Download - 1-s2.0-S0043135411002910-main.pdf

wat e r r e s e a r c h 4 5 ( 2 0 1 1 ) 4 3 9 0e4 3 9 8
Avai lab le at www.sc iencedi rect .com
journa l homepage : www.e lsev ier . com/ loca te /wat res
Analysis of the bacterial community in a laboratory-scalenitrification reactor and a wastewater treatment plant by454-pyrosequencing
Lin Ye a, Ming-Fei Shao a, Tong Zhang a,*, Amy Hin Yan Tong b, Si Lok b
aEnvironmental Biotechnology Laboratory, The University of Hong Kong, Hong Kong SAR, ChinabGenome Research Center, The Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, China
a r t i c l e i n f o
Article history:
Received 25 January 2011
Received in revised form
20 March 2011
Accepted 22 May 2011
Available online 31 May 2011
Keywords:
Activated sludge
Bacterial community
Cloning
454 High-throughput
pyrosequencing
* Corresponding author. Fax: þ86 852 2559 53E-mail address: [email protected] (T
0043-1354/$ e see front matter ª 2011 Elsevdoi:10.1016/j.watres.2011.05.028
a b s t r a c t
For full understanding of the microbial community in the wastewater treatment bioreac-
tors, one of the feasible and effective ways is to investigate the massive genetic informa-
tion contained in the activated sludge. In this study, high-throughput pyrosequencing was
applied to analyze the 16S rRNA gene of bacteria in a laboratory-scale nitrification reactor
and a full-scale wastewater treatment plant. In total, 27,458 and 26,906 effective sequence
reads of the 16S rRNA gene were obtained from the Reactor and the wastewater treatment
plant activated sludge samples respectively. The taxonomic complexities in the two
samples were compared at phylum and genus levels. According to the pyrosequencing
results, even for a laboratory-scale reactor as simple as that in this study, a small size clone
library is far from enough to reflect the whole profile of the bacterial community. In
addition, it was found that the commonly used informatics tool “RDP classifier” may
drastically assign Nitrosomonas sequences into a wrong taxonomic unit resulting in
underestimation of ammonia-oxidizing bacteria in the bioreactors. In this paper the
reasons for this mistakenly assignment were analyzed and correction methods were
proposed.
ª 2011 Elsevier Ltd. All rights reserved.
1. Introduction library (Matsumoto et al., 2009) as well as direct visualization
Nitrification is an important step for removing ammonium
nitrogen from wastewater. The key role that bacterial
communities play in wastewater treatment has been inten-
sively studied in the past decades in laboratory-scale and full-
scale bioreactors by the use of various molecular methods.
PCR-DGGE (polymerase chain reaction - denaturing gradient
gel electrophoresis) and T-RFLP (terminal restriction fragment
length polymorphism) were used to investigate the diversity
of the bacterial communities of activated sludge from
different wastewater treatment plants (WWTPs) (Boon et al.,
2002; Regan et al., 2002). More recently, analysis of clone
37.. Zhang).ier Ltd. All rights reserve
of bacterial species by FISH (fluorescent in situ hybridization)
(Hao et al., 2009) were employed to determine the community
compositions. Although bacterial species involved in nitrifi-
cation have also been characterized by the aforementioned
approaches, the extraordinary diversity of microorganisms in
the activated sludge exceeds the sensitivity and dynamic
range of those molecular methods and precludes a complete
characterization of the interplay of various components of the
microbial community in nitrification.
Pyrosequencing developed by Roche 454 Life Science
(Branford, CT, USA) is a high-throughput analytical method
that can generate huge amounts of DNA reads through
d.
wat e r r e s e a r c h 4 5 ( 2 0 1 1 ) 4 3 9 0e4 3 9 8 4391
a massively parallel sequencing-by-synthesis approach
(Margulies et al., 2005). This technology have been usedwidely
to analyze themicrobial community in various environmental
samples, such as marine water (Qian et al., 2011), soil (Roesch
et al., 2007), human distal intestine (Claesson et al., 2009),
wastewater treatment plant influent (McLellan et al., 2010)
et al.. However, few studies have been conducted on activated
sludge by this method. Kwon and colleagues (Kwon et al.,
2010) investigated the microbial diversity in an integrated
fixed-film activated sludge system. Their results showed the
bacterial abundances were quite high, totally 3034 and 1451
operational taxonomic units (OTUs) were identified at the 3%
cutoff for the suspended and attached samples, respectively.
In the present study, we characterized and compared the
bacterial communities in a laboratory-scale nitrification
reactor with that from activated sludge of a wastewater
treatment plant by 454-pyrosequencing. The diversity and
abundance of the nitrifiers in these two samples were also
investigated. Additionally, we found that RDP Classifier,
a commonly used informatics tool for pyrosequencing data
analysis, may have phylogenetically assigned Nitrosomonas
sequences into a wrong order. The key nucleotides leading to
the mistaken assignment have been identified and a method
to overcome this problem was proposed based on the BLAST
results.
