direct electrochemistry of nitrate reductase from the ...384916/uq384916_oa.pdf · electron flow...
TRANSCRIPT

�������� ����� ��
Direct Electrochemistry of Nitrate Reductase from the Fungus Neurosporacrassa
Palraj Kalimuthu, Phillip Ringel, Tobias Kruse, Paul V. Bernhardt
PII: S0005-2728(16)30081-0DOI: doi: 10.1016/j.bbabio.2016.04.001Reference: BBABIO 47650
To appear in: BBA - Bioenergetics
Received date: 14 February 2016Accepted date: 1 April 2016
Please cite this article as: Palraj Kalimuthu, Phillip Ringel, Tobias Kruse, Paul V.Bernhardt, Direct Electrochemistry of Nitrate Reductase from the Fungus Neurosporacrassa, BBA - Bioenergetics (2016), doi: 10.1016/j.bbabio.2016.04.001
This is a PDF file of an unedited manuscript that has been accepted for publication.As a service to our customers we are providing this early version of the manuscript.The manuscript will undergo copyediting, typesetting, and review of the resulting proofbefore it is published in its final form. Please note that during the production processerrors may be discovered which could affect the content, and all legal disclaimers thatapply to the journal pertain.

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
1
Direct Electrochemistry of Nitrate Reductase from the Fungus
Neurospora crassa
Palraj Kalimuthu,a Phillip Ringel,b Tobias Kruseb and Paul V. Bernhardta,*
a School of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, 4072, Australia
b Department of Plant Biology, Braunschweig University of Technology, 38106 Braunschweig, Germany
Abstract
We report the first direct (unmediated) catalytic electrochemistry of a eukaryotic nitrate reductase (NR).
NR from the filamentous fungus Neurospora crassa, is a member of the mononuclear molybdenum
enzyme family and contains a Mo, heme and FAD cofactor which are involved in electron transfer from
NAD(P)H to the (Mo) active site where reduction of nitrate to nitrite takes place. NR was adsorbed on an
edge plane pyrolytic graphite (EPG) working electrode. Non-turnover redox responses were observed in
the absence of nitrate from holo NR and three variants lacking the FAD, heme or Mo cofactor. The FAD
response is due to dissociated cofactor in all cases. In the presence of nitrate, NR shows a pronounced
cathodic catalytic wave with an apparent Michaelis constant (KM,app) of 39 μM (pH 7). The catalytic
cathodic current increases with temperature from 5 to 35 oC and an activation enthalpy of 26 kJ mol-1
was determined. In spite of dissociation of the FAD cofactor, catalytically activity is maintained.
Keywords: molybdenum, enzyme, voltammetry, nitrate reductase
*Corresponding author: E-mail: [email protected]

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
2
Introduction
The immobilization of proteins on electrode surfaces is an established methodology for
achieving direct heterogeneous electron transfer of redox proteins and larger molecular weight
oxidoreductase enzymes [1-5]. When successful, it provides a foundation for the fabrication of new
kinds of electrochemical biosensors, bio-fuel cells and biomedical devices without use of any artificial
electron transfer mediators [6, 7]. The direct electrochemistry of small redox proteins (MW < 20 kDa)
such as cytochrome c, hemoglobin and myoglobin has a long history and is now quite well understood
[8-11]. However, the successful direct electrochemistry of larger oxidoreductase enzymes is much less
common. Unless there is a specific part of the enzyme’s solvent exposed surface that has an affinity for
the electrode surface, the orientation of the protein will be unknown and most likely random. Most of
these random orientations will be inappropriate for interfacial electron transfer due to the sheer size of
the enzyme and the remoteness of the redox active cofactor(s) from the electrode surface. Another
issue is denaturation due to adsorption on the electrode surface [12-14]. These problems can be
avoided by modification of the electrode surface with (redox inert) promoters including self-assembled
monolayers [15, 16], nanoparticles [17], surfactants [18], carbon nanotubes [19, 20] and polymers [21]
which favor the orientation of enzyme on the electrode surface for electron transfer.
Figure 1. The three mononuclear Mo enzyme families.
Our focus has been on the mononuclear molybdenum oxidoreductases, which are usually
classified into three sub-families on the basis of the first coordination sphere of their Mo-containing

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
3
active site (Figure 1) [22]. However this primary classification belies the diversity of structure within each
family including the presence of other redox active cofactors that relay electrons to or from the Mo
active site during catalysis.
NR from the fungus Neurospora crassa is a complex homodimeric enzyme (MW ~200 kDa) that
catalyses the initial step in inorganic nitrogen assimilation [23]. Each NR monomer bears three redox
cofactors; the Mo cofactor (Moco), a heme and FAD (Scheme 1) [24-26]. The N terminal domain includes
the Mo cofactor active site while the C terminal domain contains the FAD and NAD(P)H binding sites.
The heme domain of NR has homology with cytochrome b5 and the FAD domain is similar to cytochrome
b5 reductase [23, 25, 27, 28].
Based on the crystal structure of NR from the yeast Pichia angusta and biochemical studies the
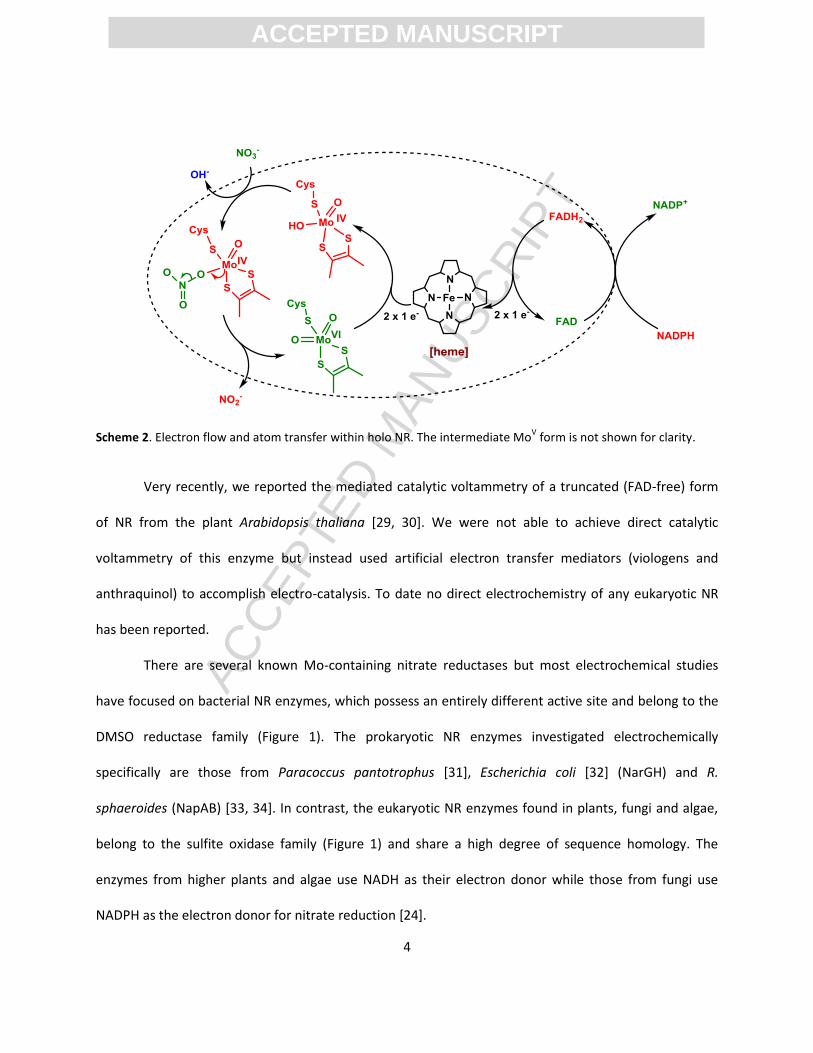
catalytic mechanism for eukaryotic nitrate reduction is as illustrated in Scheme 2 [27]. NADPH provides
the FAD cofactor with two electrons that are subsequently transferred (one at a time) via the heme to
the Mo active site. Nitrate binds to the active MoIV form of NR and O-atom transfer to Mo is coupled
with oxidation of MoIV to MoVI releasing nitrite as the product. After completion of the reductive half-
reaction, MoVI is returned to its active MoIV form by the reduced FAD and heme cofactors.
Scheme 1. Domain structure of Neurospora crassa nitrate reductase. Nitrate reductase variant K113 lacks the first N-terminal 112 amino acids. Molybdenum cofactor (Moco) and dimerization domain share a common sequence stretch comprising 11 amino acids [23].
ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
4
Scheme 2. Electron flow and atom transfer within holo NR. The intermediate MoV form is not shown for clarity.
Very recently, we reported the mediated catalytic voltammetry of a truncated (FAD-free) form
of NR from the plant Arabidopsis thaliana [29, 30]. We were not able to achieve direct catalytic
voltammetry of this enzyme but instead used artificial electron transfer mediators (viologens and
anthraquinol) to accomplish electro-catalysis. To date no direct electrochemistry of any eukaryotic NR
has been reported.
There are several known Mo-containing nitrate reductases but most electrochemical studies
have focused on bacterial NR enzymes, which possess an entirely different active site and belong to the
DMSO reductase family (Figure 1). The prokaryotic NR enzymes investigated electrochemically
specifically are those from Paracoccus pantotrophus [31], Escherichia coli [32] (NarGH) and R.
sphaeroides (NapAB) [33, 34]. In contrast, the eukaryotic NR enzymes found in plants, fungi and algae,
belong to the sulfite oxidase family (Figure 1) and share a high degree of sequence homology. The
enzymes from higher plants and algae use NADH as their electron donor while those from fungi use
NADPH as the electron donor for nitrate reduction [24].

