dissecting dopamine d2 receptor signaling · dissecting dopamine d2 receptor signaling prashant...
TRANSCRIPT

Dissecting Dopamine D2 Receptor Signaling
Prashant Donthamsetti
Submitted in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in the Graduate School of Arts and Sciences
COLUMBIA UNIVERSITY 2015

© 2015
Prashant Donthamsetti All rights reserved

ABSTRACT
Dissecting Dopamine D2 Receptor Signaling
Prashant Donthamsetti
Dopamine D2 receptor (D2R) is a G protein-coupled receptor (GPCR) that activates G protein and
arrestin signaling molecules. D2R antagonism has been a hallmark of antipsychotic medications for more
than half a century. However, this drug-class is associated with substantial side effects that decrease
quality of life and medication compliance. The development of novel antipsychotic medications with
superior therapeutic and side effect profiles has been hampered in part due to a poor understanding of
the specific D2R populations and downstream signaling molecules that must be blocked to confer
therapeutic efficacy. It has been proposed that antipsychotic medications confer their effects through the
blockade of arrestin but not G protein signaling downstream of D2R, and thus substantial efforts have
gone towards the development of ligands that selectively block arrestin signaling. However, this approach
suffers from several major limitations, namely that blockade of G protein signaling may also be important
in conferring antipsychotic effects. Moreover, currently available pharmacological and genetic tools that
have been used to probe G protein and arrestin signaling downstream of D2R in vivo suffer from on- and
off-target effects that add substantial confounds to our understanding of these processes. Herein, we
describe the development of several tools that can be used to probe G protein and arrestin-mediated
processes in vivo with high specificity, as well as mechanisms by which these processes are activated.

i
Table of contents
List of Figures ii
Acknowledgements iv
Dedication v
CHAPTER 1. Introduction 1
CHAPTER 2. The development of genetic tools to probe D2R-mediated arrestin signaling in vivo 25
CHAPTER 3. The development of genetic tools to probe D2R-mediated G protein signaling in vivo 44
CHAPTER 4. Towards understanding functional selectivity between G protein-dependent pathways
downstream of D2R 82
CHAPTER 5. Summary and Conclusions 107
Materials and Methods 110
References 134
Appendix 150

ii
List of Figures
Figure 1-1. Major dopaminergic pathways within the CNS. 2
Figure 1-2. Basal ganglia circuitry. 3
Figure 1-3. The crystal structure of the dopamine D3R with eticlopride. 5
Figure 1-4. Pharmacological agents vary in their functional properties. 6
Figure 1-5. G protein activation cycle. 7
Figure 1-6. DAR G protein signaling. 8
Figure 1-7. D2R and arrestin. 9
Figure 1-8. The DA synapse. D2R is expressed both pre- and postsynaptically within a DA synapse. 11
Figure 1-9. Classical pharmacological agents versus functional selective ligands. 15
Figure 1-10. G protein versus arrestin bias. 16
Figure 1-11. Characteristics of currently tools used to probe receptor signaling. 23
Figure 2-1. The canonical receptor-GRK-arrestin cascade 26
Figure 2-2. BRET-based cAMP sensor CAMYEL 27
Figure 2-3. Comparison of forskolin and isoproterenol stimulation of cAMP in the CAMYEL assay. 28
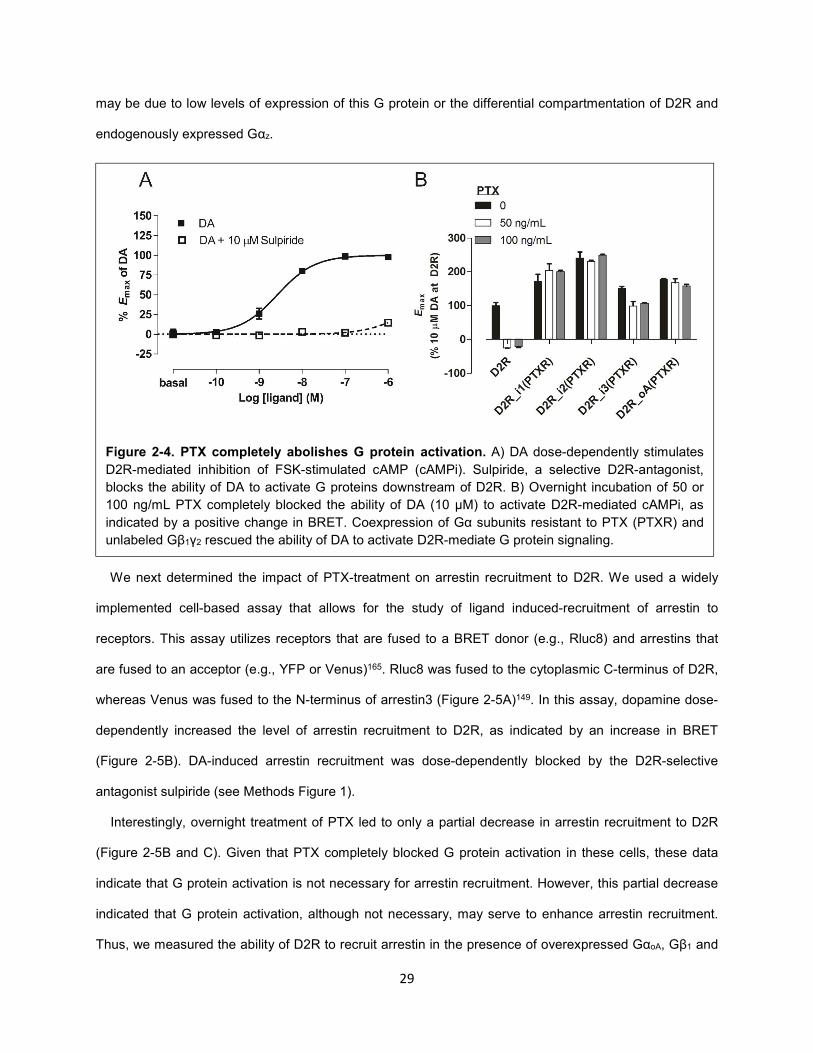
Figure 2-4. PTX completely abolishes G protein activation. 29
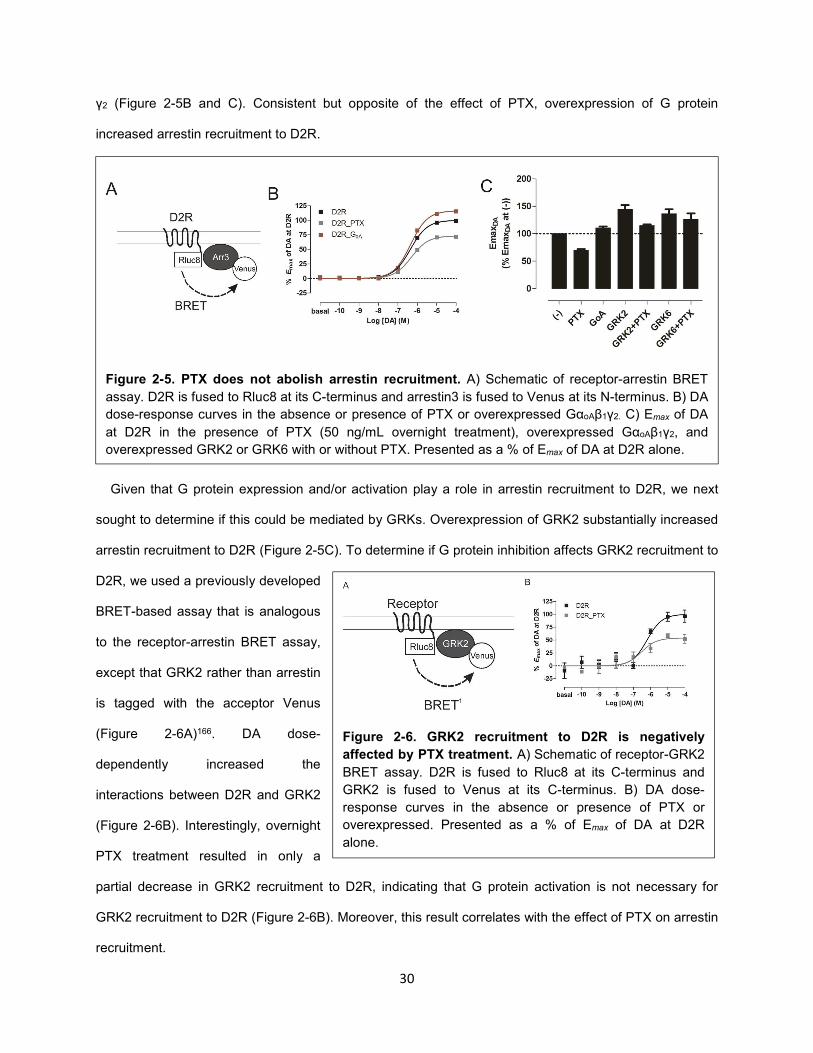
Figure 2-5. PTX does not abolish arrestin recruitment. 30
Figure 2-6. GRK2 recruitment to D2R is negatively affected by PTX treatment. 30
Figure 2-7. Arrestin biased DREADD versus D2R. 33
Figure 2-8. G protein and arrestin recruitment to D2R wildtype and mutants. 35
Figure 2-10. The kinetics of arrestin recruitment to D2(L123W:R132L) relative to wildtype D2R. 36
Figure 2-9. D2(L123W:R132L) short and long isoforms behave similarly. 36
Figure 2-11. cAMPi by D2R wildtype and its mutants. 37
Figure 2-12. Arrestin mediated-internalization of D2R and its mutants. 38
Figure 2-13. Constitutive G protein activity for D2R and its mutants. 39
Figure 2-14. Schematic of AAV viral delivery system. 40
Figure 2-15. D2R(D114A) does not recruit G protein or arrestin. 40
Figure 2-16. Expression of D2R wildtype and its mutants in D2R-expressing MSNs in the NAc
differentially affects basal locomotion and motivation tasks. 41
Figure 3-1. Comparison of modified S1 constructs. 46
Figure 3-2. S1(R9K:E129G) does not affect G protein signaling. 47
Figure 3-3. Schematic of AAV viral delivery system. 48
Figure 3-4. G protein biased D2R mutant (D2RGPB). 49
Figure 3-5. D2-A4-mediated inhibition of cyclic AMP accumulation and G protein activation. 52
Figure 3-6. D2-A4-mediated arrestin3 recruitment. (A) Schematic of the BRET biosensor used to
measure recruitment of arrestin to receptor. 53
Figure 3-7. D2-WT, D2-A3, and D2-A2-mediated G protein activation and arrestin3 recruitment. 55

iii
Figure 3-8. Agonist-induced receptor internalization. 57
Figure 3-9. Characterization of quinpirole-induced recruitment of arrestin3. 58
Figure 3-10. Agonist-induced recruitment of GRK2. 59
Figure 3-11. Influence of GRK2 on receptor recruitment of arrestin3 and internalization. 61
Figure 3-12. Clathrin- and AP-2- mediated internalization of D2-WT. 62
Figure 3-13. Receptor mediated interaction between arrestin3 and β2-adaptin. 63
Figure 3-14. Interaction between receptors and β2-adaptin. 65
Table 3-1. pEC50 values from G protein activation and recruitment assays 66
Figure 3-15. Arrestin recruitment to D2R requires the kinase activity of GRK2 or 6. 71
Figure 3-16. One hour preincubation of GRK inhibitors has no effect on arrestin recruitment. 72
Figure 3-17. Time course of arrestin recruitment to D2R and β2AR in the presence of a non-selective
GRK inhibitor. 73
Figure 3-18. D2R IL3 truncation mutant is a supersignaling receptor. 75
Figure 3-19. D2R IL1 T->V mutant is impaired at G protein and arrestin signaling. 76
Figure 3-20. G protein activation and arrestin recruitment to IL mutants of D2R. 79
Figure 4-1. Aripiprazole differentially activates G protein-mediated pathways downstream of D2R. 82
Figure 4-3. Aripiprazole and 3508 do not activate GIRK channels downstream of D2R. 84
Figure 4-4. Theoretical unbiased GIRK activity of D2R ligands. 86
Figure 4-5. 3508 is not G protein versus arrestin biased. 88
Figure 4-6. Ligand-dependent conformational changes in Gi1 upon activation of D2R. 89
Figure 4-7. D2R activation by various D2R ligands promotes the interaction between Gβγ and GRKct. 90
Figure 4-8. D2R activation by various D2R ligands promotes the interaction between Gβγ and GIRK
channels. 92
Figure 4-9. Optimization of the CODA-RET heterotrimeric G protein assay. 96
Figure 4-10. Subtype-specific G protein activation screen. 99
Figure 4-11. D2R-mediated GIRK activation is more highly correlated with Gαi1β5γ2 than Gαi1β1γ2
activation. 100
Figure 4-12. Characterization of the Gαi1β5γ2 heterotrimer. 102
Figure 4-13. Model of selective activation of cAMPi over GIRK by 3508 downstream of D2R. 104
Methods Figure 1. BRET-based arrestin assays. 133

iv
Acknowledgements
I first and foremost would like to thank my advisor, Dr. Jonathan Javitch, for years of unquantifiable
mentorship, support and friendship. I would also like to thank Drs. Hideaki Yano and Eneko Urizar for
their enthusiasm and guidance but, more importantly, their friendship. I would also like to thank the many
current and past members of the Javitch, Kellendonk, Quick and Freyberg labs for the friendship and
support. I am deeply indebted to Val Monrose for making sure the lab does not explode. I am very
thankful for Douglas Drake, who is a constant source of thoughtfulness, laughter and delicious food. I
would also like to thank Dr. Celine Gales, who was incredibly gracious for inviting me to train in her lab at
INSERM in Toulouse, France.
I would like to thank my collaborators, including Drs. Christoph Kellendonk, Zachary Freyberg, Kim
Neve, Nevin Lambert, Vsevovold Gurevich, Lei Shi, Amy Newman, Robert Lane and Arthur
Christopolous, as well as members of their labs. I would particularly like to thank Dr. Eduardo Gallo, who
performed the behavioral analyses described below, Dr. Cecilea Clayton, who contributed significantly to
the characterization of the D2-A3 mutant, and Dr. Mayako Michino for carrying modeling analyses on IL3
of D2R.
I wish to thank my thesis committee (Drs. Jonathan Javitch, Christoph Kellendonk, Richard Mailman
and Qin Fan) for their continual support throughout my graduate career. I would also like to thank my
qualifying committee (Drs. Jonathan Javitch, Susan Steinberg, Qing Fan, and Lloyd Greene) as well as
my mini-oral committee (Drs. Jonathan Javitch, Christoph Kellendonk and David Sulzer). I would also like
to thank my thesis defense committee (Drs. Jonathan Javitch, Christoph Kellendonk, Richard Mailman,
Nevin Lambert and Susan Steinberg) for their careful review of this manuscript.
Many thanks to the Columbia University Pharmacology department (especially Drs. Robert Kass,
Daniel Goldberg, Richard Robinson and Neil Harrison, and of course Karen Allis) for their support and
creating positive academic environment. I would also like to thank the Pharmacology graduate students,
especially Doug Barrows for his friendship and cat sitting support.

v
Dedication
I dedicate this thesis to many family and friends who have supported me throughout this process. This
work is most of all dedicated to my wonderful and caring fiancé Erin Turner, who has been my inspiration.
Her dedication to her work and to the people around her is awe-inspiring. I am forever in her debt for
supporting me all of these years. And of course, I would like to dedicate this work to the queen of the
household, our cat Pearl Turner-Donthamsetti.

1
CHAPTER 1. Introduction
Schizophrenia is a neurodevelopmental disorder characterized by psychosis that manifests as
hallucinations, delusions and/or disordered thought.1 Features of this disorder also include negative
symptoms (apathy, avolition, alogia) and cognitive deficits (deficits in working memory, processing speed,
social cognition). This chronic and debilitating illness affects ~24 million people worldwide2 and is
associated with substantial societal costs, estimated at ~$75 billion per year in the USA alone3.
The etiological basis of schizophrenia is not well understood4. Similar to other common diseases (e.g.,
diabetes), schizophrenia is complex, with contributions from multiple genes, as well as epigenetic and
environmental factors5. Genetic factors play a major role in the development of schizophrenia, which has
an estimated heritability of 60-85%6. A number of genetic risk factors have been identified, including rare
point mutations as well as copy-number-variants (CNVs)7. More recently, de novo CNVs such as those at
the 22q11.2 locus have been identified as important risk factors. However, individual genetic risk factors
generally account for only a small fraction of the total population, thus hampering our understanding of
the underlying basis of schizophrenia as well as the development of therapeutics targeting this disorder.
The advent of antipsychotic medications acting at the dopamine (DA) D2 receptor (D2R) revolutionized
the treatment of schizophrenia by helping to ameliorate positive symptoms8. D2R antagonism is the
unifying property of all antipsychotic drugs in use, although these compounds can produce serious side
effects that impair both quality of life and medication compliance. Remarkably, in spite of the ~60 years of
clinical development of antipsychotics, the mechanisms by which D2R blockers exert their therapeutic
actions are poorly understood, both with regards to their site of action in the brain as well as the signaling
events downstream of D2R. The overall goal of the work described herein was to better understand the
molecular basis of antipsychotic drug action, ultimately facilitating the development of more highly
efficacious therapeutics for schizophrenia with reduced side effects.
The dopamine system
The catecholamine DA is a neurotransmitter that plays a major role in the central nervous system
(CNS)9,10 Similar to other monoamines such as norepinephrine, DA generally modulates fast
2
neurotransmission mediated by glutamate and GABA10. DA is involved in the regulation of several critical
circuits, including the nigrostriatal, mesolimbic and mesocortical systems, which have been shown to
modulate learning, reward, emotional, motor and cognitive functions (Figure 1-1)10-12. The dysregulation of
DA and/or its target receptors within these circuits has been implicated in a number of other
neuropsychiatric disorders in addition to schizophrenia, including Parkinson’s disease, attention deficit
hyperactivity disorder (ADHD), Tourette’s syndrome, Huntington’s disease and drug abuse13-21.
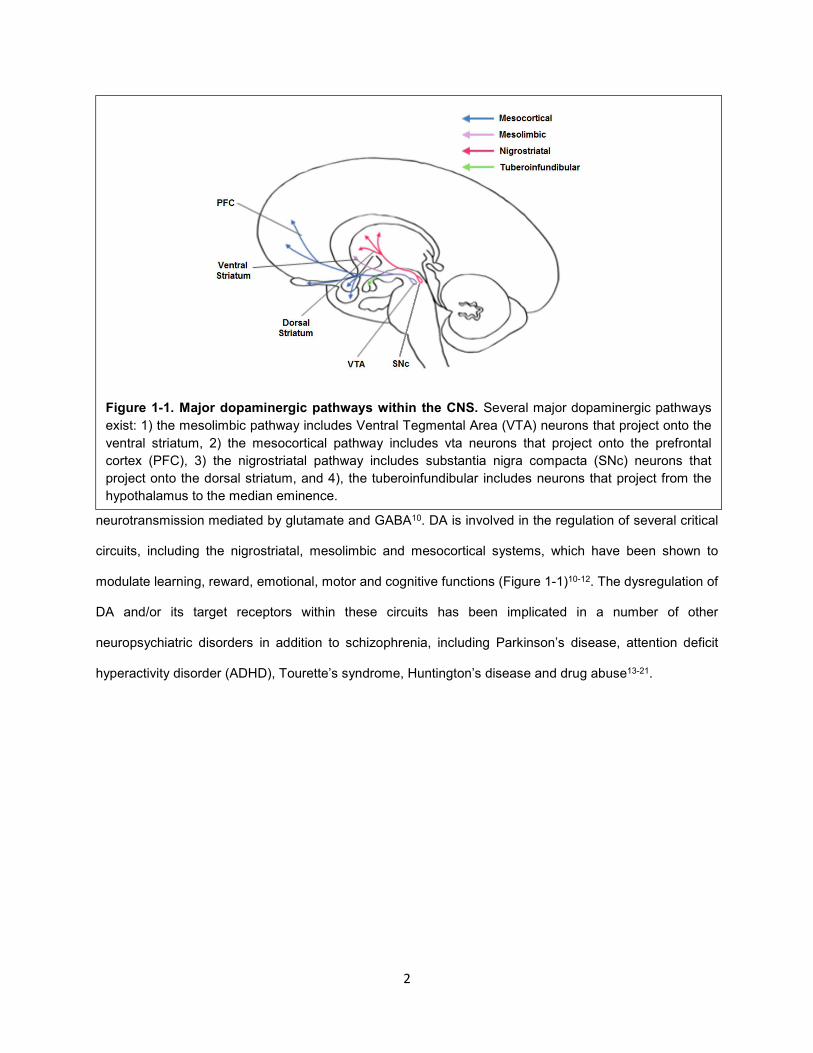
Figure 1-1. Major dopaminergic pathways within the CNS. Several major dopaminergic pathways
exist: 1) the mesolimbic pathway includes Ventral Tegmental Area (VTA) neurons that project onto the
ventral striatum, 2) the mesocortical pathway includes vta neurons that project onto the prefrontal
cortex (PFC), 3) the nigrostriatal pathway includes substantia nigra compacta (SNc) neurons that
project onto the dorsal striatum, and 4), the tuberoinfundibular includes neurons that project from the
hypothalamus to the median eminence.
3
A central component of the
nigrostriatal and mesolimbic circuits is
the striatum, which is the primary input
structure to a group of subcortical nuclei
called the basal ganglia (Figure 1-2)22.
The striatum receives dopaminergic input
from the substantia nigra compacta
(SNc) and ventral tegmental area (VTA),
which form part of the nigrostriatal and
mesolimbic systems, respectively23,24.
The SNc innervates the dorsal striatum,
including the caudate nucleus and the
putamen, whereas the VTA innervates
the ventral striatum, including the
Nucleus Accumbens (NAc). The striatum
is composed of three major classes of
cells, including medium spiny neurons
(MSNs) that represent ~95% of striatal
neurons, tonically active cholinergic
interneurons (TANs), and GABAergic
interneurons25. MSNs are GABAergic
neurons that form two populations: 1)
those that project directly onto the globus
pallidus internal segment (GPi) and the
substantia nigra reticulata (SNr),
constituting the “direct pathway”, and 2) those that project indirectly onto the GPi through the globus
pallidus external segment (GPe) and subthalamic nucleus (STN), constituting the “indirect pathway”26,27.
Recent studies, however, have indicated that these two pathways are substantially more structurally and
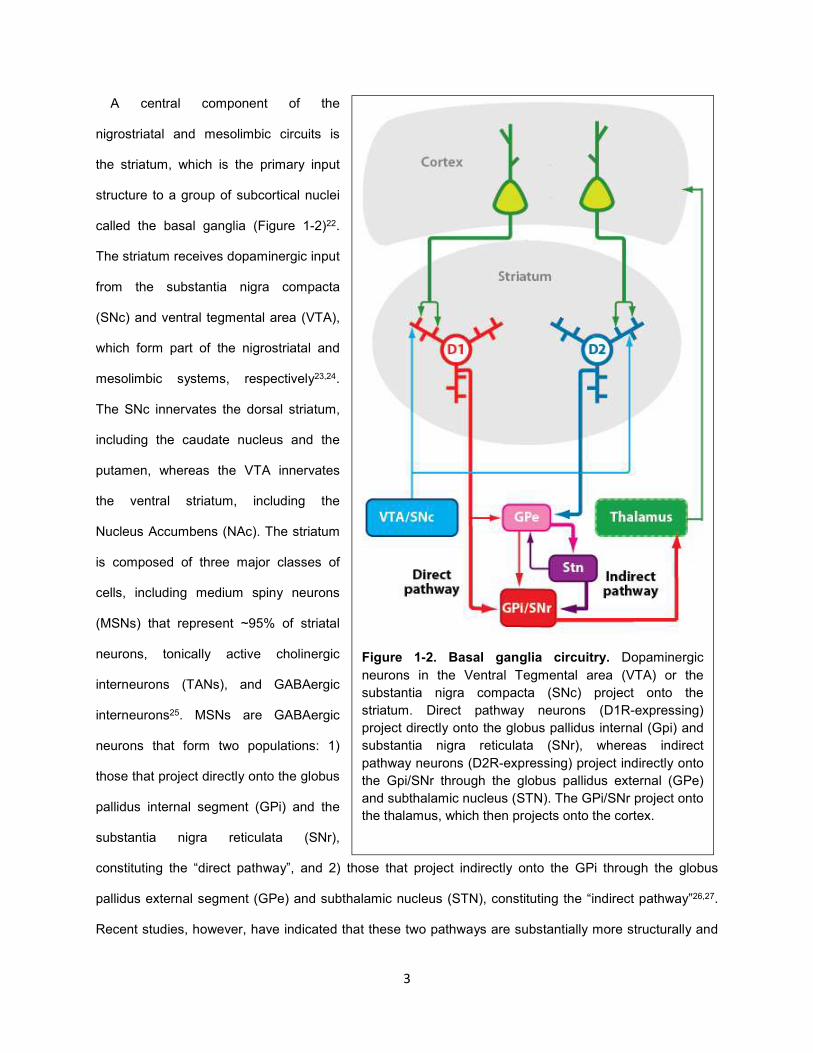
Figure 1-2. Basal ganglia circuitry. Dopaminergic
neurons in the Ventral Tegmental area (VTA) or the
substantia nigra compacta (SNc) project onto the
striatum. Direct pathway neurons (D1R-expressing)
project directly onto the globus pallidus internal (Gpi) and
substantia nigra reticulata (SNr), whereas indirect
pathway neurons (D2R-expressing) project indirectly onto
the Gpi/SNr through the globus pallidus external (GPe)
and subthalamic nucleus (STN). The GPi/SNr project onto
the thalamus, which then projects onto the cortex.

4
functionally intertwined than was thought previously, e.g., it has been shown recently that direct pathway
neurons can form highly plastic bridging collaterals that project onto the GPe (Figure 1-2)27,28. The output
structure of the basal ganglia, the thalamus, projects onto the cortex, which innervates the striatum, thus
completing both the nigrostriatal and mesolimbic circuits. Dopaminergic VTA neurons can also project
directly on the medial prefrontal cortex (mPFC), which constitutes the mesocortical system10.
Within the CNS, DA is also present in the tuberoinfundibular system, which originates in the
hypothalamus and regulates the secretion of hormones, particularly prolactin from the pituitary gland29. In
the periphery, DA regulates various physiological processes, including cardiovascular, sympathetic, and
renal functions10.
Dopamine receptor structure
DA acts through DA receptors (DARs), which are members of the G protein-coupled receptor (GPCR)
superfamily30. GPCRs constitute the largest class of membrane proteins (~800 members) and are targets
of ~25% of all currently available medications, including antipsychotic medications31,32. GPCRs respond to
a variety of extracellular signals (e.g., light, peptide hormones, neurotransmitters, and enzymes) and can
be subdivided into families based on structure and function33,34. GPCRs have seven-transmembrane (TM)
spanning segments that form the TM bundle, as well as three extracellular and intracellular loops (EL and
IL loops, respectively), which can vary in length and physical properties35. The N- and C- termini are on
the extracellular and intracellular sides of the plasma membrane, respectively, and can also vary greatly
in length and structural properties.
DARs are Family A GPCRs, which are rhodopsin-like receptors that include aminergic receptors such
as β-adrenergic receptors (β-AR) and serotonin receptors (5-HTRs)34. DARs are subgrouped into two
categories, D1-like (D1, D5) and D2-like (D2, D3, D4) receptors, based on sequence and function. D1-like
receptors are quite divergent from the D2-like receptors, and in fact are closer in sequence to the β-AR
(~43% versus ~39% sequence identity between D1-like DARs and β-AR and between D1-like and D2-like
DARs, respectively). The subtypes within the D1- and D2-like subgroup are highly homologous. D1R and
D5R are 80% homologous in their TM domain. Overall, D2R and D3R are more closely related to each
other than either is to D4R (78% identity between D2R and D3R and ~50% identity between D4R and

5
D2R/D3R in the TM domain). Several splice variants exist for D2-like receptors. Notably, splice variants of
D2R include D2-short (D2S) and D2-long (D2L), which contains an additional 29 amino acids in IL336. The
C-termini of D1-like receptors is ~7-fold greater in length than D2-like receptors37. However, IL3 is
considerably longer in D2-like receptors than D1-like receptors.
Family A GPCRs like DARs bind their
endogenous ligands through the orthosteric
binding pocket (OBS), which is formed by the
TM domain34. A high resolution crystal
structure of D3R bound to the D2R/D3R
ligand eticlopride revealed that the OBS of
this receptor is formed by TMs 3, 5, 6 and 7,
consistent with our accumulated
understanding of aminergic GPCRs based on
structure-function studies (Figure 1-3)38. Not
surprisingly, modeling studies indicate that
DA binds similarly to eticlopride within this
site in D3R39. Given the high degree of
conservation within the OBS of DARs (50-
100% identity), DA likely binds the OBS of all
DARs in a similar manner34.
Dopamine receptor pharmacology
A combination of theoretical modeling and experimental data has revealed that GPCRs exist as
collections or ensembles of interchanging tertiary conformations, consisting of both active and inactive
states.40-42 The binding of an agonist to the OBS of a GPCR stabilizes active receptor conformations that
couple to downstream transducers, including heterotrimeric G proteins, on the intracellular face of the
receptor. Agonists can vary in efficacy, i.e., maximum response achievable, and range from full to partial
agonists (Figure 1-4)43. Full agonism is most often defined by the activity of the endogenous ligand.
Figure 1-3. The crystal structure of the dopamine
D3R with eticlopride. D3R with transmembrane (TM)
segments in yellow extracellular loops (ELs) in cyan
and intracellular loops (ILs) in blue. Eticlopride (red) is
bound to the OBS site. IL3 was replaced with T4
lysozyme (not shown) to facilitate crystallization. The
N-terminus D3R was not resolved in the crystal
structure. PDB 3PBL.
6
Efficacy is dependent on several factors, including the ability of the ligand to stabilize receptor active
states as well as the kinetics of occupancy within the OBS44,45.
Orthosteric antagonists of GPCRs compete directly with endogenous ligands at the OBS43. Because
receptors can sample active states
independently of agonist binding, they
can exhibit basal or constitutive activation
of downstream transducers46. Neutral
antagonists compete with orthosteric
ligands for the OBS but do not alter the
basal equilibrium state between active
and inactive receptor states (Figure 1-4).
In contrast, inverse agonists preferentially
stabilize inactive receptor states,
decreasing constitutive activity (Figure 1-
4).
As discussed above, DA binds similarly
to DARs. However, pharmacological agents with a range of efficacies and varying degrees of affinity
across DARs have been identified34. Agonists and antagonists that display a high degree of D1- or D2-
like selectivity have been developed. Within the D2-like dopamine receptor subgroup, D3R- or D4R-
selective ligands and to a lesser degree D2R-preferential ligands have been discovered47,48. However,
within the D1-like dopamine receptor subgroup, separation of D1R- and D5R- selective ligands using
traditional medicinal chemistry efforts has thus far been unsuccessful49.
Dopamine receptor signaling
As noted above, DARs couple to G proteins, which consist of Gα, Gβ and Gγ subunits and facilitate
signal transduction upon activation50. G proteins are associated with the plasma membrane through
various post-translational lipid modifications on Gα and Gγ subunits51. In the inactive state, Gαβγ exists
as a closed complex bound to GDP within the Gα subunit (Figure 1-5). GPCRs are guanine exchange
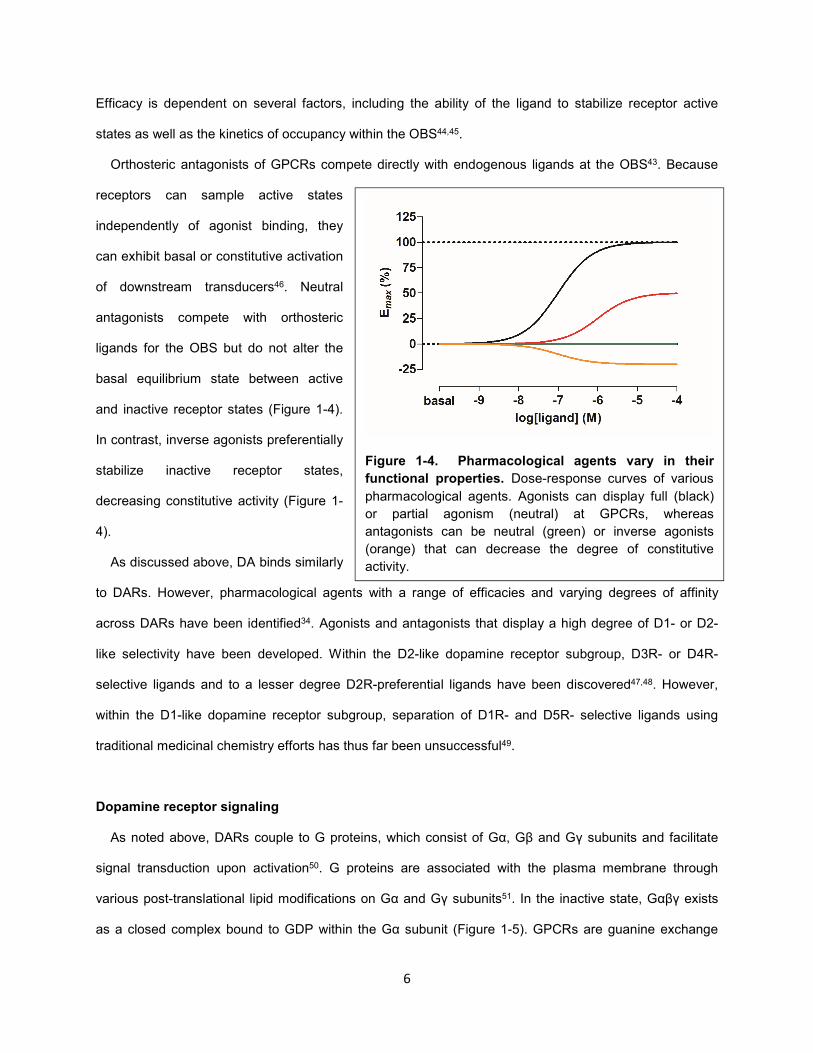
Figure 1-4. Pharmacological agents vary in their
functional properties. Dose-response curves of various
pharmacological agents. Agonists can display full (black)
or partial agonism (neutral) at GPCRs, whereas
antagonists can be neutral (green) or inverse agonists
(orange) that can decrease the degree of constitutive
activity.

7
factors (GEFs) that facilitate exchange of GDP for GTP, which leads to a conformational change between
or dissociation of the Gα and Gβγ subunits, both of which can independently bind and modulate various
downstream signaling proteins. The Gα subunit is also a GTPase that catalyzes the conversion of bound
GTP to GDP, resulting in reversion of G protein to the inactive state52. However, significant GTP to GDP
conversion requires the activity of G protein accelerating proteins (GAPs), specifically Regulator of G
protein Signaling (RGS) proteins52.
Like many GPCRs, DARs are coupled to specific G protein subtypes that have distinct functions30.
There are 23 Gα, seven Gβ, and twelve Gγ subtypes, some of which are splice variants53,54. Whereas the
function of different Gα subunits has been studied extensively, the functional differences between Gβ and
Gγ subtypes are poorly understood53. Conventionally, heterotrimeric G proteins are designated by the
identity of the Gα subunit but can contain many combinations of Gβ and Gγ53. D1-like receptors are
coupled to Gs/olf, which activate adenylate cyclase (AC), increasing cyclic AMP (cAMP) levels (Figure 1-
6A)30. Two isoforms of Gαs exist (Gαss and Gαsl)55. D2-like receptors couple to Gi/o/z, which inhibits AC,
decreasing cAMP(Figure 1-6A)30. There are three genes encoding Gαi subunits, including i1, i2, and i3.
Figure 1-5. G protein activation cycle. A) The Gα subunit of heterotrimeric G protein is bound to
GDP in the closed inactive complex. B) The G protein binds to an active receptor, mainly through the
C-terminus of the Gα subunit. C) The GPCR, which is a guanine exchange factor (GEF), catalyzes the
exchange of GDP to GTP on the Gα subunit. D) Upon GTP binding, the Gα and Gβγ subunits
dissociate or rearrange, subsequently activating various signaling pathways. E/F) RGS proteins,
which are G protein Accelerating Proteins (GAPs), enhance the GTPase function of the Gα subunit,
which converts GTP to GDP. This results in the conversion of the G protein back to its closed inactive
state (A).

8
There are also two isoforms of Gαo, oA and oB. All Gα subunits capable of coupling to D2-like receptors,
except Gαz, are sensitive to pertussis toxin (PTX), the etiological agent of whooping cough56. This protein
catalyzes the ADP-ribosylation of a cysteine within the C-terminal end of the Gα subunit, which is critical
for interaction of the G proteins with receptors57.
DAR-mediated modulation of cAMP levels affects various signaling proteins, including Protein Kinase A
(PKA), which can affect the phosphorylation state of signaling molecules such as Dopamine- and cAMP-
regulated phosphoprotein-32 (DARPP-32), a critical regulatory protein that is highly enriched in the
striatum (Figure 1-6A)58. DARs can also modulate various ion channels. D2R can modulate K+ and Ca2+
channels, either directly though the Gβγ subunits or indirectly by affecting regulators of these channel
such as phospholipase C (PLC; Figure1-6B). Gβγ release upon activation of D2-like receptors can
directly activate G protein-inwardly rectifying potassium channels (GIRKs), although Gα subunits have
also been shown to play a role in regulating GIRK activation59. D2R has been shown to potentiate the
activation of Kv1 channels through Gβγ, although the underlying mechanism is not fully understood60-62.
The activation of GIRK and Kv1 channels facilitates the inward flow of K+ ions into the cell, decreasing DA
neuron firing rates or inhibiting DA release 63.
Figure 1-6. DAR G protein signaling. A) D1-like and D2-like receptors activate and inhibit,
respectively, adenylate cyclases (AC). AC stimulates the production of cyclic AMP (cAMP), which
activates Protein Kinase A (PKA). PKA can phosphorylate various downstream substrates such as
Dopamine and cAMP-regulated Phosphoprotein-32 (DARPP-32). B) D2R can directly and indirectly
modulate various K+ and Ca2+ channels through its Gβγ subunits.

9
Like many GPCRs, D1-like and D2-like receptors have also been shown to recruit arrestins from the
cytosol to the plasma membrane upon receptor activation (Figure 1-7)8. There are four different arrestins
(arrestin-1-4)64. Whereas arrestin1 and arrestin4 are restricted to the visual system, arrestin2 and
arrestin3 are more widely expressed and are both capable of being recruited to DARs30. In general, the
time course of arrestin recruitment to GPCRs is slower than G protein coupling64.
Arrestins facilitate receptor
desensitization by binding directly to
the activated receptor, directly
competing with G protein for a
common receptor interface65. Once
recruited to receptors, arrestins can
also bind to clathrin and clathrin-
associated adaptor proteins (e.g., AP-
2), thus mediating receptor
internalization via clathrin-mediated
endocytosis (Figure 1-7)66,67. It has
been shown recently that arrestins,
once recruited to GPCRs, can also
signal by acting as scaffolds for a
variety of signaling molecules64. Recent studies indicate that arrestin, once recruited to D2R, can act as a
scaffold for protein phosphatase-2A (PP2A) and Akt, a critical signaling kinase (Figure 1-7)68. PP2A
dephosphorylates Akt, decreasing its kinase activity. This results in a decrease in the phosphorylation
state of downstream proteins such as glycogen synthase kinase-3β (GSK3β)69. Phosphorylation of
GSK3β by Akt inhibits its activity70, and thus D2R activation presumably increases GSK3β signaling.
The recruitment of arrestins to GPCRs can be enhanced by the actions of G protein receptor kinases
(GRKs)71. GRKs recognize the receptor active state and phosphorylate intracellular loops and the C-
terminus of the receptor at serine and threonine residues. This enhances the affinity of arrestin for
receptors in part due to the disruption of the so-called “polar core-phosphate sensor” of arrestin72.
Figure 1-7. D2R and arrestin. D2R (and all other DARs)
recruit arrestins, which terminate G protein signaling and
facilitate receptor internalization. Arrestin has also been
shown to facilitate signaling through D2R by acting as a
scaffold for phosphatase-2A (PP2A) and Akt. PP2A
dephosphorylates Akt, decreasing its activity. This results the
decrease in phosphorylation of glycogen synthase-3 (GSK3),
an important signaling protein.

10
Although receptor phosphorylation has been shown to increase arrestin recruitment to many GPCRs, it is
not absolutely necessary in some cases73-76. There are seven GRKs, which are divided based on
expression, structure and function. GRK1 and 7 are restricted to rods and cones of the visual
system64,71,77. GRK2 and 3 are more widely expressed and are cytosolic proteins that are recruited to the
plasma membrane upon GPCR activation. Recruitment of GRK2/3 to the plasma membrane is facilitated
by interactions of their pleckstrin homology (PH) domain with Gβγ subunits that are released upon G
protein activation. GRK4, 5 and 6 are also more widely expressed. However, these GRKs do not have PH
domains and are largely localized to the plasma membrane in part through lipid modifications such as
palmitoyl groups and interactions with lipid77. The role of phosphorylation on arrestin recruitment to DARs
is not fully understood. Although GRK2 and GRK3 have been shown to phosphorylate D2R, this is less
clear for GRK4/5/678,79.
Dopamine receptor expression and function
DARs are expressed throughout the dopamine system. D1R is highly localized to the striatum and
prefrontal cortex, whereas D5R is expressed at significantly lower levels in a number of regions including
the prefrontal cortex, substantia nigra and the MSNs of the striatum10. D2R is highly expressed in the
striatum and is also found in areas including the substantia nigra, VTA, hypothalamus and cortical areas.
D3R is overall much more limited in expression compared to D2R and is restricted to the NAc in the
striatum80. Of the DARs, D4R is expressed at the lowest level throughout the brain10.
Immunostaining81 and in situ hybridization82-84 studies indicate that within the striatum there are two
major populations of neurons, including D1R- and D2R-expressing neurons (Figure 1-2). This is
consistent studies in which mice carrying bacterial artificial chromosomes (BACs) express specific gene
reporters in these distinct neuronal populations85. Although a small percentage of MSNs coexpress both
receptors, D1R and D2R are largely separated in mice, although they have been shown recently to be
coexpressed in early development81. Interestingly, D1R-expressing neurons predominantly form the direct
pathway, whereas D2R-expressing neurons form the indirect pathway27.
11
Several populations of D2R can exist
within a DA synapse (Figure 1-8). Unlike
D1R, which is mainly expressed
postsynaptically, D2Rs are expressed
pre- and postsynaptically10. The role of
postsynaptic D2Rs is cell-type dependent.
D2R expressed in MSNs modulate
various ion channels. D2R activation in
these neurons suppresses L-type Ca2+
channels via a Gβγ-mediated mechanism,
decreasing neuronal excitability86, and it
has also been reported to affect different
K+ channel currents through PKA- and
PLC-mediated mechanisms87. The
regulation of ion channels in MSNs by
D2R is likely involved in the regulation of
behavioral processes such as locomotion.
Postsynaptic D2Rs are also expressed on
TANs within the striatum, which play a
critical role in the regulation of the
excitability of MSNs through the tonic
release of acetylcholine that acts on
receptors such as nicotinic acetylcholine
channels88. The activation of D2Rs expressed on TANS has been reported to decrease the firing rates of
these neurons in a G protein-dependent manner89.
Presynaptic D2Rs are expressed on the terminals of dopaminergic neurons and serve as part of a
negative feedback loop that decreases DA release from terminals90. D2R activates Kv1 channels in axon
terminals, shunting current and decreasing the release of DA of axon terminals60,61,90. Presynaptic D2 can
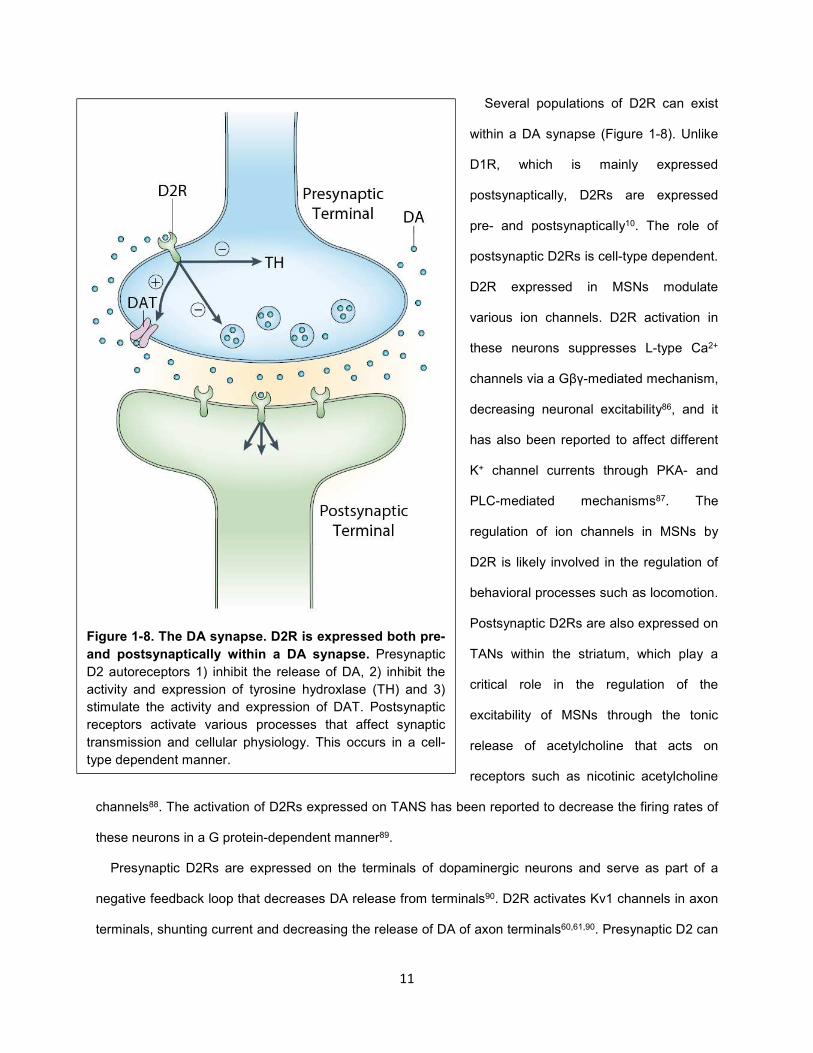
Figure 1-8. The DA synapse. D2R is expressed both pre-
and postsynaptically within a DA synapse. Presynaptic
D2 autoreceptors 1) inhibit the release of DA, 2) inhibit the
activity and expression of tyrosine hydroxlase (TH) and 3)
stimulate the activity and expression of DAT. Postsynaptic
receptors activate various processes that affect synaptic
transmission and cellular physiology. This occurs in a cell-
type dependent manner.

12
also decrease the synthesis of DA by downregulating the expression and inhibiting the enzyme activity of
tyrosine hydroxlase (TH), which is the rate limiting enzyme in the synthesis of DA that converts tyrosine to
L-DOPA, the immediate precursor of DA (Figure 1-8)90-92. D2R-mediated inhibition of PKA has been
reported to decrease TH phosphorylation and enzymatic activity92,93, suggesting the Gi/o/z mediated
inhibition of cAMP contribute to TH regulation. Presynaptic D2R activation has also been reported to
increase the surface expression and thus the activity of DA transporter (DAT), which facilitates the uptake
of DA from the synaptic space back into the dopaminergic neuron, terminating DA signaling (Figure 1-
8)94,95.
D2R autoreceptors are expressed on the soma and dendrites of dopaminergic VTA and SNc neurons.
These receptors are regulated by the somatodendritic release of DA from dopaminergic neurons96,97 and
activate GIRK channels, which decrease neuronal firing rates90.
The DA hypothesis of schizophrenia and antipsychotic medications
Our current understanding of the neurobiology and pharmacotherapy of schizophrenia has been
defined by the DA hypothesis, which was formulated based on 1) observations that DA-enhancing
compounds like amphetamine promote psychosis, and 2) the affinity of antipsychotics for D2R correlates
with clinical efficacy98. This hypothesis posits that hyperdopaminergic signaling in the mesolimbic system
facilitates positive symptoms.99 The DA hypothesis has been substantiated by imaging studies, which
have found consistently that patients with schizophrenia have enhanced dopamine storage and release in
the striatum, although more recent studies have implicated the nigrostriatal pathway projecting to the
anterior caudate of the dorsal striatum as the major area of dysregulation100,101. Thus, blockade of D2R-
mediated hyperdopaminergic transmission in the striatum in schizophrenia has formed the basis of
antipsychotic drug development1,102.
Chlorpromazine, the first antipsychotic medication, was synthesized in 1951 and was first developed as
a sedative102. However, it was later shown to be effective for treatment of positive symptoms of
schizophrenia. This and other so-called first generation (or typical) antipsychotics, notably haloperidol,
confer their antipsychotic effects through high affinity, selective antagonism of D2R102,103. Of note,
although these medications ameliorate positive symptoms, they do not improve the negative symptoms

13
and cognitive deficits associated with schizophrenia. However, blockade of D2R by these compounds is
associated with extrapyramidal side effects (EPS) such as bradykinesia and tremors as well
hyperprolactinemia. Side effects including weight gain and sedation may be associated with off-target
receptors. Second generation (or atypical) antipsychotics, e.g., clozapine, are lower potency D2R
antagonists that ameliorate positive symptoms similarly to typical antipsychotics. They were initially
reported to confer improvements in negative symptoms and cognition, although this was not verified by
recent clinical studies104-106. Relative to typical antipsychotics, atypical antipsychotics are associated with
reduced EPS and hyperprolactinemia, although long-term use is associated with metabolic side effects,
most notably substantial weight gain103. Although atypical antipsychotics have effects on multiple
receptors (including 5-HT2A and other monoaminergic receptors), D2R antagonism remains the unifying
property of all antipsychotic drugs in use. Notably, due to the high similarity between D2-like receptors,
antipsychotic medications also have substantial affinity towards D3R and D4R100,107,108.
Two major observations led to a revision of the DA hypothesis of schizophrenia: 1) D2R antagonists do
not ameliorate negative symptoms and cognitive deficits of schizophrenia, and 2) imaging studies
indicated that cortical activity, specifically blood flow in the mPFC, is reduced in patients with
schizophrenia100. It was postulated that positive symptoms were due to hyperdopaminergic transmission
in the striatum whereas negative symptoms and cognitive deficits were due to hypodopaminergic
signaling in mesocortical circuits. Thus, it was proposed that D2R partial agonists might stabilize
dopaminergic signaling by diminishing transmission in striatal areas and increasing transmission in
mesocortical circuits103.
In addition to reducing DA transmission at postsynaptic D2Rs in striatal areas, it was proposed that
partial agonists would reduce signaling by activating presynaptic D2 autoreceptors on dopaminergic
terminals103. D2R agonists have a higher potency at presynaptic autoreceptors compared to that at
postsynaptic D2R functions, although the mechanistic basis of this is not well understood. This is
highlighted by the biphasic response of D2R agonists, which at low doses preferentially stimulate D2
autoreceptor functions but only at high doses activate postsynaptic D2R mediated-behaviors such as
locomotion109.

14
3-PPP (Preclamol) was the first D2R partial agonist to be evaluated for treatment of schizophrenia
based on this rationale103. However, despite promising in vitro and animal studies, 3-PPP was
unsuccessful in clinical trials110. Subsequently, the D2R partial agonist aripiprazole (Abilify) was approved
in 2002 for treatment of schizophrenia and is the prototype for third generation antipsychotics as partial
agonists rather than antagonists of D2R (clinicaltrials.gov)103. Like atypical antipyschotics, this compound
also has affinity towards other monoaminergic receptors111. Marketed as a DA stabilizer, aripiprazole has
been proposed to be superior to 3-PPP because it provides the optimal degree of partial agonism at
D2R103.
Aripiprazole is often efficacious for treatment of positive symptoms and has reduced EPS and
endocrine side effects compared to typical antipsychotics112. Aripiprazole also has a superior metabolic
profile compared to most atypical antipsychotics103,112. However, aripiprazole fails to ameliorate negative
symptoms and cognitive deficits, calling into question the validity of treating these symptoms with D2R
partial agonists (or antagonists as discussed above). Interestingly, D2R expression in the mPFC is quite
low, whereas this region has much higher expression of D1R113. Recent imaging studies indicate that
D1R expression rather than D2R is altered in the mPFC in patients with schizophrenia100. In addition,
D2R autoreceptors are expressed in cortex, which decrease rather than increase dopaminergic
transmission in response to DA and other D2R agonists114, again consistent with a lack of therapeutic
effect on cognitive symptoms.
Despite the failure of aripiprazole to ameliorate negative symptoms and cognitive deficits of
schizophrenia, its favorable efficacy for treatment of positive symptoms and overall side effect profile
relative to both typical and atypical antipsychotics has prompted the further development of D2R partial
agonists for treatment of schizophrenia. The D2R partial agonist bifeprunox retains a similar profile as
aripiprazole towards other monoaminergic receptors103. However, although bifeprunox performed better
than placebo for treatment of positive symptoms of schizophrenia in clinical trials, it was rejected based
on poor efficacy and safety grounds (clinicaltrials.gov)115. D2R partial agonists including aplindore have
also failed in clinical trials, although others such as cariprazine are currently being investigated
(clinicaltrials.gov)116.

15
Functionally selectivity (or biased agonism)
The clinical failure of several partial agonists for treatment of schizophrenia has prompted the
reevaluation of reported mechanism of action of aripiprazole, which is the only clinically approved
antipsychotic medication that is a D2R partial agonist. Recent studies indicate that aripiprazole displays
functional selectivity (or biased agonism), which may underlie its superior clinical properties111,117,118.
Stephenson’s concept of intrinsic efficacy posits that ligands act as full agonists, partial agonists,
neutral antagonists or inverse agonists at all receptor-mediated pathways (Figure 1-4; 1-9A)43. However,
functionally selective (or biased) ligands can differentially activate pathways downstream of receptor
activation (Figure 1-9B)119,120. This has been attributed to the ability of functionally selective ligands to
stabilize conformations of a given receptor that are specifically associated with distinct downstream
transducers120. Classical agonists, in contrast, presumably stabilize a broader range of receptor active
states. The most widely reported mechanism for functional selectivity is the selective activation of either
canonical G protein or non-canonical arrestin coupling partners, which are known to differentially regulate
pathways downstream of receptor activation (Figure 1-10).121 G protein and arrestin-biased ligands have
now been reported for a number of receptors, e.g., the ligand SII selectively recruits arrestin downstream
of angiotensin receptor 1A (AT1A)122.
Figure 1-9. Classical pharmacological agents versus functional selective ligands.
A) Classical ligands like agonists or antagonists activate or block, respectively, all the pathways
downstream of a receptor. For agonists, this can result in activation of pathways associated with
therapeutic effects as well as those associated with side effects. Antagonists can block pathways
associated with pathological effects, but also those associated with normal physiological effects. B) In
contrast, functionally selective (or biased) ligands can seletively activate pathways downstream of the
receptor, offering the hope for better tuning drug action.

16
In a therapeutic context, typical
agonists or antagonists, by activating or
blocking all receptor-mediated
pathways, have the potential to elicit
both desirable and undesirable effects
(Figure 1-9A). In contrast, functionally
selective ligands, by acting as both
agonists and antagonists of different
pathways downstream of the same
receptor, provide hope for better tuning
of therapeutic and side effects of drugs
(Figure 1-9B)120. Substantial efforts have gone towards the development of functionally selective ligands
for the treatment of a number of disease states. Indeed, several functionally selective ligands are
currently being investigated in clinical trials (TRV027, TRV130; Trevena; clinicaltrials.gov)123. Functional
selectivity may also explain why some drugs from a common target class (e.g., β-blockers) display clinical
efficacy (e.g., in heart failure), while others fail, despite showing similar degrees of efficacy in preclinical
indices of biological activity124-126. To date, functional selectivity has been intensively investigated in
studies of GPCRs, but it is likely to be a more universal paradigm. For instance, the clinically relevant
phenomenon of tissue-specific agonism or antagonism mediated by selective estrogen receptor
modulators may reflect conformation-selective biased agonism at the nuclear hormone receptor
superfamily127-129.
Functional selectivity was initially controversial due to the difficulty in comparing ligand action across
different pathways mediated by a given receptor130. This was due to differences in factors including
receptor reserve, stimulus-response coupling, drug metabolites and off-target drug binding. However, the
use of heterologous expression systems to control these factors has validated the place of functional
selectivity in pharmacology. Given the emerging recognition of functional selectivity as a potentially
widespread phenomenon, recent studies have focused on various means for detecting functionally
selective ligands and quantifying their behavior in a manner that transcends the vagaries of the assay
Figure 1-10. G protein versus arrestin bias.
The most highly cited mechanism for functional selectivity
or bias is the ability of functionally selective ligands to
activate either G protein (GPB ligand; left) or arrestins
(AB ligand; right), which have been reported to mediate
different pathways.

