Download - 2015 pag-metagenome

Reconstructing metagenomes from shotgun
dataC. Titus Brown
UC Davis / School of Veterinary Medicine

Shotgun metagenomics
• Collect samples;
• Extract DNA;
• Feed into sequencer;
• Computationally analyze.
Wikipedia: Environmental shotgun sequencing.png

To assemble, or not to assemble?
Goals: reconstruct phylogenetic content and predict
functional potential of ensemble.
• Should we analyze short reads directly?
OR
• Do we assemble short reads into longer contigs first,
and then analyze the contigs?

Assembly: good.
Howe et al., 2014
Assemblies yield much more significant
homology matches.

But! Assembly is…• Morally frightening: don’t you mis-assemble
sequences?
• Computationally challenging: don’t you need big
computers?
• Technically tricky: don’t you need to be an expert?

Or… is it?• Most assembly papers analyze novel data sets and
then have to argue that their result is ok (guilty!)
• Very few assembly benchmarks have been done.
• Even fewer (trustworthy) computational
time/memory comparisons have been done.
• And even fewer “assembly recipes” have been
written down clearly.

A neat paper:
Shakya et al., 2013; pmid 23387867

A mock community!• ~60 genomes, all sequenced;
• Lab mixed with 10:1 ratio of most abundant to least
abundant;
• 2x101 reads, 107 mn reads total (Illumina);
• 10.5 Gbp of sequence in toto.
• The paper also compared16s primer sets & 454
shotgun metagenome data => reconstruction.
Shakya et al., 2013; pmid 23387867

Paper conclusions• “Metagenomic sequencing outperformed most SSU
rRNA gene primer sets used in this study.”
• “The Illumina short reads provided a very good estimates
of taxonomic distribution above the species level, with
only a two- to threefold overestimation of the actual
number of genera and orders.”
• “For the 454 data … the use of the default parameters
severely overestimated higher level diversity (~ 20- fold
for bacterial genera and identified > 100 spurious
eukaryotes).”
Shakya et al., 2013; pmid 23387867

How about assembly??• Shakya et al. did not do assembly; no standard for
analysis at the time, not experts.
• But we work on assembly!
• And we’ve been working on a tutorial/process for
doing it!
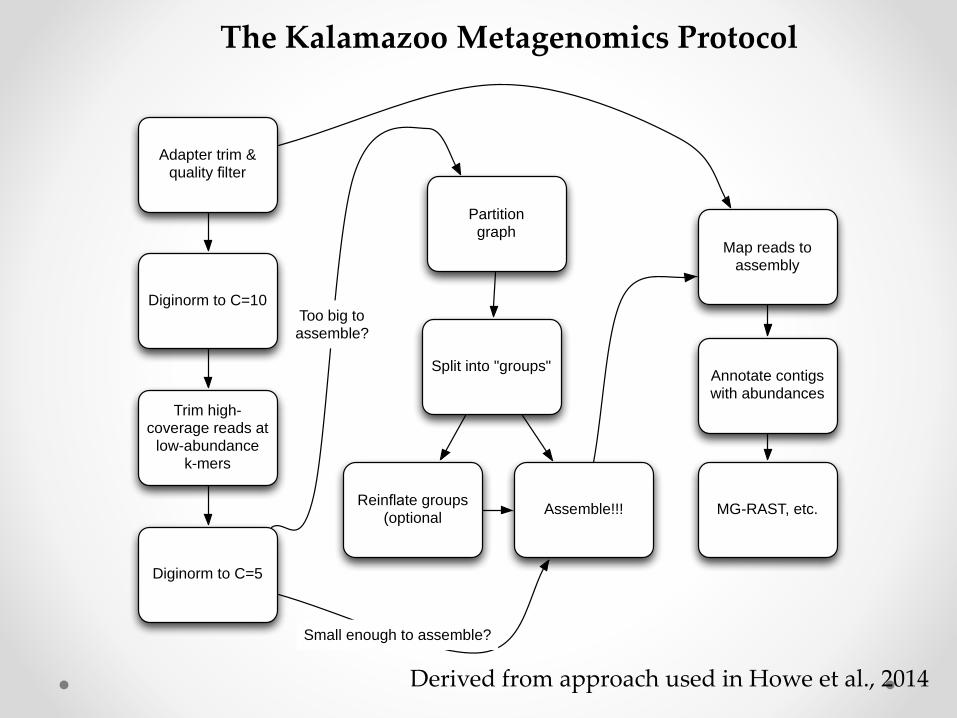
Adapter trim & quality filter
Diginorm to C=10
Trim high-coverage reads at
low-abundancek-mers
Diginorm to C=5
Partitiongraph
Split into "groups"
Reinflate groups (optional
Assemble!!!
Map reads to assembly
Too big toassemble?
Small enough to assemble?
Annotate contigs with abundances
MG-RAST, etc.
The Kalamazoo Metagenomics Protocol
Derived from approach used in Howe et al., 2014