2. Materials and methods
2.1. Reactor operation and WWTP description
In the present study, a fermentor (Sartorius Biostat� A plus)
(Goettingen, Germany) with a working volume of 2.6 L was
configured for continuous operation to conduct nitrification
studies. A pH value of 7.5 was held by automated addition of
sodium bicarbonate. Dissolved Oxygen (DO) was maintained
at 0.5 mg/L by stirring and aeration. The influent was made
with deionizedwater (67%) and seawater (33%) to simulate the
typical salinity of sewage found in Hong Kong. NH4Cl was
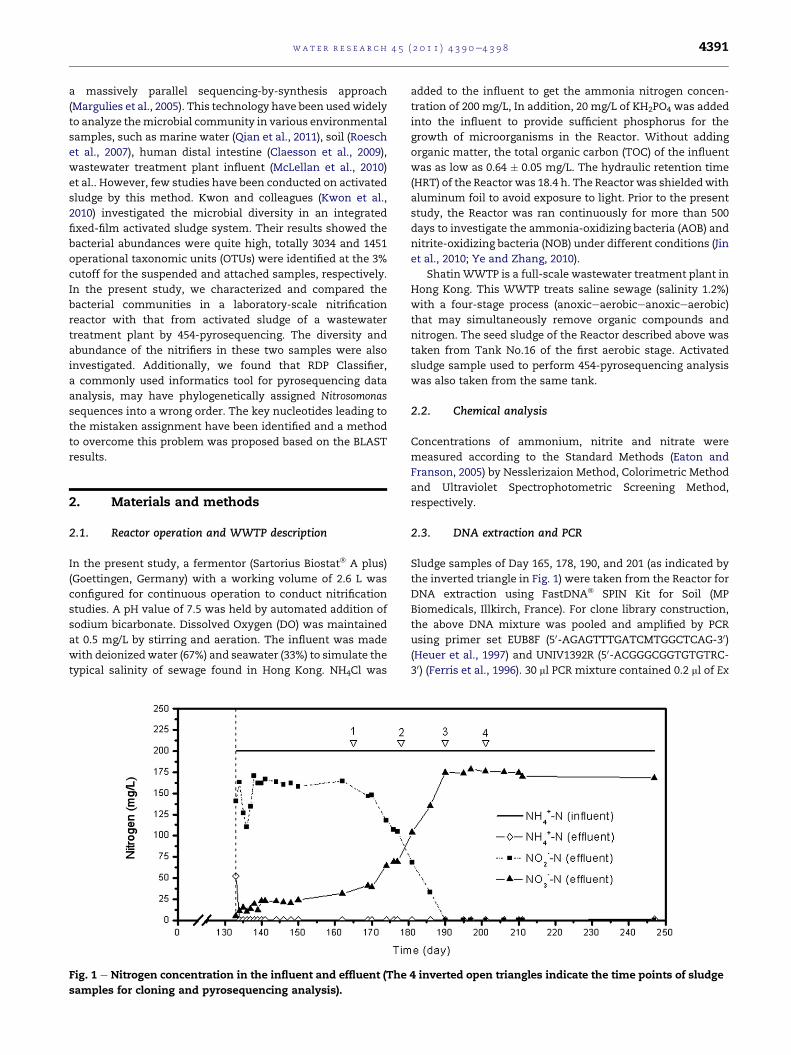
Fig. 1 e Nitrogen concentration in the influent and effluent (The
samples for cloning and pyrosequencing analysis).
added to the influent to get the ammonia nitrogen concen-
tration of 200 mg/L, In addition, 20 mg/L of KH2PO4 was added
into the influent to provide sufficient phosphorus for the
growth of microorganisms in the Reactor. Without adding
organic matter, the total organic carbon (TOC) of the influent
was as low as 0.64 � 0.05 mg/L. The hydraulic retention time
(HRT) of the Reactor was 18.4 h. The Reactor was shieldedwith
aluminum foil to avoid exposure to light. Prior to the present
study, the Reactor was ran continuously for more than 500
days to investigate the ammonia-oxidizing bacteria (AOB) and
nitrite-oxidizing bacteria (NOB) under different conditions (Jin
et al., 2010; Ye and Zhang, 2010).
ShatinWWTP is a full-scale wastewater treatment plant in
Hong Kong. This WWTP treats saline sewage (salinity 1.2%)
with a four-stage process (anoxiceaerobiceanoxiceaerobic)
that may simultaneously remove organic compounds and
nitrogen. The seed sludge of the Reactor described above was
taken from Tank No.16 of the first aerobic stage. Activated
sludge sample used to perform 454-pyrosequencing analysis
was also taken from the same tank.
2.2. Chemical analysis
Concentrations of ammonium, nitrite and nitrate were
measured according to the Standard Methods (Eaton and
Franson, 2005) by Nesslerizaion Method, Colorimetric Method
and Ultraviolet Spectrophotometric Screening Method,
respectively.
2.3. DNA extraction and PCR
Sludge samples of Day 165, 178, 190, and 201 (as indicated by
the inverted triangle in Fig. 1) were taken from the Reactor for
DNA extraction using FastDNA� SPIN Kit for Soil (MP
Biomedicals, Illkirch, France). For clone library construction,
the above DNA mixture was pooled and amplified by PCR
using primer set EUB8F (50-AGAGTTTGATCMTGGCTCAG-30)(Heuer et al., 1997) and UNIV1392R (50-ACGGGCGGTGTGTRC-30) (Ferris et al., 1996). 30 ml PCR mixture contained 0.2 ml of Ex
4 inverted open triangles indicate the time points of sludge

wat e r r e s e a r c h 4 5 ( 2 0 1 1 ) 4 3 9 0e4 3 9 84392
Taq TM(TaKaRa, Dalian, China), 3 ml of 10� Ex Taq Buffer, 3 ml of
dNTPmixture, 0.2 mMof each primer, and 20e50 ng of genomic
DNA. The thermocycling stepswere as follows: 95 �C for 7min,
followed by 35 cycles at of 95 �C for 1min, 55 �C for 1min, 72 �Cfor 1 min and a final extension step at 72 �C for 10 min.
For pyrosequencing, the above DNA mixture of the four
samples was amplified with a set of primers targeting the
hypervariable V4 region of the 16S rRNA gene (RDP’s Pyrose-
quencing Pipeline: http://pyro.cme.msu.edu/pyro/help.jsp).
The forward primer is 50-AYTGGGYDTAAAGNG-30 and the
reverse primers are the mixture of four primers, i.e. 50-TACCRGGGTHTCTAATCC-30, 50-TACCAGAGTATCTAATTC-30,50-CTACDSRGGTMTCTAATC-30, and 50-TACNVGGGTATC-
TAATCC-30 (Claesson et al., 2009). Barcodes that allow sample
multiplexing during pyrosequencing were incorporated
between the 454 adapter and the forward primers.
2.4. Cloning
PCR products were purified using PCRquick-spinTM PCR
Product Purification Kit (iNtRON Biotechnology, Sangdaewon-
Dong, Korea). The purified PCR productswere cloned using the
InsTAclone� PCR Cloning Kit (Fermentas, Burlingtong,
Ontario, Canada) following the instructions of the vendor.