17
system. For predictive and translational purposes, a critical aspect of any quantitative analysis of
functional selectivity is the need to remove the influence of the cellular background and the assay
conditions when comparing the activity of a group of agonists between pathways in the same cell type. In
this regard130-132, a number of analytical methods have recently been proposed that build upon the classic
operational model of agonism originally proposed by Black and Leff131-134.
Functional selectivity at D2R
In contrast to classical agonists and antagonists of D2R, aripiprazole displays functionally selective
properties across a number of endpoints associated with this receptor. In a behavioral assay used to
measure postsynaptic D2R activity, which utilizes 6-hydroxydopamine (6-OHDA) to lesion dopaminergic
neurons in the VTA, full and partial D2R agonists induce contralateral turning to the side of the lesion135.
However, aripiprazole induces ipsilateral turning and antagonizes the action of other agonists.
Aripiprazole also blocked apomorphine-induced stereotypy, which is also associated with D2R
antagonists136. Consistent with these actions as a postsynaptic antagonist, aripiprazole blocked D2R-
agonist induced inhibition of the firing of neurons in the NAc137. In contrast, aripiprazole has also been
reported to have partial agonist actions at other D2R-associated endpoints, decreasing prolactin
release138 and inducing yawning139. It has also been shown to have agonist effects at a number of D2
autoreceptor functions, including decreasing DA release136 and synthesis119. In contrast to the D2R
antagonist haloperidol, aripiprazole displays low cataleptogenic effects136.
In in vitro model systems, aripiprazole has also been shown to have mixed agonist/antagonists
properties. Unlike the D2R agonist quinpirole, aripiprazole did not increase K+ currents in MES23.5
cells111, which has been presumed to be a GIRK channel-mediated current103. It also did not induce D2R-
receptor internalization130. However, aripiprazole was shown, with varying degrees depending on the cell
line, to be a partial agonist for inhibition of cAMP downstream of D2R111,117. It was also a partial agonist
for increasing the phosphorylation of MAPK and the release of arachidonic acid in CHO cells118.
Interestingly, in contrast to that in MES23.5, aripiprazole was shown to be a partial agonist of GIRK
channels in Xenopus oocytes, indicating that cell line dependent difference in the ability of aripiprazole to
activate these channels140. Thus, whereas typical D2R partial agonists activate all D2R functions,

18
aripiprazole enigmatically acts as a mixed agonist/antagonist. Of note, it remains unclear which, if any, of
these actions underlie the clinical effects of aripiprazole.
Functional selectivity at D2R is not without precedent. Dihydrexidine (DHX), a D1R/D2R ligand, was
shown previously to be functionally selective. Like DA, DHX is a full agonist for the inhibition of cAMP141.
However, DHX does not facilitate D2 autoreceptor-mediated inhibition of DA release like the D2R agonist
quinpirole141. Thus, DHX acts as a preferential presynaptic antagonist and postsynaptic agonist at D2R142.
DHX is currently undergoing clinical trials for the treatment of schizophrenia; however, this trial is focused
on treating cognitive deficits related to the actions of DHX at D1R rather than D2R (clinicaltrials.gov).
The rational development of functionally selective ligands as antipsychotic medications
The discovery and ongoing characterization of functionally selective ligands at D2R suggests the
exciting possibility that compounds can be developed to both activate and inhibit different pathways
downstream of the same receptor, thus producing novel phenotypes and mechanistic insights, and
ultimately superior therapeutics for treatment of schizophrenia. However, several critical issues remain
poorly understood regarding the rational development of superior therapeutics targeting D2R, thus limiting
drug discovery efforts: 1) modeling schizophrenia in rodents, especially positive symptoms, is particularly
challenging, 2) the specific downstream effector molecule(s) to be targeted for therapeutic efficacy of
antipsychotic medications and to be avoided for side effects are still unknown and 3) the mechanisms by
which ligands, including aripiprazole, act in a functionally selective manner remain unclear.
Modeling endophenotypes of schizophrenia in rodents
A major limitation in the development of superior antipsychotic medications has been modeling
schizophrenia, particularly the positive symptoms, in preclinical animal models143. One of the primary
preclinical measures of antipsychotic efficacy is the ability of pharmacological agents or genetic
modifications to block psychostimulant (i.e., amphetamine or phencyclidine/PCP)-dependent behaviors in
rodents such as hyperlocomotion143,144. These agents are known to induce psychosis in humans, and
thus the behavioral effects of psychostimulants in rodents have been used as functional correlates of
psychosis. Moreover, antipsychotics have been shown to block the effects of psychostimulants in rodents.

19
Other models of schizophrenia include neurodevelopmental (e.g., gestational MAM, post-weaning social
isolation), lesion (e.g., neonatal ventral hippocampal lesion) and genetic (e.g., DISC1 knockout or D2R
overexpression in striatum145) models. These perturbations vary in their ability to model different aspects
of schizophrenia, including positive and negative symptoms as well as cognitive deficits. It is important to
note, however, that none of these models can accurately model psychosis directly in rodents. Further
work will be necessary to develop superior behavioral models of schizophrenia. However, this limitation is
beyond the scope of the work described herein.
Efforts to identify the signaling molecules to be targeted selectively by antipsychotic medications
The development of superior antipsychotic medications will require a firm understanding of the specific
receptor-mediated pathways that should be activated or blocked to confer therapeutic efficacy and avoid
side effects. One study recently profiled a number of clinically effective antipsychotic medications at these
endpoints and found that a common property is their ability to selectively antagonize arrestin
recruitment146. Whereas typical and atypical antipsychotic medications failed to activate G protein and
recruit arrestin downstream of D2R, aripiprazole selectively activated G protein. The authors, therefore,
proposed that arrestin blockade is the critical feature of antipsychotic drug action. This is in part in
agreement with findings that have implicated the D2R-arrestin/PP2A/Akt pathway as the primary mediator
of amphetamine-dependent behaviors68. In this study, global knockout of arrestin in mice dramatically
attenuated amphetamine-dependent effects.
Based on these studies, it has been proposed that D2R ligands like aripiprazole that antagonize
arrestin but activate G protein may be superior antipsychotic medications146. Substantial efforts have led
to the development of such ligands147, which are currently being evaluated in preclinical models of
schizophrenia. However, findings from a number of recent pharmacological studies call into the question
the validity of this approach to antipsychotic drug development. In contrast to the study described above,
aripiprazole has been shown to stimulate both D2R-mediated G protein activation and arrestin
recruitment148,149. Moreover, a novel series of selective ligands (including UNC9994) were developed that
act as antagonists of G protein but agonists of arrestin downstream of D2R148. According to the model
that arrestin blockade is necessary for antipsychotic efficacy, these compounds are not predicted to have

20
antipsychotic properties. However, these arrestin-biased compounds blocked psychostimulant-dependent
behaviors in animal models. In addition, lithium, a commonly used treatment of bipolar disorder, has been
shown to disrupt the arrestin/PP2A/Akt complex, the proposed mediator of psychostimulant-dependent
behaviors150. However, lithium does not confer antipsychotic effects in clinical settings1.
Recent genetic studies have also indicated that psychostimulant-dependent behaviors are not solely
mediated by arrestin. Although knockout of arrestin in animal models greatly attenuates amphetamine-
dependent behaviors, PCP-dependent behaviors are not significantly affected148. In addition,
psychostimulant-dependent behaviors were disrupted in animal models in which DARPP-32, a critical
signaling molecule downstream of D2R-mediated G protein activation, was selectively knocked out in
D2R-expressing MSNs151. Thus, attenuating G protein signaling downstream of D2R may also confer
antipsychotic effects, which is incompatible with the hypothesis that arrestin and not G protein blockade is
optimal for antipsychotic efficacy.
The difficulty in probing distinct D2R populations and downstream signal transduction pathways
As discussed above, substantial efforts have gone towards the development of arrestin (and G protein)
biased compounds of D2R. However, both G protein and arrestin may be involved in mediating
psychostimulant-dependent behaviors, calling into the question this approach. Further studies will be
required to better understand the interplay between these signaling molecules within different D2R-
expressing regions mediating these processes. A significant difficulty in correlating downstream signaling
of D2R to specific behavioral/therapeutic effects stems not only from limitations in modeling schizophrenia
in rodents, but just as importantly, from a lack of tools to selectively target the different effector pathways
in defined brain regions and neuronal populations.
Functionally selective ligands were recently purported as tools that can be used to probe distinct D2R-
mediated pathways (Figure 1-11)147,148. However, this approach has several limitations. Like most
pharmacological agents, these ligands have numerous off-target effects at proteins such as other
monoaminergic receptors that can add significant confounds to the analysis148. Another disadvantage is
that they indiscriminately act on all on-target D2R populations, which have divergent functions in different
regions. Not only do these ligands modulate D2Rs in the periphery, they also act on D2Rs at different

21
sites within the brain, both at the level of different neural circuits but also within a given circuit, e.g., in the
striatum, D2Rs are expressed postsynaptically on MSNs and cholinergic interneurons and as
autoreceptors and heteroreceptors on dopaminergic and corticostriatal glutamatergic terminals,
respectively. These receptor populations have opposing functions both facilitating and decreasing DA
transmission.
Moreover, even at a given D2R population, pharmacological agents like functionally selective ligands
fail to replicate the actions of the endogenous ligand. DA activates D2Rs in a highly spatially and
temporally controlled manner that may be critical for mediating psychostimulant-dependent effects. In
contrast, functionally selective ligands persistently activate/block D2R-mediated functions. These ligands
can also have differences in affinity and efficacy for a given receptor pathway relative to DA, e.g.,
UNC9994 is a partial agonist with low potency for the recruitment of arrestin downstream of D2R148. Thus,
the evaluation of functionally selective ligands with different properties in preclinical and clinical settings
can give insight onto their efficacy as therapeutics; however, these ligands exhibit poor spatio-temporal
properties for the control over distinct pathways downstream of D2R, thus limiting their ability to provide
mechanistic insights into the receptor populations and pathways that mediate psychostimulant-dependent
behaviors (and potentially psychosis).
Current genetic approaches also suffer from limitations that can hamper efforts to distinguish the roles
of signaling molecules like G proteins and arrestins downstream of specific D2R populations (Figure 1-
11). Knockout or overexpression models are subject to potential developmentally-related confounds,
although inducible models can compensate for these deficiencies. In addition, G proteins and arrestins
are composed of subtypes that may be in part functionally redundant and are often expressed in the
same cell type, i.e., D2R couples to five different Gα subunits and two different arrestin subtypes. Thus,
knockout of a single protein may be insufficient to study the role of a group of signaling molecules.
Moreover, although the global knockout or overexpression of proteins including G proteins and arrestins
can give insight into their role in mediating various processes downstream of D2R, these proteins are
known to couple to large number of GPCRs in every cell in the entire organism. Although cell-type
specific models are less invasive, non-target GPCRs are still affected. Knockout approaches also
effectively act as persistent inhibitors of a given signaling pathway, and thus have poor temporal control

22
over signaling. Thus, current genetic approaches can impair the function of a wide variety of GPCRs and
dramatically affect cellular physiology and behaviors.
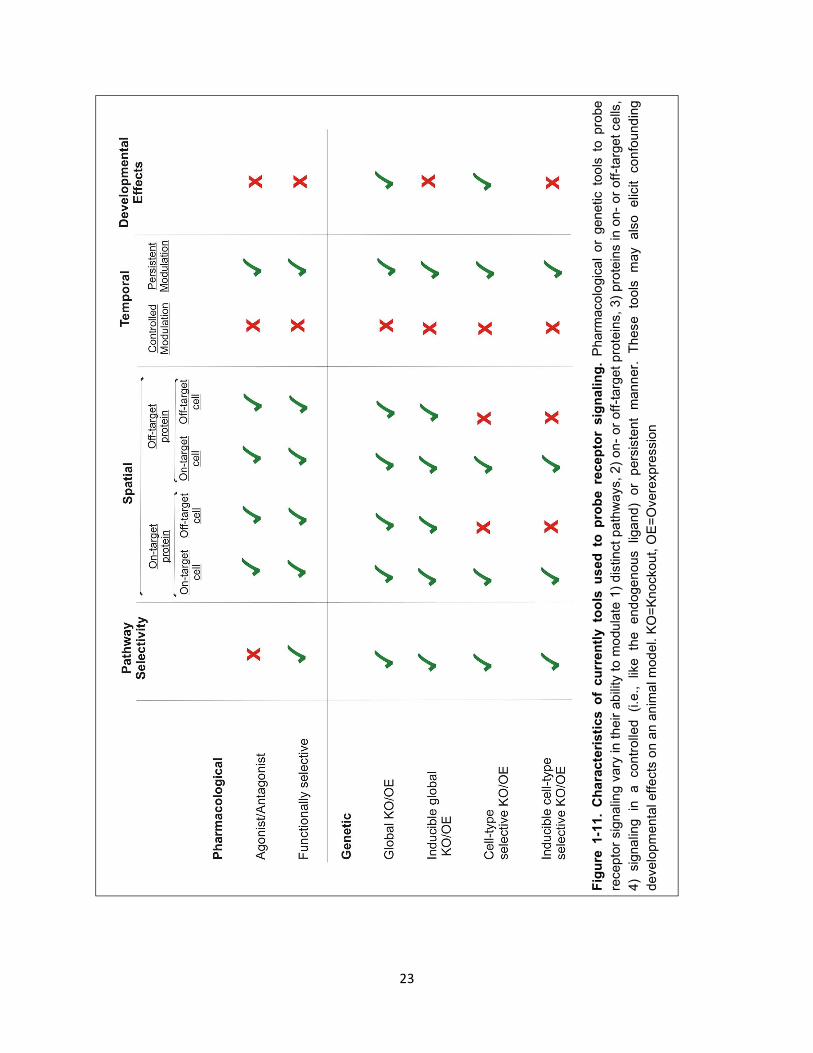
23
Fig
ure
1-1
1.
Ch
ara
cte
risti
cs o
f cu
rren
tly
to
ols
u
sed
to
p
rob
e re
cep
tor
sig
nalin
g.
Pharm
aco
logic
al
or
genetic t
oo
ls t
o p
robe
recepto
r sig
nalin
g v
ary
in t
heir a
bili
ty t
o m
odu
late
1)
dis
tinct
pa
thw
ays,
2)
on-
or
off
-targ
et
pro
tein
s,
3)
pro
tein
s in o
n-
or
off-t
arg
et
ce
lls,
4)
sig
na
ling
in
a
contr
olle
d
(i.e
.,
like
the
end
oge
nous
liga
nd)
or
pers
iste
nt
mann
er.
T
hese
tools
m
ay
als
o
elic
it con
fou
nd
ing
develo
pm
enta
l effects
on a
n a
nim
al m
od
el.
KO
=K
nocko
ut, O
E=
Overe
xpre
ssio
n

24
Bias towards either G proteins or arrestins is not sufficient to explain the properties of
functionally selective D2R ligands
As noted above, the most commonly reported mechanism of functional selectivity is the ability of
compounds to differentially activate G protein or arrestin signal transduction pathways downstream of a
given GPCR. However, functionally selective compounds of D2R, i.e., aripiprazole and DHX,
paradoxically differentially activate several G protein-mediated pathways that are thought to be
independent of arrestin recruitment, e.g., aripiprazole decreases cAMP levels111,117 (primarily a Gα-
mediated effect) but activates GIRK channels (primarily Gβγ-mediated effect) in a cell-type dependent
manner111,140. The mechanistic basis of this behavior is currently unknown. Given that 1) G proteins may
play a role in mediating psychostimulant-dependent effects in rodents and 2) aripiprazole has major
clinical relevance, further investigation is required to better understand these phenomena.
Towards the development of superior antipsychotic medications
The ability to selectively modulate specific D2R-mediated pathways offers the possibility of developing
antipsychotic agents with superior efficacy and reduced side effects. However, the specific site(s) and
pathway(s) to be targeted are unknown, in part due to limitations in currently available tools that probe
D2R-mediated signaling. Further, currently known mechanisms of functional selectivity (i.e., G protein
versus arrestin bias) are not sufficient to explain the selective properties of clinically important
antipsychotic medications. Thus, the following are described herein:
1. Novel genetically-encoded tools that can be used to probe the roles of specific D2R-mediated
pathways with high spatio-temporal specificity and minimal off-target effects.
2. The identification of transducer(s) of D2R signaling that may underlie the ability of reported
functionally selective ligands to selectively activate distinct G protein-mediated pathways.

25
CHAPTER 2. The development of genetic tools to probe D2R-mediated arrestin signaling in vivo
Currently available tools that have been used to understand the role of distinct D2R signaling pathways
in vivo suffer from several significant limitations. Pharmacological tools have significant off-target effects
and also modulate on-target receptors in both on- and off-target cell-types. Although cell-type specificity
can be achieved with current genetic strategies, off-target proteins can also be affected. Moreover, both
of these strategies modulate the receptor of interest in a manner that does not temporally mimic the
endogenous ligand. Thus, we sought to develop tools that overcome these difficulties in hopes of
advancing our understanding of the D2R populations and signaling pathways that must be targeted to
develop superior antipsychotic medications.
Receptors and the G protein-GRK-arrestin paradigm
It has been proposed that arrestins are the primary mediator of psychostimulant behaviors and that
ligands that selectively block these signaling proteins may be superior antipsychotic medications10,68,146.
Thus, we first focused on developing tools that would facilitate arrestin but not G protein signaling
downstream of D2R. However, within the current paradigm of the GPCR signaling cascade152, arrestin
recruitment to these receptors is dependent on the activation of G proteins (Figure 2-1). Arrestin
recruitment has been reported to be dependent on the activity of GRKs that phosphorylate receptors in an
agonist-dependent manner64,152. GRK2/3 are cytosolic proteins that are recruited to receptors based on
two factors: 1) the GPCR active state, and 2) binding of their PH domain to Gβγ subunits liberated from
activated heterotrimeric G protein77,153. Thus, efforts to develop genetic or pharmacological contexts
whereby a given receptor only signals through arrestins may be hampered due to the dependence of this
signaling pathway on G protein activation.

26
There are several factors that would permit arrestin recruitment to GPCRs independently of G protein
activation. The active state of the GPCR may be sufficient to recruit GRK2/3 independently of Gβγ, thus
leading to receptor phosphorylation and arrestin recruitment. In addition, GRK4/5/6, unlike GRK2/3, are
primarily embedded in the plasma membrane independently of G activation due to various lipid
modifications as well as direct interactions with phospholipids77. It has been shown that these GRKs can
phosphorylate receptors in an agonist-dependent and independent manner79. Thus, although abolishing
G protein activation may negatively affect GRK2/3 phosphorylation of GPCRs, GRK4/5/6 phosphorylation
may be sufficient to facilitate arrestin recruitment. It is also possible that D2R, like some GPCRs, does not
require GRK-dependent phosphorylation to recruit arrestins73-76.
According to in vitro studies, although GRK2 and GRK3 have been reported to phosphorylate D2R, it is
less clear if this is the case for GRK4/5/678,79. However, in vivo studies indicate that D2R may be
regulated by different GRKs in a cell-type dependent manner. GRK2 has been shown to play a functional
Figure 2-1. The canonical receptor-GRK-arrestin cascade. A) Receptor couples to G protein upon
activation by agonist. B) This leads to a dissociation of or conformational change between the Gα and
Gβγ subunits. C) GRK2/3 is recruited to the plasma membrane by binding Gβγ, facilitating the binding
and phosphorylation of the receptor by GRK2/3. This increases the affinity for the receptor for
arrestins.

27
role in DAT-expressing dopaminergic neurons but not MSNs,154 whereas GRK6 has been reported to
modulate D2Rs expressed in MSNs155
G protein activation is not necessary for arrestin recruitment to D2R
To test the dependence of G protein activation on arrestin recruitment, we took advantage of the widely
used pharmacological tool pertussis toxin (PTX)156-158. PTX is a toxin that can be purified from the
bacterium Bordetella pertussis and applied exogenously to many different cell types to ablate Gi/o
signaling156-158. Gαi and Gαo subunits are subject to inactivation by ADP-ribosylation of a critical cysteine
residue at their C-termini by PTX158. To note, Gαz, which also couples to D2R, is not subject to
inactivation by PTX because it lacks this critical cysteine residue159,160.
Before probing the effects of
PTX on arrestin, we first confirmed
that this toxin is capable of
abolishing D2R-mediated G protein
activity in a cAMP inhibition assay.
cAMP was measured using the
bioluminescence resonance energy
(BRET)-transfer based sensor
CAMYEL, which was coexpressed
heterologously with D2R in
HEK293T cells161. This sensor
detects changes in conformation in Epac, a cAMP binding protein. Binding of cAMP results in a decrease
in BRET between the donor YFP and Rluc, which are fused to Epac at its N- and C- termini, respectively
(Figure 2-2).
It has been reported previously that measuring the inhibition of cAMP levels by Gi/o/z-coupled receptors
in HEK293T cells is challenging due to low basal cAMP levels162. The application of the adenylate cyclase
(AC)-stimulating ligand forskolin (FSK) enhanced cAMP in HEK293T cells, resulting in a decrease in
BRET that stabilized after ~10 minutes (Figure 2-3). cAMP levels can also be increased by stimulating
Figure 2-2. BRET-based cAMP sensor CAMYEL. Epac, a
cAMP binding protein, is fused with an acceptor YFP and a
donor Rluc at its N- and C-termini, respectively. Upon binding
cAMP, there is a conformational change in Epac that is
associated with a decrease in bioluminescence resonance
energy transfer (BRET) between the donor and acceptor.
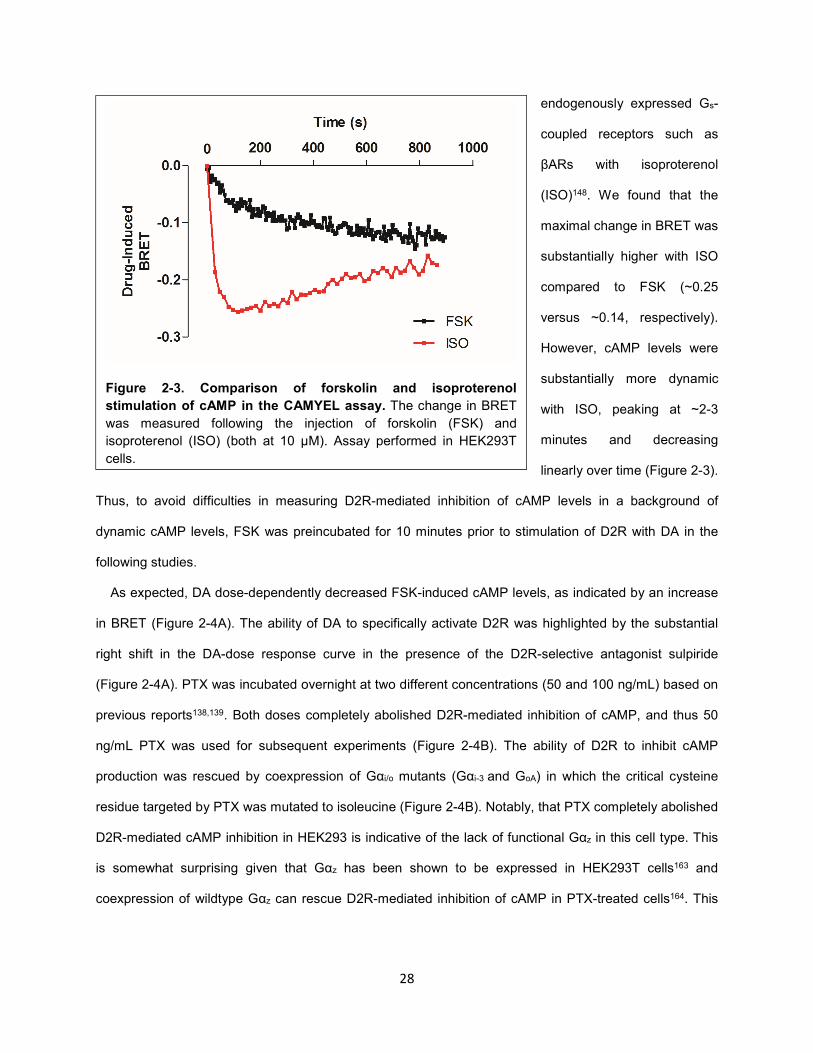
28
endogenously expressed Gs-
coupled receptors such as
βARs with isoproterenol
(ISO)148. We found that the
maximal change in BRET was
substantially higher with ISO
compared to FSK (~0.25
versus ~0.14, respectively).
However, cAMP levels were
substantially more dynamic
with ISO, peaking at ~2-3
minutes and decreasing
linearly over time (Figure 2-3).
Thus, to avoid difficulties in measuring D2R-mediated inhibition of cAMP levels in a background of
dynamic cAMP levels, FSK was preincubated for 10 minutes prior to stimulation of D2R with DA in the
following studies.
As expected, DA dose-dependently decreased FSK-induced cAMP levels, as indicated by an increase
in BRET (Figure 2-4A). The ability of DA to specifically activate D2R was highlighted by the substantial
right shift in the DA-dose response curve in the presence of the D2R-selective antagonist sulpiride
(Figure 2-4A). PTX was incubated overnight at two different concentrations (50 and 100 ng/mL) based on
previous reports138,139. Both doses completely abolished D2R-mediated inhibition of cAMP, and thus 50
ng/mL PTX was used for subsequent experiments (Figure 2-4B). The ability of D2R to inhibit cAMP
production was rescued by coexpression of Gαi/o mutants (Gαi-3 and GoA) in which the critical cysteine
residue targeted by PTX was mutated to isoleucine (Figure 2-4B). Notably, that PTX completely abolished
D2R-mediated cAMP inhibition in HEK293 is indicative of the lack of functional Gαz in this cell type. This
is somewhat surprising given that Gαz has been shown to be expressed in HEK293T cells163 and
coexpression of wildtype Gαz can rescue D2R-mediated inhibition of cAMP in PTX-treated cells164. This
Figure 2-3. Comparison of forskolin and isoproterenol
stimulation of cAMP in the CAMYEL assay. The change in BRET
was measured following the injection of forskolin (FSK) and
isoproterenol (ISO) (both at 10 μM). Assay performed in HEK293T
cells.
29
may be due to low levels of expression of this G protein or the differential compartmentation of D2R and
endogenously expressed Gαz.
We next determined the impact of PTX-treatment on arrestin recruitment to D2R. We used a widely
implemented cell-based assay that allows for the study of ligand induced-recruitment of arrestin to
receptors. This assay utilizes receptors that are fused to a BRET donor (e.g., Rluc8) and arrestins that
are fused to an acceptor (e.g., YFP or Venus)165. Rluc8 was fused to the cytoplasmic C-terminus of D2R,
whereas Venus was fused to the N-terminus of arrestin3 (Figure 2-5A)149. In this assay, dopamine dose-
dependently increased the level of arrestin recruitment to D2R, as indicated by an increase in BRET
(Figure 2-5B). DA-induced arrestin recruitment was dose-dependently blocked by the D2R-selective
antagonist sulpiride (see Methods Figure 1).
Interestingly, overnight treatment of PTX led to only a partial decrease in arrestin recruitment to D2R
(Figure 2-5B and C). Given that PTX completely blocked G protein activation in these cells, these data
indicate that G protein activation is not necessary for arrestin recruitment. However, this partial decrease
indicated that G protein activation, although not necessary, may serve to enhance arrestin recruitment.
Thus, we measured the ability of D2R to recruit arrestin in the presence of overexpressed GαoA, Gβ1 and
Figure 2-4. PTX completely abolishes G protein activation. A) DA dose-dependently stimulates
D2R-mediated inhibition of FSK-stimulated cAMP (cAMPi). Sulpiride, a selective D2R-antagonist,
blocks the ability of DA to activate G proteins downstream of D2R. B) Overnight incubation of 50 or
100 ng/mL PTX completely blocked the ability of DA (10 µM) to activate D2R-mediated cAMPi, as
indicated by a positive change in BRET. Coexpression of Gα subunits resistant to PTX (PTXR) and
unlabeled Gβ1γ2 rescued the ability of DA to activate D2R-mediate G protein signaling.
30
γ2 (Figure 2-5B and C). Consistent but opposite of the effect of PTX, overexpression of G protein
increased arrestin recruitment to D2R.
Given that G protein expression and/or activation play a role in arrestin recruitment to D2R, we next
sought to determine if this could be mediated by GRKs. Overexpression of GRK2 substantially increased
arrestin recruitment to D2R (Figure 2-5C). To determine if G protein inhibition affects GRK2 recruitment to
D2R, we used a previously developed
BRET-based assay that is analogous
to the receptor-arrestin BRET assay,
except that GRK2 rather than arrestin
is tagged with the acceptor Venus
(Figure 2-6A)166. DA dose-
dependently increased the
interactions between D2R and GRK2
(Figure 2-6B). Interestingly, overnight
PTX treatment resulted in only a
partial decrease in GRK2 recruitment to D2R, indicating that G protein activation is not necessary for
GRK2 recruitment to D2R (Figure 2-6B). Moreover, this result correlates with the effect of PTX on arrestin
recruitment.
Figure 2-5. PTX does not abolish arrestin recruitment. A) Schematic of receptor-arrestin BRET
assay. D2R is fused to Rluc8 at its C-terminus and arrestin3 is fused to Venus at its N-terminus. B) DA
dose-response curves in the absence or presence of PTX or overexpressed GαoAβ1γ2. C) Emax of DA
at D2R in the presence of PTX (50 ng/mL overnight treatment), overexpressed GαoAβ1γ2, and
overexpressed GRK2 or GRK6 with or without PTX. Presented as a % of Emax of DA at D2R alone.
Figure 2-6. GRK2 recruitment to D2R is negatively
affected by PTX treatment. A) Schematic of receptor-GRK2
BRET assay. D2R is fused to Rluc8 at its C-terminus and
GRK2 is fused to Venus at its C-terminus. B) DA dose-
response curves in the absence or presence of PTX or
overexpressed. Presented as a % of Emax of DA at D2R
alone.

31
Thus, taken together G protein activation enhances but is not necessary for the recruitment of arrestin
to D2R. Consistent with the reported role of GRK2 on arrestin recruitment to D2R167-169, this is likely due
to increased GRK2 recruitment to and the subsequent phosphorylation of D2R. Although G protein-
independent arrestin recruitment may also be dependent on GRK4/5/6, endogenous GRK2/3, which has
been shown to be expressed in HEK293 cells163, likely plays a role given that GRK2 can be recruited to
D2R in a G protein-independent manner. This is likely due to the ability of GRK2 to bind to the receptor
active state independently of Gβγ.
As discussed previously, whereas GRK2/3 are recruited to the plasma membrane through interactions
with liberated Gβγ, GRK4/5/6 are primarily localized to the plasma membrane through direct interactions
with membrane as well as lipid modifications152. Of this GRK family, GRK6 has also been reported to be
coexpressed with D2R in striatum, where it regulates D2R signaling155. Thus, we sought to determine
whether this protein enhances arrestin recruitment to D2R independently of G protein activation.
Coexpression GRK6 increased the maximal DA-induced recruitment of arrestin to D2R in a manner
similar to GRK2 (Figure 2-5C). Interestingly, enhancement of arrestin recruitment to D2R by GRK2 was
more substantially negatively affected by PTX than that for GRK6 (Figure 2-5C). Although there was a
partial decrease in arrestin recruitment to D2R coexpressed with GRK6 in the presence of PTX, it is
possible that this is due to impairment of endogenous GRK2/3 rather than GRK6 activity.
Overall, these data indicate that G protein activation can enhance but is not required for arrestin
recruitment to D2R. Thus, it is feasible to develop tools that probe arrestin signaling independently of G
protein activation downstream of D2R, especially in the striatum where GRK6 is expressed and known to
modulate D2R signaling155.
The development of an arrestin-biased D2R mutant
A major confound in studying the role of distinct signaling molecules using knockout strategies (such as
those in which arrestin3 was knocked out68,148) is that they affect signaling not only from the receptor
population of interest but also from other GPCRs that are either expressed within the same or different
cell-types. An alternative strategy involves the use of more specific genetic approaches such those
involving designer receptors (e.g., DREADDs)170. Designer receptors, which are typically mutants of

32
muscarinic acetylcholine receptors, have been engineered to selectively activate distinct signaling
proteins (Gs, Gi/o, Gq and arrestins) and are activated by otherwise inert ligands such as clozapine-N-
oxide (CNO)170 (Figure 2-7A). Most DREADDs couple both to distinct G proteins as well as arrestins.
However, more recently, an arrestin-biased DREADD mutant was developed (see below)171. Designer
receptors can also be targeted to distinct cell-types, thus conferring control over localization.
Although designer receptors can be activated in a regulated manner by controlling the administration of
the receptor agonist, they are not activated in a physiological manner, i.e., in response to the controlled
release of endogenous ligands. Moreover, these receptors are synthetic receptors that cannot fully
replicate the behavior or the receptor of interest, in this case D2R. Although GPCRs can couple to the
same G protein or arrestin pathways, they are likely differentially trafficked and compartmentalized. This
is likely related to differences in how these receptors interact with different accessory proteins that can
affect various aspects of localization and signaling172.
Although designer receptors are a significant advance over traditional knockout strategies in studying
the roles of distinct signaling molecules in defined cell-types, more refined tools will be necessary to study
the role of D2R signaling pathways in mediating psychostimulant-dependent behaviors. Thus, we sought
to develop a mutant of D2R that selectively recruits arrestins and not G proteins (Figure 2-7B). Because
this receptor would be genetically-encoded, in principle it could be targeted to distinct cell-types. In
addition, unlike DREADDs this D2R mutant would respond to endogenously released DA, thus
maintaining the normal physiological timing of receptor activation.

33
Fig
ure
2-7
. A
rresti
n b
ias
ed
DR
EA
DD
vers
us D
2R
. A
) D
RE
AD
D r
ece
pto
rs c
an b
e d
esig
ne
d t
o a
ctiv
ate
spe
cifi
c s
ign
alin
g p
rote
ins,
such a
s
arr
estin
s (
DR
EA
DD
AB).
Th
ese r
ecepto
rs a
re a
ctiv
ate
d b
y a
synth
etic
lig
and (
CN
O)
that
is o
therw
ise inert
at
oth
er
pro
tein
s.
Th
ese r
ecepto
rs a
re
activ
ate
d p
ers
iste
ntly b
y C
NO
. B
) A
n a
rrestin b
iase
d D
2R
(D
2R
AB)
can i
n p
rin
cip
le b
e a
ctiv
ate
d b
y e
ndo
geno
us D
A i
n a
hig
hly
te
mpora
lly
contr
olle
d m
ann
er.
Th
is m
uta
nt
wo
uld
als
o b
ind r
eg
ula
tory
pro
tein
s a
ssocia
ted w
ith t
he
wild
typ
e r
ece
pto
r th
at
ca
n a
ffect
rece
pto
r fu
nctio
n a
nd
traffic
kin
g.
C)
Co
mp
arison
of
diff
ere
nt
types of
rece
pto
rs to
pro
be sig
na
ling d
ow
nstr
ea
m of
rece
pto
rs.
Expre
ssio
n of
DR
EA
DD
o
r nativ
e
recepto
rs (
e.g
., D
2R
) in
sp
ecifi
c c
ell-
types a
llow
s f
or
the s
tudy o
f m
ultip
le r
ecepto
r path
ways.
How
ever,
bia
sed r
ecepto
rs o
f e
ither
DR
EA
DD
s o
r
nativ
e r
ecepto
rs c
an b
e d
evelo
ped t
hat se
lectiv
ely
activate
dis
tinct re
ce
pto
r p
ath
ways.

34
In order to develop an arrestin-biased D2R mutant, we took advantage of a recently published mutant
of the Gq-coupled, M3 muscaranic receptor-based DREADD that was reported recently to selectively
activate arrestin171. Within this receptor, the arginine in the highly conserved DRY motif found in Family A
GPCRs (R3.50 according to the Ballesteros-Weinstein GPCR numbering system) was mutated to leucine.
We introduced this mutation into the DRY motif of D2R (R132L), and similar to the case for the biased
DREADD receptor, this mutation completely abolished agonist-induced G protein activation according to
a bioluminescence resonance energy transfer (BRET)-based G protein recruitment assay (Figure 2-8A, C
and D)173. This assay resembles the receptor-arrestin or GRK BRET assays described above. However,
here a Gα subunit (in this case Gαi1) was fused to Venus in its α-helical domain (Figure 2-8A). In contrast,
according to a BRET-based arrestin recruitment assay, agonist induced-arrestin2 or arrestin3 recruitment
to D2R(R132L) was maintained, although with substantially lower maximal activation and potency
compared to that for the wildtype receptor (Figure 2-8B, C, E and F)149.
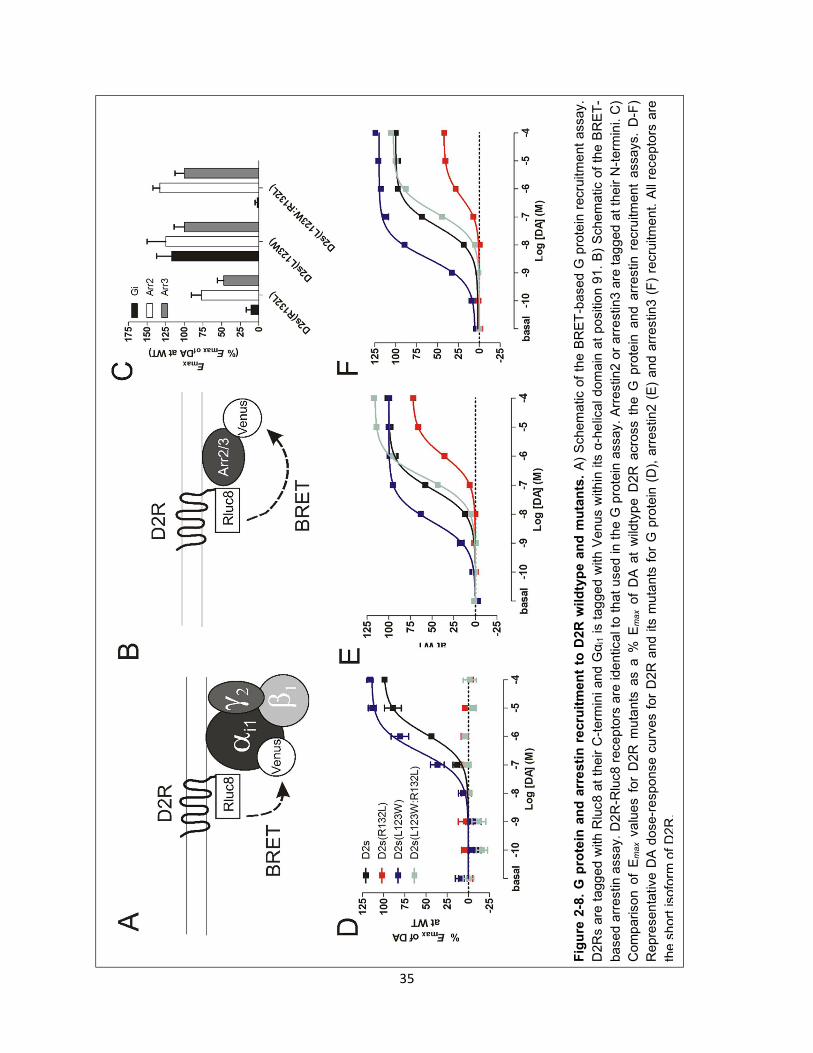
35
Fig
ure
2-8
. G
pro
tein
an
d a
rre
sti
n r
ecru
itm
en
t to
D2R
wild
typ
e a
nd
mu
tan
ts.
A)
Schem
atic
of
the B
RE
T-b
ased G
pro
tein
recru
itm
en
t assay.
D2R
s a
re t
agg
ed w
ith R
luc8 a
t th
eir C
-term
ini and G
αi1 is t
ag
ged w
ith V
enus w
ithin
its
α-h
elic
al d
om
ain
at
po
siti
on 9
1.
B)
Schem
atic
of
the B
RE
T-
based a
rrestin a
ssay.
D2
R-R
luc8 r
ece
pto
rs a
re ide
ntical to
that
used in t
he G
pro
tein
assay.
Arr
estin
2 o
r arr
estin
3 a
re t
ag
ged a
t th
eir N
-term
ini.
C)
Com
parison o
f E
max v
alu
es f
or
D2R
muta
nts
as a
% E
max o
f D
A a
t w
ildty
pe D
2R
acro
ss t
he G
pro
tein
and a
rrestin
recru
itment
assays.
D-F
)
Repre
sen
tative D
A d
ose-r
esponse c
urv
es f
or
D2R
and i
ts m
uta
nts
for
G p
rote
in (
D),
arr
estin
2 (
E)
and a
rrestin
3 (
F)
recru
itme
nt. A
ll re
cepto
rs a
re
the s
hort
isofo
rm o
f D
2R
.
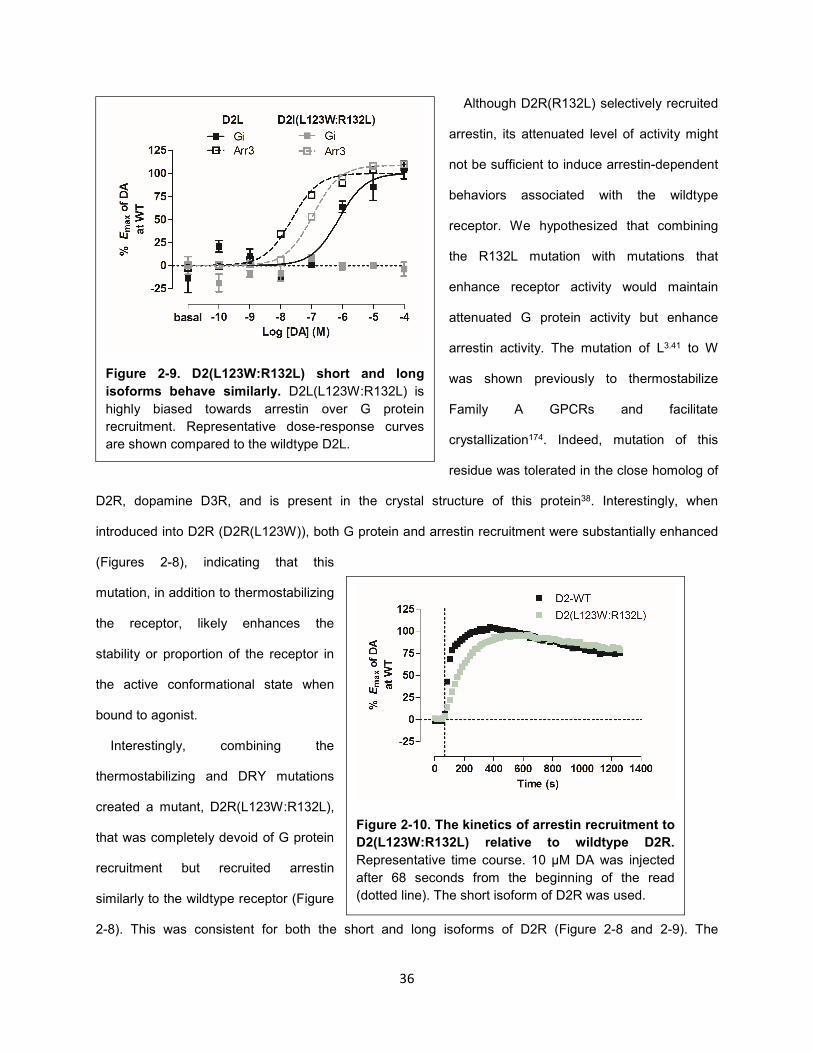
36
Although D2R(R132L) selectively recruited
arrestin, its attenuated level of activity might
not be sufficient to induce arrestin-dependent
behaviors associated with the wildtype
receptor. We hypothesized that combining
the R132L mutation with mutations that
enhance receptor activity would maintain
attenuated G protein activity but enhance
arrestin activity. The mutation of L3.41 to W
was shown previously to thermostabilize
Family A GPCRs and facilitate
crystallization174. Indeed, mutation of this
residue was tolerated in the close homolog of
D2R, dopamine D3R, and is present in the crystal structure of this protein38. Interestingly, when
introduced into D2R (D2R(L123W)), both G protein and arrestin recruitment were substantially enhanced
(Figures 2-8), indicating that this
mutation, in addition to thermostabilizing
the receptor, likely enhances the
stability or proportion of the receptor in
the active conformational state when
bound to agonist.
Interestingly, combining the
thermostabilizing and DRY mutations
created a mutant, D2R(L123W:R132L),
that was completely devoid of G protein
recruitment but recruited arrestin
similarly to the wildtype receptor (Figure
2-8). This was consistent for both the short and long isoforms of D2R (Figure 2-8 and 2-9). The
Figure 2-9. D2(L123W:R132L) short and long
isoforms behave similarly. D2L(L123W:R132L) is
highly biased towards arrestin over G protein
recruitment. Representative dose-response curves
are shown compared to the wildtype D2L.
Figure 2-10. The kinetics of arrestin recruitment to
D2(L123W:R132L) relative to wildtype D2R.
Representative time course. 10 μM DA was injected
after 68 seconds from the beginning of the read
(dotted line). The short isoform of D2R was used.
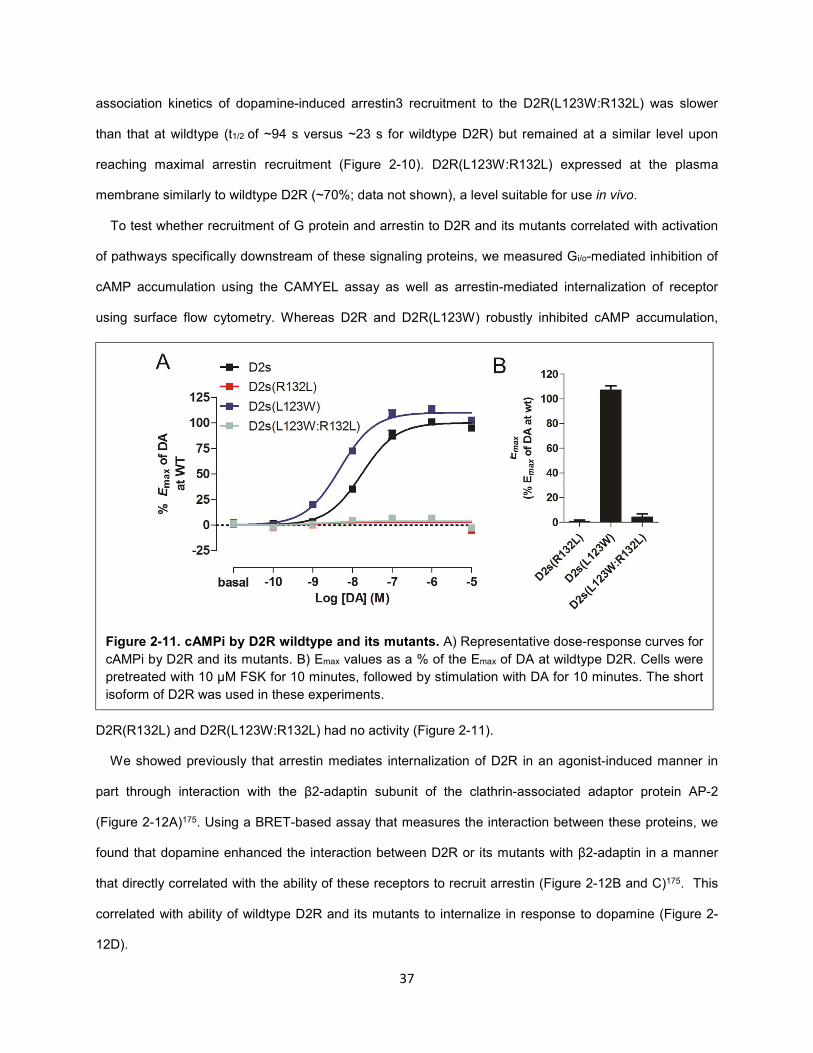
37
association kinetics of dopamine-induced arrestin3 recruitment to the D2R(L123W:R132L) was slower
than that at wildtype (t1/2 of ~94 s versus ~23 s for wildtype D2R) but remained at a similar level upon
reaching maximal arrestin recruitment (Figure 2-10). D2R(L123W:R132L) expressed at the plasma
membrane similarly to wildtype D2R (~70%; data not shown), a level suitable for use in vivo.
To test whether recruitment of G protein and arrestin to D2R and its mutants correlated with activation
of pathways specifically downstream of these signaling proteins, we measured Gi/o-mediated inhibition of
cAMP accumulation using the CAMYEL assay as well as arrestin-mediated internalization of receptor
using surface flow cytometry. Whereas D2R and D2R(L123W) robustly inhibited cAMP accumulation,
D2R(R132L) and D2R(L123W:R132L) had no activity (Figure 2-11).
We showed previously that arrestin mediates internalization of D2R in an agonist-induced manner in
part through interaction with the β2-adaptin subunit of the clathrin-associated adaptor protein AP-2
(Figure 2-12A)175. Using a BRET-based assay that measures the interaction between these proteins, we
found that dopamine enhanced the interaction between D2R or its mutants with β2-adaptin in a manner
that directly correlated with the ability of these receptors to recruit arrestin (Figure 2-12B and C)175. This
correlated with ability of wildtype D2R and its mutants to internalize in response to dopamine (Figure 2-
12D).
Figure 2-11. cAMPi by D2R wildtype and its mutants. A) Representative dose-response curves for
cAMPi by D2R and its mutants. B) Emax values as a % of the Emax of DA at wildtype D2R. Cells were
pretreated with 10 μM FSK for 10 minutes, followed by stimulation with DA for 10 minutes. The short
isoform of D2R was used in these experiments.
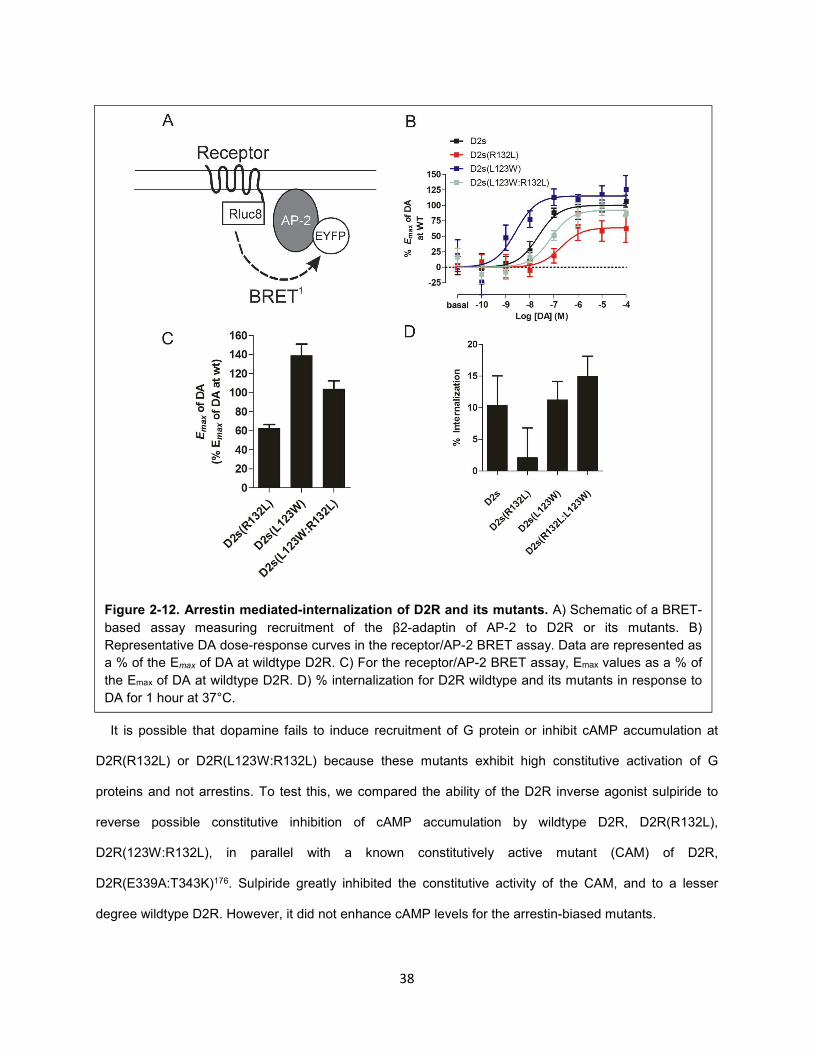
38
It is possible that dopamine fails to induce recruitment of G protein or inhibit cAMP accumulation at
D2R(R132L) or D2R(L123W:R132L) because these mutants exhibit high constitutive activation of G
proteins and not arrestins. To test this, we compared the ability of the D2R inverse agonist sulpiride to
reverse possible constitutive inhibition of cAMP accumulation by wildtype D2R, D2R(R132L),
D2R(123W:R132L), in parallel with a known constitutively active mutant (CAM) of D2R,
D2R(E339A:T343K)176. Sulpiride greatly inhibited the constitutive activity of the CAM, and to a lesser
degree wildtype D2R. However, it did not enhance cAMP levels for the arrestin-biased mutants.
Figure 2-12. Arrestin mediated-internalization of D2R and its mutants. A) Schematic of a BRET-
based assay measuring recruitment of the β2-adaptin of AP-2 to D2R or its mutants. B)
Representative DA dose-response curves in the receptor/AP-2 BRET assay. Data are represented as
a % of the Emax of DA at wildtype D2R. C) For the receptor/AP-2 BRET assay, Emax values as a % of
the Emax of DA at wildtype D2R. D) % internalization for D2R wildtype and its mutants in response to
DA for 1 hour at 37°C.
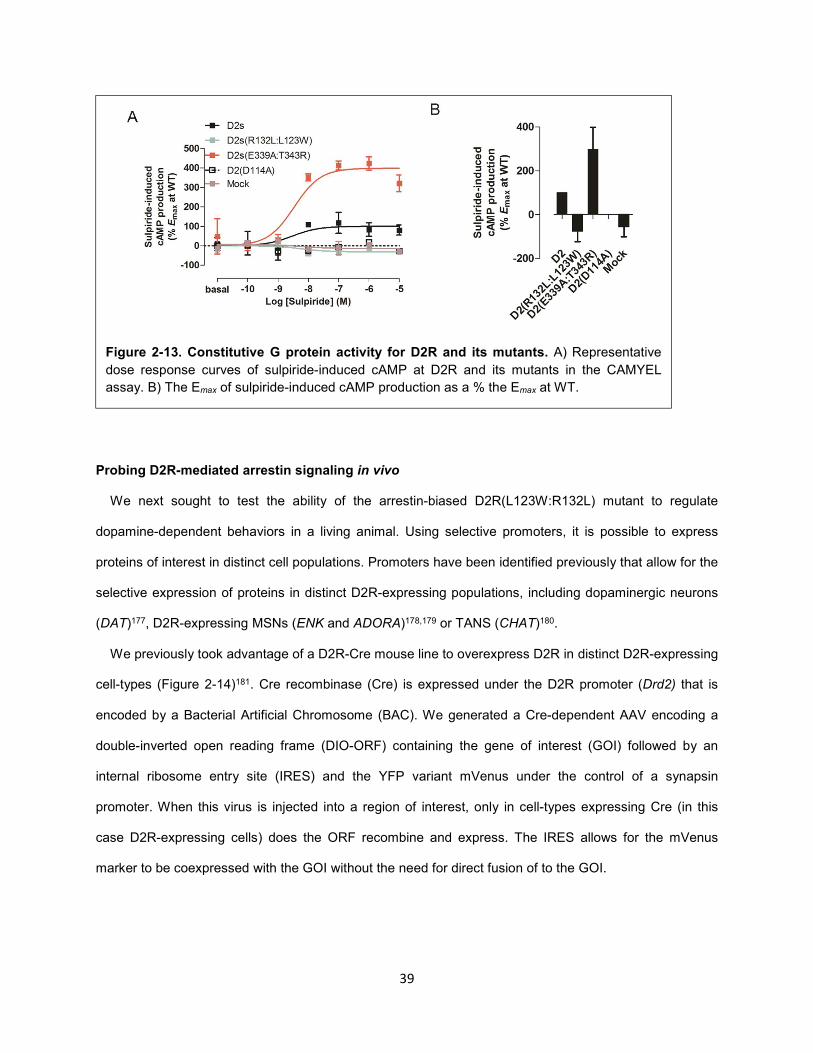
39
Probing D2R-mediated arrestin signaling in vivo
We next sought to test the ability of the arrestin-biased D2R(L123W:R132L) mutant to regulate
dopamine-dependent behaviors in a living animal. Using selective promoters, it is possible to express
proteins of interest in distinct cell populations. Promoters have been identified previously that allow for the
selective expression of proteins in distinct D2R-expressing populations, including dopaminergic neurons
(DAT)177, D2R-expressing MSNs (ENK and ADORA)178,179 or TANS (CHAT)180.
We previously took advantage of a D2R-Cre mouse line to overexpress D2R in distinct D2R-expressing
cell-types (Figure 2-14)181. Cre recombinase (Cre) is expressed under the D2R promoter (Drd2) that is
encoded by a Bacterial Artificial Chromosome (BAC). We generated a Cre-dependent AAV encoding a
double-inverted open reading frame (DIO-ORF) containing the gene of interest (GOI) followed by an
internal ribosome entry site (IRES) and the YFP variant mVenus under the control of a synapsin
promoter. When this virus is injected into a region of interest, only in cell-types expressing Cre (in this
case D2R-expressing cells) does the ORF recombine and express. The IRES allows for the mVenus
marker to be coexpressed with the GOI without the need for direct fusion of to the GOI.
Figure 2-13. Constitutive G protein activity for D2R and its mutants. A) Representative
dose response curves of sulpiride-induced cAMP at D2R and its mutants in the CAMYEL
assay. B) The Emax of sulpiride-induced cAMP production as a % the Emax at WT.
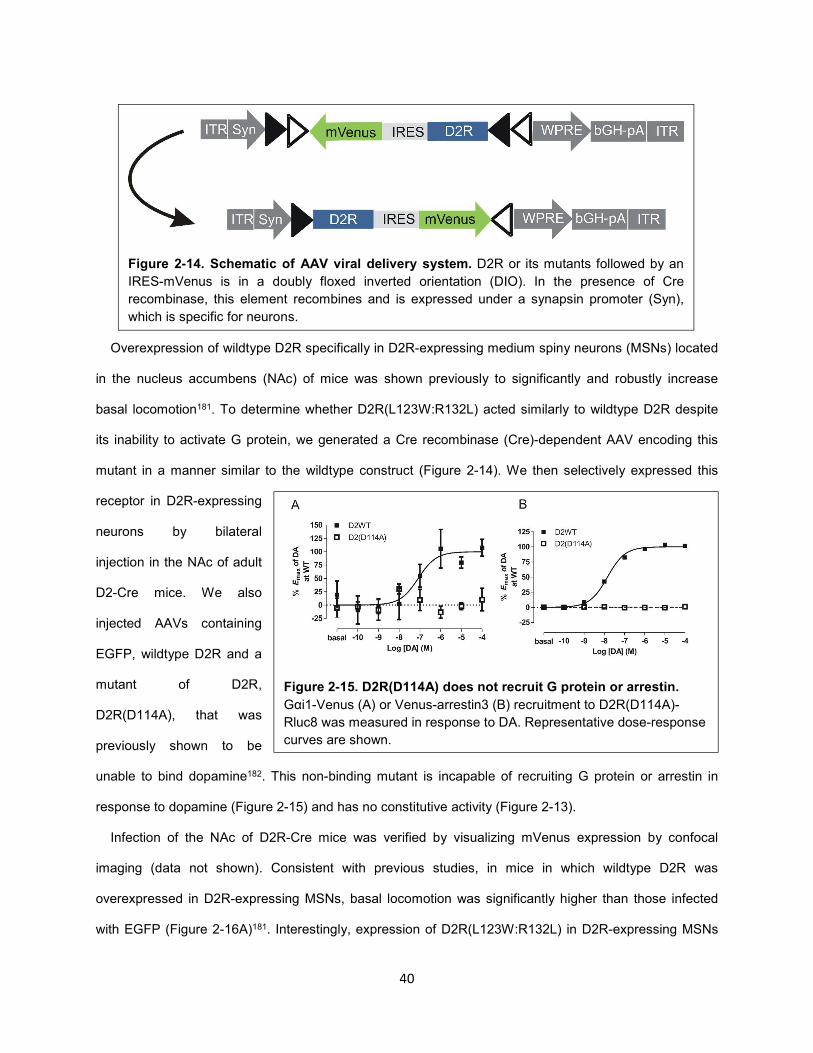
40
Overexpression of wildtype D2R specifically in D2R-expressing medium spiny neurons (MSNs) located
in the nucleus accumbens (NAc) of mice was shown previously to significantly and robustly increase
basal locomotion181. To determine whether D2R(L123W:R132L) acted similarly to wildtype D2R despite
its inability to activate G protein, we generated a Cre recombinase (Cre)-dependent AAV encoding this
mutant in a manner similar to the wildtype construct (Figure 2-14). We then selectively expressed this
receptor in D2R-expressing
neurons by bilateral
injection in the NAc of adult
D2-Cre mice. We also
injected AAVs containing
EGFP, wildtype D2R and a
mutant of D2R,
D2R(D114A), that was
previously shown to be
unable to bind dopamine182. This non-binding mutant is incapable of recruiting G protein or arrestin in
response to dopamine (Figure 2-15) and has no constitutive activity (Figure 2-13).
Infection of the NAc of D2R-Cre mice was verified by visualizing mVenus expression by confocal
imaging (data not shown). Consistent with previous studies, in mice in which wildtype D2R was
overexpressed in D2R-expressing MSNs, basal locomotion was significantly higher than those infected
with EGFP (Figure 2-16A)181. Interestingly, expression of D2R(L123W:R132L) in D2R-expressing MSNs
Figure 2-14. Schematic of AAV viral delivery system. D2R or its mutants followed by an
IRES-mVenus is in a doubly floxed inverted orientation (DIO). In the presence of Cre
recombinase, this element recombines and is expressed under a synapsin promoter (Syn),
which is specific for neurons.
Figure 2-15. D2R(D114A) does not recruit G protein or arrestin.
Gαi1-Venus (A) or Venus-arrestin3 (B) recruitment to D2R(D114A)-
Rluc8 was measured in response to DA. Representative dose-response
curves are shown.
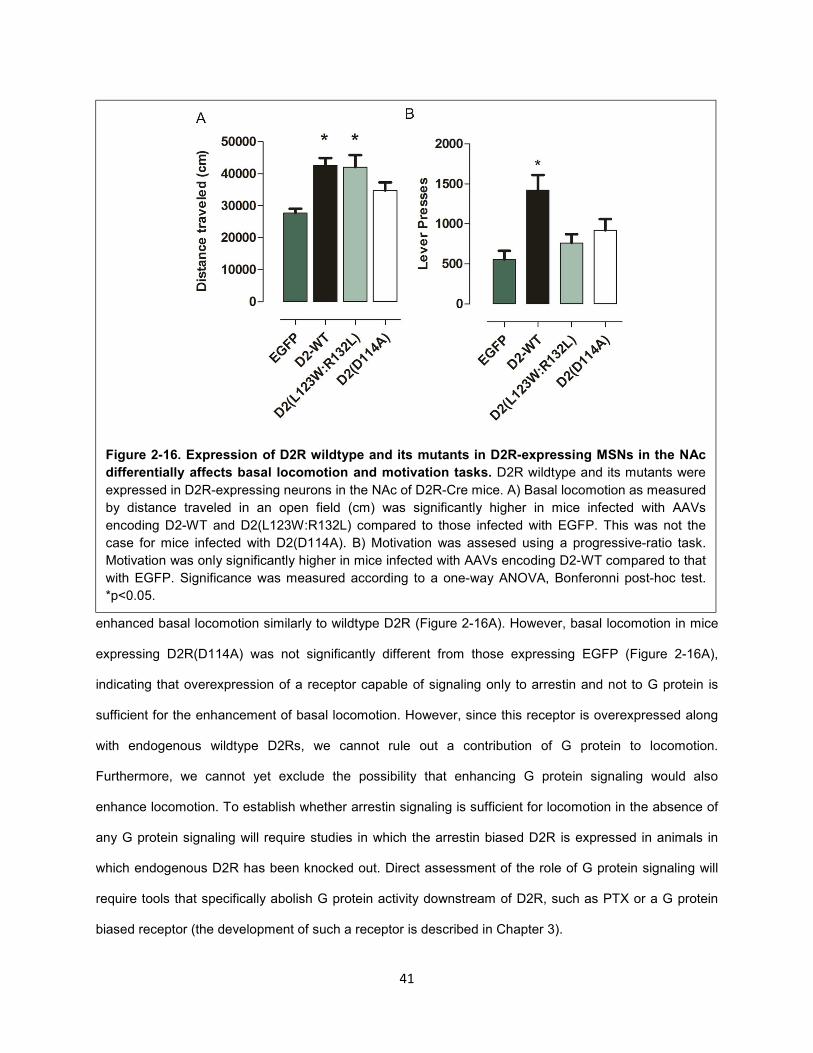
41
enhanced basal locomotion similarly to wildtype D2R (Figure 2-16A). However, basal locomotion in mice
expressing D2R(D114A) was not significantly different from those expressing EGFP (Figure 2-16A),
indicating that overexpression of a receptor capable of signaling only to arrestin and not to G protein is
sufficient for the enhancement of basal locomotion. However, since this receptor is overexpressed along
with endogenous wildtype D2Rs, we cannot rule out a contribution of G protein to locomotion.
Furthermore, we cannot yet exclude the possibility that enhancing G protein signaling would also
enhance locomotion. To establish whether arrestin signaling is sufficient for locomotion in the absence of
any G protein signaling will require studies in which the arrestin biased D2R is expressed in animals in
which endogenous D2R has been knocked out. Direct assessment of the role of G protein signaling will
require tools that specifically abolish G protein activity downstream of D2R, such as PTX or a G protein
biased receptor (the development of such a receptor is described in Chapter 3).
Figure 2-16. Expression of D2R wildtype and its mutants in D2R-expressing MSNs in the NAc
differentially affects basal locomotion and motivation tasks. D2R wildtype and its mutants were
expressed in D2R-expressing neurons in the NAc of D2R-Cre mice. A) Basal locomotion as measured
by distance traveled in an open field (cm) was significantly higher in mice infected with AAVs
encoding D2-WT and D2(L123W:R132L) compared to those infected with EGFP. This was not the
case for mice infected with D2(D114A). B) Motivation was assesed using a progressive-ratio task.
Motivation was only significantly higher in mice infected with AAVs encoding D2-WT compared to that
with EGFP. Significance was measured according to a one-way ANOVA, Bonferonni post-hoc test.
*p<0.05.

