
Advancing Science with DNA Sequence
Microbial Genome Assembly and Finishing
Alla Lapidus, Ph.D.
Microbial genomicsDOE Joint Genome Institute,
Walnut Creek, CA
Advancing Science with DNA Sequence
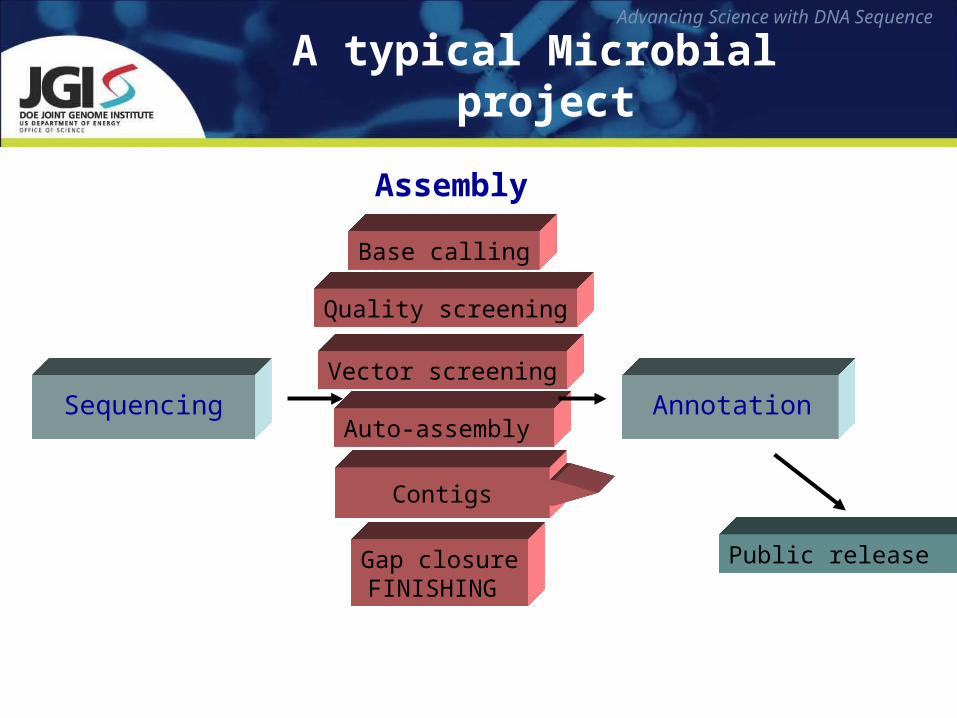
A typical Microbial project
Sequencing
Contigs
Base calling
Quality screening
Auto-assembly
Vector screening
Gap closureFINISHING
Assembly
Public release
Annotation

Advancing Science with DNA Sequence
Processing Microbial projects (Sequencing)
• Sanger only (yesterday)– 4x coverage in 3kb + 4x in 8kb + fosmids to 1x if possible– Total ~ $50k for 5mb genome draft
• Hybrid Sanger/pyrosequence/Solexa (today) – 4x coverage 8kb Sanger + 20x coverage 454 shotgun + 20x
Solexa (quality improvement)– Total ~ $35k for 5mb genome draft
• 454 + Solexa (tomorrow – starting this week)– 20x coverage 454 standard + 4x coverage 454 paired end (PE)
+ 50x coverage Solexa shotgun (quality improvement; gaps)– Total ~ $10k per 5mb genome draft

Advancing Science with DNA Sequence
Assembly (assembler)
• Sanger reads only (phrap, PGA, Arch, etc) --3kb-- --3kb-- --8kb--
--8kb-- ---------40kb--------
• Hybrid Sanger/pyrosequence/Solexa (no special assemblers; use
PGA and Arachne) 454 contig454 contig
--8kb-- --8kb-- --8kb-- --8kb--
--8kb-- --8kb-- --8kb--
454 shreds454 shreds
• 454/Solexa (Newbler, PCAP) – 454 reads only
Shotgun readsPE reads

Advancing Science with DNA Sequence
Role of Solexa data: “The Polisher”
• Align solexa reads
• Identify errors
• Automatically suggest corrections for manual curation
• Automatically suggest and implement corrections
G T A
List Disc
x1 – Gx2 – Tx3 – A
etc
x1 x2 x3