White colonies were selected for whole-cell PCR amplification
with the M13F (50-TGTAAAACGACGGCCAGT-30) and M13R (50-CAGGAAACAGCTATGAC-30) primer set. The PCR products
were purified and sequenced on the ABI 3730xl capillary
sequencer (Applied Biosystems, Foster City, CA, USA) using
M13F or M13R primers. The clone library sequences in this
study have been deposited in GenBank under accession
numbers HM117160 to HM117171.
2.5. High-throughput 454 pyrosequencing
The composition of the PCR products of V4 region of 16S rRNA
gene was determined by pyrosequencing using the Roche 454
FLX Titanium sequencer (Roche 454 Life Sciences, Branford,
CT, USA). Samples in this study were individually barcoded to
enable multiplex sequencing. The results are deposited into
the NCBI short reads archive database (Accession Number:
SRA026842.2).
2.6. Sequence analysis and phylogenetic classification
Following pyrosequencing, Python scripts were written to: 1)
remove sequences containing more than one ambiguous base
(‘N’); 2) check the completeness of the barcodes and the
adapter; 3) remove sequences shorter than 150 bps. The “RDP
Align” tool in RDP’s Pyrosequencing Pipeline was used to align
the effective sequences. A cluster filewas generatedwith “RDP
Complete Linkage Clustering” tool. From the cluster file, the
rarefaction curve was generated using the “RDP Rarefaction”
tool. Taxonomic classification of the sequences was per-
formed using the RDP Classifier (Version 2.2) with a set
confidence threshold of 50%.
Python and Biopython (Cock et al., 2009) were used to
create scripts to: 1) extract all sequences that were assigned
into the order of Burkholderiales according to the result of RDP
Classifier (the downloaded assignment detail text file); 2) use
qblast function in Biopython to run BLAST and search against
“nr” database through the internet automatically for all the
above-mentioned extracted sequences; 3) parse the BLAST
results to check the top 10 hits whether there is a hit con-
taining Nitrosomonas or Comamonas in the title; and 4) record
the maximum identities between the query sequences and
the subject sequences identified in last step.
On the other hand, all sequences obtained from pyrose-
quencing in this study were compared with Greengenes 16S
rRNA gene database (DeSantis et al., 2006) using NCBI’s
BLASTN tool (Altschul et al., 1990) and the default parameters
except for the maximum hit number of 100 (Claesson et al.,
2009). Then the sequences were assigned to NCBI taxon-
omies with MEGAN (Huson et al., 2007) by using the Lowest
Common Ancestor (LCA) algorithm and the default parame-
ters, i.e. absolute cutoff: BLAST bitscore 35, and relative cutoff:
10% of the top hits.
3. Results and discussion
3.1. Reactor performance
Prior to this study, the Reactor was operated under a very low
oxygen concentration condition (DO 0.15 mg/L). Accordingly,
the bulk of the ammoniumwas partially oxidized to nitrite. In
present study, the DO level was increased to 0.5 mg/L, the
nitrite was gradually reduced and several molecular methods
(DGGE, T-RFLP and Cloning) have been used to confirm that
nitrite-oxidizing bacteria (Nitrospira) proliferated intensively
in this period (Ye and Zhang, 2010). The operational condition
and the performance of the Reactor were described in detail in
our previous paper (Ye and Zhang, 2010).
3.2. Cloning results
Twelve OTUswere obtainedwith 3% nucleotide cutoff from 61
clones that were sequenced in 16S rRNA gene clone library.
According to RDP Classifier, the 61 sequences examined by
Sanger-dideoxy based sequencing can be assigned to 3 phyla.
Based on both RDP Classifier and BLAST analysis (Table 1),
OTU-2 and OTU-4 are Nitrosomonas and Nitrospira species,
which accounted for 21.3% and 3.2% in total bacterial
community, respectively, suggesting that these species
represent the dominant AOB and NOB in the Reactor. It should
be noted that according to the BLAST results, the most
dominant OTU, OTU-1, is probably a heterotrophic species
that is similar (max identity 99%) to an uncultured bacteria
reported in the marine sediment.
3.3. Taxonomic complexity of the bacterial community
Pyrosequencing of the Reactor sample and the WWTP sludge
sample yielded 27,458 and 26,906 effective sequence tags
respectively. The amount of sequences was comparable to
those of other studies (Kwon et al., 2010; Lee et al., 2010;
McLellan et al., 2010). RDP Classifier was firstly used to
assign these sequence tags into different phylogenetic bacte-
rial taxa. Fig. 2 and Supplementary Table S1 show the relative
bacterial community abundances on the phylum level. Except
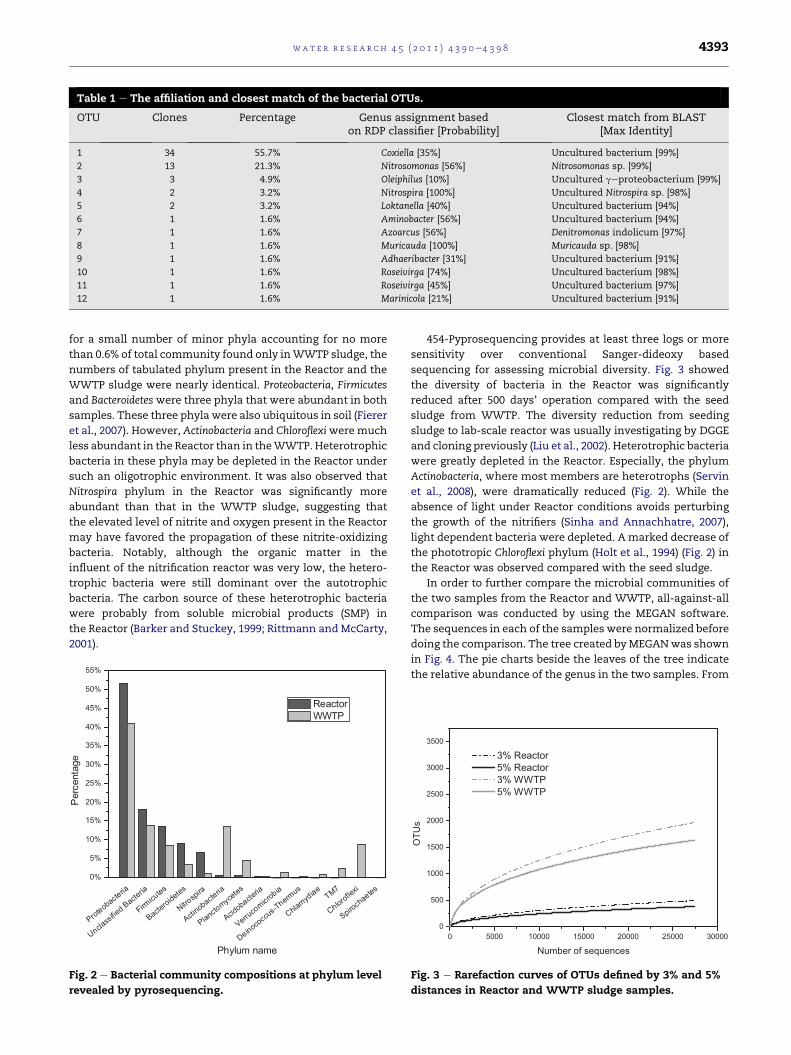
Table 1 e The affiliation and closest match of the bacterial OTUs.