42
We next investigated whether the arrestin-biased mutant D2R(L123W:R132L) acts similarly to wildtype
D2R in dopamine-dependent behaviors other than locomotion. We showed previously that
overexpression of wildtype D2Rs in MSNs of the NAc increased motivation without altering
consummatory behavior183. Using a progressive ratio lever pressing task, we measured the willingness to
expend effort to obtain a reward (evaporated milk) by mice expressing EGFP, D2R or its mutants. Lever
pressing was significantly higher in mice expressing wildtype D2R than those expressing the EGFP
control (Figure 2-16B). Remarkably, lever pressing was not significantly different between mice
expressing EGFP and D2R(D114A) or D2R(L123W:R132L) (Figure 2-16B), indicating that unlike basal
locomotion, expression of a receptor capable of recruiting arrestin and not G protein was not sufficient to
enhance motivation.
Conclusions and future directions
In this study, we developed an arrestin-biased mutant of D2R that can be selectively expressed in
distinct D2R-expressing populations. In contrast to traditional knockout approaches of GPCR signaling
proteins, this tool is less likely to affect the signaling of other receptors. Moreover, unlike DREADDs, this
receptor mutant enables the selective activation of a distinct receptor pathway in response to the
endogenous ligand in a manner that maintains the normal spatio-temporal aspects of signaling.
Selective overexpression of this mutant in D2R-expressing MSNs was sufficient to enhance basal
locomotion but not motivation in mice. Further studies will be necessary to probe additional behaviors
associated with measuring the efficacy and side effects of antipsychotics in rodents, e.g., inhibition of
psychostimulant effects on locomotion and prepulse inhibition (PPI), as well as catalepsy184,185. Moreover,
it is important to note that because this mutant receptor was expressed in a background of endogenous
wildtype D2Rs, these findings do not indicate whether arrestin recruitment independent of G protein
activation is sufficient for locomotion, or that arrestin recruitment does not facilitate reward driven
behaviors. In order to better understand the role of arrestin signaling totally independently of G protein
activation, future studies will be required that replace wildtype D2Rs with this arrestin-biased receptor,
e.g., by overexpressing this D2R mutant in D2R-Cre mice crossed with those in which D2R is knocked
out globally. In addition, overexpressing the arrestin-biased D2R receptor in other neuronal populations

43
expressing D2R will be necessary to determine the role of these receptor populations in regulating
various processes, including psychostimulant-dependent behaviors.

44
CHAPTER 3. The development of genetic tools to probe D2R-mediated G protein signaling in vivo
Previous studies have indicated that the activation of G protein signaling downstream of D2R may play
a role in mediating psychostimulant-dependent behaviors such as locomotion148,150, thus challenging the
notion that selective inhibition of the arrestin pathway will lead to the development of superior
antipsychotic medications148,150. One study implicated the signaling protein DARPP-32, which is
downstream of G protein activation, as also being involved in mediating these behaviors151. However, this
protein is regulated in a complex manner and has number of downstream substrates186, making it difficult
to interpret the specific role of G protein activation. Therefore, we sought to develop tools to probe the
role of G protein signaling downstream of D2R in a less ambiguous manner.
The development of PTX as a genetically-encoded tool to abolish G protein signaling downstream
of D2R
Global knockout of arrestin3 in mice was used previously to study the role of arrestin signaling in
mediating psychostimulant-dependent behaviors. However, D2R was shown previously to recruit
arrestin230, which may have partially overlapping functions with arrestin3. Thus, knockout of a single
arrestin protein may be insufficient to dissociate the specific roles of G protein and arrestin in mediating
D2R signaling. Although global double knockout of both arrestin2 and arrestin3 is embryonically lethal187,
it may be possible to knock out these signaling proteins simultaneously in distinct cell-types using
selective promoters. In contrast, it is not feasible to simultaneously knock out all five genes that encode G
proteins that couple to D2R, even in a cell-type specific manner30.
We sought instead to develop pertussis toxin (PTX) as a tool to probe the role of G protein signaling in
mediating psychostimulant-dependent behaviors in rodents. This toxin offers the possibility of effectively
knocking out several D2R-coupling G proteins simultaneously. Exogenously applied PTX has been used
previously to study the role of Gi/o signaling in immortalized cell lines188, primary cells189 as well as ex vivo
slices and live animals190. However, application of this toxin in live animals is technically challenging and,
more importantly, exogenous PTX inactivates Gi/o in a cell-type independent manner. Thus, PTX would

45
not only inactivate G proteins in D2R-expressing neurons, it would also affect all cell-types in the treated
area (e.g., D1R-expressing neurons).
PTX is a holotoxin composed of five subunits, S1-5, which upon binding to surface receptors on the
plasma membrane, are taken up by cells and undergo retrograde transport from endosomes to the golgi
network, endoplasmic reticulum (ER) and nucleus157. Upon activation through the actions of glutathione
and ATP, the catalytic subunit S1 (Part A of the PTX holotoxin) dissociates from S2-5 (Part B) and has
been reported to be transported from the ER lumen to the cytosol through the ER-associated-degradation
(ERAD) pathway191. This pathway facilitates the transport of misfolded proteins from the ER to the cytosol
for degradation. However, the S1 subunit of PTX avoids degradation and is transported to the cytoplasm,
localizing to the membranes where it is inactivates Gi/o.
Interestingly, the catalytic subunit of PTX, S1, can be expressed in a heterologous manner
independently of the S2-5 subunits, which are accessory proteins that traffic the S1 subunit from the
extracellular space to inside the cell rather than being directly involved in enzymatic activity188. S1 alone
has been shown to inactivate Gi/o activity both in immortalized cell lines and primary cells188,192. Because
S1 is expressed as a cytosolic protein independently of S2-5, its effects can be restricted to a given cell-
type.
S1 was recently expressed in a cell-type specific manner in live animals (drosophila and mice) to probe
the role of Gi/o signaling on various physiological processes193,194. However, these studies used
expression systems not applicable to the study of D2R signaling. Thus, we sought to adopt the cell-type
specific Cre-lox expression system (described in Chapter 2) to deliver S1 to distinct D2R-expressing
populations using viral vectors, allowing us to control both the timing, region and cell-type of Gi/o
inactivation.
Previous studies in live animals either did not directly assess S1 expression or detected it by western
blot using α-S1 antibodies193,194. We sought to tag S1 in a manner that would allow us to easily detect its
expression in D2R-expressing neurons. S1 was previously labelled at its N-terminus with GFP, which was
used as a marker for expression192. The AAV system that we used previously obviates the need to
directly label the gene of interest (GOI) with large protein fluorophores by carrying an IRES-Venus directly
after the ORF. However, although mVenus fluorescence can be used as an indicator of infection by the
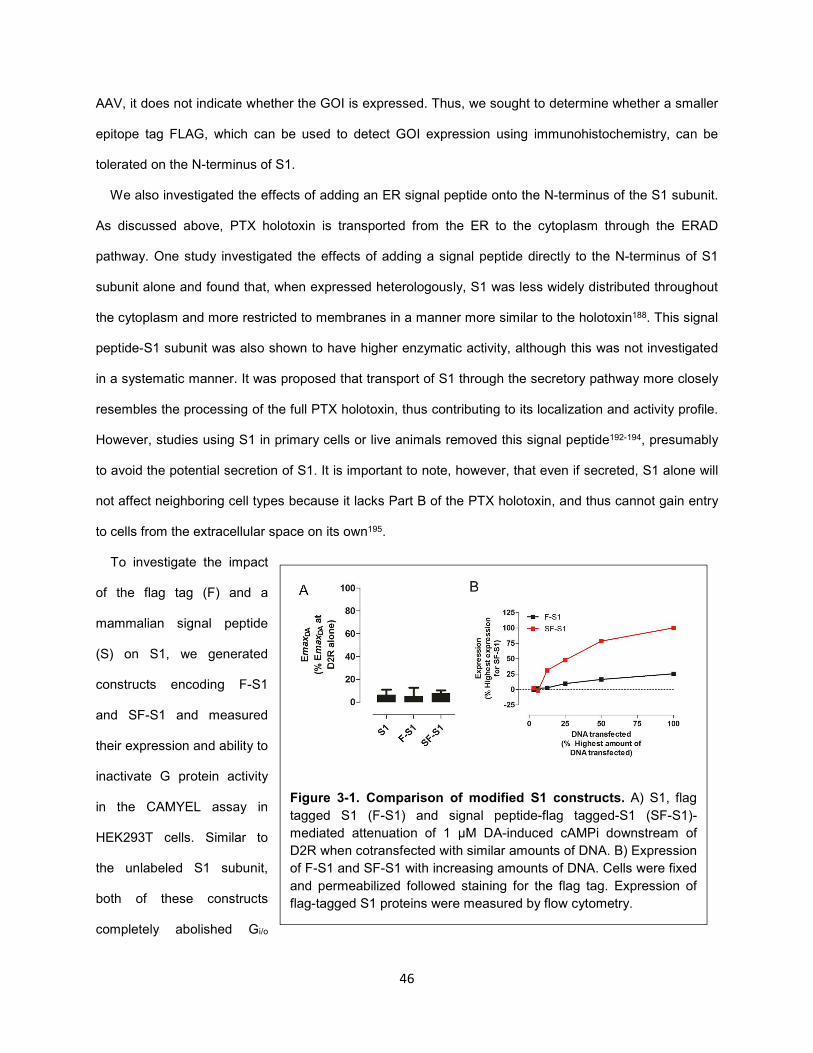
46
AAV, it does not indicate whether the GOI is expressed. Thus, we sought to determine whether a smaller
epitope tag FLAG, which can be used to detect GOI expression using immunohistochemistry, can be
tolerated on the N-terminus of S1.
We also investigated the effects of adding an ER signal peptide onto the N-terminus of the S1 subunit.
As discussed above, PTX holotoxin is transported from the ER to the cytoplasm through the ERAD
pathway. One study investigated the effects of adding a signal peptide directly to the N-terminus of S1
subunit alone and found that, when expressed heterologously, S1 was less widely distributed throughout
the cytoplasm and more restricted to membranes in a manner more similar to the holotoxin188. This signal
peptide-S1 subunit was also shown to have higher enzymatic activity, although this was not investigated
in a systematic manner. It was proposed that transport of S1 through the secretory pathway more closely
resembles the processing of the full PTX holotoxin, thus contributing to its localization and activity profile.
However, studies using S1 in primary cells or live animals removed this signal peptide192-194, presumably
to avoid the potential secretion of S1. It is important to note, however, that even if secreted, S1 alone will
not affect neighboring cell types because it lacks Part B of the PTX holotoxin, and thus cannot gain entry
to cells from the extracellular space on its own195.
To investigate the impact
of the flag tag (F) and a
mammalian signal peptide
(S) on S1, we generated
constructs encoding F-S1
and SF-S1 and measured
their expression and ability to
inactivate G protein activity
in the CAMYEL assay in
HEK293T cells. Similar to
the unlabeled S1 subunit,
both of these constructs
completely abolished Gi/o
Figure 3-1. Comparison of modified S1 constructs. A) S1, flag
tagged S1 (F-S1) and signal peptide-flag tagged-S1 (SF-S1)-
mediated attenuation of 1 μM DA-induced cAMPi downstream of
D2R when cotransfected with similar amounts of DNA. B) Expression
of F-S1 and SF-S1 with increasing amounts of DNA. Cells were fixed
and permeabilized followed staining for the flag tag. Expression of
flag-tagged S1 proteins were measured by flow cytometry.
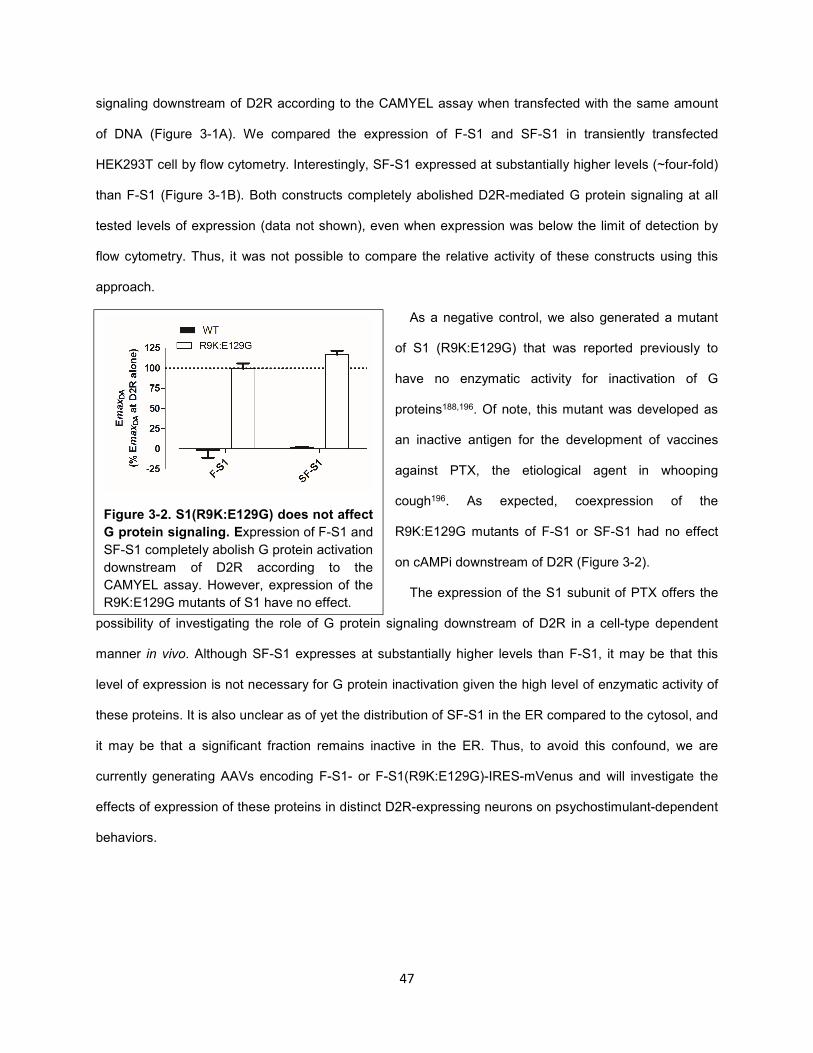
47
signaling downstream of D2R according to the CAMYEL assay when transfected with the same amount
of DNA (Figure 3-1A). We compared the expression of F-S1 and SF-S1 in transiently transfected
HEK293T cell by flow cytometry. Interestingly, SF-S1 expressed at substantially higher levels (~four-fold)
than F-S1 (Figure 3-1B). Both constructs completely abolished D2R-mediated G protein signaling at all
tested levels of expression (data not shown), even when expression was below the limit of detection by
flow cytometry. Thus, it was not possible to compare the relative activity of these constructs using this
approach.
As a negative control, we also generated a mutant
of S1 (R9K:E129G) that was reported previously to
have no enzymatic activity for inactivation of G
proteins188,196. Of note, this mutant was developed as
an inactive antigen for the development of vaccines
against PTX, the etiological agent in whooping
cough196. As expected, coexpression of the
R9K:E129G mutants of F-S1 or SF-S1 had no effect
on cAMPi downstream of D2R (Figure 3-2).
The expression of the S1 subunit of PTX offers the
possibility of investigating the role of G protein signaling downstream of D2R in a cell-type dependent
manner in vivo. Although SF-S1 expresses at substantially higher levels than F-S1, it may be that this
level of expression is not necessary for G protein inactivation given the high level of enzymatic activity of
these proteins. It is also unclear as of yet the distribution of SF-S1 in the ER compared to the cytosol, and
it may be that a significant fraction remains inactive in the ER. Thus, to avoid this confound, we are
currently generating AAVs encoding F-S1- or F-S1(R9K:E129G)-IRES-mVenus and will investigate the
effects of expression of these proteins in distinct D2R-expressing neurons on psychostimulant-dependent
behaviors.
Figure 3-2. S1(R9K:E129G) does not affect
G protein signaling. Expression of F-S1 and
SF-S1 completely abolish G protein activation
downstream of D2R according to the
CAMYEL assay. However, expression of the
R9K:E129G mutants of S1 have no effect.

48
The complete abolition of G protein activity may require that S1 be ubiquitously expressed throughout
the cell. However, this may not be the case, especially in axons and dendrites of neurons. The subcellular
distribution of S1 in neurons has not yet been investigated and may be cell-type dependent. That S1 may
be specifically localized to distinct sub-compartments may in fact be advantageous, allowing for the
investigation of the role of Gi/o signaling from specific cellular locations. This may be useful for studying
the role of G protein signaling from D2R populations that have distinct functions within a specific cell-type,
e.g., D2Rs expressed in dendrites versus those expressed in axon terminals. It may be possible to
develop more rationally subcellular-specific S1 proteins by using specific targeting sequences, e.g.,
specific 3’UTR sequences can localize mRNA to specific subcellular compartments, including dendrites197
and axons198.
An alternative approach to increase the likelihood that S1 colocalizes in neuronal sub-compartments
expressing D2R is to fuse it directly to the C-terminus of D2R (D2R-S1). We have generated this
construct and tested its activity in HEK293T cells. Interestingly, D2R-S1 is capable of inactivating
endogenous Gi/o activity, both from the fusion D2R as well as coexpressed Gi/o-coupled receptors (data
not shown). Interestingly, arrestin recruitment to this receptor is intact. We also fused the catalytically
inactive S1 to D2R (D2R-S1(R9K:E129G)). Gi/o activity from this fusion construct was maintained,
indicating that the impaired Gi/o activity of D2R-S1 is due to the catalytic activity rather than steric
impairment of receptor function by S1. We are not currently pursuing this strategy because the size of
D2R-S1 exceeds that allotted by the AAV vectors used in our expression system. These limitations can
Figure 3-3. Schematic of AAV viral delivery system. Wildtype or inactive F-S1 followed by an
IRES-mVenus is in a doubly floxed inverted orientation (DIO). In the presence of Cre recombinase,
this element recombines and is expressed under a synapsin promoter (Syn), which is specific for
neurons.

49
be addressed using different viral delivery systems, e.g., lentivirus or herpes simplex virus (HSV) as well
as stable and inducible expression systems.
In general, the use of S1 to probe G protein signaling in vivo suffers from several limitations, including
those associated with knockout strategies generally (Figure 1-11). Moreover, S1 inactivates Gi/o but not
Gz, and thus it is possible that G protein activity may not be sufficiently abolished. The role of Gαz in
mediating D2R-dependent processes was investigated previously in a global knockout mouse model199.
Quinpirole (a D2R full agonist) induced-suppression of locomotion, DA release and core body
temperature was attenuated in the Gαz knockout animal, indicating that this G protein may in fact be
involved in D2R signaling. Both D2R-induced suppression of locomotion and DA release are likely
mediated by D2 autoreceptors, which are expressed in dopaminergic terminals. It remains to be seen
what the role of Gαz in different D2R-expression cell types. Regardless, the inability of PTX to abolish the
activity of Gαz adds an additional confound for its use as a tool to probe arrestin signaling in distinct
neuronal populations. In principle, this could be addressed using several approaches, such as crossing
Gαz knockout with D2R-Cre mice or creating a knock-in mouse line in which Gαz is made sensitive to PTX
by mutating in the critical cysteine residue.
The development of a G protein-biased D2R mutant (Part I)
To probe further the differences
between G protein and arrestin
signaling downstream of D2R, we
sought to develop a G protein-biased
mutant (Figure 3-4) that could be used
in a similar manner to the arrestin-
biased mutant described in Chapter 2.
There are a number of strategies to
develop biased mutants at GPCRs.
The arrestin-biased D2R mutant was
developed by taking advantage of
Figure 3-4. G protein biased D2R mutant (D2RGPB).

50
receptor residues that are known to affect conformational states of the receptor. Thus, one possibility is
that the binding of agonists to this mutant preferentially stabilizes arrestin binding conformations in D2R
when its DRY motif is mutated to DLY. A similar approach was taken previously for the development of
arrestin-biased mutants of AT1A and β2AR200,201. Unfortunately, there are no such mutations currently
known for GPCRs, especially of the Family A type, that conformationally bias receptors towards G protein
activation.
Of note, we cannot exclude the possibility that mutating the DRY motif negatively affects the binding
site of G proteins to these receptors. An alternative strategy for the development of biased mutants of
GPCRs is to mutate receptor binding sites specific for either G protein or arrestin that do not affect the
binding and activation of the other signaling protein. Indeed, a G protein-biased mutant of D2R was
developed previously using this strategy202. Using a GST-pulldown screen between various D2R
fragments and arrestin, a four-residue segment of D2R (residues 212-215) was identified at the interface
between TM5 and IL3 of D2R that bound arrestin. When mutated to alanines, this D2R mutant (D2-A4)
selectively coupled to G protein and not arrestin. However, this mutation also greatly impaired trafficking
of the receptor to the cell membrane, thereby reducing its value as a tool to study the relationships
between specific signaling pathways and psychostimulant-dependent behaviors.
D2-A4 is not suitable for use in vivo
The goal of this work was to modify D2-A4 to maintain its G protein bias but increase its receptor
expression. We first sought to confirm these results using radioligand binding to determine D2-A4
membrane expression and BRET-based assays to monitor receptor-mediated G protein activation and
arrestin3 recruitment. When a constant amount of plasmid DNA encoding wild type D2 receptor (D2-WT)
or D2-A4 was transiently transfected into HEK293 cells, D2-A4 was expressed at approximately 10% of
the density of D2-WT (data not shown).
The ability of D2-WT and D2-A4 to inhibit forskolin-mediated activation of adenylyl cyclase was
measured to determine the capacity of each receptor to signal through G proteins. Surface expression of
these receptors was equalized to directly compare G protein activity. Quinpirole stimulation of D2-WT
produced a dose-dependent increase in the inhibition of cyclic AMP accumulation (Figure 3-5A), whereas

51
activation of D2-A4 with quinpirole resulted in less inhibition of adenylyl cyclase activity (Figure 3-5A and
B). This suggests that D2-A4 is a poor activator of G proteins. To confirm this hypothesis, D2-A4-mediated
G protein activation was measured using a BRET assay in which agonist-bound receptor induces a
separation of the Gα energy donor (Gαi1-91-Rluc8) from the complemented Gβγ acceptor (mVenus-Gβ1γ2),
thus decreasing the BRET signal (Figure 3-5C)203. Activation of D2-WT produced a dose-dependent
increase in G protein activation (Figure 3-5D), whereas activation of D2-A4 with either quinpirole or
dopamine resulted in significantly less G protein activation than seen at the wild-type receptor (Figure 3-5D
and E). In combination, the cyclic AMP and BRET data suggest that D2-A4 is a poor G protein activator
relative to D2-WT, which appears to differ from previous results using cells stably expressing D2-A4 and
D2-WT; under those conditions agonist stimulation of D2-A4 resulted in a decrease in cAMP that was
similar to that observed for the wild type receptor, but a decrease in the potency of dopamine in one
experiment in that earlier work suggests that a receptor reserve might have obscured a partial loss of
coupling efficiency202.
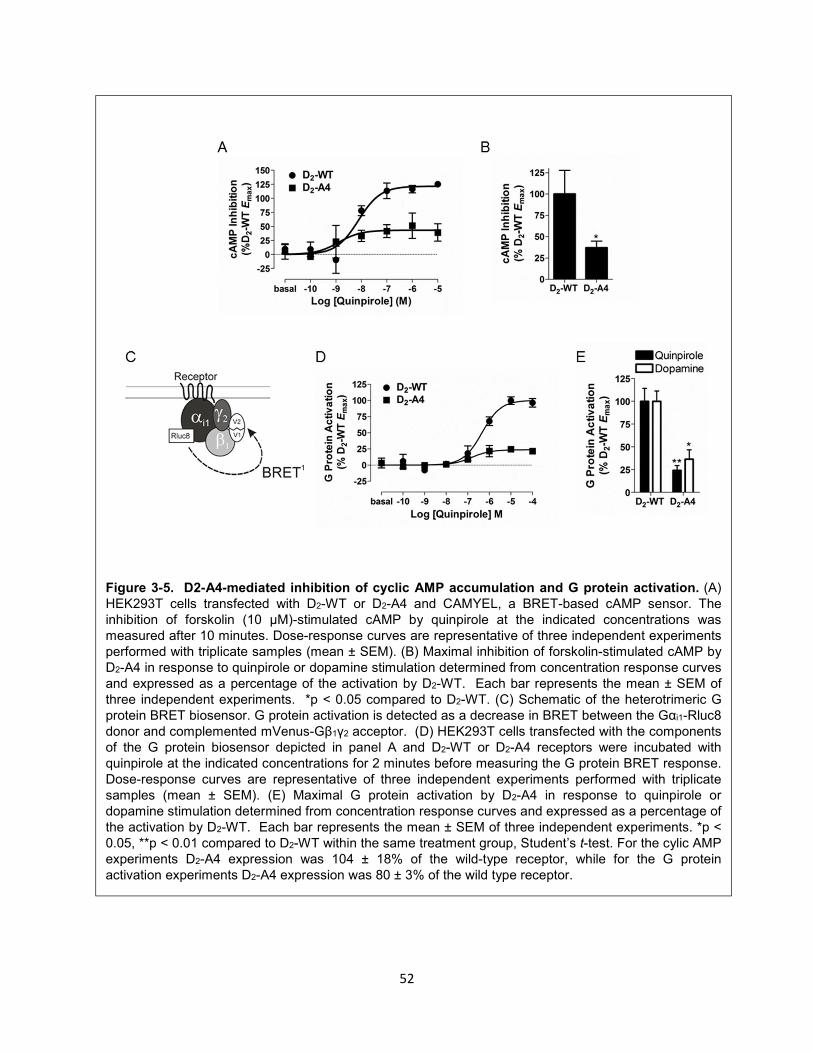
52
Figure 3-5. D2-A4-mediated inhibition of cyclic AMP accumulation and G protein activation. (A) HEK293T cells transfected with D2-WT or D2-A4 and CAMYEL, a BRET-based cAMP sensor. The inhibition of forskolin (10 µM)-stimulated cAMP by quinpirole at the indicated concentrations was measured after 10 minutes. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± SEM). (B) Maximal inhibition of forskolin-stimulated cAMP by D2-A4 in response to quinpirole or dopamine stimulation determined from concentration response curves and expressed as a percentage of the activation by D2-WT. Each bar represents the mean ± SEM of three independent experiments. *p < 0.05 compared to D2-WT. (C) Schematic of the heterotrimeric G protein BRET biosensor. G protein activation is detected as a decrease in BRET between the Gαi1-Rluc8 donor and complemented mVenus-Gβ1γ2 acceptor. (D) HEK293T cells transfected with the components of the G protein biosensor depicted in panel A and D2-WT or D2-A4 receptors were incubated with quinpirole at the indicated concentrations for 2 minutes before measuring the G protein BRET response. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± SEM). (E) Maximal G protein activation by D2-A4 in response to quinpirole or dopamine stimulation determined from concentration response curves and expressed as a percentage of the activation by D2-WT. Each bar represents the mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01 compared to D2-WT within the same treatment group, Student’s t-test. For the cylic AMP experiments D2-A4 expression was 104 ± 18% of the wild-type receptor, while for the G protein activation experiments D2-A4 expression was 80 ± 3% of the wild type receptor.
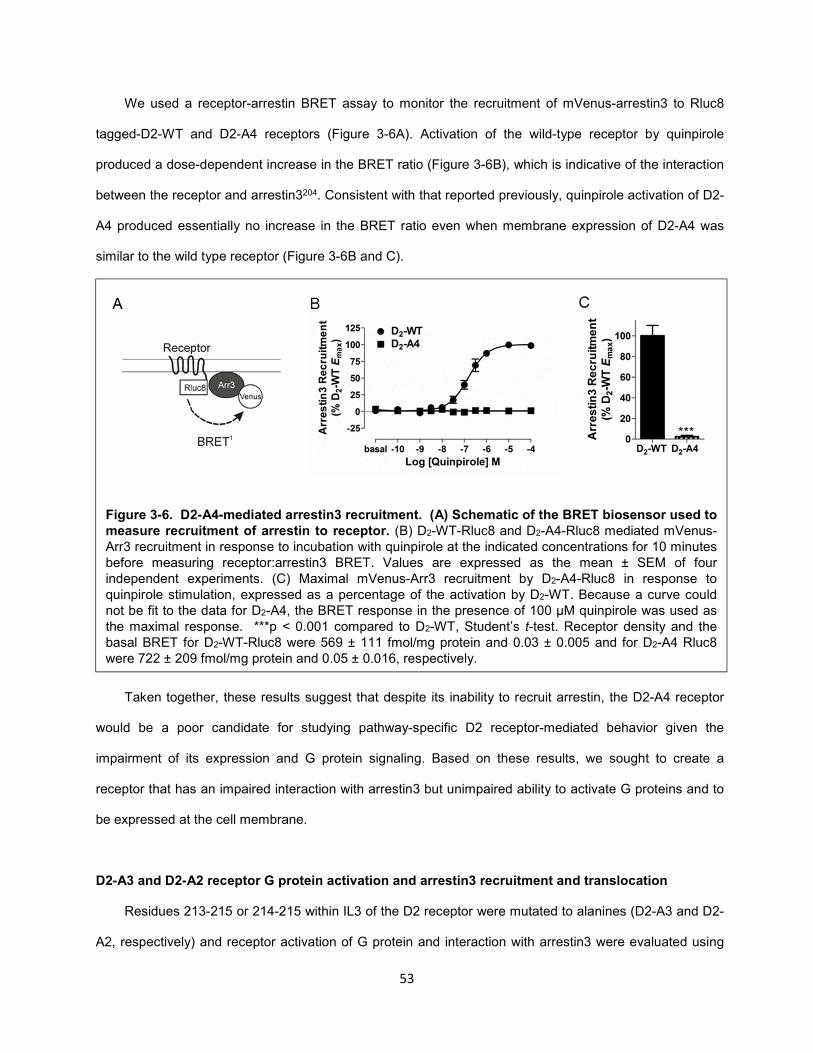
53
We used a receptor-arrestin BRET assay to monitor the recruitment of mVenus-arrestin3 to Rluc8
tagged-D2-WT and D2-A4 receptors (Figure 3-6A). Activation of the wild-type receptor by quinpirole
produced a dose-dependent increase in the BRET ratio (Figure 3-6B), which is indicative of the interaction
between the receptor and arrestin3204. Consistent with that reported previously, quinpirole activation of D2-
A4 produced essentially no increase in the BRET ratio even when membrane expression of D2-A4 was
similar to the wild type receptor (Figure 3-6B and C).
Taken together, these results suggest that despite its inability to recruit arrestin, the D2-A4 receptor
would be a poor candidate for studying pathway-specific D2 receptor-mediated behavior given the
impairment of its expression and G protein signaling. Based on these results, we sought to create a
receptor that has an impaired interaction with arrestin3 but unimpaired ability to activate G proteins and to
be expressed at the cell membrane.
D2-A3 and D2-A2 receptor G protein activation and arrestin3 recruitment and translocation
Residues 213-215 or 214-215 within IL3 of the D2 receptor were mutated to alanines (D2-A3 and D2-
A2, respectively) and receptor activation of G protein and interaction with arrestin3 were evaluated using
Figure 3-6. D2-A4-mediated arrestin3 recruitment. (A) Schematic of the BRET biosensor used to
measure recruitment of arrestin to receptor. (B) D2-WT-Rluc8 and D2-A4-Rluc8 mediated mVenus-Arr3 recruitment in response to incubation with quinpirole at the indicated concentrations for 10 minutes before measuring receptor:arrestin3 BRET. Values are expressed as the mean ± SEM of four independent experiments. (C) Maximal mVenus-Arr3 recruitment by D2-A4-Rluc8 in response to quinpirole stimulation, expressed as a percentage of the activation by D2-WT. Because a curve could not be fit to the data for D2-A4, the BRET response in the presence of 100 µM quinpirole was used as the maximal response. ***p < 0.001 compared to D2-WT, Student’s t-test. Receptor density and the basal BRET for D2-WT-Rluc8 were 569 ± 111 fmol/mg protein and 0.03 ± 0.005 and for D2-A4 Rluc8 were 722 ± 209 fmol/mg protein and 0.05 ± 0.016, respectively.

54
the techniques described above. Agonist stimulation of D2-A3 and D2-A2 produced a dose-dependent
increase (Figure 3-7A) in G protein activation that was similar to that of D2-WT (Figure 3-7B). In contrast,
the extent of quinpirole-induced recruitment of arrestin3 to the receptor was significantly less for the D2-A3
receptor than for D2-WT, with a trend for reduced recruitment of arrestin by the D2-A2 receptor mutant as
well (59 and 76% of the wild type receptor, respectively; (Figure 3-7C and D). Dopamine stimulation also
produced modestly reduced arrestin3 recruitment to the receptor by the D2-A3 and D2-A2 receptor
mutants that was not statistically different from D2-WT (Figure 3-7D). To monitor receptor induced
translocation of arrestin to the plasma membrane in a manner that does not require Rluc8 to be attached
to the C-terminus of the receptor, we developed an assay in which untagged receptors were cotransfected
with BRET sensors Rluc8-arrestin3-Sp1 and a plasma membrane marker, mem-linker-citrine-SH3 (see
methods). Relative arrestin translocation to the membrane induced by the wild type D2 receptor, D2-A3,
and D2-A2 closely matched the recruitment to the receptor observed using Rluc8-tagged receptors, except
that none of the reductions in agonist-induced translocation of arrestin to the membrane was significant
(Figure 3-7E and F). Importantly, despite modest reductions in arrestin translocation and recruitment to the
receptor, both D2-A3 and D2-A2 receptors still robustly recruited arrestin3 in response to agonist
stimulation.
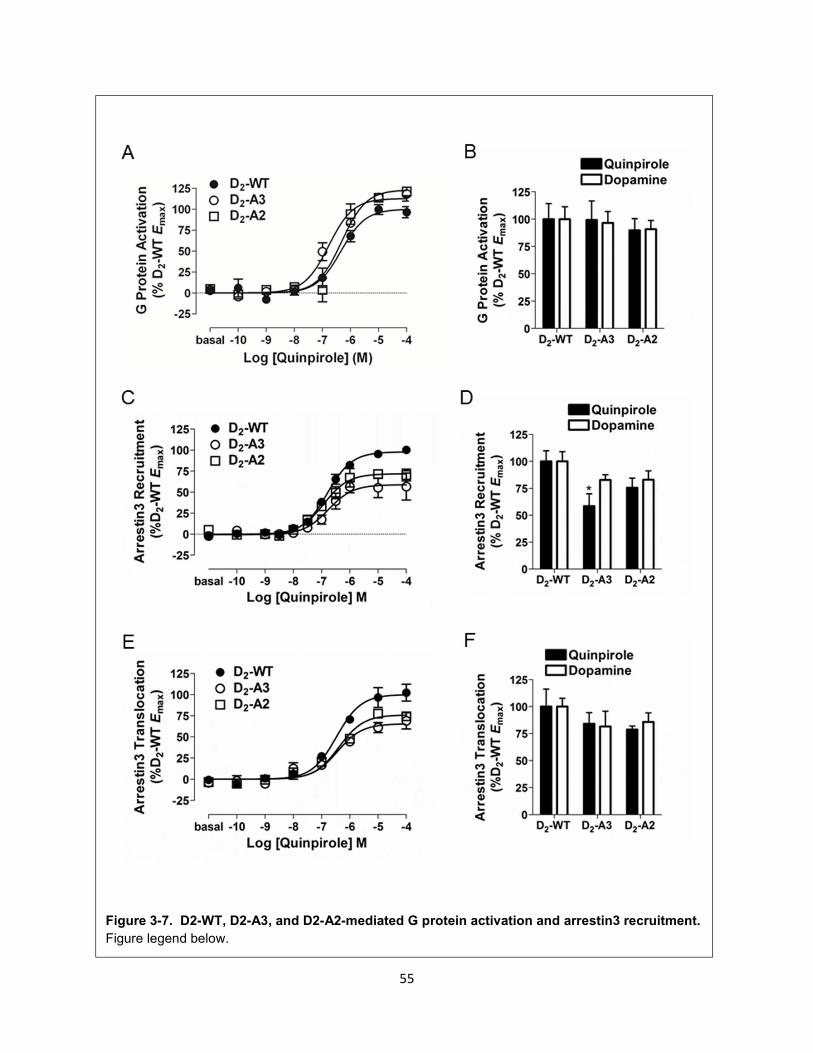
55
Figure 3-7. D2-WT, D2-A3, and D2-A2-mediated G protein activation and arrestin3 recruitment.
Figure legend below.

