dissertations in health sciences - uef electronic...
TRANSCRIPT

DIS
SE
RT
AT
ION
S | N
AG
EN
DR
A Y
AL
UR
I | DIA
BE
TO
GE
NIC
EF
FE
CT
S O
F S
TA
TIN
S | N
o 412
uef.fi
PUBLICATIONS OF THE UNIVERSITY OF EASTERN FINLAND
Dissertations in Health Sciences
ISBN 978-952-61-2465-0ISSN 1798-5706
Dissertations in Health Sciences
PUBLICATIONS OF THE UNIVERSITY OF EASTERN FINLAND
NAGENDRA YALURI
DIABETOGENIC EFFECTS OF STATINS
This study investigates the effects of statin
treatment on the risk of incident type 2 diabetes, insulin secretion and insulin
resistance in a prospective population study, and the molecular mechanisms underlying
these effects in mouse pancreatic β-cells and L6 skeletal myotubes. It shows that statin
treatment is associated with impairment in both insulin secretion and insulin sensitivity, and that the effects of simvastatin on insulin secretion and glucose uptake occur through
multiple targets.
NAGENDRA YALURI


Diabetogenic Effects of Statins


NAGENDRA YALURI
Diabetogenic Effects of Statins
To be presented by permission of the Faculty of Health Sciences, University of EasternFinland for public examination in Auditorium C100, Cathia building, University of Eastern
Finland on Saturday, 13th May 2017, at 12 noon
Publications of the University of Eastern Finland Dissertations in Health Sciences
412
Department of Medicine, Institute of Clinical Medicine, School of Medicine,Faculty of Health Sciences, University of Eastern Finland
Kuopio2017

Grano OyJyväskylä, 2017
Series Editors:Professor Tomi Laitinen, M.D., Ph.D.
Institute of Clinical Medicine, Clinical Physiology and Nuclear MedicineFaculty of Health Sciences
Professor Hannele Turunen, Ph.D.Department of Nursing Science
Faculty of Health Sciences
Professor Kai Kaarniranta, M.D., Ph.D.Institute of Clinical Medicine, Ophthalmology
Faculty of Health Sciences
Associate Professor Tarja Malm, Ph.D.A.I. Virtanen Institute for Molecular Sciences
Faculty of Health Sciences
Lecturer Veli-Pekka Ranta, Ph.D. (pharmacy)School of Pharmacy
Faculty of Health Sciences
Distributor:University of Eastern Finland
Kuopio Campus LibraryP.O. Box 1627
FI-70211 Kuopio, Finlandhttp://www.uef.fi/kirjasto
ISBN (print): 978-952-61-2465-0ISBN (pdf): 978-952-61-2466-7
ISSN (print): 1798-5706ISSN (pdf): 1798-5706
ISSN-L: 1798-5714

Author’s address: Department of Medicine, Institute of Clinical Medicine, Internal MedicineSchool of Medicine, Faculty of Health SciencesUniversity of Eastern FinlandP.O.Box 162770211 KUOPIOFINLANDE-mail: [email protected]
Supervisors: Academy Professor Markku Laakso, M.D., Ph.D.Department of Medicine, Institute of Clinical Medicine, Internal MedicineSchool of Medicine, Faculty of Health SciencesUniversity of Eastern Finland and Kuopio University Hospital
Docent Tarja Kokkola.Department of Medicine, Institute of Clinical Medicine, Internal MedicineSchool of Medicine, Faculty of Health SciencesUniversity of Eastern Finland
Professor Johanna Kuusisto, M.D., Ph.D.Department of Medicine, Institute of Clinical MedicineSchool of Medicine, Faculty of Health SciencesUniversity of Eastern FinlandKuopio University Hospital
Reviewers: Professor (Emeritus) Matti J. Tikkanen, M.D., Ph.D.Folkhälsan Research CenterUniversity of HelsinkiHELSINKIFINLAND
Professor Vesa Olkkonen, Ph.D.Minerva Foundation Institute for Medical ResearchBiomedicum 2UHELSINKIFINLAND
Opponent: Professor (Emeritus) Petri Kovanen, M.D., Ph.D.Wihuri Research InstituteBiomedicum HelsinkiHELSINKIFINLAND


V
Yaluri Nagendra, Diabetogenic Effects of Statins. Publications of the University of Eastern Finland.Dissertations in Health Sciences 412. 2017. 77 p.ISBN (print): 978-952-61-2465-0ISBN (pdf): 978-952-61-2466-7ISSN (print): 1798-5706ISSN (pdf): 1798-5706ISSN-L: 1798-5714
ABSTRACTStatins are the most effective drugs used in the treatment of hypercholesterolemia andprevention of cardiovascular disease (CVD). In spite of their benefits, statins have beenrecently shown to increase the risk of new-onset diabetes mellitus. This increase in the riskhas been reported to be the lowest for pravastatin and the highest for rosuvastatin,atorvastatin and simvastatin.
Glucose levels in the blood are mainly determined by the secretion and action of insulin.Disturbances in these processes can lead to prediabetes, and the conversion to overt diabeteshappens when increased insulin secretion from pancreatic -cells is no longer able tocompensate for increased insulin resistance. Type 2 diabetes (T2D) represents a major publichealth problem due to its increasing prevalence and vascular complications leading toincreased morbidity and mortality.
Most of the evidence on the diabetogenic effects of statins comes from population studiesand clinical trials. However, only few of them have evaluated the effects of statins on insulinsensitivity or insulin secretion. Clinical trials have selective inclusion criteria and may notreflect the risk of diabetes in the general population. The molecular mechanisms underlyingthe diabetogenic effects of statins remain unclear.
We investigated the effects of statin treatment on the risk of incident T2D, insulinsecretion and insulin resistance in the large population-based Metabolic Syndrome in Men(METSIM) study. Additionally we investigated the molecular mechanisms underlying theseeffects in mouse pancreatic -cells (MIN6) and L6 skeletal muscle cells, compared with non-diabetogenic pravastatin.
We showed that statin treatment was associated with a 46% increase in the risk ofincident T2D, a 24% decrease in insulin sensitivity and a 12% decrease in insulin secretionduring the follow-up of the METSIM study. Our in vitro experiments showed thatsimvastatin-induced decrease in insulin secretion occurred through multiple targets such asthe ATP-sensitive potassium channels, voltage-gated calcium channels, muscarinic M3receptors, GPR40 receptor, and calcium release from the endoplasmic reticulum in MIN6cells. Impaired insulin secretion caused by simvastatin was efficiently restored by GPR119 orGLP-1 receptor stimulation and by direct activation of cAMP-dependent signaling pathwayswith forskolin. Our study in L6 myotubes showed that simvastatin decreased glucoseuptake by impairing insulin signaling, resulting in decreased phosphorylation of insulinreceptor, insulin receptor substrate 1 and its downstream targets Akt kinase and glycogensynthase kinase 3 , and in decreased protein expression of glucose transporter GLUT4. Theeffects of simvastatin on insulin secretion and glucose uptake were not influenced byhyperglycemia. Pravastatin did not have adverse effects on insulin secretion or insulinsensitivity in our studies.
National Library of Medicine Classification: QU 140, QU 143, WK 810, WK 820Medical Subject Headings: Hydroxymethylglutaryl-CoA Reductase Inhibitors/adverseeffects; Simvastatin; Pravastatin; Diabetes Mellitus, Type 2; Insulin; Insulin Resistance;Insulin-Secreting Cells; Muscle Cells; Muscle Fibers, Skeletal; Mice

VI

VII
Yaluri Nagendra, Statiinien diabetesriskiä lisäävät vaikutukset. Publications of the University ofEastern Finland. Dissertations in Health Sciences 412. 2017. 77 s.ISBN (print): 978-952-61-2465-0ISBN (pdf): 978-952-61-2466-7ISSN (print): 1798-5706ISSN (pdf): 1798-5706ISSN-L: 1798-5714
ABSTRAKTIStatiinit ovat tehokkaimpia hyperkolesterolemian hoidossa sekä sydän- ja
verisuonitautien ehkäisyssä käytettäviä lääkkeitä. Hyvistä vaikutuksistaan huolimattastatiinien on viime aikoina osoitettu lisäävän riskiä sairastua diabetekseen. Pravastatiinillaon statiineista pienin vaikutus diabetesriskiin, ja rosuvastatiini, atorvastatiini ja simvastatiinilisäävät riskiä eniten.
Veren glukoositasoja säädellään insuliinin erityksen ja sen vaikutusmekanismien kautta.Häiriöt näissä mekanismeissa voivat johtaa esidiabeettiseen tilaan, ja varsinainen diabetespuhkeaa kun haiman -solujen insuliinin erityksellä ei enää pystytä kompensoimaanlisääntynyttä insuliiniresistenssiä. Tyypin 2 diabetes (T2D) on suuri kansanterveydellinenongelma, koska sen esiintyvyys on yleistymässä ja siihen liittyvät verisuonikomplikaatiotaiheuttavat lisääntynyttä sairastavuutta ja kuolleisuutta.
Statiinien diabetesriskiä lisäävät vaikutukset on havaittu enimmäkseenväestötutkimuksissa ja kliinisissä tutkimuksissa. Statiinien vaikutuksia insuliiniherkkyyteentai insuliinin eritykseen on kuitenkin arvioitu vain harvoissa tutkimuksissa. Kliinisissätutkimuksissa on valikoivat hyväksymiskriteerit ja ne eivät välttämättä kuvasta kokoväestön diabetesriskiä. Statiinien diabetesriskiä lisäävän vaikutuksen molekyylitasonmekanismeja ei toistaiseksi vielä tunneta.
Tutkimuksemme käsitteli statiinihoidon vaikutuksia tyypin 2 diabetesriskiin, insuliinineritykseen ja insuliiniresistenssiin laajassa METSIM-väestötutkimuksessa (MetabolicSyndrome in Men). Tutkimme lisäksi näiden havaintojen takana olevia molekyylitasonmekanismeja hiiren haiman -soluissa (MIN6) ja L6-luurankolihassoluissa, ja vertasimmetuloksia pravastatiiniin, joka ei lisää diabetesriskiä.
Havaitsimme, että statiinihoitoon liittyi 46% lisäys T2D:n puhkeamisessa, 24% laskuinsuliiniherkkyydessä ja 12% lasku insuliinin erityksessä METSIM-tutkimuksenseurantajakson aikana. In vitro -tutkimuksemme osoittivat että simvastatiini heikensiinsuliinin eritystä MIN6-soluissa useiden mekanismien kautta, mukaan lukien ATP-herkätkaliumkanavat, jänniteherkät kalsiumkanavat, M3-muskariinireseptorit, GPR40-reseptorit jakalsiumin vapautuminen endoplasmakalvostosta. Simvastatiinikäsittelyn insuliinin eritystäheikentävä vaikutus pystyttiin kumoamaan GPR119- tai GLP-1-reseptoreja stimuloimalla taiaktivoimalla cAMP-välitteinen signalointi forskoliinilla. L6-lihassoluilla tehdyssätutkimuksessa osoitimme, että simvastatiini vähensi glukoosin soluunottoa aiheuttamallahäiriöitä insuliinin signaloinnissa ja estämällä insuliinireseptorin,insuliinireseptorisubstraatti 1:n ja sen säätelykohteiden Akt-kinaasin jaglykogeenisyntaasikinaasi-3 :n fosforylaatiota ja vähentämällä GLUT4-glukoositransportterin määrää. Hyperglykemia ei muuttanut simvastatiinin vaikutuksiainsuliinin eritykseen ja glukoosin soluunottoon. Pravastatiinilla ei ollut tutkimuksissammehaitallisia vaikutuksia insuliiniherkkyyteen tai insuliinin eritykseen.
Luokitus: QU140, QU143, WK 810, WK 820Yleinen suomalainen asiasanasto: statiinit; diabetes; insuliini; insuliiniresistenssi; lihassolut

VIII

IX
The hallmark of science is freedom
Dedicated to science

X

XI
AcknowledgementsThis study was performed in the Department of Medicine, Institute of ClinicalMedicine, School of Medicine, Faculty of Health Sciences, University of EasternFinland.
I thank my main supervisor Professor Markku Laakso and supervisor ProfessorJohanna Kuusisto for giving me the opportunity to pursue my PhD studies, for theirguidance and support. Working in the laboratory of Professor Markku Laakso was aunique experience which will shape my future career. I am grateful for the freedomto explore new ideas and trust given to me initially, which inspired me to work hardand fueled my passion for science. I thank my second supervisor Docent TarjaKokkola for her support, guidance and contribution to my manuscripts.
I thank Docent Antero Salminen for teaching me the skills and techniques of the cellculture work when I first came to Finland, for helping me to solve technicalproblems, and for allowing me to use his cell culturing lab under his careful andkind supervision.
I thank the official reviewers of my thesis Prof. Matti Tikkanen and Prof. VesaOlkkonen for their comments and corrections which improved my thesis. I alsothank Doctor David Laaksonen for the linguistic revision of the thesis.
I thank MSc. Shalem Modi, who joined me here as my old friend from our bachelorstudies, for sharing large amount of laboratory work with me, his help in the lab wascritical for completion of my thesis work and I appreciate it very much. I thankDocent Henna Cederberg-Tamminen for sharing thoughts on our statin project andfor the enriching collaboration which allowed us to analyze the research questionfrom different angles and backgrounds. I thank her for her generosity at work aswell as in private life and for being a dear friend for me and my family. I thank MSc.Maykel Lopez-Rodriguez for his calcium experiments contributing to my 2nd paper.
For me life is like an evolution. I constantly try to learn new things from peoplearound me and my surroundings to enrich my professional and personal life. Duringthis long PhD journey I met many people whom I would like to thank for sharingtheir thoughts, knowledge and time with me: Lakshman Puli, Milka Hakkarainen,Diana Kujala, Teemu Kuulasmaa, Seija Laitinen, Leena & Jukka Uschanoff, HannaHuopio, Martin Javorsky, Jagadish Vangipurapu, Jone Salniene, Maija Tusa, DocentEija Pirinen, Marc Cerrada-Gimenez, Tapio Nuutinen, Tiina Suuronen, AssociateProfessor Anu Kauppinen, Juha Hyttinen, Minna Niittykoski, Esa Koivisto, Professor

XII
Mikko Hiltunen, Associate Professor Annakaisa Haapasalo, Mari Laitinen, AdemJemal, Professor Jukka Pelkonen, Ahmed Gazali, Anne Seppänen, Johanna Viiri,Professor Kai Kaarniranta, Mari Hytti, Eveliina Korhonen, Niina Piippo, ProfessorJorma Palvimo, Harri Makkonen, Docent Vanessa De Mello Laaksonen, AssistantProfessor Jussi Paananen, Professor Jussi Pihlajamäki, Dorota Kaminska, MaijaVaittinen, Jenna Pekkinen, Ashok Matte, Mohan Babu Budikote, Professor RashidGiniatullin, Cindy Guerrero Toro.
I thank all the laboratory and administrative personnel for helping me wheneverneeded, introducing me to Finnish culture and creating a happy workingatmosphere: Raija Räisänen, Maritta Siloaho, Katja Kostinen-Kokko, Eeva Oittinen,Sari Kärkkäinen, Kaija Eirola, Tiina Sistonen, Tarja Heikkinen, Ulla Ruotsalainen,Aija Jantunen, Leila Antikainen, Anne Toivanen, Josse Raivo, Seija Heikkinen, ArjaAfflekt, Markku Taskinen, Kalle Pasanen, Kimmo Kankkunen.
I thank my parents for their love, teaching me moral values, supporting me when Idecided to study abroad, and for giving me an example of hard work, which helpedme to achieve my goals. I thank my brother and his family for their love andsupport. I thank to my father-in-law, mother-in-law and my Anja for their trust, loveand for supporting me in difficult times. I would like to thank Mahesh, ChakraReddy, Shiva Shankar and Ram Kumar & family for their friendship.
At last I would like to give all the credit to my lovely wife Alenka for herunconditional love and support in both personal and professional life since the day Imet her. Your support and trust in me was very crucial for this achievement. Besidesplaying a key role in taking care of our family, your careful suggestions and help inpreparing the manuscripts and thesis have been vital. I could say that without youthis day would never come. At last I would like to acknowledge my boys Aman andMayan. It is a privilege and joy to have you both, you make our lives incredible and Ilove you endlessly.
This work has been supported by the Academy of Finland, the Finnish DiabetesResearch Foundation, the Finnish Cardiovascular Research Foundation, the StrategicResearch Funding from the University of Eastern Finland, and EVO grant 5263 fromthe Kuopio University Hospital.
Kuopio, March 2017
Nagendra Yaluri

XIII
List of the original publications
This dissertation is based on the following original publications:
I Cederberg H, Stan áková A, Yaluri N, Modi S, Kuusisto J, Laakso M.Increased risk of diabetes with statin treatment is associated with impairedinsulin sensitivity and insulin secretion: a 6 year follow-up study of theMETSIM cohort. Diabetologia 58: 1109-1117, 2015.
II Yaluri N‡, Modi S‡, López Rodríguez M, Stan áková A, Kuusisto J,Kokkola T, Laakso M. Simvastatin Impairs Insulin Secretion by MultipleMechanisms in MIN6 Cells. PLoS One 10: e0142902, 2015.
III Yaluri N, Modi S, Kokkola T. Simvastatin induces insulin resistance in L6skeletal muscle myotubes by suppressing insulin signaling, GLUT4expression and GSK-3 phosphorylation. Biochem Biophys Res Commun480: 194-200, 2016.
‡ equal contribution
The publications were adapted with the permission of the copyright owners.

XIV

XV
Contents
1 INTRODUCTION
2 REVIEW OF THE LITERATURE2.1 Statins and cholesterol biosynthesis
2.1.1 Statins2.1.2 Cholesterol and its biosynthesis2.1.3 Effects of statins on cholesterol biosynthesis2.1.4 Statins and their lipid lowering effects2.1.5 Statins and their cardiovascular risk lowering effects
2.2 Prediabetes and type 2 diabetes 2.2.1 Pathophysiology of prediabetes and type 2 diabetes
2.2.1.1 Insulin secretion and its regulation2.2.1.2 Insulin resistance
2.3 Diabetogenic effects of statins 2.3.1 Evidence for the diabetogenic effects of statins in population studies 2.3.2 Evidence for the diabetogenic effects of statins in clinical trials and meta- analyses 2.3.3 Effects of statins on insulin secretion and insulin sensitivity in clinical trials
and population studies2.3.4 Evidence for the diabetogenic effects of statins in genetic studies2.3.5 Mechanistic studies of the effects of statins on insulin secretion and insulin action
2.3.5.1 Effect of statins on insulin secretion2.3.5.2 Effect of statins on insulin sensitivity
3 AIMS OF THE STUDY
4 SUBJECTS AND METHODS4.1 Subjects4.2 Cell cultures4.3 Laboratory methods
5 RESULTS5.1 Association of statin treatment with the risk of diabetes, insulin sensitivity and insulin secretion (I)5.2 Molecular mechanisms of the effect of simvastatin and pravastatin on insulin secretion in MIN6 cells (II)5.3 Molecular mechanisms of the effect of simvastatin and pravastatin on insulin sensitivity in L6 myotubes (III)

XVI
6 DISCUSSION6.1 Representativeness of the study subjects (Study I)6.2 Evaluation of the methods of the in vitro studies (Studies II-III)6.3 Association of statin treatment with the risk of diabetes, insulin sensitivity and insulin secretion6.4 Molecular mechanisms of the effects of simvastatin on insulin secretion and insulin sensitivity6.5 Concluding remarks
7 SUMMARY
8 REFERENCES
Appendix: original publications

XVII
Abbreviations2hPG 2-hour plasma glucose3T3-L1 cell Murine adipocyte4S Scandinavian Simvastatin Survival Study8-pCPT-2 -O-Me-cAMP
8-(4-Chlorophenylthio)-2 -O-methyladenosine 3 ,5 -cyclicmonophosphate
ADA American Diabetes AssociationADP Adenosine diphosphateAFCAPS/TexCAPS Air Force/Texas Coronary Atherosclerosis Prevention StudyAkt Akt kinase (protein kinase B)APOB Apolipoprotein B geneAS160 Akt substrate of 160 kDaASCOT-LLA Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering
ArmASPEN Atorvastatin Study for Prevention of Coronary Heart Disease
Endpoints in Non-Insulin Dependent Diabetes MellitusATP Adenosine triphosphateAUC Area under the curveBMI Body mass indexBSA Bovine serum albuminC/EBP CCAAT-enhancer-binding protein alphaCa2+ Calcium cation[Ca2+]i Free intracellular calciumCAD Coronary artery diseasecAMP Cyclic adenosine monophosphateCARDS Collaborative Atorvastatin Diabetes StudyCARE Cholesterol and Recurrent Events TrialCHD Coronary heart diseaseCORONA Controlled Rosuvastatin Multinational Trial in Heart FailureCVD Cardiovascular diseaseDAG DiacylglycerolDI Disposition indexDIAGRAM Diabetes Genetics Replication And Meta-analysis consortiumDMAPP Dimethylallyl pyrophosphateDMEM Dulbecco’s modified Eagle’s mediumDMSO Dimethyl sulfoxideEHC Euglycemic hyperinsulinemic clampEPAC2 (cAMP-GEF)
cAMP-regulated guanine nucleotide exchange factor
ER Endoplasmic reticulum

XVIII
FFA Free fatty acidFPG Fasting plasma glucoseFPP Farnesyl pyrophosphateFTI-277 Farnesyltransferase inhibitorG6P Glucose-6-phosphateGAP GTPase-activating proteinGAPDH Glyceraldehyde 3-phosphate dehydrogenaseGENESIS Genetics of Insulin SensitivityGGPP Geranylgeranyl pyrophosphateGGTI-298 Geranylgeranyltransferase inhibitorGIP Glucose-dependent insulinotropic peptideGLP-1 Glucagon-like peptide 1GLP-1R Glucagon-like peptide 1 receptorGLUT2 Glucose transporter 2GLUT4 Glucose transporter type 4GPCR G protein coupled receptorGPP Geranyl pyrophosphateGPR119 Glucose-dependent insulinotropic receptorGPR40 (FFAR1) Free fatty acid receptor 1GRK2 G protein-coupled receptor kinase 2GS Glycogen synthaseGSIS Glucose stimulated insulin secretionGSK-3 Glycogen synthase kinase 3HbA1c Glycated hemoglobin A1cHDL High-density lipoproteinHEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acidHGP Hepatic glucose productionHMG-CoA 3-hydroxy-3-methyl-glutaryl-coenzyme AHMGCR HMG-CoA reductase geneHOMA-B Homeostasis model assessment of insulin secretionHOMA-IR Homeostasis model assessment of insulin resistanceHR Hazard ratioIDEAL Incremental Decrease in Endpoints Through Aggressive Lipid
LoweringIDL Intermediate-density lipoproteinIL-6 Interleukin 6IP3 Inositol 1,4,5-trisphosphateIP3R Inositol 1,4,5-trisphosphate receptorIPP Isopentenyl pyrophosphateIR Insulin receptorIRS Insulin receptor substrateJNK1 C-Jun N-terminal kinase 1

XIX
JUPITER Justification for the Use of Statins in Prevention: anIntervention Trial Evaluation Rosuvastatin
KATP channel ATP-sensitive potassium channelKRBH Krebs-Ringer bicarbonate HEPES BufferL6 cell Rat skeletal muscle cellLDL Low-density lipoproteinLDLR LDL receptor geneLIPID Long-term Intervention with Pravastatin in Ischaemic DiseaseLIPS Lescol intervention prevention studyM3R M3 muscarinic acetylcholine receptorMAGIC Meta-Analyses of Glucose and Insulin-related traits
ConsortiumMatsuda ISI Matsuda´s index of insulin sensitivityMETSIM Metabolic Syndrome in MenMIN6 cell Mouse pancreatic -cellMIRACL Myocardial Ischemia Reduction with Acute Cholesterol
LoweringmRNA Messenger ribonucleic acidmTOR Mammalian target of rapamycinNADPH Nicotinamide adenine dinucleotide phosphateNLRP3 NLR family pyrin domain containing 3NODM New-onset diabetes mellitusNPC1L1 NPC1 like intracellular cholesterol transporter 1 geneOGTT Oral glucose tolerance testOR Odds ratioPC1, PC2 Prohormone convertase 1, 2PCSK9 Proprotein convertase subtilisin/kexin type 9 genePDK1 Phosphoinositide-dependent protein kinase-1PH Pleckstrin homology domainPI3K Phosphoinositide 3-kinasePIP2 Phosphatidylinositol 4,5-bisphosphatePIP3 Phosphatidylinositol 3,4,5-trisphosphatePKA Protein kinase APKC Protein kinase CPLC Phospholipase CPMA Phorbol-12-myristate 13-acetatePROSPER Pravastatin in the Elderly at RiskPROVE-IT Pravastatin or Atorvastatin Evaluation and Infection TherapyRCT Randomized control trialRIPA Radioimmunoprecipitation assay bufferRyR Ryanodine receptor

XX
S6K1 Ribosomal protein S6 kinase beta-1SD Standard deviationSE Standard error of the meanSEARCH Study of the Effectiveness of Additional Reductions in
Cholesterol and HomocysteineSLC2A4 Solute carrier family 2 member 4 geneSPARCL Stroke Prevention by Aggressive Reduction in Cholesterol
LevelsSREBP Sterol regulatory element-binding proteinSTELLAR Statin Therapies for Elevated Lipid Levels compared Across
Doses of RosuvastatinSUR1 Sulphonylurea receptor 1T2D Type 2 diabetesTBC1D TBC domain family member 1DTBS-T TBS-Tween-20TNT Treating to New TargetsVGCC Voltage-gated calcium channelVLDL Very low-density lipoproteinWHO World Health OrganizationWOSCOPS West of Scotland Coronary Prevention Study

1 Introduction
Statins are the most effective drugs used in the treatment of hypercholesterolemiaand prevention of cardiovascular disease (CVD) events (Betteridge and Carmena2016, Weng et al. 2010). Statins inhibit 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase, the rate-limiting enzyme in the cholesterol biosynthesis, and thuslower the levels of plasma total and low-density lipoprotein (LDL) cholesterol,which are risk factors for CVD. Although the benefits of statin treatment in theprevention of cardiovascular outcomes are undisputable, several studies haverecently shown that treatment with statins increases the risk of new-onset diabetesmellitus (NODM) (Sattar et al. 2010). This increase in the risk of diabetes is dosedependent (Preiss and Sattar 2011a, Chogtu et al. 2015) and differs across the statins.The lowest risk of NODM was observed for pravastatin and the highest risk forrosuvastatin, atorvastatin and simvastatin (Navarese et al. 2013, Chogtu et al. 2015).
Beside statins, several other classes of drugs can affect the regulation of glucosemetabolism and induce diabetes. Among them are several antihypertensive drugs(thiazide diuretics, -blockers), glucocorticoids and immunosuppressants(ciclosporin/tacrolimus), antidepressants (fluvoxamine, venlafaxine, amitriptyline,paroxetine), atypical antipsychotics (clozapinem, olanzapine, risperidone,quetiapine), phenothiazine antipsychotics (chlorpromazine, trifluoperazine,promethazine), drugs for the treatment of HIV and epilepsy, and oral contraceptives(Davis 2010).
Glucose levels in the blood are tightly regulated within a narrow physiologicalrange, mainly by the hormones insulin and glucagon. Defects in insulin secretionand action lead to prediabetes, which is characterized by mild elevations of glucoselevels often lasting for years (Edwards and Cusi 2016). Type 2 diabetes (T2D)develops when pancreatic -cells are no longer able to secrete more insulin tocompensate for insulin resistance (Kahn et al. 2014). The incidence and prevalence ofT2D is icreasing worldwide, and longterm complications related to this diseaseincrease morbidity and mortality.
Most of the evidence on the diabetogenic effects of statins comes from populationstudies and clinical trials (Collins et al. 2016, Sattar et al. 2010, Culver et al. 2012,Rajpathak et al. 2009, Waters et al. 2011, Preiss and Sattar 2011a, Preiss et al. 2011b).However, previous population studies have not generally applied an oral glucosetolerance test (OGTT) and measured glycated hemoglobin A1c (HbA1c) as thediagnostic criteria for diabetes. Population-based studies evaluating the mechanismsunderlying the diabetogenic effects of statins are scarce and based on small samplesize. Clinical trials have been often selective and therefore may not reflect the risk ofdiabetes in the general population.
The molecular mechanisms underlying the diabetogenic effects of statins are notfully understood. Previous in vitro studies have suggested that statins impair insulinsecretion through their inhibitory effects on the voltage-gated calcium channels(VGCC) and the ATP-sensitive potassium (KATP) channels in the plasma membraneof -cells (Yada et al. 1999, Zhou et al. 2014). Insulin secretion is also modulated byseveral hormones and neurotransmitters through different but interconnectedpathways (Fu et al. 2013). The potential effects of statins on these pathways have not

2
been previously investigated. Effects of statins on insulin sensitivity have beenexplored mainly in adipose cells (Takaguri et al. 2008, Chamberlain 2001, Ganesanand Ito 2013, Nakata et al. 2006). However, skeletal muscle is the main contributor tothe whole-body insulin sensitivity, as 80% of glucose uptake occurs in skeletalmuscle under euglycemic hyperinsulinemic conditions (Cersosimo et al. 2000).Several in vitro studies have demonstrated that the treatment with different statinsdecreases glucose uptake (Kain et al. 2015, Smith et al. 2014, Chamberlain 2001,Takaguri et al. 2008). However, only few of them have been performed using skeletalmuscle cells (Kain et al. 2015, Smith et al. 2014). Additionally, these studies haveprovided conflicting results as to whether the effects of statins are dependent (Smithet al. 2014) or independent (Kain et al. 2015) on the cholesterol biosynthesis pathway.No previous in vitro study has investigated whether or not the effects of statins areinfluenced by high glucose levels.
We investigated the effects of statin treatment on the risk of incident T2D, insulinsecretion and insulin resistance in the population-based Metabolic Syndrome in Men(METSIM) study. Additionally, we investigated molecular mechanisms of impairedinsulin secretion induced by simvastatin treatment compared with non-diabetogenicpravastatin treatment in mouse pancreatic -cells (MIN6), and molecularmechanisms of insulin resistance induced by simvastatin treatment compared withpravastatin in L6 skeletal muscle cells.