OTU Clones Percentage Genus assignment basedon RDP classifier [Probability]
Closest match from BLAST[Max Identity]
1 34 55.7% Coxiella [35%] Uncultured bacterium [99%]
2 13 21.3% Nitrosomonas [56%] Nitrosomonas sp. [99%]
3 3 4.9% Oleiphilus [10%] Uncultured geproteobacterium [99%]
4 2 3.2% Nitrospira [100%] Uncultured Nitrospira sp. [98%]
5 2 3.2% Loktanella [40%] Uncultured bacterium [94%]
6 1 1.6% Aminobacter [56%] Uncultured bacterium [94%]
7 1 1.6% Azoarcus [56%] Denitromonas indolicum [97%]
8 1 1.6% Muricauda [100%] Muricauda sp. [98%]
9 1 1.6% Adhaeribacter [31%] Uncultured bacterium [91%]
10 1 1.6% Roseivirga [74%] Uncultured bacterium [98%]
11 1 1.6% Roseivirga [45%] Uncultured bacterium [97%]
12 1 1.6% Marinicola [21%] Uncultured bacterium [91%]
wat e r r e s e a r c h 4 5 ( 2 0 1 1 ) 4 3 9 0e4 3 9 8 4393
for a small number of minor phyla accounting for no more
than 0.6% of total community found only inWWTP sludge, the
numbers of tabulated phylum present in the Reactor and the
WWTP sludge were nearly identical. Proteobacteria, Firmicutes
and Bacteroidetes were three phyla that were abundant in both
samples. These three phyla were also ubiquitous in soil (Fierer
et al., 2007). However, Actinobacteria and Chloroflexiweremuch
less abundant in the Reactor than in theWWTP. Heterotrophic
bacteria in these phyla may be depleted in the Reactor under
such an oligotrophic environment. It was also observed that
Nitrospira phylum in the Reactor was significantly more
abundant than that in the WWTP sludge, suggesting that
the elevated level of nitrite and oxygen present in the Reactor
may have favored the propagation of these nitrite-oxidizing
bacteria. Notably, although the organic matter in the
influent of the nitrification reactor was very low, the hetero-
trophic bacteria were still dominant over the autotrophic
bacteria. The carbon source of these heterotrophic bacteria
were probably from soluble microbial products (SMP) in
the Reactor (Barker and Stuckey, 1999; Rittmann andMcCarty,
2001).
Proteobacteria
Unclass
ified Bacte
ria
Firmicu
tes
Bacteroidetes
Nitrosp
ira
Actinobacteria
Planctomycetes
Acidobacte
ria
Verruco
microbia
Deinococcu
s-Thermus
Chlamydiae
TM7
Chloroflexi
Spirochaetes
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
50%
55%
Perc
enta
ge
Phylum name
Reactor WWTP
Fig. 2 e Bacterial community compositions at phylum level
revealed by pyrosequencing.
454-Pyprosequencing provides at least three logs or more
sensitivity over conventional Sanger-dideoxy based
sequencing for assessing microbial diversity. Fig. 3 showed
the diversity of bacteria in the Reactor was significantly
reduced after 500 days’ operation compared with the seed
sludge from WWTP. The diversity reduction from seeding
sludge to lab-scale reactor was usually investigating by DGGE
and cloning previously (Liu et al., 2002). Heterotrophic bacteria
were greatly depleted in the Reactor. Especially, the phylum
Actinobacteria, where most members are heterotrophs (Servin
et al., 2008), were dramatically reduced (Fig. 2). While the
absence of light under Reactor conditions avoids perturbing
the growth of the nitrifiers (Sinha and Annachhatre, 2007),
light dependent bacteria were depleted. A marked decrease of
the phototropic Chloroflexi phylum (Holt et al., 1994) (Fig. 2) in
the Reactor was observed compared with the seed sludge.
In order to further compare the microbial communities of
the two samples from the Reactor and WWTP, all-against-all
comparison was conducted by using the MEGAN software.
The sequences in each of the samples were normalized before
doing the comparison. The tree created byMEGANwas shown
in Fig. 4. The pie charts beside the leaves of the tree indicate
the relative abundance of the genus in the two samples. From
0 5000 10000 15000 20000 25000 300000
500
1000
1500
2000
2500
3000
3500
3% Reactor 5% Reactor 3% WWTP 5% WWTP
OTU
s
Number of sequences
Fig. 3 e Rarefaction curves of OTUs defined by 3% and 5%
distances in Reactor and WWTP sludge samples.