56
Agonist-induced internalization of D2-WT and its mutants
One of the functional consequences of arrestin3 interaction with the D2 receptor is receptor
internalization. Agonist-induced receptor internalization was measured using a whole-cell binding assay
with the hydrophilic ligand sulpiride in HEK293 cells that stably express D2-WT, D2-A4, D2-A3, or D2-A2.
We hypothesized that the amount of agonist-induced receptor endocytosis would be positively correlated
with agonist-induced recruitment of arrestin3. Indeed, after treatment with quinpirole for 30 minutes, cell
surface expression of the D2-WT receptor was significantly decreased (17% and 30% in the absence and
presence of overexpressed arrestin3, respectively; (Figure 3-8A), whereas D2-A4 did not internalize at all
in response to agonist stimulation, even when arrestin3 was overexpressed (Figure 3-8A and B).
Quinpirole stimulation of D2-A2 receptors produced receptor internalization that was 44% of internalization
of the wild-type receptor (Figure 3-8A). Overexpression of arrestin3 with the D2-A2 receptor increased
agonist-induced receptor internalization, consistent with the ability of this receptor to recruit arrestin3,
although even in the presence of overexpressed arrestin, quinpirole-induced internalization of D2-A2 was
Figure 3-7. D2-WT, D2-A3, and D2-A2-mediated G protein activation and arrestin3 recruitment. (A) D2-WT, D2-A3 and D2-A2 mediated G protein activation was assessed in HEK293T cells co-transfected with Gαi1-Rluc8, V1-Gβ1, V2-Gγ2, and receptor, and then incubated with quinpirole at the indicated concentrations for 2 minutes before measuring the G protein BRET response. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± SEM). (B) Maximal G protein activation by D2-A3 and D2-A2 in response to quinpirole or dopamine stimulation, determined from the concentration response curves, and expressed as a percentage of the response to D2-WT. G protein activation by the mutants was not significantly different from that by WT as determined by Tukey’s multiple comparison test. D2-A3 and D2-A2 were expressed at 100 ± 5% and 78 ± 7% of the wild type receptor, respectively. (C) D2-WT, D2-A3, and D2-A2 mediated arrestin3 recruitment in HEK293 cells transfected with the indicated receptor fused to Rluc8 and mVenus-Arr3 and incubated with agonist at the indicated concentrations for 10 minutes. Values are expressed as the mean ± SEM of seven (D2-WT) or four (D2-A3 and D2-A2) independent experiments. (D) Maximal mVenus-Arr3 recruitment by D2-A3 and D2-A2 in response to quinpirole or dopamine stimulation, determined from the individual concentration-response curves and expressed as a percentage of the maximal response to D2-WT. *p < 0.05 compared to D2-WT within the same treatment group, Tukey’s multiple comparison test. Receptor density and basal BRET for D2-WT-Rluc8 were 1290 ± 151 fmol/mg protein and 0.07 ± 0.007, for D2-A3-Rluc8 were 1223 ± 160 fmol/mg protein and 0.14 ± 0.006, and for D2-A2-Rluc8 were 1300 ± 60.3 fmol/mg protein and 0.10 ± 0.008. (E) D2-WT, D2-A3, and D2-A2 mediated arrestin3 translocation to the membrane in HEK293 cells transfected with the indicated receptor, GRK2, Rluc8-arrestin3-Sp1, and mem-linker-citrine-SH3 and incubated with agonist at the indicated concentrations for 10 minutes. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± SEM). (F) Maximal Rluc8-arrestin3-SP1 membrane translocation in response to quinpirole or dopamine stimulation of untagged D2-WT, D2-A3 or D2-A2 receptors. Basal BRET for D2-WT was 0.64 ± 0.020, D2-A3 was 0.64 ± 0.017, and D2-A2 was 0.63 ± 0.023.
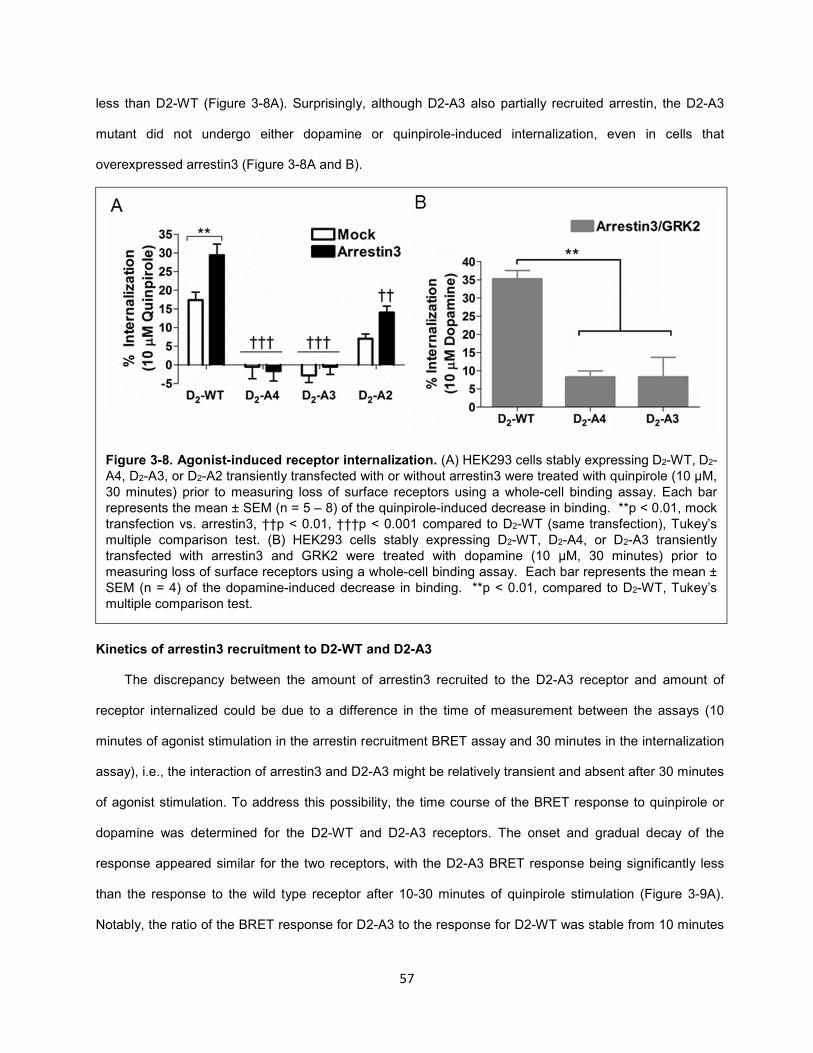
57
less than D2-WT (Figure 3-8A). Surprisingly, although D2-A3 also partially recruited arrestin, the D2-A3
mutant did not undergo either dopamine or quinpirole-induced internalization, even in cells that
overexpressed arrestin3 (Figure 3-8A and B).
Kinetics of arrestin3 recruitment to D2-WT and D2-A3
The discrepancy between the amount of arrestin3 recruited to the D2-A3 receptor and amount of
receptor internalized could be due to a difference in the time of measurement between the assays (10
minutes of agonist stimulation in the arrestin recruitment BRET assay and 30 minutes in the internalization
assay), i.e., the interaction of arrestin3 and D2-A3 might be relatively transient and absent after 30 minutes
of agonist stimulation. To address this possibility, the time course of the BRET response to quinpirole or
dopamine was determined for the D2-WT and D2-A3 receptors. The onset and gradual decay of the
response appeared similar for the two receptors, with the D2-A3 BRET response being significantly less
than the response to the wild type receptor after 10-30 minutes of quinpirole stimulation (Figure 3-9A).
Notably, the ratio of the BRET response for D2-A3 to the response for D2-WT was stable from 10 minutes
Figure 3-8. Agonist-induced receptor internalization. (A) HEK293 cells stably expressing D2-WT, D2-A4, D2-A3, or D2-A2 transiently transfected with or without arrestin3 were treated with quinpirole (10 µM, 30 minutes) prior to measuring loss of surface receptors using a whole-cell binding assay. Each bar represents the mean ± SEM (n = 5 – 8) of the quinpirole-induced decrease in binding. **p < 0.01, mock transfection vs. arrestin3, ††p < 0.01, †††p < 0.001 compared to D2-WT (same transfection), Tukey’s multiple comparison test. (B) HEK293 cells stably expressing D2-WT, D2-A4, or D2-A3 transiently transfected with arrestin3 and GRK2 were treated with dopamine (10 µM, 30 minutes) prior to measuring loss of surface receptors using a whole-cell binding assay. Each bar represents the mean ± SEM (n = 4) of the dopamine-induced decrease in binding. **p < 0.01, compared to D2-WT, Tukey’s multiple comparison test.
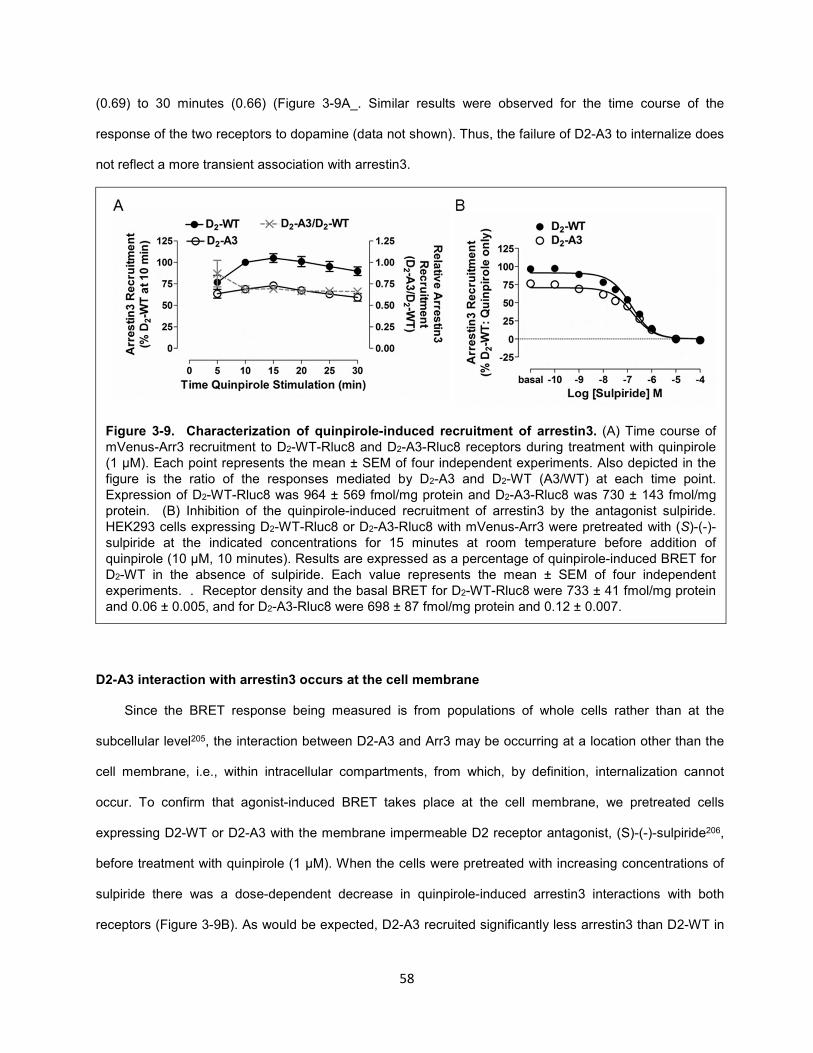
58
(0.69) to 30 minutes (0.66) (Figure 3-9A_. Similar results were observed for the time course of the
response of the two receptors to dopamine (data not shown). Thus, the failure of D2-A3 to internalize does
not reflect a more transient association with arrestin3.
D2-A3 interaction with arrestin3 occurs at the cell membrane
Since the BRET response being measured is from populations of whole cells rather than at the
subcellular level205, the interaction between D2-A3 and Arr3 may be occurring at a location other than the
cell membrane, i.e., within intracellular compartments, from which, by definition, internalization cannot
occur. To confirm that agonist-induced BRET takes place at the cell membrane, we pretreated cells
expressing D2-WT or D2-A3 with the membrane impermeable D2 receptor antagonist, (S)-(-)-sulpiride206,
before treatment with quinpirole (1 µM). When the cells were pretreated with increasing concentrations of
sulpiride there was a dose-dependent decrease in quinpirole-induced arrestin3 interactions with both
receptors (Figure 3-9B). As would be expected, D2-A3 recruited significantly less arrestin3 than D2-WT in
Figure 3-9. Characterization of quinpirole-induced recruitment of arrestin3. (A) Time course of mVenus-Arr3 recruitment to D2-WT-Rluc8 and D2-A3-Rluc8 receptors during treatment with quinpirole (1 µM). Each point represents the mean ± SEM of four independent experiments. Also depicted in the figure is the ratio of the responses mediated by D2-A3 and D2-WT (A3/WT) at each time point. Expression of D2-WT-Rluc8 was 964 ± 569 fmol/mg protein and D2-A3-Rluc8 was 730 ± 143 fmol/mg protein. (B) Inhibition of the quinpirole-induced recruitment of arrestin3 by the antagonist sulpiride. HEK293 cells expressing D2-WT-Rluc8 or D2-A3-Rluc8 with mVenus-Arr3 were pretreated with (S)-(-)-sulpiride at the indicated concentrations for 15 minutes at room temperature before addition of quinpirole (10 µM, 10 minutes). Results are expressed as a percentage of quinpirole-induced BRET for D2-WT in the absence of sulpiride. Each value represents the mean ± SEM of four independent experiments. . Receptor density and the basal BRET for D2-WT-Rluc8 were 733 ± 41 fmol/mg protein and 0.06 ± 0.005, and for D2-A3-Rluc8 were 698 ± 87 fmol/mg protein and 0.12 ± 0.007.
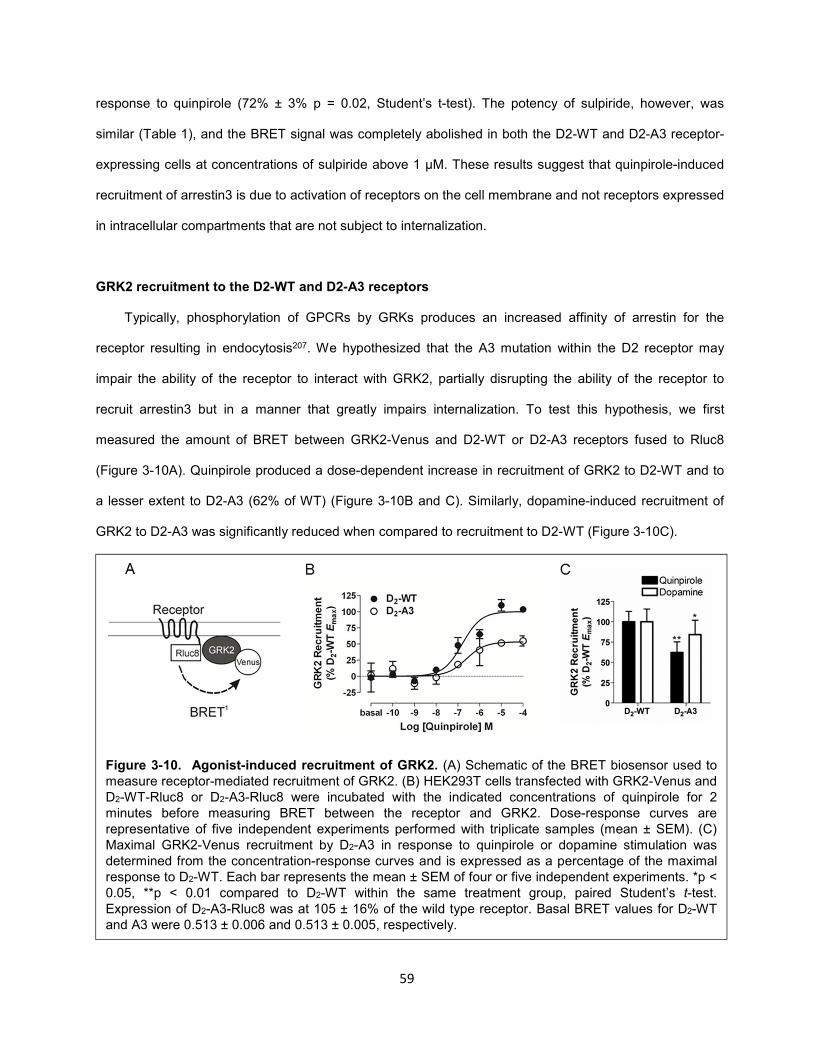
59
response to quinpirole (72% ± 3% p = 0.02, Student’s t-test). The potency of sulpiride, however, was
similar (Table 1), and the BRET signal was completely abolished in both the D2-WT and D2-A3 receptor-
expressing cells at concentrations of sulpiride above 1 μM. These results suggest that quinpirole-induced
recruitment of arrestin3 is due to activation of receptors on the cell membrane and not receptors expressed
in intracellular compartments that are not subject to internalization.
GRK2 recruitment to the D2-WT and D2-A3 receptors
Typically, phosphorylation of GPCRs by GRKs produces an increased affinity of arrestin for the
receptor resulting in endocytosis207. We hypothesized that the A3 mutation within the D2 receptor may
impair the ability of the receptor to interact with GRK2, partially disrupting the ability of the receptor to
recruit arrestin3 but in a manner that greatly impairs internalization. To test this hypothesis, we first
measured the amount of BRET between GRK2-Venus and D2-WT or D2-A3 receptors fused to Rluc8
(Figure 3-10A). Quinpirole produced a dose-dependent increase in recruitment of GRK2 to D2-WT and to
a lesser extent to D2-A3 (62% of WT) (Figure 3-10B and C). Similarly, dopamine-induced recruitment of
GRK2 to D2-A3 was significantly reduced when compared to recruitment to D2-WT (Figure 3-10C).
Figure 3-10. Agonist-induced recruitment of GRK2. (A) Schematic of the BRET biosensor used to measure receptor-mediated recruitment of GRK2. (B) HEK293T cells transfected with GRK2-Venus and D2-WT-Rluc8 or D2-A3-Rluc8 were incubated with the indicated concentrations of quinpirole for 2 minutes before measuring BRET between the receptor and GRK2. Dose-response curves are representative of five independent experiments performed with triplicate samples (mean ± SEM). (C) Maximal GRK2-Venus recruitment by D2-A3 in response to quinpirole or dopamine stimulation was determined from the concentration-response curves and is expressed as a percentage of the maximal response to D2-WT. Each bar represents the mean ± SEM of four or five independent experiments. *p < 0.05, **p < 0.01 compared to D2-WT within the same treatment group, paired Student’s t-test. Expression of D2-A3-Rluc8 was at 105 ± 16% of the wild type receptor. Basal BRET values for D2-WT and A3 were 0.513 ± 0.006 and 0.513 ± 0.005, respectively.

60
The functional consequences of GRK2 interactions with D2-WT and D2-A3 were then assessed by
overexpressing GRK2 and measuring recruitment of arrestin3 and agonist-induced receptor internalization.
GRK2 was overexpressed in cells expressing D2-WT-Rluc8 or D2-A3-Rluc8 and mVenus-Arr3 (Figure 3-
6A). Similar to previous experiments, quinpirole stimulation induced less recruitment of Arr3 to D2-A3 than
to D2-WT (Figure 3-11A). Interestingly, when GRK2 was overexpressed with D2-WT and D2-A3, at both
receptors there was an almost 5-fold leftward shift in the quinpirole dose-response curve (Figure 3-11A,
Table 3-1) and an increase in the maximal quinpirole-induced interaction with arrestin3 (53 and 37%,
respectively; Figure 3-11A and B). Furthermore, when GRK2 was overexpressed with D2-A3, the Emax for
quinpirole-induced recruitment of arrestin3 to the receptor was indistinguishable from that for D2-WT
expressed alone (i.e., without GRK2; Figure 3-11A)). These results suggest that even though the A3
mutation partially disrupts interactions with GRK2, GRK2-enhanced recruitment of arrestin3 remains intact.
Overexpression of GRK2 increased the amount of agonist-induced internalization of D2-WT (Figure 3-
11D), and internalization was further enhanced by overexpression of both GRK2 and arrestin3. Similar to
previous results, quinpirole failed to induce internalization of D2-A3 even when GRK2 and arrestin3 were
overexpressed. Although overexpression of GRK2 enhanced D2-A3-mediated recruitment of arrestin3 to a
level comparable to that of D2-WT alone (Figure 3-11A and B, Table 3-1), overexpression of GRK2 alone
or in combination with arrestin3 failed to rescue internalization of D2-A3 (Figure 3-11D).
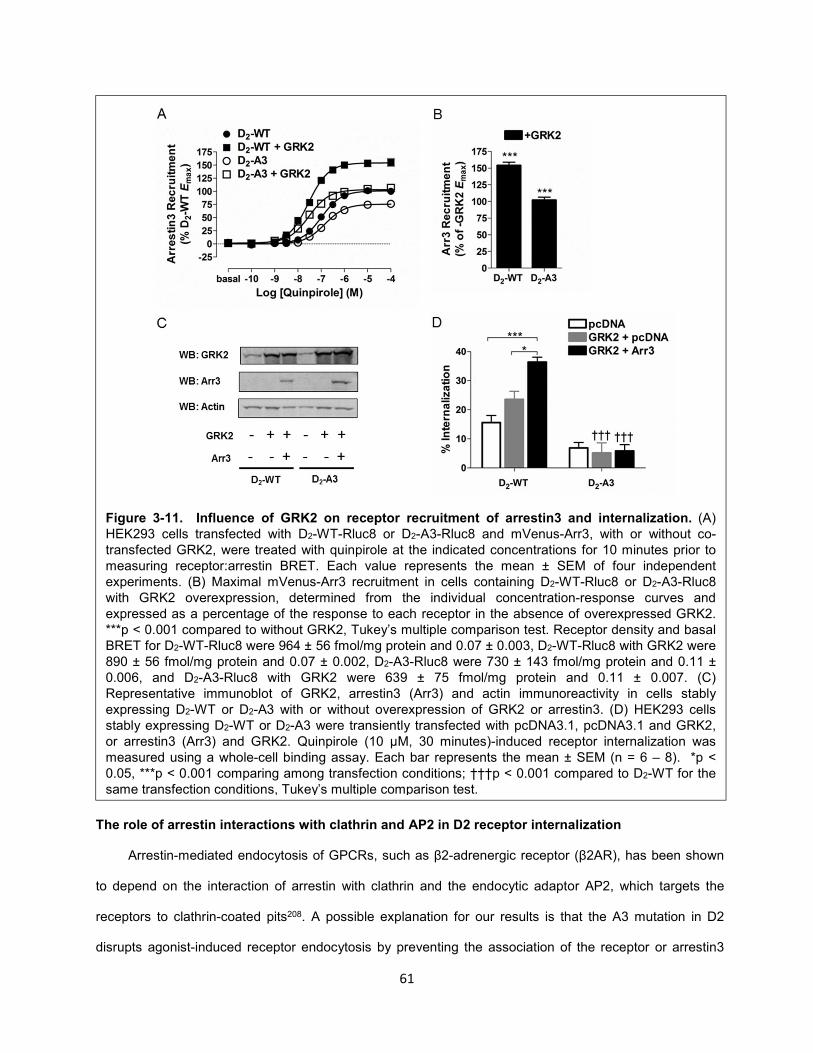
61
The role of arrestin interactions with clathrin and AP2 in D2 receptor internalization
Arrestin-mediated endocytosis of GPCRs, such as β2-adrenergic receptor (β2AR), has been shown
to depend on the interaction of arrestin with clathrin and the endocytic adaptor AP2, which targets the
receptors to clathrin-coated pits208. A possible explanation for our results is that the A3 mutation in D2
disrupts agonist-induced receptor endocytosis by preventing the association of the receptor or arrestin3
Figure 3-11. Influence of GRK2 on receptor recruitment of arrestin3 and internalization. (A) HEK293 cells transfected with D2-WT-Rluc8 or D2-A3-Rluc8 and mVenus-Arr3, with or without co-transfected GRK2, were treated with quinpirole at the indicated concentrations for 10 minutes prior to measuring receptor:arrestin BRET. Each value represents the mean ± SEM of four independent experiments. (B) Maximal mVenus-Arr3 recruitment in cells containing D2-WT-Rluc8 or D2-A3-Rluc8 with GRK2 overexpression, determined from the individual concentration-response curves and expressed as a percentage of the response to each receptor in the absence of overexpressed GRK2. ***p < 0.001 compared to without GRK2, Tukey’s multiple comparison test. Receptor density and basal BRET for D2-WT-Rluc8 were 964 ± 56 fmol/mg protein and 0.07 ± 0.003, D2-WT-Rluc8 with GRK2 were 890 ± 56 fmol/mg protein and 0.07 ± 0.002, D2-A3-Rluc8 were 730 ± 143 fmol/mg protein and 0.11 ± 0.006, and D2-A3-Rluc8 with GRK2 were 639 ± 75 fmol/mg protein and 0.11 ± 0.007. (C) Representative immunoblot of GRK2, arrestin3 (Arr3) and actin immunoreactivity in cells stably expressing D2-WT or D2-A3 with or without overexpression of GRK2 or arrestin3. (D) HEK293 cells stably expressing D2-WT or D2-A3 were transiently transfected with pcDNA3.1, pcDNA3.1 and GRK2, or arrestin3 (Arr3) and GRK2. Quinpirole (10 µM, 30 minutes)-induced receptor internalization was measured using a whole-cell binding assay. Each bar represents the mean ± SEM (n = 6 – 8). *p < 0.05, ***p < 0.001 comparing among transfection conditions; †††p < 0.001 compared to D2-WT for the same transfection conditions, Tukey’s multiple comparison test.

62
with these endocytic proteins. The primary clathrin binding site in arrestin is an LφXφ[D/E] motif209, where
X is any residue and φ is a bulky hydrophobic residue, whereas AP2 binds to a positively-charged arginine
residue; both binding sites are located in the C-terminal region of arrestin210. Dominant negative variants of
arrestin carrying either deletions or mutations of these binding sites are deficient in mediating
internalization and have been used to probe the mechanisms of arrestin-dependent internalization of
GPCRs209-211.
To investigate the role of clathrin and AP2 in arrestin-dependent internalization of the D2 receptor,
HEK293 cells stably expressing D2-WT were transfected with GRK2 and either Arr3-WT, Arr3-ΔLIEFD,
Arr3-R395E, Arr3-ΔLIEFD/R395E, or the control plasmid pcDNA3.1. Incubating cells expressing D2-WT
and Arr3-WT with quinpirole (10 µM, 30 minutes) produced robust internalization of the receptor that was
significantly greater than cells expressing the control plasmid (Figure 3-12). In contrast, none of the 3
arrestin mutants increased D2 receptor
internalization over the amount of internalization
observed in the absence of overexpressed
arrestin, indicating that, indeed, interactions with
both AP2 and clathrin are required for arrestin3-
mediated internalization of the D2 receptor.
β2-Adaptin interaction with the receptor-
arrestin complex
Adaptor protein complexes are essential for
cargo selection and subsequent clathrin-mediated
endocytosis212. For example, agonist stimulation
of some GPCRs has been shown to induce a
direct interaction between arrestin3 and β2-
adaptin to initiate clathrin-mediated receptor
endocytosis213,214, although this has not been
studied in the D2 receptor. We hypothesized that the failure of D2-A3 to internalize in response to
quinpirole may be due to its inability to induce the interaction between arrestin3 and β2-adaptin or to
Figure 3-12. Clathrin- and AP-2- mediated
internalization of D2-WT. HEK293 cells stably expressing D2-WT were transfected with GRK2 and either pcDNA3.1, wild type arrestin3 (Arr3-WT), or arrestin3 mutants: Arr3-ΔLIEFD, Arr3-R395E, or Arr3-ΔLIEFD/R395E. Cells were treated with quinpirole (10 µM) for 30 minutes and internalization was assessed using the whole-cell binding technique. Each bar represents the mean ± SEM of five independent experiments. **p < 0.01, ***p < 0.001 compared to Arr3-WT, Tukey’s multiple comparison test.
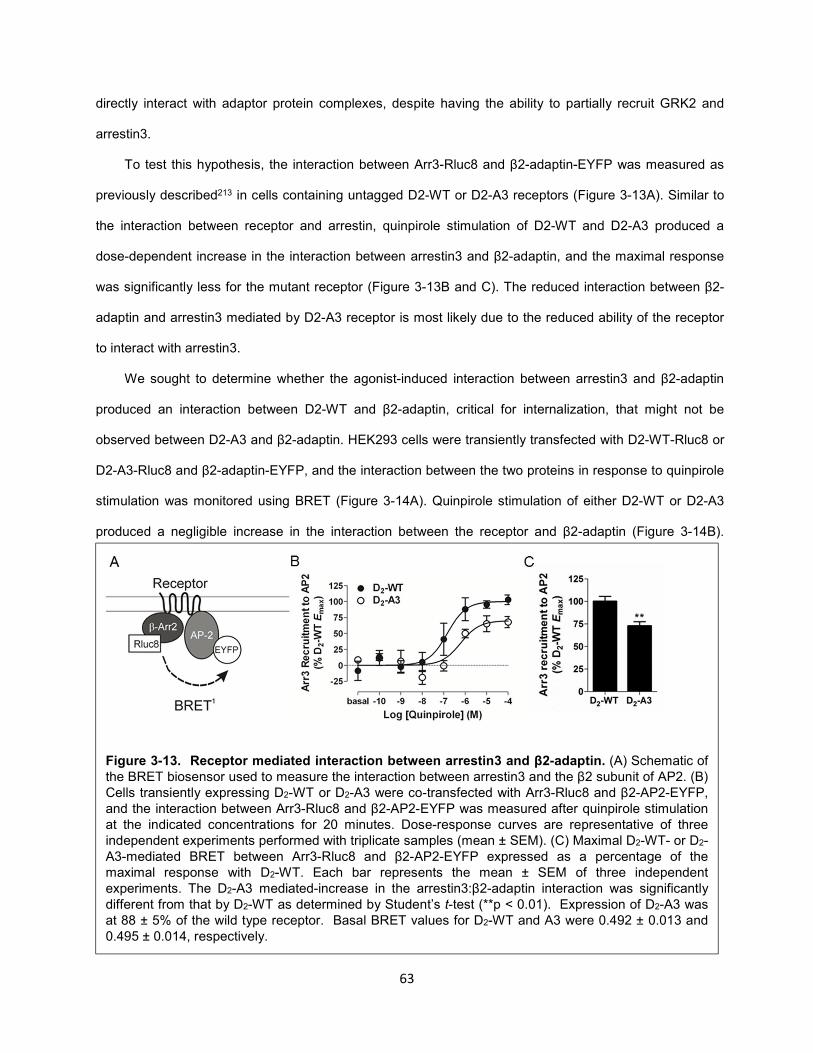
63
directly interact with adaptor protein complexes, despite having the ability to partially recruit GRK2 and
arrestin3.
To test this hypothesis, the interaction between Arr3-Rluc8 and β2-adaptin-EYFP was measured as
previously described213 in cells containing untagged D2-WT or D2-A3 receptors (Figure 3-13A). Similar to
the interaction between receptor and arrestin, quinpirole stimulation of D2-WT and D2-A3 produced a
dose-dependent increase in the interaction between arrestin3 and β2-adaptin, and the maximal response
was significantly less for the mutant receptor (Figure 3-13B and C). The reduced interaction between β2-
adaptin and arrestin3 mediated by D2-A3 receptor is most likely due to the reduced ability of the receptor
to interact with arrestin3.
We sought to determine whether the agonist-induced interaction between arrestin3 and β2-adaptin
produced an interaction between D2-WT and β2-adaptin, critical for internalization, that might not be
observed between D2-A3 and β2-adaptin. HEK293 cells were transiently transfected with D2-WT-Rluc8 or
D2-A3-Rluc8 and β2-adaptin-EYFP, and the interaction between the two proteins in response to quinpirole
stimulation was monitored using BRET (Figure 3-14A). Quinpirole stimulation of either D2-WT or D2-A3
produced a negligible increase in the interaction between the receptor and β2-adaptin (Figure 3-14B).
Figure 3-13. Receptor mediated interaction between arrestin3 and β2-adaptin. (A) Schematic of the BRET biosensor used to measure the interaction between arrestin3 and the β2 subunit of AP2. (B) Cells transiently expressing D2-WT or D2-A3 were co-transfected with Arr3-Rluc8 and β2-AP2-EYFP, and the interaction between Arr3-Rluc8 and β2-AP2-EYFP was measured after quinpirole stimulation at the indicated concentrations for 20 minutes. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± SEM). (C) Maximal D2-WT- or D2-A3-mediated BRET between Arr3-Rluc8 and β2-AP2-EYFP expressed as a percentage of the maximal response with D2-WT. Each bar represents the mean ± SEM of three independent experiments. The D2-A3 mediated-increase in the arrestin3:β2-adaptin interaction was significantly different from that by D2-WT as determined by Student’s t-test (**p < 0.01). Expression of D2-A3 was at 88 ± 5% of the wild type receptor. Basal BRET values for D2-WT and A3 were 0.492 ± 0.013 and 0.495 ± 0.014, respectively.

64
When the receptor-β2-adaptin BRET pair was co-transfected with arrestin3 and GRK2, however, robust
recruitment of β2-adaptin was seen in response to agonist stimulation (Figure 3-14B and D). The BRET
observed was significantly lower for D2-A3 than for the wild type D2 receptor (Figure 3-14C and E). The
reduced response for D2-A3 is again similar in magnitude to the mutant’s reduced ability to recruit
arrestin3 (Figure 3-7C and D) and promote the interaction between arrestin3 and β2-adaptin (Figure 3-14C
and D). These data indicate that D2-A3 has a reduced ability to recruit arrestin3, resulting in a diminished
but still quite substantial ability to bind β2-adaptin.
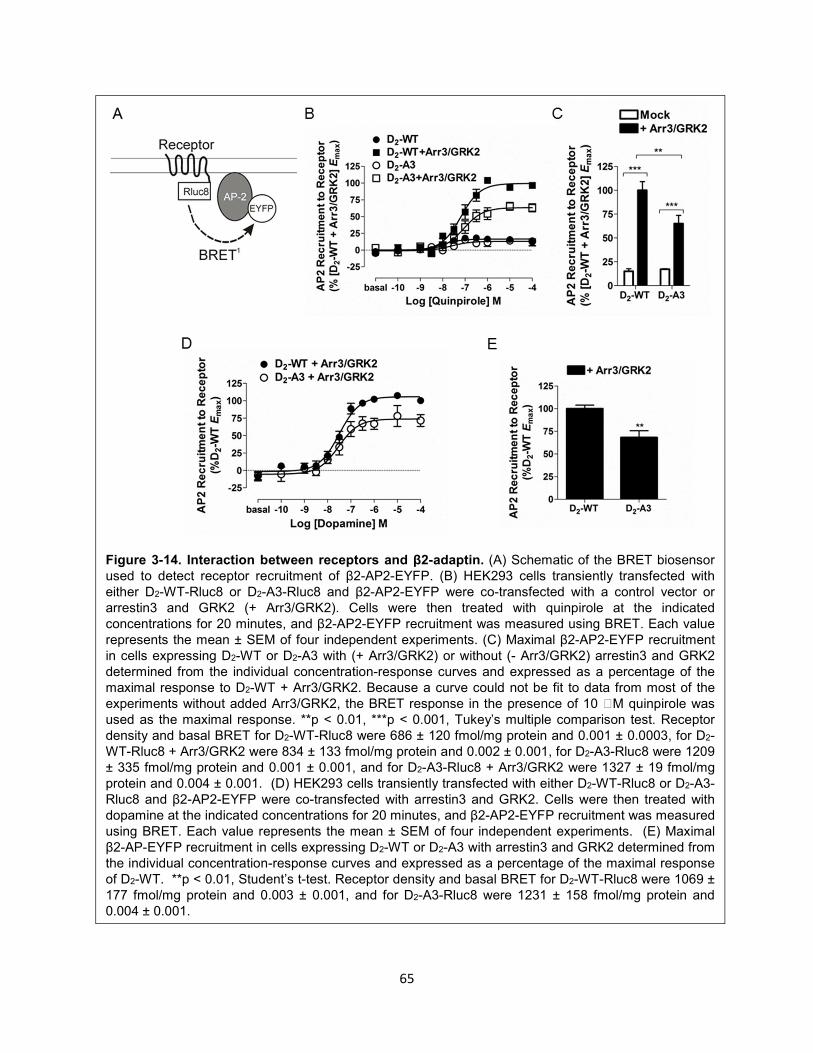
65
Figure 3-14. Interaction between receptors and β2-adaptin. (A) Schematic of the BRET biosensor used to detect receptor recruitment of β2-AP2-EYFP. (B) HEK293 cells transiently transfected with either D2-WT-Rluc8 or D2-A3-Rluc8 and β2-AP2-EYFP were co-transfected with a control vector or arrestin3 and GRK2 (+ Arr3/GRK2). Cells were then treated with quinpirole at the indicated concentrations for 20 minutes, and β2-AP2-EYFP recruitment was measured using BRET. Each value represents the mean ± SEM of four independent experiments. (C) Maximal β2-AP2-EYFP recruitment in cells expressing D2-WT or D2-A3 with (+ Arr3/GRK2) or without (- Arr3/GRK2) arrestin3 and GRK2 determined from the individual concentration-response curves and expressed as a percentage of the maximal response to D2-WT + Arr3/GRK2. Because a curve could not be fit to data from most of the experiments without added Arr3/GRK2, the BRET response in the presence of 10 M quinpirole was used as the maximal response. **p < 0.01, ***p < 0.001, Tukey’s multiple comparison test. Receptor density and basal BRET for D2-WT-Rluc8 were 686 ± 120 fmol/mg protein and 0.001 ± 0.0003, for D2-WT-Rluc8 + Arr3/GRK2 were 834 ± 133 fmol/mg protein and 0.002 ± 0.001, for D2-A3-Rluc8 were 1209 ± 335 fmol/mg protein and 0.001 ± 0.001, and for D2-A3-Rluc8 + Arr3/GRK2 were 1327 ± 19 fmol/mg protein and 0.004 ± 0.001. (D) HEK293 cells transiently transfected with either D2-WT-Rluc8 or D2-A3-Rluc8 and β2-AP2-EYFP were co-transfected with arrestin3 and GRK2. Cells were then treated with dopamine at the indicated concentrations for 20 minutes, and β2-AP2-EYFP recruitment was measured using BRET. Each value represents the mean ± SEM of four independent experiments. (E) Maximal β2-AP-EYFP recruitment in cells expressing D2-WT or D2-A3 with arrestin3 and GRK2 determined from the individual concentration-response curves and expressed as a percentage of the maximal response of D2-WT. **p < 0.01, Student’s t-test. Receptor density and basal BRET for D2-WT-Rluc8 were 1069 ± 177 fmol/mg protein and 0.003 ± 0.001, and for D2-A3-Rluc8 were 1231 ± 158 fmol/mg protein and 0.004 ± 0.001.
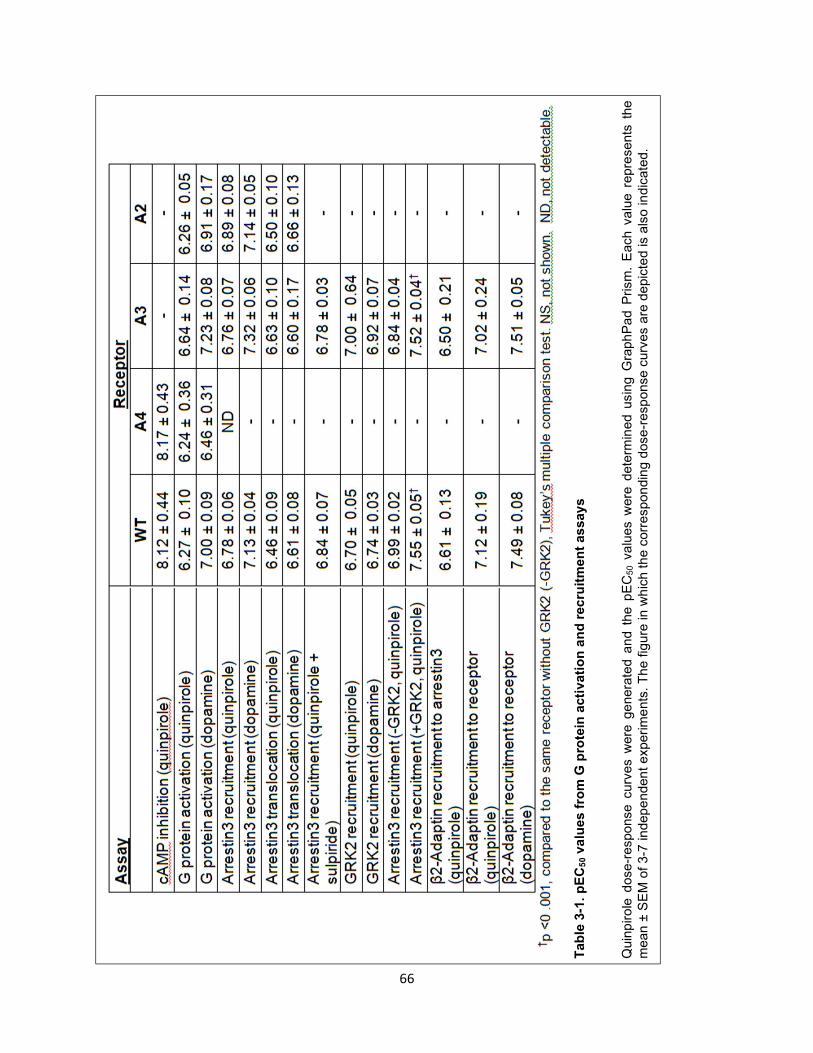
66
Tab
le 3
-1.
pE
C5
0 v
alu
es f
rom
G p
rote
in a
cti
vati
on
an
d r
ec
ruit
men
t as
say
s
Quin
piro
le d
ose-r
esponse
curv
es w
ere
ge
nera
ted a
nd t
he
pE
C5
0 v
alu
es w
ere
dete
rmin
ed u
sin
g G
raph
Pa
d P
rism
. E
ach v
alu
e r
epre
sents
the
m
ean
± S
EM
of 3-7
indep
enden
t experi
me
nts
. T
he fig
ure
in w
hic
h th
e c
orr
espon
din
g d
ose-r
esp
onse c
urv
es a
re d
epic
ted is a
lso in
dic
ate
d.

67
Recruitment of arrestin is not sufficient for the internalization of D2R
A four-residue segment of the D2 receptor, residues 212-215 in IL3, was reported previously to be
required for recruitment of arrestin and receptor internalization, but not for G protein-dependent
signaling215, a finding that we confirmed in the present study using agonist-induced BRET between the D2
receptor and arrestin3 to measure arrestin recruitment. The D2-A4 mutant also demonstrated a
considerable reduction in its ability to activate heterotrimeric G proteins, as determined by measuring the
inhibition of cyclic AMP accumulation and BRET between Gαi1 and mVenus-Gβ1γ2. Thus, we made more
limited mutations in an attempt to selectively impair recruitment of arrestin. Instead, we identified a mutant
that failed to undergo agonist-induced internalization despite retaining a substantial ability to recruit
arrestin3.
Mutants D2-A3 and D2-A2 were created with residues 213-215 or 214-215, respectively, substituted
with alanine. In contrast to D2-A4, both of these mutants had only modestly decreased ability to recruit
arrestin or induce translocation to the membrane, which was more evident with the synthetic agonist
quinpirole than with dopamine, and significantly decreased only for quinpirole at D2-A3-Rluc8. Both of the
mutants were able to fully activate Gαi.
As was reported previously215, the agonist quinpirole was unable to induce internalization of the D2-
A4 receptor, compared to robust internalization of the wild type receptor that was enhanced by
overexpression of arrestin3. Treatment with quinpirole induced partial internalization of D2-A2 that was
commensurate with the partial ability of that mutant receptor to recruit arrestin. In contrast, D2-A3
displayed little or no quinpirole- or dopamine-induced internalization, despite retaining substantial ability to
recruit arrestin.
We evaluated several hypotheses for the disjunction between the ability of D2-A3 to recruit arrestin
and its inability to be internalized. One hypothesis was that, although D2-A3 recruited a substantial amount
of arrestin3 when measured after 10 minutes of agonist treatment, the interaction between D2-A3 and
arrestin3 was less stable than that between the wild type D2 receptor and arrestin3. We determined,
however, that the time course of the recruitment of arrestin3 by the wild type and mutant receptors was
indistinguishable for both dopamine and quinpirole. A second hypothesis was that the interaction between
arrestin3 was taking place somewhere other than at the cell membrane, and therefore would not lead to

68
the receptor being internalized; however, the ability of the hydrophilic antagonist sulpiride to inhibit the
quinpirole-induced response indicated that the response came from receptors at the plasma membrane. A
third hypothesis was that the mutation impaired the ability of GRK2 to bind to the receptor and, indeed, we
determined that the BRET response between the D2 receptor and GRK2 was decreased by the A3
mutation. In vitro data using fragments of the receptor strongly support the hypothesis that the A3/A4
sequence is required for binding of arrestin3 to IL3 of the D2 receptor215; it is, however, unknown if the
same is true for GRK2. Despite the reduction in the agonist-induced BRET response, overexpression of
GRK2 enhanced quinpirole-induced arrestin3 recruitment to D2-A3 by approximately the same percentage
as recruitment of arrestin3 to the wild type D2 receptor, and quinpirole concentration-response curves for
both receptors were shifted to the left in the presence of added GRK2, suggesting that altered binding of
GRK2 to D2-A3 does not underlie its inability to be internalized.
Knockdown of either GRK2 or arrestin3 significantly reduces the amount of agonist-induced D2
receptor internalization216,217, and overexpression of either protein enhances the amount of internalization
seen in response to agonist218-221. Furthermore, this response is potentiated when GRK2 and arrestin3 are
overexpressed together218-220. Thus, it is possible that the reduced internalization of D2-A3 simply reflects
its diminished GRK2 and arrestin3 recruitment, with a partial recruitment deficit somehow translating into a
more severe internalization deficit. However, because overexpression of GRK2 increased the amount of
arrestin3 recruitment by D2-A3 to the level recruited by the wild type receptor, as quantified by the BRET
assay, overexpression of GRK2 would be predicted to increase agonist-induced internalization of D2-A3 to
the same level observed for the wild type receptor in the absence of overexpressed GRK2. This was found
not to be the case, as little or no internalization of D2-A3 was detected even when arrestin3 recruitment
was normalized by GRK2 overexpression.
Arrestin-mediated endocytosis via clathrin-coated pits requires binding of arrestin to both clathrin and
AP2, and deletion of the clathrin binding motif (Arr3-ΔLIEFD) or mutation of the arginine that is necessary
for binding to AP2 (Arr3-R395E) prevents binding to clathrin and AP2, respectively, and greatly impairs
agonist-mediated internalization of the β2AR (35). Using these mutants, we determined that arrestin-
dependent D2 receptor internalization was dependent on the interaction of arrestin3 with both AP2 and

69
clathrin and that preventing either interaction produced complete inhibition of arrestin3-mediated
internalization.
AP2 is a heterotetramer that consists of two large subunits (α2 and β2), a medium subunit (µ2) and a
small subunit (σ2); the β2 (β2-adaptin) and µ2 subunits determine cargo selection by direct interaction with
the cargo or accessory proteins222. Agonist-stimulation of GPCRs induces a direct interaction between
receptor-bound arrestin3 and β2-adaptin to initiate clathrin-mediated receptor endocytosis213,214. We tested
the hypotheses that the A3 mutation within the D2 receptor prevents the receptor from promoting the
interaction between β2-adaptin and arrestin3 or alters an arrestin-mediated interaction between the
receptor and β2-adaptin. Either effect could underlie this receptor’s inability to internalize in response to
quinpirole. Thus, we measured BRET between either arrestin3 or the D2 receptor and β2-adaptin-EYFP.
The quinpirole-induced interaction between arrestin3 and β2-adaptin was reduced but still quite substantial
in cells expressing D2-A3 compared to D2-WT. Further, although a very limited quinpirole-induced change
in BRET was observed when receptor and β2-adaptin were co-expressed, the addition of arrestin3 and
GRK2 enabled a robust quinpirole or dopamine-induced interaction between these proteins. This is the first
time, to our knowledge, that an energy transfer methodology has been used to measure the interaction
between a GPCR and β2-adaptin. The BRET response between D2-A3 and β2-adaptin was also robust,
but significantly less than the response to the wild type receptor. That this closely recapitulated the
mutation-induced decrease in arrestin3 recruitment indicates that it reflects the latter process rather than a
specific inability of D2-A3 to induce an interaction between arrestin3 and β2-adaptin. The extremely low
BRET response between the D2 receptor and β2-adaptin in the absence of overexpressed arrestin and
GRK2 suggests that functional interaction of overexpressed receptor with endogenous arrestins and GRKs
in HEK293 cells is very low; nevertheless, agonist-induced internalization of D2-WT without overexpressed
arrestin/GRK was greater than internalization of D2-A3 with overexpressed arrestin/GRK, when robust
interaction with β2-adaptin was observed. This comparison between the wild type and mutant receptors
highlights the qualitatively distinct inability of D2-A3 to undergo agonist-induced internalization.
In conclusion, D2-A3 is a partially signaling-biased receptor that fully activates G protein but has a
moderately decreased ability to recruit arrestin3 and GRK2. D2-A3 was also deficient in arrestin-
dependent interaction with β2-adaptin, most likely reflecting diminished recruitment of arrestin3 to the

70
receptor. Despite a significant capability for recruitment of arrestin and β2-adaptin, D2-A3 displayed
virtually no agonist-induced internalization even in the presence of overexpressed arrestin3 and GRK2.
Arrestin likely serves as a scaffold to bring the receptor and AP2 sufficiently close to allow BRET between
the C-terminus of the receptor and β2-adaptin. Nonetheless, in the case of D2-A3 the precise conformation
and interactions between these components may not be sufficient for internalization. Furthermore, despite
the proximity between D2-A3 and β2-adaptin, D2-A3 may not be able to directly interact with other
subunits of AP2223,224 or with another protein or lipid required for wild type receptor internalization. The
inability of D2-A3 to internalize may also reflect characteristics of the dynamic interaction among GPCR,
arrestin, and AP2 that are not captured in these BRET assays. Regardless of the molecular mechanism,
the identification of this mutant provides us with a construct suitable for exploring the physiological roles of
D2 receptor internalization in more complex systems and in vivo.
The development of a G protein-biased D2R mutant (Part II)
The D2-A3 mutant is a partially G protein-biased mutant that is attenuated at least in part in mediating
arrestin functions225. However, a fully G protein-biased D2R mutant will be necessary to fully dissociate
the role of G protein from arrestin signaling. An alternative strategy for developing such a mutant involves
the ablation of receptor phosphorylation sites that are involved in facilitating arrestin recruitment. GRK
phosphorylation of serine and threonine residues in GPCRs has been shown to enhance arrestin
recruitment71. The mutation of phosphorylation sites has led to the development of G protein-biased
mutants at a number of GPCRs75,226-228. In some cases, the deletion of the entire receptor C-tail, which
has been reported to be an important site of phosphorylation for some GPCRs, has also resulted in the
creation of G protein-biased receptors227,228.
Several studies have investigated the role of phosphorylation of D2R by GRKs on arrestin
recruitment78,229. Interestingly, these studies mutated a substantial number (but not all) of the potential
phosphorylation sites of D2R and concluded that GRK-mediated phosphorylation of D2R does not play a
major role in arrestin recruitment. However, we found this to be particularly puzzling given that
overexpression GRK2 or GRK6, which are thought to phosphorylate D2R, substantially enhances arrestin
recruitment to D2R (Figure 2-5 and 3-11).
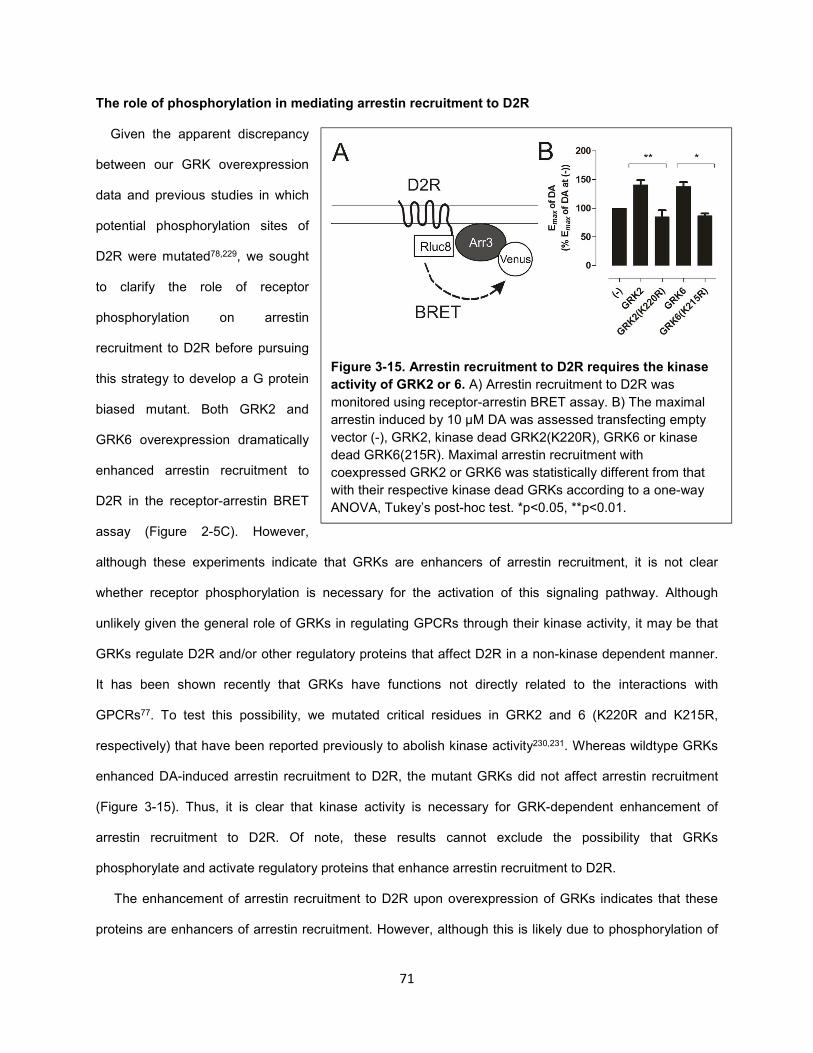
71
The role of phosphorylation in mediating arrestin recruitment to D2R
Given the apparent discrepancy
between our GRK overexpression
data and previous studies in which
potential phosphorylation sites of
D2R were mutated78,229, we sought
to clarify the role of receptor
phosphorylation on arrestin
recruitment to D2R before pursuing
this strategy to develop a G protein
biased mutant. Both GRK2 and
GRK6 overexpression dramatically
enhanced arrestin recruitment to
D2R in the receptor-arrestin BRET
assay (Figure 2-5C). However,
although these experiments indicate that GRKs are enhancers of arrestin recruitment, it is not clear
whether receptor phosphorylation is necessary for the activation of this signaling pathway. Although
unlikely given the general role of GRKs in regulating GPCRs through their kinase activity, it may be that
GRKs regulate D2R and/or other regulatory proteins that affect D2R in a non-kinase dependent manner.
It has been shown recently that GRKs have functions not directly related to the interactions with
GPCRs77. To test this possibility, we mutated critical residues in GRK2 and 6 (K220R and K215R,
respectively) that have been reported previously to abolish kinase activity230,231. Whereas wildtype GRKs
enhanced DA-induced arrestin recruitment to D2R, the mutant GRKs did not affect arrestin recruitment
(Figure 3-15). Thus, it is clear that kinase activity is necessary for GRK-dependent enhancement of
arrestin recruitment to D2R. Of note, these results cannot exclude the possibility that GRKs
phosphorylate and activate regulatory proteins that enhance arrestin recruitment to D2R.
The enhancement of arrestin recruitment to D2R upon overexpression of GRKs indicates that these
proteins are enhancers of arrestin recruitment. However, although this is likely due to phosphorylation of
Figure 3-15. Arrestin recruitment to D2R requires the kinase
activity of GRK2 or 6. A) Arrestin recruitment to D2R was
monitored using receptor-arrestin BRET assay. B) The maximal
arrestin induced by 10 µM DA was assessed transfecting empty
vector (-), GRK2, kinase dead GRK2(K220R), GRK6 or kinase
dead GRK6(215R). Maximal arrestin recruitment with
coexpressed GRK2 or GRK6 was statistically different from that
with their respective kinase dead GRKs according to a one-way
ANOVA, Tukey’s post-hoc test. *p<0.05, **p<0.01.
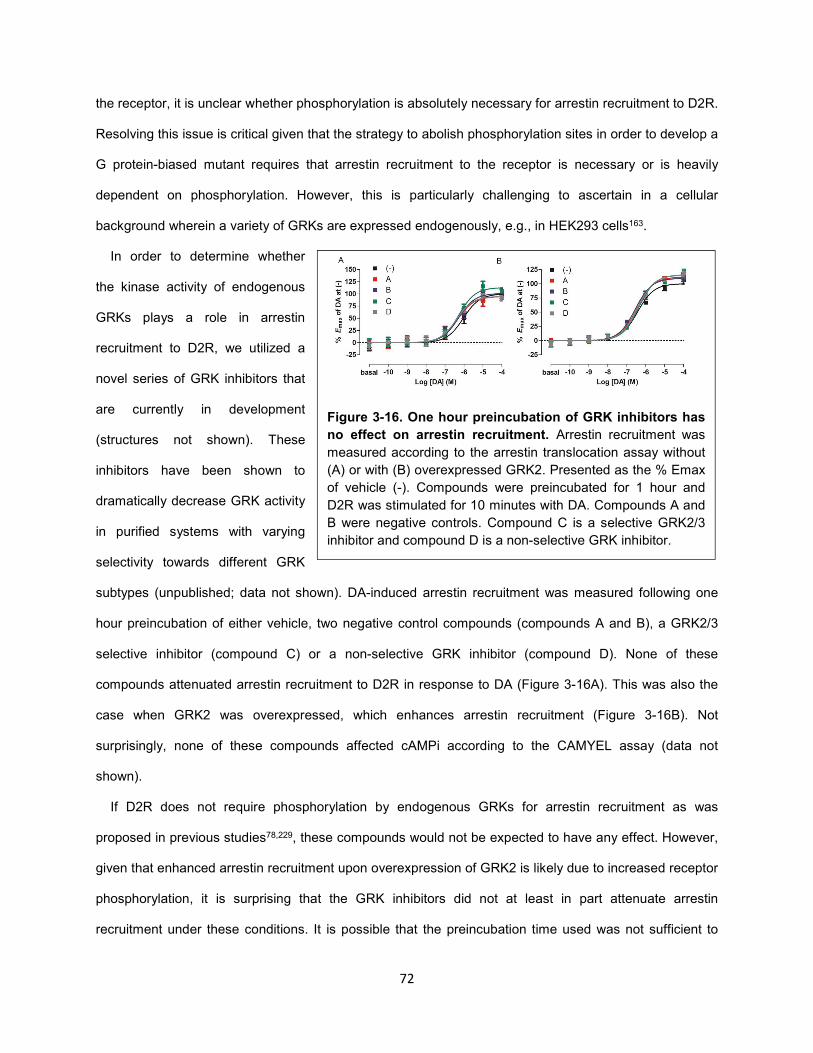
72
the receptor, it is unclear whether phosphorylation is absolutely necessary for arrestin recruitment to D2R.
Resolving this issue is critical given that the strategy to abolish phosphorylation sites in order to develop a
G protein-biased mutant requires that arrestin recruitment to the receptor is necessary or is heavily
dependent on phosphorylation. However, this is particularly challenging to ascertain in a cellular
background wherein a variety of GRKs are expressed endogenously, e.g., in HEK293 cells163.
In order to determine whether
the kinase activity of endogenous
GRKs plays a role in arrestin
recruitment to D2R, we utilized a
novel series of GRK inhibitors that
are currently in development
(structures not shown). These
inhibitors have been shown to
dramatically decrease GRK activity
in purified systems with varying
selectivity towards different GRK
subtypes (unpublished; data not shown). DA-induced arrestin recruitment was measured following one
hour preincubation of either vehicle, two negative control compounds (compounds A and B), a GRK2/3
selective inhibitor (compound C) or a non-selective GRK inhibitor (compound D). None of these
compounds attenuated arrestin recruitment to D2R in response to DA (Figure 3-16A). This was also the
case when GRK2 was overexpressed, which enhances arrestin recruitment (Figure 3-16B). Not
surprisingly, none of these compounds affected cAMPi according to the CAMYEL assay (data not
shown).
If D2R does not require phosphorylation by endogenous GRKs for arrestin recruitment as was
proposed in previous studies78,229, these compounds would not be expected to have any effect. However,
given that enhanced arrestin recruitment upon overexpression of GRK2 is likely due to increased receptor
phosphorylation, it is surprising that the GRK inhibitors did not at least in part attenuate arrestin
recruitment under these conditions. It is possible that the preincubation time used was not sufficient to
Figure 3-16. One hour preincubation of GRK inhibitors has
no effect on arrestin recruitment. Arrestin recruitment was
measured according to the arrestin translocation assay without
(A) or with (B) overexpressed GRK2. Presented as the % Emax
of vehicle (-). Compounds were preincubated for 1 hour and
D2R was stimulated for 10 minutes with DA. Compounds A and
B were negative controls. Compound C is a selective GRK2/3
inhibitor and compound D is a non-selective GRK inhibitor.

73
block endogenous GRK2/3. Thus, the inhibitors were preincubated during the entire time course of the
transient transfection (48 hours). However, even under these conditions, these compounds failed to
inhibit arrestin recruitment (data not shown).
An alternative strategy to
blocking GRKs pharmacologically is
to knockout or knockdown
genetically various GRKs. This
approach is challenging given that
there are seven GRK subtypes,
many of which have been reported
to be expressed in HEK293 cells163.
One study used this approach to
investigate the role of GRKs on
arrestin recruitment to β2AR232. Although substantial knockdown of multiple GRKs was achieved, there
was no effect on maximal arrestin recruitment. However, GRK knockdown was shown to negatively affect
the kinetics of arrestin to β2AR. We repeated the experiments using the GRK inhibitors (one hour
pretreatment) but instead monitored kinetically arrestin recruitment to D2R and β2AR (Figure 3-17A and
B, respectively). However, there was no effect of the inhibitor D, the non-selective GRK inhibitor, on
arrestin recruitment over the entire time course.
One possible explanation for these results is that arrestin recruitment to D2R does not require
phosphorylation. However, that arrestin recruitment to β2AR was not inhibited using these conditions but
was shown previously to be negatively affected by GRK knockdown232 indicates that these compounds
may not be suitable for this purpose. These compounds were tested initially in purified systems and may
not be sufficiently membrane permeant to inhibit GRKs. Initial experiments performed in culture systems
to validate these compounds did not directly monitor their effect on arrestin recruitment to GPCRs, but
instead measured an endpoint substantially downstream of receptor activation. Thus, it is possible that
these compounds bind and affect off-target proteins unrelated to GRK phosphorylation of and arrestin
recruitment to GPCRs. Another possibility is that they inhibit GRK activity in cells, but to a degree that is
Figure 3-17. Time course of arrestin recruitment to D2R and
β2AR in the presence of a non-selective GRK inhibitor.
Arrestin recruitment was measured according to the arrestin
translocation assay to D2R (A) or β2AR (B) without (-) or with
compound D, the non-selective GRK inhibitor. Presented as the
% maximal response at (-). Compound D was preincubated for
1 hour. D2R and β2AR were stimulated with 10 μM DA and
isoproterenol, respectively.