3
2 Review of the literature
2.1 STATINS AND CHOLESTEROL BIOSYNTHESIS
2.1.1 StatinsStatins were originally identified as secondary metabolites of fungi (Alberts et al.1980) which were able to inhibit cholesterol biosynthesis (Endo 1992). The first statin,mevastatin, was isolated from Pencillium citrinum (Endo et al. 1976). However, thisstatin was found to cause hepatocellular toxicity in rats. Lovastatin was isolatedfrom Aspergillus terreus by Hoffman and colleagues in 1979 (Alberts et al. 1980,Alberts 1990), and it had no hepatocellular toxicity when given to rats, and was amore potent inhibitor of HMG-CoA reductase than mevastatin. Therefore, lovastatinbecame the first statin which was approved for clinical use in humans. Since then,several natural or chemically modified statins were identified, such as pravastatin,simvastatin, fluvastatin, atorvastatin, rosuvastatin, cerivastatin and pitavastatin(Endo 2010, Stossel 2008, Hajar 2011, Tobert 2003) (Figure 1).
Figure 1. Chemical structures of different statins (Nigovi et al. 2012).
2.1.2 Cholesterol and its biosynthesisCholesterol is an important organic molecule with multiple functions. It is anessential component of all animal cell membranes and helps to maintain membranestructure integrity and fluidity (Fessler 2016). It is essential for conducting nervousimpulses, especially at the level of the synapse (Petrov et al. 2016). Cholesterol alsoserves as a precursor for the biosynthesis of bile acids (Cerqueira et al. 2016), whichare needed for the absorption of fats (Morgan et al. 2016), and for steroid hormonessuch as testosterone, estrogen, dihydroepiandrosterone, progesterone, and cortisol(Morgan et al. 2016, Cerqueira et al. 2016). Together with sun exposure, cholesterol isrequired to produce vitamin D (Cerqueira et al. 2016).

4
Approximately 80% of circulating cholesterol is derived from endogenoussynthesis, and only a small portion originates from the diet (Hegele 2009). Thesynthesis of cholesterol is initiated through a cascade of enzymatic reactions calledthe mevalonate pathway (Figure 2). The first rate-limiting step of cholesterolbiosynthesis is regulated by HMG-CoA reductase, which converts HMG-CoA intomevalonate. This step is the target for statin drugs. In the next step mevalonate isphosphorylated into pyrophosphomevalonate, which is then converted intoisopentenyl pyrophosphate (IPP). IPP can be converted reversibly to dimethylallylpyrophosphate (DMAPP), and the combination of IPP and DMAPP forms the 10-carbon isoprenoid geranyl pyrophosphate (GPP). Further addition of IPPs cangenerate the 15-carbon isoprenoid farnesyl pyrophosphate (FPP), and the 20-carbonisoprenoid geranylgeranyl pyrophosphate (GGPP). FPP can be converted intovarious other products including cholesterol, isoprenoids, dolichol, ubiquinone andisopentenyladenine (Schachter 2005, Cruz et al. 2013, Demierre et al. 2005).
Figure 2. The mammalian mevalonate pathway (Demierre et al. 2005).
Cholesterol is insoluble in water and cannot be transported alone in the circulation.Instead it is transported inside spheroidal macromolecules called lipoproteins.Lipoproteins consist of a hydrophobic core containing triacylglycerols, cholesterylesters, fat-soluble vitamins and antioxidants, and a hydrophilic coat containing freecholesterol, phospholipid and apolipoprotein molecules (Figure 3). Lipoproteins are

5
classified into 5 major classes based on their size, composition, density and function:chylomicrons, very low-density lipoprotein (VLDL), intermediate-densitylipoprotein (IDL), LDL, and high-density lipoprotein (HDL). The main cholesterol-carrying lipoprotein is LDL, which transports cholesterol from the liver to tissuesthat incorporate it into cell membranes, and HDL, which carries cholesterol that hasbeen discarded by tissues back to the liver (reverse cholesterol transport) forrecycling or excretion (Colpo et al. 2005).
Under normal conditions cholesterol levels in blood are regulated by the balancebetween its synthesis, dietary intake and the removal of extra cholesterol from theperipheral tissues (Simons and Toomre 2000). The key organ for cholesterolmetabolism and regulation of plasma levels of cholesterol is the liver (Wadhera et al.2016). Intrahepatic cholesterol, either from gut absorption or de novo synthesis, isrepackaged (along with proteins, triglycerides, and phospholipids) into VLDLparticles by the liver (Wadhera et al. 2016). VLDL particles then enter the circulationand are converted by lipoprotein lipase and cholesteryl ester transfer protein intomore cholesterol-enriched species, first IDL and then LDL. The liver regulates theconcentration of these circulating lipoprotein species primarily by their clearancethrough LDL receptors on the surface of hepatocytes (Wadhera et al. 2016).
Figure 3. Structure of a LDL particle (Nelson and Cox, 2005).
2.1.3 Effect of statins on the cholesterol biosynthesisStatins inhibit HMG-CoA reductase reversibly by binding to the active site of theenzyme through its side chains, thus blocking the interaction of HMG-CoAreductase with its natural substrate HMG-CoA and preventing activity of theenzyme (Istvan and Deisenhofer 2001). Statins bind to HMG-CoA reductase even atnanomolar concentrations, thus effectively replacing HMG-CoA, which binds atmicromolar concentrations (Moghadasian 1999).
Although all statins inhibit HMG-CoA reductase by a similar mechanism, thereare subtle differences between them (Table 1). Some statins are more effective thanother statins in their ability to bind hepatic HMG-CoA reductase at higher affinityand with a longer duration compared to other statins. Among the first and secondgeneration statins, the rank of their potency in inhibiting HMG-CoA reductase is

6
atorvastatin > simvastatin > pravastatin > lovastatin mevastatin based on in vivostudies (Dansette et al. 2000). The third generation statins which include pitavastatinand rosuvastatin are much more potent than the mevastatin derivatives (McTaggartet al. 2001). Atorvastatin and rosuvastatin have the greatest number of bondinginteractions with HMG-CoA reductase compared to other statins (Schachter 2005,Dansette et al. 2000, McTaggart et al. 2001, Maji et al. 2013, Kapur and Musunuru2008).
Table 1. Three generations of statins and their potency, LDL cholesterol lowering effects,and hydro/lipophilic nature (adapted from Kapur and Musunuru 2008).Generation Statin Potency Change
in LDLHydro/lipo-
philicity1st Pravastatin low 21%
to 42%hydrophilic
Lovastatin low lipophilicFluvastatin low lipophilic
2nd Simvastatin high 26%to 60%
lipophilicAtorvastatin high lipophilic
3rd Rosuvastatin very high 45%to 63%
hydrophilicPitavastatin high lipophilic
Statins can also inhibit extrahepatic HMG-CoA reductase depending on theirtissue permeability and metabolism. Lipophilic statins such as simvastatin are ableto enter endothelial cells by passive diffusion, unlike hydrophilic statins such aspravastatin and rosuvastatin, which are primarily targeted to liver (Dulak andJozkowicz 2005). However, the extrahepatic effect of some statins is not entirelyattributable to lipophilicity and other unknown factors may play role (Liao andLaufs 2005).
By the inhibition of L-mevalonic acid synthesis statins also prevent the synthesisof other important intermediates of cholesterol biosynthetic pathway, such as FPPand GGPP (Goldstein and Brown 1990). In a process called isoprenylation theseintermediates serve as lipid attachments and post-translationally modify a variety ofproteins, including subunit of heterotrimeric G-proteins and small G-proteins Ras,and Ras-like proteins, such as Rho, Rab, Rac, Ral, or Rap (Van Aelst and D'Souza-Schorey 1997). The isoprenylation helps different proteins to undergo covalentattachment, subcellular localization, and intracellular trafficking of membraneassociated proteins (Liao 2002, Kavalipati et al. 2015, Liao et al. 2016).
2.1.4 Statins and their lipid lowering effectsStatins have been available for clinical use since 1980s and are proved to be one ofthe most effective drugs in reducing the levels of LDL cholesterol (Betteridge andCarmena 2016). The decrease in hepatic levels of cholesterol by statins initiates aseries of coordinated cellular reactions mediated by a family of transcription factorscalled sterol regulatory element-binding proteins (SREBPs) that act through a sterolregulatory element in the genes which are targeted by SREBPs (Betteridge andCarmena 2016). This leads to upregulation of LDL receptor, which removes LDLparticles from the blood stream and is a major determinant of plasma concentrationsof LDL (Betteridge and Carmena 2016).
The first generation statins lovastatin, pravastatin and fluvastatin have the lowestpotency and they reduce LDL cholesterol levels by 30% when taken in daily doses

7
of 40-80 mg (Weng et al. 2010, Kapur and Musunuru 2008). Pravastatin has been themost rigorously tested in controlled clinical trials. Treatment with pravastatinreached 26% reduction of LDL cholesterol level in the West of Scotland CoronaryPrevention Study (WOSCOPS) (Shepherd et al. 1995), and 34% reduction in theProspective Study of Pravastatin in the Elderly at Risk (PROSPER) trial, compared toplacebo (Shepherd et al. 2002).
The second generation statins such as simvastatin and atorvastatin havesignificantly improved efficacy in reducing LDL cholesterol compared to the earlierstatins (Rozman and Monostory 2010). A daily dose of 20 mg of simvastatin or 10 mgof atorvastatin can achieve >30% reduction in LDL cholesterol levels (Weng et al.2010, Kapur and Musunuru 2008, Maji et al. 2013). In the Scandinavian SimvastatinSurvival Study (4S), treatment with simvastatin reached 35% reduction of LDLcholesterol levels, compared to placebo (Scandinavian Simvastatin Survival StudyGroup 1994). Treatment with 10 mg of atorvastatin reached 29% reduction of LDLcholesterol levels in the Anglo-Scandinavian Cardiac Outcomes Trial-LipidLowering Arm (ASCOT-LLA) prospective controlled trial (Sever et al. 2003), and40% reduction in the Collaborative Atorvastatin Diabetes Study (CARDS)prospective controlled trial (Colhoun et al. 2004).
The third generation statins such as rosuvastatin have the highest potency toinhibit HMG-CoA reductase. Rosuvastatin has multiple sites of activity againstHMG-CoA reductase (Istvan and Deisenhofer 2001), and stronger interaction withthe enzyme (Carbonell and Freire 2005). In a multicenter, double-blind, placebo-controlled trial rosuvastatin produced a 50% reduction in LDL cholesterol levelswithin 12 weeks of therapy (Olsson et al. 2002). Similarly, in another studyrosuvastatin (10–40 mg) reduced LDL cholesterol levels by 47–57% at 6 weeks oftherapy (Schneck et al. 2003).
In the study comparing the lipid lowering effects of different statins (theSTELLAR* Trial), rosuvastatin reduced LDL-C by 46–55%, atorvastatin by 37–51%,simvastatin by 28–46%, and pravastatin by 20–30% (Jones et al. 2003). Statins alsoreduced the levels of total cholesterol and triglycerides and increased HDLcholesterol levels (Jones et al. 2003).
2.1.5 Statins and their cardiovascular risk lowering effectsLDL particles are highly atherogenic and LDL cholesterol is an importantcardiovascular risk factor as demonstrated in population-based studies (Wadhera etal. 2016, Wong et al. 2016, Hegele 2009, Hu et al. 2012). In men, an increase incholesterol concentration from 5.2 to 6.2 mmol/l was associated with a threefoldincreased risk of death from coronary artery disease (myocardial infarction) (Stamleret al. 2000).
LDL cholesterol plays a fundamental role in the development of atherosclerosis(Hegele 2009). In conditions of chronically elevated levels of plasma LDL cholesterol,LDL particles which are not catabolized through regulated LDL-receptor-mediatedendocytosis extravasate through the defective endothelium into the subendothelialspace of arterial wall (Falk 2006). Retained LDL particles become oxidized andgenerate toxic intermediates which induce inflammatory responses (Lusis 2000,Rader and Daugherty 2008). They are recognized by scavenger receptors ofmacrophages and engulfed (Lusis 2000, Rader and Daugherty 2008). Macrophagesloaded with LDL become foam cells and form atherogenic plaques characterized bythe accumulation of lipids, white blood cells and cell debris in the inner layer of the

8
arterial wall (Plakkal Ayyappan et al. 2016, Bobryshev et al. 2016). Atheroscleroticplaques may protrude into the lumen of the arteries, thus limiting blood flow to thetissue. Rupture of an atherosclerotic plaque may cause thrombosis and completelyblock the blood flow in the artery, resulting in cardiovascular disease such as CADor stroke (Bentzon et al. 2014, Plakkal Ayyappan et al. 2016).
Beside the cholesterol-lowering effect, statins also reduce CVD events andcardiac-related as well as overall mortality (Slater and MacDonald 1988). A numberof reports from rigorously performed randomized controlled trials includingpatients with varying degrees of CVD risk confirmed beneficial effects of statins onreducing the CVD risk (Collins et al. 2016).Pravastatin trialsPravastatin was shown to be beneficial in both primary and secondary prevention ofCVD. In the WOSCOPS trial including men with hypercholesterolemia, pravastatinlowered primary coronary events (specified as nonfatal myocardial infarction ordeath from coronary heart disease) by 31 % compared to placebo (Shepherd et al.1995). In the PROSPER trial including elderly men and women with, or at high riskof developing, CVD and stroke, pravastatin reduced cardiac mortality by 24% andcoronary events by 19%, compared to placebo (Shepherd et al. 2002). In theCholesterol and Recurrent Events Trial (CARE) including patients with a history ofmyocardial infarction or symptomatic coronary artery disease, pravastatin (40 mgdaily) reduced coronary events by 24% compared to placebo (Sacks et al. 1996).Similarly, in the Long-term Intervention with Pravastatin in Ischaemic Disease(LIPID) study including patients with a history of myocardial infarction orhospitalization for unstable angina, pravastatin (40 mg daily) reduced the risk ofcoronary heart disease by 24% compared to placebo during the follow-up period of6.1 years (LIPID Study Group 1998).
Simvastatin trialsSimvastatin reduced cardiac mortality by 42% and coronary events by 35%compared to placebo in the 4S trial, a secondary prevention trial including patientswith CHD (Pedersen et al. 1998). The authors estimated that each additional 1%reduction in LDL cholesterol reduced the risk of major coronary events by 1.7%(Pedersen et al. 1998). In the primary prevention trial, the Heart Protection Study(HPS), simvastatin (40 mg daily) reduced cardiac mortality by 18% and majorcardiovascular events by 24% (Heart Protection Study Collaborative Group 2002). Inthis trial, the proportional reduction in the event rate was significant even inindividuals with normal cholesterol levels (LDL cholesterol below 3.0 mmol/L ortotal cholesterol below 5.0 mmol/L) (Heart Protection Study Collaborative Group2002). High-dose simvastatin therapy (80 mg daily) was shown to be more effectivethan low dose therapy (20 mg daily) in reducing LDL cholesterol and cardiovasculardeath, but was also associated with higher rate of myopathy as an adverse effect inthe A to Z trial including patients with acute coronary syndrome (de Lemos et al.2005). In the Study of the Effectiveness of Additional Reductions in Cholesterol andHomocysteine (SEARCH) trial including 12,064 survivors of myocardial infarction,80 mg simvastatin produced a 6% proportional reduction in major vascular eventscompared to 20 mg simvastatin (SEARCH Collaborative Group 2010).Atorvastatin trialsIn the primary prevention trial ASCOT-LLA including hypertensive patientswithout dyslipidemia, atorvastatin reduced the incidence of total coronary events

9
(HR 0.71) compared with placebo (Sever et al. 2003). Atorvastatin (10 mg daily)reduced acute CHD events by 36%, coronary revascularisations by 31%, and rate ofstroke by 48% in another primary prevention trial, the CARDS, including patientswith T2D without high concentrations of LDL cholesterol, (Colhoun et al. 2004). Inthe Atorvastatin Study for Prevention of Coronary Heart Disease Endpoints in Non-Insulin Dependent Diabetes Mellitus (ASPEN) trial, including subjects with T2D andLDL cholesterol levels below contemporary guideline targets, atorvastatin (10 mgdaily) produced a non-significant reduction in the relative risk of fatal and nonfatalmyocardial infarction by 27% (19% without and 36% with prior myocardialinfarction or interventional procedure) (Knopp et al. 2006). High-dose atorvastatintherapy (80 mg daily) was shown to be more effective than low-dose atorvastatintherapy (10 mg daily), producing a 22% relative reduction in the risk of majorcardiovascular events in the Treating to New Targets (TNT) study including 10,001patients with clinically evident CHD and LDL cholesterol levels of less than 3.4mmol/l (LaRosa et al. 2005). In the Myocardial Ischemia Reduction with AcuteCholesterol Lowering (MIRACL) trial, high-dose atorvastatin therapy (80 mg daily)reduced recurrent coronary events by 16% (Schwartz et al. 2001). Finally, high-doseatorvastatin therapy was shown to be more effective in reducing CVD outcomescompared to 40 mg of pravastatin daily in the Pravastatin or Atorvastatin Evaluationand Infection Therapy (PROVE-IT trial (Cannon et al. 2004) and compared to 20 mgof simvastatin daily in the Incremental Decrease in Endpoints Through AggressiveLipid Lowering (IDEAL) trial (Pedersen et al. 2005).Other trialsBeneficial effects in the prevention of CVD events were reported also by theJustification for the Use of Statins in Prevention: an Intervention Trial EvaluationRosuvastatin (JUPITER) (Ridker et al. 2008) and the Controlled RosuvastatinMultinational Trial in Heart Failure (CORONA) (Rogers et al. 2014) trials forrosuvastatin, the Air Force/Texas Coronary Atherosclerosis Prevention Study(AFCAPS/TexCAPS) trial for lovastatin (Downs et al. 1998), and the LescolIntervention Prevention Study (LIPS) trial for fluvastatin (Serruys et al. 2002).
In the JUPITER trial including 17,802 apparently healthy persons withouthyperlipidemia but with elevated high-sensitivity C-reactive protein levels, animportant risk factor for CVD, rosuvastatin (20 mg daily) significantly reduced theincidence of major cardiovascular events by 44% compared with placebo (Ridker etal. 2008). In the CORONA trial including 5,011 patients with systolic heart failureresulting from ischemia, rosuvastatin (10 mg daily) was shown to reduce the risk ofhospitalizations for heart failure by 15- 20% compared with placebo (Rogers et al.2014).
In the primary prevention AFCAPS/TexCAPS trial, lovastatin (20-40 mg daily)reduced the risk for the first acute major coronary event in men and women with theaverage levels of total and LDL cholesterol but below-average levels of HDLcholesterol (Downs et al. 1998).
In the LIPS trial, fluvastatin (80 mg daily) significantly reduced the risk of majoradverse cardiac events in patients with average cholesterol levels undergoing theirfirst successful percutaneous coronary intervention, compared with placebo (Serruyset al. 2002).
In summary, the evidence from statin trials shows that statin treatment reducesthe risk of major vascular events by 25% for each mmol/l reduction in LDLcholesterol during each year of continuous treatment (Collins et al. 2016). The

10
absolute benefits of statin treatment depend on an individual´s absolute risk ofocclusive vascular events and the absolute reduction in LDL cholesterol that isachieved. For example, lowering LDL cholesterol by 2 mmol/l with an effective low-cost statin regimen (e.g. atorvastatin 40 mg daily) for 5 years in 10,000 patientswould typically prevent major vascular events from occurring in 1,000 patientswith pre-existing occlusive vascular disease and in 500 patients who are at risk buthave not yet had a vascular event (Collins et al. 2016).
Statins have also been evaluated for potential long-term adverse effects (Maningatand Breslow 2011, Cholesterol Treatment Trialists' Collaboration 2010, Kashani et al.2006, Cholesterol Treatment Trialists' Collaboration 2012, Preiss et al. 2011b). Theonly serious adverse events associated with long-term statin treatment are myopathy(an effective regimen may cause 5 cases of myopathy in 10,000 patients treatedduring 5 years), NODM (50-100 cases) and probably hemorrhagic stroke (5-10 cases)(Collins et al. 2016). Therefore, the risk of adverse events is low in absolute terms,and the cardiovascular benefits of statin therapy likely outweigh the risk even inlow-risk patients (Sattar et al. 2010, Ridker et al. 2012).
2.2 PREDIABETES AND TYPE 2 DIABETES
Although statins are very beneficial in primary and secondary prevention of CVD,they increase the risk of NODM. Evidence from clinical trials, meta-analysis studiesand observational studies shows that patients receiving statins have a 10 to 60%increased risk of NODM (Betteridge and Carmena 2016). The risk is higher withmore intensive treatment and in patients with known risk factors for NODM (Preisset al. 2011b, Waters et al. 2013).
Diabetes mellitus is a heterogeneous chronic metabolic disease characterized bychronic hyperglycemia and disturbances in several key metabolic pathways. T2D isthe most common form of diabetes mellitus, accounting for 80-90% of all cases ofdiabetes in Europe (Läll et al. 2016). T2D develops gradually from normal toabnormal glucose tolerance, through an intermediate stage called prediabetes (Kanatet al. 2015). Prediabetes is characterized by mildly increased levels of fasting glucoselevels in the range of 5.6 – 6.9 mmol/l (impaired fasting glucose), increased post-loadglucose levels in the range of 7.8 – 11.0 mmol/l (impaired glucose tolerance), or both,according to the criteria of the American Diabetes Association (American DiabetesAssociation 2013). Diabetes is diagnosed on the basis of fasting plasma glucose (FPG)
7.0 mmol/l, 2-hour plasma glucose (2hPG) in an OGTT 11.1 mmol/l, or HbA1c
6.5% (American Diabetes Association 2013).The prevalence of prediabetes and T2D is increasing world-wide and is
considered as an epidemic in some countries (Edwards and Cusi 2016, Wild et al.2004). It is expected that the number of individuals suffering from T2D will doublein the next decade and will create a great burden on healthcare systems all over theworld (Cornell 2015, Kahn et al. 2014). The epidemic of T2D is closely associatedwith its twin epidemic of obesity due to dramatic globalization of sedentary lifestyleand erroneous nutritional habits. Obesity and T2D are increasingly affecting alsochildren and adolescents (Forbes and Cooper 2013).
Prediabetes and T2D are associated with increased morbidity and mortality,mainly attributable to the long-term macrovascular and microvascular complicationsassociated with hyperglycemia (World Health Organization 2004, Edwards and Cusi

11
2016). Microvascular complications of diabetes include diabetic retinopathy (themost common cause of acquired loss of vision), diabetic nephropathy, which canlead to chronic renal failure, and peripheral (somatic) and autonomic neuropathies(Forbes and Cooper 2013). Major macrovascular complications include acceleratedCVD resulting in myocardial infarction, cerebrovascular disease manifesting asstrokes, and peripheral arterial disease (Forbes and Cooper 2013). Very often,patients with T2D have elevated blood pressure (King et al. 1999), atherogenicdyslipidemia characterized by elevated levels of total triglycerides and low levels ofHDL cholesterol (Szalat et al. 2016), and low-grade inflammation (Pandey et al.2015).
2.2.1 Pathophysiology of prediabetes and type 2 diabetesPrediabetes and T2D are caused by impaired secretion and action of insulin.Prediabetes may precede T2D by years, and converts to T2D when pancreas is nolonger able to increase insulin secretion in a manner sufficient to compensate forinsulin resistance in peripheral insulin sensitive tissues (Edwards and Cusi 2016).
Insulin is a peptide hormone produced by the -cells of pancreatic islets. Insulin isfirst synthesized as a 110 amino acids long polypeptide called pre-proinsulin. In theendoplasmic reticulum (ER) it is cleaved to form proinsulin, folded into correctconformation and 3 disulfide bonds are formed. Proinsulin is transported into thetrans-Golgi network where immature granules are formed (Hou et al. 2009).Proinsulin matures into active insulin with the help of cellular endopeptidasesknown as prohormone convertases (PC1 and PC2), as well as the exopeptidasecarboxypeptidase E (Fu et al. 2013). The mature insulin is packed inside secretorygranules, and upon metabolic signals it is exocytosed from the cell into thecirculation (Hou et al. 2009).
Insulin plays a key role in the metabolism of carbohydrates, lipids and proteins bypromoting the uptake of glucose from the blood into skeletal muscle, adipose andliver cells where the glucose is converted into glycogen via glygogenesis or intotriglycerides via lipogenesis (Le Roith and Zick 2001, Pirola et al. 2004). Insulincirculating in the blood affects the synthesis of proteins in different tissues, acting asan anabolic hormone (Sonksen and Sonksen 2000).
The causes of prediabetes and T2D are multi-factorial and include both geneticand environmental factors (such as high-caloric diet and sedentary lifestyle),affecting -cell function and tissue insulin sensitivity (Scheen 2003). Increasingevidence from clinical, epidemiological and genetic studies indicates that -cellfailure resulting in an inappropriately low insulin secretion to glucose stimulus, andinsulin resistance are the primary defects leading to type 2 patients (AmericanDiabetes Association 2015, Morris et al. 2012).
2.2.1.1 Insulin secretion and its regulationInsulin is released from the pancreatic -cells in order to maintain normal glucosehomeostasis. -cells are the major cell type in the islets of pancreas, and the islets arescattered throughout the pancreas making approximately 1% of the total volume ofthe gland (Jiang and Morahan 2016, Fu et al. 2013). Insulin is released from -cells intwo phases. The first phase of insulin secretion is a quick release of readily availableinsulin granules which is triggered in response to increased blood glucose levels andlasts for about 10 minutes. Reduced first-phase insulin release is the hallmark of

12
T2D. The second phase of insulin secretion is independent of glucose, lasts usually 2to 3 hours where the newly formed vesicles are released slowly (Seino et al. 2011).
The most important physiological secretagogue for insulin is glucose. Thebiphasic profile of glucose stimulated insulin secretion (GSIS) from pancreatic -cellsinvolves two major signaling pathways (Röder et al. 2016a): 1) the triggeringpathway which is primarily involved in the first phase of insulin secretion throughthe canonical K+ ATP sensitive channel (KATP)-dependent pathway, and 2) theamplifying pathway, which primarily maintains the second phase of insulinsecretion and is classified into: 2a) the metabolic amplifying pathway mediated byglucose, and 2b) the neurohormonal amplifying pathway mediating the effects ofneurotransmitters and hormones. The triggering and amplifying pathways arehierarchical and not completely independent. In the absence of triggering signal (byglucose or another stimulus), the amplification pathway of -cells is functionallysilent. The triggering and amplifying pathways of insulin secretion in -cells areshown in Figure 4.
Figure 4. Regulation of glucose-stimulated insulin secretion by nutrients, hormones andneurotransmitters. Glucose-stimulated insulin secretion may be modulated by severalmechanisms. Glucose metabolism increases the ATP/ADP ratio and closes ATP-sensitivepotasium channels (KATP), depolarizing the membrane, opening voltage-dependentcalcium channels (VGCC), and thus increasing intracellular calcium ([Ca2+]i). Glucosemetabolism by the Krebs cycle also renders a series of metabolic coupling factors thatmay initiate and sustain insulin secretion. These metabolic coupling factors participate inmitochondrial shuttles, involving NADPH, pyruvate, malate, citrate, isocitrate, acyl-CoAs,and glutamate. Signaling pathways that contribute to maintaining or increasing glucose-stimulated insulin secretion include PKA and PKC. Glucagon, glucagon-like peptide 1

13
(GLP-1), and glucose-dependent insulinotropic peptide (GIP) act through the PKApathway, while acetylcholine and cholecystokinine act through the PKC pathway. Fattyacids may contribute to insulin secretion through the PKC pathway through formation ofdiacylglycerol (DAG) or through protein acylation. Amino acids may stimulate insulinrelease by increasing ATP production from the Krebs cycle, by membrane depolarization,or by participating in intracellular calcium increase. ( KG: alpha-ketoglutarate, ACC:acetyl CoA carboxylase, FAS: fatty acid synthase, GDH: glutamate dehydrogenase, GTP-SCS: GTP-succinyl CoA synthetase, ER: endoplasmic reticulum, ME: malic enzyme, MDH:malate dehydrogenase, PC: pyruvate carboxylase, PHD: pyruvate dehydrogenase, PIP2:phosphatidylinositol 4,5-bisphosphate, IP3: inositol 1,4,5-trisphosphate) (Lazo-de-la-Vega-Monroy and Fernandez-Mejia 2011).
The triggering classical pathway of the GSIS involves the following steps (Röderet al. 2016a, Henquin 2011, Wortham and Sander 2016). First the glucose enters intothe -cell through facilitated diffusion via the GLUT2 transporter. Then glucoseenters glycolysis and mitochondrial metabolism, which increases the ATP levelsinside the -cell. This induces the closure of KATP channels in the plasma membrane.The closure of KATP channels leads to the depolarization of the plasma membraneand opening of the voltage-dependent calcium channels (VGCC) allowing calcium(Ca2+) influx. This results in the raise of cytoplasmic Ca2+ which then triggers insulinexocytosis (Herrington et al. 2006).
The metabolic amplifying pathway. Glucose also activates a metabolicamplifying pathway that augments the amount of released insulin without theinvolvement of KATP channels and without further increasing Ca2+ concentration. Thispathway is also operative during glucose-potentiation of insulin secretion inducedby non-metabolized secretagogues (arginine or sulfonylureas) (Yokoi et al. 2016,Henquin 2011) in addition to stimulation of insulin secretion by glucose alone. Thecellular mechanisms underlying the metabolic amplifying pathway are still largelyelusive (Henquin 2011).
Neurohormonal amplifying pathways are activated by the binding ofneurotransmitters and hormones to receptors in the -cell membrane to potentiatenutrient-induced insulin secretion (Ahren 2009). There are numerous hormones andneurotransmitters that have receptor binding sites in the -cell plasma membrane,and the majority of these receptors belongs to the superfamily of G protein coupledreceptors (GPCRs) (Ahren 2009). The most important neurohormonal amplifyingpathways are those mediated by the parasympathetic nervous system (mediated byacetylcholine) and by incretins (e.g. GLP-1) during meals (Yokoi et al. 2016, Röder etal. 2016b).
Acetylcholine is the major neurotransmitter in pheripheral parasymphatheticnerves. Acetylcholine has a stimulatory effect on insulin secretion particularlyduring the preabsorptive phase of feeding, accompanied by an increase in theactivity of efferent vagal nerves (Ganic et al. 2016). M3 muscarinic acetylcholinereceptor (M3R) is a Gq-coupled GPCR expressed in pancreatic -cells (Yokoi et al.2016). Binding of acetylcholine to M3Rs triggers a sequence of biochemical events(Fridlyand and Philipson 2016, Yokoi et al. 2016), including the stimulation ofdistinct isoforms of phospholipase C (PLC). Activated PLC catalyzes thebreakdown of phosphatidylinositol 4,5-bisphosphate (PIP2), generating two secondmessengers diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG isimportant for the activation of different isoforms of protein kinase C (PKC), and the

