direct electrochemistry and electrocatalysis of hemoglobin on bimetallic au–pt inorganic–organic...
TRANSCRIPT

Direct Electrochemistry and Electrocatalysis of Hemoglobinon Bimetallic Au–Pt Inorganic–Organic Nanofiber HybridNanocomposite and Mesoporous Molecular Sieve MCM-41
Azadeh Azadbakht • Amir Reza Abbasi •
Mohammad Bagher Gholivand • Zohreh Derikvand
Received: 26 October 2013 / Accepted: 2 December 2013
� Springer Science+Business Media New York 2013
Abstract A glassy carbon electrode modified with
MCM-41 and bimetallic inorganic–organic nanofiber
hybrid nanocomposite was prepared and used for deter-
mination of trace levels of hydrogen peroxide (H2O2). The
direct electron transfer (DET) and electrocatalysis of
hemoglobin (Hb) entrapped in the MCM-41 modified Au–
Pt inorganic–organic nanofiber hybrid nanocomposite
electrode (Au–PtNP/NF/GCE) were investigated by using
cyclic voltammetry in 0.1 M pH 7.0 phosphate buffer
solution. Due to its uniform pore structure, high surface
area and good biocompatibility, the mesoporous silica
sieve MCM-41 provided a suitable matrix for immobili-
zation of biomolecules. The MCM-41 modified Au–Pt
inorganic–organic nanofiber hybrid nanocomposite elec-
trode showed significant promotion to DET of Hb, which
exhibited a pair of well-defined and quasi-reversible peaks
for heme Fe(III)/Fe(II) with a formal potential of -0.535 V
(vs. Ag/AgCl). Additionally, the Hb immobilized on the
MCM-41 modified electrode showed excellent electrocat-
alytic activity toward H2O2 reduction.
Keywords MCM-41 � Hydrogen peroxide � Hybrid
nanocomposite � Electrocatalytic reduction
1 Introduction
The direct electron transfer (DET) between redox proteins
and electrode surface has received widespread attention in
recent years [1]. On one hand, it can provide information
about electron transfer mechanism of proteins in biological
systems [2]. Interestingly, it is a good foundation for fabri-
cating electrochemical biosensors and bioelectroanalytical
devices [3, 4]. Hemoglobin (Hb) is a heme protein that can
store and transport oxygen in blood of vertebrate animals. It
contains two a and b subunits, each of which has one iron
bearing heme as electron-transfer center. However, Hb shows
slow rate of electron transfer on conventional electrode
because the electroactive centers are buried in the polypep-
tide. To overcome this problem, great efforts have been made
to improve the DET by immobilizing Hb within different
biocompatible materials, such as natural biomolecules [5],
biopolymers [6], hydrogels [2, 7] bioceramics [8], nanoma-
terials like carbon nanotubes [9], mesoporous materials [10,
11] and so on. Among these materials, mesoporous materials
have been actively developed to immobilize proteins or
enzymes at the surface of electrode in the recent years.
Mesoporous materials, with pore size of 2–50 nm, pos-
sess uniform pores size and large surface area
(300–1,000 m2g-1) [12] as well as mechanical and chemical
resistance. They show more effective ness in protein
immobilization, compared with conventional materials [13,
14]. In this work, the mesoporous silica sieve MCM-41 was
prepared to immobilize Hb. MCM-41 can provide favorable
microenvironments for proteins due to their large pore size,
ordered uniform pore structure, huge surface area, high
loading capacity and good biocompatibility for electron
transfer [15, 16].What is more, it can immobilize proteins or
enzymes firmly without the aid of other cross-linking
reagents. However, the conductivity of MCM-41 cannot well
A. Azadbakht (&) � A. R. Abbasi � Z. Derikvand
Department of Chemistry, Faculty of Science, Khorramabad
Branch, Islamic Azad University, Khorramabad, Iran
e-mail: [email protected]
M. B. Gholivand
Department of Analytical Chemistry, Faculty of Chemistry, Razi
University, Kermanshah, Iran
123
J Inorg Organomet Polym
DOI 10.1007/s10904-013-0012-x