74
not sufficient to substantially inhibit phosphorylation of GPCRs. Regardless, these experiments as well
those in which genetic techniques were used to inhibit GRK activity highlight the difficulty in determining
the role of endogenous GRK activity in phosphorylating GPCRs and enhancing arrestin recruitment.
Truncating IL3 of D2R does not create a G protein-biased mutant
Although it is unclear if GRK-mediated phosphorylation of D2R is necessary for arrestin recruitment, it
likely plays a stimulatory role. Therefore, we continued to pursue the strategy of mutating or truncating
various components of D2R to create a G protein-biased receptor. Most of the potential phosphorylation
sites in D2R are in its IL3 rather than its C-tail10. As discussed above, several studies have investigated
the role of phosphorylation of D2R on arrestin recruitment78,229. In one study, a subset of the potential
phosphorylation sites in the IL3 of D2R were mutated to residues that would prevent phosphorylation (13
serines (S) and threonines (T) were mutated to alanines (A) and valines (V), respectively)219. According to
a radiolabeled phosphate-incorporation assay, mutating these residues was sufficient to abolish agonist-
induced phosphorylation of the receptor. Interestingly, according the receptor-arrestin BRET assay, this
D2R mutant not only recruited arrestin but did so in a manner greater and with higher potency than the
wildtype receptor. In another study, all of the S/T residues in both IL2 and IL3 of D2R were mutated to
A/V. This mutant also recruited arrestin, although this was not measured quantitatively relative to wildtype
D2R229. Thus, both of these studies concluded that receptor phosphorylation is not necessary for arrestin
recruitment.
An alternative explanation for these results is that mutation of IL residues from S/T residues to A/V not
only impairs phosphorylation of these residues, but also alters the structure of the receptor in a manner
that facilitates arrestin recruitment. To address this possibility, we sought to truncate IL3 entirely from the
receptor. This region of the receptor has the highest number of potential phosphorylation sites and was
reported to be the critical site of agonist-induced phosphorylation78. To obtain a more accurate structural
basis for the residues constituting IL3 of D2R, we modeled D2R based on the crystal structure of D3R38
and found IL3 to span from residues 230 to 330. The two terminal residues of TM5 (amino acids 228-229)
were found to be serines, which are potential phosphorylation sites and were exposed to solvent in the
simulations, and thus potentially accessible to kinases. Using molecular dynamics simulations, we
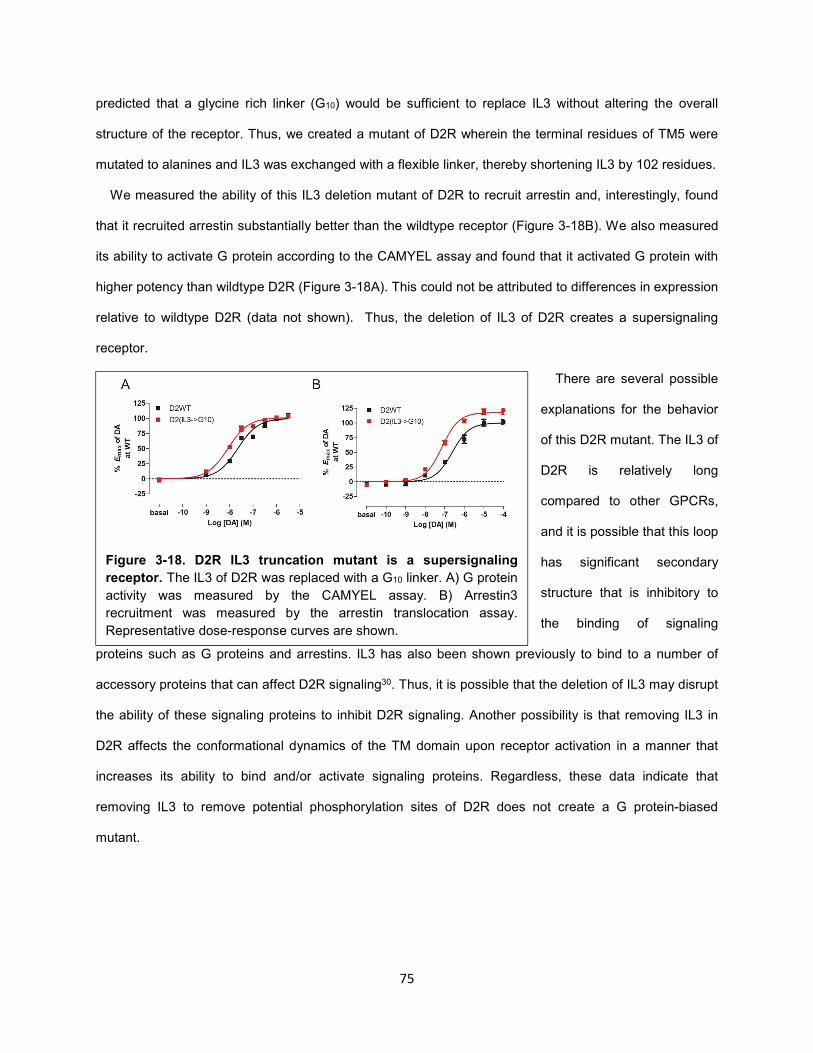
75
predicted that a glycine rich linker (G10) would be sufficient to replace IL3 without altering the overall
structure of the receptor. Thus, we created a mutant of D2R wherein the terminal residues of TM5 were
mutated to alanines and IL3 was exchanged with a flexible linker, thereby shortening IL3 by 102 residues.
We measured the ability of this IL3 deletion mutant of D2R to recruit arrestin and, interestingly, found
that it recruited arrestin substantially better than the wildtype receptor (Figure 3-18B). We also measured
its ability to activate G protein according to the CAMYEL assay and found that it activated G protein with
higher potency than wildtype D2R (Figure 3-18A). This could not be attributed to differences in expression
relative to wildtype D2R (data not shown). Thus, the deletion of IL3 of D2R creates a supersignaling
receptor.
There are several possible
explanations for the behavior
of this D2R mutant. The IL3 of
D2R is relatively long
compared to other GPCRs,
and it is possible that this loop
has significant secondary
structure that is inhibitory to
the binding of signaling
proteins such as G proteins and arrestins. IL3 has also been shown previously to bind to a number of
accessory proteins that can affect D2R signaling30. Thus, it is possible that the deletion of IL3 may disrupt
the ability of these signaling proteins to inhibit D2R signaling. Another possibility is that removing IL3 in
D2R affects the conformational dynamics of the TM domain upon receptor activation in a manner that
increases its ability to bind and/or activate signaling proteins. Regardless, these data indicate that
removing IL3 to remove potential phosphorylation sites of D2R does not create a G protein-biased
mutant.
Figure 3-18. D2R IL3 truncation mutant is a supersignaling
receptor. The IL3 of D2R was replaced with a G10 linker. A) G protein
activity was measured by the CAMYEL assay. B) Arrestin3
recruitment was measured by the arrestin translocation assay.
Representative dose-response curves are shown.
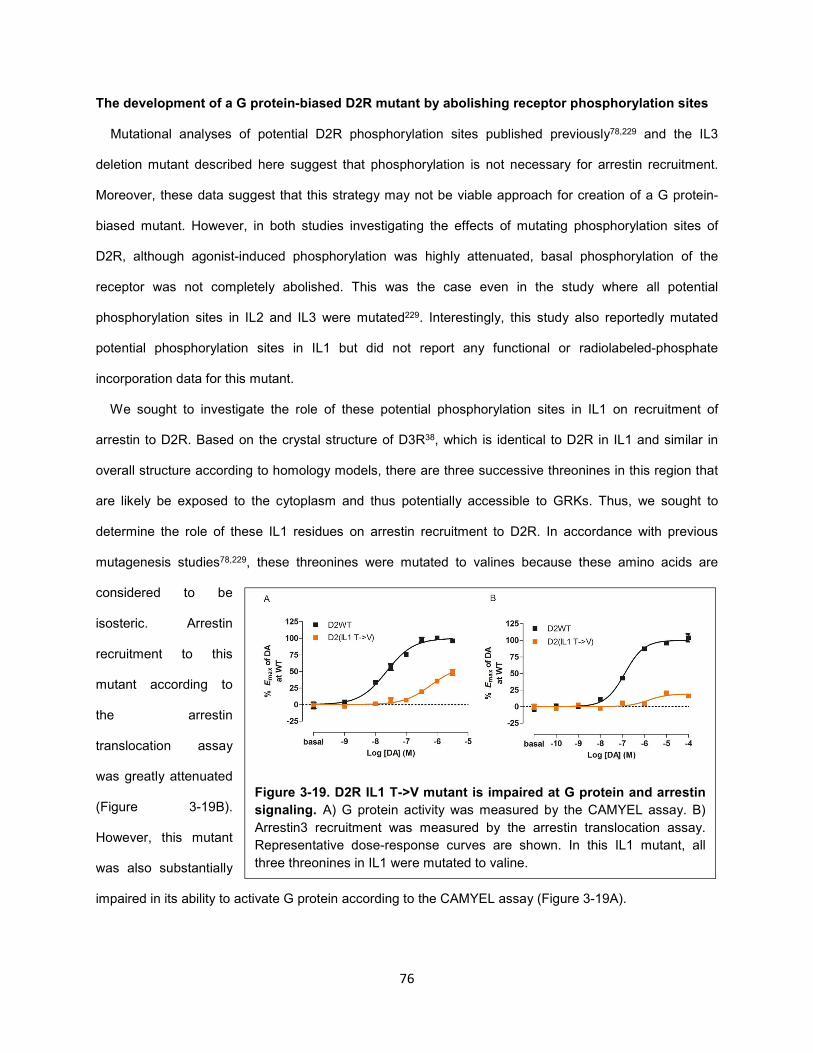
76
The development of a G protein-biased D2R mutant by abolishing receptor phosphorylation sites
Mutational analyses of potential D2R phosphorylation sites published previously78,229 and the IL3
deletion mutant described here suggest that phosphorylation is not necessary for arrestin recruitment.
Moreover, these data suggest that this strategy may not be viable approach for creation of a G protein-
biased mutant. However, in both studies investigating the effects of mutating phosphorylation sites of
D2R, although agonist-induced phosphorylation was highly attenuated, basal phosphorylation of the
receptor was not completely abolished. This was the case even in the study where all potential
phosphorylation sites in IL2 and IL3 were mutated229. Interestingly, this study also reportedly mutated
potential phosphorylation sites in IL1 but did not report any functional or radiolabeled-phosphate
incorporation data for this mutant.
We sought to investigate the role of these potential phosphorylation sites in IL1 on recruitment of
arrestin to D2R. Based on the crystal structure of D3R38, which is identical to D2R in IL1 and similar in
overall structure according to homology models, there are three successive threonines in this region that
are likely be exposed to the cytoplasm and thus potentially accessible to GRKs. Thus, we sought to
determine the role of these IL1 residues on arrestin recruitment to D2R. In accordance with previous
mutagenesis studies78,229, these threonines were mutated to valines because these amino acids are
considered to be
isosteric. Arrestin
recruitment to this
mutant according to
the arrestin
translocation assay
was greatly attenuated
(Figure 3-19B).
However, this mutant
was also substantially
impaired in its ability to activate G protein according to the CAMYEL assay (Figure 3-19A).
Figure 3-19. D2R IL1 T->V mutant is impaired at G protein and arrestin
signaling. A) G protein activity was measured by the CAMYEL assay. B)
Arrestin3 recruitment was measured by the arrestin translocation assay.
Representative dose-response curves are shown. In this IL1 mutant, all
three threonines in IL1 were mutated to valine.

77
It is possible that the receptor is sensitive to the mutation of these IL1 residues given that they are
located so close to the plasma membrane. Therefore, we mutated these threonines to either glycines (G),
alanines (A) or cysteines (C) rather than valines. Mutants carrying either T to G or C were impaired in
their expression relative to wildtype D2R (~30%; data not shown). In contrast, the T to C mutant, D2R(IL1
T->C), expressed similarly to wildtype. Interestingly, whereas its ability to activate G protein was not
impaired, its arrestin recruitment was attenuated (~64% of that at wildtype; Figure 3-20A).
In an effort to bias the receptor further towards G protein, we investigated whether this modification in
IL1 would confer additional G protein bias when combined with possible phosphorylation site mutations in
other ILs of D2R. However, we first probed the effect of mutating these sites in IL2 or IL3 alone. As in
previous studies78,229, S/T residues were mutated to A/V in these regions. Interestingly, the mutation of all
potential phosphorylation sites in IL2 did not substantially affect either G protein or arrestin recruitment
(Figure 3-20A). However, whereas arrestin recruitment to the IL3 mutant was impaired relative to wildtype
D2R (~30%), G protein activation was not affected (Figure 3-20A).
The results seen for the IL3 mutant are partially inconsistent with previous studies, in particular that in
which mutation of 13 potential phosphorylation sites in IL3 resulted in increased arrestin recruitment
compared to wildtype D2R78. One possible difference between these two studies is the species of D2R
used (in that study rat D2R was used instead of human D2R used here). We generated a mutant of
human D2R wherein we mutated 13 residues homologous to those mutated previously in the rat D2R.
Arrestin recruitment to this mutant was similar to wildtype (data not shown), which was inconsistent with
the previous study that found that arrestin recruitment to the rat D2R mutant was elevated. Regardless,
these data indicate that additional mutations in IL3 beyond these 13 residues are required to impair
arrestin recruitment to D2R.
We next evaluated G protein and arrestin activation by receptors with all potential phosphorylation sites
mutated in two ILs. All receptors had wildtype-like levels of G protein activation (Figure 3-20A).
Interestingly, in the combined IL1:IL2 mutant, there was no loss in arrestin recruitment as seen in the IL1
alone mutant (Figure 3-20A). The decrease in arrestin recruitment to the IL1 or IL3 mutants alone were
approximately additive when combined (~60% decrease in arrestin recruitment to IL1:IL3 relative to
wildtype D2R). Surprisingly, although 1) mutating phosphorylation sites in IL2 alone had no effect on

78
arrestin recruitment relative to wildtype and 2) that in IL3 led to a decrease of ~30%, the combined IL2:IL3
mutant was greatly impaired in arrestin recruitment (70% decrease relative to wildtype) (Figure 3-20A).
This is suggestive of potentially complex interactions between IL2 and IL3.
We combined mutations in all ILs (IL1:IL2:IL3). This mutant exhibited extreme G protein bias. Arrestin
recruitment to this mutant was ~10% of that for wildtype D2R, whereas G protein activation of this mutant
was substantially higher than expected (~180% of wildtype) (Figure 3-20-C). Increased G protein
activation may be due to the loss of arrestin-mediated termination of G protein signaling. However, we
cannot exclude the possibility that these combined mutations positively affect G protein binding and
activation. Regardless, the IL1:IL2:IL3 mutant exhibits unprecedented G protein bias. Therefore, we are
currently generating an AAV encoding this receptor mutant to investigate the role of G protein in
mediating psychostimulant-dependent behaviors in rodents in a manner analogous to that described in
Chapter 2.
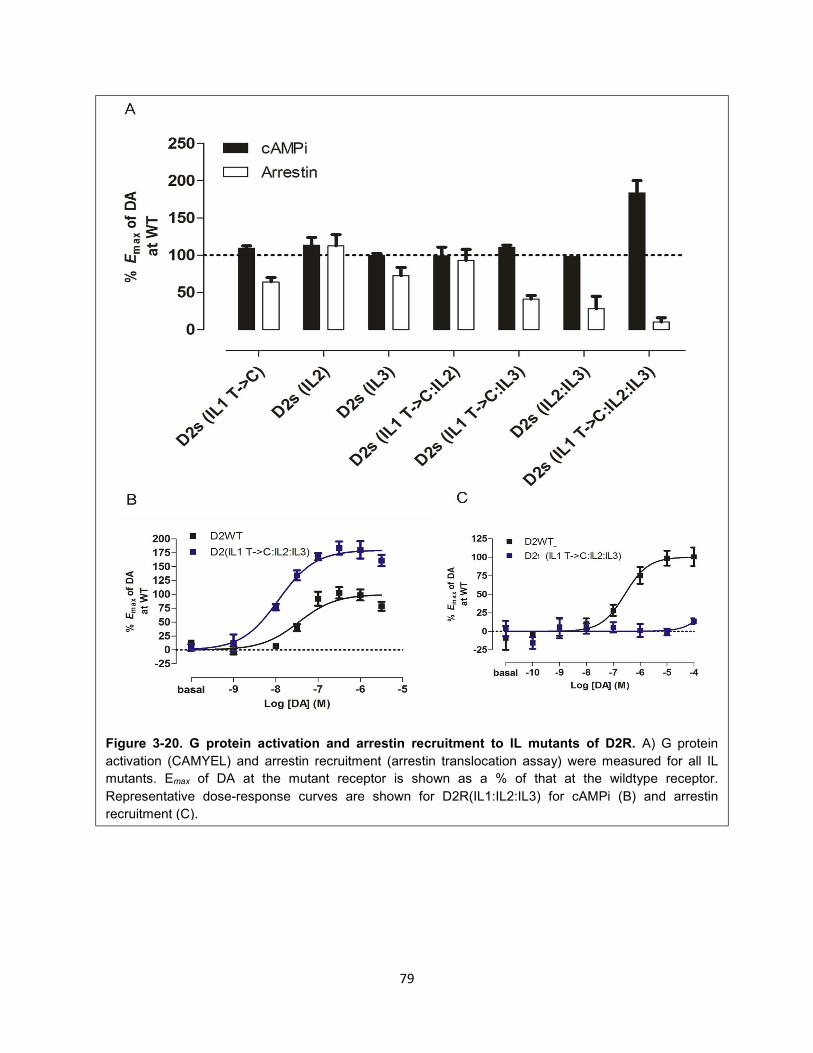
79
Figure 3-20. G protein activation and arrestin recruitment to IL mutants of D2R. A) G protein
activation (CAMYEL) and arrestin recruitment (arrestin translocation assay) were measured for all IL
mutants. Emax of DA at the mutant receptor is shown as a % of that at the wildtype receptor.
Representative dose-response curves are shown for D2R(IL1:IL2:IL3) for cAMPi (B) and arrestin
recruitment (C).

80
Conclusions and future directions
It has been proposed that antipsychotic medications that specifically block arrestin but spare G protein-
mediated pathways may be superior antipsychotic medications. However, G protein activation
downstream of D2R may play a role in facilitating psychostimulant-dependent behaviors in rodents
according DARPP-32 knockout studies suggesting that blockade of G protein signaling might also be
necessary. That this protein is substantially downstream of G protein activation prompted us to develop
tools to probe the role of this signaling pathway downstream of D2R in a less ambiguous manner.
We are now currently generating genetically-encoded AAV vectors for delivery of the catalytically active
subunit of PTX S1 to distinct cell-types, specifically D2R-expression neurons. This toxin is capable of
inactivating Gi/o proteins by ADP-ribosylation of a cysteine residue on the C-terminus of these G proteins.
Notably, according to studies described above (Figure 2-5), the inactivation of G proteins may result in a
partial decrease in the ability of D2R to recruit arrestins depending on if arrestin recruitment is dependent
of GRK2/3 activity, e.g., in dopaminergic neurons. However, this will likely not be the case in MSNs where
GRK6 rather than GRK2 plays a major role in the regulation of D2R. S1 is also incapable of inactivating
Gz proteins, which have been shown to be involved in mediating D2R signaling based on knockout
studies. Regardless, such an approach allows for the first time to potentially knockout a substantial
component of G protein signaling downstream of D2R in specific cell-types. Unlike arrestins expressed in
D2R-expressing neurons, which are encoded by two genes, G proteins are encoded by five genes. Thus,
using traditional knockout strategies are not feasible.
As with knockout strategies generally, S1 of PTX will negatively affect G protein signaling from other
GPCR coexpressed with D2R. In an attempt to avoid this confound, we developed two biased D2Rs. By
ablating all potential phosphorylation sites of D2R, we created a receptor that is highly biased towards G
protein and is incapable of recruiting arrestin. This receptor is complementary to the arrestin-biased D2R
described in Chapter 2, and will be useful in differentiating between G protein and arrestin-mediated
processes, especially with respect to psychostimulant-dependent behaviors. We also developed a
partially biased D2R with G protein activation and arrestin recruitment intact but agonist-induced
internalization impaired. Although agonist-induced internalization is an arrestin-mediated process, the
recruitment of arrestin to the receptor is not sufficient to facilitate this process. It remains to be determined

81
if other arrestin-mediated processes are intact, e.g., Akt signaling. If this is the case, it will be interesting
to probe the activity of this biased receptor in vivo, given that the completely G protein or arrestin-biased
receptors are incapable of differentiating between distinct processes downstream of a single signaling
protein like arrestin (or G protein; see Chapter 4).

82
CHAPTER 4. Towards understanding functional selectivity between G protein-dependent
pathways downstream of D2R
Aripiprazole (Abilify) is a highly clinically relevant compound due to its therapeutic efficacy and reduced
side effects for treatment of the positive symptoms of schizophrenia. It has been reported as the first D2R
partial agonist approved clinically as an antipsychotic medication. This is in contrast to all other approved
antipsychotics, which are antagonists of D2R. However, the failure of a number of other partial agonists in
clinical trials has led to a reevaluation of molecular mechanism of action of aripiprazole.
Recent studies have indicated that this ligand is functionally selective or biased, and it has been
proposed that this may underlie its superior clinical profile103,111,130. Unlike classical agonists and
antagonists that activate or block, respectively, all pathways downstream of a receptor, aripiprazole is a
mixed D2R agonist/antagonist at a number of different endpoints in animals as well as in in vitro model
systems. The identification of how this behavior relates to the clinical efficacy of aripiprazole as well as
mechanisms by which aripiprazole selectively activates distinct D2R-mediated pathways may provide
insights into the development of superior antipsychotic medications.
Interestingly, whereas
aripiprazole has been
reported to be a partial
agonist for the inhibition of
cAMP downstream of
D2R103,111,130, it was reported
to be an antagonist111 or
agonist140 for activation of
GIRK channels in heterologous systems using different cell types (Figure 4-1). As discussed above, the
most highly cited mechanism of functional selectivity is the selective activation of G proteins and arrestins,
which have been shown to couple to different pathways in a receptor-dependent manner123. However, the
ability of aripiprazole to differentially mediate cAMP inhibition (cAMPi) and GIRK activation is paradoxical
given that both of these pathways are thought to be G protein- but not arrestin-mediated D2R pathways.
Figure 4-1. Aripiprazole differentially activates G protein-mediated
pathways downstream of D2R. Whereas DA inhibits cAMP
accumulation (cAMPi) and activates GIRKs downstream of D2R (left),
aripiprazole facilitates cAMPi but activates or inhibits GIRK activity
depending on the cell-type.
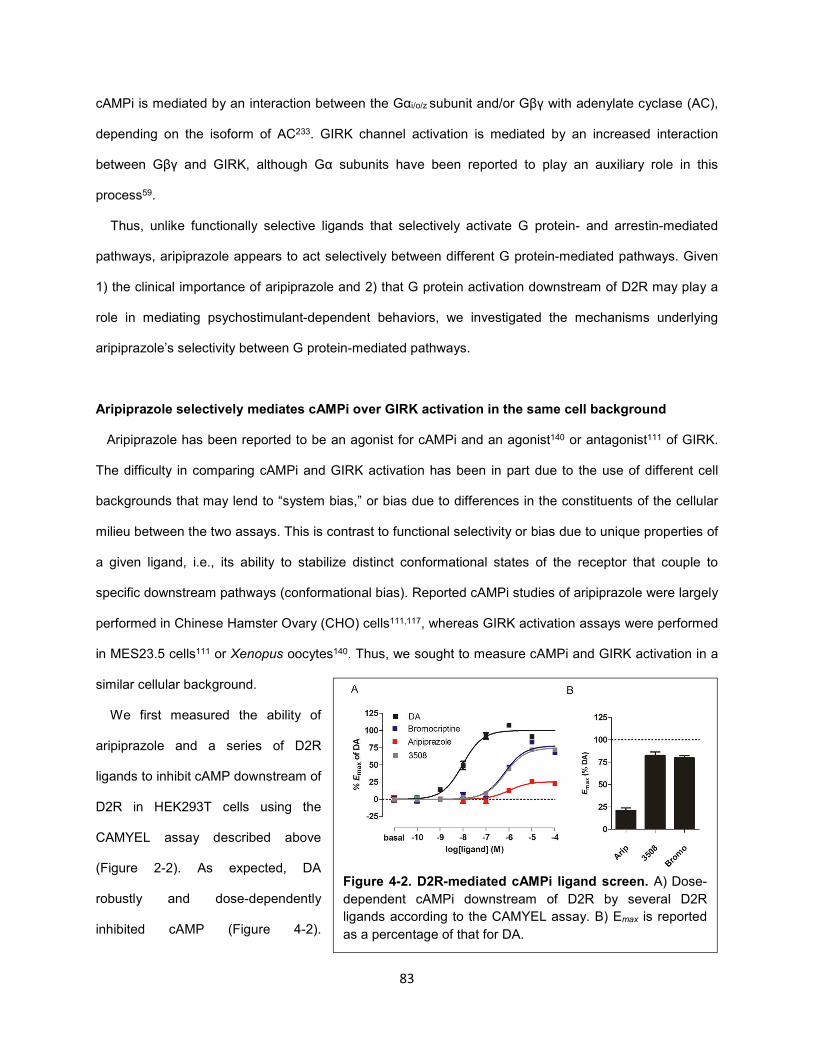
83
cAMPi is mediated by an interaction between the Gαi/o/z subunit and/or Gβγ with adenylate cyclase (AC),
depending on the isoform of AC233. GIRK channel activation is mediated by an increased interaction
between Gβγ and GIRK, although Gα subunits have been reported to play an auxiliary role in this
process59.
Thus, unlike functionally selective ligands that selectively activate G protein- and arrestin-mediated
pathways, aripiprazole appears to act selectively between different G protein-mediated pathways. Given
1) the clinical importance of aripiprazole and 2) that G protein activation downstream of D2R may play a
role in mediating psychostimulant-dependent behaviors, we investigated the mechanisms underlying
aripiprazole’s selectivity between G protein-mediated pathways.
Aripiprazole selectively mediates cAMPi over GIRK activation in the same cell background
Aripiprazole has been reported to be an agonist for cAMPi and an agonist140 or antagonist111 of GIRK.
The difficulty in comparing cAMPi and GIRK activation has been in part due to the use of different cell
backgrounds that may lend to “system bias,” or bias due to differences in the constituents of the cellular
milieu between the two assays. This is contrast to functional selectivity or bias due to unique properties of
a given ligand, i.e., its ability to stabilize distinct conformational states of the receptor that couple to
specific downstream pathways (conformational bias). Reported cAMPi studies of aripiprazole were largely
performed in Chinese Hamster Ovary (CHO) cells111,117, whereas GIRK activation assays were performed
in MES23.5 cells111 or Xenopus oocytes140. Thus, we sought to measure cAMPi and GIRK activation in a
similar cellular background.
We first measured the ability of
aripiprazole and a series of D2R
ligands to inhibit cAMP downstream of
D2R in HEK293T cells using the
CAMYEL assay described above
(Figure 2-2). As expected, DA
robustly and dose-dependently
inhibited cAMP (Figure 4-2).
Figure 4-2. D2R-mediated cAMPi ligand screen. A) Dose-
dependent cAMPi downstream of D2R by several D2R
ligands according to the CAMYEL assay. B) Emax is reported
as a percentage of that for DA.
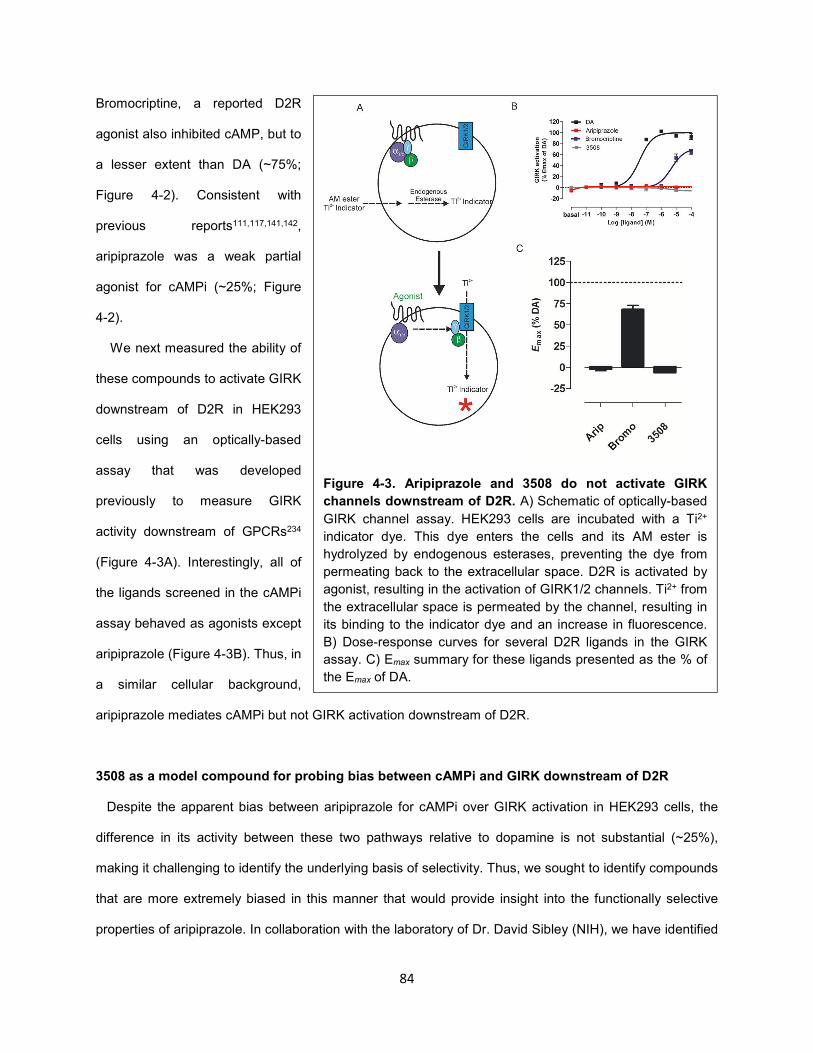
84
Bromocriptine, a reported D2R
agonist also inhibited cAMP, but to
a lesser extent than DA (~75%;
Figure 4-2). Consistent with
previous reports111,117,141,142,
aripiprazole was a weak partial
agonist for cAMPi (~25%; Figure
4-2).
We next measured the ability of
these compounds to activate GIRK
downstream of D2R in HEK293
cells using an optically-based
assay that was developed
previously to measure GIRK
activity downstream of GPCRs234
(Figure 4-3A). Interestingly, all of
the ligands screened in the cAMPi
assay behaved as agonists except
aripiprazole (Figure 4-3B). Thus, in
a similar cellular background,
aripiprazole mediates cAMPi but not GIRK activation downstream of D2R.
3508 as a model compound for probing bias between cAMPi and GIRK downstream of D2R
Despite the apparent bias between aripiprazole for cAMPi over GIRK activation in HEK293 cells, the
difference in its activity between these two pathways relative to dopamine is not substantial (~25%),
making it challenging to identify the underlying basis of selectivity. Thus, we sought to identify compounds
that are more extremely biased in this manner that would provide insight into the functionally selective
properties of aripiprazole. In collaboration with the laboratory of Dr. David Sibley (NIH), we have identified
Figure 4-3. Aripiprazole and 3508 do not activate GIRK
channels downstream of D2R. A) Schematic of optically-based
GIRK channel assay. HEK293 cells are incubated with a Ti2+
indicator dye. This dye enters the cells and its AM ester is
hydrolyzed by endogenous esterases, preventing the dye from
permeating back to the extracellular space. D2R is activated by
agonist, resulting in the activation of GIRK1/2 channels. Ti2+ from
the extracellular space is permeated by the channel, resulting in
its binding to the indicator dye and an increase in fluorescence.
B) Dose-response curves for several D2R ligands in the GIRK
assay. C) Emax summary for these ligands presented as the % of
the Emax of DA.

85
a number of compounds with unique pharmacological phenotypes, some of which are functionally
selective147,235. We found one such compound, 3508, to be highly biased towards inhibition of cAMPi over
GIRK activation (unpublished; Figure 4-3A and B). This compound inhibits cAMP similarly to
bromocriptine but does not activate GIRK channels, thus displaying more extreme bias than aripiprazole
for cAMPi over GIRK activation. Of note, we found that DA and 3508 act as an agonist and antagonist,
respectively, of GIRK channels in dopaminergic neurons from the VTA (unpublished; data not shown),
highlighting that our heterologous system for measuring GIRK activity displays physiological relevance.
The apparent functionally selective properties of aripiprazole but not 3508 are due to differences
in assay sensitivity
One possible explanation for the difference in the abilities of aripiprazole and 3508 to differentially
inhibit cAMP and activate GIRK channels downstream of D2R is differential assay sensitivity, or so-called
observational bias. Although these two assays were performed in the same cell background, several
factors can differentially affect assay sensitivity, including differences in the levels of D2R or the strength
of coupling between the D2R and downstream assay components, i.e., stimulus-response coupling. It is
critical to distinguish this type of bias from conformational bias, which is dependent on the unique ability
of a given ligand to stabilize distinct sets of active receptor states rather the general differences between
assays that uniformly affect the activity of all receptor ligands.
Several approaches have been developed to distinguish conformational bias from observational bias.
One such widely used approach relies on the Black-Leff operational model of agonism and has been
described in detail131,133. In order to factor out differences in sensitivity in assessing functional selectivity,
this approach normalizes the effect of a test ligand to a reference ligand (typically the endogenous ligand)
that is assumed to be unbiased at two distinct pathways. Dose-response curves for the test agonist and a
reference agonist are fitted to the operational model, which measures the affinity (KA-1) for the receptor
and efficacy (τ) in activating a particular signaling pathway. For each pathway, a "transduction coefficient"
term, log(τ/KA), is calculated for each compound. The relative efficiency of the test agonist to activate a
given pathway is quantified by the difference in log(τ/KA) between the test and reference agonists, namely
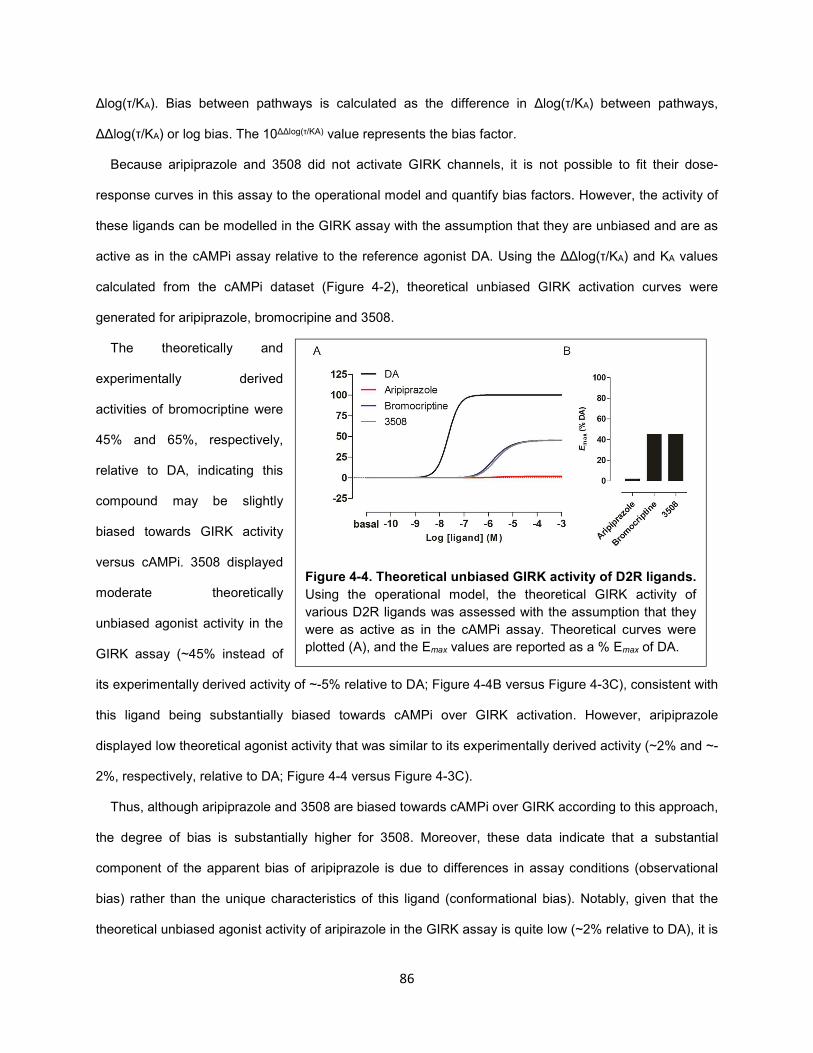
86
Δlog(τ/KA). Bias between pathways is calculated as the difference in Δlog(τ/KA) between pathways,
ΔΔlog(τ/KA) or log bias. The 10ΔΔlog(τ/KA) value represents the bias factor.
Because aripiprazole and 3508 did not activate GIRK channels, it is not possible to fit their dose-
response curves in this assay to the operational model and quantify bias factors. However, the activity of
these ligands can be modelled in the GIRK assay with the assumption that they are unbiased and are as
active as in the cAMPi assay relative to the reference agonist DA. Using the ΔΔlog(τ/KA) and KA values
calculated from the cAMPi dataset (Figure 4-2), theoretical unbiased GIRK activation curves were
generated for aripiprazole, bromocripine and 3508.
The theoretically and
experimentally derived
activities of bromocriptine were
45% and 65%, respectively,
relative to DA, indicating this
compound may be slightly
biased towards GIRK activity
versus cAMPi. 3508 displayed
moderate theoretically
unbiased agonist activity in the
GIRK assay (~45% instead of
its experimentally derived activity of ~-5% relative to DA; Figure 4-4B versus Figure 4-3C), consistent with
this ligand being substantially biased towards cAMPi over GIRK activation. However, aripiprazole
displayed low theoretical agonist activity that was similar to its experimentally derived activity (~2% and ~-
2%, respectively, relative to DA; Figure 4-4 versus Figure 4-3C).
Thus, although aripiprazole and 3508 are biased towards cAMPi over GIRK according to this approach,
the degree of bias is substantially higher for 3508. Moreover, these data indicate that a substantial
component of the apparent bias of aripiprazole is due to differences in assay conditions (observational
bias) rather than the unique characteristics of this ligand (conformational bias). Notably, given that the
theoretical unbiased agonist activity of aripirazole in the GIRK assay is quite low (~2% relative to DA), it is
Figure 4-4. Theoretical unbiased GIRK activity of D2R ligands.
Using the operational model, the theoretical GIRK activity of
various D2R ligands was assessed with the assumption that they
were as active as in the cAMPi assay. Theoretical curves were
plotted (A), and the Emax values are reported as a % Emax of DA.

87
possible that its agonist activity was not detected experimentally in the GIRK assay. These data may
underlie the apparent discrepancy in the literature, wherein aripiprazole was reported to be both an
agonist and antagonist of GIRK activation downstream of D2R111,140. Specifically, the study reporting
agonist activity of aripiprazole140 may have utilized a GIRK assay with sufficiently high sensitivity to detect
agonist activity of weak partial agonists like aripiprazole.
The functionally selective properties of 3508 cannot be attributed to G protein versus arrestin bias
Although the apparent bias of aripiprazole is largely due to observational bias, the analysis described
above (Figure 4-4) indicates that 3508 may display substantial conformational bias. Thus, we sought to
investigate further the unprecedented functional selectivity of this compound. The most highly cited
mechanism underlying functional selectivity is the ability of ligands to selectively activate G protein or
arrestin. It is unlikely that the functionally selective behavior of 3508 towards cAMPi can be attributed to G
protein or arrestin bias given that cAMPi and GIRK activation are G protein-mediated pathways and are
not thought to be directly modulated by arrestin. However, it is possible that arrestin can indirectly and
differentially modulate these pathways by scaffolding associated regulatory proteins. Thus, we measured
the ability of this ligand to activate G protein and recruit arrestin.
We first measured the ability of a series of D2R ligands to induce a conformational change between or
dissociation of the heterotrimeric G proteins using a BRET-based assay in which the Gαi1 subunit is
labeled with the donor Rluc8 and the Gβ1γ2 subunits are labeled with the split acceptor mVenus (Figure 4-
5A). As expected, all ligands tested behaved as agonists, including 3508 (Figure 4-5C). We next tested
the ability of these compounds to recruit arrestin using the BRET-based receptor-arrestin recruitment
assay (Figure 4-5B). As in the G protein activation assay, all ligands acted as agonists (Figure 4-5C).
Moreover, these ligands acted with similar efficacy relative to DA between the two pathways (Figure 4-
5C). These data indicate that 3508 does not differentially activate G protein and arrestin, and thus this
type of behavior likely does not underlie the ability of 3508 to selectivity mediate cAMPi over GIRK
activation.
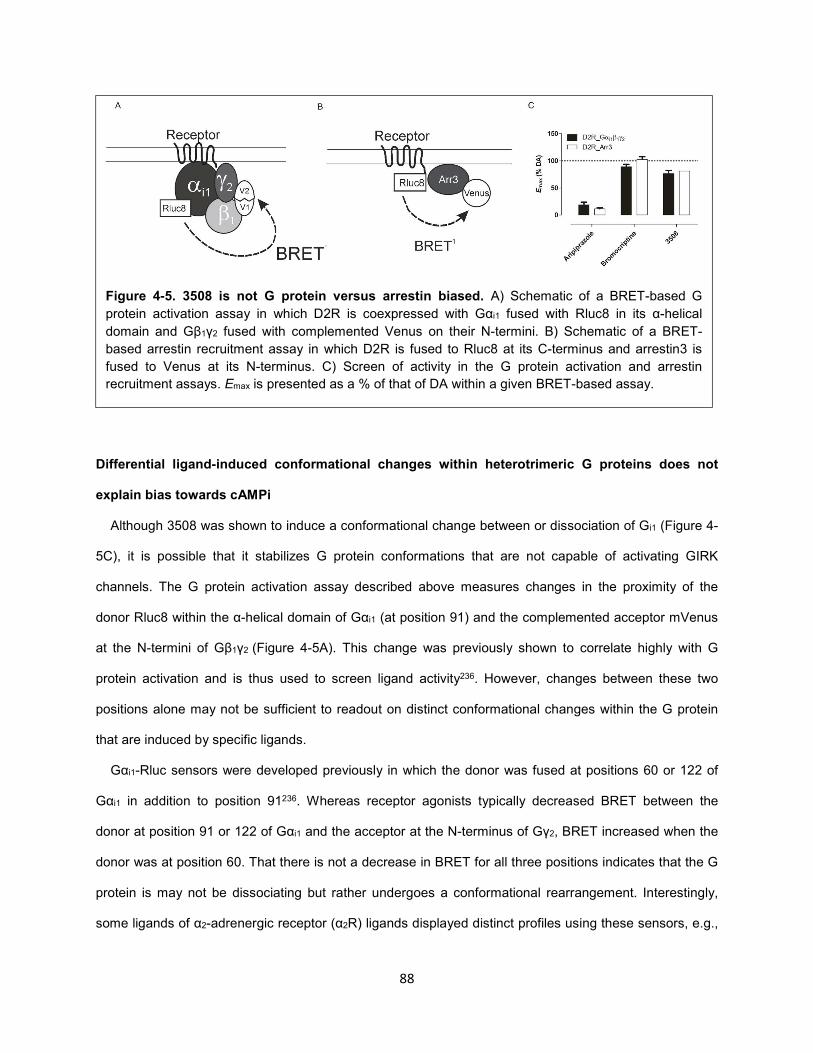
88
Differential ligand-induced conformational changes within heterotrimeric G proteins does not
explain bias towards cAMPi
Although 3508 was shown to induce a conformational change between or dissociation of Gi1 (Figure 4-
5C), it is possible that it stabilizes G protein conformations that are not capable of activating GIRK
channels. The G protein activation assay described above measures changes in the proximity of the
donor Rluc8 within the α-helical domain of Gαi1 (at position 91) and the complemented acceptor mVenus
at the N-termini of Gβ1γ2 (Figure 4-5A). This change was previously shown to correlate highly with G
protein activation and is thus used to screen ligand activity236. However, changes between these two
positions alone may not be sufficient to readout on distinct conformational changes within the G protein
that are induced by specific ligands.
Gαi1-Rluc sensors were developed previously in which the donor was fused at positions 60 or 122 of
Gαi1 in addition to position 91236. Whereas receptor agonists typically decreased BRET between the
donor at position 91 or 122 of Gαi1 and the acceptor at the N-terminus of Gγ2, BRET increased when the
donor was at position 60. That there is not a decrease in BRET for all three positions indicates that the G
protein is may not be dissociating but rather undergoes a conformational rearrangement. Interestingly,
some ligands of α2-adrenergic receptor (α2R) ligands displayed distinct profiles using these sensors, e.g.,
Figure 4-5. 3508 is not G protein versus arrestin biased. A) Schematic of a BRET-based G
protein activation assay in which D2R is coexpressed with Gαi1 fused with Rluc8 in its α-helical
domain and Gβ1γ2 fused with complemented Venus on their N-termini. B) Schematic of a BRET-
based arrestin recruitment assay in which D2R is fused to Rluc8 at its C-terminus and arrestin3 is
fused to Venus at its N-terminus. C) Screen of activity in the G protein activation and arrestin
recruitment assays. Emax is presented as a % of that of DA within a given BRET-based assay.
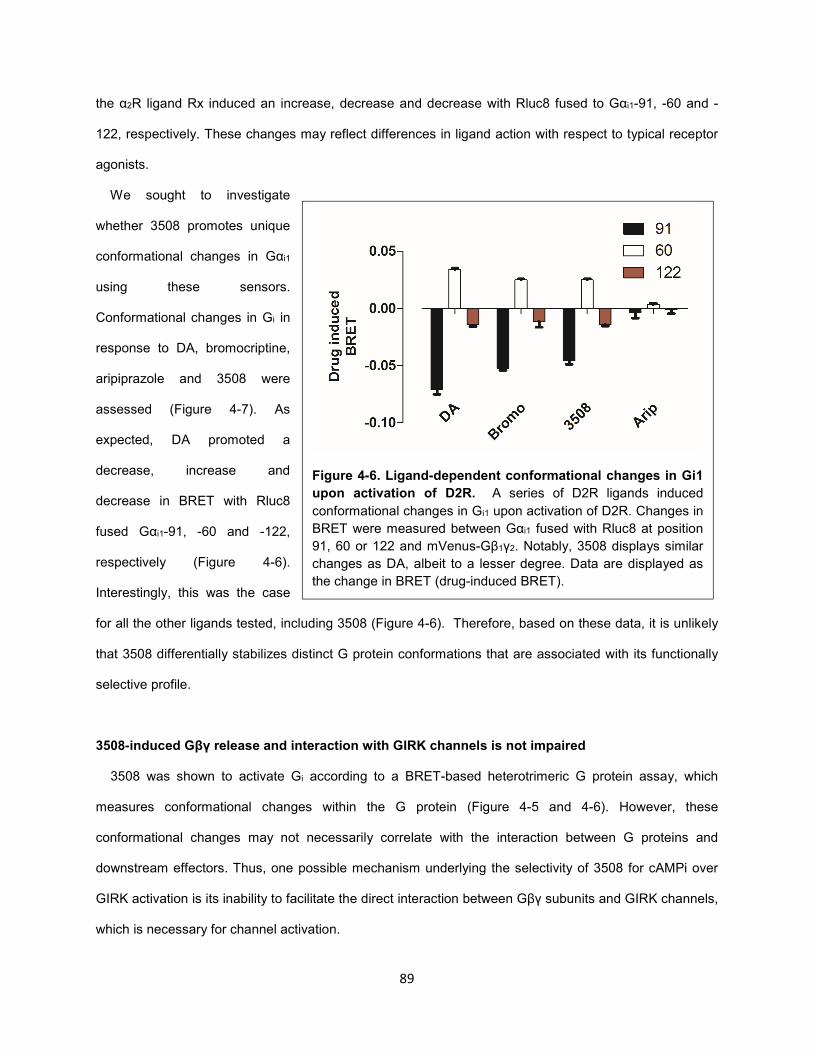
89
the α2R ligand Rx induced an increase, decrease and decrease with Rluc8 fused to Gαi1-91, -60 and -
122, respectively. These changes may reflect differences in ligand action with respect to typical receptor
agonists.
We sought to investigate
whether 3508 promotes unique
conformational changes in Gαi1
using these sensors.
Conformational changes in Gi in
response to DA, bromocriptine,
aripiprazole and 3508 were
assessed (Figure 4-7). As
expected, DA promoted a
decrease, increase and
decrease in BRET with Rluc8
fused Gαi1-91, -60 and -122,
respectively (Figure 4-6).
Interestingly, this was the case
for all the other ligands tested, including 3508 (Figure 4-6). Therefore, based on these data, it is unlikely
that 3508 differentially stabilizes distinct G protein conformations that are associated with its functionally
selective profile.
3508-induced Gβγ release and interaction with GIRK channels is not impaired
3508 was shown to activate Gi according to a BRET-based heterotrimeric G protein assay, which
measures conformational changes within the G protein (Figure 4-5 and 4-6). However, these
conformational changes may not necessarily correlate with the interaction between G proteins and
downstream effectors. Thus, one possible mechanism underlying the selectivity of 3508 for cAMPi over
GIRK activation is its inability to facilitate the direct interaction between Gβγ subunits and GIRK channels,
which is necessary for channel activation.
Figure 4-6. Ligand-dependent conformational changes in Gi1
upon activation of D2R. A series of D2R ligands induced
conformational changes in Gi1 upon activation of D2R. Changes in
BRET were measured between Gαi1 fused with Rluc8 at position
91, 60 or 122 and mVenus-Gβ1γ2. Notably, 3508 displays similar
changes as DA, albeit to a lesser degree. Data are displayed as
the change in BRET (drug-induced BRET).
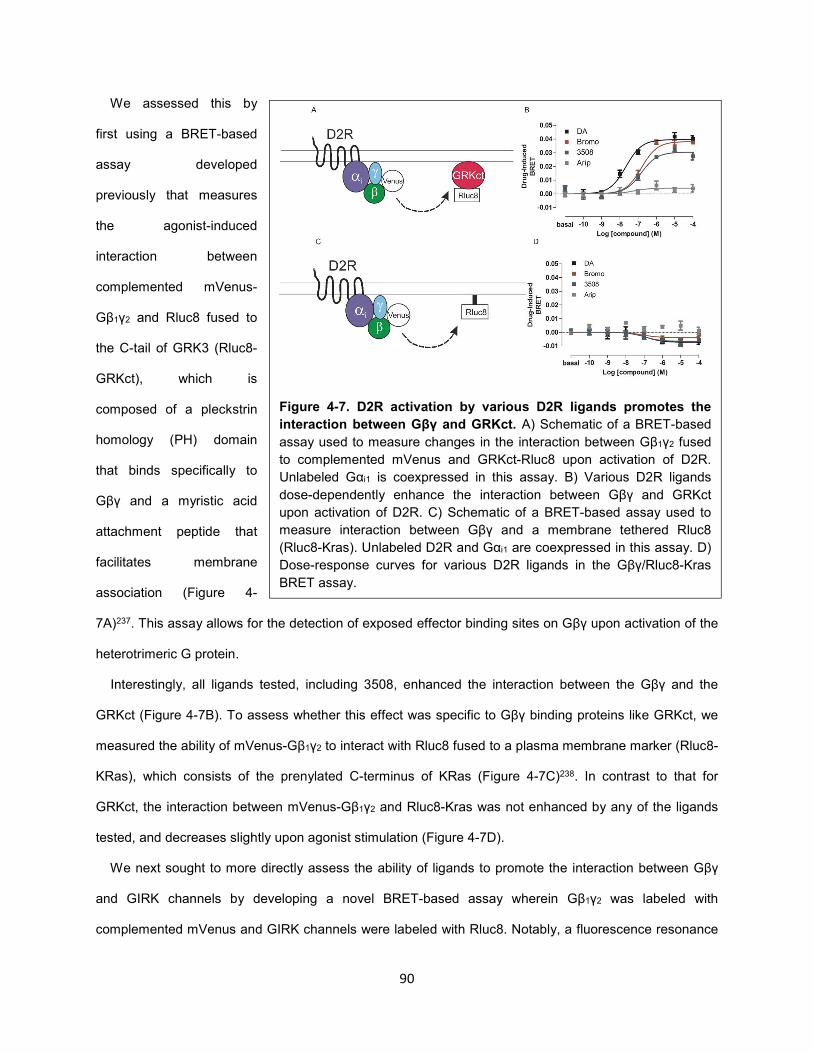
90
We assessed this by
first using a BRET-based
assay developed
previously that measures
the agonist-induced
interaction between
complemented mVenus-
Gβ1γ2 and Rluc8 fused to
the C-tail of GRK3 (Rluc8-
GRKct), which is
composed of a pleckstrin
homology (PH) domain
that binds specifically to
Gβγ and a myristic acid
attachment peptide that
facilitates membrane
association (Figure 4-
7A)237. This assay allows for the detection of exposed effector binding sites on Gβγ upon activation of the
heterotrimeric G protein.
Interestingly, all ligands tested, including 3508, enhanced the interaction between the Gβγ and the
GRKct (Figure 4-7B). To assess whether this effect was specific to Gβγ binding proteins like GRKct, we
measured the ability of mVenus-Gβ1γ2 to interact with Rluc8 fused to a plasma membrane marker (Rluc8-
KRas), which consists of the prenylated C-terminus of KRas (Figure 4-7C)238. In contrast to that for
GRKct, the interaction between mVenus-Gβ1γ2 and Rluc8-Kras was not enhanced by any of the ligands
tested, and decreases slightly upon agonist stimulation (Figure 4-7D).
We next sought to more directly assess the ability of ligands to promote the interaction between Gβγ
and GIRK channels by developing a novel BRET-based assay wherein Gβ1γ2 was labeled with
complemented mVenus and GIRK channels were labeled with Rluc8. Notably, a fluorescence resonance
Figure 4-7. D2R activation by various D2R ligands promotes the
interaction between Gβγ and GRKct. A) Schematic of a BRET-based
assay used to measure changes in the interaction between Gβ1γ2 fused
to complemented mVenus and GRKct-Rluc8 upon activation of D2R.
Unlabeled Gαi1 is coexpressed in this assay. B) Various D2R ligands
dose-dependently enhance the interaction between Gβγ and GRKct
upon activation of D2R. C) Schematic of a BRET-based assay used to
measure interaction between Gβγ and a membrane tethered Rluc8
(Rluc8-Kras). Unlabeled D2R and Gαi1 are coexpressed in this assay. D)
Dose-response curves for various D2R ligands in the Gβγ/Rluc8-Kras
BRET assay.