14
activated PKC increases the effect of free intracellular calcium [Ca2+]i concentrationon exocytosis of insulin granules (Ruiz de Azua et al. 2011). IP3 binds to IP3receptors of the ER to stimulate the release of Ca2+ from ER stores which results inrapid elevation of [Ca2+]i. The increase in the levels of [Ca2+]i and the PKC mediatedeffects on exocytosis are the two main insulinotropic effects of acetylcholine(Fridlyand and Philipson 2016, Yokoi et al. 2016). In addition, the acetylcholinepathway also plays a role in other cellular activities involving the activation ofinward Na+ current which depolarizes the membrane and facilitates calcium influx,which likely facilitates insulin release (Swayne et al. 2009, Ruiz de Azua et al. 2011).
Glucagon-like peptide 1 receptor (GLP-1R) is a GS-coupled GPCR expressed in -cells, and its activation with GLP-1 or agonists such as exenatide facilitate GSIS(Fridlyand and Philipson 2016, Yokoi et al. 2016). GLP-1 is a peptide hormone fromthe glucagon family, produced by L cells of the intestine in response to the presenceof nutrients in the lumen of the small intestine. Activation of the GLP-1R in the -cellactivates plasma membrane bound adenylate cyclase, which catalyzes theconversion of ATP to produce cAMP (Fridlyand and Philipson 2016). Increasedproduction of cAMP activates two signaling pathways: a protein kinase A (PKA)pathway and a cAMP-regulated guanine nucleotide exchange factor (cAMP-GEF orEPAC2) pathway (Campbell and Drucker 2013, Yamada et al. 2016, Meloni et al.2013). EPAC2 functions as a guanine nucleotide exchange factor for small RAS-likeG-proteins and regulates their activity (Almahariq et al. 2014, Meloni et al. 2013), andalso regulates KATP channel activity by facilitating inhibition of the channel throughATP binding (Shibasaki et al. 2014). PKA phosphorylates SUR1 subunits of the KATP
channels, disrupts the binding of ADP and leads to KATP channel closure anddepolarization of the cell (Meloni et al. 2013). Thus the activities of both PKA andEPAC2 result in a coordinated potentiation of glucose-initiated cell membranedepolarization, facilitating the next step in the cascade towards insulin secretion, theopening of VGCC (Meloni et al. 2013). This increases the magnitude of the inwardcalcium current produced by VGCCs and stimulates insulin secretion (Meloni et al.2013). Furthermore, activation of GLP-1R via both PKA and EPAC2 participates incalcium induced calcium release from intracellular stores such as the ER andsecretory granules (Graves and Hinkle 2003, Kang et al. 2005), which also enhancesgranule exocytosis and insulin secretion. The release of calcium from intracellularstores is controlled by inositol trisphosphate receptor (IP3R) and the ryanodinereceptor (RyR) (Röder et al. 2016a, Meloni et al. 2013). Finally, the activation of PKAand EPAC2 through GLP-1R modulates the refilling of the readily-releasable pools,an event important for the second phase of insulin secretion and granular priming(Röder et al. 2016a, Meloni et al. 2013).
GPR119 (glucose-dependent insulinotropic receptor) belongs to the group of Gs-coupled receptors, similarly to GLP-1R. It is a lipid-responsive GPCR activated bysome lysophospholipids that contain well-defined fatty acids at the C-1 position (Im2013, Oh da and Olefsky 2016), and is expressed in -cells and in gastrointestinalenteroendocrine cells (Parker et al. 2009, Chu et al. 2007, Oh da and Olefsky 2016).Similarly to GLP-1R, activation of GPR119 stimulates insulin secretion in a glucose-dependent manner by stimulation of adenylate cyclase and an increase inintracellular cAMP levels (Chu et al. 2007, Yoshida et al. 2010, Oh da and Olefsky2016). In addition to the direct stimulation of insulin secretion, activation of GPR119can augment insulin secretion also indirectly via multiple pathways: through GLP-1by stimulating its biosynthesis and secretion from the intestinal L cells (Chepurny et

15
al. 2016, Ekberg et al. 2016, Oh da and Olefsky 2016, Lan et al. 2012), and by thestimulation of -cell regeneration (Ansarullah et al. 2013).
GPR40, also known as free fatty acid receptor 1, is highly expressed in pancreatic-cells (Briscoe et al. 2003, Itoh et al. 2003). It is a Gq-coupled protein receptor
belonging to the same family of GPCRs as the acetylcholine receptors (Blad et al.2012), and its ligands are unsaturated and saturated medium- or long-chain free fattyacids such as oleic and linoleic acid. GPR40 can mediate fatty-acid-inducedenhancement of GSIS from pancreatic -cells (Itoh et al. 2003, Tomita et al. 2006), andthis effect has been observed only in the presence of elevated glucose levels(Hamdouchi et al. 2016). Activation of GPR40 increases cytosolic calciumconcentrations via phospholipase C (PLC) and L-type Ca2+ channel-mediatedpathway (Fujiwara et al. 2005, Zhou et al. 2012, Yamada et al. 2016, Shapiro et al.2005, Yang et al. 2010), and may increase cAMP concentrations according to some(Hauge et al. 2014, Kotarsky et al. 2003, Gromada 2006, Feng et al. 2006) but not all(Yamada et al. 2016, Song et al. 2013, Welters et al. 2006, Yang et al. 2010) reports.Furthermore, the activation of GPR40 inhibits the opening of voltage-gated K+
channels which in turn facilitates the increase of calcium influx through L-typecalcium channels, thereby augmenting GSIS (Feng et al. 2006). Although themechanism of GPR40-stimulated insulin secretion is similar to that of cholinergicstimulation of insulin release via the M3R (Gilon and Henquin 2001, Mancini andPoitout 2013), FFA activation of GPR40 leads predominantly to influx of extracellularCa2+ with minimal cytosolic ingress from ER Ca2+ stores (Mancini and Poitout 2013,Fujiwara et al. 2005, Zhao et al. 2008) in contrast to acetylcholine, which actsprimarily through IP3-mediated Ca2+ release from the ER (Mancini and Poitout 2013,Vettor et al. 2008). In addition to pancreatic -cells, GPR40 is also expressed in theenteroendocrine cells of the gastrointestinal tract, where upon activation itstimulates the secretion of incretins such as GLP-1 and glucose-dependentinsulinotropic polypetide (GIP) (Hamdouchi et al. 2016, Edfalk et al. 2008, Xiong etal. 2013). Therefore, GPR40 has the potential to modulate insulin secretion bothdirectly in pancreatic -cells and indirectly through the regulation of incretinsecretion.
Cholesterol biosynthesis pathway and insulin secretionThe HMG CoA reductase inhibitors not only inhibit the cholesterol biosynthesis butalso decrease the synthesis of isoprenoid intermediates which are derived frommevalonate (Cerqueira et al. 2016, Mullen et al. 2016, Wood et al. 2014), such asGGPP and FPP. Many proteins which interact with membrane-bound receptorsundergo posttranslational modifications by isoprenoids (Wang and Casey 2016,Mullen et al. 2016, Hooff et al. 2010). These include heterotrimeric G-proteins andsmall G-proteins belonging to the family of Ras, Rho, Rap, and Rab GTPases, whichplay roles in various steps needed for insulin secretion (Kowluru 2010). In general,modification with FPP is necessary for proper localization of Ras family proteins,whereas GGPP is required for Rho, Rab, and Rap family proteins (Tsuchiya et al.2010), although some Rho GTPases require both farnesylation andgeranylgeranylation for proper function and intracellular localization (Rikitake andLiao 2005). The small GTPases are important for mobilization of insulin granules andtheir fusion to the plasma membrane (e.g., Rap1, Rac1, and Cdc42), they are involvedin vesicle docking, cytoskeletal remodeling, and granule priming and fusion(Kowluru and Kowluru 2015).

16
2.2.1.2 Insulin resistanceInsulin resistance can be defined as an attenuated biological response to normal orelevated plasma levels of insulin (Cefalu 2001). Insulin resistance translates intoimpaired insulin-mediated glucose disposal in insulin-sensitive tissues, such asskeletal muscle, liver and adipose tissue. Additionally, insulin resistance in theskeletal muscle leads to a decline in muscle glycogen synthesis. In the insulinresistant liver, insulin fails to suppress gluconeogenesis, but continues to stimulatefatty acid synthesis. In adipose tissue, insulin resistance leads to impaired inhibitionof lipolysis (Hardy et al. 2012).
There are numerous causes and mechanisms of insulin resistance. In rare cases,the cause is genetic, but in most cases (including T2D), insulin resistance is triggeredby cellular perturbations, such as lipotoxicity, inflammation, glucotoxicity,mitochondrial dysfunction, and ER stress (Boucher et al. 2014), leading to post-receptor defects in insulin signaling. Such defects may include down-regulation ordeficiencies of the insulin receptor, IRS proteins, or PI3K, and abnormalities ofglucose transporter 4 (GLUT4) function (Wilcox 2005). Several environmental andphysiological factors can contribute to insulin resistance, such as diet, stress,immobilization, obesity, sleep deprivation, pregnancy, exercise and physical activity(Sah et al. 2016). Insulin resistance often co-exists with obesity (Moscavitch et al.2016), and clusters with several metabolic cardiovascular risk factors, commonlycalled the metabolic syndrome (Reaven 2004).
Insulin resistance typically precedes the development of diabetes and iscommonly found in non-diabetic first-degree relatives of diabetic patients (Vaag etal. 1992). Insulin resistance is associated with compensatory hyperinsulinemia,whereby the pancreas compensates for impaired insulin action by secretingincreased amounts of insulin to maintain normal glucose levels (Sung et al. 2016).
Insulin signaling pathwayThe following chapter will describe insulin signaling pathway in the skeletal muscle.However, the early signaling events are common to all insulin sensitive tissues. Theinsulin signaling pathway, regulation of glucose uptake and glycogen synthesis in askeletal muscle cell are shown in Figure 5.
The insulin signaling pathway starts with binding of insulin to the insulinreceptor (IR). Insulin receptor is a heterotetrameric glycoprotein with twoextracellular -subunits and two transmembrane subunits with tyrosine kinaseactivity. Insulin first binds to the subunit, which activates the intrinsic kinaseactivity in the subunit of IR. The activation of one subunit creates a trans-autophosphorylation reaction in which tyrosine phosphorylation of one subunitphosphorylates the adjacent subunit. The phosphorylated IR specifically interactswith insulin receptor substrate (IRS) family of proteins (mainly IRS1) andphosphorylates IRS at a number of tyrosine residues (Sesti et al. 2001, DeFronzo andTripathy 2009, Sah et al. 2016). IRS1 can also be phosphorylated on its serineresidues. While the importance of tyrosine phosphorylation of IRS1 in insulinsignaling is undisputed, serine phosphorylation is subjected to complex regulation,as several pathways and kinases can phosphorylate IRS1 at serine residues,including JNK1 (c-Jun N-terminal kinase 1), mTOR (mammalian target ofrapamycin) - S6K1 (ribosomal protein S6 kinase beta-1) kinase pathway, GRK2 (Gprotein-coupled receptor kinase 2) and some PKC isoforms (Copps and White 2012).IRS1 serine phosphorylation has been linked to both insulin resistance (Copps and

17
White 2012) and insulin sensitivity (Boucher et al. 2014, Luo et al. 2007, Weigert et al.2008, Paz et al. 1999, Danielsson et al. 2005, Giraud et al. 2004).
Figure 5. Insulin signaling pathway in the skeletal muscle cell (Gonzalez-Franquesa et al.2012).IRS, insulin receptor substrate; S, serine; Y, tyrosine; PH, pleckstrin homology domainof the IRS1; SHC, Src homology 2 domain; GRB2, growth factor receptor-bound protein2; ERK, extracellular-signal-regulated kinases or classical MAP kinases; PIP2,phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate;PDK1, phosphoinositide-dependent protein kinase 1; Akt, Akt kinase; mTOR, mammaliantarget of rapamycin; GSK-3, glycogen synthase kinase 3; GS, glycogen synthase;AS160, 160 kDa Akt substrate; GDP, guanosine diphosphate; GTP, guanosinetriphosphate; aPKC, atypical protein kinase C.
The critical pathway linking the IRS proteins to the metabolic actions of insulin is thePI3K and Akt pathway. The PI3Ks (class I) are heterodimers consisting of aregulatory and a catalytic subunit, each of which occurs as several isoforms (Vadaset al. 2011). Two SH2 domains in the regulatory subunits of PI3K bind to tyrosinephosphorylated IRS1/2, which leads to the recruitment of PI3K at the plasmamembrane (Boucher et al. 2014, Shaw 2011). This results in the activation of thecatalytic subunit of PI3K, which rapidly phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to generate the lipid second messenger phosphatidylinositol3,4,5-trisphosphate (PIP3). PIP3 activates a serine/threonine phosphorylation cascadeof pleckstrin homology (PH)-domain containing proteins, such as phosphoinositidedependent protein kinase-1 (PDK1), the serine/threonine protein kinase Akt, and theatypical PKC and isoforms (Boucher et al. 2014, Standaert et al. 2001,Bandyopadhyay et al. 1997).

18
Akt is a central molecule for many cellular and metabolic functions (HäggbladSahlberg et al. 2016, Poloz and Stambolic 2015, Mackenzie and Elliott 2014). The Aktfamily of proteins consists of three different isoforms of serine/threonine proteinkinases encoded by different genes (Schultze et al. 2011). All isoforms possess a PHdomain, allowing interaction with PIP3 and recruitment to the plasma membrane.After recruitment to the plasma membrane, Akt is phosphorylated and activated(Mora et al. 2004) by two further kinases, PDK1 and mTOR complex 2, within the T-loop of the catalytic domain (Thr308) and the carboxyl terminal hydrophobic domain(Ser473), resulting in phosphorylation of many downstream targets involved incellular growth and metabolism (Noguchi et al. 2014, Zoncu et al. 2011, Zinzalla et al.2011, Gao et al. 2014, Mackenzie and Elliott 2014).
The major targets of activated Akt mediating metabolic functions are glycogensynthase kinase 3 (GSK-3) (Chen et al. 2016, Mackenzie and Elliott 2014, Bond 2016,Lai et al. 2012) and the Akt substrate of 160 kDa (AS160) (Guridi et al. 2016, Shao andTian 2015, Mik osz et al. 2016, Quan et al. 2015, Lai et al. 2012). AS160, also known asTBC domain family member 4 (TBC1D4), is a factor in Akt-induced GLUT4translocation in skeletal muscle. AS160 acts as a GTPase activating protein (GAP)which maintains Rab-GTPase(s) in an inactive form by inducing their conversion tothe GDP-bound form, thereby retaining GLUT4 within storage vesicles (Quan et al.2015, Lai et al. 2012). Rab proteins are critical organizers of intracellular membranetrafficking (Zerial and McBride 2001). Activated Akt phosphorylates AS160, leadingto a reduction in Rab-GTP activity, promoting GLUT4 translocation to plasmamembrane and glucose uptake (Howlett et al. 2008, Ishikura and Klip 2008, Tan et al.2012, Miinea et al. 2005, Sano et al. 2003). Thus, any defects in the PI3K/Akt/AS160transduction pathway would ultimately reduce glucose uptake in skeletal muscle(Mik osz et al. 2016, Consitt et al. 2013, Klip et al. 2014, Lansey et al. 2012, Vind et al.2011). However, AS160 knockdown was shown to only partially release the pool ofintracellular GLUT4 mobilized by insulin, suggesting that other unknown Aktsubstrate proteins must make major contributions to overall GLUT4 regulation byinsulin (Lansey et al. 2012, Osorio-Fuentealba et al. 2013, Bai et al. 2007). aPKCs alsoplay role in controlling GLUT4 translocation, in addition to Akt (Nishizaki et al.2016, Shao and Tian 2015).
Out of the three Akt isoforms, Akt2 is most abundant in insulin-sensitive tissuesand seems to play a predominant role in controlling GLUT4 trafficking in adipose(Takenaka et al. 2016, Xu et al. 2016) and muscle cells (Gonzalez et al. 2011, Consitt etal. 2013, Jain et al. 2015, Mik osz et al. 2016) as well as in mediating insulin signalingto control glucose output in liver (Han et al. 2016, Pauta et al. 2016, Teixeira et al.2016, Perry et al. 2014). Akt1 isoform appears to control cell and body size (Miao etal. 2016, Shearin et al. 2016, Wittenberg et al. 2016) and Akt3 controls brain size(Miao et al. 2016, Easton et al. 2005). Akt2 knock-out mice are insulin resistant anddevelop diabetes (Cho et al. 2001), whereas Akt1 and Akt3 knock-out mice do not(Bunner et al. 2014, Mackenzie and Elliott 2014, Guo 2014, Boucher et al. 2014, Choet al. 2001). Furthermore, hepatic deletion of the Akt1 and Akt2 isoform in micecauses glucose intolerance, insulin resistance, and a defective transcriptionalresponse to feeding in the liver (Lu et al. 2012). In humans, a mutation in the geneencoding Akt2 results in severe insulin resistance (George et al. 2004), establishingAkt2 as a key protein in the maintenance of euglycemia (Thauvin-Robinet et al. 2013,Hussain et al. 2011, Wan et al. 2011, Garofalo et al. 2003).

19
Besides the metabolic functions, Akt also mediates other cellular events such ascellular growth, apoptosis and protein synthesis, and controls the expression ofseveral genes (Boucher et al. 2014, Thauvin-Robinet et al. 2013).
GLUT4 is a product of the gene SLC2A4 and functions as a facilitative glucosetransport protein. GLUT4 comprises 12 transmembrane domains. Characteristicsequences in the COOH- and NH2-terminal domains of GLUT4 are importantdeterminants of its intracellular localization and trafficking (Huang and Czech 2007).GLUT4 is most abundantly expressed in adipose tissue, cardiac and skeletal muscle(Mueckler 2001, Maria et al. 2015, Richter and Hargreaves 2013, Sylow et al. 2016).
Activation of the insulin signaling pathway in skeletal muscle leads totranslocation of a large intracellular pool of GLUT4 (associated with low-densitymicrosomes) to the plasma membrane and their subsequent activation after insertioninto the cell membrane (Lee-Young et al. 2016, Jensen et al. 2014, Leto and Saltiel2012, Sylow et al. 2016). The Rab GAPs TBC domain family member 1 (TBC1D1) andAS160 play important roles in this insulin-stimulated translocation of GLUT4, and anew substrate Rab28 for the GAP domains of both TBC1D1 and AS160 has beenidentified recently (Zhou et al. 2016).
The decrease in GLUT4 expression or decreased translocation from the cytoplasmto the plasma membrane causes a decrease in glucose uptake, which leads to insulinresistance and T2D (Xu et al. 2015, Olson 2012, Khalique et al. 2016). Glucosetransport activity was found to be severely impaired in insulin resistant individualswith T2D (Leto and Saltiel 2012, Goodpaster et al. 2014), and mouse models withdisrupted GLUT4 in muscle or adipose tissue show insulin resistance and glucoseintolerance (Postic et al. 2004, Herman and Kahn 2006, Abel et al. 2001, Minokoshi etal. 2003, Zisman et al. 2000, Li et al. 2000). Furthermore, low levels of GLUT4 due todefective AS160 in skeletal muscle also caused postprandial hyperglycemia andhyperinsulinemia in the AS160R917K mutant mice (Xie et al. 2016). These findingsconfirm the major role of GLUT4 in glucose disposal in insulin sensitive tissues.
Glucose metabolism. After glucose is transported into the cell, the metabolicpathway of glycolysis converts glucose first into glucose-6-phosphate (G6P) by anenzyme hexokinase, and finally into pyruvate via a series of intermediate metabolites(Han et al. 2016, Jensen et al. 2011, Li et al. 2015, Lunt et al. 2011). Pyruvate enters themitochondria where it is converted into acetyl-CoA by the enzyme pyruvatedehydrogenase complex, a process called pyruvate decarboxylation (Han et al. 2016,Lunt et al. 2011). The acetyl-CoA is the starting point for the tricarboxylic acid (TCA)cycle, in which acetyl-CoA is coverted to citrate (Lunt et al. 2011).The TCA cycle releases stored energy through the oxidation of acetyl-CoA derivedfrom carbohydrates, fats and proteins into carbon dioxide, and chemical energy inthe form of adenosine triphosphate (ATP) (Han et al. 2016, Li et al. 2015, Lunt et al.2011). Alternatively, G6P can be converted into glucose-1-phosphate for thesynthesis of glycogen. Glycogen is a readily mobilized storage form of glucose,which can be stored in the liver or skeletal muscle (Han et al. 2016, Jensen et al. 2011,Li et al. 2015). Glycogen synthesis is regulated by the enzyme glycogen synthase(GS) (DeFronzo and Tripathy 2009). At low plasma insulin concentration, forexample in fasting, glycogen synthesis and glucose oxidation contribute equally toglucose disposal. However, with increasing plasma insulin concentration, GS isactivated by insulin, and glycogen synthesis accounts for 70% of glucose disposal(Yang and Yang 2016, Jensen et al. 2011). The predominant site of glycogen synthesisis the skeletal muscle (Adeva-Andany et al. 2016, Jensen et al. 2011). Impaired

20
glycogen synthesis is considered to be one of the earliest metabolic defects seen inthe pathogenesis of T2D (Xirouchaki et al. 2016, Wood and O'Neill 2012).
GSK-3 is a serine/threonine kinase required for the regulation of glycogenmetabolism. Two isoforms of human GSK-3, with molecular weights of 51 kDa and46 kDa have been identified (Pandey and DeGrado 2016). Both isoforms share 85%homology at the amino acid level and are expressed ubiquitously, but functionaldifferences between them are still unclear (Nikoulina et al. 2000, McCubrey et al.2014, Maqbool et al. 2016, Carnagarin et al. 2015, Takahashi-Yanaga 2013).Unphosphorylated GSK-3 is active and inhibits the activity of its substrate GS. Aktactivated by insulin phosphorylates GSK-3 on Ser9, thus inactivating GSK-3 (Carnagarin et al. 2015, Copps and White 2012), the inhibitory phosphorylation ofGS is relieved, and the activated GS promotes glycogen synthesis (Jensen andRichter 2012, Jensen et al. 2011, Pandey and DeGrado 2016). Studies in cells over-expressing GSK-3 have shown that twofold activation of GSK-3 is sufficient toinhibit GS (Nikoulina et al. 2000). It has been shown that the activity of GS is reducedin T2D (Qi et al. 2016, Beck-Nielsen 2012, Prats et al. 2009, Pedersen et al. 2015), andGS mRNA and protein expression may be reduced in muscle from diabetic subjects(Park et al. 2000, Motoyama et al. 2003, Gaster et al. 2002, Huang et al. 2000). A novelinhibitor of GSK-3 has been shown to have glucose-lowering and anti-diabeticeffects in three animal models (Kim et al. 2015).
Insulin signaling in the liverThe main role of insulin in the liver is to suppress hepatic glucose production (HGP)when serum glucose levels are high. In the liver, insulin activates IR, whichphosphorylates IRS1 and IRS2, leading to activation of PI3K and ultimately Akt2.Activation of Akt2 promotes glycogen synthesis and inhibits gluconeogenesis(Zhang and Liu 2014). Insulin achieves these effects by directly regulating theactivities of several enzymes (e.g. GSK-3), or indirectly by regulating the expressionof genes encoding hepatic enzymes such phosphoenolpyruvate carboxylase, whichis the rate-limiting enzyme in gluconeogenesis, glucose 6-phosphatase, fructose 1,6-bisphosphatase, glycolytic enzymes (glucokinase and pyruvate kinase), andlipogenic enzymes (fatty acid synthase, acetyl CoA carboxylase) (Saltiel et al. 2016).Various transcription factors are involved in this regulation, such as hepatic nuclearfactor 3 (HNF-3), HNF-4, SREBP-1c, forkhead transcription factor FOXO1, and PGC-1 (Yoon et al. 2001). Defects in insulin signaling and action in the liver lead toincreased HGP and impaired glucose disposal contributing to hyperglycemia(Zhang and Liu 2014), whereas insulin continues to stimulate fatty acid synthesis(Brown and Goldstein 2008).
Insulin signaling in the adipose tissueAdipose tissue is an important energy storage organ as well as an endocrine organ.Insulin stimulates the uptake of glucose into adipocytes by up-regulation of GLUT4,and then glucose is converted to lipids by activation of lipid synthetic enzymes suchas pyruvate dehydrogenase, fatty acid synthase, and acetyl CoA carboxylase. Insulinalso inhibits lipolysis in adipocytes through several mechanisms (Zhang and Liu2014) including inhibition of phosphodiesterase 3b mediated by Aktphosphorylation, leading to reduced intracellular cAMP levels and PKA activity(Ahmad et al. 2009), Akt-dependent PKA-mediated inhibition of hormone-sensitivelipase (Choi et al. 2010), and Akt-independent PKA-mediated phosphorylation of a

21
lipid droplet-associated substrate perilipin (Choi et al. 2010). Insulin resistance inadipose tissue is manifested by impaired insulin-stimulated glucose transport andimpaired inhibition of lipolysis (Hardy et al. 2012).
Expansion of visceral (but not subcutaneous) adipose tissue in obesity results ininsulin resistance in other tissues and increased risk of T2D through severalmechanisms (Hardy et al. 2012). Visceral adipocytes secrete adipose-specificcytokines (such as leptin) and inflammatory cytokines (such as tumor necrosis factor
and interleukin 6 (IL-6) which are delivered via the portal vein to the liver and cancause liver and systemic insulin resistance (Rytka et al. 2011). Excess lipid contentcan result in insulin resistance of enlarged adipocytes (Lee et al. 2011). Excess fattyacids released from insulin resistant adipose tissue can lead to their ectopicaccumulation in liver and muscle, resulting in their insulin resistance via theformation of metabolically toxic products such as ceramides (Holland et al. 2011).
Plasma membrane cholesterol and insulin resistance of adipocytesOne of the causes of insulin resistance of adipose cells is the reduction in cholesterolcontent of their plasma membrane which increases its fluidity (Pilon 2016, Paniagua2016). The reduction of cholesterol in the plasma membrane increases the activationof SREBP2 and its target genes involved in cholesterol homeostasis, and increases theadipocytic factors such as IL-6, tumor necrosis factor and angiotensinogen (vanHarmelen et al. 2000, Hotamisligil et al. 1993, Fried et al. 1998). However, it was alsoshown that the depletion of plasma membrane cholesterol in 3T3-L1 cells leads todecreased gene and protein expression of GLUT4 (Le Lay et al. 2001) and decreasestyrosine-specific phosphorylation of IRS1 in response to insulin, thus affecting theinsulin signaling pathway (Betteridge and Carmena 2016).
2.3 DIABETOGENIC EFFECTS OF STATINS
Statins are well tolerated with a low prevalence of adverse effects (Collins et al.2016). In spite of that new data have emerged linking statin therapy to an increasedrisk of NODM.
2.3.1 Evidence for the diabetogenic effects of statins in population studiesIn the Women's Health Initiative study including 153,840 postmenopausal women(50-79 years), statin use at baseline was associated with an increased risk of self-reported NODM by 48% over 1,004,466 person-years of follow-up, and thisassociation was observed for all types of statins (Culver et al. 2012).
In the general population of Taiwan including 42,060 men ( 45 years) and women( 55 years) from the Taiwan National Health Insurance Research Database, annualrates of diabetes were significantly higher in statin users than in non-users (2.4% vs.2.1%, p<0.001) over a median of 7.2 years among non-diabetic individuals at thecohort entry (Wang et al. 2012).
In a retrospective cohort study performed using the Irish national pharmacyclaims database including 239,628 patients newly treated with statins, statin use wasassociated with an increased risk of NODM by 18% (41% for rosuvastatin, 23% foratorvastatin and 15% for simvastatin, no significant effect of fluvastatin andpravastatin) (Zaharan et al. 2013). The authors observed a significant linear

22
association between NODM and the duration and cumulative dose of the treatmentwith all types of statins except for fluvastatin which demonstrated only anassociation of NODM with the duration of treatment (Zaharan et al. 2013).
In a Canadian population study based on prescription records of 471,250 newusers of statins without diabetes ( 66 years), atorvastatin, rosuvastatin andsimvastatin were associated with a 22%, 18% and 10% increased risk of NODMcompared with the reference drug pravastatin over the 14-year study period (Carteret al. 2013). There was no significantly increased risk of diabetes among people whoreceived fluvastatin or lovastatin treatment (Carter et al. 2013). The absolute risk forNODM was about 31 and 34 events per 1000 person years for atorvastatin androsuvastatin, compared with 26 per 1000 person years for pravastatin (Carter et al.2013).
In an Italian health-care utilization database including 115,709 individualswithout diabetes at entry (40-80 years), statin treatment was associated with a 12-32% increased risk of NODM during a 6.4 year follow-up, depending on theadherence with the treatment (Corrao et al. 2014). There was a significant associationof an increased risk of diabetes with higher adherence to statin therapy (Corrao et al.2014).
A meta-analysis of eight population cohorts based on the administrativedatabases from six Canadian provinces and two international databases from the UKand US, including 136,966 patients newly treated with statins for secondaryprevention of CVD (aged 40 years, follow-up from 1997 to 2011), evaluated anincrease in NODM from higher potency statins compared with lower potency statins(Dormuth et al. 2014). It showed that higher potency statins (rosuvastatin 10 mg,atorvastatin 20 mg, and simvastatin 40 mg) were associated with a 15% increase inthe risk of NODM, compared with lower potency statins (Dormuth et al. 2014).
A Dutch prospective cohort study including 4,645 patients with establishedvascular disease without T2D at baseline (age 18-80 years, follow-up from 1996 to2011) investigated whether an increased risk of NODM with statin treatment onlyapplies to patients at high risk of T2D or for all patients (van de Woestijne et al.2015). This study found that statin therapy was associated with a 63% increased riskof NODM. The risk was independent of the number of metabolic syndrome criteria(van de Woestijne et al. 2015). Additionally, the increased risk of NODM was morepronounced in patients with low baseline glucose levels (<5.6 mmol/L) (van deWoestijne et al. 2015).
In a 3-year follow-up of an observational “The Cooper Center LongitudinalStudy”, including 8,853 generally healthy men and women (aged 20-90 years), statinuse was associated with a two-fold increased risk of incident T2D in individualswith impaired fasting glucose at baseline, but not among those with normal glucoselevels (Radford et al. 2015). Additionally, the risk of incident T2D was substantiallyreduced by increasing fitness (Radford et al. 2015).
2.3.2 Evidence for the diabetogenic effects of statins in clinical trials and meta-analysesThe first study reporting on the effect of statin treatment on glucose homeostasis wasthe WOSCOPS, a primary prevention trial including middle-aged men withhypercholesterolemia, where a significant 30% reduction in the incidence of NODMwas recorded in the pravastatin-treated(40 mg daily) group compared to theplacebo group after 5 years of treatment (Freeman et al. 2001). In contrast, the

