
S1
Supporting Information for
Adsorption of Hydrogen Sulfide on Metal-Organics
Frameworks
Juan José Gutiérrez-Sevillano,† Ana Martín-Calvo,
† David Dubbeldam,
‡ Sofía Calero,
† and Said
Hamad†,*
† Department of Physical, Chemical and Natural Systems, University Pablo de Olavide, Ctra. Utrera
km. 1, 41013, Seville, Spain.
‡ Van 't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park, 9041098 XH
Amsterdam, Netherlands.
Electronic Supplementary Material (ESI) for RSC AdvancesThis journal is © The Royal Society of Chemistry 2013

S2
Table of Contents
S1. Influence of the molecular dipole on the adsorption .................................................. 3
S2. Variation of the average occupation profile with the molecular dipole ..................... 4
S3. Snapshots of ab initio Molecular Dynamics simulations of H2S and H2O adsorbed on a cluster
model of Cu-BTC ............................................................................................................. 7
Electronic Supplementary Material (ESI) for RSC AdvancesThis journal is © The Royal Society of Chemistry 2013
S3
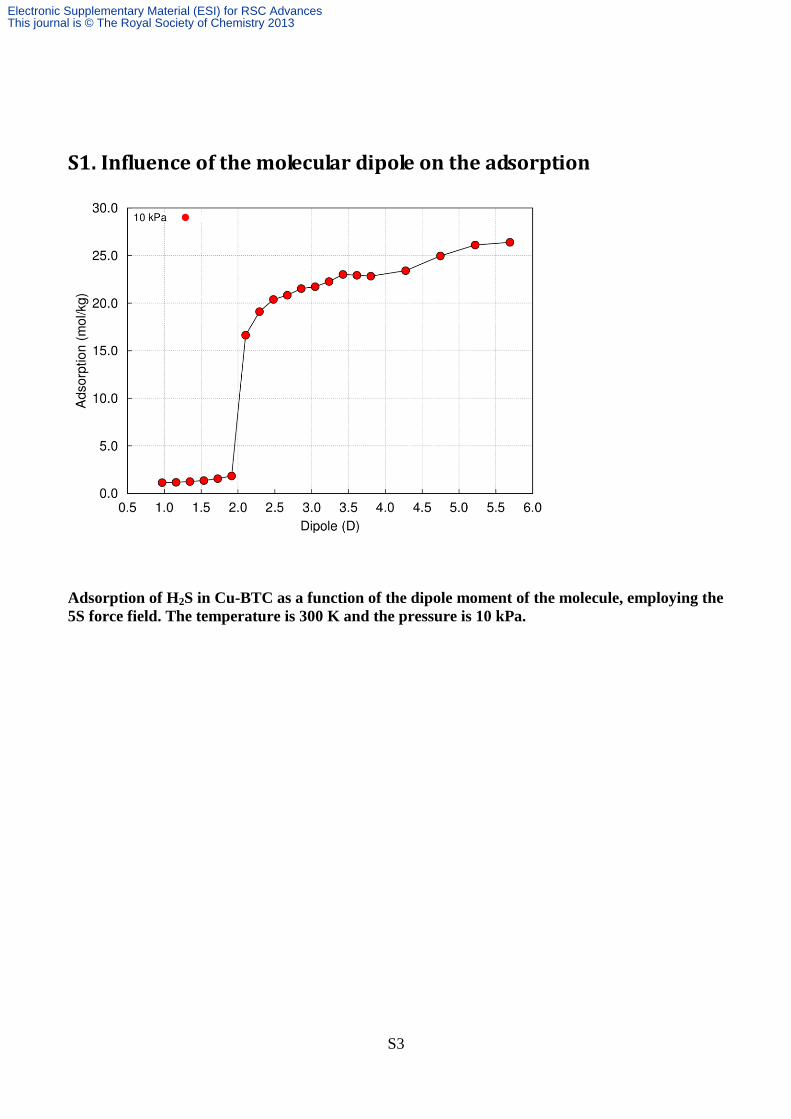
S1. Influence of the molecular dipole on the adsorption
Adsorption of H2S in Cu-BTC as a function of the dipole moment of the molecule, employing the
5S force field. The temperature is 300 K and the pressure is 10 kPa.