91
energy transfer (FRET)-based assay was developed previously that measures interactions between Gβγ
and GIRKs239. Mammals express four GIRK channel subunits, GIRK1-4240. These channels are homo- or
heterotetrameric proteins that are composed of either all GIRK2, GIRK3 or GIRK4 subunits, or any of
these subunits complexed with GIRK1. Dopaminergic neurons that express D2 autoreceptors have been
shown to be express both GIRK2 homotetramers and GIRK1/2 heterotetramers241,242, and more recently
GIRK2/3243. We sought to develop sensors that could be used to detect interactions between GIRK2 or
GIRK1/2 channels and Gβγ (Figure 4-8A).
GIRK channel subunits were shown previously to tolerate fusion proteins on their N- and C-termini,
which are facing intracellularly239. Thus, we fused Rluc8 to either termini of GIRK1 and GIRK2. Because
GIRK1 has been reported to require GIRK2 to form a functional channel, we coexpressed D2R with both
GIRK1 fusion proteins, unfused GIRK2 and complemented mVenus-Gβ1γ2 (Figure 4-8B). DA enhanced
the interaction between mVenus-Gβ1γ2 and Rluc8-GIRK1/GIRK2 but not GIRK1-Rluc8/GIRK2 (Figure 4-
8B). However, DA enhanced the interaction of between mVenus-Gβ1γ2 and both Rluc8-GIRK2 and
GIRK2-Rluc8 when coexpressed with unfused GIRK1 (Figure 4-8B). The greatest DA-induced BRET was
seen for the BRET assay involving GIRK2-Rluc8/GIRK1.
Interestingly, DA did not substantially enhance the interaction between mVenus-Gβ1γ2 and Rluc8-
GIRK2 expressed without unfused GIRK1 (Figure 4-8B). We hypothesized that four Rluc8 proteins within
a single GIRK homotetrameric channel may interfere with Gβ1γ2 binding. Thus, we coexpressed fused
channels with unfused GIRK2 channels. Under these conditions, DA-induced BRET increased relative to
GIRK2-Rluc8 expressed alone, albeit to a much lower level than in the GIRK2-Rluc8/GIRK1 assay
(Figure 4-8B). These data indicate that the placement of the donor Rluc8 on the GIRK subunit and the
stoichiometry of GIRK channels to Rluc8 has a substantial effect on the degree of DA-induced BRET.
We next screened the ability of a series of D2R ligands to enhance the interaction between Gβ1γ2 and
GIRK1/2. All compounds tested, including 3508, behaved as agonists at all GIRK1/2 channel
combinations (Figure 4-8C). Interactions between Gβ1γ2 and GIRK2 have not yet been screened.
Although 3508 enhances the interaction between Gβ1γ2 and GIRK1/2, it is possible that the binding mode
of the Gβ1γ2 on GIRK1/2 promoted by this ligand may not permit channel activation. However, 3508
enhanced uniformly the interaction between Venus-Gβ1γ2 and GIRK1/2 channels fused with Rluc8 at
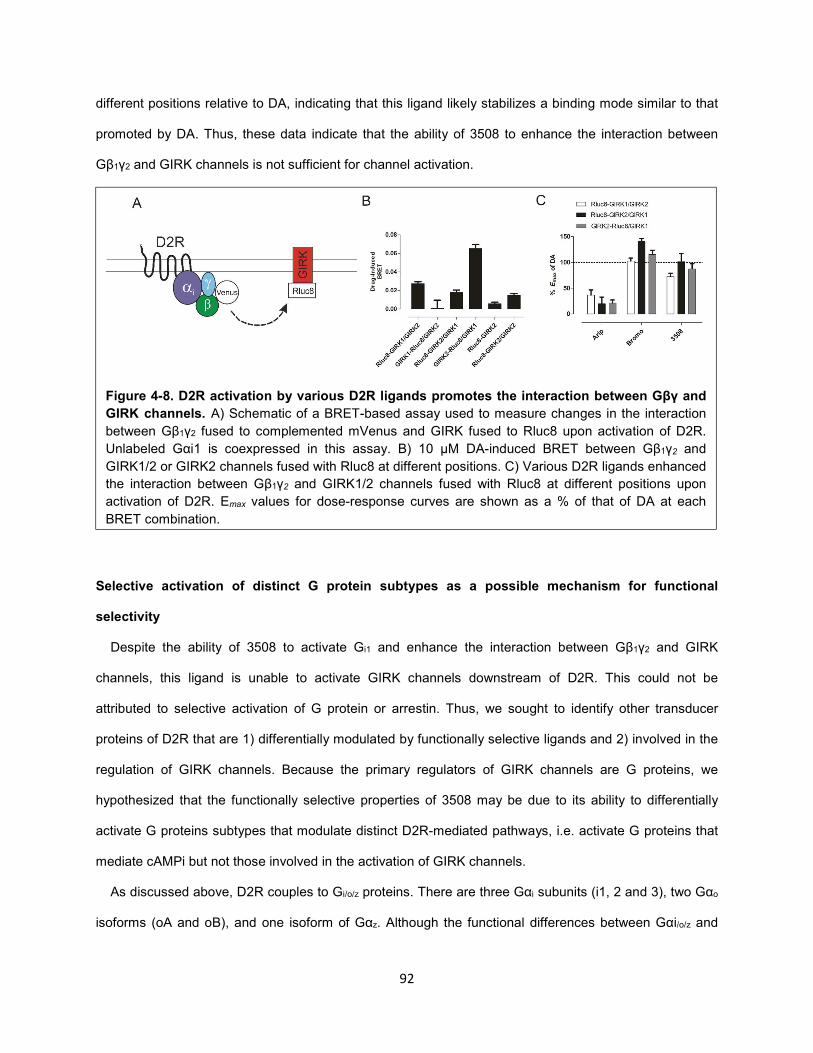
92
different positions relative to DA, indicating that this ligand likely stabilizes a binding mode similar to that
promoted by DA. Thus, these data indicate that the ability of 3508 to enhance the interaction between
Gβ1γ2 and GIRK channels is not sufficient for channel activation.
Selective activation of distinct G protein subtypes as a possible mechanism for functional
selectivity
Despite the ability of 3508 to activate Gi1 and enhance the interaction between Gβ1γ2 and GIRK
channels, this ligand is unable to activate GIRK channels downstream of D2R. This could not be
attributed to selective activation of G protein or arrestin. Thus, we sought to identify other transducer
proteins of D2R that are 1) differentially modulated by functionally selective ligands and 2) involved in the
regulation of GIRK channels. Because the primary regulators of GIRK channels are G proteins, we
hypothesized that the functionally selective properties of 3508 may be due to its ability to differentially
activate G proteins subtypes that modulate distinct D2R-mediated pathways, i.e. activate G proteins that
mediate cAMPi but not those involved in the activation of GIRK channels.
As discussed above, D2R couples to Gi/o/z proteins. There are three Gαi subunits (i1, 2 and 3), two Gαo
isoforms (oA and oB), and one isoform of Gαz. Although the functional differences between Gαi/o/z and
Figure 4-8. D2R activation by various D2R ligands promotes the interaction between Gβγ and
GIRK channels. A) Schematic of a BRET-based assay used to measure changes in the interaction
between Gβ1γ2 fused to complemented mVenus and GIRK fused to Rluc8 upon activation of D2R.
Unlabeled Gαi1 is coexpressed in this assay. B) 10 μM DA-induced BRET between Gβ1γ2 and
GIRK1/2 or GIRK2 channels fused with Rluc8 at different positions. C) Various D2R ligands enhanced
the interaction between Gβ1γ2 and GIRK1/2 channels fused with Rluc8 at different positions upon
activation of D2R. Emax values for dose-response curves are shown as a % of that of DA at each
BRET combination.

93
other Gα subunits have been well established, differences within the Gαi/o/z family are not well
understood. Gβγ subunits are encoded by five Gβ and 12 Gγ genes. Generally, these Gβ and Gγ
subunits are thought to be functionally redundant and have been shown to associate combinatorially53.
However, several knockdown studies have indicated that specific G protein subtypes are critical for
regulating distinct physiological processes in a context-dependent manner, e.g., within the same cell type,
muscarinic receptor 4 (M4R) and somatostatin receptor 1 (SSTR1) preferentially couple to distinct sets of
Gα, Gβ, and Gγ subtypes for inhibition of Ca2+ currents, indicating that selectivity is not due to levels of
expression but is specific to the identity of the receptor244-246. These effects may be due to differences in
sequence and structure between different G protein subtypes53. In addition, D1R has been shown to
preferentially couple to Gαolf, Gβ1, and Gγ7 for the activation of cAMP production in striatal tissue,
although this may be due to the enrichment of these G protein subtypes in this brain region247. Which G
protein subtypes, if any, are critical for activation of distinct D2R mediated-pathways is not well
understood. Moreover, whether specific G protein subtypes play a role in functional selectivity has yet to
be determined.
Given that GIRK channels are regulated primarily by Gβγ subunits and to some degree Gα, we sought
to probe the ability of 3508 to modulate defined heterotrimeric G proteins. However, the difficulty in
studying functional selectivity at this level is in part due to the paucity of tools that can discriminate
between distinct G protein subtypes. One commonly used approach to study G protein activation involves
the detection of agonist-dependent incorporation of GTPγS into the Gα subunit of a heterotrimeric G
protein. For pertussis toxin (PTX) sensitive Gα subunits such as Gαi/o, it is possible to differentiate
between subtypes in heterologous systems by inactivating endogenous Gαi/o and heterologously
expressing specific PTX-resistant Gαi or Gαo subtypes. Interestingly, using this method, the D2R partial
agonist 3-PPP was shown previously to selectively activate Gαo over Gαi-containing G proteins248. This
assay, however, is unable to differentiate between PTX-insensitive Gα subunits as well as distinct
subtypes of Gβ and Gγ. Thus, this methodology lacks subtype specificity and therefore cannot
differentiate between the combinatorial diversity of heterotrimeric G proteins.

94
Development of an assay to probing subtype-specific G protein activation
In an effort to address the limitations in studying G protein activation in a subtype-dependent manner, a
resonance energy transfer (RET)-based method has been developed that can differentiate between
distinct Gα subtypes249. In this system, the donor molecule Rluc8 was inserted at a conserved position
within the α-helical domain of a number of Gα subunits, and ligand-induced BRET was measured
between the Gα-Rluc8 and Gγ2 fused with the acceptor GFP2 on its N-terminus (GFP2-Gγ2). In addition
to the PTX-sensitive Gα subtypes (Gαi1-3 and GαoA/B), Gαq/11, Gα12/13 and Gαs sensors were also
developed. These sensors were validated for their ability to couple specifically to target receptors. Similar
to that described above (Figure 4-6A), there is a decrease in BRET upon addition of agonist, which is
indicative of a disassociation of or conformational change between the Gα and Gγ sensors.
Although the system described above confers Gα subtype specificity249, it is limited to a single Gγ
subtype (Gγ2) and lacks subtype specificity for Gβ. To address this issue, we combined the current BRET
system with bimolecular fluorescence or luminescence complementation (BiFc or BiLc) to create a
Complemented Donor Acceptor-RET (CODA-RET) assay250 in which all G protein subunits are labeled.
BiFc and BiLc have been used previously to investigate protein-protein interactions251. In BiFc,
fluorescent molecules are split into two non-fluorescent components (e.g., Venus is split into V1 and V2).
Similarly, in BiLc, luminescent molecules are split into non-luminescent components (e.g., Rluc8 is split
into L1 and L2). When fused to two interacting proteins, these inactive halves fold into a functional
chromophore. In a previous study, split BiFc fragments V1 and V2 fused to Gβ and Gγ subtypes were
used to study localization and trafficking of distinct Gβγ dimers in live cells, indicating that these strategies
are suitable to labeling both the Gβ and Gγ subunits simultaneously252. Moreover, the combination of
BRET and BiFc was implemented above (Figure 4-6A) to probe G protein activation with subtype
specificity for all three G protein subunits.
Before extending this system to G protein subtypes other than Gαi1β1γ2, we first sought to identify the
optimal configuration of combining BRET and BiFc or BiLc. The heterotrimer consisting of Gαi1β1γ2
coupling to D2R was used as a model system. Split chromophores were tagged to Gβ and Gγ rather than
Gα for a several reasons: 1) a number of Gβγ combinations have already been shown to be functional
when fused to BiFc fragments252, and 2) a split chromophore within a Gα subunit would presumably

95
disrupt G protein activation by preventing or interfering with the conformational change between or
disassociation of Gα and Gβγ. We used splits of the BRET acceptors Venus (V1/V2) and GFP2 (G1/G2)
as well as the BRET donor Rluc8 (L1/L2 and F1/F2253, which are irreversible and reversible BiLc systems,
respectively).
The possible combinations of BRET and BiFc or BiLc within a heterotrimeric G protein include: 1)
D2R/Gα-Chromophore1/Gβγ-split Chromophore2 or 2) D2R-Chromophore1/PTX-insensitive Gα/Gβγ-split
Chromophore2 (Figure 4-9A). The first configuration largely resembles the orientation of the BRET system
developed previously249. Several permutations of this configuration utilize BiLc-Gβγ fusions proteins,
which require the use of Gα fused to an acceptor molecule (e.g., Venus) rather than Rluc8. The second
configuration involves D2R fused to either a donor or acceptor molecule that can undergo BRET with Gβγ
fused to a complemented molecule. To achieve selectivity for Gα, PTX must be used to inactivate
endogenous Gαi/o, and thus only coexpressed PTX-insensitive Gα subtypes will be active.
To identify the optimal BRET configuration, D2R was stimulated with a saturating concentration of DA
(10 μM; Figure 4-9B). For comparison, we also measured BRET between full length donor and acceptors
at different positions within the heterotrimeric G protein. Generally, assay configurations utilizing BiFc
were more sensitive than those containing BiLc. The BRET configuration requiring the use of PTX was
not sufficiently sensitive relative to other configurations to merit the use of PTX in all future experiments.
Not surprisingly, BRET2 configurations (those using GFP2) were more sensitive compared to BRET1
(those using Venus; refer to the following protocol254 for a detailed description of BRET1 and BRET2).
However, because of the relative ease of use of BRET1 compared to BRET2, BRET1/BiFc assays with Gα
subunits fused to the donor molecule and Gβγ fused to the split acceptor molecule were developed to
probe the activation of G proteins in addition to Gαi1β1γ2. Notably, like the BRET-based G protein
activation assay developed previously, agonist-induced activation in this configuration led to a decrease
in BRET between the donor and acceptor molecules.
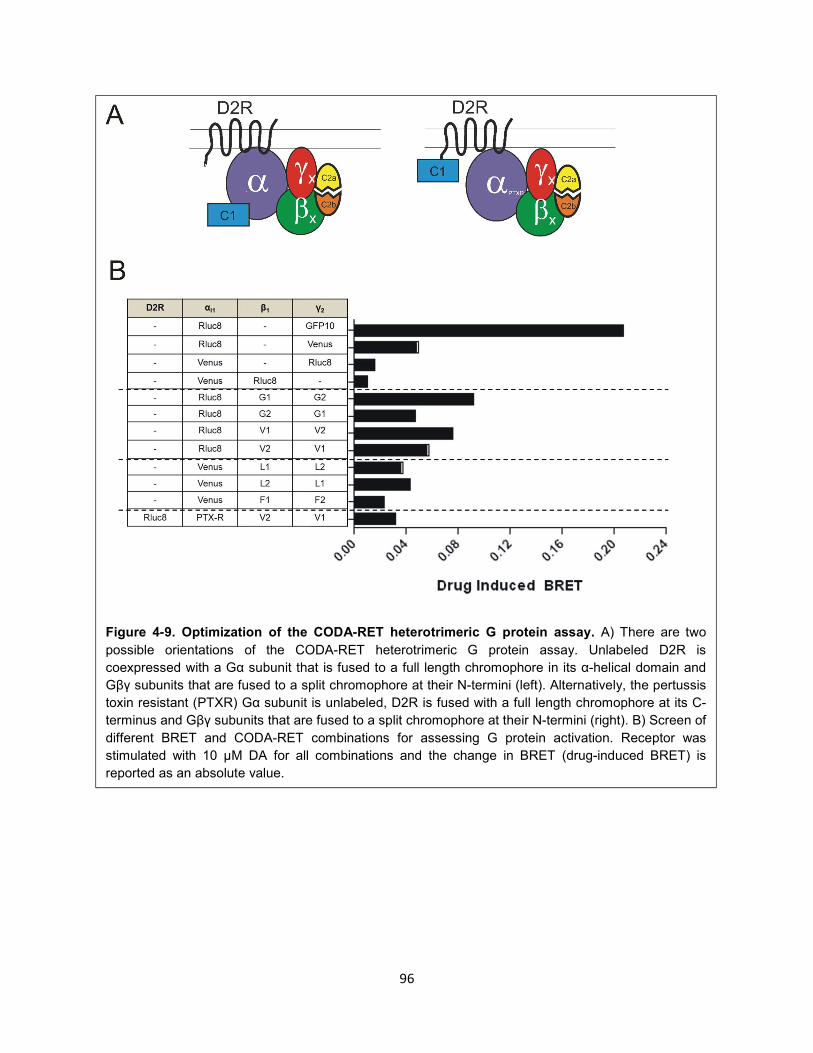
96
Figure 4-9. Optimization of the CODA-RET heterotrimeric G protein assay. A) There are two
possible orientations of the CODA-RET heterotrimeric G protein assay. Unlabeled D2R is
coexpressed with a Gα subunit that is fused to a full length chromophore in its α-helical domain and
Gβγ subunits that are fused to a split chromophore at their N-termini (left). Alternatively, the pertussis
toxin resistant (PTXR) Gα subunit is unlabeled, D2R is fused with a full length chromophore at its C-
terminus and Gβγ subunits that are fused to a split chromophore at their N-termini (right). B) Screen of
different BRET and CODA-RET combinations for assessing G protein activation. Receptor was
stimulated with 10 μM DA for all combinations and the change in BRET (drug-induced BRET) is
reported as an absolute value.

97
Probing subtype-specific G protein activation
Based on the optimization of the CODA-RET G protein activation system (Figure 4-9), we sought to
extend this assay to different G protein subtypes, inserting Rluc8 in the α-helical domain of Gα subunits
and fusing split Venus at the N-termini of Gβγ subunits. We assumed that inserting the donor and
acceptor at these positions would be tolerated structurally and would be amenable to CODA-RET given
the similar overall structure of different G protein subtypes53. Fusions of Gαi1,i2,i3 and GαoA/B with Rluc8
were developed previously249. We also inserted Rluc8 in a homologous position of Gαz (Gαz-Rluc8).
Venus split fragments were fused to all Gβ subtypes (Gβ1-5) and split fragments of five of the 12 Gγ
subtypes (Gγ1,2,3,7,11) were generated.
All of these G protein subtypes have been shown to be expressed in human striatum, albeit to varying
degrees (www.brain-map.org), e.g., Gγ7 is expressed more highly than Gγ1 in this region. Of note, it is
unclear which of these G proteins are specifically expressed in D2R-expressing neurons within the
striatum. More importantly, it is also unclear if these G protein subtypes are expressed in dopaminergic
neurons, where D2R regulates GIRK channel activity. Moreover, even if this is the case it is possible that
some of these G proteins do not couple functionally to D2R in these neurons. Despite these limitations,
we sought to establish proof of principle that ligands can differentially activate G protein subtypes in this
CODA-RET system, thus providing insight into a novel form of functional selectivity and potentially the
mechanism of action of 3508 at D2R. The use of the split acceptor allows us to study, for the first time,
signaling by defined Gα, Gβ, and Gγ subunits, filtering out contributions from endogenous G proteins that
otherwise complicate analysis.
A number of D2R ligands were screened for their ability to modulate various combinations of G protein
subtypes, wherein several patterns emerged (Figure 4-10). Generally, there were only minor differences
in the ability of ligands to activate G proteins containing different Gαi subunits (i1-i3) when Gβ1 and Gγ2
were fixed (Figure 4-10). Ligands activated G proteins containing GαoA and Gαz more robustly than those
containing Gαi subunits when Gβ1 and Gγ2 were fixed (Figure 4-10). Ligand activity was strikingly similar
across different Gγ subtypes when Gαi1 and Gβ1 were fixed (Figure 4-10). Interestingly, when Gαi1 and
Gγ2 were fixed, whereas ligand activity was similar between G proteins containing Gβ1-4, the activity of
some ligands relative to DA was reduced at those G proteins containing Gβ5 (Figure 4-10). This was most

98
apparent for 3508, which robustly activated Gβ1-4-containing heterotrimeric G proteins but had no
detectable activity at those containing Gβ5. Thus, these data indicate that although ligand activity is
largely homogenous across different G protein subtypes, several unique G proteins emerge, in particular
Gβ5.
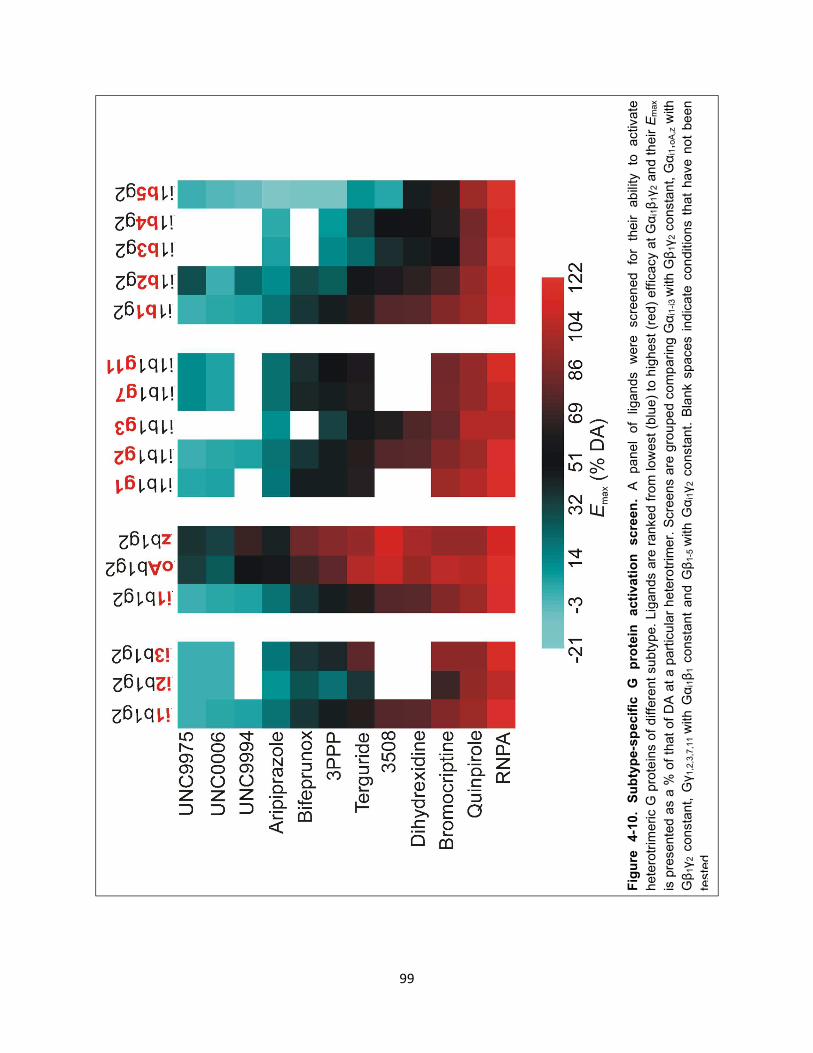
99
Fig
ure
4-1
0.
Su
bty
pe-s
pecif
ic
G
pro
tein
a
cti
vati
on
scre
en
. A
pa
ne
l of
ligands
were
scre
en
ed
for
their
ab
ility
to
activ
ate
hete
rotr
imeric G
pro
tein
s o
f diff
ere
nt
su
bty
pe.
Lig
an
ds a
re r
anked
fro
m low
est
(blu
e)
to h
igh
est
(red)
eff
icacy a
t G
αi1β
1γ
2 a
nd t
heir E
ma
x
is p
resente
d a
s a
% o
f th
at
of
DA
at
a p
art
icu
lar
hete
rotr
imer.
Scre
ens a
re g
rou
pe
d c
om
paring
Gα
i1-i
3 w
ith G
β1γ
2 c
onsta
nt, G
αi1, o
A,z w
ith
Gβ
1γ
2 c
onsta
nt,
Gγ
1,2
,3,7
,11
with
Gα
i1β
1 c
onsta
nt
an
d G
β1
-5 w
ith G
αi1γ
2 c
onsta
nt.
Bla
nk s
paces i
nd
icate
con
ditio
ns t
hat
ha
ve
not
be
en
teste
d.
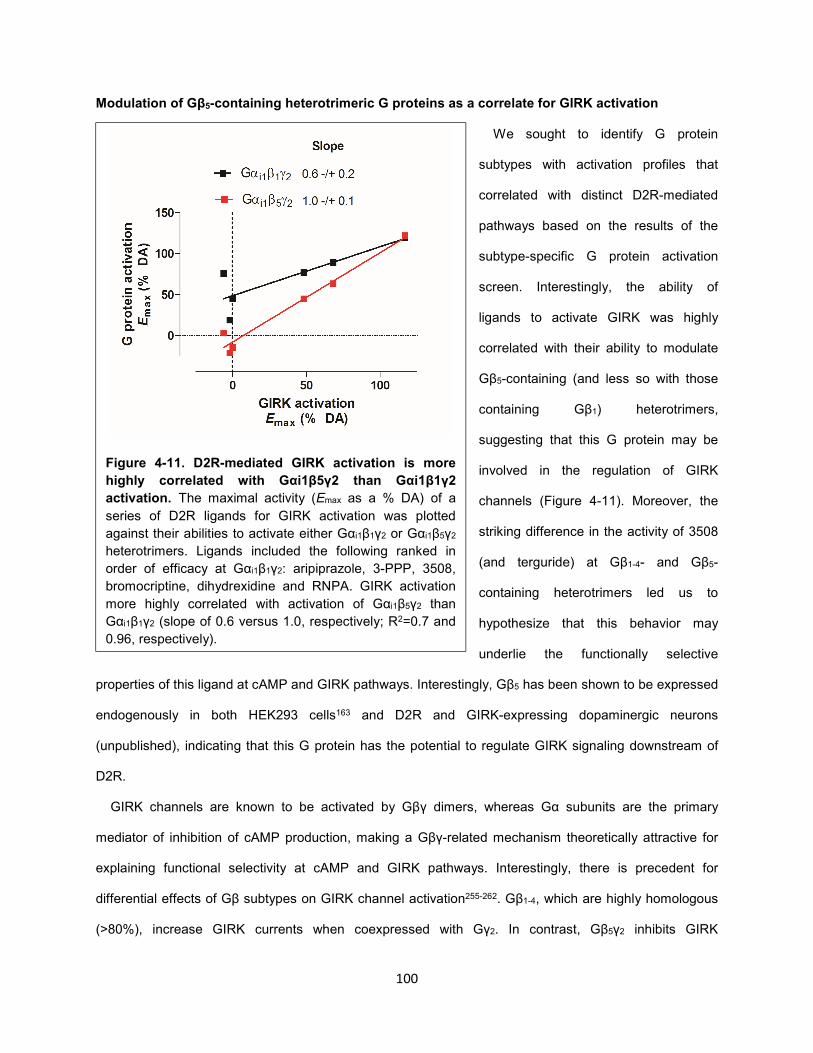
100
Modulation of Gβ5-containing heterotrimeric G proteins as a correlate for GIRK activation
We sought to identify G protein
subtypes with activation profiles that
correlated with distinct D2R-mediated
pathways based on the results of the
subtype-specific G protein activation
screen. Interestingly, the ability of
ligands to activate GIRK was highly
correlated with their ability to modulate
Gβ5-containing (and less so with those
containing Gβ1) heterotrimers,
suggesting that this G protein may be
involved in the regulation of GIRK
channels (Figure 4-11). Moreover, the
striking difference in the activity of 3508
(and terguride) at Gβ1-4- and Gβ5-
containing heterotrimers led us to
hypothesize that this behavior may
underlie the functionally selective
properties of this ligand at cAMP and GIRK pathways. Interestingly, Gβ5 has been shown to be expressed
endogenously in both HEK293 cells163 and D2R and GIRK-expressing dopaminergic neurons
(unpublished), indicating that this G protein has the potential to regulate GIRK signaling downstream of
D2R.
GIRK channels are known to be activated by Gβγ dimers, whereas Gα subunits are the primary
mediator of inhibition of cAMP production, making a Gβγ-related mechanism theoretically attractive for
explaining functional selectivity at cAMP and GIRK pathways. Interestingly, there is precedent for
differential effects of Gβ subtypes on GIRK channel activation255-262. Gβ1-4, which are highly homologous
(>80%), increase GIRK currents when coexpressed with Gγ2. In contrast, Gβ5γ2 inhibits GIRK
Figure 4-11. D2R-mediated GIRK activation is more
highly correlated with Gαi1β5γ2 than Gαi1β1γ2
activation. The maximal activity (Emax as a % DA) of a
series of D2R ligands for GIRK activation was plotted
against their abilities to activate either Gαi1β1γ2 or Gαi1β5γ2
heterotrimers. Ligands included the following ranked in
order of efficacy at Gαi1β1γ2: aripiprazole, 3-PPP, 3508,
bromocriptine, dihydrexidine and RNPA. GIRK activation
more highly correlated with activation of Gαi1β5γ2 than
Gαi1β1γ2 (slope of 0.6 versus 1.0, respectively; R2=0.7 and
0.96, respectively).

101
currents257,263. Gβ5 is unique in both sequence and function53. Unlike other Gβ subunits, Gβ5 can dimerize
not only with Gγ but also with Regulator of G protein Signaling (RGS) family 7 (R7) proteins, important
regulators of a range of neuronal processes, including motor control and reward behavior264. Of note, the
ability of Gβ5 to inhibit channel activity was dependent on coexpression of Gγ257, highlighting the
importance of Gβ5γ dimers and not Gβ5-RGS7 dimers for this effect.
Validation and characterization of the Gαi1β5γ2 heterotrimer
There has been considerable debate as to whether Gβ5γ dimers exist in native systems53,265. Although
these dimers can be detected when expressed heterologously in cells257, it is unclear whether
endogenous Gβ5 and Gγ subunits can associate. This has been partly due to difficulty in purifying
endogenously expressed Gβ5γ dimers from cells, which has been attributed to the instability of these
dimers in detergents266. Interestingly, one recent comprehensive analysis of overexpressed G proteins
detected such dimers and found evidence for the existence of a Gαiβ5γ heterotrimeric G proteins267.
Given the controversy in the existence of Gβ5γ dimers, we sought to exclude the possibility that using
BiFc with split Venus at the N-termini of Gβ5 and Gγ2 forces these two subunits together and thus that the
ligand-induced conformational changes seen in Gαi1β5γ2 CODA-RET assay are artifactual. Thus, we
fused full length Venus to Gβ5 at its N-terminus and performed BRET between Gαi1-Rluc8 and this fusion
protein. Saturating concentrations of DA (10 μM) failed to promote a change in BRET as seen in the
CODA-RET assay (data not shown). However, when unfused Gγ2 was coexpressed in this assay, DA
promoted a robust change in BRET, similar to that in the CODA-RET assay (data not shown). These data
indicate that this conformational change between Gαi1 and Gβ5 requires Gγ and that these changes can
occur even when Gβ5γ are not artificially fused together. It is important to note, however, that these data
do not exclude the possibility that Gβ5γ dimers are not present in native tissues.
In order to gain insight into the potential mechanism of regulation of GIRK channels by Gβ5- containing
heterotrimers, we probed ligand-induced conformational changes within these G proteins using the Gαi1-
Rluc8 biosensors described above (Figure 4-6). The preliminary ligand screens at different G protein
subtypes were conducted with Gαi1 containing Rluc8 within the α-helical domain at position 91 (Figure 4-
10), the sensor with the largest dynamic range. As discussed above, when this sensor is coexpressed
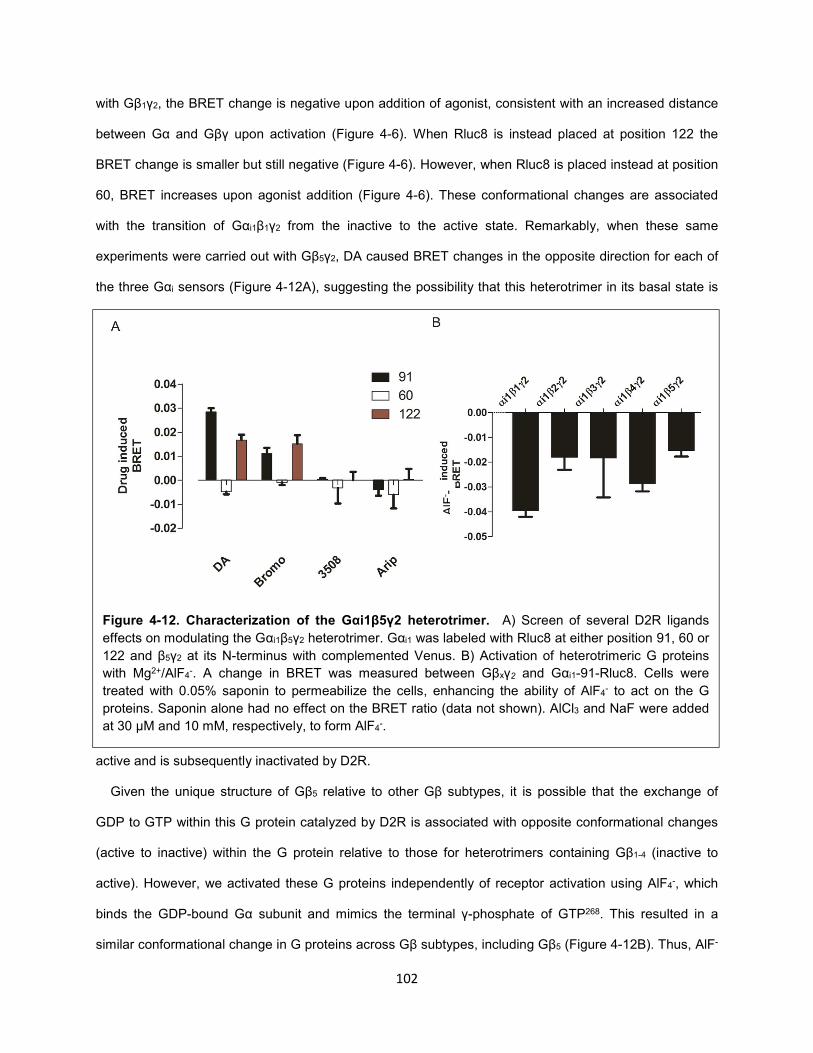
102
with Gβ1γ2, the BRET change is negative upon addition of agonist, consistent with an increased distance
between Gα and Gβγ upon activation (Figure 4-6). When Rluc8 is instead placed at position 122 the
BRET change is smaller but still negative (Figure 4-6). However, when Rluc8 is placed instead at position
60, BRET increases upon agonist addition (Figure 4-6). These conformational changes are associated
with the transition of Gαi1β1γ2 from the inactive to the active state. Remarkably, when these same
experiments were carried out with Gβ5γ2, DA caused BRET changes in the opposite direction for each of
the three Gαi sensors (Figure 4-12A), suggesting the possibility that this heterotrimer in its basal state is
active and is subsequently inactivated by D2R.
Given the unique structure of Gβ5 relative to other Gβ subtypes, it is possible that the exchange of
GDP to GTP within this G protein catalyzed by D2R is associated with opposite conformational changes
(active to inactive) within the G protein relative to those for heterotrimers containing Gβ1-4 (inactive to
active). However, we activated these G proteins independently of receptor activation using AlF4-, which
binds the GDP-bound Gα subunit and mimics the terminal γ-phosphate of GTP268. This resulted in a
similar conformational change in G proteins across Gβ subtypes, including Gβ5 (Figure 4-12B). Thus, AlF-
Figure 4-12. Characterization of the Gαi1β5γ2 heterotrimer. A) Screen of several D2R ligands
effects on modulating the Gαi1β5γ2 heterotrimer. Gαi1 was labeled with Rluc8 at either position 91, 60 or
122 and β5γ2 at its N-terminus with complemented Venus. B) Activation of heterotrimeric G proteins
with Mg2+/AlF4-. A change in BRET was measured between Gβxγ2 and Gαi1-91-Rluc8. Cells were
treated with 0.05% saponin to permeabilize the cells, enhancing the ability of AlF4- to act on the G
proteins. Saponin alone had no effect on the BRET ratio (data not shown). AlCl3 and NaF were added
at 30 μM and 10 mM, respectively, to form AlF4-.
AlF
- -

103
-incorporation into Gαi1β5γ2-GDP, as is the case for other G proteins, is likely associated with the
formation of the active conformation.
There are several possible explanations for the effects of activated D2R on Gαi1β5γ2. Activated D2R
may catalyze the reverse exchange of GTP to GDP within the Gαi1β5γ2 heterotrimer, i.e., these G proteins
are in the active state in their basal state, and activated D2R promotes the conversion from the active to
the inactive state by exchanging GTP for GDP. Alternatively, D2R may catalyze the release of GTP from
Gαi1β5γ2, resulting in a nucleotide-free G protein. The ability of GPCRs to catalyze the release of GTP
from the G proteins has been previously reported, albeit not for Gβ5-containing heterotrimers269.
A model of functional selectivity between G protein-mediated pathways
Because Gβ5γ2 was found to inhibit GIRK currents when expressed heterologously257,263, this led us to
hypothesize that DA activates GIRK not only through activation and release of Gβ1γ2 (as seen in Figure 4-
8) but also through a release of inhibition mediated by inactivation of Gβ5γ2 . Within this model (Figure 4-
13), both DA and 3508 promote the inhibition of cAMP by activating Gαi and Gβ1-4-containing
heterotrimers. Furthermore, GIRK channels are blocked by constitutively active Gαi1β5γ2 heterotrimers.
Activation of D2R (and possibly other GPCRs coupled to GIRK channels) results in the simultaneous
release of Gβ1γ2 that activate GIRK channels and the inactivation of Gαi1β5γ2, thus disinhibiting the
channel. Of note, 3508 is inactive at each of the three biosensors (Figure 4-12A). Thus, we propose that
since 3508 is unable to modulate the activity state of Gβ5γ2, inhibition is maintained.
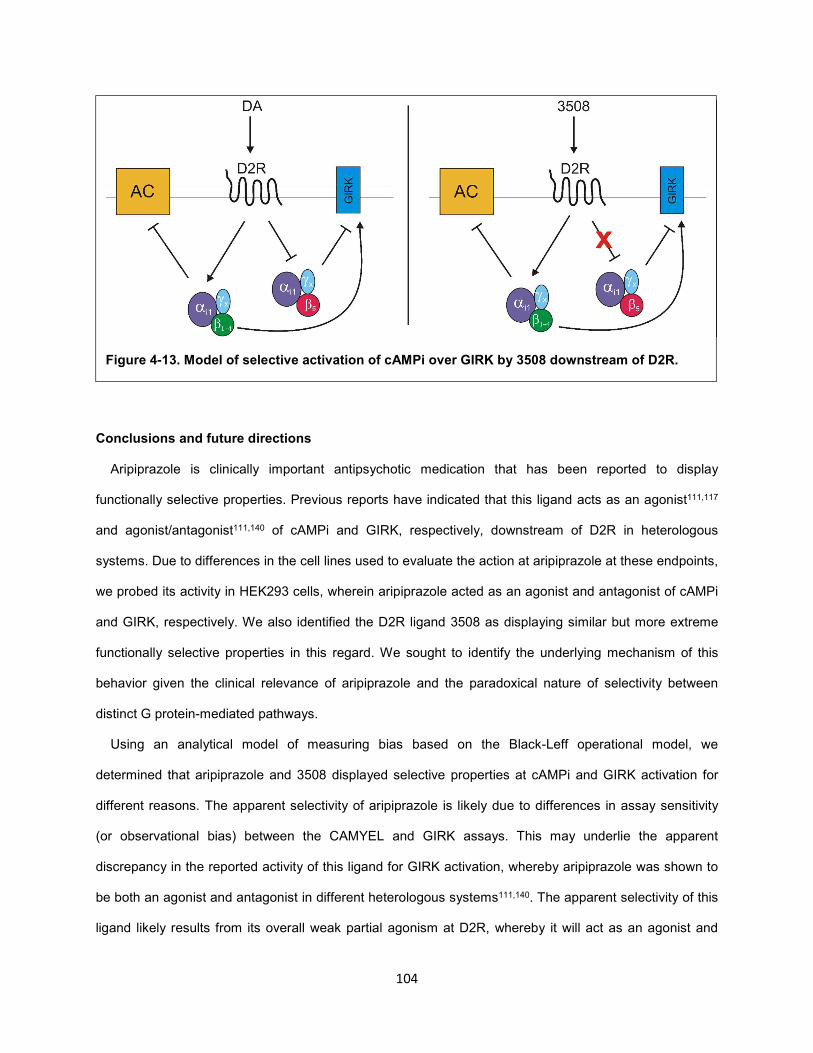
104
Conclusions and future directions
Aripiprazole is clinically important antipsychotic medication that has been reported to display
functionally selective properties. Previous reports have indicated that this ligand acts as an agonist111,117
and agonist/antagonist111,140 of cAMPi and GIRK, respectively, downstream of D2R in heterologous
systems. Due to differences in the cell lines used to evaluate the action at aripiprazole at these endpoints,
we probed its activity in HEK293 cells, wherein aripiprazole acted as an agonist and antagonist of cAMPi
and GIRK, respectively. We also identified the D2R ligand 3508 as displaying similar but more extreme
functionally selective properties in this regard. We sought to identify the underlying mechanism of this
behavior given the clinical relevance of aripiprazole and the paradoxical nature of selectivity between
distinct G protein-mediated pathways.
Using an analytical model of measuring bias based on the Black-Leff operational model, we
determined that aripiprazole and 3508 displayed selective properties at cAMPi and GIRK activation for
different reasons. The apparent selectivity of aripiprazole is likely due to differences in assay sensitivity
(or observational bias) between the CAMYEL and GIRK assays. This may underlie the apparent
discrepancy in the reported activity of this ligand for GIRK activation, whereby aripiprazole was shown to
be both an agonist and antagonist in different heterologous systems111,140. The apparent selectivity of this
ligand likely results from its overall weak partial agonism at D2R, whereby it will act as an agonist and
Figure 4-13. Model of selective activation of cAMPi over GIRK by 3508 downstream of D2R.

105
antagonist at sensitive and insensitive systems, respectively. Thus, aripiprazole is likely not
conformationally biased with respect to cAMPi and GIRK activation, which may have significant
implications on drug design of novel antipsychotics based on this drug. Aripiprazole was shown
previously to have selective properties at a variety of different endpoints in addition to cAMP and GIRK
pathways, and further studies will be required to investigate aripiprazole’s conformationally bias at other
endpoints.
Although aripiprazole likely does not conformationally bias D2R towards cAMPi over GIRK activation,
this ligand may still behave in a selective manner in vivo, e.g., D2R-mediated cAMPi may be more
sensitive than GIRK activation, resulting in this ligand behaving as an agonist and antagonist,
respectively, of these pathways. Thus, it will be critical to determine the activity of aripiprazole on GIRK
channels in native systems, specifically in the soma and dendrites of dopaminergic neurons. The
evaluation of aripiprazole in native systems may also give insight onto the level of sensitivity that
heterologous systems (e.g., in HEK293 cells) must be adjusted for them to be physiologically relevant,
i.e., assay systems that are overly or under sensitive will not be predictive of ligand activity in vivo.
In contrast to aripiprazole, 3508 was shown to be functionally selective independent of the sensitivity of
the cAMPi and GIRK assays according to the operational model analysis. This could not be attributed to
the selective activation of either the generic G protein Gαi1β1γ2 or arrestin. This was also not due to
differential conformational changes within the G protein or the inability of 3508 to promote the interaction
between β1γ2 and GIRK channels. Using a novel CODA-RET assay for probing G protein activation in a
subtype-specific manner, we found that activation of Gβ5-containing heterotrimers highly correlated with
GIRK activation. It is possible that these G proteins are not involved directly in the functionally selective
properties of 3508 and instead reflect the ability of 3508 to stabilize distinct conformations of D2R.
However, given that Gβ5γ was shown previously to be an inhibitor of GIRK channels, we hypothesized
that this G protein may underlie the inability of 3508 to activate GIRK channels downstream of D2R.
Interestingly, using conformational biosensors we found that D2R appears to catalyze the transition of
Gβ5-containing heterotrimers from the active to the inactive state. It is unclear if this is due to the
transition of the GTP-bound state to the GDP- or apo-state of the G protein. Moreover, it is also unclear
as of yet if the inactivation of Gβ5-containing heterotrimers by D2R results in the decreased interaction

106
between this G protein and GIRK channels. Further studies will be required similar to those probing
agonist-induced changes in interactions between Gβ1γ2 and GIRK (Figure 4-8). Interestingly, a previous
study found that agonist-stimulation of mGluR2 promoted an decrease and increase, respectively, in the
interaction between Gβ1-4γ and Gβ5γ with GIRK1/4 channels239. It is interesting to note that these changes
are opposite than what is expected and may relate to differences in assay conditions such as GIRK
channel type and labeling positions. Regardless, these data are consistent with the opposing actions of
Gβ1-4γ and Gβ5γ on GIRK channels.
Further studies will also be necessary to probe the functional role of Gβ5 in regulating GIRK channels.
In previous studies, Gβ5γ overexpression decreased GIRK activity. According to the model described
above (Figure 4-13), knockdown or knockout of Gβ5 should result in the disinhibition of GIRK channels,
which would either increase the basal activity of the channel and/or the maximal activity in response to
agonists like DA. Moreover, knockdown of Gβ5 may allow 3508 to act as an agonist for GIRK channels by
obviating the need for this ligand to disinhibit basal actions of Gβ5 on these channels. Consistent with this
model, GABAB-mediated GIRK signaling was enhanced in mice in which Gβ5 was knocked out globally270,
although the authors attributed this to a loss in activity of Gβ5-RGS7 rather than Gβ5γ. Future studies will
be required to investigate the effect of Gβ5 knockout on D2R-mediated GIRK signaling, in particular on
3508 activity.
Overall, these data indicate that 3508 displays unprecedented functional selectivity between G protein-
mediated pathways downstream of D2R and does so to a greater degree than aripiprazole. It will be
interesting to probe the effects of this ligand in vivo in rodents, e.g., the ability of this ligand to block
psychostimulant-mediated effects. These studies may provide insight onto the role of D2R-mediated
GIRK activation in regulating these behaviors.

107
CHAPTER 5. Summary and Conclusions
D2R antagonism has been a hallmark of antipsychotic medications for more than half a century.
However, this drug-class is associated with substantial side effects that decrease quality of life and
medication compliance. The development of novel antipsychotic medications with superior therapeutic
and side effect profiles has been hampered in part due to a poor understanding of the specific D2R
populations and downstream signaling molecules that must be blocked to confer therapeutic efficacy.
It has been proposed that antipsychotic medications confer their effects through the blockade of
arrestin but not G protein signaling downstream of D2R. This hypothesis was formulated based on
observations that whereas most antipsychotic medications block both G proteins and arrestin, the highly
clinically relevant antipsychotic aripiprazole activates G proteins but not arrestins. Moreover, genetic
studies in rodents implicated arrestin signaling as being necessary for mediating psychostimulant-
dependent behaviors in rodents. Substantial efforts have now gone towards the development of
functionally selective or biased ligands of D2R that selectively activate G protein and block arrestin
signaling.
The hypothesis that selective blockade of arrestin signaling downstream of D2R will lead to superior
antipsychotic medications suffers from several major limitations, namely that blockade of G protein
signaling may also be important in conferring antipsychotic effects. Moreover, currently available
pharmacological and genetic tools that have been used to probe G protein and arrestin signaling
downstream of D2R in vivo suffer from on- and off-target effects that add substantial confounds to our
understanding of these processes. Thus, we sought to develop novel tools to probe these processes in
an unambiguous manner.
We have now developed a mutant receptor of D2R that is capable of activating arrestins but not G
proteins (Chapter 2). This receptor was expressed in specific D2R-expressing populations, i.e., MSNs in
the NAc of mice, facilitating enhanced arrestin signaling. Whereas overexpression of wildtype D2R
enhanced both basal locomotion and motivation in these mice, the mutant receptor only enhanced the
basal locomotion, indicating that these behaviors are likely differentially regulated. Further studies in
which wildtype receptors are replaced with this biased receptor will provide additional insights onto the

108
role of arrestin signaling downstream of D2R. These studies, combined with analogous studies using the
G protein-biased receptor as well as those using S1 to abolish G protein activity (Chapter 3), will give
insight into the role of G protein versus arrestin signaling downstream of D2R in mediating a number
dopamine-dependent behaviors, including locomotion, motivation and psychostimulant-dependent
behaviors. Moreover, expression of these tools in other D2R-expressing cell-types, e.g., dopaminergic
neurons, TANs or D2R-expressing neurons in the mPFC, will provide additional insight onto the role of G
protein and arrestin signaling pathways downstream of D2R.
Of note, it is possible that D2R-mediated G protein and arrestin signaling are not independent of one
another, i.e. the activation of psychostimulant-dependent behaviors may require the coactivation of both
of these signaling molecules. In this case, the strategy to develop functionally selective or biased ligands
as superior antipsychotic medications will require reevaluation. It is also important to note that G proteins
and arrestins each mediate a variety of downstream processes. It will be critical to develop tools that
differentiate between these signaling pathways, e.g., D2-A3, which recruits arrestin but does not
internalize in an agonist-induced manner, or 3508, which selectively activates distinct G protein-mediated
pathways.
It is important to note that although G protein- or arrestin-biased D2Rs offer unprecedented control in
understanding D2R-mediated processes, their utility is currently limited to the predictive value of animal
models of the positive symptoms of schizophrenia, such as psychostimulant-dependent behaviors in
rodents. Moreover, even if the specific D2R-expressing populations and signaling pathways that must be
blocked to ameliorate psychosis are identified, current pharmacological agents that modulate GPCRs,
even functionally selective ligands, have major limitations, such as off-target effects, on-target effects in
off-target cell-types and poor spatio-temporal control of signaling processes.
An alternative approach that confers substantially higher specificity is the use of gene therapies.
Although these approaches are challenging, particular for CNS applications, gene therapies have recently
become more viable. Indeed, virally-mediated overexpression of tyrosine hydroxylase (TH) in
dopaminergic neurons is currently being evaluated for the treatment Parkinson’s disease (ProSavin;
Phase II clinical trials completed; clinicaltrials.gov). Thus, it is conceivable that such approaches may one
day be effective for the treatment of the positive symptoms (as well as other symptoms) of schizophrenia.

109
In particular, the cell-type specific expression or knock-in of biased D2Rs like those described above
would provide unprecedented control over D2R-mediated processes, potentially ameliorating psychosis
with minimal side effects.

110
Materials and Methods
Note: A significant portion of the experimental procedures focus on experiments relating to the
characterization of D2-A3. However, these methods are applicable to other studies described in
this work.
Materials
Quinpirole, dopamine hydrochloride, (+)-butaclamol, haloperidol, (S)-(-)-sulpiride, and G418 were
purchased from Sigma-Aldrich (St. Louis, MO). Phosphatase inhibitor cocktail set II and protease inhibitor
cocktail set III were purchased from Calbiochem (Billerica, MA) and coelenterazine h from Dalton Chemical
Laboratories (Toronto, Ontario, Canada). The radioligands [3H](-)-sulpiride and [3H]YM-09151-2, were
purchased from Perkin Elmer (Waltham, MA). Rabbit anti-actin, mouse anti-arrestin3, rabbit anti-GRK2,
and mouse anti-c-myc antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and
mouse anti-FLAG M2 was purchased from Sigma-Aldrich (St. Louis, MO). Alexa Fluor® 488 goat anti-
mouse IgG, Alexa Fluor® 647 goat anti-mouse IgG, and Alexa Fluor® 568 goat anti-rabbit IgG were
purchased from Life Technologies (Grand Island, NY).
Plasmids
For the G protein activation BRET assay, pcDNA3.1 plasmids were used carrying c-myc tagged wild
type or mutant rat D2L (D2-WT, A2, A3, or A4), Renilla luciferase 8 (Rluc8) inserted at the 91st position of
Gαi1 (Gαi1-91-Rluc8), and mVenus fragments V1 and V2 fused to Gβ1 and Gγ2, respectively (V1-β1 and
V2-γ2). For the BRET based-inhibition of cAMP assay, pcDNA3.1 plasmids carrying D2-WT or A4 and
CAMYEL (ATCC) were used. For the recruitment BRET assays, FLAG-tagged wild type human D2L
receptors fused to Renilla luciferase 8 (D2-Rluc8), mVenus fused to human arrestin3 (mVenus-Arr3),
mVenus fused to bovine GRK2 (GRK2-Venus), human β2-Adaptin-EYFP (β2-AP-EYFP) and pcDNA3.1
plasmids were used. Human arrestin3 fused to Rluc8 at its C-terminus with a SRPPVAT amino acid linker
(Arr3-Rluc8) was created in pcDNA3.1. The arrestin translocation assay used Rluc8-arrestin3 with an SH3-
binding peptide (Sp1) at its C-terminus (Rluc8-arrestin3-Sp1) and a doubly palmitoylated fragment of

111
GAP43 linked to citrine and an SH3 domain through a serine- and glycine-rich linker (mem-linker-citrine-
SH3). For internalization and immunoblotting assays, plasmids carrying rat D2L receptor, wild type and
mutants of arrestin3, and GRK2 were used.
Mutagenesis
Multiple point mutations of both the D2L receptor, D2-Rluc8, and arrestin were constructed using the
QuikChange® Lightning site-directed mutagenesis kit (Stratagene, La Jolla, CA) per the manufacturer’s
instructions to introduce alanine substitution of residues 214 and 215 (A2), 213-215 (A3) or 212-215 (A4)
within IL3. Residues of arrestin3 were deleted (373-LIEFD-377) or mutated (R395E) alone or in
combination using this method. All mutations were confirmed by DNA sequencing.
Cell Culture and Transfections
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 5% bovine
calf serum (Thermo Scientific, Logan, UT), 5% fetal clone serum (Thermo Scientific), and 1% penicillin-
streptomycin solution (Thermo Scientific) at 37˚C with 10% CO2. HEK293T cells were maintained in
DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin
(Corning) at 37˚C with 10% CO2. For the internalization and western blot studies, stable expression of
either wild type or mutant D2L receptors in HEK293 cells was obtained by transfecting 6 µg of D2 receptor
cDNA using Lipofectamine 2000 (Invitrogen, Grand Island, NY) per the manufacturer’s recommended
protocol. Pools of stably transfected cells were then maintained under selective pressure using 600 µg/ml
G418 for 3 weeks, after which the amount of G418 in the growth medium was decreased to 300 µg/ml.
Transient expression of arrestin3, GRK2, and/or pcDNA3.1 into HEK293 cells stably expressing wild type
or mutant D2L receptors was obtained by transfecting 3 µg of cDNA using Lipofectamine 2000
(Invitrogen). After overnight incubation at 37˚C transfection complexes were rinsed off, the cells were split
into new plates and incubated for an additional 48 hours before conducting experiments. Protein
expression was confirmed by immunoblotting.
For all BRET studies, constructs were transiently transfected into either HEK293 or HEK293T cells
using polyethylenimine (PEI; Polysciences, Inc., Warrington, PA). The quantity of transfected DNA was

112
adjusted so that the levels of membrane expression of wild type and mutant D2 receptors were similar as
determined by radioligand binding assay or fluorescence-activated cell sorting (FACS) as described below.
For the arrestin3 mutant internalization studies, 2 µg of GRK2 were contransfected with 2 µg of
pcDNA3.1, Arr3-WT, Arr3-ΔLIEFD, Arr3-R395E or Arr3-ΔLIEFD/R395E into HEK293 cells stably
expressing wild-type D2L receptors using PEI at a ratio of 2 µl of PEI (1 µg/µl) per 1 µg of cDNA. After
overnight incubation at 37° C cells were washed with PBS, split into new plates and incubated for an
additional 48 hours before conducting experiments.
D2L Receptor Radioligand Binding
To compare membrane expression of D2 receptors, cells were lysed in ice-cold hypotonic buffer (1 mM
HEPES, 2 mM EDTA, pH 7.4) for 20 min at 4˚C, scraped from the plate, and centrifuged at 17,000 g at 4˚C
for 20 min. The resulting pellet was resuspended in Tris-buffered saline (TBS; 50 mM Tris, 120 mM NaCl,
pH 7.4) and homogenized for 10 sec using a Polytron homogenizer (Brinkmann Instruments, Westbury,
NY). Protein concentration of the membranes was determined using a bicinchoninic acid (BCA) protein
assay kit (Thermo Scientific) and 5 µg of protein was incubated in TBS containing 0.02% bovine serum
albumin (BSA; Fisher Scientific, Waltham, MA) at a volume of 1 ml with [3H]YM-09151-2 for 1 hr at 25˚C.
Non-specific binding was assessed using (+)-butaclamol (2 µM). Membranes were then harvested and
radioactivity measured as described previously. Data were analyzed by nonlinear regression using
GraphPad Prism to determine Kd and Bmax values.
FACS
Transfected cells expressing N-terminal FLAG- or c-myc-tagged D2 receptor were dissociated and
surface receptors labeled using mouse anti-FLAG or anti-c-myc antibodies and goat anti-mouse-Alexa647
antibodies diluted 1/400 in PBS (with 0.1% of BSA and 0.1% of NaN3), and quantitated using a C6 Flow
Cytometer (Accuri). This FACS assay was used to equalize receptor expression in the assays for G protein
activation (Fig. 1 and Fig. 3A & B), inhibition of cAMP (Fig. 1D), GRK2-Venus recruitment, and the
interaction between arrestin and β2-adaptin.