23
PROSPER trial including elderly men and women recorded a significant 32%increase in the risk of NODM with pravastatin treatment (40 mg daily for 3.2 years)compared with placebo (Shepherd et al. 2002). In the LIPID trial including patientswith T2D or IFG, pravastatin treatment (40 mg daily for 6 years) did not affectsignificantly the incidence of T2D (Keech et al. 2003).
JUPITER, a double-blind randomized study including 17,802 individuals withelevated levels of C-reactive protein, showed that rosuvastatin treatment (20 mgdaily) was associated with a significant 25% increase in the incidence of NODM anda small but significant 0.3% increase in the median HbA1C levels during the follow-up of 1.9 years, compared with placebo (Ridker et al. 2008). In the sex-stratifiedanalysis, the risk of NODM was significantly increased in women but not in men(Mora et al. 2010).
A meta-analysis of five prospective randomized control trials (RCTs) including39,791 participants suggested that statins as a class did not significantly increase therisk of NODM, but indicated potential differences between the different types ofstatins (Coleman et al. 2008). Pravastatin showed a non-significant trend towards adecreased risk of NODM whereas atorvastatin, rosuvastatin and simvastatinevaluated together were associated with a significant 14% increase in the risk ofNODM (Coleman et al. 2008).
A meta-analysis of six RCTs including 57,593 patients with a mean follow-up of3.9 years reported a 13% significant increase in the risk of NODM with statintreatment (Rajpathak et al. 2009). However, after the inclusion of the WOSCOPS trialin the analysis the risk of NODM was no longer statistically significant (Rajpathak etal. 2009).
A larger meta-analysis of 13 RCTs including 91,140 participants without T2D(Sattar et al. 2010) showed that the treatment with atorvastatin (10 mg), pravastatin(40 mg), simvastatin (40 mg) or rosuvastatin (20 mg) was associated with a 9%increase in the risk of NODM over 4 years with little (11%) heterogeneity betweenthe trials. The risk of NODM was highest among the participants >70 years. Neitherbaseline BMI nor changes in LDL cholesterol concentrations accounted for theobserved variation in the risk of NODM (Sattar et al. 2010).
A meta-analysis of five RCTs including 32,752 participants compared thedifference between an intensive-dose and moderate-dose statin therapy on the riskof NODM, and demonstrated that individuals who received intensive-dose therapywith high-potency statins (simvastatin 80 mg, atorvastatin 80 mg) had a 12%increased risk of developing NODM compared with those receiving moderate-dosetherapy (Preiss et al. 2011b).
An analysis of three large RCTs using a high-dose atorvastatin treatment (80 mgdaily) showed that this treatment was associated with a 37% increased risk ofNODM compared with placebo in the Stroke Prevention by Aggressive Reduction inCholesterol Levels (SPARCL) trial, and a non-significant trend towards an increasedrisk of NODM compared to a low-dose atorvastatin treatment in the TNT andIDEAL trials (Waters et al. 2011). The subsequent analysis showed that the risk ofdeveloping NODM with high-dose atorvastatin treatment was dependent on thebaseline risk factors for diabetes in these trials (Waters et al. 2013). In this studyincluding 15,056 patients with CVD, atorvastatin treatment did not increase theincidence of NODM in patients with 0 to 1 diabetes risk factors but increased theincidence of NODM by 24% in patients with 2 to 4 risk factors, compared withlower-dose statin therapy (Waters et al. 2013). Nevertheless, the number of CVD

24
events was significantly reduced with atorvastatin treatment in both diabetes riskgroups (Waters et al. 2013).
The largest meta-analysis of 17 RCTs including a total of 113,394 patients showedthat pravastatin (40 mg daily) was associated with the lowest risk for NODM [oddsratio (OR) 1.07, 95% confidence interval (CI) 0.86 to 1.30], rosuvastatin (20 mg daily)with the highest risk (OR 1.25, 95% CI 0.82 to 1.90), and atorvastatin (80 mg daily)with an intermediate risk (OR 1.15, 95% CI 0.90 to 1.50), compared with placebo(Navarese et al. 2013). Furthermore, for each statin, increased doses were associatedwith a numerically higher risk for NODM compared with moderate doses(pravastatin 10-20 mg, rosuvastatin 10 mg, or atorvastatin 10 mg, once a day)(Navarese et al. 2013). These findings suggest that different types and doses ofstatins show different potential to increase the incidence of NODM (Navarese et al.2013).
In addition to the risk of NODM associated with statin treatment, several studieshave investigated the effects of statin treatment on the glycemic control amongpatients with pre-existing T2D. In a systematic review and meta-analysis of ninetrials involving >9,000 patients with a history of preexisting diabetes, the individualswho were receiving various doses of atorvastatin, pravastatin or simvastatin hadhigher levels of HbA1c (mean difference 0.12%) compared to the control groups(Erqou et al. 2014). These results suggest that statins may also worsen the glycemiccontrol of diabetic patients.
In summary, several studies suggest that statins differ in their diabetogenic effect(Sattar et al. 2014, Arnaboldi and Corsini 2015, Chan et al. 2015, Sabatine et al. 2004).Atorvastatin, rosuvastatin, simvastatin, lovastatin and fluvastatin were found toworsen glycemic parameters in both diabetic and non-diabetic individuals, whereaspravastatin and pitavastatin seem to have no effect on glycemic parameters (Sasakiet al. 2006, Chapman et al. 2014). The risk of developing NODM during statintreatment seems to be higher in individuals with risk factors for T2D (Waters et al.2013), the elderly (Sattar et al. 2010), and women (Mora et al. 2010).
2.3.3 Effects of statins on insulin secretion and insulin sensitivity in clinical trialsand population studiesSeveral studies have investigated the effects of statins on insulin sensitivity inindividuals without diabetes, but the evidence on the effects of statins on -cellfunction is limited.
Beta-cell function assessed by the homeostasis model assessment of insulinsecretion (HOMA-B), calculated from fasting insulin and glucose levels, was notaffected by the treatment with simvastatin 40 mg or rosuvastatin 10 mg orsimvastatin/ezetimibe 10/10 mg for 12 weeks in 153 patients with elevated levels ofLDL cholesterol (Moutzouri et al. 2011). Treatment with cerivastatin (0.4 mg/day) for3 months improved first-phase insulin secretion measured with the euglycemichyperinsulinemic clamp (EHC) method in a double-blind, placebo-controlled,randomized crossover study including 15 patients with mild hyperglycemia(Paniagua et al. 2002).
Effects of statins on insulin sensitivity determined by the "gold standard" EHCmethod were investigated in several small-sized studies. In a study of 18 patientswith primary hypercholesterolemia, short-term simvastatin treatment (20 mg/day for2 months) did not have significant effect on insulin sensitivity (Altunba et al. 2003).Similarly in a double-blind randomized crossover study including 18 subjects with

25
familial combined hyperlipidemia, a high dose of rosuvastatin (40 mg daily for 12weeks) did not change insulin sensitivity compared to placebo (ter Avest et al. 2005).In contrast, the aforementioned study by Paniagua including 15 patients with mildhyperglycemia showed that treatment with cerivastatin (0.4 mg/day) for 3 monthsincreased insulin-mediated glucose uptake measured with the EHC (Paniagua et al.2002).
In a study including 153 patients with elevated levels of LDL cholesterol whereinsulin resistance was assessed by the homeostasis model assessment of insulinresistance (HOMA-IR) calculated from fasting insulin and glucose levels, threetreatment regimens were compared: simvastatin 40 mg or rosuvastatin 10 mg orsimvastatin/ezetimibe 10/10 mg for 12 weeks (Moutzouri et al. 2011). All threetreatment regimens had significant increases in HOMA-IR and fasting insulin levelscompared to baseline, with no change in FPG, HbA1c and HOMA-B. No significantdifference was observed between the treatment regimens (Moutzouri et al. 2011).
A systematic review and meta-analysis of 16 statin trials (Baker et al. 2010) foundthat statins had no significant impact on insulin sensitivity when pooled as a class,compared with placebo/control groups. However, pooled pravastatin studiesshowed that pravastatin increased insulin sensitivity, whereas pooled simvastatinstudies showed a statistically significant decrease in insulin sensitivity, and nosignificant effect was observed in rosuvastatin or atorvastatin pooled studies (Bakeret al. 2010). These results suggest that the effects of statins on insulin sensitivity mayvary by the statin type.
2.3.4 Evidence for the diabetogenic effects of statins in genetic studiesThe results from a large Mendelian randomization study suggested that theincreased risk of NODM observed with statins was at least partially explained by theinhibition of HMG-CoA reductase (Swerdlow et al. 2015). In this study including upto 223,463 individuals, two single nucleotide polymorphisms (SNPs) in the geneencoding HMG-CoA reductase (HMGCR) were used as proxies for the inhibition ofHMG-CoA reductase by statins. The rs17238484-G allele and rs12916-T allele wereeach associated with lower LDL cholesterol concentration and were designated theeffect alleles, to facilitate direct comparison with statin treatment (Swerdlow et al.2015). These LDL cholesterol lowering alleles were found to be associated withhigher risk of T2D, higher plasma glucose and insulin concentrations, and higherbody weight and waist circumference (Swerdlow et al. 2015). Additionally, adirectionally consistent association of statin treatment with lower LDL cholesterol,increased body weight and increased risk of NODM was observed in a meta-analysisof included clinical trials (Swerdlow et al. 2015).
This finding posed a question, whether the diabetes risk is limited to theinhibition of HMG-CoA reductase, or whether other pathways regulating the levelsof cholesterol or other lipoproteins have a similar effect. A Dutch study including63,320 individuals with familial hypercholesterolemia, characterized by high plasmalevels of LDL cholesterol because of a mutation in the genes encoding LDL receptor(LDLR), apolipoprotein B (APOB) or proprotein convertase subtilisin/kexin type 9(PCSK9) showed a significantly lower prevalence of T2D in patients than theirunaffected relatives (Besseling et al. 2015). Furthermore, an inverse dose-responserelationship between the severity of the familial mutation and prevalence of T2Dwas observed, with more severe mutations in LDLR gene (especially receptor-negative mutations) associated with lower prevalence of T2D than milder APOB

26
mutations (Besseling et al. 2015). These findings raise the possibility of a causalrelationship between LDL receptor-mediated transmembrane cholesterol transportand T2D (Besseling et al. 2015).
Another large meta-analysis investigating LDL cholesterol lowering geneticvariants in or near several genes encoding molecular targets for lipid-lowering drugsshowed that variants at NPC1L1 (encoding target for ezetimibe), PCSK9 (encodingthe target for PCSK9 inhibitors) and HMGCR were associated with a higher risk ofT2D and lower levels of LDL cholesterol (Lotta et al. 2016). This study suggests thatmultiple cholesterol-lowering mechanisms by distinct pathways are associated withan increased risk of T2D, including cholesterol absorption (NPC1L1), endogenouscholesterol synthesis (HMGCR) and internalization of cholesterol-rich particles intothe cell (PCSK9) (Lotta et al. 2016). Additional evidence for the link between geneticvariants associated with lipids and T2D comes from an analysis of the largestavailable genetic datasets (DIAGRAM, MAGIC, GENESIS), which showed thatgenetically determined lower LDL cholesterol was associated with a higher risk ofT2D, however, it did not identify consistent evidence for a causal role of circulatinglipids in T2D (Fall et al. 2015).
2.3.5 Mechanistic studies of the effects of statins on insulin secretion and insulinactionThe mechanisms by which statins increase the risk of NODM are largely unknown.The following section reviews potential mechanisms suggested by the experimentalin vitro and in vivo studies by which statins could increase the risk of NODM. Theeffects of statins on both insulin secretion and insulin sensitivity will be reviewed.
2.3.5.1 Effect of statins on insulin secretionLovastatinA study investigating the effects of lovastatin (15-30 µM) in rat pancreatic islets(Metz et al. 1993) showed that lovastatin decreased glucose-induced insulin secretionby approximately 50%, and this effect was reversed by co-incubating the islets withmevalonate. Lovastatin treatment also resulted in the accumulation of G proteins inthe cytoplasm and the authors suggested that that inhibition of post-translationalprenylation of G proteins impedes their maturation, association to the plasmamembrane and biological effect (Metz et al. 1993). G proteins play a key role intrafficking secretory granules from the cell. Therefore this effect may also contributeto decreased insulin secretion (Metz et al. 1993). Similar results were obtained frominsulin secreting HIT-T15 cells (Li et al. 1993).PravastatinPravastatin (100 mg/ml) did not have any effect on insulin secretion in rat islet -cells (Yada et al. 1999).SimvastatinSimvastatin (0.1-3.0 µg/ml) was shown to decrease dose-dependently the glucose-induced secretion of insulin in rat islet -cells by decreasing the first phase insulinsecretion and calcium oscillations (Yada et al. 1999). Simvastatin was also shown toinhibit L-type calcium channels which resulted in the inhibition of the rise ofcytosolic calcium inside the cell, induced by L-arginine and KCl (potassium chloride)which are known to act by mediating the opening of L-type calcium channels (Yadaet al. 1999).

27
ATP plays a key role in insulin secretion by binding to the KATP channels leadingto depolarization of the plasma membrane and subsequent opening of the calciumchannels. In a study using a mouse -cell line, simvastatin dose-dependentlydecreased ATP production in -cells, and the protein and mRNA expression ofGLUT2, voltage-dependent calcium channels, and increased the expression of Kir6.2potassium channel subunits (Zhou et al. 2014). These results indicate thatsimvastatin may affect insulin secretion by reducing GLUT2 expression andinhibiting membrane depolarization and calcium influx.
AtorvastatinIn diet-induced obese C57BL/6J mice, atorvastatin (30 mg/kg daily) treatmentpreserved -cell sensitivity to glucose assessed by hyperglycemic clamp, improvedplasma lipid profiles and pancreatic weight (Chen et al. 2014). Additionally,atorvastatin treated mice had larger insulin positive -cell area, reduced ER stressand reduced apoptosis, suggesting that atorvastatin preserved -cell function byenhancing -cell proliferation and amelioration of pancreatic ER stress (Chen et al.2014).
2.3.5.2 Effect of statins on insulin sensitivitySeveral studies examining the effects of different statins on insulin sensitivity andinsulin signaling in various cell models have been published. The effects ofsimvastatin were examined in L6 rat myotubes (Kain et al. 2015), mouse C2C12myotubes (Li et al. 2016), human myotubes (Smith et al. 2014), and 3T3-L1adipocytes (Maeda and Horiuchi 2009, Ganesan and Ito 2013, Araki et al. 2007). Theeffects of atorvastatin and pravastatin were studied in 3T3-L1 adipocytes (Nakata etal. 2006, Takaguri et al. 2008), and the effects of lovastatin were studied in 3T3-L1adipocytes (Chamberlain 2001) and fibroblasts (McGuire et al. 1994).
LovastatinIn 3T3-L1 adipocytes, lovastatin treatment was found to down-regulate proteinexpression of GLUT4, whereas GLUT1 protein expression was up-regulated(Chamberlain 2001). These changes were associated with a significant inhibition ofinsulin-stimulated glucose transport. The effects of lovastatin were reversed bymevalonate, suggesting that inhibition of isoprenoid biosynthesis causes insulinresistance in 3T3-L1 adipocytes (Chamberlain 2001).
In rat fibroblasts, lovastatin treatment was shown to inhibit insulin-stimulatedPI3K activity by 75%, to inhibit the association of its regulatory subunit p85 withtyrosine phosphorylated IRS1 and the beta subunit of the IR, and to decrease thelevel of tyrosine phosphorylated IR beta subunit after insulin stimulation (McGuireet al. 1994). These results demonstrate that lovastatin disrupts early events of insulinmitogenic signaling, which could explain the anti-mitogenic effects of lovastatin(McGuire et al. 1994).PravastatinIn 3T3-L1 adipocytes, pravastatin did not have a significant effect on the GLUT4protein expression (Ganesan and Ito 2013). In another study in 3T3-L1 adipocytes,pravastatin did not affect insulin-stimulated glucose uptake or the translocation ofGLUT4 into the plasma membrane (Takaguri et al. 2008).

28
Simvastatin In human myotubes from healthy donors, treatment with simvastatin caused adose-dependent decrease in glucose uptake and oxidation (Smith et al. 2014).Simvastatin also caused a decrease in maturation and activity of legumain, a cysteineprotease involved in apoptosis, which could contribute to the observed myotoxicityof statins (Smith et al. 2014).
A study using L6 myotubes reported that simvastatin induced insulin resistancethrough a mechanism independent of the inhibition of cholesterol biosynthesis, by afatty acid mediated effect on insulin signaling pathway (Kain et al. 2015).
A study using 3T3-L1 adipocytes showed that simvastatin but not pravastatinsignificantly decreased GLUT4 protein expression, and this effect seemed to beprevented by co-treatment with coenzyme Q10 which plays a key role inmitochondrial oxidative phosphorylation (Ganesan and Ito 2013). Synthesis of Q10requires the isoprene residues generated from the cholesterol biosynthetic pathway(Ganesan and Ito 2013).AtorvastatinAtorvastatin treatment of 3T3-L1 adipocytes was shown to inhibit adipocytematuration, to attenuate the expression of SLC2A4 (gene encoding GLUT4) andC/EBP , and to impair insulin action (Nakata et al. 2006). These effects werereversed by mevalonate or GGPP, suggesting that they are due to isoprenoiddepletion. In addition, in NSY mice, a model for type 2 diabetes, atorvastatinaccelerated glucose intolerance as a result of insulin resistance, and decreasedSLC2A4 expression in white adipose tissue (Nakata et al. 2006).
In another study in 3T3-L1 adipocytes, atorvastatin significantly decreasedinsulin-stimulated glucose uptake and prevented the translocation of GLUT4 intothe plasma membrane (Takaguri et al. 2008). Insulin-induced tyrosinephosphorylation of IRS1 and serine/threonine phosphorylation of Akt were alsoreduced by atorvastatin. In addition, atorvastatin decreased the amounts of the smallGTPases Rab4 and RhoA requiring lipid modification with FPP or GGPP in themembrane fraction (Takaguri et al. 2008). Rab4 plays a key role in GLUT4translocation form cytoplasm to cell membrane and, in combination with RhoA, inthe insulin signal transduction pathway, suggesting that atorvastatin decreasesglucose uptake by preventing the isoprenyl modification of these proteins (Takaguriet al. 2008).FluvastatinIn obese mice, long-term fluvastatin treatment was shown to impair insulin-stimulated glucose uptake in adipose tissue (Henriksbo et al. 2014). The authorsobserved in adipose tissue explants that the fluvastatin-induced activation of theNLRP3/caspase-1 pathway was required for the development of insulin resistance(Henriksbo et al. 2014). Furthermore, four different statins were found to increase ina dose-dependent manner interleukin-1 secretion from macrophages which ischaracteristic of NLRP3 inflammasome activation (Henriksbo et al. 2014). This studysuggests an NLRP3/caspase-1-mediated mechanism of statin-induced insulinresistance (Henriksbo et al. 2014).
In summary, these studies using different statins in various cell and animal modelsshow that simvastatin, atorvastatin, lovastatin and fluvastatin may affect varioussteps in insulin action, and the effects in some, but not in all studies wereattributable to inhibition of isoprenoid synthesis by statin treatment.

29
3. Aims of the study
The main aims of the study were to investigate the effects of statin treatment on therisk of T2D, insulin secretion and insulin sensitivity in a population-based study,and to investigate the mechanisms underlying these effects by employing in vitromodels for insulin secretion and insulin resistance.
The specific aims of the study were:
1. To investigate the effects of statin treatment on the risk of incident T2D, worseningof hyperglycemia, and on insulin sensitivity and insulin secretion in the population-based METSIM study;
2. To investigate the effects of simvastatin and pravastatin on insulin secretion inmouse pancreatic MIN6 -cells by systematic assessment of all major pathwaysregulating insulin secretion;
3. To investigate the effects of simvastatin and pravastatin on insulin sensitivity in L6myotubes, and the mechanisms underlying these effects.

30

31
4. Subjects and Methods
4.1 SUBJECTS
The METSIM Study (Study I)The cross-sectional METSIM study was performed in 2005 – 2010 at the ClinicalResearch Unit of the University of Kuopio and included 10,197 men, aged 45–73 years, randomly selected from the population register of Kuopio, Eastern Finland(population 95,000) (Stancakova et al. 2009). An OGTT (75 g of glucose, glucose andinsulin measurements at 0, 30 and 120 min) was performed, and glucose tolerancewas classified according to the ADA (American Diabetes Association 2006).Participants with previously diagnosed type 1 diabetes (n 25), newly (n 649) orpreviously diagnosed T2D (n 763) or those without OGTT data (n 11) wereexcluded. A total of 8,749 men without diabetes at baseline were included instatistical analyses (age 57 7 years, BMI 26.8 3.8 kg/m2, mean SD). An ongoingfollow-up study started in 2010 and so far 5,419 individuals have participated. Thestudy protocol and measurements are identical to those of the baseline study. Thestudy was approved by the Ethics Committee of the University of Eastern Finlandand Kuopio University Hospital and conducted in accordance with the HelsinkiDeclaration. All study participants gave written informed consent.
Out of the 8,749 non-diabetic participants at baseline, 625 developed T2D during a5.9 year follow-up study. Diagnosis of T2D was based on the following criteria: (1)FPG 7.0 mmol/l, 2hPG 11.1 mmol/l in an OGTT or HbA1c 6.5% among 4,806 non-diabetic individuals who participated in the ongoing METSIM follow-up study in2010 – 2014; (2) glucose-lowering medication started after the baseline study(information obtained from the National Drug Reimbursement registry for all 8,749non-diabetic participants); (3) T2D diagnosed by physician as per medical records orin outpatient/primary care laboratory measurements. Of the diabetes diagnoses inthe METSIM follow-up study, 22.6% were based on FPG alone, 24.9% on 2hPGalone, 31.6% on HbA1c alone and 20.8% on different combinations of these criteria.
Methods and calculations used in the METSIM studyBMI was calculated as weight (kg) divided by height (m) squared. Waistcircumference was measured at the midpoint between the lateral iliac crest andlowest rib. Smoking status was defined as current smoking (yes vs. no). Familyhistory of diabetes (yes vs. no) was defined as a first-degree or second-degreerelative having diabetes vs. no family history of diabetes. Physical activity(physically active vs. inactive) refers to leisure-time exercise (physically active,regular exercise [at least 30 min once or twice a week] vs. physically inactive,occasional exercise or no exercise). Alcohol intake was defined as total alcohol intakein grams per week. The use of beta-blockers and diuretics at baseline was recorded(yes vs. no). CVD at baseline was defined as a history of non-fatal myocardialinfarction or stroke. Plasma glucose was measured by enzymatic hexokinasephotometric assay (Konelab Systems reagents; Thermo Fisher Scientific, Vantaa,Finland). HbA1c was analysed with a Tosoh G7 glycohemoglobin analyser (TosohBioscience, San Francisco, CA, USA). Plasma insulin concentrations were measured

32
by a luminometric immunoassay measurement (ADVIA Centaur Insulin IRI, no.02230141; Siemens Medical Solutions Diagnostics, Tarrytown, NY, USA). LDLcholesterol, HDL cholesterol and total triacylglycerols were measured by enzymaticcolorimetric tests (Konelab Systems reagents). The trapezoidal method was used tocalculate the glucose and insulin area under the curve (AUC) in an OGTT based onsamples collected at 0, 30 and 120 min. The Matsuda index of insulin sensitivity (ISI)was calculated as 10,000 / (fasting insulin fasting glucose mean insulin duringOGTT mean glucose in an OGTT) (Matsuda 1999). The disposition index (DI) wascalculated as a product of insulin sensitivity and insulin secretion (MatsudaISI insulin AUC0–30 min/glucose AUC0–30 min) as previously reported (Stancakova et al.2009).
4.2 Cell cultures
MIN6 cell culture (Study II)Mouse pancreatic MIN6 -cells (Miyazaki et al. 1990) were obtained from MerjaRoivainen, National Institute for Health and Welfare, Helsinki, Finland (originallyobtained from Prof. Jun-ichi Miyazaki, Osaka University, Japan). The cells werecultured at 37°C in a humidified atmosphere of 5% CO2 in DMEM containing 25 mMglucose supplemented with 15% heat inactivated fetal bovine serum (Gibco), 2 mML-glutamine and 100 units/ml penicillin, 100 µg/ml streptomycin, 5 µl/l -mercaptoethanol and 3.4 g/l NaHCO3.
L6 cell culture (Study III)L6 cells (CLR-1458, ATCC) were maintained as myoblasts in DMEM, 4.5 g/l glucose,supplemented with 10% fetal bovine serum (Gibco), 2 mM L-glutamine, 100 units/mlpenicillin and 100 µg/ml streptomycin, at 37°C in an atmosphere of 5% CO2. Afterthe myoblasts reached confluence, the media was switched to differentiation mediacontaining DMEM, 1 g/l glucose, 2% horse serum (Gibco), 2 mM L-glutamine, 100units/ml penicillin and 100 µg/ml streptomycin. The media was changed every 48hours until the cells were differentiated judged from the morphology under themicroscope. All the experiments were conducted in differentiated L6 myotubes.
4.3 Laboratory methods
Insulin Secretion Assay (Study II)MIN6 cells were washed with glucose-free KRBH (Krebs-Ringer bicarbonate HEPESBuffer) (119 mM NaCl, 4.74 mM KCl, 2.54 mM CaCl2, 1.19 mM MgSO4, 1.19 mMKH2PO4, 10 mM HEPES, 25 mM NaHCO3 and 0.1% BSA) at pH 7.4 thrice beforeincubating them in the same buffer at 37°C for 1 hour. Later, cells were washed oncewith glucose free KRBH and pre-incubated with either 14.3 µM simvastatin orpravastatin in the presence of activators and inhibitors in KRBH for 30 minutes at37°C. The cells were later treated with compounds in KRBH containing either 5.5mM or 16.7 mM glucose at 37°C for 1 hour. Ca2+- free KRBH Buffer (135 mM NaCl,3.6 mM KCl, 0.5 mM NaH2PO4, 0.5 mM MgCl2, 10 mM HEPES, 0.1 mM EGTA, 2 mMNaHCO3 and 0.1% BSA) at pH 7.4 was used in the insulin secretion experimentsassessing the influence of the extracellular calcium. In these experiments, the cellswere treated with normal KRBH with compounds for 30 minutes, washed twice withCa2+- free KRBH and later treated with compounds in Ca2+- free KRBH for 45

33
minutes. KRBH buffer was collected for insulin assay and cells were washed withphosphate buffered saline (PBS) once. Cells were lysed with RIPA buffer containingprotease and phosphatase inhibitors, collected for protein estimation and westernblotting and stored at -70°C until further analysis. Proteins were estimated with BCAprotein assay (Pierce). Insulin was measured with AlphaLISA Insulin Kit(PerkinElmer) according to the manufacturer’s instructions.Glucose uptake assay (Studies II-III)MIN6 cells were washed thrice with glucose-free KRBH buffer, pre-incubated for 1hour and were later treated with 14.3 µM simvastatin in the same buffer for 30minutes, washed once with glucose free KRBH buffer and treated with simvastatinin KRBH buffer containing 5.5 or 16.7 mM glucose for 1 hour. L6 myotubes wereexposed to test compounds for 24 hours in differentiation media supplemented with5.5 mM or 16.7 mM glucose. The control cells were treated with DMSO. Aftertreatments, the myotubes were washed with PBS and incubated with HEPES-buffered saline (140 mM NaCl, 20 mM HEPES, 5 mM KCl, 2.5 mM MgSO4, 1 mMCaCl2, pH 7.4) for 1 hour with the test compounds. 100 nM human insulin (Sigma-Aldrich) was added to the cells for the last 30 minutes. KRBH buffer from MIN6 cellsand HEPES-buffered saline from L6 myotubes were removed and the cells weretreated with respective buffers containing 10 µM 2-deoxy-D-glucose and 1 µCi 2-deoxy-D-[2,6-3H] glucose (PerkinElmer) for 15 minutes at room temperature. Cellswere washed with ice cold PBS while the plates were on ice. 200 µl-500 µl of 0.2 NNaOH was added to each well and the plate was incubated for 90 minutes at roomtemperature with constant shaking. Collected samples were stored at -70°C.OptiPhase HiSafe 2 (PerkinElmer) was added to the samples and radioactivity wasmeasured using 1450 MicroBeta Trilux (Wallac). The protein content of the lysateswas measured using the BCA Protein Assay (Pierce).
Immunoblotting (Studies II-III)After the incubation according to the specific experiment the cells were washed oncewith PBS and lysed with RIPA buffer along with protease and phosphataseinhibitors. Protein concentrations were measured by BCA protein assay (Pierce).Protein samples were subjected to gel electrophoresis and transferred topolyvinylidene fluoride (PVDF) membranes. The membranes were blocked for 1hour at room temperature and incubated overnight at +4°C with primary antibodies.After the primary antibody incubations, the membranes were incubated withhorseradish peroxidase conjugated secondary antibodies for 1 hour at roomtemperature. Membranes were washed with TBS-T for 3x5 minutes between everystep. Membranes were finally washed in TBS, the bands were visualized usingchemiluminescence (ECL Plus, Pierce) and images were captured in Image QuantRT-ECL equipment (GE Healthcare). Bands were quantified with Quantity Onesoftware (Bio-Rad).cAMP and PKC assay (Study II)GPR119-overexpressing CHO-K1 cell lines (Euroscreen) were used to determinecAMP using cAMP HTRF™ functional assay (FAST-0370C). The cells were firsttreated with simvastatin and pravastatin for 30 minutes and then stimulated withthe GPR119 agonist oleoylethanolamide, which increases cAMP levels. The totalPKC activity in MIN6 cells was analyzed with PKC kinase activity kit (Enzo)according to the manufacturer’s instructions.