Electronic Supplementary Material (ESI) for RSC AdvancesThis journal is © The Royal Society of Chemistry 2013

S4
S2. Variation of the average occupation profile with the molecular dipole
0.95 D 1.15 D 1.35 D
1.55 D 1.75 D 1.95 D
2.1 D 2.3 D 2.45 D
Electronic Supplementary Material (ESI) for RSC AdvancesThis journal is © The Royal Society of Chemistry 2013

S5
2.65 D 2.85 D 3.05 D
3.25 D 3.45 D 3.65 D
3.8 D 4.3 D 4.8 D
Electronic Supplementary Material (ESI) for RSC AdvancesThis journal is © The Royal Society of Chemistry 2013
S6
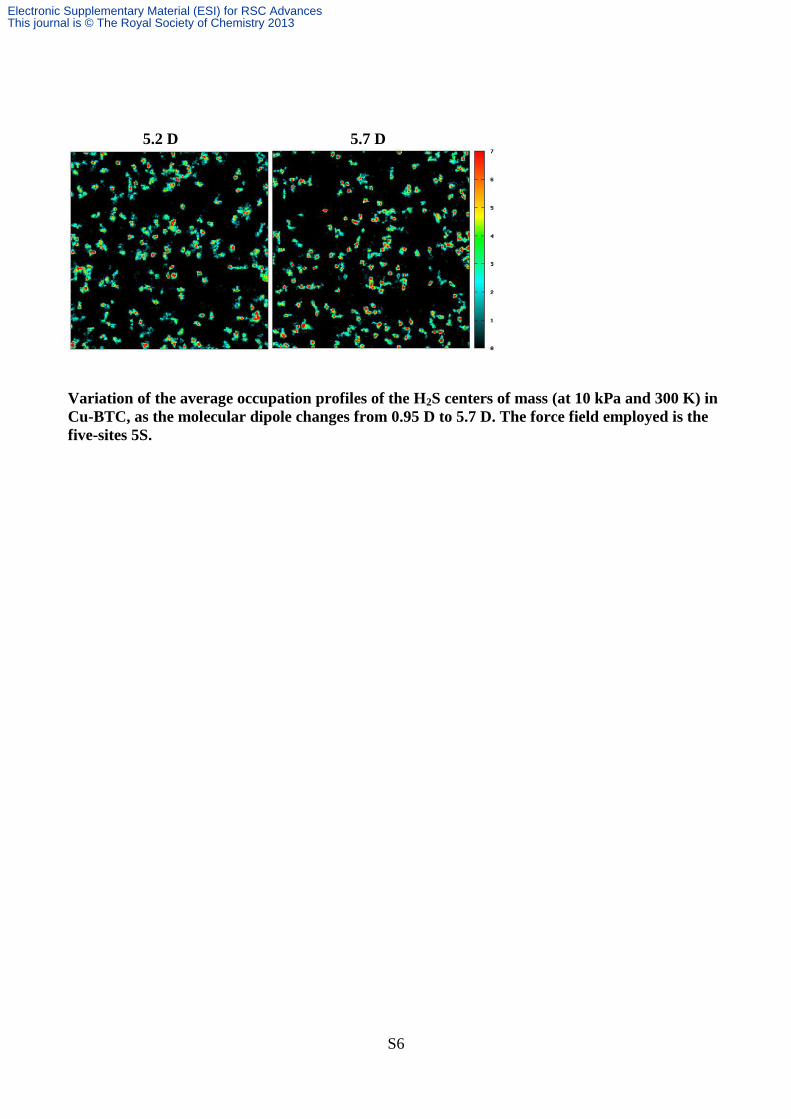
5.2 D 5.7 D
Variation of the average occupation profiles of the H2S centers of mass (at 10 kPa and 300 K) in
Cu-BTC, as the molecular dipole changes from 0.95 D to 5.7 D. The force field employed is the
five-sites 5S.