Advancing Science with DNA Sequence
Errors corrected by Solexa
CCTCTTTGATGGAAATGATA**TCTTCGAGCATCGCCTC**GGGTTTTCCATACAGAGAACCTTTGATGATGAACCGGTTGAAGATCTGCGGGTCAAA CCTCTTTGATGGAAATAATA**TATTCGAGCATC TTAGTGGAAATGATA**TCTTCGAGCATCGCCTC CGAGCNTCGCCTC**GGGCTTTCCCT CGAGCATCGCCTC**GGGTTCTCCATACACAGA GCATCGCCTC**GGGTTTTCAATACAGAGAACCT CAGCGCCTC**GGGTTTTCCATACAGAGAACCTT ATCGCCTC**GGGTTTTCCAGACAGAGAACCTTT GGTTC**GGGTTTTCCATACAGAGAACCTTTGAT GTTTTCCATACAGAGAACATTTGATGATGAAC GTTGTCCATACAGAGAACTTTTGATGATGAAC TATANCATACAGAGAACCTTTGATGATGAACC ATTTCCAGACAGAGAACCNTTGATGATGAACC CAAACAGAGAACCTTTGAGGATGAACCGGTTG ACAGGGAACCTTAGATGATGAACCGGTTGAAG ACAGAGAACCTTAGATGATGAACCGGTTGAAG ACCGTTGATGATGAACCGGTTGAAGATCTGCG GATGGTGAACGGGTTGAAGATCTGCGGGTCAA GGTTTGAAGATCTGCGGGTCAAACCAGTCCTC GGTGGAAGATCTGCGGGTAAAACCAGTCCTCT GGT.GNAGAGCTGCGGGTCAAACCAGTCCTCTG TGAAGATCTGCGGTTCAAACCAGTCCTCTCCC GATCGGCGTGTCAAACCAGTCCTCTGCCTCGT TCTGCGGGTCAAACCAGTACTCTGCCTCGTTC
Frame shift detected (454 contig)
454 contig
Finished consensus
Sanger reads

Advancing Science with DNA Sequence
Assembly: unordered set of contigs
What we get
10 16 21
10 21
Clone walk(Sanger lib)
Ordered sets of contigs (scaffolds)
New technologies: no clones to walk off
16
PCR - sequence
pri1 pri2
PCR product

Advancing Science with DNA Sequence
Why do we have gaps
•Sequencing coverage may not span all regions of the genome, thus producing gaps in the assembly.•Assembly results of the shotgun reads may produce misassembled regions due to repetitive sequences.•A biased base content (this can result in failure to be cloned, poor stability in the chosen host-vector system, or inability of the polymerase to reliably copy the sequence): ~ AT-rich DNA clones poorly in bacteria (cloning bias; promoters like structures )=> uncaptured gaps ~GC rich DNA is difficult to PCR and to sequence and often requires the use of special chemistry => captured gaps
What are gaps (Sanger)?- Genome areas not covered by
random shotgun

Advancing Science with DNA Sequence
Low GC project and 454Thermotoga lettingae TMO (JGI ID 4002278)
Draft assembly: - 55 total contigs; 41 contigs >2kb- 38GC% - biased Sanger libraries
Draft assembly +454- 2 total contigs; 1 contigs >2kb- 454 – no cloning
6810 bases 454 only out of 2,170,737bp
<166bp> - average length of gaps

Advancing Science with DNA Sequence
High GC stops (Sanger and Hybrid)
• The presence of small hairpins (inverted repeat sequences) in the DNA that re anneal ether during sequencing or electrophoresis resulting in failed sequencing reactions or unreadable electrophoresis results. (This can be aided by adding modifiers to the reaction, sequencing smaller clones and running gels at higher temperatures in the presence of stronger denaturants).