34
Intracellular calcium measurements (Study II)Intracellular calcium [Ca2+]i was measured using the ratiometric Ca2+ probe Fura-2with an IX81-ZDC inverted microscope (Olympus) controlled by Cell^R software(Olympus). MIN6 cells grown on poly-L lysine coated coverslips were equilibratedin glucose-free KRBH buffer and loaded with Fura-2-AM at 37°C with 5% CO2 for 30min. All used compounds, alone or combined, were pre-incubated in the sameconditions together with Fura-2-AM. After pre-incubation, the coverslips weremounted in a custom made perfusion chamber placed in an environmentallycontrolled culture chamber for perfusion. The cells were perfused for 2–3 min withglucose-free KRBH buffer (+/- testing compounds) for baseline detection followed bythe effective stimulation (glucose +/- testing compounds). The imaging paradigmconsisted of alternating excitation at 340 nm and 380 nm (300 ms each) and emissiondetection at 510 nm with a CCD camera (Hamamatsu). Images were taken everysecond and analyzed offline using Cell^R software. Individual cells were manuallysegmented and the 340/380 nm ratio of emission at 510 nm was calculated for eachtime point as indicative of [Ca2+]i levels.Statistical analysisStudy I: Statistical analyses were conducted using the SPSS version 19. BMI, waistcircumference, total cholesterol, LDL cholesterol, HDL cholesterol, totaltriacylglycerols, glucose and HbA1c levels, Matsuda insulin sensitivity index (ISI)and DI were log-transformed to correct for their skewed distribution. HRs for therisk of diabetes were calculated with Cox regression. Risk of diabetes with statintreatment (yes/no) or according to the type of statin (simvastatin, atorvastatin, orother statins [including rosuvastatin, pravastatin, fluvastatin and lovastatin] vs. nostatin), the dose of statin, and changes in statin treatment during the study werecalculated with Cox regression. The association of statin treatment with FPG, 2hPGand glucose AUC at follow-up was evaluated with linear regression analysis(N 4,679 non-diabetic participants at baseline had follow-up data available,excluding individuals diagnosed with diabetes between baseline and follow-up)adjusted for age, BMI, waist circumference, current smoking, physical activity,alcohol intake, family history of diabetes, use of beta-blockers and use of diuretics, aswell as for the length of follow-up time (in months), and additionally for FPG, 2hPG,Matsuda ISI, DI, LDL cholesterol, HDL cholesterol, total triacylglycerols and CVDand the changes in LDL cholesterol, HDL cholesterol, total triacylglycerols and BMI.P 0.004 was considered as statistically significant given the 12 different modelstested (Bonferroni correction for multiple testing) and p 0.05 was considerednominally significant. Differences in Matsuda ISI and DI (Table 3) in non-diabeticindividuals at baseline treated with simvastatin or atorvastatin vs. no statin and inindividuals receiving low-dose or high-dose atorvastatin or simvastatin vs. no statinwere compared with the ANOVA post hoc tests. Matsuda ISI and DI between theindividuals with and without statin therapy in categories of FPG and 2hPG werecompared using the t test.Studies II, III: Data were collected from independent experiments, with three or morereplicates per experiment. All data were analyzed with Mann-Whitney test or one-way ANOVA with Bonferroni post-hoc test or Student’s t-test and P < 0.05 wasconsidered statistically significant. Data are presented as values relative to the meanof control (control = 100%). Error bars represent standard error of the mean (SEM).Statistical analyses were conducted using the SPSS or GraphPad Prism software.

35
5. Results
5.1 ASSOCIATION OF STATIN TREATMENT WITH THE RISK OFDIABETES, INSULIN SENSITIVITY AND INSULIN SECRETION (I)
Effect of statin treatment on the risk of type 2 diabetesA total of 2,142 (24.5%) of the 8,749 non-diabetic men were on statin medication atbaseline (65.9% on simvastatin, 18.1% on atorvastatin, 8.6% on rosuvastatin, 3.8% onfluvastatin, 2.3% on lovastatin and 1.3% on pravastatin).
At entry, the individuals who developed diabetes on statin treatment had asimilar metabolic risk factor profile to those who developed diabetes without statintreatment. Compared to those who did not develop diabetes they were older, moreobese, physically less active, had lower levels of HDL cholesterol and higher levelsof total triacylglycerols, FPG, 2hPG and HbA1c, were more insulin resistant and hadlower insulin secretion.
Participants treated with statins developed diabetes more often than participantswithout statin treatment (11.2% vs. 5.8%, p 0.001). Statin treatment increased therisk of T2D by twofold during the follow-up [hazard ratio (HR) 2.01 (95% CI 1.71,2.36)] (Figure 6a). After adjustment for age, BMI, waist circumference, physicalactivity, smoking, alcohol intake, family history of diabetes and beta-blocker anddiuretic treatment, the HR was 1.46 (1.22, 1.74).
Both simvastatin and atorvastatin increased the risk of T2D compared with nostatin treatment [HR 2.11 (95% CI 1.76, 2.54) and 1.50 (95% CI 1.30, 1.730],respectively), and these associations remained significant after the adjustment forconfounding factors. Other statins did not increase the risk of diabetes (Figure 6b).The risk of diabetes was dose dependent for both simvastatin and atorvastatin(Figure 6c, d).
Statin treatment significantly increased the levels of 2hPG and the glucose AUCat follow-up (p 0.001 and p 0.001, respectively), and nominally the levels of FPG atfollow-up (p 0.037) after adjustment for confounding factors (I, Table 2). Theassociation of statin treatment with 2hPG at the follow-up study was stronger thanthat with FPG; the association remained nominally significant after the adjustmentfor FPG at baseline, Matsuda ISI and DI but was abolished after the adjustment for2hPG at baseline. Adjustment for CVD, LDL cholesterol, HDL cholesterol and totaltriacylglycerols and change in BMI did not attenuate the associations of statintreatment with glycaemia.
Effect of statin treatment on insulin secretion and insulin sensitivityStatin treatment was associated with a 24.3% reduction of insulin sensitivity(Matsuda ISI) in the lowest category of FPG (<5.0 mmol/l) and with a 19.5%reduction of insulin sensitivity in the lowest category of 2hPG (<5.0 mmol/l)compared with individuals without statin treatment (p 0.001). Statin treatmentreduced insulin secretion (DI) by 12.0% in the lowest category of FPG comparedwith individuals without statin treatment (p 0.01).

36
Treatment with either simvastatin or atorvastatin was associated with significantreduction in Matsuda ISI (21.9 and 24.4%, respectively) and DI (7.6 and 7.4%,respectively) compared with individuals without statin treatment. There was asignificant decrease in insulin sensitivity with an increasing dose of simvastatin (lowdose, 20.8%; high dose, 25.4%) and atorvastatin (16.6% and 30.2%, respectively).Corresponding decreases in insulin secretion were considerably smaller for bothsimvastatin (low dose, 6.6%; high dose, 9.8%) and atorvastatin (3.4% and 10.5%,respectively).
Figure 6. Risk of T2D by statin treatment during the 5.9 year follow-up. (a) Total cohort(625 cases of new T2D and 8,124 non-diabetic controls). Black line, statin treatment atbaseline (n=2,141); grey line, no statin treatment at baseline (n=6,607). (b) Risk bydifferent statins. Black continuous line, atorvastatin (n=388); black dotted line,simvastatin (n=1,409); grey dotted line, other statins (including rosuvastatin,pravastatin, fluvastatin and lovastatin, n=342); grey continuous line, no statintreatment. (c) Risk by dose of simvastatin. Black line, high dose (40 or 80 mg/day,n=385); dotted line, low dose (10 or 20 mg/day, n=971); grey line, no statin treatment.(d) Risk by dose of atorvastatin. Black line, high dose (20 or 40 mg/day, n=197); dottedline, low dose (10 mg/day, n=175); grey line, no statin treatment.

37
5.2 MOLECULAR MECHANISMS OF THE EFFECT OF SIMVASTATINAND PRAVASTATIN ON INSULIN SECRETION IN MIN6 CELLS (II)
In all experiments described here, MIN6 -cells were treated with 6 µg/ml (14.3 µM)simvastatin or 12 µg/ml (26.3 µM) pravastatin at normal (5.5 mM) glucoseconcentration. Experiments were also performed at 16.7 mM glucose with similarresults.
Simvastatin, but not pravastatin, decreased insulin secretion. Simvastatindecreased insulin secretion by 59% (p<0.01), whereas treatment with pravastatinincreased insulin secretion nonsignificantly by 71% compared to control (II, Figure1A).
Effect of simvastatin on insulin secretion via the cholesterol biosyntheticpathway. MIN6 -cells were treated with simvastatin and DL-mevalolactone (1mM), an activator of mevalonate synthesis (intermediate in cholesterol biosynthesis),isoprenoids GGPP and FPP (both 20 µM), and inhibitors of geranylgeranyltransferase 1 (GGTI-298) and farnesyltransferase (FTI-277) (both 20 µM). Simvastatinsignificantly decreased insulin secretion stimulated by the activators of DL-mevalolactone (p=0.002) (II, Figure 1C), GGPP and FPP (p=0.0002) (II, Figure 1E). Thedecrease in insulin secretion by simvastatin was comparable to the effects of GGTI-298 (decrease by 59%) and FTI-277 (decrease by 64%) (p<0.01) (II, Figure 1G).
Simvastatin impairs tolbutamide-, KCl-, acetylcholine- and PMA-mediatedinsulin secretion. MIN6 -cells were treated with simvastatin, tolbutamide (100µM), potassium chloride (KCl, 40 mM), acetylcholine (10 µM), and phorbol myristateacetate (PMA, 0.5 µM), an activator of PKC. Treatment with tolbutamide, KCl,acetylcholine and PMA significantly increased insulin secretion by 150%, 444%,1194%, and 572% (p<0.05), respectively. Simvastatin decreased insulin secretionstimulated by tolbutamide, KCl, acetylcholine (all p=0.004) and PMA (non-significantly). Simvastatin had no effect on PKC activity (II, Figure 2A, E-G).
Simvastatin impairs insulin secretion mediated by the GPR40 signalingpathway. MIN6 -cells were treated with simvastatin and the GPR40 agonistsTAK875 and GW9508 (both 40 µM). Treatment with TAK875 and GW9508 increasedinsulin secretion by 538% and 596% (p<0.05). Simvastatin decreased TAK875- andGW9508-induced insulin secretion (p=0.002 and 0.010, respectively) to 33 and 77% ofcontrol (II, Figure 3A). Natural agonists of GPR40 receptor, oleic acid (OA) andlinoleic acid (LA), also increased insulin secretion dose-dependently, and treatmentwith simvastatin significantly decreased insulin secretion stimulated by OA and LA(p<0.05) (II, Figure 3E).
Simvastatin slightly impairs GLP-1-mediated and GPR119-mediated insulinsecretion. MIN6 -cells were treated with simvastatin, glucagon like peptide-1 (GLP-1) amide (100 nM) and a GPR119 agonist AS-1269574 (40 µM). Treatment with GLP-1amide or AS-1269574 increased insulin secretion by 139% and 80% (p<0.01),respectively. Treatment with simvastatin decreased GLP-1- and AS-1269574-stimulated insulin secretion only slightly, and insulin secretion still remained higherthan in the control (p<0.05) or simvastatin treatment (p<0.01). This suggests thatGLP-1 and AS-1269574 can partially restore simvastatin-induced defect in insulinsecretion. Similar results were observed with another GLP-1 receptor agonist,exendin-4 (II, Figure 4A, C).
Simvastatin decreases intracellular cAMP levels. Treatment with simvastatindecreased the intracellular concentration of cAMP, the downstream target of the

38
GLP-1R and GPR119 pathways, slightly by 22% in GPR119-overexpressing CHO-K1cells (II, Figure 4E). The membrane-permeable adenylate cyclase activator forskolin(10 µM) increased insulin secretion by 166% (p<0.05) and simvastatin decreasedforskolin-stimulated insulin secretion to a level 46% higher than control (II, Figure4F), suggesting that cAMP elevation can restore simvastatin-induced decrease ininsulin secretion similarly as GLP-1R and GPR119 agonists.
Simvastatin decreases PKA- and EPAC2-mediated insulin secretion. MIN6 -cells were treated with simvastatin, with 1 mM 8-bromo-cAMP, which activates bothPKA and exchange protein activated by cAMP 2 (EPAC2), and with 50 µM 8-pCPT-
-O-Me-cAMP, a cAMP analog specific for activation of EPACs. Treatment with 8-bromo-cAMP or 8-pCPT-2 -O-Me-cAMP increased insulin secretion by 593% and69% (p<0.05), respectively. Simvastatin decreased both 8-bromo-cAMP- and 8-pCPT-
-O-Me-cAMP-induced insulin secretion (p<0.05) (II, Figure 4G).Effect of simvastatin on the intracellular Ca2+ levels. Simvastatin at 7 different
concentrations (0.5 – 12.5 µM) dose-dependently inhibited the increase in [Ca2+]i
levels stimulated by 16.7 mM glucose (II, Figure 5A). Treatments with GLP-1 or AS-1269574 were not able to restore the inhibitory effect of simvastatin on [Ca2+]i levels(II, Figure 5H).
Simvastatin decreases calcium release from endoplasmic reticulum. Treatmentwith 10 mM caffeine, an activator of ryanodine receptors (RyR) of the ER, increasedthe insulin secretion by 394% (p<0.01), and simvastatin almost completely abolishedcaffeine-stimulated insulin secretion (to 18% of control, p<0.01). 5 µM of ryanodine, aRyR inhibitor, decreased insulin secretion by 18%, and a combination of ryanodinewith simvastatin by 57% compared to control (p<0.05) (II, Figure 6B).
Effects of simvastatin on glucose uptake and glycolysis pathway. Simvastatinhad no significant effect on glucose uptake (II, Figure 6C-D), GLUT2 proteinexpression (II, Figure 6E), pyruvate levels (II, Figure 6F) or the ADP/ATP ratio (II,Figure 6G-H).
All experiments mentioned above were performed also at high glucoseconcentration (16.7 mM), and we obtained essentially the same results.
Figure 7 summarizes the effects of simvastatin on the insulin secretion pathways.

39
Figure 7. Different pathways of insulin secretion, links between them and effects ofsimvastatin on these pathways. Simvastatin (Simva) decreases insulin secretionmediated by ATP-sensitive potassium (KATP) channels (A), membrane depolarization andvoltage-gated calcium channels (B), acetylcholine and GPR40 receptors (C and D), andcholesterol biosynthetic pathway (E). These effects may be attributable to the actions ofsimvastatin on KATP channels, calcium channels, or Ca2+ release from endoplasmicreticulum via inositol 3-phophate (IP3) and ryanodine receptors (RyR) or insulin granuleexocytosis. Activation of GLP-1R (F) and GPR119 (G) pathways restores simvastatin-decreased insulin secretion. DAG - diacylglycerol, EPAC2 - exchange protein activated bycAMP 2, ER - endoplasmic reticulum, IP3 - inositol 1,4,5-trisphosphate, PKA - proteinkinase A, PLC - phospholipase C beta, PIP2 - phosphatidylinositol 4,5-bisphosphate.

40
5.3 MOLECULAR MECHANISMS OF THE EFFECT OF SIMVASTATINAND PRAVASTATIN ON INSULIN SENSITIVITY IN L6 MYOTUBES(III)
In all experiments described here, L6 myotubes were treated with 6 µg/ml (14.3 µM)simvastatin or 12 µg/ml (26.3 µM) pravastatin at normal (5.5 mM) glucoseconcentration.
Simvastatin, but not pravastatin, decreased glucose uptake. Treatment withsimvastatin significantly decreased basal glucose uptake by 46% (p<0.05) andinsulin-stimulated glucose uptake by 58% (p<0.001) (III, Figure 1B-C). Treatmentwith pravastatin had a non-significant trend to increase the basal glucose uptake (III,Figure 1D).
Effect of simvastatin on glucose uptake via the cholesterol biosyntheticpathway. L6 myotubes were treated with simvastatin, DL-mevalolactone (0.5 mM or1 mM), GGPP and FPP (both 20 µM). Treatment with DL-mevalolactone or FPP didnot affect glucose uptake, and GGPP increased glucose uptake by 49% (p<0.01). DL-mevalolactone and GGPP restored the simvastatin-decreased glucose uptake (p<0.01compared to simvastatin alone), but FPP did not have the same effect (III, Figure 2A-C).
Effect of simvastatin on the insulin signaling pathway. Treatment withsimvastatin decreased phosphorylation of insulin receptor (pIR) non-significantly by34%, phosphorylation of insulin receptor substrate 1 (pIRS1) at Ser307, Ser612 andTyr608 by 50% (p<0.001), 40% (p<0.01), and 59% (p<0.01) in basal conditions, and by49% (p<0.01), 33% (p<0.05) and non-significantly by 11% in the insulin-stimulatedconditions. Tyrosine phosphorylation of IRS1 was strongly stimulated by insulintreatment (III, Figure 3A, C, E, G).
Effect of simvastatin on the Akt signaling and glycogen synthesis pathway.Treatment with simvastatin decreased Ser473 phosphorylation of Akt (pAkt(Ser473))by 29% in basal conditions (p<0.01 compared to control) and non-significantly by27% in insulin-stimulated conditions (compared to insulin alone). Treatment withinsulin increased pAkt(Ser473) by 292% (p<0.01) compared to control (III, Figure 4A).Treatment with simvastatin decreased phosphorylation of GSK-3 (pGSK-3 ) by50% in basal conditions (p<0.05 compared to control) and non-significantly by 12%in insulin-stimulated conditions (compared to insulin alone). Treatment with insulinincreased pGSK-3 by 73% (p<0.05) compared to control (III, Figure 4C).
Effect of simvastatin on GLUT4 protein expression. Treatment with simvastatindecreased GLUT4 protein expression by 28% in basal conditions (p<0.01 comparedto control), and non-significantly by 20% in insulin-stimulated conditions (comparedto insulin alone). Treatment with insulin alone did not have a significant effect onGLUT4 protein expression (III, Figure 4E).
Figure 8 summarizes the effects of simvastatin on the insulin signaling pathwayand its downstream target pathways.
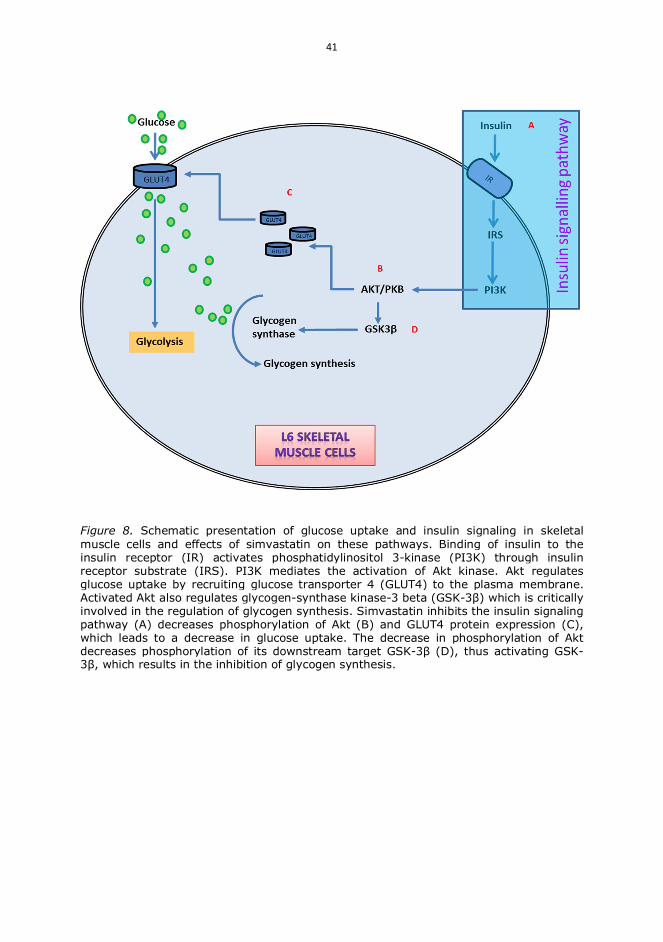
41
Figure 8. Schematic presentation of glucose uptake and insulin signaling in skeletalmuscle cells and effects of simvastatin on these pathways. Binding of insulin to theinsulin receptor (IR) activates phosphatidylinositol 3-kinase (PI3K) through insulinreceptor substrate (IRS). PI3K mediates the activation of Akt kinase. Akt regulatesglucose uptake by recruiting glucose transporter 4 (GLUT4) to the plasma membrane.Activated Akt also regulates glycogen-synthase kinase-3 beta (GSK-3 ) which is criticallyinvolved in the regulation of glycogen synthesis. Simvastatin inhibits the insulin signalingpathway (A) decreases phosphorylation of Akt (B) and GLUT4 protein expression (C),which leads to a decrease in glucose uptake. The decrease in phosphorylation of Aktdecreases phosphorylation of its downstream target GSK-3 (D), thus activating GSK-
, which results in the inhibition of glycogen synthesis.

42

43
6. Discussion
6.1 REPRESENTATIVENESS OF THE STUDY SUBJECTS
The METSIM Study (Study I)The METSIM Study included 8,479 non-diabetic Finnish men, randomly selectedfrom the population of Kuopio, Eastern Finland, aged between 45-73 years who werefollowed up for 5.9 years. New diabetes was diagnosed in 625 men by means of anOGTT, HbA1c 6.5%, or glucose-lowering medication started during the follow-up.
The large sample size of this study and careful phenotyping of the participantsallowed us to investigate the mechanisms underlying the risk of T2D associated withstatin treatment. Previous studies evaluating the diabetogenic effects of statintreatment have been selective, especially the statin trials which included participantsat high risk of CVD and hypercholesterolemia (Rajpathak et al. 2009, Waters et al.2011, Sattar et al. 2010, Preiss et al. 2011b). Therefore, the risk of diabetes in clinicaltrials is likely to differ from that in the general population, including the METSIMstudy. Furthermore, in previous studies the diagnosis of diabetes has often beenbased on self-reported diabetes or fasting glucose measurement (Culver et al. 2012,Rajpathak et al. 2009, Sattar et al. 2010, Preiss et al. 2011b) underestimating the trueincidence of T2D. No previous population-based study has applied the currentdiagnostic criteria for diabetes, namely FPG, 2hPG and HbA1c, or evaluated insulinsecretion and insulin sensitivity, potential mechanisms underlying the diabetogeniceffects of statins. In our study, insulin secretion and insulin sensitivity wereevaluated with previously validated OGTT-derived indices, which allowed us toassess the effects of statin treatment on two main pathophysiological mechanisms inthe development of T2D.
The METSIM study has limitations. It includes only Finnish men, and thereforethe validity of the results for women or for other populations remains uncertain. Dueto the large size of the METSIM study we could not use the most accurate methodsto evaluate insulin sensitivity (clamp) and insulin secretion (IVGTT orhyperglycemic clamp). Although our cohort was large, the power of our study todemonstrate significant associations of less frequently used statins with the risk ofT2D and underlying mechanisms was limited.
6.2 EVALUATION OF THE METHODS OF in vitro STUDIES (StudiesII-III)
Study II was performed using MIN6 cells as an in vitro model to investigate insulinsecretion. The use of primary -cells (from perfused pancreas or intact isolated islets)in biochemical and molecular research is limited by the availability of pancreaticendocrine tissue, the technically demanding isolation of individual pancreatic cells,cell purification and maintenance of native characteristics, as well as by cellular andhormonal heterogeneity among different individuals (Skelin et al. 2010). Severalinsulin-secreting cell lines retaining the characteristic features of -cells have been

44
developed using different techniques such as irradiation, viral transformation, andtransgenic technology. The most widely used -cell lines are RINm5F, HIT-T15,MIN6, INS-1, and BRIN-BD11 cells (Poitout 1996).
MIN6 cells originate from a transgenic C57BL/6 mouse insulinoma expressing aninsulin-promoter/T-antigen construct, and form islet-like aggregates (Poitout et al.1996). The advantage of MIN6 cells is that they express GLUT-2 and glucokinase andrespond to glucose within the physiological range (Miyazaki et al. 1990). None of thecell lines perfectly mimics primary -cell physiology. Insulin secretion from MIN6cells, cultured as monolayers, differs substantially from pancreatic islets, at leastpartly due to missing cell-to-cell contacts. It is well established that during the courseof passaging a loss of glucose-induced insulin secretion from MIN6 cells occurs(Dowling et al. 2006, O’Driscoll et al. 2006), possibly due to an outgrowth of cellswith a poor response to glucose or a reduced expression of the genes responsible forglucose-induced insulin secretion (Skelin et al. 2010, Miyazaki et al. 1990). Tominimize this problem we have not cultured MIN6 cells beyond 20 passages.
Insulin release is usually measured by radioimmunoassay (RIA) or enzyme-linkedimmunoassay (ELISA). The ELISA technique is highly sensitive and shows highspecificity and less variation compared to RIA, even at low insulin concentrations(Webster et al. 1990). In the present study we used an AlphaLISA immunoassay, achemiluminescent assay with high sensitivity, wide dynamic range and robustperformance compared to ELISA (Bielefeld-Sévigny 2009, Ullman et al. 1996).
Study III was performed using L6 cells as an in vitro model to investigate glucoseuptake and insulin signaling pathway. Skeletal muscle is the major insulin-targettissue responsible for the maintenance of whole body glucose homeostasis. The mostfrequently used cell lines to investigate insulin signaling in skeletal muscle cells arethe rat-derived muscle cell line L6 and mouse-derived muscle cell line C2C12, whichcan be differentiated into myotubes with morphological and physiologicalproperties resembling muscle fibers, and show insulin-dependent glucose uptakeand translocation of GLUT4 (Nedachi and Kanzaki 2006). When stimulated byinsulin, L6 myotubes appeared to exhibit larger glucose uptake than C2C12 cells(Sarabia et al. 1990), suggesting that L6 myotubes are the most promising candidateas an in vitro model to investigate glucose uptake in muscle cells. The advantages ofusing these cell lines include reducing the use of animals, a shorter experimentalperiod, homogeneous and easy culturing, and avoidance of the influence of systemicfactors from other organs. A major shortcoming of these muscle cell lines is adeficiency in contractibility (Manabe et al. 2012), but this is not a significantlimitation in our study.
To measure glucose uptake we used the radio-labeled analog 3H-2-deoxy-D-glucose and measured accumulated 3H-2-deoxy-D-glucose-6-phosphate, which is thegold standard method with high sensitivity, providing reproducible results(Yamamoto et al. 2015).
Pharmacological approach in Studies II-III. We used a pure pharmacologicalapproach to identify the mechanisms of statin-induced impairment in insulinsecretion in MIN6 cells and insulin sensitivity in L6 cells. Most of the compoundsthat we used affect a particular pathway or they are specific to a particular receptoractivating the pathway. Several of them (e.g. tolbutamide, GLP-1 analogs) were orare well-established drugs for treatment of patients with T2D. The dosages which we

45
used for treating the cells have been established in the same or different cell lines.The advantage of the pharmacological approach to investigate molecularmechanisms is that it is fast, reliable, and allows changing the dosage of thecompounds easily to see the effect range in order to decrease cytotoxicity. Limitationof this approach is a possibility of unknown off-target effects which may mask thepositive effects of the compound and confound the results. Although the drugs usedin our study are specific, they may have other unknown targets due to structuralsimilarities with the receptors or other proteins which the compounds are known totarget. To minimize possible off-target effects we examined each pathway with twodifferent activators or inhibitors.
6.3 ASSOCIATION OF STATIN TREATMENT WITH THE RISK OFDIABETES, INSULIN SENSITIVITY AND INSULIN SECRETION
Our study reported two main findings: 1) statin treatment was associated with anincreased risk of incident T2D and worsening of hyperglycemia during the follow-up, and 2) statin treatment was associated with a reduction in both insulin secretionand insulin sensitivity during the follow-up compared with individuals withoutstatin treatment.
The increase in the risk of incident T2D was 46% in our study. Several previousstudies using registry or prescription-based data have reported a relatively smaller(10-22%) increase in the risk of T2D than our study (Carter et al. 2013, Wang et al.2012, Zaharan et al. 2013). A similar increase in the risk of T2D by 48% with statintreatment was reported in the Women’s Health Initiative study. However, in thisstudy the diagnosis of diabetes was self-reported (Culver et al. 2012). In a meta-analysis of 13 randomized statin trials including > 91,000 participants, the diagnosisof T2D was based on the levels of FPG or a physician´s report, and statin treatmentwas associated with a 9% increased risk of T2D (Sattar et al. 2010). In our study, T2Dwas diagnosed based on FPG, 2hPG and HbA1c (in 56.5% of the diabetes diagnoses),in addition to the prescription-based data, hospital registry data and physician´sreports. Our observation of the higher risk of T2D with statin treatment suggests thatprevious studies using FPG or physician´s reports as a sole criterion for thediagnosis of T2D may have underestimated incident diabetes in individuals treatedwith statin. Furthermore, statin trials in particular have selective inclusion criteria(especially hypercholesterolemia), and therefore they do not represent the generalpopulation which likely contributes to differences in the rates of incident T2D.Importantly, the individuals who developed diabetes on statin treatment in ourstudy had a similar metabolic risk factor profile at baseline to those who developeddiabetes without statin treatment, suggesting that statins increased the risk ofdiabetes independently of the risk profile of the participants at baseline.
We observed differences in the diabetogenic effects of different statins.Atorvastatin and simvastatin were the most diabetogenic statins, whereaspravastatin, fluvastatin and lovastatin were less diabetogenic, similarly as reportedin a previous study (Navarese et al. 2013). The risk of incident T2D with simvastatinand atorvastatin treatment increased in a dose-dependent manner in our study, inagreement with a meta-analysis including five statin trials (Preiss et al. 2011b).
In addition to the increased risk of incident T2D, we report the novel finding thatstatin treatment was associated with an increase in the levels of 2hPG during the