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
5
Herein we demonstrate direct catalytic electrochemistry of recombinant NR from the
filamentous fungus N. crassa [23]. The holo NR employed in this study lacks the first 112 amino acids
from its N-terminus (K113-holo NR, Scheme 3A) and this deletion enhances protein yield and purity
without loss of functionality relative to full-length holo NR [23]. Furthermore this recombinant system is
well understood and a number of key variants may be expressed which lack each of the three cofactors
(Mo, heme and FAD). The H654A/H677A double mutation (heme-free NR, Scheme 3B) lacks the heme
binding histidine and so the heme cofactor is absent altogether from this variant. Similarly the R778E
mutant (FAD-free NR, Scheme 3C) does not bind FAD due to the loss of this key arginine residue. The so-
called cytochrome c reducing fragment only contains amino acids 618–984 and lacks the Mo cofactor
(Mo-free NR, Scheme 3D). This fragment has been shown to catalytically reduce cytochrome c (a non-
physiological function in this case) using NADPH as the reductant [35] but in this truncated form it has
no capacity to reduce nitrate [23].
Scheme 3: Neurospora crassa nitrate reductase variants examined in this study. The first and last residues of the NR domains are indicated. An asterisk indicates the position of modified amino acids and missing cofactors.
A
B
C
D
holo-NR
heme-free NR
FAD-free NR
Mo-free NR

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
6
The four proteins shown in Scheme 3 provide a unique opportunity to study the electro-catalytic
properties of these variants and to gauge the relative importance of each redox active cofactor. As will
be shown it has also enabled a systematic study of the redox cofactors under non-turnover conditions.

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
7
Materials and Methods
Cloning of Neurospora crassa K113 nitrate reductase and variants. For creation of N terminal
truncated nitrate reductase variant K113, the first 336 codons were deleted. PCR-based amplification of
NR variant K113 coding sequence was carried out using Phusion High-Fidelity polymerase (NEB) with
primers (5´-ATACACGTGAAACCAGCCTACCCCCTCCC-3´) and (5´-
ATTACTAGTTCAAAAAACTAATACATCCTCATCCTTCC-3. As a template the readily available full length N.
crassa nitrate reductase coding sequence [23] was used. As templates for PCR based cloning of NR
variants K113 H654A/H677A (hereafter referred to as heme-free NR) and K113 R778E (FAD-free NR) we
used the respective coding sequences described earlier [23]. The CloneJET™ PCR Cloning Kit (Thermo
Scientific) has been used for subcloning according to the manufacturer's instructions. Upon sequence
confirmation, SpeI and PmlI based subcloning into the expression vector was performed. As expression
vector we chose a pQE-80L (Qiagen GmbH) based vector, allowing the N-terminal fusion of a 6xHis-tag
and the C-terminal fusion of a Twin-Strep-tag® [23, 36] to NR K113 variants.
Enzymes and Materials. K113 nitrate reductase variants were expressed and purified from E.
coli strains TP1000 [37] and BL21, respectively. Heterologous gene expression and affinity purification
was performed as described earlier [23]. SDS-PAGE analyses of all proteins studied herein are shown in
the Supporting Information. Sodium nitrate, sodium sulfite, polyethyleneimine and polymyxin B were
purchased from Aldrich and were used as received. All other reagents used were of analytical grade
purity and used without any further purification. All solutions were prepared in purified water
(Millipore, resistivity 18.2 MΩ.cm). Phosphate buffer was prepared using equal amounts of
Na2HPO4/NaH2PO4 to give a total phosphate concentration of 100 mM. The mixture of buffers (20 mM
citric acid, 20 mM MES, 20 mM Bis-Tris, 20 mM Tris and 20 mM CHES) was used for pH dependent

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
8
experiments in the range 4.5 < pH < 10 and the desired pH was obtained by addition of dilute acetic acid
or NaOH.
Electrochemical Measurements and Electrode Cleaning. Cyclic voltammetry (CV) was
carried out with a BAS 100B/W electrochemical workstation using a three-electrode system consisting of
edge-plane pyrolytic graphite (EPG) working electrode, a platinum wire counter electrode, and a
Ag/AgCl reference electrode (+196 mV vs the normal hydrogen electrode, NHE). All potentials are cited
versus NHE. Unless otherwise stated, electrochemical solutions were purged with argon for at least 30
min. prior to the series of experiments and all experiments were performed under a blanket of argon
gas. Voltammetry carried out at different temperatures ranging from 5 to 35 oC was achieved using a
Huber, Ministat-125 temperature controlled water bath connected to a water jacketed electrochemical
cell.
The variation of the observed limiting catalytic current (ilim) as a function of nitrate
concentration followed Michaelis-Menten kinetics and the data were fit to eq. (1)
(1)
where imax is the limiting current at saturating concentration of nitrate and KM is the apparent Michaelis
constant.
The pH dependence of the catalytic current was modeled with eq. (2), which is applicable for an
enzyme that is deactivated by either deprotonation at high pH (pKa1) or protonation at low pH (pKa2) and
iopt is the maximum current at the pH optimum [38].
(2)
Enzyme Electrode Preparation. The EPG working electrode (surface area 0.1 cm2) was
prepared by cleaving several layers from the face of the electrode with a microtome and then cleaning
by sonication in Milli-Q water. No abrasives were used. The cleaned electrode was dried in a nitrogen

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
9
atmosphere. A 3 µL droplet of NR solution (141 µM in Tris-HCl buffer pH 8) was added to the conducting
surface of the inverted EPG electrode surface along with a mixture of 1.5 µL polyethyleneimine (PEI, 5
%) and 1.5 µL polymyxin B (PM, 5 %). This mixture was allowed to dry to a thin film over ca. 1 h at 4 oC
(in a refrigerator). In some cases (as indicated in the text and figure captions) the enzyme coated
electrode was covered with a dialysis membrane (MW cutoff 3.5 kDa), presoaked in water, to prevent
protein loss to the bulk solution. The dialysis membrane was carefully pressed onto the electrode with a
Teflon cap and fastened with a rubber O-ring to prevent leakage of the protein solution under the
membrane. The electro-active enzyme was always confined to the electrode surface (not under
diffusion control) while nitrate and nitrite were able to diffuse to and from the electrode surface (across
the membrane if present). The resulting enzyme-modified electrode was stored at 4 oC in 100 mM
phosphate buffer solution (pH 7.0) when not in use.