Advancing Science with DNA Sequence
High GC project and 454Xylanimonas cellulosilytica DSM 15894 (3.8 MB; 72.1% GC)
PGA assembly - 9x of 8kb PGA assembly - 9x of 8kb +454
Assembly Total contigs Major contigs Scaffolds Misassenblies* N50
PGA-8kb 210 166 4 165 41,048
PGA-8kb+454 33 23 2 14 288,369

Advancing Science with DNA Sequence
What is Finishing?
The process of taking a rough draft assembly composed of
shotgun sequencing reads, identifying and resolving miss
assemblies, sequence gaps and regions of low quality to
produce a highly accurate finished DNA sequence.
1. All low quality areas in consensus (<Q30) should be reviewed and re-
sequenced.
2. No single clone coverage, i.e. minimum of 2X depth everywhere.
3. Final error rate should be less than 1 per 50 Kb.
Current standards:

Advancing Science with DNA Sequence
Genome closure issues
• Resolve repeats and mis-assemblies– Repeats within or in vicinity of other repeats
– Large repetitive regions
– Complex repetitive regions (tandems)
• Fill in gaps– DNA region lethal to E.coli (Sanger libraries)
– Hairpins, GC rich, hard stops or other 2° structure/physical premature termination
– Hard to PCR (new technologies)
• Other issues– Homopolymeric tracts and other
polymorphisms (SNPs, VNTRs, indels)

Advancing Science with DNA Sequence
JGI Microbial FinishingCurrently: >250 individual microbes
“I am all for finished genomes! It will serve us best in the long run.. Unfinished ones are
likely to contribute to some chaos” – Proff. Sallie W. Chisholm. MIT

Advancing Science with DNA Sequence
Metagenomic assembly
• Typically size of metagenomic sequencing project is very large
• Different organisms have different coverage. Non-uniform sequence coverage results in significant under- and over-representation of certain community members
• Low coverage for the majority of organisms in highly complex communities leads to poor (if any) assemblies
• Chimerical contigs produced by co-assembly of sequencing reads originating from different species.
• Genome rearrangements and the presence of mobile genetic elements (phages, transposons) in closely related organisms further complicate assembly.
• No assemblers developed for metagenomic data sets
The whole-genome shotgun sequencing approach was used for a number of
microbial community projects, however useful quality control and assembly
of these data require reassessing methods developed to handle relatively
uniform sequences derived from isolate microbes.

Advancing Science with DNA Sequence
QC: Annotation of poor quality sequence
To avoid this:
make sure you use high quality sequence;
choose proper assembler

Advancing Science with DNA Sequence
Recommendations for metagenomic assembly
- Use Trimmer (Lucy etc) to treat reads PRIOR to assembly
- Do not use PHRAP for metagenomic projects- None of the existing assemblers designed for metagenomic
data but assemblers like PGA work better with paired reads information and produce better assemblies

Advancing Science with DNA Sequence
Metagenomic finishing: projects
Completed Projects:
Candidatus Korarchaeum cryptofilum OPF8 - is the first of this apparently ancient hyperthermophilic phyletic group to be sequenced
Desulforudis audaxviator - isolated from old water in fissures of a South African gold mine at a depth of 3000 meters. Finished with Sanger and 454
Candidatus Accumulibacter phosphatis Type IIA (CAP) - from EBPR sludge community, US
In progress:
Candidatus Endomicrobium trichonymphae - an intracellular symbiont of a flagellate protist, itself part of the hindgut community of a termite host. It is of interest in the pursuit of the efficient breakdown of cellulose and lignin necessary in the hoped-for conversion of bulk plant materials to CO2-neutral fuel

Advancing Science with DNA Sequence
Metagenomic finishing: approach
Binning:Binning: Which DNA fragment
derived from which phylotype?
(BLAST; GC%; read depth)
Non-CAP readsNon-CAP reads
CAP readsCAP reads
++
Complete genome of Complete genome of Candidatus Accumulibacter
phosphatis
Lucy/PGALucy/PGA
Candidatus Accumulibacter phosphatis (CAP)
~ 45%

Advancing Science with DNA Sequence
The end

![MICHEL L. LAPIDUS lapidusmath.ucr.edu/~lapidus/papers/papers/Lapidus Website.pdf · (Eds.), World Scientific, Singapore, 1988, pp. 327-335. ... [CP10] “The Vibrations of Fractal](https://cdn.vdocuments.us/doc/165x107/5b7106aa7f8b9a66338e0c1f/michel-l-lapidus-lapiduspaperspaperslapidus-websitepdf-eds-world-scientific.jpg)