46
follow-up in participants without previously diagnosed diabetes. This suggests thatstatin treatment may increase the risk of T2D mainly through the elevation of 2hPGlevels. Previous studies have reported increased levels of FPG in non-diabeticindividuals receiving statin treatment (Sukhija et al. 2009, Rautio et al. 2012),whereas others did not confirm such an association (Moutzouri et al. 2011).
To investigate the mechanisms underlying statin-induced diabetes, we analyzedthe association of simvastatin and atorvastatin treatment with changes in the OGTT-based indices of insulin sensitivity (Matsuda ISI) and insulin secretion (Dispositionindex) during the follow-up. Our study demonstrated a significant dose-dependentdecrease in Matsuda ISI during the follow-up in participants receiving simvastatinor atorvastatin. This is in agreement with the elevation in 2hPG, which is determinedpredominantly by insulin resistance (Stancakova et al. 2009). Our results are also inagreement with a meta-analysis of 16 statin trials showing that simvastatin treatmentwas associated with increased insulin resistance (Baker et al. 2010). Furthermore, inour study simvastatin and atorvastatin treatments were associated with a significantdecrease in the DI reflecting insulin secretion relative to insulin sensitivity. In a smalltrial including 24 Japanese patients with T2D, atorvastatin treatment resulted in alower OGTT-based DI than pravastatin treatment during 12 weeks (Mita et al. 2007).Our results suggest that impaired ability of -cells to respond adequately todecreased insulin sensitivity is probably the mechanism underlying thehyperglycemic and diabetogenic effects of simvastatin and atorvastatin.
6.4 MOLECULAR MECHANISMS OF THE EFFECTS OF SIMVASTATINON INSULIN SECRETION AND INSULIN SENSITIVITY
Effects of simvastatin and pravastatin on insulin secretion and insulin sensitivity.Our METSIM study gave a good starting point to investigate in detail themechanisms of insulin secretion and insulin resistance with in vitro models. Ourstudies showed that the treatment with simvastatin decreased both glucose-stimulated insulin secretion in MIN6 -cells and insulin-stimulated glucose uptakein L6 myotubes. These effects were observed at both normal (5.5 mM) and high (16.7mM) glucose concentrations indicating that the effects of simvastatin are notsubstantially affected by the presence of hyperglycemia. This finding agrees withstudies showing that statin treatment does not essentially worsen glucose control inindividuals with diabetes (Erqou et al. 2014, Colhoun et al. 2004).
In contrast to simvastatin, pravastatin did not decrease either insulin secretion inMIN6 cells or glucose uptake in L6 myotubes. Our observation is in agreement withprevious reports showing that simvastatin, but not pravastatin, was associated withan increased risk of incident diabetes (Sattar et al. 2010). As simvastatin is lipophilicand pravastatin hydrophilic, it has been proposed that the difference in their effectsstems from differences in lipophilicity (Kahn et al. 2014). However, rosuvastatin ishydrophilic but increases substantially the risk of diabetes (Navarese et al. 2013), andtherefore lipophilicity alone cannot explain the differences in the diabetogenic effectsof statins.
Some (Smith et al. 2014, Metz et al. 1993, Nakata et al. 2006), but not all (Kain et al.2015) studies suggest that the effects of statins on insulin secretion and insulinsensitivity are mediated by their inhibitory effect on cholesterol biosynthesis and thesynthesis of isoprenoid intermediates derived from mevalonate. The isoprenoids

47
GGPP and FPP act as substrates for prenylation of numerous cellular proteinsplaying a role in insulin secretion (Tsuchiya et al. 2010) and insulin signaling (Xu etal. 2015). In our study, GGPP and FPP inhibitors decreased insulin secretion inaccordance with previous studies (Tsuchiya et al. 2010, Kowluru 2003), and alsodecreased glucose uptake in L6 myotubes. However, addition of mevalonate, GGPPor FPP, did not restore the simvastatin-induced decrease in insulin secretion,whereas mevalonate and GGPP, but not FPP, did restore simvastatin-decreasedglucose uptake in L6 myotubes. Therefore, it is apparent that the effect ofsimvastatin on insulin secretion is mediated by mechanisms other than the inhibitionof the cholesterol biosynthetic pathway. In contrast, decreased glucose uptake bysimvastatin in L6 myotubes may be partly mediated by an inhibition of themevalonate pathway of cholesterol synthesis leading to an alteration of post-translational prenylation of proteins by GGPP. In previous studies, the treatmentwith mevalolactone restored simvastatin-decreased glucose uptake in humanmyotubes (Smith et al. 2014), but not in another study using L6 myotubes (Kain et al.2015).
To conclude, our in vitro studies contributed several novel findings andmechanisms to the understanding of simvastatin-induced impairment in insulinsecretion and insulin sensitivity.
Molecular mechanisms of simvastatin-induced impairment in insulin secretionWe confirmed previously reported results that simvastatin has inhibitory effects oninsulin secretion via KATP and VGCC channels (Yada et al. 1999, Zhou et al. 2014).We demonstrated that simvastatin inhibited insulin secretion stimulated bytolbutamide (closes the KATP channels), and KCl (directly depolarizes plasmamembrane and leads to the opening of VGCCs) by inhibiting the glucose-inducedraise of intracellular Ca2+ in a dose-dependent manner. Furthermore, wedemonstrated that simvastatin decreased insulin secretion via additional pathwaysand receptors such as acetylcholine receptor, GPR40, and by inhibition of Ca2+
release from intracellular stores in MIN6 cells.The inhibitory effects of simvastatin on insulin secretion mediated by
acetylcholine and GPR40 are novel findings. Both acetylcholine M3R and GPR40receptors belong to the same family of G-protein coupled receptors (Blad et al. 2012)present on the plasma membrane of -cells. Insulin secretion mediated by the M3Rand GPR40 receptors is dependent on Ca2+ influx into the -cell (Feng et al. 2006),and Ca2+ release from intracellular stores, such as the ER (Itoh et al. 2003). Our resultsshowing that simvastatin abolished insulin secretion stimulated by caffeine, anactivator of ryanodine receptor RyR on the ER, suggest that simvastatin decreasesCa2+ release from the ER. M3Rs activate insulin secretion also by the formation ofDAG, which activates PKC (Gautam et al. 2006). Our experiments with the DAGanalog PMA showed that although a direct stimulation of PKC activity partiallyreversed the simvastatin-induced decrease in insulin secretion, the activity of PKCwas not affected by simvastatin.
Another novel finding was that decreased insulin secretion by simvastatin waspartially restored via the activation of GPR119 or GLP-1R signaling. Agonists of bothGLP-1R and GPR119 increase insulin secretion by stimulating adenylate cyclasewhich catalyzes the conversion of ATP to cAMP (Doyle and Egan 2007, Sato et al.2014). In our study a direct activation of adenylate cyclase by forskolin also restoredinsulin secretion decreased by simvastatin. The downstream targets of cAMP, PKA

48
and EPAC2, are crucial in the GLP-1R-mediated insulin secretion (Chepurny et al.2009, Kelley et al. 2009, Seino et al. 2009), opening of VGCCs, and closure of KATP
channels. Our experiments with EPAC2 and PKA inhibitors also suggest a role ofPKA and EPAC2 in GPR119-mediated insulin secretion. Using cAMP analogsactivating both PKA and EPAC2 (8-bromo-cAMP), or a specific activator of theEPACs (8-pCPT-2 -O-Me-cAMP), we showed for the first time that simvastatinreduced insulin secretion via both EPAC2 and PKA pathways.
The GPR119 and GLP-1R pathways require the coupling of glucose-induced Ca2+
influx through VGCCs to enhance insulin exocytosis (Ning et al. 2008). The GLP-1Rpathway participates in Ca2+ release where Ca2+ influx through VGCCs facilitates therelease of Ca2+ from intracellular stores (Graves and Hinkle 2003, Nakagaki et al.2000) via IP3R and RyR (Kang et al. 2001, Kang et al. 2005). GRP119 likely exertssimilar effects since forskolin has been found to potentiate caffeine-induced Ca2+
spikes in cultured -cells (Islam et al. 1992). However, in our study GLP-1R andGPR119 agonists were not able to counteract the inhibitory effect of simvastatin onintracellular Ca2+ levels after glucose stimulation. This suggests that the Ca2+-independent effects of cAMP (Yajima et al. 1999, Ammälä et al. 1993) might beresponsible for the restoration of simvastatin-decreased insulin secretion by theactivation of GLP-1R and GPR119 receptors. One such possibility is a directinteraction of cAMP with the secretory machinery (Ammälä et al. 1993).
Molecular mechanisms of simvastatin-induced impairment in insulin sensitivityIn our study simvastatin decreased both basal and insulin-stimulated glucoseuptake, in contrast to an earlier report (Kain et al. 2015) where simvastatin andatorvastatin decreased only insulin-stimulated glucose uptake with no significanteffect on basal glucose uptake in L6 myotubes. Similar to our results, a dose-dependent effect of simvastatin on glucose uptake in the absence of insulin has beenreported in human myotubes (Smith et al. 2014).
Glucose uptake results from a series of events in the insulin signaling cascadeleading to the phosphorylation of Akt, which then promotes glucose uptake byfacilitating GLUT4 translocation from intracellular stores to the plasma membrane(Wang et al. 1999). We systematically investigated the effects of simvastatin on theinsulin signaling pathway and found that simvastatin inhibited tyrosinephosphorylation of IR (Tyr1361) and IRS1 (Tyr698) in basal and insulin-stimulatedconditions, indicating that simvastatin has a direct inhibitory effect on the pathway.Furthermore, simvastatin decreased the phosphorylation of Akt at both basal andinsulin-stimulated conditions. In previous studies, atorvastatin was shown to down-regulate the IR beta-subunit and IRS1, and to suppress the insulin-induced activationof IRS1 and Akt in adipocytes (Takaguri et al. 2008, Nakata et al. 2006). Lovastatindecreased the phosphorylation of the IR beta-subunit in fibroblasts (McGuire et al.1994). In L6 myotubes (Kain et al. 2015) simvastatin decreased the phosphorylationof IRS1 and Akt in insulin-stimulated conditions, consistent with the presentfindings.
GLUT4 is required for both insulin-stimulated and basal glucose uptake (Zismanet al. 2000). We showed that the treatment of L6 myotubes with simvastatindecreased protein expression of GLUT4 at both basal and insulin-stimulatedconditions. Similar inhibitory effects on GLUT4 protein expression have beenreported in 3T3-L1 adipocytes with simvastatin (Ganesan and Ito 2013), lovastatin(Chamberlain 2001) and atorvastatin (Takaguri et al. 2008, Nakata et al. 2006).

49
Defects in GLUT4 trafficking or protein expression are known to cause insulinresistance (Bogan 2012). Our results add further evidence that decreased proteinexpression of GLUT4 contributes to simvastatin-decreased glucose uptake in L6myotubes.
A downstream target of Akt playing a role in muscle insulin resistance is GSK-3 ,a serine/threonine kinase essential for the regulation of glycogen synthesis.Unphosphorylated GSK-3 inhibits the activity of GS, and Akt directlyphosphorylates and inactivates GSK-3 (Roach and Larner 1977). Impaired insulin-stimulated glycogen synthesis has been found in patients with T2D and persons atthe risk of this disease (DeFronzo 2009). We report a novel finding that the treatmentwith simvastatin decreased GSK-3 phosphorylation, thus activating GSK-3 , whichcould result in the inhibition of GS. Our results suggest that simvastatin may affectglucose utilization in skeletal muscle, in addition to altered glucose uptake. Weobserved that the effects of simvastatin on glucose uptake and insulin signaling wereessentially similar at normal and high glucose, suggesting that these mechanisms areindependent of glucose levels.
6.5 CONCLUDING REMARKS
Our population-based METSIM study demonstrated that a widely used statin,simvastatin, was associated with impairment of both insulin secretion and insulinsensitivity. Our in vitro studies extended the previous findings on molecularmechanisms underlying the diabetogenic effects of statins. We showed thatsimvastatin decreased insulin secretion and insulin sensitivity via several pathwaysand targets, suggesting multiple mechanisms for statin-induced diabetes. Theseeffects were not entirely explained by the inhibited biosynthesis of cholesterol andisoprenoid intermediates in skeletal muscle cells or by impaired Ca2+ flux into the -cell and reduced intracellular calcium stores. In contrast, pravastatin had no adverseeffects on insulin secretion or insulin sensitivity.
Important novel findings of our study were that the simvastatin-reduced insulinsecretion was partially restored by the activation of GPR119 or GLP-1R signaling.These results suggest that the agonists of GLP-1R and GPR119 could be especiallybeneficial for individuals with or at risk of developing T2D treated with simvastatin.Further studies are needed to test this hypothesis.
There are several other possible mechanisms of statin-induced diabetes andimpairment in insulin secretion and insulin sensitivity which we did not investigate.Statins may cause a depletion in membrane cholesterol, which has been shown to beassociated with insulin resistance (Le Lay et al. 2001, Parpal et al. 2001). Statins maycause insulin resistance also by immune mechanisms, since they were shown toactivate NLRP3, a pyrin-like protein playing a crucial role in innate immunity andinflammation (Henriksbo et al. 2014). Statins are associated with reduced levels ofCoenzyme Q10 (Larsen et al. 2013), a molecule synthesized from isoprene residuesand playing a central role in mitochondrial oxidative phosphorylation, and thus maycontribute to both insulin resistance (Ganesan and Ito 2013) and –cell dysfunction.Up-regulation of LDL receptors by statins leading to cholesterol accumulation insidethe cell may also lead to -cell dysfunction (Besseling et al. 2015).
In spite of the diabetogenic effects of statins, these drugs are generally well-tolerated and their benefits in preventing cardiovascular outcomes outweigh the risk

50
of NODM according to many large cholesterol lowering trials. Therefore, if LDLlowering treatment is indicated, treatment with statins should be continued for theprevention of cardiovascular events in people without diabetes, with monitoring ofglucose tolerance and implementation of lifestyle changes to prevent the worseningof glucose tolerance. More research is needed to understand detailed molecularmechanisms of the harmful effects of statins to help physicians to select the besttreatment option for an individual patient with hypercholesterolemia and a high riskof T2D.

51
7. Summary
The main findings of the studies I-III were:
Study I: Treatment with statins was associated with a 46% increase in the risk ofincident T2D during a 6-year follow-up. Statin treatment was also associated with a24% decrease in insulin sensitivity and a 12% decrease in insulin secretion comparedto individuals without statin treatment. These effects were dose dependent forsimvastatin and atorvastatin.
Study II: Simvastatin, but not pravastatin, decreased glucose-stimulated insulinsecretion in MIN6 -cells. Simvastatin induced a decrease in insulin secretionthrough multiple targets such as the ATP-sensitive potassium channels, voltage-gated calcium channels, muscarinic M3 receptors, GPR40 receptor, and calciumrelease from the endoplasmic reticulum. Impaired insulin secretion caused bysimvastatin was effectively restored by GPR119 or GLP-1 receptor stimulation andby direct activation of cAMP-dependent signaling pathways with forskolin.
Study III: Simvastatin, but not pravastatin, decreased glucose uptake in both basaland insulin-stimulated conditions in L6 myotubes. The effect of simvastatin wasattributable to impaired insulin signaling due to decreased phosphorylation of IR,IRS1 and its downstream targets Akt and GSK-3 , as well as to decreased proteinexpression of glucose transporter GLUT4. The effect of simvastatin on glucoseuptake was reversed with geranylgeranyl pyrophosphate, implying the role ofprotein geranylgeranylation in maintaining insulin sensitivity.

52

53
References
Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E et al. Adipose-selectivetargeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature2001; 409: 729-733.
Adeva-Andany MM, Gonzalez-Lucan M, Donapetry-Garcia C, Fernandez-FernandezC, Ameneiros-Rodriguez E. Glycogen metabolism in humans. BBA Clin 2016; 5:85-100.
Ahmad F, Lindh R, Tang Y, Ruishalme I, Ost A, Sahachartsiri B et al. Differentialregulation of adipocyte PDE3B in distinct membrane compartments by insulinand the beta3-adrenergic receptor agonist CL316243: effects of caveolin-1knockdown on formation/maintenance of macromolecular signalling complexes.Biochem J 2009; 424: 399-410.
Ahren B. Islet G protein-coupled receptors as potential targets for treatment of type 2diabetes. Nat Rev Drug Discov 2009; 8: 369-385.
Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C et al. Mevinolin: a highlypotent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductaseand a cholesterol-lowering agent. Proc Natl Acad Sci U S A 1980; 77: 3957-3961.
Alberts AW. Lovastatin and simvastatin--inhibitors of HMG CoA reductase andcholesterol biosynthesis. Cardiology 1990; 77 Suppl 4: 14-21.
Almahariq M, Mei FC, Cheng X. Cyclic AMP sensor EPAC proteins and energyhomeostasis. Trends Endocrinol Metab 2014; 25: 60-71.
Altunba H, Balci MK, Karayalcin U. No effect of simvastatin treatment on insulinsensitivity in patients with primary hypercholesterolemia. Endocr Res 2003; 29:265-275.
American Diabetes Association. Standards of Medical Care in Diabetes—2006.Diabetes Care 2006; 29: S4-S42.
American Diabetes Association. Standards of Medical Care in Diabetes—2013.Diabetes Care 2013; 36: S11-S66.
American Diabetes Association. Standards of Medical Care in Diabetes—2015.Diabetes Care 2015; 38: S1-S93.
Ammälä C, Ashcroft FM, Rorsman P. Calcium-independent potentiation of insulinrelease by cyclic AMP in single beta-cells. Nature 1993; 363: 356-358.
Ansarullah, Lu Y, Holstein M, DeRuyter B, Rabinovitch A, Guo Z. Stimulating beta-cell regeneration by combining a GPR119 agonist with a DPP-IV inhibitor. PLoSOne 2013; 8: e53345.
Araki S, Dobashi K, Asayama K, Shirahata A. Simvastatin enhances induction ofinducible nitric oxide synthase in 3T3-L1 adipocytes. Free Radic Res 2007; 41:1028-1034.

54
Arnaboldi L, Corsini A. Could changes in adiponectin drive the effect of statins onthe risk of new-onset diabetes? The case of pitavastatin. Atheroscler Suppl 2015;16: 1-27.
Bai L, Wang Y, Fan J, Chen Y, Ji W, Qu A et al. Dissecting multiple steps of GLUT4trafficking and identifying the sites of insulin action. Cell Metab 2007; 5: 47-57.
Baker WL, Talati R, White CM, Coleman CI. Differing effect of statins on insulinsensitivity in non-diabetics: a systematic review and meta-analysis. Diabetes ResClin Pract 2010; 87: 98-107.
Bandyopadhyay G, Standaert ML, Galloway L, Moscat J, Farese RV. Evidence forinvolvement of protein kinase C (PKC)-zeta and noninvolvement ofdiacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6myotubes. Endocrinology 1997; 138: 4721-4731.
Beck-Nielsen H. The role of glycogen synthase in the development of hyperglycemiain type 2 diabetes: 'To store or not to store glucose, that's the question'. DiabetesMetab Res Rev 2012; 28: 635-644.
Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation andrupture. Circ Res 2014; 114: 1852-1866.
Besseling J, Kastelein JJ, Defesche JC, Hutten BA, Hovingh GK. Association betweenfamilial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA2015; 313: 1029-1036.
Betteridge DJ, Carmena R. The diabetogenic action of statins - mechanisms andclinical implications. Nat Rev Endocrinol 2016; 12: 99-110.
Bielefeld-Sévigny M. AlphaLISA immunoassays platform - the no-wash high-throughput alternative to ELISA. Assay Drug Development Technology 2009; 7:90-92.
Blad CC, Tang C, Offermanns S. G protein-coupled receptors for energy metabolitesas new therapeutic targets. Nat Rev Drug Discov 2012; 11: 603-619.
Bobryshev YV, Ivanova EA, Chistiakov DA, Nikiforov NG, Orekhov AN.Macrophages and Their Role in Atherosclerosis: Pathophysiology andTranscriptome Analysis. Biomed Res Int 2016; 2016: 9582430.
Bogan JS. Regulation of glucose transporter translocation in health and diabetes.Annu Rev Biochem 2012; 81: 507-532.
Bond P. Regulation of mTORC1 by growth factors, energy status, amino acids andmechanical stimuli at a glance. J Int Soc Sports Nutr 2016; 13: 8-016-0118-y.eCollection 2016.
Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal andinsulin-resistant states. Cold Spring Harb Perspect Biol 2014; 6:10.1101/cshperspect.a009191.
Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM et al.The orphan G protein-coupled receptor GPR40 is activated by medium and longchain fatty acids. J Biol Chem 2003; 278: 11303-11311.
Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenicparadox. Cell Metab 2008; 7: 95-96.

55
Bunner AE, Chandrasekera PC, Barnard ND. Knockout mouse models of insulinsignaling: Relevance past and future. World J Diabetes 2014; 5: 146-159.
Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretinhormone action. Cell Metab 2013; 17: 819-837.
Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R et al.Intensive versus moderate lipid lowering with statins after acute coronarysyndromes. N Engl J Med 2004; 350: 1495-1504.
Carbonell T, Freire E. Binding thermodynamics of statins to HMG-CoA reductase.Biochemistry 2005; 44: 11741-11748.
Carnagarin R, Dharmarajan AM, Dass CR. Molecular aspects of glucose homeostasisin skeletal muscle--A focus on the molecular mechanisms of insulin resistance.Mol Cell Endocrinol 2015; 417: 52-62.
Carter AA, Gomes T, Camacho X, Juurlink DN, Shah BR, Mamdani MM. Risk ofincident diabetes among patients treated with statins: population based study.BMJ 2013; 346: f2610.
Cefalu WT. Insulin resistance: cellular and clinical concepts. Exp Biol Med(Maywood) 2001; 226: 13-26.
Cerqueira NM, Oliveira EF, Gesto DS, Santos-Martins D, Moreira C, Moorthy HN etal. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016; 55:5483-5506.
Cersosimo E, Triplitt C, Mandarino LJ, DeFronzo RA. Pathogenesis of Type 2Diabetes Mellitus. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR,Grossman A, Hershman JM et al, eds. Endotext. South Dartmouth (MA):MDText.com, Inc, 2000.
Chamberlain LH. Inhibition of isoprenoid biosynthesis causes insulin resistance in3T3-L1 adipocytes. FEBS Lett 2001; 507: 357-361.
Chan DC, Pang J, Watts GF. Pathogenesis and management of the diabetogeniceffect of statins: a role for adiponectin and coenzyme Q10? Curr Atheroscler Rep2015; 17: 472.
Chapman MJ, Orsoni A, Robillard P, Hounslow N, Sponseller CA, Giral P. Effect ofhigh-dose pitavastatin on glucose homeostasis in patients at elevated risk ofnew-onset diabetes: insights from the CAPITAIN and PREVAIL-US studies.Curr Med Res Opin 2014; 30: 775-784.
Chen H, Fajol A, Hoene M, Zhang B, Schleicher ED, Lin Y et al. PI3K-resistant GSK3controls adiponectin formation and protects from metabolic syndrome. ProcNatl Acad Sci U S A 2016; 113: 5754-5759.
Chen ZY, Liu SN, Li CN, Sun SJ, Liu Q, Lei L et al. Atorvastatin helps preservepancreatic beta cell function in obese C57BL/6 J mice and the effect is related toincreased pancreas proliferation and amelioration of endoplasmic-reticulumstress. Lipids Health Dis 2014; 13: 98-511X-13-98.
Chepurny OG, Leech CA, Kelley GG, Dzhura I, Dzhura E, Li X et al. Enhanced Rap1activation and insulin secretagogue properties of an acetoxymethyl ester of an

56
Epac-selective cyclic AMP analog in rat INS-1 cells: studies with 8-pCPT-2'-O-Me-cAMP-AM. J Biol Chem 2009; 284: 10728-10736.
Chepurny OG, Holz GG, Roe MW, Leech CA. GPR119 Agonist AS1269574 ActivatesTRPA1 Cation Channels to Stimulate GLP-1 Secretion. Mol Endocrinol 2016; 30:614-629.
Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB,3rd et al. Insulinresistance and a diabetes mellitus-like syndrome in mice lacking the proteinkinase Akt2 (PKB beta). Science 2001; 292: 1728-1731.
Chogtu B, Magazine R, Bairy KL. Statin use and risk of diabetes mellitus. World JDiabetes 2015; 6: 352-357.
Choi SM, Tucker DF, Gross DN, Easton RM, DiPilato LM, Dean AS et al. Insulinregulates adipocyte lipolysis via an Akt-independent signaling pathway. MolCell Biol 2010; 30: 5009-5020.
Cholesterol Treatment Trialists' (CTT) Collaboration, Baigent C, Blackwell L,Emberson J, Holland LE, Reith C et al. Efficacy and safety of more intensivelowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in26 randomised trials. Lancet 2010; 376: 1670-1681.
Cholesterol Treatment Trialists' (CTT) Collaboration, Emberson JR, Kearney PM,Blackwell L, Newman C, Reith C et al. Lack of effect of lowering LDL cholesterolon cancer: meta-analysis of individual data from 175,000 people in 27randomised trials of statin therapy. PLoS One 2012; 7: e29849.
Chu ZL, Jones RM, He H, Carroll C, Gutierrez V, Lucman A et al. A role for beta-cell-expressed G protein-coupled receptor 119 in glycemic control by enhancingglucose-dependent insulin release. Endocrinology 2007; 148: 2601-2609.
Coleman CI, Reinhart K, Kluger J, White CM. The effect of statins on thedevelopment of new-onset type 2 diabetes: a meta-analysis of randomizedcontrolled trials. Curr Med Res Opin 2008; 24: 1359-1362.
Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ etal. Primary prevention of cardiovascular disease with atorvastatin in type 2diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentrerandomised placebo-controlled trial. Lancet 2004; 364: 685-696. Collins R, ReithC, Emberson J, Armitage J, Baigent C, Blackwell L et al. Interpretation of theevidence for the efficacy and safety of statin therapy. Lancet 2016; 388: 2532-2561.
Colpo A. LDL cholesterol: Bad cholesterol, or bad science? Journal of AmericanPhysicians and Surgeons 2005: 10: 83-89.
Consitt LA, Van Meter J, Newton CA, Collier DN, Dar MS, Wojtaszewski JF et al.Impairments in site-specific AS160 phosphorylation and effects of exercisetraining. Diabetes 2013; 62: 3437-3447.
Copps KD, White MF. Regulation of insulin sensitivity by serine/threoninephosphorylation of insulin receptor substrate proteins IRS1 and IRS2.Diabetologia 2012; 55: 2565-2582.
Cornell S. Continual evolution of type 2 diabetes: an update on pathophysiology andemerging treatment options. Ther Clin Risk Manag 2015; 11: 621-632.

57
Corrao G, Ibrahim B, Nicotra F, Soranna D, Merlino L, Catapano AL et al. Statins andthe risk of diabetes: evidence from a large population-based cohort study.Diabetes Care 2014; 37: 2225-2232.
Cruz PM, Mo H, McConathy WJ, Sabnis N, Lacko AG. The role of cholesterolmetabolism and cholesterol transport in carcinogenesis: a review of scientificfindings, relevant to future cancer therapeutics. Front Pharmacol 2013; 4: 119.
Culver AL, Ockene IS, Balasubramanian R, Olendzki BC, Sepavich DM, Wactawski-Wende J et al. Statin use and risk of diabetes mellitus in postmenopausal womenin the Women's Health Initiative. Arch Intern Med 2012; 172: 144-152.
Danielsson A, Ost A, Nystrom FH, Stralfors P. Attenuation of insulin-stimulatedinsulin receptor substrate-1 serine 307 phosphorylation in insulin resistance oftype 2 diabetes. J Biol Chem 2005; 280: 34389-34392.
Dansette PM, Jaoen M, Pons C. HMG-CoA reductase activity in human livermicrosomes: comparative inhibition by statins. Exp Toxicol Pathol 2000; 52: 145-148.
Davis S. Diabetogenic drugs: Treating chronic conditions to minimise new onsetdiabetes. SA Pharmaceutical Journal 2010; 77: 22-33.
de Lemos JA, Zirlik A, Schonbeck U, Varo N, Murphy SA, Khera A et al. Associationsbetween soluble CD40 ligand, atherosclerosis risk factors, and subclinicalatherosclerosis: results from the Dallas Heart Study. Arterioscler Thromb VascBiol 2005; 25: 2192-2196.
DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect intype 2 diabetes. Diabetes Care 2009; 32 Suppl 2: S157-63.
DeFronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a newparadigm for the treatment of type 2 diabetes mellitus. Diabetes 2009; 58: 773-795.
Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancerprevention. Nat Rev Cancer 2005; 5: 930-942.
Dormuth CR, Filion KB, Paterson JM, James MT, Teare GF, Raymond CB et al.Higher potency statins and the risk of new diabetes: multicentre, observationalstudy of administrative databases. BMJ 2014; 348: g3244.
Dowling P, O'Driscoll L, O'Sullivan F, Dowd A, Henry M, Jeppesen PB et al.Proteomic screening of glucose-responsive and glucose non-responsive MIN-6beta cells reveals differential expression of proteins involved in protein folding,secretion and oxidative stress. Proteomics 2006; 6: 6578-6587.
Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA et al. Primaryprevention of acute coronary events with lovastatin in men and women withaverage cholesterol levels: results of AFCAPS/TexCAPS. Air Force/TexasCoronary Atherosclerosis Prevention Study. JAMA 1998; 279: 1615-1622.
Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in thepancreas. Pharmacol Ther 2007; 113: 546-593.
Dulak J, Jozkowicz A. Anti-angiogenic and anti-inflammatory effects of statins:relevance to anti-cancer therapy. Curr Cancer Drug Targets 2005; 5: 579-594.

