direct self-consistent field computations on gpu clusters
DESCRIPTION
Direct Self-Consistent Field Computations on GPU Clusters. Ivan Ufimtsev, Todd Martinez Department of Chemistry Stanford University. Guochun Shi, Volodymyr Kindratenko National Center for Supercomputing Applications University of Illinois at Urbana-Champaign. Presentation Outline. - PowerPoint PPT PresentationTRANSCRIPT

National Center for Supercomputing ApplicationsUniversity of Illinois at Urbana-Champaign
Direct Self-Consistent Field Computations on GPU Clusters
Guochun Shi,Volodymyr Kindratenko
National Center for Supercomputing Applications
University of Illinois at Urbana-Champaign
Ivan Ufimtsev,Todd Martinez
Department of Chemistry
Stanford University

IPDPS 200
Presentation Outline
• GPU computing• NCSA’s Lincoln GPU cluster
• SCF theory in Quantum Chemistry• Implementation on a GPU cluster• Kernels for J and K matrices• Parallelization strategy for GPU cluster• Performance
• Conclusions and future work

9/22/02 2/4/04 6/18/05 10/31/06 3/14/080
200
400
600
800
1000
1200
NVIDIA GPU
Intel CPU
GF
lop/
s
7900 GTX
8800 GTX
8800 Ultra
GTX 285
Intel Xeon Quad-core 3 GHz
GTX 280
Why GPUs?
5800 5950 Ultra6800 Ultra
7800 GTX
IPDPS 200
GPU Performance Trends

IPDPS 200
NVIDIA Tesla T10 GPU Architecture
• T10 architecture• 240 streaming
processors arranged as 30 streaming multiprocessors
• At 1.3 GHz this provides• 1 TFLOP SP• 86.4 GFLOP DP
• 512-bit interface to off-chip GDDR3 memory• 102 GB/s bandwidth
TPC 1
Geometry controller
SMC
SM
Shared memory
SFU SFU
SP SP
SP SP
SP SP
SP SP
C cache
MT issue
I cache
SM
Shared memory
SFU SFU
SP SP
SP SP
SP SP
SP SP
C cache
MT issue
I cache
SM
Shared memory
SFU SFU
SP SP
SP SP
SP SP
SP SP
C cache
MT issue
I cache
Texture units
Texture L1
TPC 10
Geometry controller
SMC
SM
Shared memory
SFU SFU
SP SP
SP SP
SP SP
SP SP
C cache
MT issue
I cache
SM
Shared memory
SFU SFU
SP SP
SP SP
SP SP
SP SP
C cache
MT issue
I cache
SM
Shared memory
SFU SFU
SP SP
SP SP
SP SP
SP SP
C cache
MT issue
I cache
Texture units
Texture L1
Thread execution managerInput assemblerPCIe interface
L2ROPL2 ROP
512-bit memory interconnect
DRAM DRAM DRAM DRAM DRAM DRAM DRAM DRAM

IPDPS 200
Intel 64 Tesla Linux Cluster Lincoln• Dell PowerEdge 1955
server• Intel 64 (Harpertown) 2.33
GHz dual socket quad core• 16 GB DDR2• Infiniband SDR
• Tesla S1070 1U GPU Computing Server• 1.3 GHz Tesla T10 processors• 4x4 GB GDDR3 SDRAM
• Cluster• Servers: 192• Accelerator Units: 96
• Two Compute Nodes
Dell PowerEdge 1955 server
IB
Tesla S1070
T10 T10
PCIe interface
DRAM DRAM
T10 T10
PCIe interface
DRAM DRAM
Dell PowerEdge 1955 server
PCIe x8 PCIe x8
SDR IB SDR IB

IPDPS 200
HPL Benchmark for Lincoln
1 node 2 nodes 4 nodes 8 nodes 16 nodes 32 nodes0
500
1000
1500
2000
2500
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
Lincoln (GFLOPS)
Lincoln (% of peak)
system size
achi
eved
GF
LO
PS
% o
f pe
ak
We used Massimiliano Fatica(nvidia)’s GPU enabled HPL package.

IPDPS 200
Why do we need to deal with…
Energy (H = E):
Quantifies intra/intermolecular interactions
Drives chemistry, little interesting happens on flat surface
Geometry optimization (RE = 0)
Searches for stable atomic arrangements (molecular shapes)
Molecular dynamics (∂2R/ ∂t2 = -1/M RE)
The chemistry itself (at some, sometimes crude, approximation)
Studies system at atomistic time, and length scales
Quantum Chemistry

IPDPS 200
Exact energy is a hard problem
1
2
2
xi22
yi22
zi2
i
ZA
ri RAi,A
1
ri rji, j
ri E ri
ri ?
E ?