Computational protocol for assembly
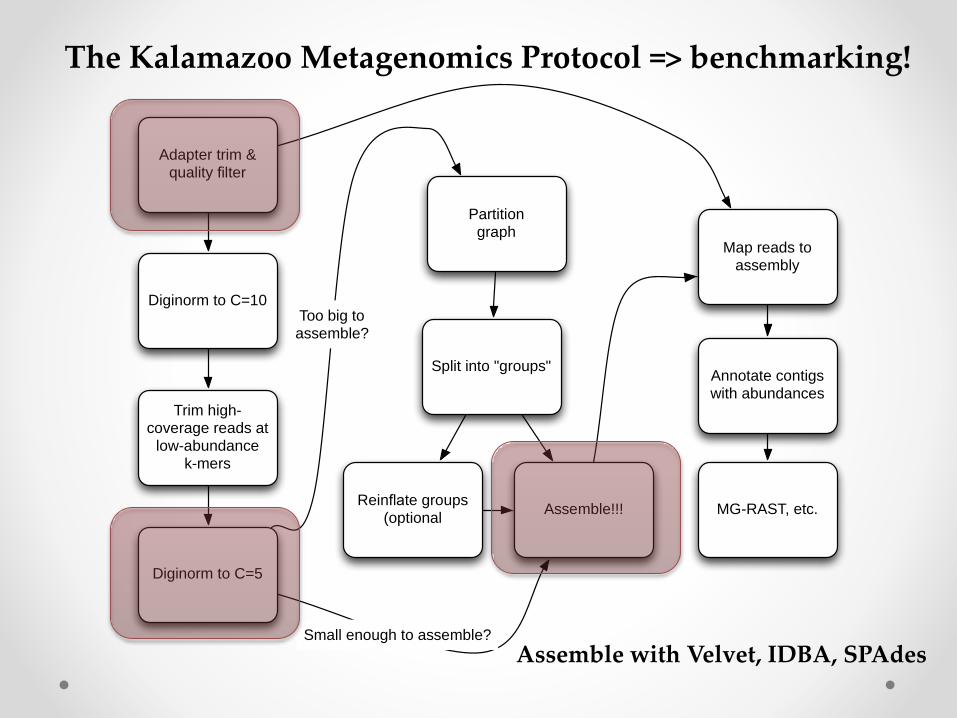
Adapter trim & quality filter
Diginorm to C=10
Trim high-coverage reads at
low-abundancek-mers
Diginorm to C=5
Partitiongraph
Split into "groups"
Reinflate groups (optional
Assemble!!!
Map reads to assembly
Too big toassemble?
Small enough to assemble?
Annotate contigs with abundances
MG-RAST, etc.
The Kalamazoo Metagenomics Protocol => benchmarking!
Assemble with Velvet, IDBA, SPAdes

Benchmarking process• Apply various filtering treatments to the data
(x3)o Basic quality trimming and filtering
o + digital normalization
o + partitioning
• Apply different assemblers to the data for each treatment (x3)o IDBA
o SPAdes
o Velvet
• Measure compute time/memory req’d.
• Compare assembly results to “known” answer with Quast.

Recovery, by assembler
Velvet IDBA Spades
Quality Quality Quality
Total length (>= 0 bp) 1.6E+08 2.0E+08 2.0E+08
Total length (>= 1000 bp) 1.6E+08 1.9E+08 1.9E+08
Largest contig 561,449 979,948 1,387,918
# misassembled contigs 631 1032 752
Genome fraction (%) 72.949 90.969 90.424
Duplication ratio 1.004 1.007 1.004
Conclusion: SPAdes and IDBA achieve similar results.
Dr. Sherine Awad