58
Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS et al. Role forAkt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol2005; 25: 1869-1878.
Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells andmediates free fatty acid stimulation of incretin secretion. Diabetes 2008; 57: 2280-2287.
Edwards CM, Cusi K. Prediabetes: A Worldwide Epidemic. Endocrinol Metab ClinNorth Am 2016; 45: 751-764.
Ekberg JH, Hauge M, Kristensen LV, Madsen AN, Engelstoft MS, Husted AS et al.GPR119, a Major Enteroendocrine Sensor of Dietary Triglyceride MetabolitesCoacting in Synergy With FFA1 (GPR40). Endocrinology 2016; 157: 4561-4569.
Endo A, Kuroda M, Tanzawa K. Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase by ML-236A and ML-236B fungalmetabolites, having hypocholesterolemic activity. FEBS Lett 1976; 72: 323-326.
Endo A. The discovery and development of HMG-CoA reductase inhibitors. J LipidRes 1992; 33: 1569-1582.
Endo A. A historical perspective on the discovery of statins. Proc Jpn Acad Ser BPhys Biol Sci 2010; 86: 484-493.
Erqou S, Lee CC, Adler AI. Statins and glycaemic control in individuals withdiabetes: a systematic review and meta-analysis. Diabetologia 2014; 57: 2444-2452.
Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol 2006; 47: C7-12.Fall T, Xie W, Poon W, Yaghootkar H, Magi R, GENESIS Consortium et al. Using
Genetic Variants to Assess the Relationship Between Circulating Lipids andType 2 Diabetes. Diabetes 2015; 64: 2676-2684.
Feng DD, Luo Z, Roh SG, Hernandez M, Tawadros N, Keating DJ et al. Reduction involtage-gated K+ currents in primary cultured rat pancreatic beta-cells bylinoleic acids. Endocrinology 2006; 147: 674-682.
Fessler MB. The Intracellular Cholesterol Landscape: Dynamic Integrator of theImmune Response. Trends Immunol 2016; 37: 819-830.
Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev 2013; 93:137-188.
Freeman DJ, Norrie J, Sattar N, Neely RD, Cobbe SM, Ford I et al. Pravastatin and thedevelopment of diabetes mellitus: evidence for a protective treatment effect inthe West of Scotland Coronary Prevention Study. Circulation 2001; 103: 357-362.
Fridlyand LE, Philipson LH. Pancreatic Beta Cell G-Protein Coupled Receptors andSecond Messenger Interactions: A Systems Biology Computational Analysis.PLoS One 2016; 11: e0152869.
Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues ofobese subjects release interleukin-6: depot difference and regulation byglucocorticoid. J Clin Endocrinol Metab 1998; 83: 847-850.
Fu Z, Gilbert ER, Liu D. Regulation of insulin synthesis and secretion and pancreaticBeta-cell dysfunction in diabetes. Curr Diabetes Rev 2013; 9: 25-53.

59
Fujiwara K, Maekawa F, Yada T. Oleic acid interacts with GPR40 to induce Ca2+signaling in rat islet beta-cells: mediation by PLC and L-type Ca2+ channel andlink to insulin release. Am J Physiol Endocrinol Metab 2005; 289: E670-7.
Ganesan S, Ito MK. Coenzyme Q10 ameliorates the reduction in GLUT4 transporterexpression induced by simvastatin in 3T3-L1 adipocytes. Metab Syndr RelatDisord 2013; 11: 251-255.
Ganic E, Singh T, Luan C, Fadista J, Johansson JK, Cyphert HA et al. MafA-Controlled Nicotinic Receptor Expression Is Essential for Insulin Secretion and IsImpaired in Patients with Type 2 Diabetes. Cell Rep 2016; 14: 1991-2002.
Gao Y, Moten A, Lin HK. Akt: a new activation mechanism. Cell Res 2014; 24: 785-786.
Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL et al. Severediabetes, age-dependent loss of adipose tissue, and mild growth deficiency inmice lacking Akt2/PKB beta. J Clin Invest 2003; 112: 197-208.
Gaster M, Petersen I, Hojlund K, Poulsen P, Beck-Nielsen H. The diabetic phenotypeis conserved in myotubes established from diabetic subjects: evidence forprimary defects in glucose transport and glycogen synthase activity. Diabetes2002; 51: 921-927.
Gautam D, Han SJ, Hamdan FF, Jeon J, Li B, Li JH et al. A critical role for beta cell M3muscarinic acetylcholine receptors in regulating insulin release and bloodglucose homeostasis in vivo. Cell Metab 2006; 3: 449-461.
George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC et al. A familywith severe insulin resistance and diabetes due to a mutation in AKT2. Science2004; 304: 1325-1328.
Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergiccontrol of pancreatic beta-cell function. Endocr Rev 2001; 22: 565-604.
Giraud J, Leshan R, Lee YH, White MF. Nutrient-dependent and insulin-stimulatedphosphorylation of insulin receptor substrate-1 on serine 302 correlates withincreased insulin signaling. J Biol Chem 2004; 279: 3447-3454.
Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990; 343:425-430.
Gonzalez E, Flier E, Molle D, Accili D, McGraw TE. Hyperinsulinemia leads touncoupled insulin regulation of the GLUT4 glucose transporter and the FoxO1transcription factor. Proc Natl Acad Sci U S A 2011; 108: 10162-10167.
Gonzalez-Franquesa A, De Nigris V, Lerin C, Garcia-Roves PM. Skeletal MuscleMitochondrial Function/Dysfunction and Type 2 Diabetes, Skeletal Muscle -From Myogenesis to Clinical Relations. In: Cseri J (Ed) 2012, InTech, DOI:10.5772/50130.
Goodpaster BH, Bertoldo A, Ng JM, Azuma K, Pencek RR, Kelley C et al. Interactionsamong glucose delivery, transport, and phosphorylation that underlie skeletalmuscle insulin resistance in obesity and type 2 Diabetes: studies with dynamicPET imaging. Diabetes 2014; 63: 1058-1068.

60
Graves TK, Hinkle PM. Ca(2+)-induced Ca(2+) release in the pancreatic beta-cell:direct evidence of endoplasmic reticulum Ca(2+) release. Endocrinology 2003;144: 3565-3574.
Gromada J. The free fatty acid receptor GPR40 generates excitement in pancreaticbeta-cells. Endocrinology 2006; 147: 672-673.
Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights frommouse models into disease mechanisms. J Endocrinol 2014; 220: T1-T23.
Guridi M, Kupr B, Romanino K, Lin S, Falcetta D, Tintignac L et al. Alterations tomTORC1 signaling in the skeletal muscle differentially affect whole-bodymetabolism. Skelet Muscle 2016; 6: 13.
Häggblad Sahlberg S, Mortensen AC, Haglöf J, Engskog MK, Arvidsson T,Pettersson C et al. Different functions of AKT1 and AKT2 in molecularpathways, cell migration and metabolism in colon cancer cells. Int J Oncol 2016.
Hajar R. Statins: past and present. Heart Views 2011; 12: 121-127.
Hamdouchi C, Kahl SD, Patel Lewis A, Cardona GR, Zink RW, Chen K et al. TheDiscovery, Preclinical and Early Clinical Development of Potent and SelectiveGPR40 Agonists for the Treatment of Type 2 Diabetes Mellitus (LY2881835,LY2922083 and LY2922470). J Med Chem 2016; 59: 10891-10916.
Han HS, Kang G, Kim JS, Choi BH, Koo SH. Regulation of glucose metabolism froma liver-centric perspective. Exp Mol Med 2016; 48: e218.
Hardy OT, Czech MP, Corvera S. What causes the insulin resistance underlyingobesity? Curr Opin Endocrinol Diabetes Obes 2012; 19: 81-87.
Hauge M, Vestmar MA, Husted AS, Ekberg JP, Wright MJ, Di Salvo J et al. GPR40(FFAR1) - Combined Gs and Gq signaling in vitro is associated with robustincretin secretagogue action ex vivo and in vivo. Mol Metab 2014; 4: 3-14.
Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study ofcholesterol lowering with simvastatin in 20,536 high-risk individuals: arandomised placebo-controlled trial. Lancet 2002; 360: 7-22.
Hegele RA. Plasma lipoproteins: genetic influences and clinical implications. NatRev Genet 2009; 10: 109-121.
Henquin JC. The dual control of insulin secretion by glucose involves triggering andamplifying pathways in beta-cells. Diabetes Res Clin Pract 2011; 93 Suppl 1: S27-31.
Henriksbo BD, Lau TC, Cavallari JF, Denou E, Chi W, Lally JS et al. Fluvastatincauses NLRP3 inflammasome-mediated adipose insulin resistance. Diabetes2014; 63: 3742-3747.
Herman MA, Kahn BB. Glucose transport and sensing in the maintenance of glucosehomeostasis and metabolic harmony. J Clin Invest 2006; 116: 1767-1775.
Herrington J, Zhou YP, Bugianesi RM, Dulski PM, Feng Y, Warren VA et al. Blockersof the delayed-rectifier potassium current in pancreatic beta-cells enhanceglucose-dependent insulin secretion. Diabetes 2006; 55: 1034-1042.
Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4

61
requires saturated fatty acid-induced ceramide biosynthesis in mice. J ClinInvest 2011; 121: 1858-1870.
Hooff GP, Wood WG, Muller WE, Eckert GP. Isoprenoids, small GTPases andAlzheimer's disease. Biochim Biophys Acta 2010; 1801: 896-905.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosisfactor-alpha: direct role in obesity-linked insulin resistance. Science 1993; 259:87-91.
Hou JC, Min L, Pessin JE. Insulin granule biogenesis, trafficking and exocytosis.Vitam Horm 2009; 80: 473-506.
Howlett KF, Mathews A, Garnham A, Sakamoto K. The effect of exercise and insulinon AS160 phosphorylation and 14-3-3 binding capacity in human skeletalmuscle. Am J Physiol Endocrinol Metab 2008; 294: E401-7.
Hu J, La Vecchia C, de Groh M, Negri E, Morrison H, Mery L et al. Dietarycholesterol intake and cancer. Ann Oncol 2012; 23: 491-500.
Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab 2007; 5: 237-252.Huang X, Vaag A, Hansson M, Weng J, Laurila E, Groop L. Impaired insulin-
stimulated expression of the glycogen synthase gene in skeletal muscle of type 2diabetic patients is acquired rather than inherited. J Clin Endocrinol Metab 2000;85: 1584-1590.
Hussain K, Challis B, Rocha N, Payne F, Minic M, Thompson A et al. An activatingmutation of AKT2 and human hypoglycemia. Science 2011; 334: 474.
Im DS. Intercellular Lipid Mediators and GPCR Drug Discovery. Biomol Ther(Seoul) 2013; 21: 411-422.
Ishikura S, Klip A. Muscle cells engage Rab8A and myosin Vb in insulin-dependentGLUT4 translocation. Am J Physiol Cell Physiol 2008; 295: C1016-25.
Islam MS, Rorsman P, Berggren PO. Ca(2+)-induced Ca2+ release in insulin-secretingcells. FEBS Lett 1992; 296: 287-291.
Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoAreductase. Science 2001; 292: 1160-1164.
Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S et al. Free fattyacids regulate insulin secretion from pancreatic beta cells through GPR40.Nature 2003; 422: 173-176.
Jain SS, Luiken JJ, Snook LA, Han XX, Holloway GP, Glatz JF et al. Fatty acidtransport and transporters in muscle are critically regulated by Akt2. FEBS Lett2015; 589: 2769-2775.
Jensen J, Rustad PI, Kolnes AJ, Lai YC. The role of skeletal muscle glycogenbreakdown for regulation of insulin sensitivity by exercise. Front Physiol 2011; 2:112.
Jensen TE, Richter EA. Regulation of glucose and glycogen metabolism during andafter exercise. J Physiol 2012; 590: 1069-1076.
Jensen TE, Angin Y, Sylow L, Richter EA. Is contraction-stimulated glucose transportfeedforward regulated by Ca2+? Exp Physiol 2014; 99: 1562-1568.

62
Jiang FX, Morahan G. Insulin-secreting beta cells require a post-genomic concept.World J Diabetes 2016; 7: 198-208.
Jones PH, Davidson MH, Stein EA, Bays HE, McKenney JM, Miller E et al.Comparison of the efficacy and safety of rosuvastatin versus atorvastatin,simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol 2003;92: 152-160.
Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes:perspectives on the past, present, and future. Lancet 2014; 383: 1068-1083.
Kain V, Kapadia B, Misra P, Saxena U. Simvastatin may induce insulin resistancethrough a novel fatty acid mediated cholesterol independent mechanism. SciRep 2015; 5: 13823.
Kanat M, DeFronzo RA, Abdul-Ghani MA. Treatment of prediabetes. World JDiabetes 2015; 6: 1207-1222.
Kang G, Chepurny OG, Holz GG. cAMP-regulated guanine nucleotide exchangefactor II (Epac2) mediates Ca2+-induced Ca2+ release in INS-1 pancreatic beta-cells. J Physiol 2001; 536: 375-385.
Kang G, Chepurny OG, Rindler MJ, Collis L, Chepurny Z, Li WH et al. A cAMP andCa2+ coincidence detector in support of Ca2+-induced Ca2+ release in mousepancreatic beta cells. J Physiol 2005; 566: 173-188.
Kapur NK, Musunuru K. Clinical efficacy and safety of statins in managingcardiovascular risk. Vasc Health Risk Manag 2008; 4: 341-353.
Kashani A, Phillips CO, Foody JM, Wang Y, Mangalmurti S, Ko DT et al. Risksassociated with statin therapy: a systematic overview of randomized clinicaltrials. Circulation 2006; 114: 2788-2797.
Kavalipati N, Shah J, Ramakrishan A, Vasnawala H. Pleiotropic effects of statins.Indian J Endocrinol Metab 2015; 19: 554-562.
Keech A, Colquhoun D, Best J, Kirby A, Simes RJ, Hunt D et al. Secondaryprevention of cardiovascular events with long-term pravastatin in patients withdiabetes or impaired fasting glucose: results from the LIPID trial. Diabetes Care2003; 26: 2713-2721.
Kelley GG, Chepurny OG, Schwede F, Genieser HG, Leech CA, Roe MW et al.Glucose-dependent potentiation of mouse islet insulin secretion by Epacactivator 8-pCPT-2'-O-Me-cAMP-AM. Islets 2009; 1: 260-265.
Khalique A, Sarwade RD, Pandey PR, Vijayakumar MV, Bhat MK, Seshadri V.Prolonged exposure to insulin with insufficient glucose leads to impaired Glut4translocation. Biochem Biophys Res Commun 2016; 474: 64-70.
Kim KM, Lee KS, Lee GY, Jin H, Durrance ES, Park HS et al. Anti-diabetic efficacy ofKICG1338, a novel glycogen synthase kinase-3beta inhibitor, and its molecularcharacterization in animal models of type 2 diabetes and insulin resistance. MolCell Endocrinol 2015; 409: 1-10.
King P, Peacock I, Donnelly R. The UK prospective diabetes study (UKPDS): clinicaland therapeutic implications for type 2 diabetes. Br J Clin Pharmacol 1999; 48:643-648.

63
Klip A, Sun Y, Chiu TT, Foley KP. Signal transduction meets vesicle traffic: thesoftware and hardware of GLUT4 translocation. Am J Physiol Cell Physiol 2014;306: C879-86.
Knopp RH, d'Emden M, Smilde JG, Pocock SJ. Efficacy and safety of atorvastatin inthe prevention of cardiovascular end points in subjects with type 2 diabetes: theAtorvastatin Study for Prevention of Coronary Heart Disease Endpoints in non-insulin-dependent diabetes mellitus (ASPEN). Diabetes Care 2006; 29: 1478-1485.
Kotarsky K, Nilsson NE, Olde B, Owman C. Progress in methodology. Improvedreporter gene assays used to identify ligands acting on orphan seven-transmembrane receptors. Pharmacol Toxicol 2003; 93: 249-258.
Kowluru A. Regulatory roles for small G proteins in the pancreatic beta-cell: lessonsfrom models of impaired insulin secretion. Am J Physiol Endocrinol Metab 2003;285: E669-84.
Kowluru A. Small G proteins in islet beta-cell function. Endocr Rev 2010; 31: 52-78.
Kowluru A, Kowluru RA. Protein prenylation in islet beta-cell function in health anddiabetes: Putting the pieces of the puzzle together. Biochem Pharmacol 2015; 98:363-370.
Lai YC, Liu Y, Jacobs R, Rider MH. A novel PKB/Akt inhibitor, MK-2206, effectivelyinhibits insulin-stimulated glucose metabolism and protein synthesis in isolatedrat skeletal muscle. Biochem J 2012; 447: 137-147.
Läll K, Mägi R, Morris A, Metspalu A, Fischer K. Personalized risk prediction fortype 2 diabetes: the potential of genetic risk scores. Genet Med 2016.
Lan H, Lin HV, Wang CF, Wright MJ, Xu S, Kang L et al. Agonists at GPR119mediate secretion of GLP-1 from mouse enteroendocrine cells through glucose-independent pathways. Br J Pharmacol 2012; 165: 2799-2807.
Lansey MN, Walker NN, Hargett SR, Stevens JR, Keller SR. Deletion of Rab GAPAS160 modifies glucose uptake and GLUT4 translocation in primary skeletalmuscles and adipocytes and impairs glucose homeostasis. Am J PhysiolEndocrinol Metab 2012; 303: E1273-86.
LaRosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC et al. Intensivelipid lowering with atorvastatin in patients with stable coronary disease. N EnglJ Med 2005; 352: 1425-1435.
Larsen S, Stride N, Hey-Mogensen M, Hansen CN, Bang LE, Bundgaard H et al.Simvastatin effects on skeletal muscle: relation to decreased mitochondrialfunction and glucose intolerance. J Am Coll Cardiol 2013; 61: 44-53.
Lazo-de-la-Vega-Monroy ML, Fernandez-Mejia C. Beta-Cell Function and Failure inType 1 Diabetes, Type 1 Diabetes - Pathogenesis, Genetics and Immunotherapy.In: Wagner D (Ed) 2011, InTech, DOI: 10.5772/22089.
Le Lay S, Krief S, Farnier C, Lefrere I, Le Liepvre X, Bazin R et al. Cholesterol, a cellsize-dependent signal that regulates glucose metabolism and gene expression inadipocytes. J Biol Chem 2001; 276: 16904-16910.
Le Roith D, Zick Y. Recent advances in our understanding of insulin action andinsulin resistance. Diabetes Care 2001; 24: 588-597.

64
Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI et al. Inflammation is necessary forlong-term but not short-term high-fat diet-induced insulin resistance. Diabetes2011; 60: 2474-2483.
Lee-Young RS, Hoffman NJ, Murphy KT, Henstridge DC, Samocha-Bonet D, SiebelAL et al. Glucose-6-phosphate dehydrogenase contributes to the regulation ofglucose uptake in skeletal muscle. Mol Metab 2016; 5: 1083-1091.
Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control ofGLUT4. Nat Rev Mol Cell Biol 2012; 13: 383-396.
Li G, Regazzi R, Balch WE, Wollheim CB. Stimulation of insulin release frompermeabilized HIT-T15 cells by a synthetic peptide corresponding to the effectordomain of the small GTP-binding protein rab3. FEBS Lett 1993; 327: 145-149.
Li J, Houseknecht KL, Stenbit AE, Katz EB, Charron MJ. Reduced glucose uptakeprecedes insulin signaling defects in adipocytes from heterozygous GLUT4knockout mice. FASEB J 2000; 14: 1117-1125.
Li W, Liang X, Zeng Z, Yu K, Zhan S, Su Q et al. Simvastatin inhibits glucose uptakeactivity and GLUT4 translocation through suppression of the IR/IRS-1/Aktsignaling in C2C12 myotubes. Biomed Pharmacother 2016; 83: 194-200.
Li XB, Gu JD, Zhou QH. Review of aerobic glycolysis and its key enzymes - newtargets for lung cancer therapy. Thorac Cancer 2015; 6: 17-24.
Liao JK. Isoprenoids as mediators of the biological effects of statins. J Clin Invest2002; 110: 285-288.
Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 2005; 45:89-118.
Liao P, Hemmerlin A, Bach TJ, Chye ML. The potential of the mevalonate pathwayfor enhanced isoprenoid production. Biotechnol Adv 2016; 34: 697-713.
LIPID Study Group (The Long-Term Intervention with Pravastatin in IschaemicDisease Study Group). Prevention of cardiovascular events and death withpravastatin in patients with coronary heart disease and a broad range of initialcholesterol levels. N Engl J Med 1998; 339: 1349-1357.
Lotta LA, Sharp SJ, Burgess S, Perry JR, Stewart ID, Willems SM et al. AssociationBetween Low-Density Lipoprotein Cholesterol-Lowering Genetic Variants andRisk of Type 2 Diabetes: A Meta-analysis. JAMA 2016; 316: 1383-1391.
Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S et al. Insulin regulatesliver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med 2012;18: 388-395.
Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolicrequirements of cell proliferation. Annu Rev Cell Dev Biol 2011; 27: 441-464.
Luo M, Langlais P, Yi Z, Lefort N, De Filippis EA, Hwang H et al. Phosphorylation ofhuman insulin receptor substrate-1 at Serine 629 plays a positive role in insulinsignaling. Endocrinology 2007; 148: 4895-4905.
Lusis AJ. Atherosclerosis. Nature 2000; 407: 233-241.

65
Mackenzie RW, Elliott BT. Akt/PKB activation and insulin signaling: a novel insulinsignaling pathway in the treatment of type 2 diabetes. Diabetes Metab SyndrObes 2014; 7: 55-64.
Maeda T, Horiuchi N. Simvastatin suppresses leptin expression in 3T3-L1 adipocytesvia activation of the cyclic AMP-PKA pathway induced by inhibition of proteinprenylation. J Biochem 2009; 145: 771-781.
Maji D, Shaikh S, Solanki D, Gaurav K. Safety of statins. Indian J Endocrinol Metab2013; 17: 636-646.
Manabe Y, Miyatake S, Takagi M, Nakamura M, Okeda A, Nakano T et al.Characterization of an acute muscle contraction model using cultured C2C12myotubes. PLoS One 2012; 7: e52592.
Mancini AD, Poitout V. The fatty acid receptor FFA1/GPR40 a decade later: howmuch do we know? Trends Endocrinol Metab 2013; 24: 398-407.
Maningat P, Breslow JL. Needed: pragmatic clinical trials for statin-intolerantpatients. N Engl J Med 2011; 365: 2250-2251.
Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucosetolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care1999; 22: 1462-1470.
Maqbool M, Mobashir M, Hoda N. Pivotal role of glycogen synthase kinase-3: Atherapeutic target for Alzheimer's disease. Eur J Med Chem 2016; 107: 63-81.
Maria Z, Campolo AR, Lacombe VA. Diabetes Alters the Expression andTranslocation of the Insulin-Sensitive Glucose Transporters 4 and 8 in the Atria.PLoS One 2015; 10: e0146033.
McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL et al.GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014;5: 2881-2911.
McGuire TF, Xu XQ, Corey SJ, Romero GG, Sebti SM. Lovastatin disrupts earlyevents in insulin signaling: a potential mechanism of lovastatin's anti-mitogenicactivity. Biochem Biophys Res Commun 1994; 204: 399-406.
McTaggart F, Buckett L, Davidson R, Holdgate G, McCormick A, Schneck D et al.Preclinical and clinical pharmacology of Rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87: 28B-32B.
Meloni AR, DeYoung MB, Lowe C, Parkes DG. GLP-1 receptor activated insulinsecretion from pancreatic beta-cells: mechanism and glucose dependence.Diabetes Obes Metab 2013; 15: 15-27.
Metz SA, Rabaglia ME, Stock JB, Kowluru A. Modulation of insulin secretion fromnormal rat islets by inhibitors of the post-translational modifications of GTP-binding proteins. Biochem J 1993; 295 ( Pt 1): 31-40.
Miao L, Yang L, Huang H, Liang F, Ling C, Hu Y. mTORC1 is necessary butmTORC2 and GSK3beta are inhibitory for AKT3-induced axon regeneration inthe central nervous system. Elife 2016; 5: e14908.

66
Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J et al. AS160, the Aktsubstrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J 2005; 391: 87-93.
Miklosz A, Lukaszuk B, Zendzian-Piotrowska M, Kurek K, Chabowski A. TheEffects of AS160 Modulation on Fatty Acid Transporters Expression and LipidProfile in L6 Myotubes. Cell Physiol Biochem 2016; 38: 267-282.
Minokoshi Y, Kahn CR, Kahn BB. Tissue-specific ablation of the GLUT4 glucosetransporter or the insulin receptor challenges assumptions about insulin actionand glucose homeostasis. J Biol Chem 2003; 278: 33609-33612.
Mita T, Watada H, Nakayama S, Abe M, Ogihara T, Shimizu T et al. Preferable effectof pravastatin compared to atorvastatin on beta cell function in Japanese early-state type 2 diabetes with hypercholesterolemia. Endocr J 2007; 54: 441-447.
Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y et al. Establishmentof a pancreatic beta cell line that retains glucose-inducible insulin secretion:special reference to expression of glucose transporter isoforms. Endocrinology1990; 127: 126-132.
Moghadasian MH. Clinical pharmacology of 3-hydroxy-3-methylglutaryl coenzymeA reductase inhibitors. Life Sci 1999; 65: 1329-1337.
Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator ofAGC kinase signal transduction. Semin Cell Dev Biol 2004; 15: 161-170.
Mora S, Glynn RJ, Hsia J, MacFadyen JG, Genest J, Ridker PM. Statins for theprimary prevention of cardiovascular events in women with elevated high-sensitivity C-reactive protein or dyslipidemia: results from the Justification forthe Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin(JUPITER) and meta-analysis of women from primary prevention trials.Circulation 2010; 121: 1069-1077.
Morgan AE, Mooney KM, Wilkinson SJ, Pickles NA, Mc Auley MT. Cholesterolmetabolism: A review of how ageing disrupts the biological mechanismsresponsible for its regulation. Ageing Res Rev 2016; 27: 108-124.
Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V et al.Large-scale association analysis provides insights into the genetic architectureand pathophysiology of type 2 diabetes. Nat Genet 2012; 44: 981-990.
Moscavitch SD, Kang HC, Filho RA, Mesquita ET, Neto HC, Rosa ML. Comparisonof adipokines in a cross-sectional study with healthy overweight, insulin-sensitive and healthy lean, insulin-resistant subjects, assisted by a family doctorprimary care program. Diabetol Metab Syndr 2016; 8: 9-016-0125-9. eCollection2016.
Motoyama K, Emoto M, Tahara H, Komatsu M, Shoji T, Inaba M et al. Association ofmuscle glycogen synthase polymorphism with insulin resistance in type 2diabetic patients. Metabolism 2003; 52: 895-899.
Moutzouri E, Liberopoulos E, Mikhailidis DP, Kostapanos MS, Kei AA, Milionis H etal. Comparison of the effects of simvastatin vs. rosuvastatin vs.simvastatin/ezetimibe on parameters of insulin resistance. Int J Clin Pract 2011;65: 1141-1148.

67
Mueckler M. Insulin resistance and the disruption of Glut4 trafficking in skeletalmuscle. J Clin Invest 2001; 107: 1211-1213.
Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signallingand the mevalonate pathway in cancer. Nat Rev Cancer 2016; 16: 718-731.
Nakagaki I, Sasaki S, Hori S, Kondo H. Ca2+ and electrolyte mobilization followingagonist application to the pancreatic beta cell line HIT. Pflugers Arch 2000; 440:828-834.
Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statinson the adipocyte maturation and expression of glucose transporter 4 (SLC2A4):implications in glycaemic control. Diabetologia 2006; 49: 1881-1892.
Navarese EP, Buffon A, Andreotti F, Kozinski M, Welton N, Fabiszak T et al. Meta-analysis of impact of different types and doses of statins on new-onset diabetesmellitus. Am J Cardiol 2013; 111: 1123-1130.
Nedachi T, Kanzaki M. Regulation of glucose transporters by insulin andextracellular glucose in C2C12 myotubes. Am J Physiol Endocrinol Metab 2006;291: E817-28.
Nelson D, Cox M. Lehninger principles of biochemistry. New York: WH Freemanand Company, 2005.
Nigovi B, Mornar A, Sertic M. A Review of Current Trends and Advances inAnalytical Methods for Determination of Statins: Chromatography andCapillary Electrophoresis, Chromatography - The Most Versatile Method ofChemical Analysis. In: Calderon L (Ed) 2012, InTech, DOI: 10.5772/48694.
Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potentialrole of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2diabetes. Diabetes 2000; 49: 263-271.
Ning Y, O'Neill K, Lan H, Pang L, Shan LX, Hawes BE et al. Endogenous andsynthetic agonists of GPR119 differ in signalling pathways and their effects oninsulin secretion in MIN6c4 insulinoma cells. Br J Pharmacol 2008; 155: 1056-1065.
Nishizaki T, Gotoh A, Shimizu T, Tanaka A. The phosphatidylethanolaminederivative diDCP-LA-PE mimics intracellular insulin signaling. Sci Rep 2016; 6:27267.
Noguchi M, Hirata N, Suizu F. The links between AKT and two intracellularproteolytic cascades: ubiquitination and autophagy. Biochim Biophys Acta 2014;1846: 342-352.
O'Driscoll L, Gammell P, McKiernan E, Ryan E, Jeppesen PB, Rani S et al. Phenotypicand global gene expression profile changes between low passage and highpassage MIN-6 cells. J Endocrinol 2006; 191: 665-676.
Oh da Y, Olefsky JM. G protein-coupled receptors as targets for anti-diabetictherapeutics. Nat Rev Drug Discov 2016; 15: 161-172.
Olson AL. Regulation of GLUT4 and Insulin-Dependent Glucose Flux. ISRN MolBiol 2012; 2012: 856987.