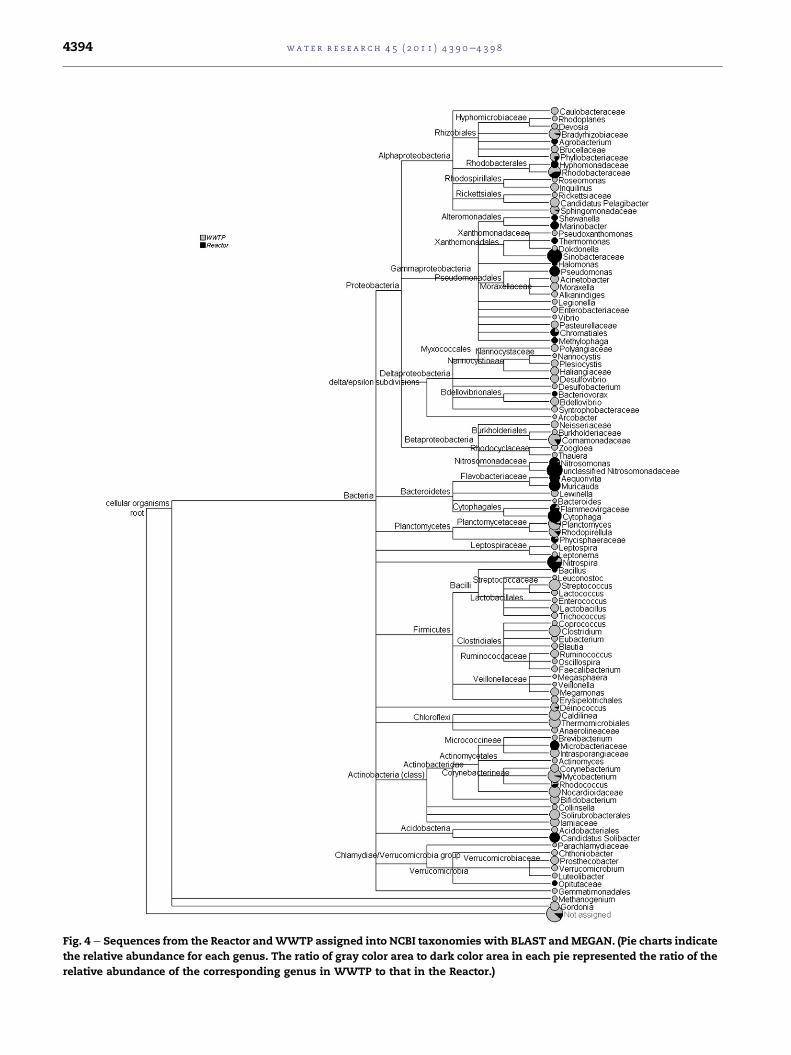
Fig. 4 e Sequences from the Reactor andWWTP assigned into NCBI taxonomies with BLAST andMEGAN. (Pie charts indicate
the relative abundance for each genus. The ratio of gray color area to dark color area in each pie represented the ratio of the
relative abundance of the corresponding genus in WWTP to that in the Reactor.)
wat e r r e s e a r c h 4 5 ( 2 0 1 1 ) 4 3 9 0e4 3 9 84394

wat e r r e s e a r c h 4 5 ( 2 0 1 1 ) 4 3 9 0e4 3 9 8 4395
Fig. 4, it could be seen that at the genus level the microbial
communities of the Reactor and the WWTP were quite
different. There were some genera (such as Marinobacter,
Pseudomonas, Aequorivita, Muricauda, etc.) appearing only in
the Reactor. According to the previous reports (Bowman
and Nichols, 2002; Gauthier et al., 1992; Yoon et al., 2005),
these genera are usually halotolerant bacteria and exist in
the marine environment. These bacteria may come from the
seawater of the influent and could adapt themselves to the
conditions of the Reactor. Many other genera, which were
marked by gray color in Fig. 4, exist only in the WWTP. That
indicates the diversity of the bacteria in the WWTP was much
more complex than that in the Reactor. Also, there are some
genera, including Phyllobacteriaceae, Rhodobacteraceae, Chroma-
tiales, Comamonadaceae, Nitrospira, Rhodococcus, etc., exist both
in the Reactor and the WWTP.
According to the cluster files produced by the “RDP
Complete Linkage Clustering” tool, there were 494 and 381
OTUs in Reactor sludge using two cutoffs levels of 3% and 5%,
respectively. By contrast, WWTP sludge had 1986 and 1648
OTUs using the same cutoffs. According to the rarefaction
curve (Fig. 3), the species complexity in the Reactor was six-
fold less than that in WWTP. Judging from the numbers of
OTUs obtained by pyrosequencing in this study, not to
mention the WWTP, even for a laboratory reactor as simple
as that in our study, a small scale clone library was not
sufficient to reflect the whole profile of the bacterial
community, especially for those minor populations.
Furthermore, it can be deduced that the commonly used
molecular methods (such as DGGE and T-RFLP) in environ-
mental biotechnology may also have insufficient resolutions
to characterize the microbial communities in the wastewater
treatment bioreactors. High-throughput sequencing methods
have the potential to be effective means for better under-
standing of the microorganisms in various environmental
engineering facilities.
3.4. Diversity and abundances of AOB and NOB
It was found that in the results of RDP Classifier (Table 2),
the sequences that were assigned into Nitrosomonadales
order were quite few. For the Reactor sludge sample, only
0.65% of the sequences were classified into this order, which
was inconsistent with the limited results from Sanger-
dideoxy sequencing of the clone library. Such low abun-
dance of AOB also conflicted with the performance (high
ammonium removing rate) of the Reactor. A further check
showed that there were large numbers of Nitrosomonadales
Table 2 e Relative abundance of dominant AOB and NOB in th
Sample AOB/NOB RDP Classifier Clone Library
Reads % Reads %
Reactor Nitrosomonadales 179 0.65% 13 21.3%
Nitrospirales 1782 6.49% 2 3.2%
WWTP Nitrosomonadales 14 0.05% e e
Nitrospirales 273 1.01% e e
sequences that were wrongly assigned by the RDP Classifier
into the order of Burkholderiales, a neighbor of Nitro-
somonadales in Proteobacteria phylum. Most of these mis-
assigned sequences were closely related (identity>97%) to
Nitrosomonas species as shown by BLAST analysis. Following
reclassification, 15.54% of the sequences from the reactor
sludge sample were Nitrosomonas related, indicating that
Nitrosomonas-like AOB were remarkably enriched in the
reactor (Table 2).