IPDPS 200
Hartree-Fock approximation is one of the simplest
A 1 r1 2 r2 ... N rN
is an antisymmetrized product of N 1-electron orbitals
i r Cij j r j1
K
Expand over predefined basis set
Cij ?

IPDPS 200
Hartree-Fock Self Consistent Field (SCF) procedure
F C CESC
Fk1 C F Ck Fk1Ck1 ESCk1
Repeat until Ck+1 more or less equals Ck

IPDPS 200
Hartree-Fock equations
F C CESC
• All matrices are of NN size (N ~ 1,000 … 10,000)• N3 operations to solve HF equations (need to deal with diagonalization)• N4 operations to get F
Fij C H ijcore Jij C 1
2Kij C
Jij [ij | kl]Pkl C k ,l
Kij [ik | jl]Pkl C k ,l
[ij | kl] i r1 j r1 1
r1 r2k r2 l r2 dr1dr2
IPDPS 200
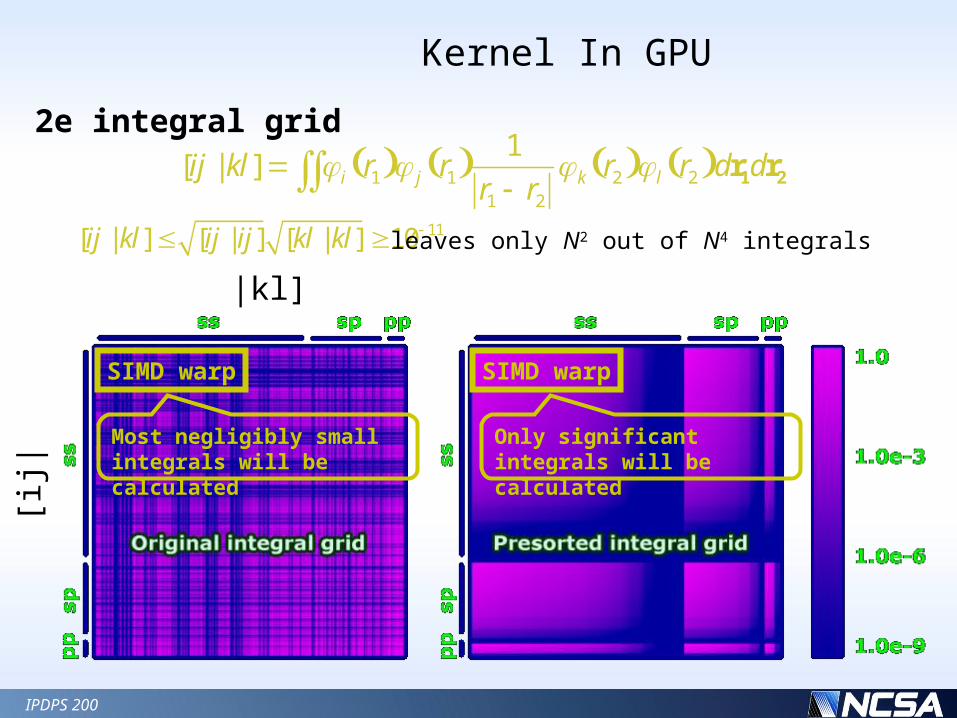
2e integral grid
SIMD warp
Most negligibly small integrals will be calculated
SIMD warp
Only significant integrals will be calculated
[ij|
|kl]
[ij | kl] [ij | ij] [kl | kl] 10 11leaves only N2 out of N4 integrals
[ij | kl] i r1 j r1 1
r1 r2k r2 l r2 dr1dr2
Kernel In GPU

IPDPS 200
Kernel in GPU: J-matrix implementation
Jij [ij | kl]Pklk ,l
[ij | ij]
[kl | kl]
[ij | kl] [ij | ij] [kl | kl]

IPDPS 200
Kernels in GPU: K-matrix implementation
Kij [ik | jl]Pklk ,l
[ik | ik]
[ jl | jl]

IPDPS 200
Singe node execution time breakdown
olestra0
2
4
6
8
10
12
14
16
18
J
K
J/K reduction
LA
uncounted
KPQ
JPQ
runtime (seconds)
bpti0.00
50.00
100.00
150.00
200.00
250.00
J
K
LA
uncounted
J/K reduction
KPQ
JPQ
runtime (seconds)
• The J and K matrices computation and Linear Algebra (LA) computation dominate the overall execution time
• Pair quantity computations can be significant

IPDPS 200
GPU cluster parallelization strategy
• Each GPU has a global id• nodeid * num_gpu_per_node + local_gpu_index
• J/K matrices work distribution• Computations for elements in J and K matrices are not
even. • Sort pre-computed pair quantities and choose every one
element in N to compute for each GPU
• LA using intel MKL