68
Olsson AG, Istad H, Luurila O, Ose L, Stender S, Tuomilehto J et al. Effects ofrosuvastatin and atorvastatin compared over 52 weeks of treatment in patientswith hypercholesterolemia. Am Heart J 2002; 144: 1044-1051.
Osorio-Fuentealba C, Contreras-Ferrat AE, Altamirano F, Espinosa A, Li Q, Niu W etal. Electrical stimuli release ATP to increase GLUT4 translocation and glucoseuptake via PI3Kgamma-Akt-AS160 in skeletal muscle cells. Diabetes 2013; 62:1519-1526.
Pandey A, Chawla S, Guchhait P. Type-2 diabetes: Current understanding andfuture perspectives. IUBMB Life 2015; 67: 506-513.
Pandey MK, DeGrado TR. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapyand Imaging. Theranostics 2016; 6: 571-593.
Paniagua JA, Lopez-Miranda J, Escribano A, Berral FJ, Marin C, Bravo D et al.Cerivastatin improves insulin sensitivity and insulin secretion in early-stateobese type 2 diabetes. Diabetes 2002; 51: 2596-2603.
Paniagua JA. Nutrition, insulin resistance and dysfunctional adipose tissuedetermine the different components of metabolic syndrome. World J Diabetes2016; 7: 483-514.
Park KS, Ciaraldi TP, Carter L, Mudaliar S, Nikoulina SE, Webster NJ et al. Inductionof insulin resistance in human skeletal muscle cells by downregulation ofglycogen synthase protein expression. Metabolism 2000; 49: 962-968.
Parker HE, Habib AM, Rogers GJ, Gribble FM, Reimann F. Nutrient-dependentsecretion of glucose-dependent insulinotropic polypeptide from primary murineK cells. Diabetologia 2009; 52: 289-298.
Parpal S, Karlsson M, Thorn H, Stralfors P. Cholesterol depletion disrupts caveolaeand insulin receptor signaling for metabolic control via insulin receptorsubstrate-1, but not for mitogen-activated protein kinase control. J Biol Chem2001; 276: 9670-9678.
Pauta M, Rotllan N, Fernandez-Hernando A, Langhi C, Ribera J, Lu M et al. Akt-mediated foxo1 inhibition is required for liver regeneration. Hepatology 2016;63: 1660-1674.
Paz K, Liu YF, Shorer H, Hemi R, LeRoith D, Quan M et al. Phosphorylation ofinsulin receptor substrate-1 (IRS-1) by protein kinase B positively regulates IRS-1function. J Biol Chem 1999; 274: 28816-28822.
Pedersen AJ, Hingst JR, Friedrichsen M, Kristensen JM, Hojlund K, Wojtaszewski JF.Dysregulation of muscle glycogen synthase in recovery from exercise in type 2diabetes. Diabetologia 2015; 58: 1569-1578.
Pedersen TR, Kjekshus J, Pyörälä K, Olsson AG, Cook TJ, Musliner TA et al. Effect ofsimvastatin on ischemic signs and symptoms in the Scandinavian simvastatinsurvival study (4S). Am J Cardiol 1998; 81: 333-335.
Pedersen TR, Faergeman O, Kastelein JJ, Olsson AG, Tikkanen MJ, Holme I et al.High-dose atorvastatin vs usual-dose simvastatin for secondary prevention aftermyocardial infarction: the IDEAL study: a randomized controlled trial. JAMA2005; 294: 2437-2445.

69
Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepaticinsulin resistance and type 2 diabetes. Nature 2014; 510: 84-91.
Petrov AM, Kasimov MR, Zefirov AL. Brain Cholesterol Metabolism and Its Defects:Linkage to Neurodegenerative Diseases and Synaptic Dysfunction. ActaNaturae 2016; 8: 58-73.
Pilon M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis 2016;15: 167.
Pirola L, Johnston AM, Van Obberghen E. Modulation of insulin action. Diabetologia2004; 47: 170-184.
Plakkal Ayyappan J, Paul A, Goo YH. Lipid droplet-associated proteins inatherosclerosis (Review). Mol Med Rep 2016; 13: 4527-4534.
Poitout V, Olson LK, Robertson RP. Insulin-secreting cell lines: classification,characteristics and potential applications. Diabetes Metab 1996; 22: 7-14.
Poloz Y, Stambolic V. Obesity and cancer, a case for insulin signaling. Cell Death Dis2015; 6: e2037.
Postic C, Mauvais-Jarvis F, Girard J. Mouse models of insulin resistance and type 2diabetes. Ann Endocrinol (Paris) 2004; 65: 51-59.
Prats C, Helge JW, Nordby P, Qvortrup K, Ploug T, Dela F et al. Dual regulation ofmuscle glycogen synthase during exercise by activation andcompartmentalization. J Biol Chem 2009; 284: 15692-15700.
Preiss D, Sattar N. Statins and the risk of new-onset diabetes: a review of recentevidence. Curr Opin Lipidol 2011a; 22: 460-466.
Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD et al. Risk of incidentdiabetes with intensive-dose compared with moderate-dose statin therapy: ameta-analysis. JAMA 2011b; 305: 2556-2564.
Qi Z, Xia J, Xue X, He Q, Ji L, Ding S. Long-term treatment with nicotinamideinduces glucose intolerance and skeletal muscle lipotoxicity in normal chow-fedmice: compared to diet-induced obesity. J Nutr Biochem 2016; 36: 31-41.
Quan C, Xie B, Wang HY, Chen S. PKB-Mediated Thr649 Phosphorylation ofAS160/TBC1D4 Regulates the R-Wave Amplitude in the Heart. PLoS One 2015;10: e0124491.
Rader DJ, Daugherty A. Translating molecular discoveries into new therapies foratherosclerosis. Nature 2008; 451: 904-913.
Radford NB, DeFina LF, Barlow CE, Kerr A, Chakravorty R, Khera A et al. Effect offitness on incident diabetes from statin use in primary prevention.Atherosclerosis 2015; 239: 43-49.
Rajpathak SN, Kumbhani DJ, Crandall J, Barzilai N, Alderman M, Ridker PM. Statintherapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care2009; 32: 1924-1929.
Rautio N, Jokelainen J, Oksa H, Saaristo T, Peltonen M, Vanhala M et al. Do statinsinterfere with lifestyle intervention in the prevention of diabetes in primaryhealthcare? One-year follow-up of the FIN-D2D project. BMJ Open 2012; 2:10.1136/bmjopen-2012-001472.

70
Reaven G. The metabolic syndrome or the insulin resistance syndrome? Differentnames, different concepts, and different goals. Endocrinol Metab Clin North Am2004; 33: 283-303.
Richter EA, Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake.Physiol Rev 2013; 93: 993-1017.
Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM,Jr, Kastelein JJ et al.Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359: 2195-2207.
Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefitsand diabetes risks of statin therapy in primary prevention: an analysis from theJUPITER trial. Lancet 2012; 380: 565-571.
Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res 2005; 97: 1232-1235.
Roach RJ, Larner J. Covalent phosphorylation in the regulation glycogen synthaseactivity. Mol Cell Biochem 1977; 15: 179-200.
Röder PV, Wong X, Hong W, Han W. Molecular regulation of insulin granulebiogenesis and exocytosis. Biochem J 2016a; 473: 2737-2756.
Röder PV, Wu B, Liu Y, Han W. Pancreatic regulation of glucose homeostasis. ExpMol Med 2016b; 48: e219.
Rogers JK, Jhund PS, Perez AC, Bohm M, Cleland JG, Gullestad L et al. Effect ofrosuvastatin on repeat heart failure hospitalizations: the CORONA Trial(Controlled Rosuvastatin Multinational Trial in Heart Failure). JACC Heart Fail2014; 2: 289-297.
Rozman D, Monostory K. Perspectives of the non-statin hypolipidemic agents.Pharmacol Ther 2010; 127: 19-40.
Ruiz de Azua I, Gautam D, Guettier JM, Wess J. Novel insights into the function ofbeta-cell M3 muscarinic acetylcholine receptors: therapeutic implications.Trends Endocrinol Metab 2011; 22: 74-80.
Rytka JM, Wueest S, Schoenle EJ, Konrad D. The portal theory supported by venousdrainage-selective fat transplantation. Diabetes 2011; 60: 56-63.
Sabatine MS, Wiviott SD, Morrow DA, McCabe CH, Cannon CP. High-doseatorvastatin associated with worse glycemic control: A PROVE-IT TIMI 22Substudy. Circulation 2004; 110: 843.
Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG et al. The effectof pravastatin on coronary events after myocardial infarction in patients withaverage cholesterol levels. Cholesterol and Recurrent Events Trial investigators.N Engl J Med 1996; 335: 1001-1009.
Sah SP, Singh B, Choudhary S, Kumar A. Animal models of insulin resistance: Areview. Pharmacol Rep 2016; 68: 1165-1177.
Saltiel AR. Insulin Signaling in the Control of Glucose and Lipid Homeostasis.Handb Exp Pharmacol 2016; 233: 51-71.

71
Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS et al. Insulin-stimulatedphosphorylation of a Rab GTPase-activating protein regulates GLUT4translocation. J Biol Chem 2003; 278: 14599-14602.
Sarabia V, Ramlal T, Klip A. Glucose uptake in human and animal muscle cells inculture. Biochem Cell Biol 1990; 68: 536-542.
Sasaki J, Iwashita M, Kono S. Statins: beneficial or adverse for glucose metabolism. JAtheroscler Thromb 2006; 13: 123-129.
Sato K, Sugimoto H, Rikimaru K, Imoto H, Kamaura M, Negoro N et al. Discovery ofa novel series of indoline carbamate and indolinylpyrimidine derivatives aspotent GPR119 agonists. Bioorg Med Chem 2014; 22: 1649-1666.
Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ et al. Statins andrisk of incident diabetes: a collaborative meta-analysis of randomised statintrials. Lancet 2010; 375: 735-742.
Sattar NA, Ginsberg H, Ray K, Chapman MJ, Arca M, Averna M et al. The use ofstatins in people at risk of developing diabetes mellitus: evidence and guidancefor clinical practice. Atheroscler Suppl 2014; 15: 1-15.
Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterollowering in 4444 patients with coronary heart disease: the ScandinavianSimvastatin Survival Study (4S). Lancet 1994; 344: 1383-1389.
Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties ofstatins: an update. Fundam Clin Pharmacol 2005; 19: 117-125.
Scheen AJ. Pathophysiology of type 2 diabetes. Acta Clin Belg 2003; 58: 335-341.Schneck DW, Knopp RH, Ballantyne CM, McPherson R, Chitra RR, Simonson SG.
Comparative effects of rosuvastatin and atorvastatin across their dose ranges inpatients with hypercholesterolemia and without active arterial disease. Am JCardiol 2003; 91: 33-41.
Schultze SM, Jensen J, Hemmings BA, Tschopp O, Niessen M. Promiscuous affairs ofPKB/AKT isoforms in metabolism. Arch Physiol Biochem 2011; 117: 70-77.
Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D et al. Effectsof atorvastatin on early recurrent ischemic events in acute coronary syndromes:the MIRACL study: a randomized controlled trial. JAMA 2001; 285: 1711-1718.
SEARCH Collaborative Group (Study of the Effectiveness of Additional Reductionsin Cholesterol and Homocysteine Collaborative Group), Armitage J, Bowman L,Wallendszus K, Bulbulia R, Rahimi K et al. Intensive lowering of LDLcholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors ofmyocardial infarction: a double-blind randomised trial. Lancet 2010; 376: 1658-1669.
Seino S, Takahashi H, Fujimoto W, Shibasaki T. Roles of cAMP signalling in insulingranule exocytosis. Diabetes Obes Metab 2009; 11 Suppl 4: 180-188.
Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinicalimplications for obesity and diabetes. J Clin Invest 2011; 121: 2118-2125.

72
Serruys PW, de Feyter P, Macaya C, Kokott N, Puel J, Vrolix M et al. Fluvastatin forprevention of cardiac events following successful first percutaneous coronaryintervention: a randomized controlled trial. JAMA 2002; 287: 3215-3222.
Sesti G, Federici M, Hribal ML, Lauro D, Sbraccia P, Lauro R. Defects of the insulinreceptor substrate (IRS) system in human metabolic disorders. FASEB J 2001; 15:2099-2111.
Sever PS, Dahlof B, Poulter NR, Wedel H, Beevers G, Caulfield M et al. Prevention ofcoronary and stroke events with atorvastatin in hypertensive patients who haveaverage or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial--Lipid Lowering Arm (ASCOT-LLA): amulticentre randomised controlled trial. Lancet 2003; 361: 1149-1158.
Shao D, Tian R. Glucose Transporters in Cardiac Metabolism and Hypertrophy.Compr Physiol 2015; 6: 331-351.
Shapiro H, Shachar S, Sekler I, Hershfinkel M, Walker MD. Role of GPR40 in fattyacid action on the beta cell line INS-1E. Biochem Biophys Res Commun 2005;335: 97-104.
Shaw LM. The insulin receptor substrate (IRS) proteins: at the intersection ofmetabolism and cancer. Cell Cycle 2011; 10: 1750-1756.
Shearin AL, Monks BR, Seale P, Birnbaum MJ. Lack of AKT in adipocytes causessevere lipodystrophy. Mol Metab 2016; 5: 472-479.
Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW et al.Prevention of coronary heart disease with pravastatin in men withhypercholesterolemia. West of Scotland Coronary Prevention Study Group. NEngl J Med 1995; 333: 1301-1307.
Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM et al.Pravastatin in elderly individuals at risk of vascular disease (PROSPER): arandomised controlled trial. Lancet 2002; 360: 1623-1630.
Shibasaki T, Takahashi T, Takahashi H, Seino S. Cooperation between cAMPsignalling and sulfonylurea in insulin secretion. Diabetes Obes Metab 2014; 16Suppl 1: 118-125.
Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol2000; 1: 31-39.
Skelin M, Rupnik M, Cencic A. Pancreatic beta cell lines and their applications indiabetes mellitus research. ALTEX 2010; 27: 105-113.
Slater EE, MacDonald JS. Mechanism of action and biological profile of HMG CoAreductase inhibitors. A new therapeutic alternative. Drugs 1988; 36 Suppl 3: 72-82.
Smith R, Solberg R, Jacobsen LL, Voreland AL, Rustan AC, Thoresen GH et al.Simvastatin inhibits glucose metabolism and legumain activity in humanmyotubes. PLoS One 2014; 9: e85721.
Song WJ, Mondal P, Li Y, Lee SE, Hussain MA. Pancreatic beta-cell response toincreased metabolic demand and to pharmacologic secretagogues requiresEPAC2A. Diabetes 2013; 62: 2796-2807.

73
Sonksen P, Sonksen J. Insulin: understanding its action in health and disease. Br JAnaesth 2000; 85: 69-79.
Stamler J, Daviglus ML, Garside DB, Dyer AR, Greenland P, Neaton JD. Relationshipof baseline serum cholesterol levels in 3 large cohorts of younger men to long-term coronary, cardiovascular, and all-cause mortality and to longevity. JAMA2000; 284: 311-318.
Stancakova A, Javorsky M, Kuulasmaa T, Haffner SM, Kuusisto J, Laakso M.Changes in insulin sensitivity and insulin release in relation to glycemia andglucose tolerance in 6,414 Finnish men. Diabetes 2009; 58: 1212-1221.
Standaert ML, Bandyopadhyay G, Kanoh Y, Sajan MP, Farese RV. Insulin and PIP3activate PKC-zeta by mechanisms that are both dependent and independent ofphosphorylation of activation loop (T410) and autophosphorylation (T560) sites.Biochemistry 2001; 40: 249-255.
Stossel TP. The discovery of statins. Cell 2008; 134: 903-905.
Sukhija R, Prayaga S, Marashdeh M, Bursac Z, Kakar P, Bansal D et al. Effect ofstatins on fasting plasma glucose in diabetic and nondiabetic patients. J InvestigMed 2009; 57: 495-499.
Sung KC, Park HY, Kim MJ, Reaven G. Metabolic markers associated with insulinresistance predict type 2 diabetes in Koreans with normal blood pressure orprehypertension. Cardiovasc Diabetol 2016; 15: 47-016-0368-7.
Swayne LA, Mezghrani A, Varrault A, Chemin J, Bertrand G, Dalle S et al. TheNALCN ion channel is activated by M3 muscarinic receptors in a pancreaticbeta-cell line. EMBO Rep 2009; 10: 873-880.
Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T et al.HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight:evidence from genetic analysis and randomised trials. Lancet 2015; 385: 351-361.
Sylow L, Kleinert M, Richter EA, Jensen TE. Exercise-stimulated glucose uptake -regulation and implications for glycaemic control. Nat Rev Endocrinol 2016.
Szalat A, Durst R, Leitersdorf E. Managing dyslipidaemia in type 2 diabetes mellitus.Best Pract Res Clin Endocrinol Metab 2016; 30: 431-444.
Takaguri A, Satoh K, Itagaki M, Tokumitsu Y, Ichihara K. Effects of atorvastatin andpravastatin on signal transduction related to glucose uptake in 3T3L1adipocytes. J Pharmacol Sci 2008; 107: 80-89.
Takahashi-Yanaga F. Activator or inhibitor? GSK-3 as a new drug target. BiochemPharmacol 2013; 86: 191-199.
Takenaka N, Nihata Y, Satoh T. Rac1 Activation Caused by Membrane Translocationof a Guanine Nucleotide Exchange Factor in Akt2-Mediated Insulin Signaling inMouse Skeletal Muscle. PLoS One 2016; 11: e0155292.
Tan SX, Ng Y, Burchfield JG, Ramm G, Lambright DG, Stockli J et al. The RabGTPase-activating protein TBC1D4/AS160 contains an atypicalphosphotyrosine-binding domain that interacts with plasma membranephospholipids to facilitate GLUT4 trafficking in adipocytes. Mol Cell Biol 2012;32: 4946-4959.

74
Teixeira SD, Panveloski-Costa AC, Carvalho A, Monteiro Schiavon FP, Ruiz MarqueAC, Campello RS et al. Thyroid hormone treatment decreases hepatic glucoseproduction and renal reabsorption of glucose in alloxan-induced diabetic Wistarrats. Physiol Rep 2016; 4: e12961.
ter Avest E, Abbink EJ, Holewijn S, de Graaf J, Tack CJ, Stalenhoef AF. Effects ofrosuvastatin on endothelial function in patients with familial combinedhyperlipidaemia (FCH). Curr Med Res Opin 2005; 21: 1469-1476.
Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J etal. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am JHum Genet 2013; 93: 141-149.
Tomita T, Masuzaki H, Iwakura H, Fujikura J, Noguchi M, Tanaka T et al. Expressionof the gene for a membrane-bound fatty acid receptor in the pancreas and isletcell tumours in humans: evidence for GPR40 expression in pancreatic beta cellsand implications for insulin secretion. Diabetologia 2006; 49: 962-968.
Tsuchiya M, Hosaka M, Moriguchi T, Zhang S, Suda M, Yokota-Hashimoto H et al.Cholesterol biosynthesis pathway intermediates and inhibitors regulate glucose-stimulated insulin secretion and secretory granule formation in pancreatic beta-cells. Endocrinology 2010; 151: 4705-4716.
Ullman EF, Kirakossian H, Switchenko AC, Ishkanian J, Ericson M, Wartchow CA etal. Luminescent oxygen channeling assay (LOCI): sensitive, broadly applicablehomogeneous immunoassay method. Clin Chem 1996; 42: 1518-1526.
Vaag A, Henriksen JE, Beck-Nielsen H. Decreased insulin activation of glycogensynthase in skeletal muscles in young nonobese Caucasian first-degree relativesof patients with non-insulin-dependent diabetes mellitus. J Clin Invest 1992; 89:782-788.
Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activationand inhibition of class I phosphoinositide 3-kinases. Sci Signal 2011; 4: re2.
Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev1997; 11: 2295-2322.
van de Woestijne AP, van der Graaf Y, Westerink J, Nathoe HM, Visseren FL. Effectof statin therapy on incident type 2 diabetes mellitus in patients with clinicallymanifest vascular disease. Am J Cardiol 2015; 115: 441-446.
van Harmelen V, Elizalde M, Ariapart P, Bergstedt-Lindqvist S, Reynisdottir S,Hoffstedt J et al. The association of human adipose angiotensinogen geneexpression with abdominal fat distribution in obesity. Int J Obes Relat MetabDisord 2000; 24: 673-678.
Vettor R, Granzotto M, De Stefani D, Trevellin E, Rossato M, Farina MG et al. Loss-of-function mutation of the GPR40 gene associates with abnormal stimulatedinsulin secretion by acting on intracellular calcium mobilization. J ClinEndocrinol Metab 2008; 93: 3541-3550.
Vind BF, Pehmoller C, Treebak JT, Birk JB, Hey-Mogensen M, Beck-Nielsen H et al.Impaired insulin-induced site-specific phosphorylation of TBC1 domain family,member 4 (TBC1D4) in skeletal muscle of type 2 diabetes patients is restored byendurance exercise-training. Diabetologia 2011; 54: 157-167.

75
Wadhera RK, Steen DL, Khan I, Giugliano RP, Foody JM. A review of low-densitylipoprotein cholesterol, treatment strategies, and its impact on cardiovasculardisease morbidity and mortality. J Clin Lipidol 2016; 10: 472-489.
Wan M, Leavens KF, Saleh D, Easton RM, Guertin DA, Peterson TR et al.Postprandial hepatic lipid metabolism requires signaling through Akt2independent of the transcription factors FoxA2, FoxO1, and SREBP1c. CellMetab 2011; 14: 516-527.
Wang KL, Liu CJ, Chao TF, Huang CM, Wu CH, Chen SJ et al. Statins, risk ofdiabetes, and implications on outcomes in the general population. J Am CollCardiol 2012; 60: 1231-1238.
Wang M, Casey PJ. Protein prenylation: unique fats make their mark on biology. NatRev Mol Cell Biol 2016; 17: 110-122.
Wang Q, Somwar R, Bilan PJ, Liu Z, Jin J, Woodgett JR et al. Protein kinase B/Aktparticipates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol1999; 19: 4008-4018.
Waters DD, Ho JE, DeMicco DA, Breazna A, Arsenault BJ, Wun CC et al. Predictorsof new-onset diabetes in patients treated with atorvastatin: results from 3 largerandomized clinical trials. J Am Coll Cardiol 2011; 57: 1535-1545.
Waters DD, Ho JE, Boekholdt SM, DeMicco DA, Kastelein JJ, Messig M et al.Cardiovascular event reduction versus new-onset diabetes during atorvastatintherapy: effect of baseline risk factors for diabetes. J Am Coll Cardiol 2013; 61:148-152.
Webster HV, Bone AJ, Webster KA, Wilkin TJ. Comparison of an enzyme-linkedimmunosorbent assay (ELISA) with a radioimmunoassay (RIA) for themeasurement of rat insulin. J Immunol Methods 1990; 134: 95-100.
Weigert C, Kron M, Kalbacher H, Pohl AK, Runge H, Haring HU et al. Interplay andeffects of temporal changes in the phosphorylation state of serine-302, -307, and -318 of insulin receptor substrate-1 on insulin action in skeletal muscle cells. MolEndocrinol 2008; 22: 2729-2740.
Welters HJ, Diakogiannaki E, Mordue JM, Tadayyon M, Smith SA, Morgan NG.Differential protective effects of palmitoleic acid and cAMP on caspaseactivation and cell viability in pancreatic beta-cells exposed to palmitate.Apoptosis 2006; 11: 1231-1238.
Weng TC, Yang YH, Lin SJ, Tai SH. A systematic review and meta-analysis on thetherapeutic equivalence of statins. J Clin Pharm Ther 2010; 35: 139-151.
Wilcox G. Insulin and insulin resistance. Clin Biochem Rev 2005; 26: 19-39.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates
for the year 2000 and projections for 2030. Diabetes Care 2004; 27: 1047-1053.
Wittenberg AD, Azar S, Klochendler A, Stolovich-Rain M, Avraham S, Birnbaum L etal. Phosphorylated Ribosomal Protein S6 Is Required for Akt-DrivenHyperplasia and Malignant Transformation, but Not for Hypertrophy,Aneuploidy and Hyperfunction of Pancreatic beta-Cells. PLoS One 2016; 11:e0149995.

76
Wong ND, Zhao Y, Patel R, Patao C, Malik S, Bertoni AG et al. Cardiovascular RiskFactor Targets and Cardiovascular Disease Event Risk in Diabetes: A PoolingProject of the Atherosclerosis Risk in Communities Study, Multi-Ethnic Study ofAtherosclerosis, and Jackson Heart Study. Diabetes Care 2016; 39: 668-676.
Wood RJ, O'Neill EC. Resistance Training in Type II Diabetes Mellitus: Impact onAreas of Metabolic Dysfunction in Skeletal Muscle and Potential Impact onBone. J Nutr Metab 2012; 2012: 268197.
Wood WG, Li L, Muller WE, Eckert GP. Cholesterol as a causative factor inAlzheimer's disease: a debatable hypothesis. J Neurochem 2014; 129: 559-572.
World Health Organization. Diabetes action now booklet: a life-threateningcondition. Geneva: The World Health Organization 2004; 1-17.
Wortham M, Sander M. Mechanisms of beta-cell functional adaptation to changes inworkload. Diabetes Obes Metab 2016; 18 Suppl 1: 78-86.
Xie B, Chen Q, Chen L, Sheng Y, Wang HY, Chen S. The Inactivation of RabGAPFunction of AS160 Promotes Lysosomal Degradation of GLUT4 and CausesPostprandial Hyperglycemia and Hyperinsulinemia. Diabetes 2016; 65: 3327-3340.
Xiong Y, Swaminath G, Cao Q, Yang L, Guo Q, Salomonis H et al. Activation of FFA1mediates GLP-1 secretion in mice. Evidence for allosterism at FFA1. Mol CellEndocrinol 2013; 369: 119-129.
Xirouchaki CE, Mangiafico SP, Bate K, Ruan Z, Huang AM, Tedjosiswoyo BW et al.Impaired glucose metabolism and exercise capacity with muscle-specificglycogen synthase 1 (gys1) deletion in adult mice. Mol Metab 2016; 5: 221-232.
Xu PT, Song Z, Zhang WC, Jiao B, Yu ZB. Impaired translocation of GLUT4 results ininsulin resistance of atrophic soleus muscle. Biomed Res Int 2015; 2015: 291987.
Xu Y, Nan D, Fan J, Bogan JS, Toomre D. Optogenetic activation reveals distinct rolesof PIP3 and Akt in adipocyte insulin action. J Cell Sci 2016; 129: 2085-2095.
Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but notpravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretiondue to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol1999; 126: 1205-1213.
Yajima H, Komatsu M, Schermerhorn T, Aizawa T, Kaneko T, Nagai M et al. cAMPenhances insulin secretion by an action on the ATP-sensitive K+ channel-independent pathway of glucose signaling in rat pancreatic islets. Diabetes 1999;48: 1006-1012.
Yamada H, Yoshida M, Ito K, Dezaki K, Yada T, Ishikawa SE et al. Potentiation ofGlucose-stimulated Insulin Secretion by the GPR40-PLC-TRPC Pathway inPancreatic beta-Cells. Sci Rep 2016; 6: 25912.
Yamamoto N, Ueda-Wakagi M, Sato T, Kawasaki K, Sawada K, Kawabata K et al.Measurement of Glucose Uptake in Cultured Cells. Curr Protoc Pharmacol 2015;71: 12.14.1-26.
Yang H, Yang L. Targeting cAMP/PKA pathway for glycemic control and type 2diabetes therapy. J Mol Endocrinol 2016; 57: R93-R108.

77
Yang M, Chisholm JW, Soelaiman S, Shryock JC. Sulfonylureas uncouple glucose-dependence for GPR40-mediated enhancement of insulin secretion from INS-1Ecells. Mol Cell Endocrinol 2010; 315: 308-313.
Yokoi N, Gheni G, Takahashi H, Seino S. beta-Cell glutamate signaling: Its role inincretin-induced insulin secretion. J Diabetes Investig 2016; 7 Suppl 1: 38-43.
Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J et al. Control of hepaticgluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001;413: 131-138.
Yoshida S, Ohishi T, Matsui T, Tanaka H, Oshima H, Yonetoku Y et al. NovelGPR119 agonist AS1535907 contributes to first-phase insulin secretion in ratperfused pancreas and diabetic db/db mice. Biochem Biophys Res Commun2010; 402: 280-285.
Zaharan NL, Williams D, Bennett K. Statins and risk of treated incident diabetes in aprimary care population. Br J Clin Pharmacol 2013; 75: 1118-1124.
Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol2001; 2: 107-117.
Zhang J, Liu F. Tissue-specific insulin signaling in the regulation of metabolism andaging. IUBMB Life 2014; 66: 485-495.
Zhao YF, Pei J, Chen C. Activation of ATP-sensitive potassium channels in ratpancreatic beta-cells by linoleic acid through both intracellular metabolites andmembrane receptor signalling pathway. J Endocrinol 2008; 198: 533-540.
Zhou J, Li W, Xie Q, Hou Y, Zhan S, Yang X et al. Effects of simvastatin on glucosemetabolism in mouse MIN6 cells. J Diabetes Res 2014; 2014: 376570.
Zhou YJ, Song YL, Zhou H, Li Y. Linoleic acid activates GPR40/FFA1 andphospholipase C to increase [Ca2+]i release and insulin secretion in islet beta-cells. Chin Med Sci J 2012; 27: 18-23.
Zhou Z, Menzel F, Benninghoff T, Chadt A, Du C, Holman GD et al. Rab28 is aTBC1D1/TBC1D4 substrate involved in GLUT4 trafficking. FEBS Lett 2016.
Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by associationwith the ribosome. Cell 2011; 144: 757-768.
Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB et al.Targeted disruption of the glucose transporter 4 selectively in muscle causesinsulin resistance and glucose intolerance. Nat Med 2000; 6: 924-928.
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senseslysosomal amino acids through an inside-out mechanism that requires thevacuolar H(+)-ATPase. Science 2011; 334: 678-683.

78

DIS
SE
RT
AT
ION
S | N
AG
EN
DR
A Y
AL
UR
I | DIA
BE
TO
GE
NIC
EF
FE
CT
S O
F S
TA
TIN
S | N
o 412
uef.fi
PUBLICATIONS OF THE UNIVERSITY OF EASTERN FINLAND
Dissertations in Health Sciences
ISBN 978-952-61-2465-0ISSN 1798-5706
Dissertations in Health Sciences
PUBLICATIONS OF THE UNIVERSITY OF EASTERN FINLAND
NAGENDRA YALURI
DIABETOGENIC EFFECTS OF STATINS
This study investigates the effects of statin
treatment on the risk of incident type 2 diabetes, insulin secretion and insulin
resistance in a prospective population study, and the molecular mechanisms underlying
these effects in mouse pancreatic β-cells and L6 skeletal myotubes. It shows that statin
treatment is associated with impairment in both insulin secretion and insulin sensitivity, and that the effects of simvastatin on insulin secretion and glucose uptake occur through
multiple targets.
NAGENDRA YALURI