Treatments: some effect
IDBA
Quality Diginorm Partition
Total length (>= 0 bp) 2.0E+08 2.0E+08 2.0E+08
Total length (>= 1000 bp) 1.9E+08 2.0E+08 1.9E+08
Largest contig 979,948 1,469,321 551,171
# misassembled contigs 1032 916 828
Unaligned length 10,709,716 10,637,811 10,644,357
Genome fraction (%) 90.969 91.003 90.082
Duplication ratio 1.007 1.008 1.007
Conclusion: Treatments do not alter results much.
Dr. Sherine Awad
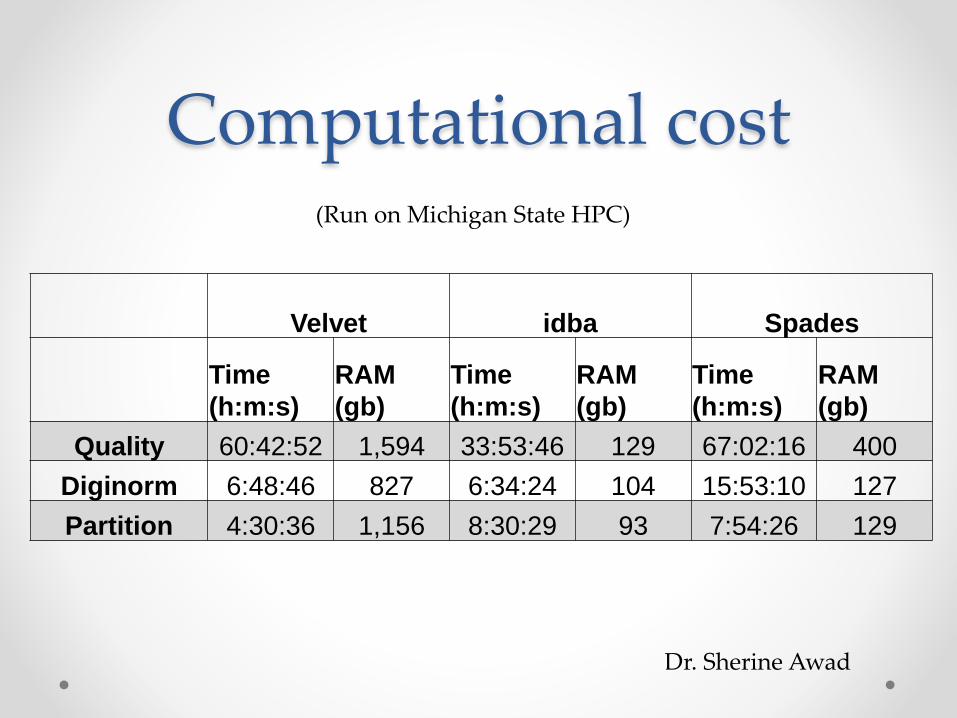
Computational cost
Velvet idba Spades
Time
(h:m:s)
RAM
(gb)
Time
(h:m:s)
RAM
(gb)
Time
(h:m:s)
RAM
(gb)
Quality 60:42:52 1,594 33:53:46 129 67:02:16 400
Diginorm 6:48:46 827 6:34:24 104 15:53:10 127
Partition 4:30:36 1,156 8:30:29 93 7:54:26 129
(Run on Michigan State HPC)
Dr. Sherine Awad

Need to understand:• What is not being assembled and why?
o Low coverage?
o Strain variation?
o Something else?
• Effects of strain variation
• Additional contigs being assembled –contamination? Spurious assembly?
• Performance of MEGAHIT assembler (a new assembler that is very fast but still young).

Other observations• 90% recovery is not bad; relatively few
misassemblies, too.
• This was not a highly polymorphic community BUT it
did have several closely related strains; more
generally, we see that strains do generate
chimeras, but not different species gen’ly.
• Challenging to execute even with a
tutorial/protocol :(

But! Assembly is…• Morally frightening: don’t you mis-assemble
sequences? NO. (Or at least, not systematically.)
• Computationally challenging: don’t you need big
computers? YES. (But that’s changing.)
• Technically tricky: don’t you need to be an expert?
UNFORTUNATELY STILL YES BUT THERE’S HOPE.

Benchmarking & protocols
• Our work is completely reproducible and open.
• You can re-run our benchmarks yourself if you want!
• We will be adding new assemblers in as time
permits.
• Protocol is open, versioned, citable… but also still a
work in progress :)

Using shotgun sequence to cross-
validate amplicon predictions
0.00%
5.00%
10.00%
15.00%
20.00%
25.00%
30.00%
35.00%
40.00%
AMP/RDP AMP/SILVA WGS/RDP WGS/SILVA WGS/SILVA(LSU)
Amplicon seq missing Verrucomicrobia
Jaron Guo

Primer bias against Verrucomicrobia
Check taxonomy of reads causingmismatch (A)
Verrucomicrobia cause 70% (117/168) of
mismatch
Current primers are not effective at amplifying Verrucomicrobia
Jaron Guo

Thanks!Please contact me at [email protected]!
Everything I talked about is freely available.
Search for ‘khmer protocols’.