In both the Reactor and the WWTP sludge, the dominant
AOB species was Nitrosomonas and the dominant NOB species
was Nitrospira, which were affiliated to Nitrosomonadales and
Nitrospirales order, respectively. This result was consistent
with the previous reports of activated sludge from other
researchers (Layton et al., 2005; Logemann et al., 1998).
Except Nitrosomonas and Nitrospira, the other AOB and NOB
genus were very rare in the reactor. Nitrosospira, another
genus belonging to b-Proteobacteria AOB, accounted for only
0.6% and 0.056% in the reactor and the WWTP sludge
samples, respectively. Only one sequence and seven
sequences of Nitrosococcus were found in 27,458 and 26,906
sequences in the Reactor and WWTP sludge samples,
respectively. For NOB, only 13 Nitrobacter sequences were
found in the WWTP sludge and none was found in the
reactor. The present results suggested that except Nitro-
somonas and Nitrospira, all other species of bacterial nitrifiers
play only a very small role in nitrification process in the
wastewater treatment reactors.
3.5. Mistakenly classified Nitrosomonas sequences
In this study, as aforementioned, RDP Classifier may wrongly
assign sequences inNitrosomonadales order into another order,
consequently, leading to an underestimation of the abun-
dance of AOB in the samples. Except for the samples in this
study, we also investigated the pyrosequencing results of
other 14 samples from WWTPs in another study, a lot of
sequences were mistakenly assigned to Burkholderiales.
Thirty sequences, including 10 from mistakenly assigned
Nitrosomonas sequences, 10 from correctly assigned Nitro-
somonas sequences, and 10 from Comamonas (a genus in Bur-
kholderiales) sequences, were selected from the sequences
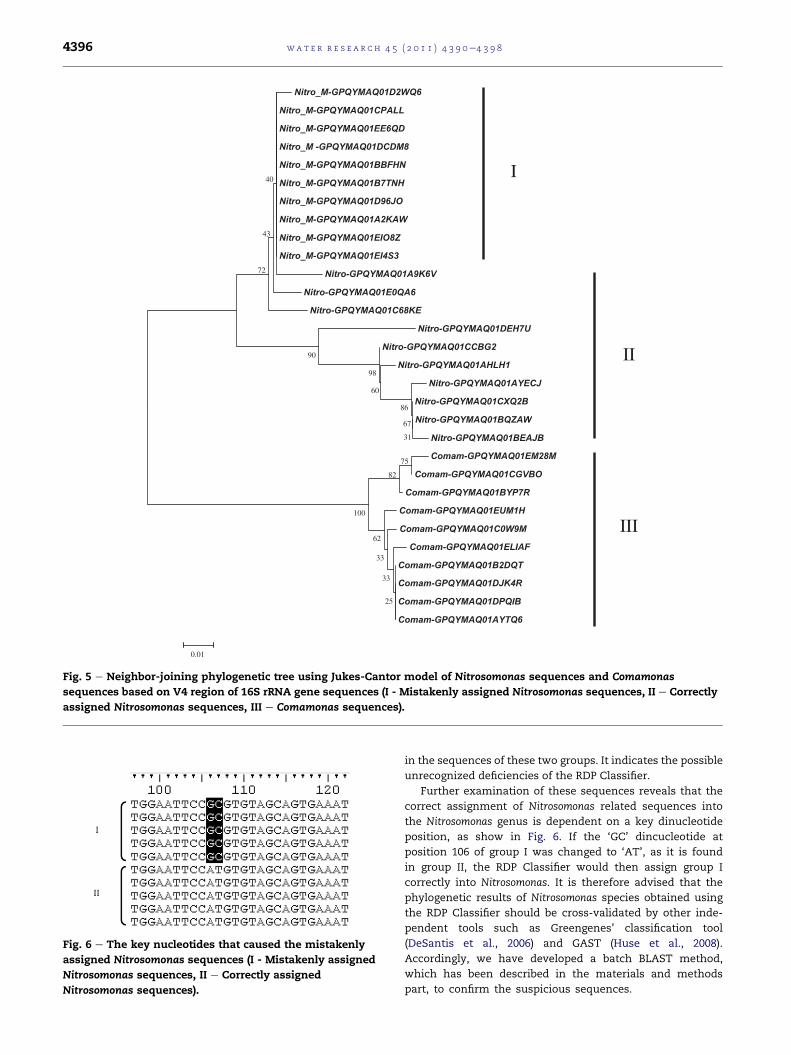
obtained by the pyrosequencing, and used to draw
a Neighbor-joining phylogenetic tree using Jukes-Cantor
model (Fig. 5). It was found that the Nitrosomonas sequences
and Comamonas sequences can be clearly classified into
different groups, indicating that there are marked differences
e Reactor and WWTP sludge.