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
10
Results and Discussion
Enzyme film composition. The greatest challenge in direct enzyme electrochemistry is
establishing heterogeneous (interfacial) electron transfer; electrochemical communication with the
redox active cofactors. This requires deliberate adsorption of the enzyme onto the working electrode to
remove the restriction of enzyme diffusion becoming rate limiting. The enzyme film must fulfil three
requirements: (i) retention of native enzyme function; (ii) enable the redox cofactors to be in proximity
of the electrode surface and (iii) physi-sorption must be sufficiently strong that the film is in contact with
the electrode. Several different enzyme films were examined before the combination of polymyxin and
polyethyleneimine was chosen; both cationic hydrophilic compounds that have an affinity for the edge-
plane pyrolytic graphite surface, which is negatively charged at neutral pH due to surface carboxylate
and phenolic functional groups. Although heterogeneous electron transfer from the NR enzyme film was
demonstrated (see below) desorption of the film does occur over time with the enzyme eventually being
lost to the bulk solution. To avoid this, the enzyme may be entrapped beneath a dialysis membrane. This
ensures that enzyme film desorption is a reversible process (dynamic equilibrium) and under these
conditions electrochemical activity was sustained.
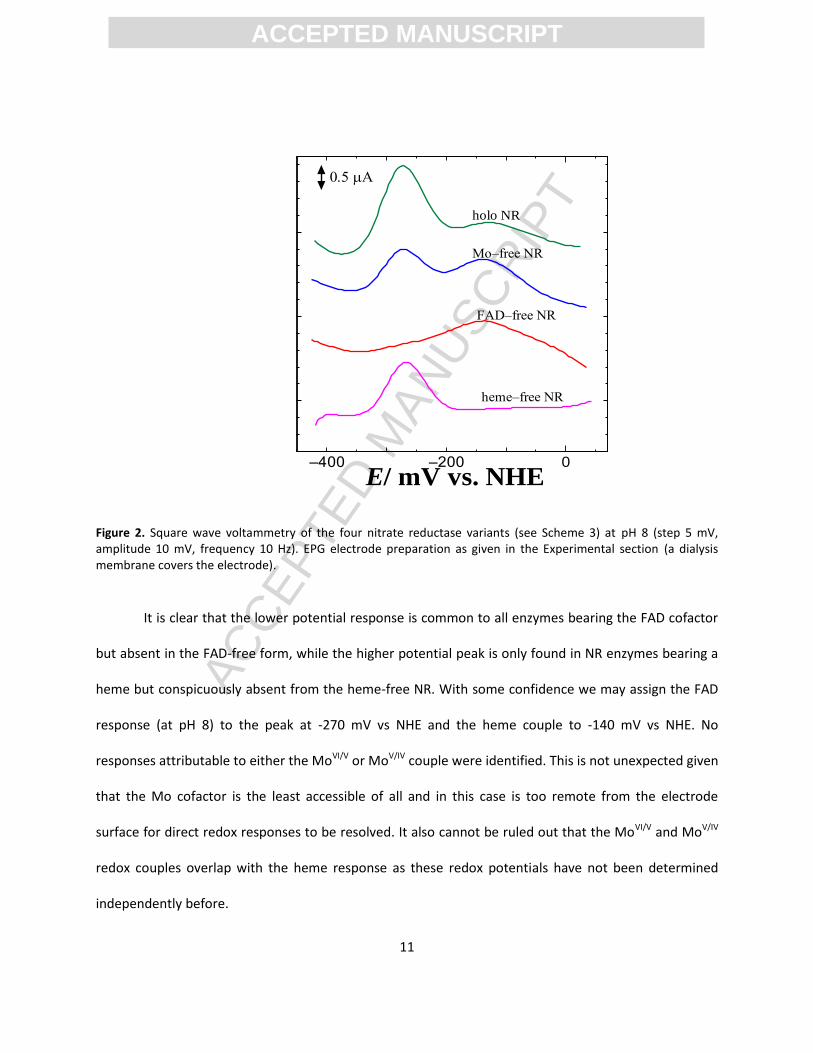
Non-turnover redox responses. We firstly examined the voltammetry of holo-NR, FAD-free NR;
heme-free NR and Mo-free NR (see Scheme 3) in the absence of nitrate. Under ideal conditions
electrochemical responses from the individual cofactors may be identified. Although weak responses
were identified by conventional cyclic voltammetry, square wave voltammetry, a more sensitive
experiment, was employed to better resolve Faradaic responses above the background charging current
(Figure 2).
ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
11
–400 –200 0
/ mV vs. NHEE
holo NR
FAD–free NR
Mo–free NR
heme–free NR
Figure 2. Square wave voltammetry of the four nitrate reductase variants (see Scheme 3) at pH 8 (step 5 mV, amplitude 10 mV, frequency 10 Hz). EPG electrode preparation as given in the Experimental section (a dialysis membrane covers the electrode).
It is clear that the lower potential response is common to all enzymes bearing the FAD cofactor
but absent in the FAD-free form, while the higher potential peak is only found in NR enzymes bearing a
heme but conspicuously absent from the heme-free NR. With some confidence we may assign the FAD
response (at pH 8) to the peak at -270 mV vs NHE and the heme couple to -140 mV vs NHE. No
responses attributable to either the MoVI/V or MoV/IV couple were identified. This is not unexpected given
that the Mo cofactor is the least accessible of all and in this case is too remote from the electrode
surface for direct redox responses to be resolved. It also cannot be ruled out that the MoVI/V and MoV/IV
redox couples overlap with the heme response as these redox potentials have not been determined
independently before.