Electronic Supplementary Material (ESI) for RSC AdvancesThis journal is © The Royal Society of Chemistry 2013
S7
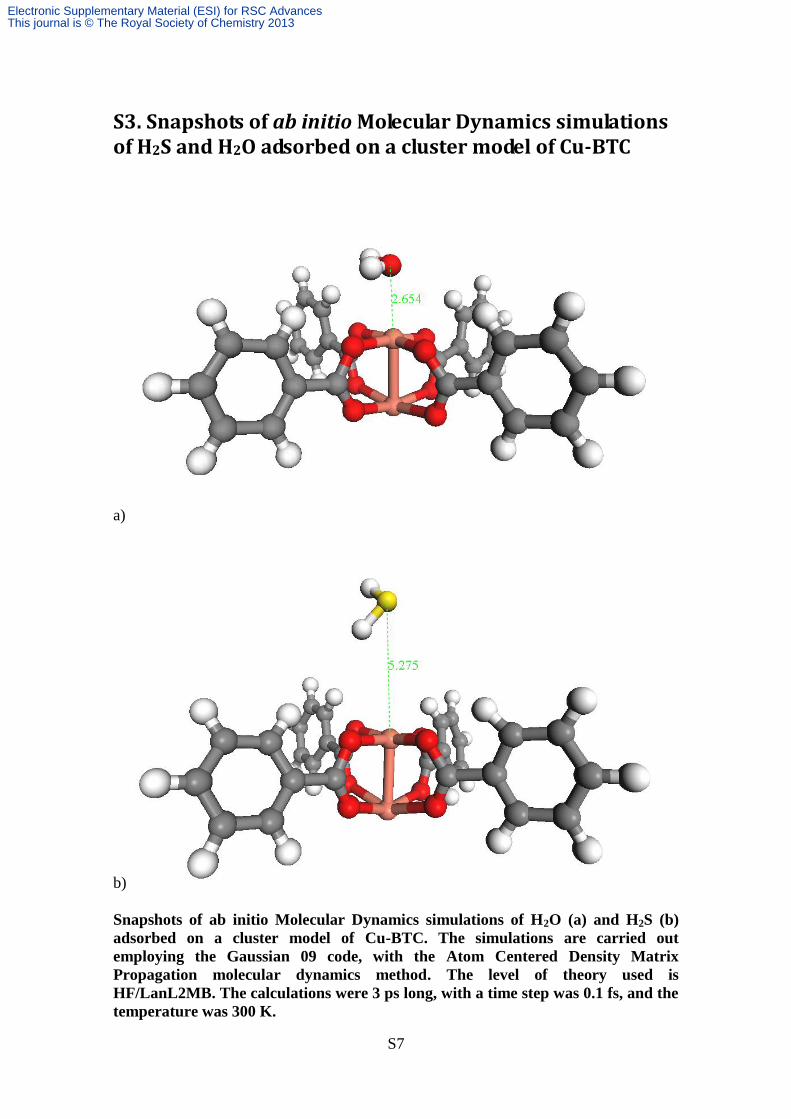
S3. Snapshots of ab initio Molecular Dynamics simulations of H2S and H2O adsorbed on a cluster model of Cu-BTC
a)
b)
Snapshots of ab initio Molecular Dynamics simulations of H2O (a) and H2S (b)
adsorbed on a cluster model of Cu-BTC. The simulations are carried out
employing the Gaussian 09 code, with the Atom Centered Density Matrix
Propagation molecular dynamics method. The level of theory used is
HF/LanL2MB. The calculations were 3 ps long, with a time step was 0.1 fs, and the
temperature was 300 K.
Electronic Supplementary Material (ESI) for RSC AdvancesThis journal is © The Royal Society of Chemistry 2013