BLAST-corrected RDP Classifier MEGAN Assignment
Reads % Reads %
4059 14.78% 4268 15.54%
1782 6.49% 1817 6.61%
14 0.05% 13 0.05%
273 1.01% 273 1.01%
Nitro_M-GPQYMAQ01D2WQ6
Nitro_M-GPQYMAQ01CPALL
Nitro_M-GPQYMAQ01EE6QD
Nitro_M -GPQYMAQ01DCDM8
Nitro_M-GPQYMAQ01BBFHN
Nitro_M-GPQYMAQ01B7TNH
Nitro_M-GPQYMAQ01D96JO
Nitro_M-GPQYMAQ01A2KAW
Nitro_M-GPQYMAQ01EIO8Z
Nitro_M-GPQYMAQ01EI4S3
Nitro-GPQYMAQ01A9K6V
Nitro-GPQYMAQ01E0QA6
Nitro-GPQYMAQ01C68KE
Nitro-GPQYMAQ01DEH7U
Nitro-GPQYMAQ01CCBG2
Nitro-GPQYMAQ01AHLH1
Nitro-GPQYMAQ01AYECJ
Nitro-GPQYMAQ01CXQ2B
Nitro-GPQYMAQ01BQZAW
Nitro-GPQYMAQ01BEAJB
Comam-GPQYMAQ01EM28M
Comam-GPQYMAQ01CGVBO
Comam-GPQYMAQ01BYP7R
Comam-GPQYMAQ01EUM1H
Comam-GPQYMAQ01C0W9M
Comam-GPQYMAQ01ELIAF
Comam-GPQYMAQ01B2DQT
Comam-GPQYMAQ01DJK4R
Comam-GPQYMAQ01DPQIB
Comam-GPQYMAQ01AYTQ6
75
82
25
33
33
62
100
31
67
86
60
98
90
72
43
40
0.01
I
II
III
Fig. 5 e Neighbor-joining phylogenetic tree using Jukes-Cantor model of Nitrosomonas sequences and Comamonas
sequences based on V4 region of 16S rRNA gene sequences (I - Mistakenly assigned Nitrosomonas sequences, II e Correctly
assigned Nitrosomonas sequences, III e Comamonas sequences).
Fig. 6 e The key nucleotides that caused the mistakenly
assigned Nitrosomonas sequences (I - Mistakenly assigned
Nitrosomonas sequences, II e Correctly assigned
Nitrosomonas sequences).
wat e r r e s e a r c h 4 5 ( 2 0 1 1 ) 4 3 9 0e4 3 9 84396
in the sequences of these two groups. It indicates the possible
unrecognized deficiencies of the RDP Classifier.
Further examination of these sequences reveals that the
correct assignment of Nitrosomonas related sequences into
the Nitrosomonas genus is dependent on a key dinucleotide
position, as show in Fig. 6. If the ‘GC’ dincucleotide at
position 106 of group I was changed to ‘AT’, as it is found
in group II, the RDP Classifier would then assign group I
correctly into Nitrosomonas. It is therefore advised that the
phylogenetic results of Nitrosomonas species obtained using
the RDP Classifier should be cross-validated by other inde-
pendent tools such as Greengenes’ classification tool
(DeSantis et al., 2006) and GAST (Huse et al., 2008).
Accordingly, we have developed a batch BLAST method,
which has been described in the materials and methods
part, to confirm the suspicious sequences.

wat e r r e s e a r c h 4 5 ( 2 0 1 1 ) 4 3 9 0e4 3 9 8 4397
4. Conclusions
The diversity of the bacterial community in the nitrification
reactor was significantly reduced compared with seeding
sludge from the WWTP after 500 days’ operation. While, the
bacteria both in the reactor and the seeding sludge distributed
almost over the same phyla.
RDP classifier is a powerful tool for pyrosequencing data
analysis but it appears to misassign Nitrosomonas sequences
into wrong taxonomic rank. Some other tools, such as Blast
and Greengenes classification tool, can be used to correct the
results of the RDP classifier. We also developed a batch Blast
method to confirm the suspicious sequences.
According to the pyrosequencing results, for such
a reactor, a small scale clone library is not enough to reflect
the profile of the bacterial community.
Although the influent of the nitrification reactor contained
nearly no organic matter, the heterotrophic bacteria were still
dominant and much more than autotrophic bacteria in the
reactor.
Acknowledgments
Dr. Ming-Fei Shao thanks HKU for the postdoctoral fellowship.
Lin Ye thanks HKU for the postgraduate studentship. We also
would like to thank Hong Kong General Research Fund
(HKU7197-08E) for financial support of this study and W Chan
and CK Wong for technical help in pyrosequencing.
Appendix. Supplementary data
Supplementary data associated with this article can be found,
in the online version, at doi:10.1016/j.watres.2011.05.028.
r e f e r e n c e s
Altschul, S., Gish, W., Miller, W., Myers, E., Lipman, D., 1990. Basiclocal alignment search tool. J. Mol. Biol. 215, 403e410.
Barker, D.J., Stuckey, D.C., 1999. A review of soluble microbialproducts (SMP) in wastewater treatment systems. Water Res.33, 3063e3082.
Boon, N., Windt, W., Verstraete, W., Top, E., 2002. Evaluation ofnested PCReDGGE (denaturing gradient gel electrophoresis)with group-specific 16S rRNA primers for the analysis ofbacterial communities from different wastewater treatmentplants. FEMS Microbiol. Ecol. 39, 101e112.
Bowman, J., Nichols, D., 2002. Aequorivita gen. nov., a member ofthe family Flavobacteriaceae isolated from terrestrial andmarine Antarctic habitats. Int. J. Syst. Evol. Microbiol. 52, 1533.
Claesson, M., O’Sullivan, O., Wang, Q., Nikkila, J., Marchesi, J.,Smidt, H., De Vos, W., Ross, R., O’Toole, P., 2009. Comparativeanalysis of pyrosequencing and a phylogenetic microarray forexploring microbial community structures in the humandistal intestine. PloS One 4, e6669.
Cock, P., Antao, T., Chang, J., Chapman, B., Cox, C., Dalke, A.,Friedberg, I., Hamelryck, T., Kauff, F., Wilczynski, B., 2009.Biopython: freely available Python tools for computationalmolecular biology and bioinformatics. Bioinformatics 25, 1422.
DeSantis,T.,Hugenholtz, P., Larsen,N.,Rojas,M.,Brodie,E.,Keller,K., Huber, T., Dalevi, D., Hu, P., Andersen, G., 2006. Greengenes,a chimera-checked 16S rRNA gene database and workbenchcompatible with ARB. Appl. Environ. Microbiol. 72, 5069.
Eaton, A., Franson, M., 2005. Standard methods for theexamination of water & wastewater. Am Public Health Assn.
Ferris, M., Muyzer, G., Ward, D., 1996. Denaturing gradient gelelectrophoresis profiles of 16S rRNA-defined populationsinhabiting a hot spring microbial mat community. Appl.Environ. Microbiol. 62, 340e346.