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
12
–400 –200 0
/ mV vs. NHEE
a
l4 5 6 7 8 9 10
-200
-180
-160
-140
-120
-100
-80
-60
-40
-20
0
Slope = -32 mV
E /
mV
vs
NH
EpH
Figure 3. (A) Square wave voltammetry of FAD-free NR at different pH values (a) 4, (b) 4.54, (c) 4.98, (d) 5.48, (e) 6, (f) 6.46, (g) 6.93, (h) 7.44, (i) 7.96, (j) 8.54, (k) 9 and (l) 9.49; step 5 mV, amplitude 10 mV, frequency 10 Hz) and (B) the redox potentials as a function of pH. EPG electrode preparation as given in the Experimental section.
The pH dependence of the heme redox potential of FAD-free NR varies linearly with pH (Figure
3) but with a slope of only -32 mV/pH which, given the obligate one electron stoichiometry, is too small
to be consistent with proton associated electron transfer reaction.
The pH-dependence of the FAD response in both Mo-free NR (Supporting Information Figure S1)
and holo NR (Figure S2) shows two distinct linear regions. Below pH 7 the slope of the plot is -59 mV/pH
which is consistent with a 2e-/2H+ reaction (FAD/FADH2) while above pH 7 the slope is about -26 mV/pH,
which is consistent with a 2e-/1H+ reaction (FAD/FADH-) i.e. the hydroquinone is singly deprotonated in
this range. The break in the plot yields a pKa of 6.7 for the hydroquinone from this analysis. However,
the FAD cofactor is weakly (non-covalently) bound to NR and a dissociation constant of Kd(FAD) = 0.61
M has been reported [23]. The published assay for NR [23] is conducted in the presence of excess FAD
ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
13
–400 –200 0
–2
–1
0
/ mV vs. NHEE
I
a
b
to ensure that the enzyme has a full complement of cofactors so it was important to examine whether
the FAD electrochemical response is from enzyme-bound FAD or simply from dissociated FAD. A control
experiment was carried out using only FAD (enzyme free) and the same electrode modification protocols
used in Figures 2 and 3 (and Figures S1 and S2). The data are shown in Supporting Information Figure S3
and clearly the behavior of the enzyme-free FAD response is indistinguishable from that seen in Figures
S1 and S2; the only discriminating feature is the weaker heme response which naturally is absent from
Figure S3. So we conclude that dissociation of the FAD cofactor is facile under these conditions and the
low potential couple is simply of free FAD, not enzyme bound FAD.
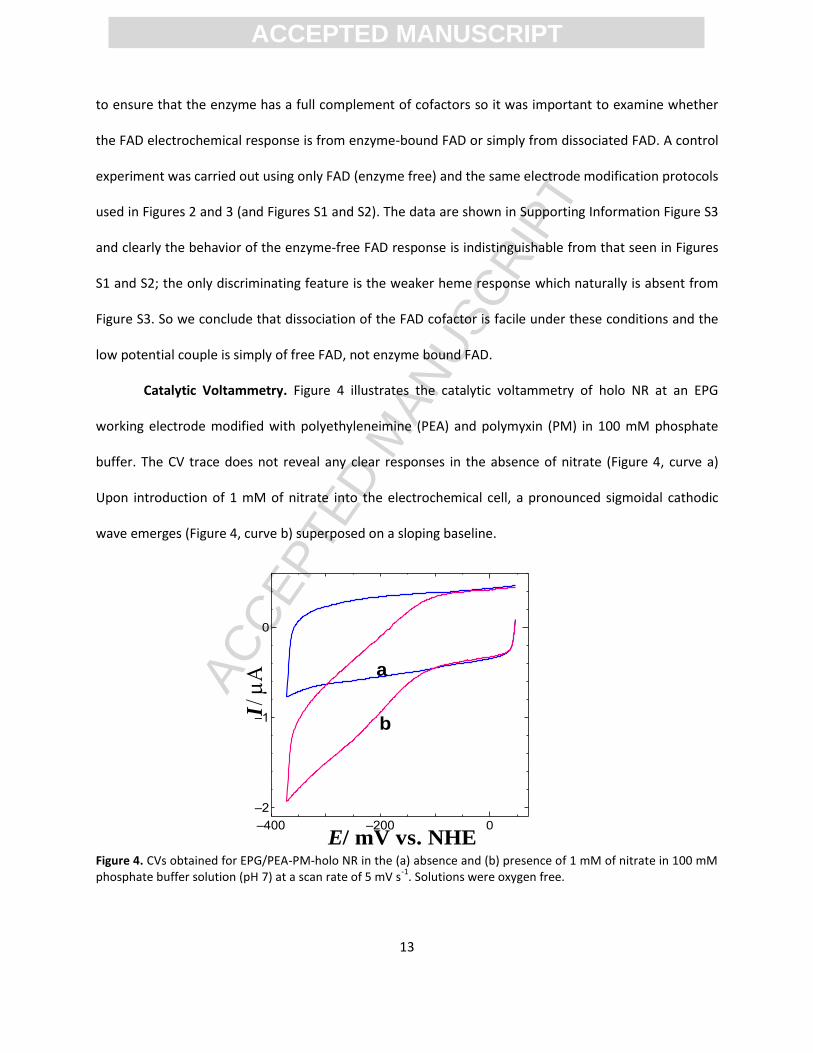
Catalytic Voltammetry. Figure 4 illustrates the catalytic voltammetry of holo NR at an EPG
working electrode modified with polyethyleneimine (PEA) and polymyxin (PM) in 100 mM phosphate
buffer. The CV trace does not reveal any clear responses in the absence of nitrate (Figure 4, curve a)
Upon introduction of 1 mM of nitrate into the electrochemical cell, a pronounced sigmoidal cathodic
wave emerges (Figure 4, curve b) superposed on a sloping baseline.
Figure 4. CVs obtained for EPG/PEA-PM-holo NR in the (a) absence and (b) presence of 1 mM of nitrate in 100 mM phosphate buffer solution (pH 7) at a scan rate of 5 mV s
-1. Solutions were oxygen free.

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
14
Both holo NR and the PEI/PM promotors must be present for catalysis. Omission of the enzyme
(Supporting Information, Figure S4) or the PEI/PM promotors (Figure S5) leads to no nitrate reduction
current. It is possible that NR denatures at the unmodified graphite electrode surface while the
PEI/polymyxin coating on the electrode provides a more protein compatible interface. Alternatively it
may be that NR alone does not adsorb sufficiently strongly to the EPG electrode, a prerequisite for
electrochemical activity, without the PEI and polymyxin co-adsorbates. The observed cathodic current is
due to electrochemically driven enzymatic reduction of nitrate by NR adsorbed on the electrode. It is
noted that the composition of promoters (5% polyethyleneimine and 5% polymyxin B) plays a vital role
facilitating electron transfer between NR and the EPG electrode and the relative amounts of PEI and
polymyxin are important. Many combinations were tried and we found that doubling the amounts of
the two promoters led to a 30% decrease in catalytic activity which perhaps indicates electrode fouling
by excess promoter. Interestingly catalytic activity is still seen if only PEI or only PM is used as a
promoter (supporting information, Figure S6) but the combination of the two gives superior catalysis.
Figure 5. CVs obtained for the increasing concentration of nitrate (a) 0, (b) 20, (c) 40, (d) 80, (e) 160, (f) 320, (g) 640, (h) 960 and (i) 1280 µM at the EPG/PEI-PM-holo NR electrode (no membrane) in 100 mM phosphate buffer solution (pH 7) at a scan rate of 5 mV s
-1. (B) Plot of the nitrate concentration dependence of the cathodic current
at -250 mV vs NHE and curve fit to equation 1.
–300 –200 –100 0
–1
0
/ mV vs. NHEE
I
a
i
A
0 200 400 600 800 1000 1200 1400 1600
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
B
I /
A
Concentration (M)
KM
= 39 M