113
Internalization Assay - D2 receptor internalization was measured by using a whole-cell [3H]sulpiride
binding assay. HEK293 cells stably expressing wild-type or mutant D2 receptors were transiently
transfected with pcDNA3.1, wild type or mutant arrestin3, and/or GRK2 as described above. After
overnight incubation with the transfection complexes, cells were harvested and split into three plates, one
for confirmation of arrestin3 and/or GRK2 protein expression (described below), and two for the
internalization assay. Forty-eight hours after transfection, cells were incubated with either a vehicle control,
quinpirole (10 µM), or dopamine (10 µM) for 30 min at 37˚C, washed three times with ice-cold PBS and
scraped into 2 ml of cold assay buffer (PBS containing 2 mM EDTA and 0.01% BSA). The total number of
cells in each condition was determined using a NucleoCounter (Chemometec, Denmark) and 300,000 cells
were incubated in assay buffer at a volume of 1 ml with the membrane impermeant antagonist [3H](-)-
sulpiride (final concentration, 5 nM) for 3 hrs at 4˚C with or without unlabeled haloperidol (10 µM).
Membranes were harvested and radioactivity was measured.
Immunoblot
To confirm expression of arrestin3 and/or GRK2, cells were washed twice with cold PBS and lysed in
ice-cold RIPA lysis buffer (Millipore, Billerica, MA) containing phosphatase inhibitor cocktail set II (1:100)
and protease inhibitor cocktail set III (1:200) for 20 min with gentle agitation at 4˚C. Samples were then
centrifuged for 15 min (14,000g, 4˚C) and the protein concentration of the supernatant was determined
using a BCA protein assay. Proteins (30-40 µg total) were separated by SDS-PAGE on CriterionTM 10%
Tris-HCl precast gels (Bio-Rad, Hercules, CA) and transferred onto PVDF membranes (Millipore).
Membranes were washed once in TBS, blocked in blocking buffer (TBS containing 5% BSA) for 1 hr at
room temperature, and incubated in mouse anti-β-arrestin2 (1:300) and rabbit anti-GRK2 (1:1000)
antibodies overnight at 4˚C. After primary antibody incubation, membranes were washed three times for 5
min in TBS containing 0.1% Tween-20 (TBST; Sigma-Aldrich), incubated with Alexa Fluor® 488 goat anti-
mouse (1:500) and Alexa Fluor® 568 goat anti-rabbit (1:500) secondary antibodies diluted in blocking
buffer for 2 hrs at room temperature, then washed three times for 5 min in TBST and once for 5 min in
TBS. Fluorescence was detected using a Typhoon 9410 variable mode imager (GE Healthcare,
Piscataway, NJ). Antibody was then stripped off the membrane using RestoreTM western blot stripping

114
buffer (Thermo Scientific) per the manufacturer’s protocol and vigorously washed four times for 5 min in
TBS. The membrane was then blocked in blocking buffer for 1 hr at room temperature and incubated with
rabbit anti-actin (1:500) diluted in blocking buffer overnight at 4˚C. Membranes were then washed,
incubated in Alexa Flour® 568 goat anti-rabbit, washed again and fluorescence was detected as described
above.
Bioluminescence Resonance Energy Transfer (BRET)
BRET-based assays were used heavily in this manuscript. The following is (1) brief technical description
of BRET used in these studies, followed by (2) an in depth background and protocol comparing two types
of BRET-based assays used to measure arrestin recruitment:
1. HEK293 or HEK293T cells were transiently co-transfected with plasmids as described above. For
mVenus-Arr3 and β2-AP-EYFP recruitment to the receptor, cells were harvested and split into two plates,
one for radioligand binding assays and one for BRET assays. Forty-eight hours after transfection cells
were harvested, resuspended in assay buffer (phosphate-buffered saline (PBS) with Ca2+, MgCl2, and 0.2
µM ascorbic acid) and plated at approximately 150,000 cells per well in 96-well OptiPlates (Perkin Elmer).
Vehicle or quinpirole was added with 2 µM coelenterazine h and incubated at 25˚C. Emission of the donor
(460 nm) and the acceptor (535 nm) was then measured once at 10 min or every 5 min for 30 min using a
VictorTM X Light luminescence reader (Perkin Elmer) and the BRET ratio was calculated as described
previously. Cells used in the G protein activation, GRK2 recruitment to receptor, and arrestin3 recruitment
to β2-AP-EYFP BRET assays were prepared and assayed as described previously and were measured 2,
2, and 20 minutes, respectively, after addition of quinpirole.
For the arrestin translocation assay, pcDNA3.1 encoding wild type or mutant D2 receptors was
cotransfected with plasmids containing GRK2, Rluc8-arrestin3-Sp1, and a membrane marker mem-linker-
citrine-SH3. The Sp1 and SH3 helper peptides, adapted from the helper-interaction FRET (hiFRET)
system, were used to enhance the interaction between arrestin recruited to the plasma membrane by
receptor and the membrane marker, thus increasing the dynamic range of the assay. Dopamine and
quinpirole induced-arrestin recruitment to the membrane was measured after 10 minutes of stimulation.

115
D2-mediated inhibition of cAMP was measured using a BRET-based cAMP sensor, CAMYEL. Cells
transiently transfected with D2-WT or A4 receptor and CAMYEL were preincubated for 10 minutes with 10
μM forskolin (Sigma) to stimulate cAMP production, followed by addition of quinpirole. Quinpirole-
mediated inhibition of forskolin-induced cAMP was measured after 10 minutes.
For all BRET studies, the vehicle BRET ratio was subtracted from the quinpirole BRET ratio and
presented as a percent of the Emax fit of quinpirole at the wild type receptor. Data were analyzed by
nonlinear regression using Prism (GraphPad, San Diego, CA). To depict the variability in the activity of D2-
WT and mutant receptors across replicate experiments, we calculated the mean of the Emax values for
D2-WT, converted the Emax value for each replicate to a percentage of that value, and then determined
the mean ± SEM for all replicates.
2. Using bioluminescent resonance energy transfer (BRET) to characterize agonist-induced
arrestin recruitment to modified and unmodified G protein-coupled receptors (GPCRs)
INTRODUCTION
G protein-coupled receptors (GPCRs), which are targeted by ~25% of drugs currently on the market,
signal differentially through G proteins and arrestins. In certain disease contexts it can be desirable to
selectivity activate a single pathway 32,123. Therefore, significant efforts were invested into the discovery of
compounds that selectively activate either G proteins or arrestins (so called functionally-selective or
biased ligands). To screen for bias it is critical to accurately probe the ability of compounds to activate
these pathways. High-throughput assays that are currently commercially available for screening of drug
dependent-arrestin recruitment to GPCRs (e.g., PathHunter, DiscoveRx; Tango, Life Technologies)
require the use of modified receptors and arrestins that are fused to accessory protein fragments that
complement upon receptor activation271,272. These modifications have the potential to alter normal
signaling, and the complementation-based assays also may artificially augment the pharmacological
activity of the compounds tested (e.g., efficacy, kinetic profile).
Bioluminescence resonance energy transfer (BRET) has been used to dynamically monitor arrestin
recruitment to GPCRs. It is widely used to measure protein-protein interactions in living cells254,273-275. To

116
study protein-protein interactions in living cells using BRET, one protein is genetically fused with a donor
protein, typically a variant of Renilla luciferase, and the other is fused to an acceptor protein, typically a
variant of green fluorescent protein (GFP). When the donor and acceptor are within ~10 nm of each other
the donor non-radioactively transfers energy to and excites the acceptor, which results in emission from
the acceptor protein. Here we describe two BRET-based assays that are capable of monitoring subtype-
specific arrestin recruitment to a wide variety of GPCRs: the traditional approach in which the receptor is
labeled with a donor molecule (referred to here as the receptor-arrestin BRET assay; see Basic Protocol
1), and a novel approach that does not require receptor labeling (referred to here as the arrestin
translocation BRET assay; see Basic Protocol 2).
BASIC PROTOCOL 1
Receptor-arrestin BRET assay
A widely used cell-based assay to study ligand induced-recruitment of arrestin to receptors utilizes
receptors that are fused to a BRET donor (typically Renilla luciferase - Rluc or Rluc8) and arrestins that
are fused to an acceptor (typically variants of GFP, e.g., YFP or Venus)165 (MethodsFigure 1A). Rluc must
be fused to the cytoplasmic C-terminus of receptors, either directly or through linkers (e.g., the amino
acids SGGGS) that facilitate proper folding of both proteins and prevent steric hindrance between the
receptor and the sensor. Receptors are generally linked to the donor, e.g., Rluc8 276). N- or C- terminally
acceptor fused-arrestins have been described previously 149,165,277.
The receptor-arrestin BRET assay can be used to screen compounds in a medium- to high-throughput
manner in immortalized mammalian cell lines including HEK293 and CHO 165. Receptor and arrestin
biosensors can be transiently or stably transfected, and can be co-expressed with G protein receptor
kinases (GRKs) to enhance agonist-dependent phosphorylation of the receptor and subsequent arrestin
recruitment 225. This protocol can be applied to a wide variety of GPCRs. Here, as an example, we
describe the use for recruitment BRET of Venus fused to the N-terminus of arrestin-3 (Venus-arrestin3) to
dopamine D2 receptor (D2R) fused with Rluc8 at its C-terminus (D2R-Rluc8) in transiently transfected
HEK293T cells (Methods Figure 1A).

117
Materials
Mammalian expression plasmids (these plasmids that are not commercially available and can be
obtained from authors for non-profit use):
Plasmid encoding donor-fused GPCR, e.g., pcDNA3.1-D2R-linker-Rluc8
Plasmid encoding acceptor-fused arrestin, e.g., pIRES-puro-Venus-linker-arrestin-3
Plasmid encoding GRK, e.g., pcDNA3.1-GRK2
Empty vector plasmid, e.g., pcDNA3.1 (Life Technologies)
HEK293T cells (ATCC, CRL-3216)
HEK293T culture medium (see recipe below)
Dulbecco’s Phosphate buffered saline, e.g., DPBS; Cellgro (21-031-CV)
DPBS, pH 7.4, 40mg/L sodium bisulfite (sodium bisulfite is used in the assay buffer to reduce the level of
dopamine oxidation and is not universally required)
Glucose stock (0.5 M)
DPBS, pH 7.4, 5 mM glucose
Trypsin, e.g., Cellgro, 25-052-Cl
Polyethyleimine, 1 µg/µL stock, e.g., PEI; Linear, MW 25,000, Polysciences (23966-2); see supporting
protocols for details on resuspension
DMEM, e.g., Gibco, 11965-092)
Dimethyl Sulfoxide
Dopamine hydrochloride, e.g., Sigma-Aldrich, H8502
S(-)-Sulpiride, e.g., Sigma RBI (S7771)
22 mM or 44 mM stocks of compounds of interest (e.g., dopamine, sulpiride) in appropriate solvent (e.g.,
distilled water, DMSO), depending on whether screening is planned in agonist or antagonist mode,
respectively (see below)
10 cm tissue culture plates, e.g., BD Falcon (353003)
Compound plate, e.g., Greiner 96-well V-bottom plates (651101)
BRET assay plate, e.g., Greiner 96-well white plates, Flat bottom (655075) or PerkinElmer Black/White
96-well Isoplate (6005030)

118
Pipette basin, e.g., USA scientific (2320-2620)
12-channel multichannel pipettor, e.g., Labsystems Finnpipette 50-300 µL (Z368989), or 5-50 µL
(Z678031)
Repeater pipettor, e.g. Eppendorf Repeater Plus Pipettor (022260201)
5 mM coelenterazine H in absolute ethanol, e.g., Dalton (DC001437) or NanoLight (301); see supporting
protocols for details on resuspension
DPBS, pH 7.4, 40 mg/L sodium bisulfite, 50 µM coelenterazine H
Plate reader for luminescence, fluorescence and BRET detection, e.g., Pherastar FS, BMG; Tecan F500
Software data for analysis (Microsoft Excel and GraphPad Prism)
HEK293T Culture Medium
DMEM (Gibco, 11965-092) supplemented with:
10% fetal bovine serum, e.g., FBS, Atlanta Biologics (D13056)
100 U/ml penicillin-streptomycin, e.g., Cellgro (30-002-Cl)
Protocol steps
The following protocol is described for either a single dopamine agonist curve or sulpiride antagonist
curve at a single dose of dopamine. However, this procedure can be scaled up, depending on the number
of compounds to be screened.
Note: All mammalian tissue culture should be carried out using aseptic technique in a laminar flow hood.
Cells should be maintained in an incubator at 37°C at 5% CO2.
Day 1. Cell seeding
The following is described for the use of HEK293T cells, in this case with a pre-prepared, fully confluent
10 cm plate of cells.
1. Aspirate media. Wash plates with 3 mL sterile DPBS and aspirate.
2. Add 1 mL trypsin. Incubate at room temperature for 30 seconds to 1 minute.
3. Detach cells with 5 mL HEK293T culture medium and place in 15 mL conical tube.
4. Spin for 3 minutes at 600 rcf at room temperature.
5. Aspirate media. Resuspend in 15 mL HEK293T culture medium.
6. Count cells using a hemocytometer or cell counter.

119
7. Seed 3-4 million HEK293T cells into a 10 cm tissue culture plate in a total volume of 10 mL.
Day 2. Transfection
8. After 24 hours, prepare plasmids for transient transfection. Combine the appropriate amounts of
each plasmid, including the donor-fused receptor, the acceptor-fused arrestin, and if desired, the
appropriate amount of GRK, in a 1.5 mL microfuge tube. The amount of each plasmid will need to
be optimized to obtain the appropriate expression level as indicated by luminescence and
fluorescence on the day of the assay. The amounts of DNA will vary, depending on the identity of
the receptor-donor construct and the efficiency of the transient transfection. In general, the
highest signal-to-noise ratio will be obtained when the acceptor is in stoichiometric excess. Adjust
the total amount of plasmid DNA to 20 µg using the empty vector plasmid. As an example, for the
D2R-arrestin-3 BRET, use the following amounts of plasmid DNA [D2R-linker-Rluc8 (0.2 µg);
Venus-linker-arrestin-3 (8 µg); if desired, GRK2 (5 µg); pCDNA3.1 (6.8 or 11.8 µg the empty
vector depending on whether GRK2 is included)].
9. In a tissue culture hood, add non-supplemented DMEM to the tube containing the plasmid DNA
so that the final volume is 500 µL.
10. Vortex the 1 µg/µL PEI stock solution.
11. In a 1.5 mL microfuge tube, add non-supplemented DMEM, followed by PEI pipetted directly into
the DMEM. Use optimized PEI ratio as determined by PEI optimization (see support protocol).
The final volume of the DMEM/PEI solution should be 500 µL.
12. Vortex the DMEM/PEI solution well.
13. Add 500 µL of DMEM/PEI solution to 500 µL of DMEM/DNA solution.
14. Vortex. Incubate at room temperature for 15 minutes.
15. Drip pipette the 1 mL mixture onto the media of the 10 cm plate containing HEK293T cells. Gently
rock the 10 cm plate back and forth to mix, and return the plate to the incubator.
Day 3.
16. After 24 hours, aspirate the media and replace with 10 mL fresh HEK293T cell culture media.
Day 4. BRET assay

120
All subsequent steps do not require aseptic technique and can be performed outside of the tissue culture
hood.
Compound preparation
Agonist mode
The following is described for screening the activity of an agonist in a dose-dependent manner. In a 96-
well, V-bottom compound plate, prepare in triplicate an appropriate dilution series of agonist depending
on its expected activity range. In each well of a 96-well BRET assay plate, 45 µL of compound will be
added to a final volume of 100 µL. Therefore, each concentration needs to be 2.22-fold higher than the
desired final concentration to adjust for this dilution. The preparation of a dilution series of dopamine for a
single curve ranging from 10 pM to 100 µM is described below as an example:
a) Dilute 3 µL of the freshly made 22 mM dopamine stock 100-fold into a final volume of 300 µL
DPBS with sodium bisulfite for a maximum concentration of 222 µM.
b) From this high concentration, carry out a 10-fold dilution series to a minimum concentration of
22 pM (a total of seven dilutions).
c) Aliquot 60 µL of each individual dilution into three wells of the V-bottom plate, with an
additional three wells for the vehicle (DPBS with sodium bisulfite). This dilution series (24
wells in total) can either be aliquoted into three columns or two rows of the V-bottom
compound plate 96-well plate.
Antagonist mode
To screen the activity of an antagonist in a dose-dependent manner, a series of appropriate dilutions of
the antagonist (to yield final concentrations that range from ineffective to those that completely suppress
receptor activity) are prepared in triplicate in a 96-well, V-bottom compound plate, together with a single
dose of agonist (typically at its EC80 concentration, i.e., the concentration that elicits 80% of the maximum
response). If the inhibitory constant (Ki) of a particular compound at the receptor of interest is known, the
dilution series can be adjusted appropriately around this concentration. Otherwise, a broad concentration
range of 10 pM to 100 µM should be used initially to ensure that antagonist activity is detected. In each
well of a 96-well BRET assay plate, 22.5 µL of antagonist and agonist will be injected into final volume of
100 µL. Therefore, each concentration needs to be set to 4.44-fold higher than the desired final

121
concentration. The preparation of a dilution series of sulpiride for a single curve ranging from 10 pM to
100 µM, as well as a single dose of dopamine at 1 µM is described below as an example:
a) Dilute 1.5 µL of the 44 mM sulpiride stock 100-fold into a final volume of 150 µL DPBS with
sodium bisulfite for a maximum concentration of 444 µM.
b) From this high concentration carry out a 10-fold dilution series to a minimum concentration of
44 pM (a total of seven dilutions).
c) Aliquot 30 µL of each individual dilution into three wells of the V-bottom plate, with an
additional three wells for the vehicle to serve as a negative control (DPBS with sodium
bisulfite). This dilution series (24 wells in total) can either be aliquoted into three columns or
two rows of a 96-well plate.
d) Prepare 1 mL of 4.44 µM dopamine in a 1.5 mL microfuge tube. Aliquot 30 µL into 24 wells of
a 96-well plate in a similar format as that performed for the sulpiride antagonist dilution
series.
Note: For compound preparation in both agonist and antagonist mode, some compounds are
dissolved in DMSO or other solvents that may affect the BRET assay, and therefore it is
critical to adjust each dilution so that the concentration of solvent is equivalent throughout the
entire concentration curves.
Cell preparation
17. Aspirate the media from the transfected 10 cm plate.
18. Wash the plate with 3 mL DPBS. Be careful not to detach the cells.
19. Aspirate the DPBS.
20. Detach by scrapping with a pipettor using a volume of 2.5 mL DPBS with 5 mM glucose and
transfer to a 15 mL conical tube.
21. Using a multichannel pipette, aliquot 45 µL of cells into wells from either three columns or two
rows of the BRET assay plate, depending on the format chosen for the compound plate.
Perform BRET experiment
22. Dilute 10 µL of 5 mM coelenterazine stock in DPBS 100-fold to a concentration of 50 µM (final
volume of 1 mL). Because coelenterazine is light-sensitive, store it in a dark tube.

122
Agonist mode
23. At time 0 minutes, use a repeater pipettor to inject 10 µL of 50 µM coelenterazine into each well
of the 96-well BRET assay plate containing cells. Stagger the injection time for each well to
account for the amount of time it will take the plate reader to read one well (~1-2 seconds). Inject
in the pattern that the plate reader will read across the plate (e.g., in a serpentine fashion across
columns or rows).
24. After 8 minutes, inject 45 µL of agonist one row or column at a time using a multipipettor.
Stagger the injection to account for the amount of time it will take the plate reader to read each
entire row or column (~10-20 seconds).
25. Read the plate after 2, 10, and 20 minutes from the start of agonist injection using a BRET plate
reader with detection filters or monochrometers set for Rluc8 (~485 nm) and Venus (~525nm).
Antagonist mode
1. Pre-incubate the antagonist for the desired amount of time prior to the addition of agonist (10
minutes for sulpiride) by injecting 22.5 µL of antagonist one row or column at a time using a
multichannel pipettor. Stagger the injection into each row or column ~10-20 seconds to account
for the amount of time it will take the plate reader to read each well.
2. Eight minutes prior to the injection of agonist, using a repeater pipettor inject 10 µL of 50 µM
coelenterazine into each well of the 96-well BRET assay plate containing cells. Stagger the
injection time for each well to account for the amount of time it will take the plate reader to read
one well (~1-2 seconds). Inject in the pattern that the plate reader will read across the plate (e.g.,
in a serpentine fashion across columns or rows).
3. Inject 22.5 µL of agonist one row or column at a time using a multichannel pipettor. Stagger the
injection to account for the amount of time it will take the plate reader to read each row or column
(~10-20 seconds).
4. Read the plate after 2, 10, and 20 minutes from the start of agonist injection using a BRET plate
reader with detection filters set for Rluc8 (~485 nm) and Venus (~525nm).

123
Data Analysis
5. Export the raw data for each filter set. Ensure that the luminescence counts for the 485 nm filter
(which corresponds to emission from Rluc8) is not saturated. If it is saturated, reduce the amount
of plasmid DNA encoding for the donor-fused protein. Generally these counts should be in the
range of ~100,000-1,000,000. However, this will depend greatly on the instrument and exact filter
sets used, and will need to be determined empirically.
6. In Microsoft Excel, calculate the BRET ratio by dividing the raw counts from the 525 nm filter set
by that of the 485 nm readings.
7. Organize the data with the appropriate compound concentrations.
8. Import into a points-only, XY plot in GraphPad Prism, with three replicate values.
9. Fit the data to a non-linear regression curve. For fitting an agonist curve or antagonist curves, use
the log(agonist) vs. response or log(inhibitor) vs. response fits, respectively.
To calculate the compound-induced effect, transform the Y-values using the Y=Y-K function with the K-
value being the bottom fit of the non-linear regression. Examples of dopamine agonist and sulpiride
antagonist curves are shown in Methods Figure 1B.
ALTERNATE PROTOCOL
Arrestin translocation BRET assay
Like the commercially available PathHunter and Tango assays, fusion of a protein directly to the C-
terminus of the receptor in the receptor-arrestin BRET assay may affect receptor function. Therefore, we
have developed a novel BRET based-assay to overcome this problem 225. Rather than being directly
fused to the receptor, the BRET sensor is instead fused to an unrelated plasma membrane marker.
Translocation of arrestin to the plasma membrane by activated receptor increases the proximity of
arrestin to this marker, resulting in increased resonance energy transfer (RET). Arrestin translocation to
the plasma membrane is dynamically regulated as receptor antagonists can reverse agonist induced-

124
arrestin translocation. Moreover, this assay is capable of characterizing GPCR ligands with varying
efficacy, potency, and kinetic profiles, potentially in a high-throughput manner.
Specifically, in immortalized mammalian cells lines, e.g., human embryonic kidney cells (HEK293),
unlabeled GPCRs are coexpressed with Rluc8-arrestin-3 labeled with the weak helper peptide Sp1 at its
C-terminus (Rluc8-arrestin3-Sp1) and a membrane marker composed of a doubly palmitoylated fragment
of GAP43 linked to citrine and the weak helper peptide SH3 through a serine and glycine rich linker
(mem-linker-citrine-SH3). Although not strictly necessary for the detection of arrestin translocation to the
plasma membrane, the Sp1 and SH3 helper peptides, adapted from the helper-interaction FRET
(hiFRET) system described recently 278, are used to enhance the interaction between arrestin recruited to
the plasma membrane by receptor and the membrane marker. Although this modification moderately
increases the basal signal between the plasma membrane marker and arrestin, it also increases the
dynamic range of the assay. As in the receptor-arrestin BRET assay, G protein receptor kinases (GRKs)
can be coexpressed to enhance receptor phosphorylation and arrestin recruitment.
Unlike the commercially available assays and the receptor-arrestin BRET assay, this assay can be
used to screen any GPCR by simply co-expressing the unlabeled receptor with the arrestin and plasma
membrane biosensors. Therefore, this assay does not require the generation and optimization of a fusion
receptor. Here we use translocation of arrestin-3 to the plasma membrane by D2R as an example
(Methods Figure 1C).
Materials
Mammalian expression plasmids:
Plasmid encoding for GPCR (e.g., pcDNA3.1-D2R)
Plasmid encoding for Rluc8-Arrestin-3-Sp1
Plasmid encoding for mem-linker-citrine-SH3
All other reagents and protocol steps are identical to that described for the receptor-arrestin BRET assay
outlined above, except for plasmid DNA preparation described below.
The procedure for the transient transfection of HEK293T cells for the arrestin translocation assay is
identical to the procedure described above except for the plasmids used. In this case, the donor is the
Rluc8-Arrestin-3-Sp1 and the acceptor is mem-linker-citrine-SH3. However, unlike in the receptor-

125
arrestin BRET assay, the receptor is not fused with a BRET sensor. The following amounts of plasmid
DNA is an example of the amounts used for transfection [Rluc8-Arrestin-3-Sp1 (0.25 µg); mem-linker-
citrine-SH3 (5 µg); if desired, GRK2 (5 µg); D2R (2µg); pCDNA3.1 (4.8 or 9.8 µg the empty vector
depending on whether GRK2 is included)].
For the rest of the protocol, see direct arrestin-receptor BRET protocol above. Examples of
dopamine agonist and sulpiride antagonist curves are shown in Methods Figure 1D.
SUPPORT PROTOCOLS
Support Protocol 1
Polyethylenimine (PEI) preparation and optimization
Different lots of PEI, even if from the same manufacturer, vary in activity (the efficiency of transfection).
PEI is also cytotoxic to cells. Therefore, the DNA:PEI ratio must be optimized for each lot to maximize
transfection efficiency and reduce cytotoxicity.
PEI Stock (1 µg /µL) stock preparation
1. Add 25 mg of PEI to 25 mL dH20 and stir continuously with a stir bar.
2. Add concentrated HCl dropwise to the solution to adjust the pH to less than 2.0.
3. Stir until the PEI is dissolved (30 minutes to 1 hour). Maintain a pH of less than 2.0 throughout
this step.
4. Add concentrated NaOH dropwise to the solution to adjust the pH to 7.2.
5. Sterile-filter solution in a tissue culture hood using a 0.2 micron filter.
6. Aliquot the solution into 1.5 mL microfuge tubes (1 mL/tube).
7. Store at -20° C.
PEI transfection optimization
To optimize that DNA:PEI ratio, utilize a similar protocol as that performed in the arrestin BRET assays
described above (Day 1-3), except with the following modifications:
Day 1. Cell seeding
1. Seed six 10 cm plates with 3-4 million cells (e.g., HEK293T).

126
Day 2. Transfection
2. Prepare six 1.5 mL microfuge tubes, each containing the 20 µg of plasmid DNA described in the
arrestin BRET assays.
3. Prepare mixtures of a 1:1 to 1:6 ratio of DNA:PEI (either 20, 40, 60, 80, 100, or 120 µL PEI to 20
µg of plasmid DNA) and transfect into 10 cm plates as described in the protocol above for the
arrestin BRET assays.
Day 3.
4. After 24 hours, aspirate the media and replace with 10 mL fresh HEK293T cell culture media.
Day 4. Assessment of transfection efficiency
5. Before preparation of cells, take note of the cell health for each condition. At higher PEI levels,
cells may noticeably be unhealthy or detached from the 10 cm plate.
6. Prepare cells as described above and aliquot 45 µL of cells in triplicate from each transfection
into a 96-well BRET plate (a total of 18 wells).
7. Adjust the volume in each well to a final volume of 90 µL using 45 µL DPBS with sodium bisulfite.
8. Inject 10 µL of DPBS with sodium bisulfite, 50 µM coelenterazine H.
9. Read the plate after 8 minutes from the start of coelenterazine H injection using a BRET plate
reader with detection filters set for Rluc8 (~485 nm) and Venus (~525 nm).
10. Compare the luminescence counts from the 485 nm filter, using these data as a measure of
transfection efficiency across the PEI titration range. Select the ratio that results in the highest
luminescence with the least cell death for all further experiments using this lot of PEI.
Support Protocol 2
Coelenterazine H preparation
Coelenterazine H is temperature, light and oxygen sensitive and will lose activity over time. Because the
amount of coelenterzine H purchased is typically in great excess of the amount required for a single
experiment, it must be stored properly to avoid decomposition. Take note of the following upon
preparation of the stock:

127
1. Dissolve the coelenterzine H in absolute ethanol to the appropriate concentration (5 mM) under
low light conditions, and aliquot into 1.5 mL microfuge tubes.
2. After aliquoting, fill each tube with enough argon or nitrogen gas to cover the solution and
displace atmospheric air.
3. Seal each tube with Parafilm and store at -20° C in the dark. This reagent is typically stable for at
least 3 years under these conditions.
COMMENTARY
Background Information
Arrestin and drug development
GPCRs canonically couple to heterotrimeric G proteins, to initiate a variety of downstream signaling
processes. These receptors also recruit arrestins, which terminate G protein signaling and facilitate
receptor internalization 64. It has been shown recently that arrestins, once recruited to GPCRs, can signal
independently of G proteins by acting as scaffolds for a variety of signaling molecules 64. Some ligands
(so called functionally selective or biased ligands) can selectivity activate either G protein or arrestin, and
may be superior medications for diseases in which one pathway downstream of a target receptor is
associated with efficacy and the other is associated with side effects 123. Indeed, substantial efforts have
gone towards the development of these ligands, some of which are currently in clinical trials (TRV027,
TRV130; Trevena; clinicaltrials.gov).
There are a number of commercially available assays for screening G protein activity, both at the
receptor-G protein level and downstream of the activation of G protein 272. However, screening for arrestin
activity is more limited due to a paucity of reliable readouts specifically downstream of arrestin. Thus,
most available assays for screening arrestin activity (BRET based-assays; PathHunter, DiscoveRx;
Tango, Life Technologies) rely on monitoring arrestin recruitment to receptor 272. Commercially available
assays, including PathHunter and Tango, require the use of modified receptors that are fused to
accessory proteins that pseudo-irreversibly complement upon recruitment: e.g., in the PathHunter assay,
two inactive fragments of β-galactosidase recombine to form an active enzyme upon arrestin recruitment

128
to the receptor 271. These accessory proteins in and of themselves have affinity for each other, and thus
may artificially enhance the stability the receptor-arrestin complex. Additionally, protein complementation
usually is not reversible. Therefore, it is difficult to accurately characterize pharmacological properties
such as efficacy, potency, and kinetics relative to reference agonists because these assays are inherently
at non-equilibrium conditions.
BRET as a tool to study GPCR pharmacology
Bioluminescence resonance energy transfer (BRET) is a photophysical phenomenon that involves non-
radiative transfer of energy from a donor (usually the product of a luciferase-catalyzed oxidation reaction)
to a nearby acceptor (most often a fluorescent protein). This is seen in nature where BRET allows marine
organisms to emit bright green rather than blue light 279,280. In pharmacological research, BRET has been
widely applied to detect dynamic protein-protein interactions, to monitor conformational changes in
individual proteins and within multi-protein complexes, and to report on changes in cellular activity and
metabolism 281. BRET has been especially useful for studying the structure and function of G protein-
coupled receptors (GPCRs), as well as their interactions with G proteins and arrestins, and is suitable for
medium- to high-throughput drug screening.
Generally, implementation of BRET assays is straightforward, as both donor and acceptor can be
genetically encoded as fusion proteins, and because BRET assays are relatively simple to perform and
quantify without specialized equipment. One of the advantages of BRET is that this assay is extremely
sensitive. Because BRET is based on luminescence, there is no background signal from cellular
autofluorescence and photons can be collected for long periods of time without a significant accumulation
of noise. Another advantage is that unlike Förster (or fluorescence) resonance energy transfer (FRET),
there is no possibility of unintended direct excitation of the acceptor; therefore, error is not introduced by
correction procedures. Additionally, BRET is ratiometric and therefore insensitive to variables such as cell
number and transfection efficiency. As a consequence, photon emission ratios can be measured with
very high precision and reproducibility, allowing detection of very weak BRET efficiency.

129
Critical Parameters and Troubleshooting
Generating Fusion Proteins
The most widely used BRET based-assay described above (Basic Protocol) requires the fusion of a
BRET sensor at the C-terminus of receptors. As in the commercially available PathHunter and Tango
assays, this may affect receptor function. BRET requires the fusion of relatively large probes (luciferases
and fluorescent proteins) to proteins of interest. The presence of these probes can easily change the
behavior of the proteins being studied in an unpredictable manner. Thus, it is necessary to ascertain that
luciferase-fused receptors are still active, i.e., can activate G proteins comparably to unfused WT
receptors 282. To achieve this, receptors are often connected to luciferase via a flexible linker 254.
Poor expression or disruption of the activity of receptor-Rluc fusions requires further optimization, either
by extending the linker length, introducing a linker with different physical properties, e.g., a rigid linker
composed of one or more repeats of EAAAK, or by exchanging the donor and acceptor on the receptor
and arrestin. These modifications may help facilitate the proper folding of the receptor and BRET sensor
domains of the fusion protein. However, they can also affect both the proximity and orientation of donor
and acceptor moieties relative to one another, which dictate the degree of RET between the receptor and
arrestin fusion proteins and, thus, the dynamic range of the assay.
N- or C-terminal acceptor-fused arrestin constructs have been developed previously, which allow for
characterization of arrestin recruitment in a subtype-specific manner 149,165,277. Although arrestins with C-
terminally fused GFP or Venus are recruited to receptors, caution must be taken given that this region of
the protein plays an important role in arrestin response to the receptor-attached phosphates 283-288.
Disruption of this phosphate sensor may increase basal interactions between arrestin and clathrin or AP-2
66,289.
Specific and non-specific BRET
In order for BRET to occur, the energy donor and acceptor must be in close proximity (~10 nm).
Therefore, as a general rule BRET signals can be generated in cells when either: 1) donor- and acceptor-
fused proteins are specifically associated, either directly or as part of a macromolecular complex, or 2)
when both donor and acceptor are associated with the same membrane, even without a stable direct or
indirect interaction between the two proteins. BRET signals produced by these two mechanisms are

130
referred to as “specific” and “nonspecific” BRET, respectively, although the latter is also sometimes
referred to as “bystander” BRET. Specific and nonspecific signals are not mutually exclusive, and both
can be useful for pharmacological assays.
In the case of the receptor-arrestin BRET assay (Basic Protocol), the increase in BRET seen upon
activation of the receptor is presumably due to an increase in the direct interaction between donor-fused
receptor and acceptor-fused arrestin. However, it is also possible that the increase in BRET is
nonspecific, due to activation of endogenous receptors that are activated by the compound of interest.
For example, HEK cells express high levels of chemokine receptor CXCR4 163, and off-target activation of
this receptor may increase the nonspecific interaction between arrestin recruited to the plasma membrane
by CXCR4 and the fused receptor of interest. Therefore, compounds identified as hits in drug screens
must be validated by control experiments using specific antagonists of the fused receptor of interest that
fully inhibit agonist signaling via that particular receptor.
The arrestin translocation assay (Alternate Protocol) takes advantage of nonspecific BRET in order to
obviate the use of fused receptors. Donor-fused arrestin is recruited to the membrane by an unfused
receptor of interest from the cytosol. Due to random proximity, the increased localization of arrestin in this
compartment can be sufficient to increase the nonspecific BRET between this sensor and many proteins
localized in the plasma membrane (in this case a fragment of GAP43 that is doubly palmitoylated). This is
not the case if the two proteins are localized in different micro-compartments of the plasma membrane
(e.g., clathrin coated pits).
The magnitude of the nonspecific interaction can be increased in several ways: 1) increasing the
expression level of the unfused receptor of interest, which increases the total amount of cytosolic arrestin
that can be recruited to the membrane upon receptor activation, 2) overexpression of GRKs, which
phosphorylate the receptor and increase the affinity of the receptor for arrestin, 3) increasing the
expression level of the acceptor-fused plasma membrane marker, which increases the probability that
arrestins recruited to the plasma membrane will be in close proximity to or randomly collide with the
membrane marker. This assay is also subject to nonspecific BRET associated with off-target activation of
endogenous receptors, and thus it is critical to use an appropriate antagonist control to ensure agonist
activity is solely mediated by the receptor of interest.

131
Negative controls
Regardless of the assay used, it is helpful to include negative controls. Arrestin-KNC mutants that
have substitutions of 12 key receptor-binding residues on the surface and thus do not bind GPCRs were
developed recently 277. The use of these control arrestins N-terminally fused with Venus, which are the
same size as wildtype arrestin, allow for the detection with reasonable confidence of “basal” (e.g.,
agonist-independent) interaction of arrestin-3 with GPCRs, which KNC and some other arrestin-3 mutants
do not demonstrate 75,282,290. Ideally, to have full set of controls, one would also require receptor mutants
that do not bind either G proteins or arrestins. In some cases, such mutants have recently become
available (reviewed in 291).
Interpretation of arrestin recruitment
The commercially available PathHunter and Tango assays, as well as both BRET-based arrestin
assays, measure the degree of recruitment of arrestin from the cytosol to the plasma membrane.
However once recruited to the receptor, arrestin can adopt multiple conformations that may be involved
differentially in various arrestin fuctions, i.e., inhibition of G protein-mediated signaling, internalization, and
activation of downstream signaling pathways 64,292. These conformations can be differentially stabilized in
a ligand-specific manner, and this has been shown using an alternative BRET assay in which arrestin is
doubly fused with both a donor and acceptor sensor 293. Upon activation of the receptor, arrestin is
recruited to the receptor and undergoes a conformational change, which is indicated by a change in
BRET. Unfortunately, this assay has a poor dynamic range relative to both BRET-based arrestin assays,
and is not suitable for robust drug screening. Additionally, there is a paucity of available screening assays
that read out the signaling that is unambiguously downstream of arrestin. For example, phospho-ERK
(pERK) assays are commonly used to study arrestin activity, but interpretation of the results of this assay
is complicated by the contribution of G protein to this endpoint 294-296. Thus, caution must be applied in
interpreting arrestin recruitment as a readout of arrestin activity because arrestin-dependent inhibition of
G protein signaling, internalization of the receptor, and activation of downstream signaling proteins are
not measured.

132
Anticipated Results
The background BRET ratio seen in the arrestin translocation is generally higher than that in the receptor-
arrestin BRET assay, which is indicative of higher basal interaction between arrestin and the plasma
membrane assay (~0.5-0.7 and ~0.4-0.5, respectively, depending on the expression level of the
biosensors and BRET filter sets specific to each plate reader). This is due the hiBRET peptides (Sp1 and
SH3) used in this assay. The maximal drug-induced BRET values (ranging from 0 to ~0.3) depend on the
ability of the receptor to recruit arrestin, the expression level of receptor and the other components of the
assay, and the presence or absence of overexpressed GRKs. These values are not consistently higher in
one arrestin BRET assay versus another, and therefore these assays must be compared empirically.
Time Considerations
For transient transfections, both assays are generally performed over a period of four days and include
cell seeding, transfection and expression over a period of 24-48 hours, and processing and assaying.
When stable cell lines are available, only cell seeding, processing and assaying are needed, which
reduces the overall time by 24-48 hours. Overall, assaying can take anywhere from 10 minutes to two
hours depending on the number of samples and plates being run. The time it takes to read a full 96-well
plate can take anywhere from one to five minutes depending on the plate reader.
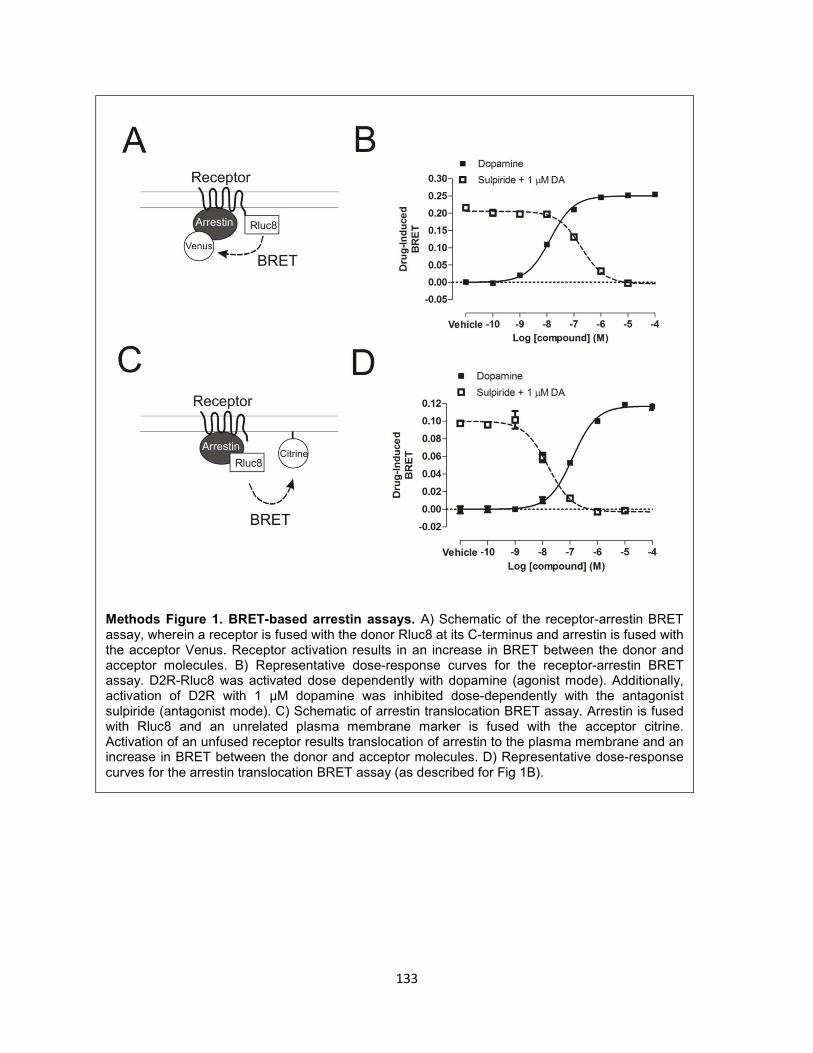
133
Methods Figure 1. BRET-based arrestin assays. A) Schematic of the receptor-arrestin BRET assay, wherein a receptor is fused with the donor Rluc8 at its C-terminus and arrestin is fused with the acceptor Venus. Receptor activation results in an increase in BRET between the donor and acceptor molecules. B) Representative dose-response curves for the receptor-arrestin BRET assay. D2R-Rluc8 was activated dose dependently with dopamine (agonist mode). Additionally, activation of D2R with 1 μM dopamine was inhibited dose-dependently with the antagonist sulpiride (antagonist mode). C) Schematic of arrestin translocation BRET assay. Arrestin is fused with Rluc8 and an unrelated plasma membrane marker is fused with the acceptor citrine. Activation of an unfused receptor results translocation of arrestin to the plasma membrane and an increase in BRET between the donor and acceptor molecules. D) Representative dose-response curves for the arrestin translocation BRET assay (as described for Fig 1B).

134
References
1 Goodman, L. S., Brunton, L. L., Chabner, B. & Knollmann, B. r. C. Goodman & Gilman's pharmacological basis of therapeutics. 12th edn, (McGraw-Hill, 2011).
2 Abi-Dargham, A. Schizophrenia: overview and dopamine dysfunction. The Journal of clinical psychiatry 75, e31, doi:10.4088/JCP.13078tx2c (2014).
3 Kennedy, J. L., Altar, C. A., Taylor, D. L., Degtiar, I. & Hornberger, J. C. The social and economic burden of treatment-resistant schizophrenia: a systematic literature review. International clinical psychopharmacology 29, 63-76, doi:10.1097/YIC.0b013e32836508e6 (2014).
4 Escudero, I. & Johnstone, M. Genetics of schizophrenia. Current psychiatry reports 16, 502, doi:10.1007/s11920-014-0502-8 (2014).
5 Ayhan, Y., Sawa, A., Ross, C. A. & Pletnikov, M. V. Animal models of gene-environment interactions in schizophrenia. Behavioural brain research 204, 274-281, doi:10.1016/j.bbr.2009.04.010 (2009).
6 Lichtenstein, P. et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 373, 234-239, doi:10.1016/S0140-6736(09)60072-6 (2009).
7 Rodriguez-Murillo, L., Gogos, J. A. & Karayiorgou, M. The genetic architecture of schizophrenia: new mutations and emerging paradigms. Annual review of medicine 63, 63-80, doi:10.1146/annurev-med-072010-091100 (2012).
8 Karam, C. S. et al. Signaling pathways in schizophrenia: emerging targets and therapeutic strategies. Trends in pharmacological sciences 31, 381-390, doi:10.1016/j.tips.2010.05.004 (2010).
9 Carlsson, A., Lindqvist, M. & Magnusson, T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 180, 1200 (1957).
10 Beaulieu, J. M. & Gainetdinov, R. R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacological reviews 63, 182-217, doi:10.1124/pr.110.002642 (2011).
11 Anden, N. E. et al. Demonstration and Mapping out of Nigro-Neostriatal Dopamine Neurons. Life sciences 3, 523-530 (1964).
12 Dahlstroem, A. & Fuxe, K. Evidence for the Existence of Monoamine-Containing Neurons in the Central Nervous System. I. Demonstration of Monoamines in the Cell Bodies of Brain Stem Neurons. Acta physiologica Scandinavica. Supplementum, SUPPL 232:231-255 (1964).
13 Ehringer, H. & Hornykiewicz, O. [Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system]. Klinische Wochenschrift 38, 1236-1239 (1960).
14 Mink, J. W. Neurobiology of basal ganglia and Tourette syndrome: basal ganglia circuits and thalamocortical outputs. Advances in neurology 99, 89-98 (2006).
15 Swanson, J. M. et al. Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychology review 17, 39-59, doi:10.1007/s11065-007-9019-9 (2007).
16 Gizer, I. R., Ficks, C. & Waldman, I. D. Candidate gene studies of ADHD: a meta-analytic review. Human genetics 126, 51-90, doi:10.1007/s00439-009-0694-x (2009).
17 Jakel, R. J. & Maragos, W. F. Neuronal cell death in Huntington's disease: a potential role for dopamine. Trends in neurosciences 23, 239-245 (2000).
18 Cyr, M., Sotnikova, T. D., Gainetdinov, R. R. & Caron, M. G. Dopamine enhances motor and neuropathological consequences of polyglutamine expanded huntingtin. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 20, 2541-2543, doi:10.1096/fj.06-6533fje (2006).
19 Hyman, S. E., Malenka, R. C. & Nestler, E. J. Neural mechanisms of addiction: the role of reward-related learning and memory. Annual review of neuroscience 29, 565-598, doi:10.1146/annurev.neuro.29.051605.113009 (2006).
20 Di Chiara, G. & Bassareo, V. Reward system and addiction: what dopamine does and doesn't do. Current opinion in pharmacology 7, 69-76, doi:10.1016/j.coph.2006.11.003 (2007).

135
21 Koob, G. F. & Volkow, N. D. Neurocircuitry of addiction. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 35, 217-238, doi:10.1038/npp.2009.110 (2010).
22 Gerfen, C. R. & Surmeier, D. J. Modulation of striatal projection systems by dopamine. Annual review of neuroscience 34, 441-466, doi:10.1146/annurev-neuro-061010-113641 (2011).
23 Tritsch, N. X., Ding, J. B. & Sabatini, B. L. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature 490, 262-266, doi:10.1038/nature11466 (2012).
24 Tritsch, N. X. & Sabatini, B. L. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76, 33-50, doi:10.1016/j.neuron.2012.09.023 (2012).
25 Tepper, J. M., Tecuapetla, F., Koos, T. & Ibanez-Sandoval, O. Heterogeneity and diversity of striatal GABAergic interneurons. Frontiers in neuroanatomy 4, 150, doi:10.3389/fnana.2010.00150 (2010).
26 Albin, R. L., Young, A. B. & Penney, J. B. The functional anatomy of basal ganglia disorders. Trends in neurosciences 12, 366-375 (1989).
27 Calabresi, P., Picconi, B., Tozzi, A., Ghiglieri, V. & Di Filippo, M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nature neuroscience 17, 1022-1030, doi:10.1038/nn.3743 (2014).
28 Cazorla, M. et al. Dopamine D2 receptors regulate the anatomical and functional balance of basal ganglia circuitry. Neuron 81, 153-164, doi:10.1016/j.neuron.2013.10.041 (2014).
29 MacLeod, R. M. & Login, I. Control of prolactin secretion by the hypothalamic catecholamines. Advances in sex hormone research 2, 211-231 (1976).
30 Neve, K. A. The dopamine receptors. 2nd edn, (Humana Press, 2010). 31 Stevens, R. C. et al. The GPCR Network: a large-scale collaboration to determine human GPCR
structure and function. Nature reviews. Drug discovery 12, 25-34, doi:10.1038/nrd3859 (2013). 32 Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there? Nature
reviews. Drug discovery 5, 993-996, doi:10.1038/nrd2199 (2006). 33 Ritter, S. L. & Hall, R. A. Fine-tuning of GPCR activity by receptor-interacting proteins. Nature
reviews. Molecular cell biology 10, 819-830, doi:10.1038/nrm2803 (2009). 34 Michino, M. et al. What can crystal structures of aminergic receptors tell us about designing
subtype-selective ligands? Pharmacological reviews 67, 198-213, doi:10.1124/pr.114.009944 (2015).
35 Kobilka, B. K. G protein coupled receptor structure and activation. Biochim Biophys Acta 1768, 794-807, doi:10.1016/j.bbamem.2006.10.021 (2007).
36 Giros, B. et al. Alternative splicing directs the expression of two D2 dopamine receptor isoforms. Nature 342, 923-926, doi:10.1038/342923a0 (1989).
37 Missale, C., Nash, S. R., Robinson, S. W., Jaber, M. & Caron, M. G. Dopamine receptors: from structure to function. Physiological reviews 78, 189-225 (1998).
38 Chien, E. Y. et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091-1095, doi:10.1126/science.1197410 (2010).
39 Newman, A. H. et al. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. Journal of medicinal chemistry 55, 6689-6699, doi:10.1021/jm300482h (2012).
40 Spijker, P., Vaidehi, N., Freddolino, P. L., Hilbers, P. A. & Goddard, W. A., 3rd. Dynamic behavior of fully solvated beta2-adrenergic receptor, embedded in the membrane with bound agonist or antagonist. Proceedings of the National Academy of Sciences of the United States of America 103, 4882-4887, doi:10.1073/pnas.0511329103 (2006).
41 Xiao, X. et al. Understanding the conformation transition in the activation pathway of beta2 adrenergic receptor via a targeted molecular dynamics simulation. Physical chemistry chemical physics : PCCP 17, 2512-2522, doi:10.1039/c4cp04528a (2015).
42 Nygaard, R. et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell 152, 532-542, doi:10.1016/j.cell.2013.01.008 (2013).
43 Kenakin, T. P. A pharmacology primer : techniques for more effective and strategic drug discovery. 4th edn, (Academic Press, 2014).
44 Guo, D., Mulder-Krieger, T., AP, I. J. & Heitman, L. H. Functional efficacy of adenosine A(2)A receptor agonists is positively correlated to their receptor residence time. British journal of pharmacology 166, 1846-1859, doi:10.1111/j.1476-5381.2012.01897.x (2012).

136
45 Sykes, D. A., Dowling, M. R. & Charlton, S. J. Exploring the mechanism of agonist efficacy: a relationship between efficacy and agonist dissociation rate at the muscarinic M3 receptor. Molecular pharmacology 76, 543-551, doi:10.1124/mol.108.054452 (2009).
46 Khilnani, G. & Khilnani, A. K. Inverse agonism and its therapeutic significance. Indian journal of pharmacology 43, 492-501, doi:10.4103/0253-7613.84947 (2011).
47 Heidbreder, C. A. & Newman, A. H. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Annals of the New York Academy of Sciences 1187, 4-34, doi:10.1111/j.1749-6632.2009.05149.x (2010).
48 Simpson, M. M. et al. Dopamine D4/D2 receptor selectivity is determined by A divergent aromatic microdomain contained within the second, third, and seventh membrane-spanning segments. Molecular pharmacology 56, 1116-1126 (1999).
49 Giorgioni, G., Piergentili, A., Ruggieri, S. & Quaglia, W. Dopamine D5 receptors: a challenge to medicinal chemists. Mini reviews in medicinal chemistry 8, 976-995 (2008).
50 Chung, K. Y. et al. Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature 477, 611-615, doi:10.1038/nature10488 (2011).
51 Vogler, O., Barcelo, J. M., Ribas, C. & Escriba, P. V. Membrane interactions of G proteins and other related proteins. Biochim Biophys Acta 1778, 1640-1652, doi:10.1016/j.bbamem.2008.03.008 (2008).
52 De Vries, L., Zheng, B., Fischer, T., Elenko, E. & Farquhar, M. G. The regulator of G protein signaling family. Annual review of pharmacology and toxicology 40, 235-271, doi:10.1146/annurev.pharmtox.40.1.235 (2000).
53 McIntire, W. E. Structural determinants involved in the formation and activation of G protein betagamma dimers. Neuro-Signals 17, 82-99, doi:10.1159/000186692 (2009).
54 Hurowitz, E. H. et al. Genomic characterization of the human heterotrimeric G protein alpha, beta, and gamma subunit genes. DNA research : an international journal for rapid publication of reports on genes and genomes 7, 111-120 (2000).
55 O'Donnell, J. K., Sweet, R. W. & Stadel, J. M. Expression and characterization of the long and short splice variants of GS alpha in S49 cyc- cells. Molecular pharmacology 39, 702-710 (1991).
56 Gagnon, A. W. et al. Identification of Gz alpha as a pertussis toxin-insensitive G protein in human platelets and megakaryocytes. Blood 78, 1247-1253 (1991).
57 Katada, T. & Ui, M. ADP ribosylation of the specific membrane protein of C6 cells by islet-activating protein associated with modification of adenylate cyclase activity. The Journal of biological chemistry 257, 7210-7216 (1982).
58 Neve, K. A., Seamans, J. K. & Trantham-Davidson, H. Dopamine receptor signaling. Journal of receptor and signal transduction research 24, 165-205 (2004).
59 Berlin, S. et al. G alpha(i) and G betagamma jointly regulate the conformations of a G betagamma effector, the neuronal G protein-activated K+ channel (GIRK). The Journal of biological chemistry 285, 6179-6185, doi:10.1074/jbc.M109.085944 (2010).
60 Fulton, S. et al. Contribution of Kv1.2 voltage-gated potassium channel to D2 autoreceptor regulation of axonal dopamine overflow. The Journal of biological chemistry 286, 9360-9372, doi:10.1074/jbc.M110.153262 (2011).
61 Martel, P., Leo, D., Fulton, S., Berard, M. & Trudeau, L. E. Role of Kv1 potassium channels in regulating dopamine release and presynaptic D2 receptor function. PloS one 6, e20402, doi:10.1371/journal.pone.0020402 (2011).
62 Yang, J. et al. Dopaminergic modulation of axonal potassium channels and action potential waveform in pyramidal neurons of prefrontal cortex. The Journal of physiology 591, 3233-3251, doi:10.1113/jphysiol.2013.251058 (2013).
63 Einhorn, L. C., Gregerson, K. A. & Oxford, G. S. D2 dopamine receptor activation of potassium channels in identified rat lactotrophs: whole-cell and single-channel recording. The Journal of neuroscience : the official journal of the Society for Neuroscience 11, 3727-3737 (1991).
64 Lefkowitz, R. J. Arrestins come of age: a personal historical perspective. Progress in molecular biology and translational science 118, 3-18, doi:10.1016/B978-0-12-394440-5.00001-2 (2013).
65 Szczepek, M. et al. Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nature communications 5, 4801, doi:10.1038/ncomms5801 (2014).

