Consider peptide SAM• Mass = 308.12802 • You should see:
• Instead, you see 308.13
308.13 310.13

Fa 06 CSE182
Isotopes
• C-12 is the most common. Suppose C-13 occurs with probability 1%
• EX: SAM – Composition: C11 H22 N3 O5 S1
• What is the probability that you will see a single C-13?
• Note that C,S,O,N all have isotopes. Can you compute the isotopic distribution?
€
11
1
⎛
⎝ ⎜
⎞
⎠ ⎟⋅0.01⋅(0.99)10

Fa 06 CSE182
All atoms have isotopes
• Isotopes of atoms– O16,18, C-12,13, S32,34….– Each isotope has a frequency of occurrence
• If a molecule (peptide) has a single copy of C-13, that will shift its peak by 1 Da
• With multiple copies of a peptide, we have a distribution of intensities over a range of masses (Isotopic profile).
• How can you compute the isotopic profile of a peak?

Fa 06 CSE182
Isotope Calculation
• Denote:– Nc : number of carbon atoms in the peptide
– Pc : probability of occurrence of C-13 (~1%)
– Then
€
Pr[Peak at M] =NC0
⎛ ⎝ ⎜
⎞ ⎠ ⎟pc
0 1− pc( )NC
Pr[Peak at M +1] =NC1
⎛ ⎝ ⎜
⎞ ⎠ ⎟pc
1 1− pc( )NC −1
+1
Nc=50
+1
Nc=200

Fa 06 CSE182
Isotope Calculation Example • Suppose we consider Nitrogen, and Carbon• NN: number of Nitrogen atoms• PN: probability of occurrence of N-15• Pr(peak at M)• Pr(peak at M+1)?• Pr(peak at M+2)?
€
Pr[Peak at M] =NC0
⎛ ⎝ ⎜
⎞ ⎠ ⎟pc
0 1− pc( )NC NN
0 ⎛ ⎝ ⎜
⎞ ⎠ ⎟pN
0 1− pN( )NN
Pr[Peak at M +1] =NC1
⎛ ⎝ ⎜
⎞ ⎠ ⎟pc
1 1− pc( )NC −1 NN
0 ⎛ ⎝ ⎜
⎞ ⎠ ⎟pN
0 1− pN( )NN
+NC0
⎛ ⎝ ⎜
⎞ ⎠ ⎟pc
0 1− pc( )NC NN
1 ⎛ ⎝ ⎜
⎞ ⎠ ⎟pN
1 1− pN( )NN −1
How do we generalize? How can we handle Oxygen (O-16,18)?

Fa 06 CSE182
General isotope computation
• Definition:– Let pi,a be the abundance of the isotope with mass i Da
above the least mass– Ex: P0,C : abundance of C-12, P2,O: O-18 etc.
• Characteristic polynomial
• Prob{M+i}: coefficient of xi in (x) (a binomial convolution)
€
φ(x) = p0,a + p1,ax + p2,ax2 +L( )
a∏
Na

Fa 06 CSE182
Isotopic Profile Application
• In DxMS, hydrogen atoms are exchanged with deuterium• The rate of exchange indicates how buried the peptide is (in
folded state)• Consider the observed characteristic polynomial of the isotope
profile t1, t2, at various time points. Then
• The estimates of p1,H can be obtained by a deconvolution
• Such estimates at various time points should give the rate of incorporation of Deuterium, and therefore, the accessibility.
€
φt2 (x) = φt1(x)(p0,H + p1,H )N H

Fa 06 CSE182
Quiz
How can you determine the charge on a peptide?
Difference between the first and second isotope peak is 1/Z
Proposal: Given a mass, predict a composition, and the isotopic profile Do a ‘goodness of fit’ test to isolate the peaks corresponding to the isotope Compute the difference
Fa 06 CSE182
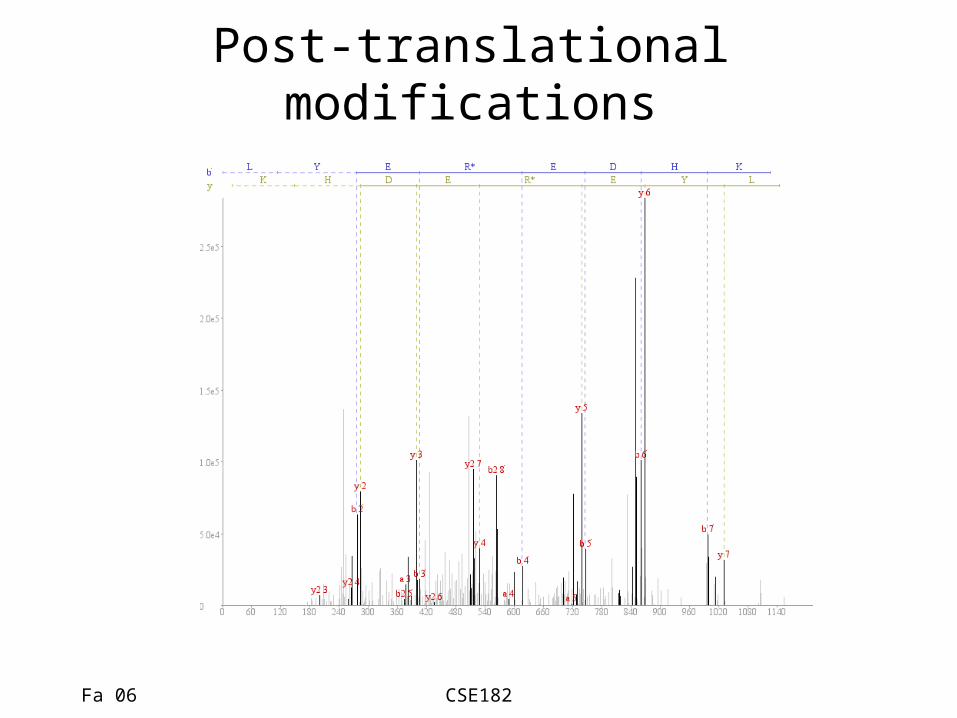
Post-translational modifications

Fa 06 CSE182
Tandem MS summary
• The basics of peptide ID using tandem MS is simple. – Correlate experimental with theoretical spectra
• In practice, there might be many confounding problems.– Isotope peaks, noise peaks, varying charges, post-
translational modifications, no database.
• Recall that we discussed how peptides could be identified by scanning a database.
• What if the database did not contain the peptide of interest?

Fa 06 CSE182
De novo analysis basics
• Suppose all ions were prefix ions? Could you tell what the peptide was?
• Can post-translational modifications help?

Fa 06 CSE182