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
15
Substrate Concentration Dependence. Figure 5A illustrates the effect of increasing nitrate
concentration at the EPG/PEI-PM-holo NR electrode in 100 mM phosphate buffer solution (pH 7). In this
case no dialysis membrane is present and nitrate may diffuse freely to enzyme film on the electrode. A
well-defined sigmoidal cathodic wave is observed around -175 mV vs NHE upon increasing additions of
nitrate into the electrochemical cell (Figure 5A). This wave grows in amplitude with nitrate
concentration and saturates around 400 μM nitrate (Figure 5B). Figure 5B displays the plot obtained for
the concentration of nitrate vs cathodic reduction current at -250 mV where the current becomes
potential independent after compensation for the sloping baseline. An apparent Michaelis constant of
KM,NO3- = 39 µM was obtained from a fit to eq. 1. This value is significantly lower than reported from a
conventional solution assay (290 µM – 440 µM) with NADPH the reductant [23]. The system here is
inherently simple and only comprises two reacting components; holo NR continuously reactivated by
the working electrode and nitrate diffusing to the electrode. The higher apparent KM value seen in the
NADPH assay (a ternary system where holo NR, nitrate and NADPH must all combine) may reflect rate
limitations involving the NADPH reductant.
If the same electrochemical experiment is carried out with a holo NR modified EPG electrode
covered with a dialysis membrane then very similar results are obtained (Supporting Information Figure
S7). The only notable difference is a slight increase in the apparent Michaelis constant from 39 µM (no
membrane) to 72 µM (membrane). Assuming that the membrane has no specific interaction with the
enzyme this observation is consistent with mass transport of nitrate to the enzyme film being slowed
due to its passage across the membrane. Under these conditions equation 1 does not strictly apply and
the apparent KM value is no longer the true Michaelis constant. We recently reported a similar
membrane influence on the current/substrate concentration profile of a different Mo enzyme in a
mediated electrochemical system [39]. The presence of the membrane does enhance the lifetime of the
ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
16
–400 –200 0
–2
–1
0
/ mV vs. NHEE
I
holo NR
FAD free
Mo free
Heme free
enzyme electrode as any NR which dissociates from the electrode surface is retained under the
membrane rather than being irreversibly lost into the bulk solution. This is illustrated in Supporting
Information Figure S8 where some loss of activity is seen in the absence of a membrane over periods of
hours.
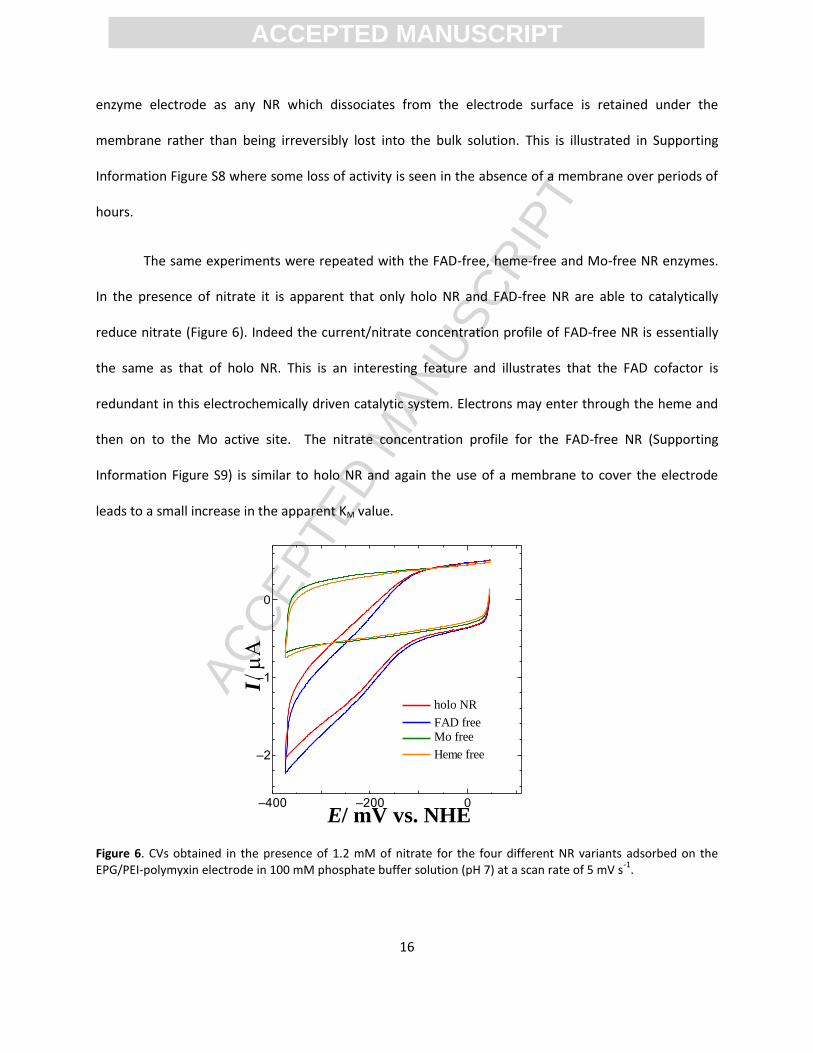
The same experiments were repeated with the FAD-free, heme-free and Mo-free NR enzymes.
In the presence of nitrate it is apparent that only holo NR and FAD-free NR are able to catalytically
reduce nitrate (Figure 6). Indeed the current/nitrate concentration profile of FAD-free NR is essentially
the same as that of holo NR. This is an interesting feature and illustrates that the FAD cofactor is
redundant in this electrochemically driven catalytic system. Electrons may enter through the heme and
then on to the Mo active site. The nitrate concentration profile for the FAD-free NR (Supporting
Information Figure S9) is similar to holo NR and again the use of a membrane to cover the electrode
leads to a small increase in the apparent KM value.
Figure 6. CVs obtained in the presence of 1.2 mM of nitrate for the four different NR variants adsorbed on the EPG/PEI-polymyxin electrode in 100 mM phosphate buffer solution (pH 7) at a scan rate of 5 mV s
-1.
ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
17
0.00320 0.00325 0.00330 0.00335 0.00340 0.00345 0.00350 0.00355 0.00360
-14.2
-14.0
-13.8
-13.6
-13.4
-13.2
-13.0 B
ln (
i) (
i in
A)
1/T (T in K)–400 –200 0
–3
–2
–1
0
/ mV vs. NHEE
I
5 Co
10 Co
15 Co
20 Co
25 Co
30 Co
35 Co
A
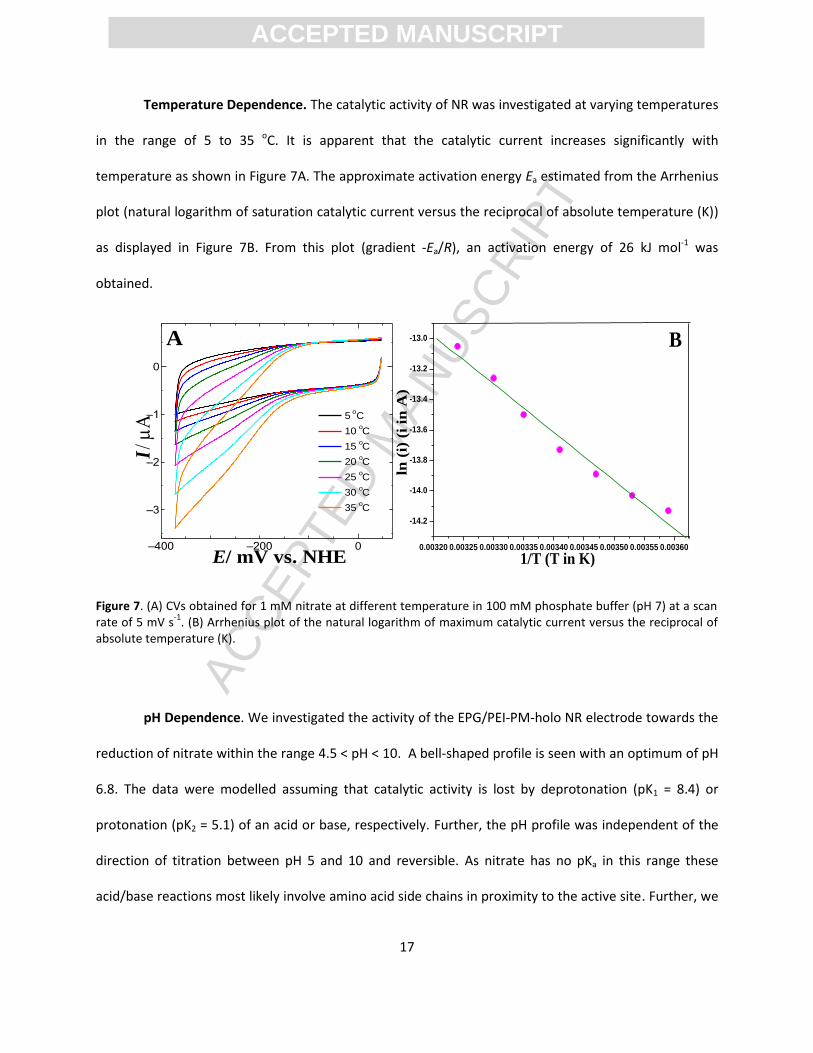
Temperature Dependence. The catalytic activity of NR was investigated at varying temperatures
in the range of 5 to 35 oC. It is apparent that the catalytic current increases significantly with
temperature as shown in Figure 7A. The approximate activation energy Ea estimated from the Arrhenius
plot (natural logarithm of saturation catalytic current versus the reciprocal of absolute temperature (K))
as displayed in Figure 7B. From this plot (gradient -Ea/R), an activation energy of 26 kJ mol-1 was
obtained.
Figure 7. (A) CVs obtained for 1 mM nitrate at different temperature in 100 mM phosphate buffer (pH 7) at a scan rate of 5 mV s
-1. (B) Arrhenius plot of the natural logarithm of maximum catalytic current versus the reciprocal of
absolute temperature (K).
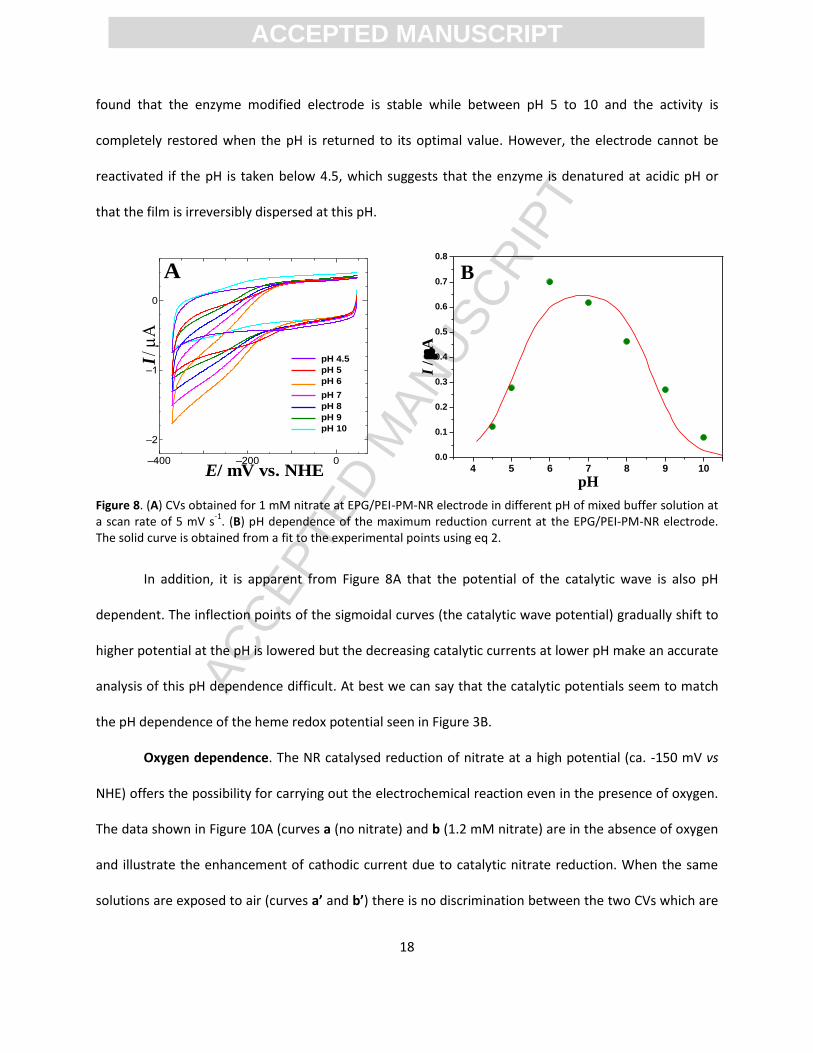
pH Dependence. We investigated the activity of the EPG/PEI-PM-holo NR electrode towards the
reduction of nitrate within the range 4.5 < pH < 10. A bell-shaped profile is seen with an optimum of pH
6.8. The data were modelled assuming that catalytic activity is lost by deprotonation (pK1 = 8.4) or
protonation (pK2 = 5.1) of an acid or base, respectively. Further, the pH profile was independent of the
direction of titration between pH 5 and 10 and reversible. As nitrate has no pKa in this range these
acid/base reactions most likely involve amino acid side chains in proximity to the active site. Further, we
ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
18
–400 –200 0
–2
–1
0
/ mV vs. NHEE
I
pH 5
pH 6
pH 7
pH 8
pH 9
pH 10
pH 4.5
A
4 5 6 7 8 9 10
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
B
I /
A
pH
found that the enzyme modified electrode is stable while between pH 5 to 10 and the activity is
completely restored when the pH is returned to its optimal value. However, the electrode cannot be
reactivated if the pH is taken below 4.5, which suggests that the enzyme is denatured at acidic pH or
that the film is irreversibly dispersed at this pH.
Figure 8. (A) CVs obtained for 1 mM nitrate at EPG/PEI-PM-NR electrode in different pH of mixed buffer solution at a scan rate of 5 mV s
-1. (B) pH dependence of the maximum reduction current at the EPG/PEI-PM-NR electrode.
The solid curve is obtained from a fit to the experimental points using eq 2.
In addition, it is apparent from Figure 8A that the potential of the catalytic wave is also pH
dependent. The inflection points of the sigmoidal curves (the catalytic wave potential) gradually shift to
higher potential at the pH is lowered but the decreasing catalytic currents at lower pH make an accurate
analysis of this pH dependence difficult. At best we can say that the catalytic potentials seem to match
the pH dependence of the heme redox potential seen in Figure 3B.
Oxygen dependence. The NR catalysed reduction of nitrate at a high potential (ca. -150 mV vs
NHE) offers the possibility for carrying out the electrochemical reaction even in the presence of oxygen.
The data shown in Figure 10A (curves a (no nitrate) and b (1.2 mM nitrate) are in the absence of oxygen
and illustrate the enhancement of cathodic current due to catalytic nitrate reduction. When the same
solutions are exposed to air (curves a’ and b’) there is no discrimination between the two CVs which are
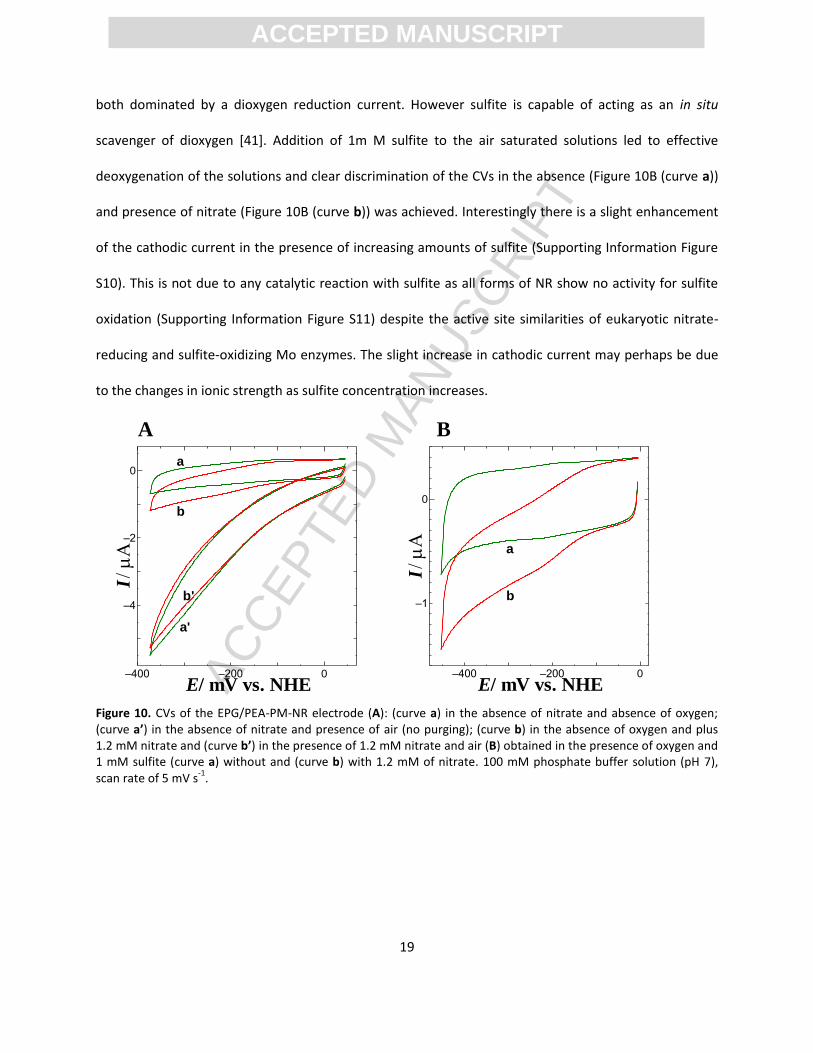
ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
19
–400 –200 0
–4
–2
0
/ mV vs. NHEE
I
a
a'
b
b'
–400 –200 0
–1
0
/ mV vs. NHEE
I
a
b
both dominated by a dioxygen reduction current. However sulfite is capable of acting as an in situ
scavenger of dioxygen [41]. Addition of 1m M sulfite to the air saturated solutions led to effective
deoxygenation of the solutions and clear discrimination of the CVs in the absence (Figure 10B (curve a))
and presence of nitrate (Figure 10B (curve b)) was achieved. Interestingly there is a slight enhancement
of the cathodic current in the presence of increasing amounts of sulfite (Supporting Information Figure
S10). This is not due to any catalytic reaction with sulfite as all forms of NR show no activity for sulfite
oxidation (Supporting Information Figure S11) despite the active site similarities of eukaryotic nitrate-
reducing and sulfite-oxidizing Mo enzymes. The slight increase in cathodic current may perhaps be due
to the changes in ionic strength as sulfite concentration increases.
Figure 10. CVs of the EPG/PEA-PM-NR electrode (A): (curve a) in the absence of nitrate and absence of oxygen; (curve a’) in the absence of nitrate and presence of air (no purging); (curve b) in the absence of oxygen and plus 1.2 mM nitrate and (curve b’) in the presence of 1.2 mM nitrate and air (B) obtained in the presence of oxygen and 1 mM sulfite (curve a) without and (curve b) with 1.2 mM of nitrate. 100 mM phosphate buffer solution (pH 7), scan rate of 5 mV s
-1.
A B