IPDPS 200
pre-compute pair-wise quantities
compute J and K (Eq. 8, 9)
form Fock sub-matrices (Eq. 7)
gather complete Fock matrix F
scatter F
compute matrix C (Eq. 5)
gather and broadcast P
Converge?
Guess initial molecular orbital coefficients matrix Cand compute density matrix P (Eq.10)
done
start
pre-
com
pute
Com
pute
Jand
Kga
ther
Fock
mat
rix
Dis
trib
ute
Fock
mat
rix
Solv
eei
genv
alue
prob
lem
final
gath
er
1
2
3
4
5
6
yes
no
master MPI processes, multiple POSIX threads
master MPI processes,multiple POSIX threads,
GPUs
master MPI processes,rank 0 MPI process
all MPI processes
all MPI processes
all MPI processes,rank 0 MPI process
Parallelization strategy (II)
• Start as MPI program, each node has as many MPI processes as CPU cores
• One MPI process per node is designated as “master”
• The master MPI processes create threads for controlling GPUs as well as CPU work threads
• MPI processes/GPU management threads/CPU work threads are awaken or put to sleep as needed

IPDPS 200
node 0
Computing J and K matrices on GPUs
Reduction of J and K matrices, form the Fock matrix
Pair-quantity computing on CPUUsing density matrices P
Distribute the Fock matrix, do linear algebra, compute matrix C and P, gather P
Broadcast P
node 1
node 2
node 3
MPI process
CPU work thread
CPU thread for managing GPU kernels
Fock matrix
Distr-ed fork matrix
Distr-ed P matrix
P matrix
Partial J and K
Generated guess matrix C and compute matrix P
IPDPS 200
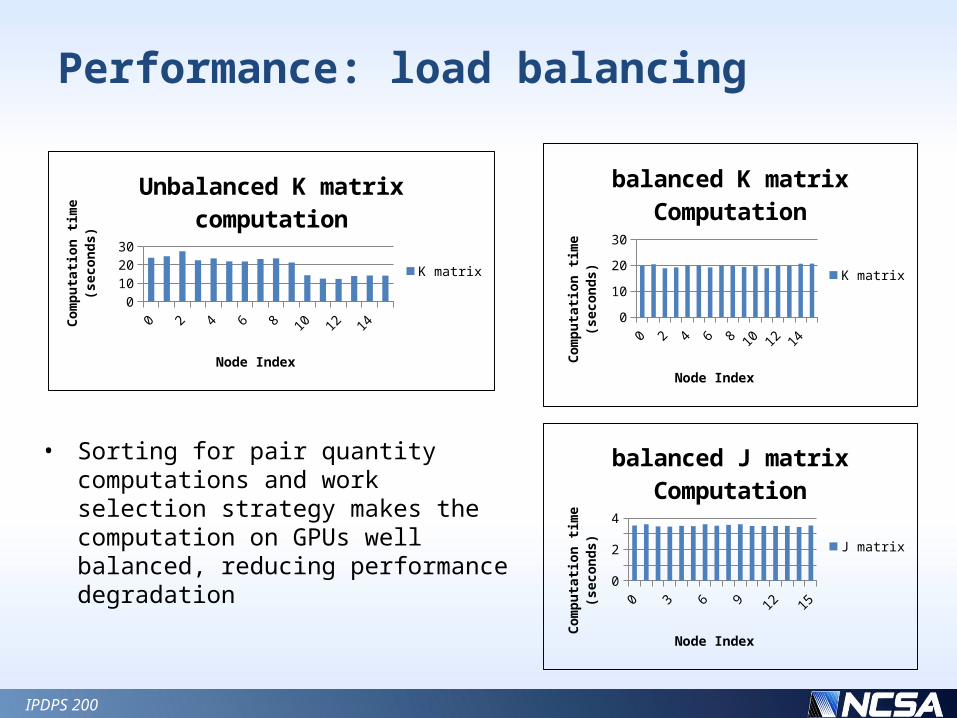
Performance: load balancing
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 150
10
20
30
Unbalanced K matrix computa-tion
K matrix
Node Index
Com
puta
tion
tim
e (s
econ
ds)
0 2 4 6 8 10 12 140
1
2
3
4
balanced J matrix Compu-tation
J matrix
Node IndexCom
puta
tion
tim
e (s
econ
ds)
0 2 4 6 8 10 12 1405
1015202530
balanced K matrix Compu-tation
K matrix
Node Index
Com
puta
tion
tim
e (s
econ
ds)
• Sorting for pair quantity computations and work selection strategy makes the computation on GPUs well balanced, reducing performance degradation