137
66 Kim, Y. M. & Benovic, J. L. Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. The Journal of biological chemistry 277, 30760-30768, doi:10.1074/jbc.M204528200 (2002).
67 Laporte, S. A., Miller, W. E., Kim, K. M. & Caron, M. G. beta-Arrestin/AP-2 interaction in G protein-coupled receptor internalization: identification of a beta-arrestin binging site in beta 2-adaptin. The Journal of biological chemistry 277, 9247-9254, doi:10.1074/jbc.M108490200 (2002).
68 Beaulieu, J. M. et al. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261-273, doi:10.1016/j.cell.2005.05.012 (2005).
69 Urs, N. M., Snyder, J. C., Jacobsen, J. P., Peterson, S. M. & Caron, M. G. Deletion of GSK3beta in D2R-expressing neurons reveals distinct roles for beta-arrestin signaling in antipsychotic and lithium action. Proceedings of the National Academy of Sciences of the United States of America 109, 20732-20737, doi:10.1073/pnas.1215489109 (2012).
70 Cross, D. A. et al. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. The Biochemical journal 303 ( Pt 1), 21-26 (1994).
71 Ribas, C. et al. The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta 1768, 913-922, doi:10.1016/j.bbamem.2006.09.019 (2007).
72 Vishnivetskiy, S. A. et al. How does arrestin respond to the phosphorylated state of rhodopsin? The Journal of biological chemistry 274, 11451-11454 (1999).
73 Chen, C. H., Paing, M. M. & Trejo, J. Termination of protease-activated receptor-1 signaling by beta-arrestins is independent of receptor phosphorylation. The Journal of biological chemistry 279, 10020-10031, doi:10.1074/jbc.M310590200 (2004).
74 Jala, V. R., Shao, W. H. & Haribabu, B. Phosphorylation-independent beta-arrestin translocation and internalization of leukotriene B4 receptors. The Journal of biological chemistry 280, 4880-4887, doi:10.1074/jbc.M409821200 (2005).
75 Gimenez, L. E. et al. Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. The Journal of biological chemistry 287, 9028-9040, doi:10.1074/jbc.M111.311803 (2012).
76 Richardson, M. D. et al. Human substance P receptor lacking the C-terminal domain remains competent to desensitize and internalize. Journal of neurochemistry 84, 854-863 (2003).
77 Gurevich, E. V., Tesmer, J. J., Mushegian, A. & Gurevich, V. V. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacology & therapeutics 133, 40-69, doi:10.1016/j.pharmthera.2011.08.001 (2012).
78 Namkung, Y., Dipace, C., Javitch, J. A. & Sibley, D. R. G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. The Journal of biological chemistry 284, 15038-15051, doi:10.1074/jbc.M900388200 (2009).
79 Li, L. et al. G protein-coupled receptor kinases of the GRK4 subfamily phosphorylate inactive GPCRs. The Journal of biological chemistry, doi:10.1074/jbc.M115.644773 (2015).
80 Sokoloff, P. et al. Localization and function of the D3 dopamine receptor. Arzneimittel-Forschung 42, 224-230 (1992).
81 Biezonski, D. K., Trifilieff, P., Meszaros, J., Javitch, J. A. & Kellendonk, C. Evidence for limited D1 and D2 receptor coexpression and colocalization within the dorsal striatum of the neonatal mouse. The Journal of comparative neurology 523, 1175-1189, doi:10.1002/cne.23730 (2015).
82 Gerfen, C. R. et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429-1432 (1990).
83 Le Moine, C. & Bloch, B. D1 and D2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. The Journal of comparative neurology 355, 418-426, doi:10.1002/cne.903550308 (1995).
84 Aubert, I., Ghorayeb, I., Normand, E. & Bloch, B. Phenotypical characterization of the neurons expressing the D1 and D2 dopamine receptors in the monkey striatum. The Journal of comparative neurology 418, 22-32 (2000).

138
85 Thibault, D., Loustalot, F., Fortin, G. M., Bourque, M. J. & Trudeau, L. E. Evaluation of D1 and D2 dopamine receptor segregation in the developing striatum using BAC transgenic mice. PloS one 8, e67219, doi:10.1371/journal.pone.0067219 (2013).
86 Hernandez-Lopez, S. et al. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. The Journal of neuroscience : the official journal of the Society for Neuroscience 20, 8987-8995 (2000).
87 Perez, M. F., White, F. J. & Hu, X. T. Dopamine D(2) receptor modulation of K(+) channel activity regulates excitability of nucleus accumbens neurons at different membrane potentials. J Neurophysiol 96, 2217-2228, doi:10.1152/jn.00254.2006 (2006).
88 Lim, S. A., Kang, U. J. & McGehee, D. S. Striatal cholinergic interneuron regulation and circuit effects. Frontiers in synaptic neuroscience 6, 22, doi:10.3389/fnsyn.2014.00022 (2014).
89 Chuhma, N., Mingote, S., Moore, H. & Rayport, S. Dopamine neurons control striatal cholinergic neurons via regionally heterogeneous dopamine and glutamate signaling. Neuron 81, 901-912, doi:10.1016/j.neuron.2013.12.027 (2014).
90 Ford, C. P. The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 282C, 13-22, doi:10.1016/j.neuroscience.2014.01.025 (2014).
91 Niquet, J. & Charli, J. In vitro expression of tyrosine hydroxylase by a subpopulation of rat melanotrophs is down-regulated by dopamine. Brain research bulletin 51, 479-484 (2000).
92 Lindgren, N. et al. Dopamine D(2) receptors regulate tyrosine hydroxylase activity and phosphorylation at Ser40 in rat striatum. The European journal of neuroscience 13, 773-780 (2001).
93 Lew, J. Y. et al. Increased site-specific phosphorylation of tyrosine hydroxylase accompanies stimulation of enzymatic activity induced by cessation of dopamine neuronal activity. Molecular pharmacology 55, 202-209 (1999).
94 Mayfield, R. D. & Zahniser, N. R. Dopamine D2 receptor regulation of the dopamine transporter expressed in Xenopus laevis oocytes is voltage-independent. Molecular pharmacology 59, 113-121 (2001).
95 Cass, W. A. & Gerhardt, G. A. Direct in vivo evidence that D2 dopamine receptors can modulate dopamine uptake. Neuroscience letters 176, 259-263 (1994).
96 Adell, A. & Artigas, F. The somatodendritic release of dopamine in the ventral tegmental area and its regulation by afferent transmitter systems. Neuroscience and biobehavioral reviews 28, 415-431, doi:10.1016/j.neubiorev.2004.05.001 (2004).
97 Bjorklund, A. & Lindvall, O. Dopamine in dendrites of substantia nigra neurons: suggestions for a role in dendritic terminals. Brain research 83, 531-537 (1975).
98 Creese, I., Burt, D. R. & Snyder, S. H. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. The Journal of neuropsychiatry and clinical neurosciences 8, 223-226 (1996).
99 Carlsson, A. Does dopamine play a role in schizophrenia? Psychological medicine 7, 583-597 (1977).
100 Kuepper, R., Skinbjerg, M. & Abi-Dargham, A. The dopamine dysfunction in schizophrenia revisited: new insights into topography and course. Handbook of experimental pharmacology, 1-26, doi:10.1007/978-3-642-25761-2_1 (2012).
101 Lyon, G. J. et al. Presynaptic regulation of dopamine transmission in schizophrenia. Schizophrenia bulletin 37, 108-117, doi:10.1093/schbul/sbp010 (2011).
102 Howes, O. D. & Kapur, S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophrenia bulletin 35, 549-562, doi:10.1093/schbul/sbp006 (2009).
103 Mailman, R. B. & Murthy, V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Current pharmaceutical design 16, 488-501 (2010).
104 Lieberman, J. A. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia: efficacy, safety and cost outcomes of CATIE and other trials. The Journal of clinical psychiatry 68, e04 (2007).
105 Lieberman, J. A. et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. The New England journal of medicine 353, 1209-1223, doi:10.1056/NEJMoa051688 (2005).
106 Harvey, P. D. Cognitive Outcomes in the CATIE Schizophrenia Trial: Why do they seem different from previous results? Psychiatry 4, 20-23 (2007).

139
107 Girgis, R. R. et al. In vivo binding of antipsychotics to D3 and D2 receptors: a PET study in baboons with [11C]-(+)-PHNO. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 36, 887-895, doi:10.1038/npp.2010.228 (2011).
108 Ginovart, N. & Kapur, S. Role of dopamine D(2) receptors for antipsychotic activity. Handbook of experimental pharmacology, 27-52, doi:10.1007/978-3-642-25761-2_2 (2012).
109 Meller, E., Bohmaker, K., Namba, Y., Friedhoff, A. J. & Goldstein, M. Relationship between receptor occupancy and response at striatal dopamine autoreceptors. Molecular pharmacology 31, 592-598 (1987).
110 Tamminga, C. A., Cascella, N. G., Lahti, R. A., Lindberg, M. & Carlsson, A. Pharmacologic properties of (-)-3PPP (preclamol) in man. Journal of neural transmission. General section 88, 165-175 (1992).
111 Shapiro, D. A. et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 28, 1400-1411, doi:10.1038/sj.npp.1300203 (2003).
112 Stroup, T. S. et al. Results of phase 3 of the CATIE schizophrenia trial. Schizophrenia research 107, 1-12, doi:10.1016/j.schres.2008.10.011 (2009).
113 Gaspar, P., Bloch, B. & Le Moine, C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. The European journal of neuroscience 7, 1050-1063 (1995).
114 Fedele, E., Andrioli, G. C., Ruelle, A. & Raiteri, M. Release-regulating dopamine autoreceptors in human cerebral cortex. British journal of pharmacology 110, 20-22 (1993).
115 Casey, D. E., Sands, E. E., Heisterberg, J. & Yang, H. M. Efficacy and safety of bifeprunox in patients with an acute exacerbation of schizophrenia: results from a randomized, double-blind, placebo-controlled, multicenter, dose-finding study. Psychopharmacology 200, 317-331, doi:10.1007/s00213-008-1207-7 (2008).
116 Heinrich, J. N. et al. Aplindore (DAB-452), a high affinity selective dopamine D2 receptor partial agonist. European journal of pharmacology 552, 36-45, doi:10.1016/j.ejphar.2006.08.063 (2006).
117 Lawler, C. P. et al. Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 20, 612-627, doi:10.1016/S0893-133X(98)00099-2 (1999).
118 Urban, J. D., Vargas, G. A., von Zastrow, M. & Mailman, R. B. Aripiprazole has functionally selective actions at dopamine D2 receptor-mediated signaling pathways. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 32, 67-77, doi:10.1038/sj.npp.1301071 (2007).
119 Kenakin, T. Functional selectivity and biased receptor signaling. The Journal of pharmacology and experimental therapeutics 336, 296-302, doi:10.1124/jpet.110.173948 (2011).
120 Kenakin, T. & Miller, L. J. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacological reviews 62, 265-304, doi:10.1124/pr.108.000992 (2010).
121 DeWire, S. M., Ahn, S., Lefkowitz, R. J. & Shenoy, S. K. Beta-arrestins and cell signaling. Annual review of physiology 69, 483-510, doi:10.1146/annurev.ph.69.013107.100021 (2007).
122 Kendall, R. T. et al. The beta-arrestin pathway-selective type 1A angiotensin receptor (AT1A) agonist [Sar1,Ile4,Ile8]angiotensin II regulates a robust G protein-independent signaling network. The Journal of biological chemistry 286, 19880-19891, doi:10.1074/jbc.M111.233080 (2011).
123 Violin, J. D., Crombie, A. L., Soergel, D. G. & Lark, M. W. Biased ligands at G-protein-coupled receptors: promise and progress. Trends in pharmacological sciences 35, 308-316, doi:10.1016/j.tips.2014.04.007 (2014).
124 Wisler, J. W. et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proceedings of the National Academy of Sciences of the United States of America 104, 16657-16662, doi:10.1073/pnas.0707936104 (2007).
125 Drake, M. T. et al. beta-arrestin-biased agonism at the beta2-adrenergic receptor. The Journal of biological chemistry 283, 5669-5676, doi:10.1074/jbc.M708118200 (2008).
126 Galandrin, S. & Bouvier, M. Distinct signaling profiles of beta1 and beta2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the

140
pluridimensionality of efficacy. Molecular pharmacology 70, 1575-1584, doi:10.1124/mol.106.026716 (2006).
127 Burris, T. P. et al. Nuclear receptors and their selective pharmacologic modulators. Pharmacological reviews 65, 710-778, doi:10.1124/pr.112.006833 (2013).
128 Bruning, J. B. et al. Coupling of receptor conformation and ligand orientation determine graded activity. Nature chemical biology 6, 837-843, doi:10.1038/nchembio.451 (2010).
129 Srinivasan, S. et al. Ligand-binding dynamics rewire cellular signaling via estrogen receptor-alpha. Nature chemical biology 9, 326-332, doi:10.1038/nchembio.1214 (2013).
130 Urban, J. D. et al. Functional selectivity and classical concepts of quantitative pharmacology. The Journal of pharmacology and experimental therapeutics 320, 1-13, doi:10.1124/jpet.106.104463 (2007).
131 Kenakin, T. & Christopoulos, A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nature reviews. Drug discovery 12, 205-216, doi:10.1038/nrd3954 (2013).
132 Rajagopal, S. et al. Quantifying ligand bias at seven-transmembrane receptors. Molecular pharmacology 80, 367-377, doi:10.1124/mol.111.072801 (2011).
133 Kenakin, T., Watson, C., Muniz-Medina, V., Christopoulos, A. & Novick, S. A simple method for quantifying functional selectivity and agonist bias. ACS chemical neuroscience 3, 193-203, doi:10.1021/cn200111m (2012).
134 Griffin, M. T., Figueroa, K. W., Liller, S. & Ehlert, F. J. Estimation of agonist activity at G protein-coupled receptors: analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. The Journal of pharmacology and experimental therapeutics 321, 1193-1207, doi:10.1124/jpet.107.120857 (2007).
135 Kikuchi, T. et al. 7-(4-[4-(2,3-Dichlorophenyl)-1-piperazinyl]butyloxy)-3,4-dihydro-2(1H)-quinolinon e (OPC-14597), a new putative antipsychotic drug with both presynaptic dopamine autoreceptor agonistic activity and postsynaptic D2 receptor antagonistic activity. The Journal of pharmacology and experimental therapeutics 274, 329-336 (1995).
136 Semba, J., Watanabe, A., Kito, S. & Toru, M. Behavioural and neurochemical effects of OPC-14597, a novel antipsychotic drug, on dopaminergic mechanisms in rat brain. Neuropharmacology 34, 785-791 (1995).
137 Amano, T. et al. Antagonizing effects of a novel antipsychotic quinolinone derivative (OPC-14597) on dopaminergic inhibition of neuronal activities in the nucleus accumbens. Progress in neuro-psychopharmacology & biological psychiatry 19, 105-116 (1995).
138 Inoue, T., Domae, M., Yamada, K. & Furukawa, T. Effects of the novel antipsychotic agent 7-(4-[4-(2,3-dichlorophenyl)-1-piperazinyl]butyloxy)-3,4-dihydro -2(1H)-quinolinone (OPC-14597) on prolactin release from the rat anterior pituitary gland. The Journal of pharmacology and experimental therapeutics 277, 137-143 (1996).
139 Fujikawa, M., Nagashima, M., Inoue, T., Yamada, K. & Furukawa, T. Partial agonistic effects of OPC-14597, a potential antipsychotic agent, on yawning behavior in rats. Pharmacology, biochemistry, and behavior 53, 903-909 (1996).
140 Heusler, P., Newman-Tancredi, A., Loock, T. & Cussac, D. Antipsychotics differ in their ability to internalise human dopamine D2S and human serotonin 5-HT1A receptors in HEK293 cells. European journal of pharmacology 581, 37-46, doi:10.1016/j.ejphar.2007.11.046 (2008).
141 Mottola, D. M. et al. Functional selectivity of dopamine receptor agonists. I. Selective activation of postsynaptic dopamine D2 receptors linked to adenylate cyclase. The Journal of pharmacology and experimental therapeutics 301, 1166-1178 (2002).
142 Kilts, J. D. et al. Functional selectivity of dopamine receptor agonists. II. Actions of dihydrexidine in D2L receptor-transfected MN9D cells and pituitary lactotrophs. The Journal of pharmacology and experimental therapeutics 301, 1179-1189 (2002).
143 Jones, C. A., Watson, D. J. & Fone, K. C. Animal models of schizophrenia. British journal of pharmacology 164, 1162-1194, doi:10.1111/j.1476-5381.2011.01386.x (2011).
144 Sun, T., Hu, G. & Li, M. Repeated antipsychotic treatment progressively potentiates inhibition on phencyclidine-induced hyperlocomotion, but attenuates inhibition on amphetamine-induced hyperlocomotion: relevance to animal models of antipsychotic drugs. European journal of pharmacology 602, 334-342, doi:10.1016/j.ejphar.2008.11.036 (2009).

141
145 Kellendonk, C. et al. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron 49, 603-615, doi:10.1016/j.neuron.2006.01.023 (2006).
146 Masri, B. et al. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proceedings of the National Academy of Sciences of the United States of America 105, 13656-13661, doi:10.1073/pnas.0803522105 (2008).
147 Free, R. B. et al. Discovery and characterization of a G protein-biased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Molecular pharmacology 86, 96-105, doi:10.1124/mol.113.090563 (2014).
148 Allen, J. A. et al. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proceedings of the National Academy of Sciences of the United States of America 108, 18488-18493, doi:10.1073/pnas.1104807108 (2011).
149 Klewe, I. V. et al. Recruitment of beta-arrestin2 to the dopamine D2 receptor: insights into anti-psychotic and anti-parkinsonian drug receptor signaling. Neuropharmacology 54, 1215-1222, doi:10.1016/j.neuropharm.2008.03.015 (2008).
150 Beaulieu, J. M. et al. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell 132, 125-136, doi:10.1016/j.cell.2007.11.041 (2008).
151 Bateup, H. S. et al. Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proceedings of the National Academy of Sciences of the United States of America 107, 14845-14850, doi:10.1073/pnas.1009874107 (2010).
152 Reiter, E., Ahn, S., Shukla, A. K. & Lefkowitz, R. J. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annual review of pharmacology and toxicology 52, 179-197, doi:10.1146/annurev.pharmtox.010909.105800 (2012).
153 Huang, C. C. & Tesmer, J. J. Recognition in the face of diversity: interactions of heterotrimeric G proteins and G protein-coupled receptor (GPCR) kinases with activated GPCRs. The Journal of biological chemistry 286, 7715-7721, doi:10.1074/jbc.R109.051847 (2011).
154 Daigle, T. L. et al. Selective deletion of GRK2 alters psychostimulant-induced behaviors and dopamine neurotransmission. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 39, 2450-2462, doi:10.1038/npp.2014.97 (2014).
155 Gainetdinov, R. R. et al. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron 38, 291-303 (2003).
156 Mangmool, S. & Kurose, H. G(i/o) protein-dependent and -independent actions of Pertussis Toxin (PTX). Toxins 3, 884-899, doi:10.3390/toxins3070884 (2011).
157 Locht, C., Coutte, L. & Mielcarek, N. The ins and outs of pertussis toxin. The FEBS journal 278, 4668-4682, doi:10.1111/j.1742-4658.2011.08237.x (2011).
158 Fields, T. A. & Casey, P. J. Signalling functions and biochemical properties of pertussis toxin-resistant G-proteins. The Biochemical journal 321 ( Pt 3), 561-571 (1997).
159 Fong, H. K., Yoshimoto, K. K., Eversole-Cire, P. & Simon, M. I. Identification of a GTP-binding protein alpha subunit that lacks an apparent ADP-ribosylation site for pertussis toxin. Proceedings of the National Academy of Sciences of the United States of America 85, 3066-3070 (1988).
160 Matsuoka, M., Itoh, H., Kozasa, T. & Kaziro, Y. Sequence analysis of cDNA and genomic DNA for a putative pertussis toxin-insensitive guanine nucleotide-binding regulatory protein alpha subunit. Proceedings of the National Academy of Sciences of the United States of America 85, 5384-5388 (1988).
161 Jiang, L. I. et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. The Journal of biological chemistry 282, 10576-10584, doi:10.1074/jbc.M609695200 (2007).
162 Bailes, H. J. & Lucas, R. J. Human melanopsin forms a pigment maximally sensitive to blue light (lambdamax approximately 479 nm) supporting activation of G(q/11) and G(i/o) signalling cascades. Proceedings. Biological sciences / The Royal Society 280, 20122987, doi:10.1098/rspb.2012.2987 (2013).
163 Atwood, B. K., Lopez, J., Wager-Miller, J., Mackie, K. & Straiker, A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC genomics 12, 14, doi:10.1186/1471-2164-12-14 (2011).

142
164 Obadiah, J. et al. Adenylyl cyclase interaction with the D2 dopamine receptor family; differential coupling to Gi, Gz, and Gs. Cellular and molecular neurobiology 19, 653-664 (1999).
165 Hamdan, F. F., Audet, M., Garneau, P., Pelletier, J. & Bouvier, M. High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. Journal of biomolecular screening 10, 463-475, doi:10.1177/1087057105275344 (2005).
166 Namkung, Y., Dipace, C., Urizar, E., Javitch, J. A. & Sibley, D. R. G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. The Journal of biological chemistry 284, 34103-34115, doi:10.1074/jbc.M109.055707 (2009).
167 Cho, D. I., Beom, S., Van Tol, H. H., Caron, M. G. & Kim, K. M. Characterization of the desensitization properties of five dopamine receptor subtypes and alternatively spliced variants of dopamine D2 and D4 receptors. Biochemical and biophysical research communications 350, 634-640, doi:10.1016/j.bbrc.2006.09.090 (2006).
168 Ito, K., Haga, T., Lameh, J. & Sadee, W. Sequestration of dopamine D2 receptors depends on coexpression of G-protein-coupled receptor kinases 2 or 5. European journal of biochemistry / FEBS 260, 112-119 (1999).
169 Kim, K. M. et al. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. The Journal of biological chemistry 276, 37409-37414, doi:10.1074/jbc.M106728200 (2001).
170 Urban, D. J. & Roth, B. L. DREADDs (Designer Receptors Exclusively Activated by Designer Drugs): Chemogenetic Tools with Therapeutic Utility. Annual review of pharmacology and toxicology 55, 399-417, doi:10.1146/annurev-pharmtox-010814-124803 (2015).
171 Nakajima, K. & Wess, J. Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Molecular pharmacology 82, 575-582, doi:10.1124/mol.112.080358 (2012).
172 Magalhaes, A. C., Dunn, H. & Ferguson, S. S. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. British journal of pharmacology 165, 1717-1736, doi:10.1111/j.1476-5381.2011.01552.x (2012).
173 Gales, C. et al. Real-time monitoring of receptor and G-protein interactions in living cells. Nature methods 2, 177-184, doi:10.1038/nmeth743 (2005).
174 Roth, C. B., Hanson, M. A. & Stevens, R. C. Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu122(3.41), a critical residue in GPCR structure. Journal of molecular biology 376, 1305-1319, doi:10.1016/j.jmb.2007.12.028 (2008).
175 Clayton, C. C., Donthamsetti, P., Lambert, N. A., Javitch, J. A. & Neve, K. A. Mutation of three residues in the third intracellular loop of the dopamine D2 receptor creates an internalization-defective receptor. The Journal of biological chemistry 289, 33663-33675, doi:10.1074/jbc.M114.605378 (2014).
176 Wilson, J., Lin, H., Fu, D., Javitch, J. A. & Strange, P. G. Mechanisms of inverse agonism of antipsychotic drugs at the D(2) dopamine receptor: use of a mutant D(2) dopamine receptor that adopts the activated conformation. Journal of neurochemistry 77, 493-504 (2001).
177 Zhuang, X., Masson, J., Gingrich, J. A., Rayport, S. & Hen, R. Targeted gene expression in dopamine and serotonin neurons of the mouse brain. Journal of neuroscience methods 143, 27-32, doi:10.1016/j.jneumeth.2004.09.020 (2005).
178 Hikida, T., Kimura, K., Wada, N., Funabiki, K. & Nakanishi, S. Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron 66, 896-907, doi:10.1016/j.neuron.2010.05.011 (2010).
179 Gangarossa, G. et al. Spatial distribution of D1R- and D2R-expressing medium-sized spiny neurons differs along the rostro-caudal axis of the mouse dorsal striatum. Frontiers in neural circuits 7, 124, doi:10.3389/fncir.2013.00124 (2013).
180 Warner-Schmidt, J. L. et al. Cholinergic interneurons in the nucleus accumbens regulate depression-like behavior. Proceedings of the National Academy of Sciences of the United States of America 109, 11360-11365, doi:10.1073/pnas.1209293109 (2012).
181 Gallo, E. F. et al. Upregulation of Dopamine D2 Receptors in the Nucleus Accumbens Indirect Pathway Increases Locomotion but Does Not Reduce Alcohol Consumption.

143
Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology, doi:10.1038/npp.2015.11 (2015).
182 Han, Y., Moreira, I. S., Urizar, E., Weinstein, H. & Javitch, J. A. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nature chemical biology 5, 688-695, doi:10.1038/nchembio.199 (2009).
183 Trifilieff, P. et al. Increasing dopamine D2 receptor expression in the adult nucleus accumbens enhances motivation. Molecular psychiatry 18, 1025-1033, doi:10.1038/mp.2013.57 (2013).
184 Powell, S. B., Zhou, X. & Geyer, M. A. Prepulse inhibition and genetic mouse models of schizophrenia. Behavioural brain research 204, 282-294, doi:10.1016/j.bbr.2009.04.021 (2009).
185 Hoffman, D. C. & Donovan, H. Catalepsy as a rodent model for detecting antipsychotic drugs with extrapyramidal side effect liability. Psychopharmacology 120, 128-133 (1995).
186 Svenningsson, P. et al. DARPP-32: an integrator of neurotransmission. Annual review of pharmacology and toxicology 44, 269-296, doi:10.1146/annurev.pharmtox.44.101802.121415 (2004).
187 Li, H. et al. beta-arrestin 2 regulates Toll-like receptor 4-mediated apoptotic signalling through glycogen synthase kinase-3beta. Immunology 130, 556-563, doi:10.1111/j.1365-2567.2010.03256.x (2010).
188 Castro, M. G., McNamara, U. & Carbonetti, N. H. Expression, activity and cytotoxicity of pertussis toxin S1 subunit in transfected mammalian cells. Cellular microbiology 3, 45-54 (2001).
189 Maus, M., Homburger, V., Bockaert, J., Glowinski, J. & Premont, J. Pretreatment of mouse striatal neurons in primary culture with 17 beta-estradiol enhances the pertussis toxin-catalyzed ADP-ribosylation of G alpha o,i protein subunits. Journal of neurochemistry 55, 1244-1251 (1990).
190 Knott, C., Maguire, J. J., Moratalla, R. & Bowery, N. G. Regional effects of pertussis toxin in vivo and in vitro on GABAB receptor binding in rat brain. Neuroscience 52, 73-81 (1993).
191 Hazes, B. & Read, R. J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 36, 11051-11054, doi:10.1021/bi971383p (1997).
192 Ikeda, S. R., Jeong, S. W., Kammermeier, P. J., Ruiz-Velasco, V. & King, M. M. Heterologous expression of a green fluorescent protein-pertussis toxin S1 subunit fusion construct disrupts calcium channel modulation in rat superior cervical ganglion neurons. Neuroscience letters 271, 163-166 (1999).
193 Ferris, J., Ge, H., Liu, L. & Roman, G. G(o) signaling is required for Drosophila associative learning. Nature neuroscience 9, 1036-1040, doi:10.1038/nn1738 (2006).
194 Regard, J. B. et al. Probing cell type-specific functions of Gi in vivo identifies GPCR regulators of insulin secretion. The Journal of clinical investigation 117, 4034-4043, doi:10.1172/JCI32994 (2007).
195 Bartley, T. D., Whiteley, D. W., Mar, V. L., Burns, D. L. & Burnette, W. N. Pertussis holotoxoid formed in vitro with a genetically deactivated S1 subunit. Proceedings of the National Academy of Sciences of the United States of America 86, 8353-8357 (1989).
196 Loosmore, S. et al. Characterization of pertussis toxin analogs containing mutations in B-oligomer subunits. Infection and immunity 61, 2316-2324 (1993).
197 Rook, M. S., Lu, M. & Kosik, K. S. CaMKIIalpha 3' untranslated region-directed mRNA translocation in living neurons: visualization by GFP linkage. The Journal of neuroscience : the official journal of the Society for Neuroscience 20, 6385-6393 (2000).
198 Vuppalanchi, D. et al. Conserved 3'-untranslated region sequences direct subcellular localization of chaperone protein mRNAs in neurons. The Journal of biological chemistry 285, 18025-18038, doi:10.1074/jbc.M109.061333 (2010).
199 Leck, K. J. et al. Gz proteins are functionally coupled to dopamine D2-like receptors in vivo. Neuropharmacology 51, 597-605, doi:10.1016/j.neuropharm.2006.05.002 (2006).
200 Wei, H. et al. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proceedings of the National Academy of Sciences of the United States of America 100, 10782-10787, doi:10.1073/pnas.1834556100 (2003).
201 Shenoy, S. K. et al. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. The Journal of biological chemistry 281, 1261-1273, doi:10.1074/jbc.M506576200 (2006).

144
202 Lan, H., Liu, Y., Bell, M. I., Gurevich, V. V. & Neve, K. A. A dopamine D2 receptor mutant capable of G protein-mediated signaling but deficient in arrestin binding. Molecular pharmacology 75, 113-123, doi:10.1124/mol.108.050534 (2009).
203 Newman, A. H. et al. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J.Med.Chem. 55, 6689-6699, doi:10.1021/jm300482h [doi] (2012).
204 Pfleger, K. D., Seeber, R. M. & Eidne, K. A. Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat.Protoc. 1, 337-345, doi:nprot.2006.52 [pii];10.1038/nprot.2006.52 [doi] (2006).
205 Coulon, V. et al. Subcellular imaging of dynamic protein interactions by bioluminescence resonance energy transfer. Biophys.J. 94, 1001-1009, doi:S0006-3495(08)70696-9 [pii];10.1529/biophysj.107.117275 [doi] (2008).
206 Guo, N. et al. Impact of D2 receptor internalization on binding affinity of neuroimaging radiotracers. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 35, 806-817, doi:npp2009189 [pii];10.1038/npp.2009.189 [doi] (2010).
207 Reiter, E. & Lefkowitz, R. J. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol.Metab 17, 159-165, doi:S1043-2760(06)00056-7 [pii];10.1016/j.tem.2006.03.008 [doi] (2006).
208 Kang, D. S., Tian, X. & Benovic, J. L. Role of beta-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr.Opin.Cell Biol. 27, 63-71, doi:S0955-0674(13)00181-6 [pii];10.1016/j.ceb.2013.11.005 [doi] (2014).
209 Krupnick, J. G., Goodman, O. B., Jr., Keen, J. H. & Benovic, J. L. Arrestin/clathrin interaction. Localization of the clathrin binding domain of nonvisual arrestins to the carboxy terminus. J.Biol.Chem. 272, 15011-15016 (1997).
210 Laporte, S. A., Oakley, R. H., Holt, J. A., Barak, L. S. & Caron, M. G. The interaction of beta-arrestin with the AP-2 adaptor is required for the clustering of beta 2-adrenergic receptor into clathrin-coated pits. J.Biol.Chem. 275, 23120-23126, doi:10.1074/jbc.M002581200 [doi];M002581200 [pii] (2000).
211 Kim, Y. M. & Benovic, J. L. Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. J.Biol.Chem. 277, 30760-30768, doi:10.1074/jbc.M204528200 [doi];M204528200 [pii] (2002).
212 McMahon, H. T. & Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat.Rev.Mol.Cell Biol. 12, 517-533, doi:nrm3151 [pii];10.1038/nrm3151 [doi] (2011).
213 Hamdan, F. F. et al. Unraveling G protein-coupled receptor endocytosis pathways using real-time monitoring of agonist-promoted interaction between beta-arrestins and AP-2. J.Biol.Chem. 282, 29089-29100, doi:M700577200 [pii];10.1074/jbc.M700577200 [doi] (2007).
214 Laporte, S. A., Miller, W. E., Kim, K. M. & Caron, M. G. beta-Arrestin/AP-2 interaction in G protein-coupled receptor internalization: identification of a beta-arrestin binging site in beta 2-adaptin. J.Biol.Chem. 277, 9247-9254, doi:10.1074/jbc.M108490200 [doi];M108490200 [pii] (2002).
215 Lan, H., Liu, Y., Bell, M. I., Gurevich, V. V. & Neve, K. A. A dopamine D2 receptor mutant capable of G protein-mediated signaling but deficient in arrestin binding. Mol.Pharmacol. 75, 113-123, doi:mol.108.050534 [pii];10.1124/mol.108.050534 [doi] (2009).
216 Namkung, Y., Dipace, C., Urizar, E., Javitch, J. A. & Sibley, D. R. G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J.Biol.Chem. 284, 34103-34115, doi:M109.055707 [pii];10.1074/jbc.M109.055707 [doi] (2009).
217 Macey, T. A., Gurevich, V. V. & Neve, K. A. Preferential Interaction between the dopamine D2 receptor and Arrestin2 in neostriatal neurons. Mol.Pharmacol. 66, 1635-1642, doi:10.1124/mol.104.001495 [doi];mol.104.001495 [pii] (2004).
218 Kim, K. M. et al. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J.Biol.Chem. 276, 37409-37414, doi:10.1074/jbc.M106728200 [doi];M106728200 [pii] (2001).

145
219 Namkung, Y., Dipace, C., Javitch, J. A. & Sibley, D. R. G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J.Biol.Chem. 284, 15038-15051, doi:M900388200 [pii];10.1074/jbc.M900388200 [doi] (2009).
220 Cho, D. I. et al. ARF6 and GASP-1 are post-endocytic sorting proteins selectively involved in the intracellular trafficking of dopamine D(2) receptors mediated by GRK and PKC in transfected cells. Br.J.Pharmacol. 168, 1355-1374, doi:10.1111/bph.12025 [doi] (2013).
221 Iwata, K., Ito, K., Fukuzaki, A., Inaki, K. & Haga, T. Dynamin and rab5 regulate GRK2-dependent internalization of dopamine D2 receptors. Eur.J.Biochem. 263, 596-602 (1999).
222 Robinson, M. S. & Bonifacino, J. S. Adaptor-related proteins. Curr.Opin.Cell Biol. 13, 444-453, doi:S0955-0674(00)00235-0 [pii] (2001).
223 Diviani, D., Lattion, A. L., Abuin, L., Staub, O. & Cotecchia, S. The adaptor complex 2 directly interacts with the alpha 1b-adrenergic receptor and plays a role in receptor endocytosis. J.Biol.Chem. 278, 19331-19340, doi:10.1074/jbc.M302110200 [doi];M302110200 [pii] (2003).
224 Paing, M. M., Temple, B. R. & Trejo, J. A tyrosine-based sorting signal regulates intracellular trafficking of protease-activated receptor-1: multiple regulatory mechanisms for agonist-induced G protein-coupled receptor internalization. J.Biol.Chem. 279, 21938-21947, doi:10.1074/jbc.M401672200 [doi];M401672200 [pii] (2004).
225 Clayton, C. C., Donthamsetti, P., Lambert, N. A., Javitch, J. A. & Neve, K. A. Mutation of Three Residues in the Third Intracellular Loop of the Dopamine D2 Receptor Creates an Internalization-Defective Receptor. The Journal of biological chemistry, doi:10.1074/jbc.M114.605378 (2014).
226 Kong, K. C. et al. M3-muscarinic receptor promotes insulin release via receptor phosphorylation/arrestin-dependent activation of protein kinase D1. Proceedings of the National Academy of Sciences of the United States of America 107, 21181-21186, doi:10.1073/pnas.1011651107 (2010).
227 Thompson, D., McArthur, S., Hislop, J. N., Flower, R. J. & Perretti, M. Identification of a novel recycling sequence in the C-tail of FPR2/ALX receptor: association with cell protection from apoptosis. The Journal of biological chemistry 289, 36166-36178, doi:10.1074/jbc.M114.612630 (2014).
228 Butcher, A. J. et al. Concomitant action of structural elements and receptor phosphorylation determines arrestin-3 interaction with the free fatty acid receptor FFA4. The Journal of biological chemistry 289, 18451-18465, doi:10.1074/jbc.M114.568816 (2014).
229 Cho, D. et al. Agonist-induced endocytosis and receptor phosphorylation mediate resensitization of dopamine D(2) receptors. Molecular endocrinology 24, 574-586, doi:10.1210/me.2009-0369 (2010).
230 Freedman, N. J. et al. Phosphorylation and desensitization of the human beta 1-adrenergic receptor. Involvement of G protein-coupled receptor kinases and cAMP-dependent protein kinase. The Journal of biological chemistry 270, 17953-17961 (1995).
231 Simon, V., Robin, M. T., Legrand, C. & Cohen-Tannoudji, J. Endogenous G protein-coupled receptor kinase 6 triggers homologous beta-adrenergic receptor desensitization in primary uterine smooth muscle cells. Endocrinology 144, 3058-3066, doi:10.1210/en.2002-0138 (2003).
232 Violin, J. D., Ren, X. R. & Lefkowitz, R. J. G-protein-coupled receptor kinase specificity for beta-arrestin recruitment to the beta2-adrenergic receptor revealed by fluorescence resonance energy transfer. The Journal of biological chemistry 281, 20577-20588, doi:10.1074/jbc.M513605200 (2006).
233 Hanoune, J. & Defer, N. Regulation and role of adenylyl cyclase isoforms. Annual review of pharmacology and toxicology 41, 145-174, doi:10.1146/annurev.pharmtox.41.1.145 (2001).
234 Niswender, C. M. et al. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Molecular pharmacology 73, 1213-1224, doi:10.1124/mol.107.041053 (2008).
235 Free B.R., C. J. L., Roof R., Doyle T., Han Y., Donthamsetti P., Javitch J.A., Southall N., Emmitte K., Lindsley C., Ferrer M., Sibley D.R. High-Throughput Screening for Modulators of the D2 Dopamine Receptor Yields Unique and Selective Pharmacological Chemotypes. The Tenth Annual Catecholamine Symposium. (2012).

146
236 Gales, C. et al. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nature structural & molecular biology 13, 778-786, doi:10.1038/nsmb1134 (2006).
237 Hollins, B., Kuravi, S., Digby, G. J. & Lambert, N. A. The c-terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cellular signalling 21, 1015-1021, doi:10.1016/j.cellsig.2009.02.017 (2009).
238 Lan, T. H., Liu, Q., Li, C., Wu, G. & Lambert, N. A. Sensitive and high resolution localization and tracking of membrane proteins in live cells with BRET. Traffic 13, 1450-1456, doi:10.1111/j.1600-0854.2012.01401.x (2012).
239 Riven, I., Iwanir, S. & Reuveny, E. GIRK channel activation involves a local rearrangement of a preformed G protein channel complex. Neuron 51, 561-573, doi:10.1016/j.neuron.2006.08.017 (2006).
240 Luscher, C. & Slesinger, P. A. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nature reviews. Neuroscience 11, 301-315, doi:10.1038/nrn2834 (2010).
241 Thompson, L., Barraud, P., Andersson, E., Kirik, D. & Bjorklund, A. Identification of dopaminergic neurons of nigral and ventral tegmental area subtypes in grafts of fetal ventral mesencephalon based on cell morphology, protein expression, and efferent projections. The Journal of neuroscience : the official journal of the Society for Neuroscience 25, 6467-6477, doi:10.1523/JNEUROSCI.1676-05.2005 (2005).
242 Chung, H. J., Qian, X., Ehlers, M., Jan, Y. N. & Jan, L. Y. Neuronal activity regulates phosphorylation-dependent surface delivery of G protein-activated inwardly rectifying potassium channels. Proceedings of the National Academy of Sciences of the United States of America 106, 629-634, doi:10.1073/pnas.0811615106 (2009).
243 Kotecki, L. et al. GIRK Channels Modulate Opioid-Induced Motor Activity in a Cell Type- and Subunit-Dependent Manner. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 7131-7142, doi:10.1523/JNEUROSCI.5051-14.2015 (2015).
244 Kleuss, C. et al. Assignment of G-protein subtypes to specific receptors inducing inhibition of calcium currents. Nature 353, 43-48, doi:10.1038/353043a0 (1991).
245 Kleuss, C., Scherubl, H., Hescheler, J., Schultz, G. & Wittig, B. Different beta-subunits determine G-protein interaction with transmembrane receptors. Nature 358, 424-426, doi:10.1038/358424a0 (1992).
246 Kleuss, C., Scherubl, H., Hescheler, J., Schultz, G. & Wittig, B. Selectivity in signal transduction determined by gamma subunits of heterotrimeric G proteins. Science 259, 832-834 (1993).
247 Schwindinger, W. F. et al. Loss of G protein gamma 7 alters behavior and reduces striatal alpha(olf) level and cAMP production. The Journal of biological chemistry 278, 6575-6579, doi:10.1074/jbc.M211132200 (2003).
248 Lane, J. R., Powney, B., Wise, A., Rees, S. & Milligan, G. Protean agonism at the dopamine D2 receptor: (S)-3-(3-hydroxyphenyl)-N-propylpiperidine is an agonist for activation of Go1 but an antagonist/inverse agonist for Gi1,Gi2, and Gi3. Molecular pharmacology 71, 1349-1359, doi:10.1124/mol.106.032722 (2007).
249 Sauliere, A. et al. Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nature chemical biology 8, 622-630, doi:10.1038/nchembio.961 (2012).
250 Urizar, E. et al. CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nature chemical biology 7, 624-630, doi:10.1038/nchembio.623 (2011).
251 Ciruela, F., Vilardaga, J. P. & Fernandez-Duenas, V. Lighting up multiprotein complexes: lessons from GPCR oligomerization. Trends in biotechnology 28, 407-415, doi:10.1016/j.tibtech.2010.05.002 (2010).
252 Hynes, T. R. et al. Visualization of G protein betagamma dimers using bimolecular fluorescence complementation demonstrates roles for both beta and gamma in subcellular targeting. The Journal of biological chemistry 279, 30279-30286, doi:10.1074/jbc.M401432200 (2004).
253 Stefan, E. et al. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proceedings of the National Academy of Sciences of the United States of America 104, 16916-16921, doi:10.1073/pnas.0704257104 (2007).

147
254 Hamdan, F. F., Percherancier, Y., Breton, B. & Bouvier, M. Monitoring protein-protein interactions in living cells by bioluminescence resonance energy transfer (BRET). Current protocols in neuroscience / editorial board, Jacqueline N. Crawley ... [et al.] Chapter 5, Unit 5 23, doi:10.1002/0471142301.ns0523s34 (2006).
255 Bayewitch, M. L. et al. Inhibition of adenylyl cyclase isoforms V and VI by various Gbetagamma subunits. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 12, 1019-1025 (1998).
256 Lei, Q., Jones, M. B., Talley, E. M., Garrison, J. C. & Bayliss, D. A. Molecular mechanisms mediating inhibition of G protein-coupled inwardly-rectifying K+ channels. Molecules and cells 15, 1-9 (2003).
257 Lei, Q. et al. Activation and inhibition of G protein-coupled inwardly rectifying potassium (Kir3) channels by G protein beta gamma subunits. Proceedings of the National Academy of Sciences of the United States of America 97, 9771-9776 (2000).
258 Lindorfer, M. A. et al. Differential activity of the G protein beta5 gamma2 subunit at receptors and effectors. The Journal of biological chemistry 273, 34429-34436 (1998).
259 Mirshahi, T., Robillard, L., Zhang, H., Hebert, T. E. & Logothetis, D. E. Gbeta residues that do not interact with Galpha underlie agonist-independent activity of K+ channels. The Journal of biological chemistry 277, 7348-7355, doi:10.1074/jbc.M109999200 (2002).
260 Watson, A. J., Katz, A. & Simon, M. I. A fifth member of the mammalian G-protein beta-subunit family. Expression in brain and activation of the beta 2 isotype of phospholipase C. The Journal of biological chemistry 269, 22150-22156 (1994).
261 Zhang, S., Coso, O. A., Lee, C., Gutkind, J. S. & Simonds, W. F. Selective activation of effector pathways by brain-specific G protein beta5. The Journal of biological chemistry 271, 33575-33579 (1996).
262 Zhou, J. Y., Siderovski, D. P. & Miller, R. J. Selective regulation of N-type Ca channels by different combinations of G-protein beta/gamma subunits and RGS proteins. The Journal of neuroscience : the official journal of the Society for Neuroscience 20, 7143-7148 (2000).
263 Mirshahi, T. & Logothetis, D. E. GIRK channel trafficking: different paths for different family members. Mol Interv 2, 289-291, doi:10.1124/mi.2.5.289 (2002).
264 Anderson, G. R., Posokhova, E. & Martemyanov, K. A. The R7 RGS protein family: multi-subunit regulators of neuronal G protein signaling. Cell biochemistry and biophysics 54, 33-46, doi:10.1007/s12013-009-9052-9 (2009).
265 Berman, D. M. & Gilman, A. G. Mammalian RGS proteins: barbarians at the gate. The Journal of biological chemistry 273, 1269-1272 (1998).
266 Jones, M. B., Siderovski, D. P. & Hooks, S. B. The G betagamma dimer as a novel source of selectivity in G-protein signaling: GGL-ing at convention. Mol Interv 4, 200-214, doi:10.1124/mi.4.4.4 (2004).
267 Hillenbrand, M., Schori, C., Schoppe, J. & Pluckthun, A. Comprehensive analysis of heterotrimeric G-protein complex diversity and their interactions with GPCRs in solution. Proceedings of the National Academy of Sciences of the United States of America 112, E1181-1190, doi:10.1073/pnas.1417573112 (2015).
268 Vincent, S., Brouns, M., Hart, M. J. & Settleman, J. Evidence for distinct mechanisms of transition state stabilization of GTPases by fluoride. Proceedings of the National Academy of Sciences of the United States of America 95, 2210-2215 (1998).
269 Hommers, L. G., Klenk, C., Dees, C. & Bunemann, M. G proteins in reverse mode: receptor-mediated GTP release inhibits G protein and effector function. The Journal of biological chemistry 285, 8227-8233, doi:10.1074/jbc.M109.015388 (2010).
270 Xie, K. et al. Gbeta5 recruits R7 RGS proteins to GIRK channels to regulate the timing of neuronal inhibitory signaling. Nature neuroscience 13, 661-663, doi:10.1038/nn.2549 (2010).
271 van der Lee, M. M. et al. Pharmacological characterization of receptor redistribution and beta-arrestin recruitment assays for the cannabinoid receptor 1. Journal of biomolecular screening 14, 811-823, doi:10.1177/1087057109337937 (2009).
272 Zhang, R. & Xie, X. Tools for GPCR drug discovery. Acta pharmacologica Sinica 33, 372-384, doi:10.1038/aps.2011.173 (2012).
273 Salahpour, A. et al. BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Frontiers in endocrinology 3, 105, doi:10.3389/fendo.2012.00105 (2012).

148
274 Pfleger, K. D., Seeber, R. M. & Eidne, K. A. Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nature protocols 1, 337-345, doi:10.1038/nprot.2006.52 (2006).
275 Kocan, M. & Pfleger, K. D. Study of GPCR-protein interactions by BRET. Methods in molecular biology 746, 357-371, doi:10.1007/978-1-61779-126-0_20 (2011).
276 Loening, A. M., Fenn, T. D., Wu, A. M. & Gambhir, S. S. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein engineering, design & selection : PEDS 19, 391-400, doi:10.1093/protein/gzl023 (2006).
277 Vishnivetskiy, S. A. et al. Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. The Journal of biological chemistry 286, 24288-24299, doi:10.1074/jbc.M110.213835 (2011).
278 Grunberg, R. et al. Engineering of weak helper interactions for high-efficiency FRET probes. Nature methods 10, 1021-1027, doi:10.1038/nmeth.2625 (2013).
279 Morin, J. G. & Hastings, J. W. Energy transfer in a bioluminescent system. Journal of cellular physiology 77, 313-318, doi:10.1002/jcp.1040770305 (1971).
280 Morise, H., Shimomura, O., Johnson, F. H. & Winant, J. Intermolecular energy transfer in the bioluminescent system of Aequorea. Biochemistry 13, 2656-2662 (1974).
281 Marullo, S. & Bouvier, M. Resonance energy transfer approaches in molecular pharmacology and beyond. Trends in pharmacological sciences 28, 362-365, doi:10.1016/j.tips.2007.06.007 (2007).
282 Gimenez, L. E., Babilon, S., Wanka, L., Beck-Sickinger, A. G. & Gurevich, V. V. Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell Signal 26, 1523-1531 (2014).
283 Shukla, A. K. et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137-141, doi:10.1038/nature12120 (2013).
284 Gurevich, V. V. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. The Journal of biological chemistry 273, 15501-15506 (1998).
285 Hirsch, J. A., Schubert, C., Gurevich, V. V. & Sigler, P. B. The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell 97, 257-269 (1999).
286 Celver, J., Vishnivetskiy, S. A., Chavkin, C. & Gurevich, V. V. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 277, 9043-9048 (2002).
287 Kovoor, A., Celver, J., Abdryashitov, R. I., Chavkin, C. & Gurevich, V. V. Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 274, 6831-6834 (1999).
288 Vishnivetskiy, S. A. et al. How does arrestin respond to the phosphorylated state of rhodopsin? J. Biol. Chem. 274, 11451-11454 (1999).
289 Nobles, K. N., Guan, Z., Xiao, K., Oas, T. G. & Lefkowitz, R. J. The active conformation of beta-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of beta-arrestins1 and -2. The Journal of biological chemistry 282, 21370-21381, doi:10.1074/jbc.M611483200 (2007).
290 Gimenez, L. E., Vishnivetskiy, S. A., Baameur, F. & Gurevich, V. V. Manipulation of Very Few Receptor Discriminator Residues Greatly Enhances Receptor Specificity of Non-visual Arrestins. The Journal of biological chemistry 287, 29495-29505, doi:10.1074/jbc.M112.366674 (2012).
291 Wess, J., Nakajima, K. & Jain, S. Novel designer receptors to probe GPCR signaling and physiology. Trends in pharmacological sciences 34, 385-392 (2013).
292 Gurevich, V. V. & Gurevich, E. V. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharm Ther 110, 465-502 (2006).
293 Charest, P. G., Terrillon, S. & Bouvier, M. Monitoring agonist-promoted conformational changes of beta-arrestin in living cells by intramolecular BRET. EMBO reports 6, 334-340, doi:10.1038/sj.embor.7400373 (2005).
294 Ahn, S., Shenoy, S. K., Wei, H. & Lefkowitz, R. J. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. The Journal of biological chemistry 279, 35518-35525, doi:10.1074/jbc.M405878200 (2004).
295 Eishingdrelo, H. & Kongsamut, S. Minireview: Targeting GPCR Activated ERK Pathways for Drug Discovery. Current chemical genomics and translational medicine 7, 9-15, doi:10.2174/2213988501307010009 (2013).

149
296 Coffa, S., Breitman, M., Spiller, B. W. & Gurevich, V. V. A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry 50, 6951-6958 (2011).
297 Michino, M. et al. A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Molecular pharmacology 84, 854-864, doi:10.1124/mol.113.087833 (2013).
298 Lane, J. R. et al. A new mechanism of allostery in a G protein-coupled receptor dimer. Nature chemical biology 10, 745-752, doi:10.1038/nchembio.1593 (2014).

150
Appendix
In addition to the work described above, the following manuscripts relating to dopamine D2-like receptor
pharmacology were published by Prashant Donthamsetti during his pre-doctoral period. Topics include 1)
determining the molecular determinants of selectivity between the close receptor homologs, D2R and
D3R, and 2) identifying the molecular mechanism of action of the first reported drug-like allosteric
modulator of D2R.
Section I. Determining the molecular determinants of pharmacological selectivity at close receptor
homologs D2R and D3R
Manuscript 1: Molecular determinants of selectivity and efficacy at the dopamine D3 receptor39.
The dopamine D3 receptor (D3R) has been implicated in substance abuse and other neuropsychiatric
disorders. The high sequence homology between the D3R and D2R, especially within the orthosteric
binding site (OBS) that binds dopamine, has made the development of D3R-selective compounds
challenging. Here, we deconstruct into pharmacophoric elements a series of D3R-selective substituted-4-
phenylpiperazine compounds and use computational simulations and binding and activation studies to
dissect the structural bases for D3R selectivity and efficacy. We find that selectivity arises from divergent
interactions within a second binding pocket (SBP) separate from the OBS, whereas efficacy depends on
the binding mode in the OBS. Our findings reveal structural features of the receptor that are critical to
selectivity and efficacy that can be used to design highly D3R-selective ligands with targeted efficacies.
These findings are generalizable to other GPCRs in which the SBP can be targeted by bitopic or allosteric
ligands.
Manuscript 2: A single glycine in extracellular loop 1 is the critical determinant for
pharmacological specificity of dopamine D2 and D3 receptors297.
Subtype-selective agents for the dopamine D3 receptor (D3R) have been considered as potential
medications for drug addiction and other neuropsychiatric disorders. Medicinal chemistry efforts have led
to the discovery of 4-phenylpiperazine derivatives that are >100-fold selective for

151
the dopamine D3 receptor over dopamine D2 receptor (D2R), despite high sequence identity (78% in the
transmembrane domain). Based on the recent crystal structure of D3R, we demonstrated that the 4-
phenylpiperazine moiety in this class of D3R-selective compounds binds to the conserved orthosteric
binding site, whereas the extended aryl amide moiety is oriented toward a divergent secondary binding
pocket (SBP). In an effort to further characterize molecular determinants of the selectivity of these
compounds, we modeled their binding modes in D3R and D2R by comparative ligand docking
and molecular dynamics simulations. We found that the aryl amide moiety in the SBP differentially
induces conformational changes in transmembrane segment 2 and extracellular loop 1 (EL1), which
amplify the divergence of the SBP in D3R and D2R.Receptor chimera and site-directed mutagenesis
studies were used to validate these binding modes and to identify a divergent glycine in EL1 as critical to
D3R over D2R subtype selectivity. A better understanding of drug-dependent receptor conformations
such as these is key to the rational design of compounds targeting a specific receptor among closely
related homologs, and may also lead to discovery of novel chemotypes that exploit subtle differences in
protein conformations.
Manuscript 3. What can crystal structures of aminergic receptors tell us about designing subtype-
selective ligands34?
G protein-coupled receptors (GPCRs) are integral membrane proteins that represent an important class
of drug targets. In particular, aminergic GPCRs interact with a significant portion of drugs currently on the
market. However, most drugs that target these receptors are associated with undesirable side effects,
which are due in part to promiscuous interactions with close homologs of the intended target receptors.
Here, based on a systematic analysis of all 37 of the currently available high-resolution crystal structures
of aminergic GPCRs, we review structural elements that contribute to and can be exploited for designing
subtype-selective compounds. We describe the roles of secondary binding pockets (SBPs), as well as
differences in ligand entry pathways to the orthosteric binding site, in determining selectivity. In addition,
using the available crystal structures, we have identified conformational changes in the SBPs that are
associated with receptor activation and explore the implications of these changes for the rational
development of selective ligands with tailored efficacy.

152
Section II. Determining the mechanism of allosteric modulation of D2R by SB269652
Manuscript 1. A new mechanism of allostery in a G protein-coupled receptor dimer298.
SB269652 is to our knowledge the first drug-like allosteric modulator of the dopamine D2 receptor (D2R),
but it contains structural features associated with orthosteric D2R antagonists. Using a functional
complementation system to control the identity of individual protomers within a dimeric D2R complex, we
converted the pharmacology of the interaction between SB269652 and dopamine from allosteric to
competitive by impairing ligand binding to one of the protomers, indicating that the allostery requires D2R
dimers. Additional experiments identified a 'bitopic' pose for SB269652 extending from the orthosteric site
into a secondary pocket at the extracellular end of the transmembrane (TM) domain, involving TM2 and
TM7. Engagement of this secondary pocket was a requirement for the allosteric pharmacology of
SB269652. This suggests a new mechanism whereby a bitopic ligand binds in an extended pose on one
G protein-coupled receptor protomer to allosterically modulate the binding of a ligand to the orthosteric
site of a second protomer.