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
20
Conclusions
The unmediated catalytic electrochemistry of eukaryotic NR from the fungus Neurospora crassa
was demonstrated for the first time on an EPG electrode with the mixture of promoters
polyethyleneimine and polymyxin B. The enzyme modified electrode doesn’t show any catalytic current
in the absence of promoters and the amount of promoter was also vital in terms of maintaining keeping
native function and electrical communication with the electrode. The catalytic nitrate reduction current
increased non-linearly and an apparent Michaelis constant (KM,app) was found to be 39 μM. A bell shaped
pH profile was obtained with pH optimum of pH 6.8. The catalytic cathodic wave is significantly
increased upon increasing the temperature and an activation energy of 26 kJ mol-1 was obtained. The
FAD cofactor is shown to be nonessential for electrocatalysis and evidently direct reduction of the heme
cofactor is sufficient for catalytic activity. The electrode could be used to detect nitrate even without
purging the solution with an inert gas but the use of sulfite to deoxygenate the solution was necessary.
Acknowledgements. Support from the Australian Research Council (DP150103345) is
gratefully acknowledged.
Supporting Information Available. FAD voltammetry (in the absence and presence of
NR). Control experiments for catalytic activity towards the nitrate reduction at EPG/PEA-PM and
unmodified EPG/NR electrodes as well as sulfite oxidation experiments in the absence or presence of
NR. Catalytic voltammograms comparing nitrate reduction activity at EPG/PEA-NR, EPG/PM-NR and
EPG/PEA-PM-NR electrodes. SDS-PAGE analysis of all recombinant NR forms are also provided.