IPDPS 200
Atoms Electrons Orbitals S shells P shells
Olestra 453 1366 2131 1081 350
BPTI 875 3400 4893 2202 897
CspA 1732 6290 8753 4220 1511
Performance
0 1 2 4 8 16 320.00
10.00
20.00
30.00
40.00
50.00
60.00
Olestra
1 2 4 8 16 32 64 1280.00
50.00
100.00
150.00
200.00
250.00
BPTI
2 4 8 16 32 64 1280.00
100.00
200.00
300.00
400.00
500.00
600.00
CspA
# of nodes
Ru
ntim
e (
s)
Using 321g basis set
IPDPS 200
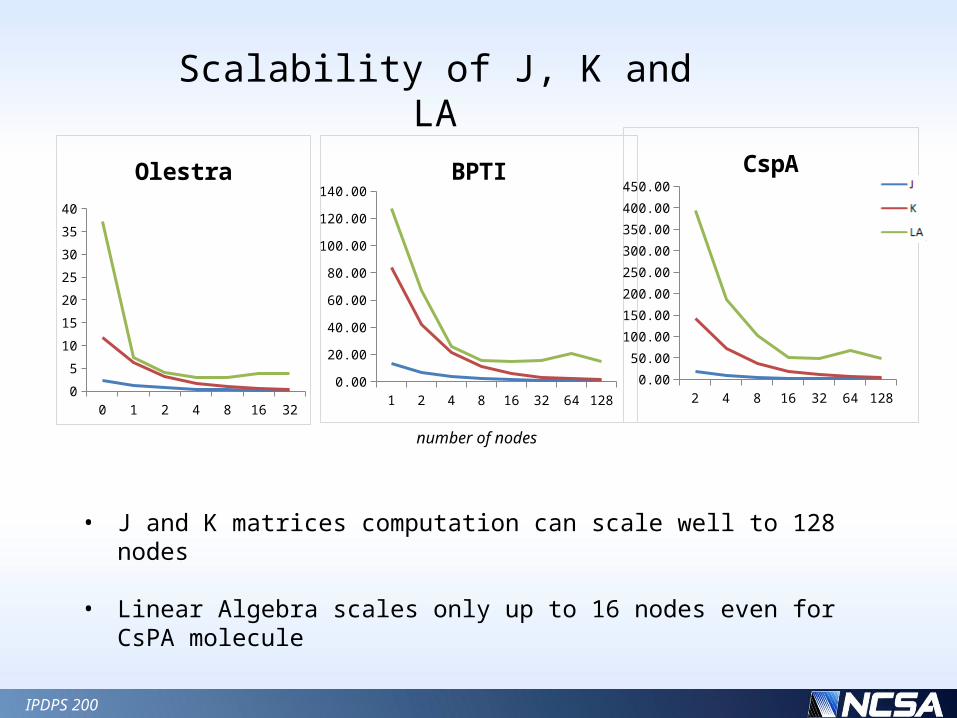
Scalability of J, K and LA
0 1 2 4 8 16 320
5
10
15
20
25
30
35
40
Olestra
1 2 4 8 16 32 64 1280.00
20.00
40.00
60.00
80.00
100.00
120.00
140.00
BPTI
2 4 8 16 32 64 1280.00
50.00
100.00
150.00
200.00
250.00
300.00
350.00
400.00
450.00
CspA
• J and K matrices computation can scale well to 128 nodes
• Linear Algebra scales only up to 16 nodes even for CsPA molecule
number of nodes

IPDPS 200
2 4 8 16 32 64 128
0
50
100
150
200
250
300
350
400
450
P matrix assembly diagonalization dgemm
# of cluster nodes
tim
e p
er i
tera
tio
n (
secs
)
Performance: Linear Algebra breakdown
• Diagonization scales the worst, dgemm is also important
• A fast, scalable GPU based SCALAPACK is needed• Magma from UTK?• Cula?

IPDPS 200
Results: Olestra molecule
Olestra molecule consisting of 453 atoms (a small example model used of testing the developed software) can be computed by the state-of-the-art quantum chemistry software package GAMESS running on an Intel Pentium D 3 GHz processor in over 12,408 seconds whereas our 8-node GPU cluster implementation performs the same computation in just over 5 seconds, a 2,452× speedup.

IPDPS 200
Example: CspA molecule
For larger models, one SCF iteration for Cold shock protein A (CspA) molecule consisting of 1,732 atoms can be done in 88 seconds on a 16 node GPU cluster.

IPDPS 200
Conclusions and future work
• GPU computing brings Quantum Chemistry computing to a new level
• Parallelization enables computing of large molecules in shorter time• J and K matrices show good scalability• Linear Algebra can only scale up to 16 nodes
• Linear Algebra becomes a major bottleneck• A linear algebra package using GPUs with good scalability is
needed• Matrix multiplication and eigenvalue solver
• Only S and P orbitals are supported at this moment• Alexey Titov (NCSA) is working on D orbitals

IPDPS 200
Acknowledgement
• This work was supported by the National Science Foundation grant CHE-06-26354.