Fierer, N., Bradford, M.A., Jackson, R.B., 2007. Toward anecological classification of soil bacteria. Ecology 88, 1354e1364.
Gauthier, M., Lafay, B., Christen, R., Fernandez, L., Acquaviva, M.,Bonin, P., Bertrand, J., 1992. Marinobacterhydrocarbonoclasticus gen. nov., sp. nov., a new, extremelyhalotolerant, hydrocarbon-degrading marine bacterium. Int. J.Syst. Evol. Microbiol. 42, 568.
Hao, X., Wang, Q., Zhang, X., Cao, Y., Mark Loosdrecht, C.M., 2009.Experimental evaluation of decrease in bacterial activity dueto cell death and activity decay in activated sludge. Water Res.43, 3604e3612.
Holt, J., Krieg, N., Sneath, P., Staley, J., Williams, S., 1994. Bergey’sManual of Systematic Bacteriology, nineth ed. Williams andWilkins.
Heuer, H., Krsek, M., Baker, P., Smalla, K., Wellington, E., 1997.Analysis of actinomycete communities by specificamplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl.Environ. Microbiol. 63, 3233e3241.
Huse, S., Dethlefsen, L., Huber, J., Welch, D., Relman, D.,Sogin, M., 2008. Exploring microbial diversity and taxonomyusing SSU rRNA hypervariable tag sequencing. PLoS Genet. 4,e1000255.
Huson, D., Auch, A., Qi, J., Schuster, S., 2007. MEGAN analysis ofmetagenomic data. Genome Res. 17, 377e386.
Jin, T., Zhang, T., Yan, Q., 2010. Characterization andquantification of ammonia-oxidizing archaea (AOA) andbacteria (AOB) in a nitrogen-removing reactor using T-RFLPand qPCR. Appl. Microbiol. Biotechnol. 87, 1167e1176.
Kwon, S., Kim, T., Yu, G., Jung, J., Park, H., 2010. Bacterialcommunity composition and diversity of a full-scaleintegrated fixed-film activated sludge system as investigatedby pyrosequencing. J. Microbiol. Biotechnol. 20, 1717e1723.
Layton, A., Dionisi, H., Kuo, H., Robinson, K., Garrett, V.,Meyers, A., Sayler, G., 2005. Emergence of competitivedominant ammonia-oxidizing bacterial populations in a full-scale industrial wastewater treatment plant. Appl. Environ.Microbiol. 71, 1105.
Lee, T.K., Van Doan, T., Yoo, K., Choi, S., Kim, C., Park, J., 2010.Discovery of commonly existing anode biofilm microbes intwo different wastewater treatment MFCs using FLX Titaniumpyrosequencing. Appl. Microbiol. Biotechnol., 1e9.
Liu, W.T., Chan, O.C., Fang, H.H.P., 2002. Microbial communitydynamics during start-up of acidogenic anaerobic reactors.Water Res. 36, 3203e3210.
Logemann, S., Schantl, J., Bijvank, S., Loosdrecht, M., Kuenen, J.,Jetten, M., 1998. Molecular microbial diversity in a nitrifyingreactor system without sludge retention. FEMS Microbiol. Ecol.27, 239e249.
Margulies, M., Egholm, M., Altman, W., Attiya, S., Bader, J.,Bemben, L., Berka, J., Braverman, M., Chen, Y., Chen, Z., 2005.Genome sequencing in microfabricated high-density picolitrereactors. Nature 437, 376e380.
Matsumoto, S., Katoku, M., Saeki, G., Terada, A., Aoi, Y.,Tsuneda, S., Picioreanu, C., van Loosdrecht, M., 2009. Microbialcommunity structure in autotrophic nitrifying granulescharacterized by experimental and simulation analyses.Environ. Microbiol. 12, 192e206.

wat e r r e s e a r c h 4 5 ( 2 0 1 1 ) 4 3 9 0e4 3 9 84398
McLellan, S., Huse, S., Mueller Spitz, S., Andreishcheva, E.,Sogin, M., 2010. Diversity and population structure of sewagederived microorganisms in wastewater treatment plantinfluent. Environ. Microbiol. 12, 378e392.
Qian, P., Wang, Y., Lee, O., Lau, S., Yang, J., Lafi, F., Al-Suwailem, A., Wong, T., 2011. Vertical stratification ofmicrobial communities in the Red Sea revealed by 16S rDNApyrosequencing. ISME J. 5, 507e518.
Regan, J.M., Harrington, G.W., Noguera, D.R., 2002. Ammonia-andnitrite-oxidizing bacterial communities in a pilot-scalechloraminated drinking water distribution system. Appl.Environ. Microbiol. 68, 73e81.
Rittmann, B., McCarty, P., 2001. EnvironmentalBiotechnology: Principles and Applications. McGraw-Hill,New York.
Roesch, L., Fulthorpe, R., Riva, A., Casella, G., Hadwin, A., Kent, A.,Daroub, S., Camargo, F., Farmerie, W., Triplett, E., 2007.
Pyrosequencing enumerates and contrasts soil microbialdiversity. ISME J. 1, 283e290.
Servin, J., Herbold, C., Skophammer, R., Lake, J., 2008. EvidenceExcluding the Root of the tree of Life from the Actinobacteria.Mol. Biol. Evol. 25, 1e4.
Sinha, B., Annachhatre, A., 2007. Partial nitrificationdoperationalparameters and microorganisms involved. Rev. Environ. Sci.Biotechnol. 6, 285e313.
Ye, L., Zhang, T., 2010. Estimation of nitrifier abundances ina partial nitrification reactor treating ammonium-rich salinewastewater using DGGE, T-RFLP and mathematical modeling.Appl. Microbiol. Biotechnol. 88 (6), 1403e1412.
Yoon, J., Lee, M., Oh, T., Park, Y., 2005. Muricauda flavescens sp.nov. and Muricauda aquimarina sp. nov., isolated from a saltlake near Hwajinpo Beach of the East Sea in Korea, andemended description of the genus Muricauda. Int. J. Syst. Evol.Microbiol. 55, 1015.