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
21
References
[1] J. Hirst, Elucidating the mechanisms of coupled electron transfer and catalytic reactions by protein film voltammetry, Biochim. Biophys. Acta, Bioenerg. 1757 (2006) 225-239.
[2] F.A. Armstrong, G.S. Wilson, Recent developments in faradaic bioelectrochemistry, Electrochim. Acta 45 (2000) 2623-2645.
[3] Y. Degani, A. Heller, Direct electrical communication between chemically modified enzymes and metal electrodes. 2. Methods for bonding electron-transfer relays to glucose oxidase and D-amino-acid oxidase, J. Am. Chem. Soc. 110 (1988) 2615-2620.
[4] E. Katz, I. Willner, Nanobiotechnology: integrated nanoparticle-biomolecule hybrid systems: Synthesis, properties, and applications, Angew. Chem., Int. Ed. 43 (2004) 6042-6108.
[5] P.V. Bernhardt, Enzyme Electrochemistry - Biocatalysis on an Electrode, Aust. J. Chem. 59 (2006) 233-256.
[6] D. Zheng, S.K. Vashist, K. Al-Rubeaan, J.H.T. Luong, F.-S. Sheu, Mediatorless amperometric glucose biosensing using 3-aminopropyltriethoxysilane-functionalized graphene, Talanta 99 (2012) 22-28.
[7] Y. Wang, L. Ge, C. Ma, Q. Kong, M. Yan, S. Ge, J. Yu, Self-powered and sensitive DNA detection in a three-dimensional origami-based biofuel cell based on a porous Pt-paper cathode, Chem. - Eur. J. 20 (2014) 12453-12462.
[8] C. Wang, C. Yang, Y. Song, W. Gao, X. Xia, Adsorption and direct electron transfer from hemoglobin into a three-dimensionally ordered macroporous gold film, Adv. Funct. Mater. 15 (2005) 1267-1275.
[9] C. Mu, Q. Zhao, D. Xu, Q. Zhuang, Y. Shao, Silicon Nanotube Array/Gold Electrode for Direct Electrochemistry of Cytochrome c, J. Phys. Chem. B 111 (2007) 1491-1495.
[10] S.M. Dengale, A.K. Yagati, Y.-H. Chung, J. Min, J.-W. Choi, An electrochemical H2O2 detection method based on direct electrochemistry of myoglobin immobilized on gold deposited ITO electrode, J. Nanosci. Nanotechnol. 13 (2013) 6424-6428.
[11] T.d.F. Paulo, I.C.N. Diógenes, H.D. Abruña, Direct Electrochemistry and Electrocatalysis of Myoglobin Immobilized on l-Cysteine Self-Assembled Gold Electrode, Langmuir 27 (2011) 2052-2057.
[12] G.-X. Ma, Y.-G. Wang, C.-X. Wang, T.-H. Lu, Y.-Y. Xia, Hemoglobin immobilized on whisker-like carbon composites and its direct electrochemistry, Electrochim. Acta 53 (2008) 4748-4753.
[13] R. Huang, N. Hu, Direct electrochemistry and electrocatalysis with horseradish peroxidase in Eastman AQ films, Bioelectrochemistry 54 (2001) 75-81.
[14] X. Chen, X. Peng, J. Kong, J. Deng, Facilitated electron transfer from an electrode to horseradish peroxidase in a biomembrane-like surfactant film, J. Electroanal. Chem. 480 (2000) 26-33.
[15] P. Kalimuthu, J. Tkac, U. Kappler, J.J. Davis, P.V. Bernhardt, Highly Sensitive and Stable Electrochemical Sulfite Biosensor Incorporating a Bacterial Sulfite Dehydrogenase, Anal. Chem. 82 (2010) 7374-7379.
[16] P. Kalimuthu, M.D. Heath, J.M. Santini, U. Kappler, P.V. Bernhardt, Electrochemically driven catalysis of Rhizobium sp. NT-26 arsenite oxidase with its native electron acceptor cytochrome c552, Biochim. Biophys. Acta, Bioenerg. 1837 (2014) 112-120.
[17] S. Li, J. Xia, C. Liu, W. Cao, J. Hu, Q. Li, Direct electrochemistry of cytochrome c at a novel gold nanoparticles-attached NH2+ ions implantation-modified indium tin oxide electrode, J. Electroanal. Chem. 633 (2009) 273-278.

ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
22
[18] K.-F. Aguey-Zinsou, P.V. Bernhardt, A.G. McEwan, J.P. Ridge, The first non-turnover voltammetric response from a molybdenum enzyme: direct electrochemistry of dimethylsulfoxide reductase from Rhodobacter capsulatus, J. Biol. Inorg. Chem. 7 (2002) 879-883.
[19] J.J. Gooding, Nanostructuring electrodes with carbon nanotubes: A review on electrochemistry and applications for sensing, Electrochim. Acta 50 (2005) 3049-3060.
[20] C. Gao, Z. Guo, J.-H. Liu, X.-J. Huang, The new age of carbon nanotubes: An updated review of functionalized carbon nanotubes in electrochemical sensors, Nanoscale 4 (2012) 1948-1963.
[21] G.-X. Wang, Y. Qian, X.-X. Cao, X.-H. Xia, Direct electrochemistry of cytochrome c on a graphene/poly (3,4-ethylenedioxythiophene) nanocomposite modified electrode, Electrochem. Commun. 20 (2012) 1-3.
[22] R. Hille, J. Hall, P. Basu, The Mononuclear Molybdenum Enzymes, Chem. Rev. 114 (2014) 3963-4038.
[23] P. Ringel, J. Krausze, J. van den Heuvel, U. Curth, A.J. Pierik, S. Herzog, R.R. Mendel, T. Kruse, Biochemical Characterization of Molybdenum Cofactor-free Nitrate Reductase from Neurospora crassa, J. Biol. Chem. 288 (2013) 14657-14671.
[24] W.H. Campbell, Nitrate reductase structure, function and regulation: bridging the gap between biochemistry and physiology, Annu. Rev. Plant Physiol. Plant Mol. Biol. 50 (1999) 277-303.
[25] W.H. Campbell, J.R. Kinghorn, Functional domains of assimilatory nitrate reductases and nitrite reductases, Trends Biochem. Sci. 15 (1990) 315-319.
[26] C. Gonzalez, N. Brito, G.A. Marzluf, Functional analysis by site-directed mutagenesis of individual amino acid residues in the flavin domain of Neurospora crassa nitrate reductase, Mol. Gen. Genet. 249 (1995) 456-464.
[27] K. Fischer, G.G. Barbier, H.-J. Hecht, R.R. Mendel, W.H. Campbell, G. Schwarz, Structural basis of eukaryotic nitrate reduction: Crystal structures of the nitrate reductase active site, Plant Cell 17 (2005) 1167-1179.
[28] C. Probst, P. Ringel, V. Boysen, L. Wirsing, M.M. Alexander, R.R. Mendel, T. Kruse, Genetic characterization of the Neurospora crassa molybdenum cofactor biosynthesis, Fungal Genet. Biol. 66 (2014) 69-78.
[29] P. Kalimuthu, K. Fischer-Schrader, G. Schwarz, P.V. Bernhardt, Mediated Electrochemistry of Nitrate Reductase from Arabidopsis thaliana, J. Phys. Chem. B 117 (2013) 7569-7577.
[30] P. Kalimuthu, K. Fischer-Schrader, G. Schwarz, P.V. Bernhardt, A sensitive and stable amperometric nitrate biosensor employing Arabidopsis thaliana nitrate reductase, J. Biol. Inorg. Chem. 20 (2015) 385-393.
[31] L.J. Anderson, D.J. Richardson, J.N. Butt, Catalytic protein film voltammetry from a respiratory nitrate reductase provides evidence for complex electrochemical modulation of enzyme activity, Biochemistry 40 (2001) 11294-11307.
[32] S.J. Elliott, K.R. Hoke, K. Heffron, M. Palak, R.A. Rothery, J.H. Weiner, F.A. Armstrong, Voltammetric Studies of the Catalytic Mechanism of the Respiratory Nitrate Reductase from Escherichia coli: How Nitrate Reduction and Inhibition Depend on the Oxidation State of the Active Site, Biochemistry 43 (2004) 799-807.
[33] S. Dementin, P. Arnoux, B. Frangioni, S. Grosse, C. Leger, B. Burlat, B. Guigliarelli, M. Sabaty, D. Pignol, Access to the Active Site of Periplasmic Nitrate Reductase: Insights from Site-Directed Mutagenesis and Zinc Inhibition Studies, Biochemistry 46 (2007) 9713-9721.
[34] V. Fourmond, M. Sabaty, P. Arnoux, P. Bertrand, D. Pignol, C. Leger, Reassessing the Strategies for Trapping Catalytic Intermediates during Nitrate Reductase Turnover, J. Phys. Chem. B 114 (2010) 3341-3347.
ACC
EPTE
D M
ANU
SCR
IPT
ACCEPTED MANUSCRIPT
23
[35] R.H. Garrett, A. Nason, Involvement of a B-type cytochrome in the assimilatory nitrate reductase of Neurospora crassa, Proceedings of the National Academy of Sciences of the United States of America 58 (1967) 1603-1610.
[36] T.G.M. Schmidt, L. Batz, L. Bonet, U. Carl, G. Holzapfel, K. Kiem, K. Matulewicz, D. Niermeier, I. Schuchardt, K. Stanar, Development of the Twin-Strep-tag® and its application for purification of recombinant proteins from cell culture supernatants, Protein Expression Purif. 92 (2013) 54-61.
[37] T. Palmer, C.-L. Santini, C. Iobbi-Nivol, D.J. Eaves, D.H. Boxer, G. Giordano, Involvement of the narJ and mob gene products in distinct steps in the biosynthesis of the molybdoenzyme nitrate reductase in Escherichia coli, Mol. Microbiol. 20 (1996) 875-884.
[38] M.S. Brody, R. Hille, The Kinetic Behavior of Chicken Liver Sulfite Oxidase, Biochemistry 38 (1999) 6668-6677.
[39] P. Kalimuthu, U. Kappler, P.V. Bernhardt, Catalytic voltammetry of the molybdoenzyme sulfite dehydrogenase from Sinorhizobium meliloti, J. Phys. Chem. B 118 (2014) 7091-7099.
[40] R.H. Garrett, P. Greenbaum, Inhibition of the Neurospora crassa nitrate reductase complex by metal-binding agents, Biochim. Biophys. Acta, Enzymol. 302 (1973) 24-32.
[41] D. Quan, J.H. Shim, J.D. Kim, H.S. Park, G.S. Cha, H. Nam, Electrochemical determination of nitrate with nitrate reductase-immobilized electrodes under ambient air, Anal. Chem. 77 (2005) 4467-4473.
Fig. 9
–400 –200 0
–2
–1
0
/ mV vs. NHEE
I abc
d