satisfy its application as protein supports of biosensors, the
Au and Pt nanoparticle can make up for this drawback.
Over the past decade, one dimensional (1D) inorganic–
organic hybrid nanomaterials have received much interest
because of their intriguing properties and potential applica-
tions in chemical or biochemical sensors, catalysis and
nanodevices [17–19]. These hybrid materials that are based
on the combination of organic and inorganic species exhibit
advantages over organic materials such as light weight,
flexibility and good mold ability. Moreover, these hybrid
materials have many advantages over inorganic materials in
different aspects such as high strength, heat stability and
chemical resistance [20–22]. Such features of (1D) organic–
inorganic hybrid nanomaterials make them ideal building
blocks for a new generation of electrochemical sensors.
Recently, Gong et al. developed a hybrid of bimetallic–
inorganic–organic nanofibers (NFs) for the stripping assay of
Hg(II) [23]. Decoration of organic nanowires with metal NPs
could be an attractive route to fabricate inorganic–organic
hybrid nanomaterials without compromising the functions of
nanowires or nanoparticles [19]. The nanoparticles fre-
quently display unusual physical and chemical properties
depending upon their size, shape, and stabilizing agents.
Nanoparticles also facilitate electron transfer and can be
easily modified in a wide range of biomolecules and chem-
ical ligands. Therefore, combinations of nanowires and
metal nanoparticles have received much interest, owing to
their intriguing properties and potential applications in
chemical sensing [24]. 3,30,5,50-Tetramethylbenzidine
(TMB), as being much less hazardous than benzidine and
more sensitive as a chromogenic reagent, has been investi-
gated for many years [25]. Doping of TMB-based organic
NFs by incorporation of metal ions is of particular interest.
In this paper, we immobilized Hb on MCM-41 bimetallic
Au–Pt inorganic–organic hybrid nanocomposite glassy car-
bon electrode to form stable modified electrodes that cata-
lyze the reduction of hydrogen peroxide (H2O2).
Considering the good performance of MCM-41, an attempt
to use it to immobilize Hb and to realize the direct electro-
chemistry of Hb had been carried out. The results showed
that the immobilized Hb almost retained its native structure
and displayed high electroactivity and electrocatalysis for
H2O2. This study demonstrated that MCM-41 bimetallic
Au–Pt inorganic–organic hybrid nanocomposite glassy car-
bon electrode was suitable for the immobilization of protein.
2 Experimental
2.1 Chemicals and Reagents
H2O2, ascorbic acids (AA), uric acids (UA) and Hb (from
bovine blood) were available from Sigma. TMB, H2PtCl6,
HNO3, HCl and sodium citrate were purchased from
Merck. Cetyltrimethylammonium bromide (CTAB), tetra-
ethyl orthosilicate (TEOS) and HAuCl4 were purchased
from Aldrich. All other chemicals were of analytical
reagent grade and used without further purification.
2.2 Instrumentation
Electrochemical experiments were performed via using an
Autolab modular electrochemical system (Eco Chem.
Utrecht, the Netherlands) equipped with PSTA 20 model
and driven by GPES software (Eco Chem.). A conventional
three-electrode cell was used with a saturated Ag|AgCl as
reference electrode, a Pt wire as counter electrode, and a
modified glassy carbon as working electrode. All experi-
ments were carried out at ambient temperature of
25 ± 1 �C. A Metrohm pH-meter (model 691) was also
employed for pH measurements. The surface morphology
of modified electrodes was characterized with a scanning
electron microscope (SEM) (PhilipsXL 30) with gold
coating.
2.3 Preparation of MCM-41
The MCM-41 was synthesized as follows: The molar ratio
of synthesis was accorded with previous literatures [26].
2.3689 g CTAB was added to a basic solution of (0.48 g
NaOH in 35 mL H2O) under stirring. When the solution
became homogeneous, TEOS was added, giving rise to
coloured slurry. The mixture was stirred unceasingly at
85 �C for 24 h and then the sample was filtrated. Finally,
MCM-41 was obtained after calcining at 540 �C for 5 h.
2.4 Electrode Modification
To prepare a modified electrode glassy carbon electrode
was polished with emery paper followed by alumina (1.0
and 0.05 lm) and then thoroughly washed with twice-
distilled water. Then, the electrode was placed in an eth-
anol container and used bath ultrasonic cleaner to remove
adsorbed particles. TMB-based NFs were prepared
according to the previous work [27]. Briefly, 2.5 mL of
2 mM aqueous H2PtCl6 solution was added into 4 mL of
1.25 mM ethanol TMB solution at the room temperature.
Several minutes later, doped NFs were formed with a large
amount of blue–violet precipitate observed. Then, the
precipitate was collected by centrifugation, washed several
times with water, and dried at 60 �C. The suspension of
TMB-based NFs (0.75 mg mL-1) was prepared by dis-
persing the resulting powdered NFs into the ethanol solu-
tion under ultrasonication for 2 h. Subsequently, 10 lL of
the NFs dispersion was dropped onto the surface of the
GCE and was kept at the room temperature until dry.
J Inorg Organomet Polym
123

Further modification of Au nanoparticles (AuNPs) onto
NFs/GCE was conducted by cyclic voltammetry (CV)
scanning from 0.2 to -1.0 V in 0.1 M KCl solution con-
taining 0.125 mM HAuCl4 at a scan rate of 50 mV s-1 for
16 cycles. During the electrode position process, a part of
doped Pt(II) ions was simultaneously reduced to Pt atoms,
thus leading to a bimetallic Au–PtNPs inorganic–organic
hybrid nanocomposite [27]. After that, the electrode
(denoted as Au–PtNPs/GCE) was thoroughly rinsed with
water and kept at the room temperature for further use.
A standard Hb solution was prepared by dissolving Hb
in acetate buffer (pH 5.0) solution. In each adsorption
experiment, 0.1 g of MCM-41 sample was added to 5 mL
of Hb solutions with different concentrations. After stirred
for 5 h, the mixtures were centrifuged at 10,000 rpm for
30 min. Then, the concentration of Hb in the supernatants
was determined by means of UV spectra at 405.4 nm. The
binding amounts of Hb onto MCM-41 supports were cal-
culated by subtracting the free Hb concentration from the
total Hb concentration. After this process, the Hb/MCM-41
was rinsed with water and then with acetate buffer (pH 5.0)
solution. Then, 100 lL of the Hb/MCM-41 suspension was
mixed with 10 lL of 5 wt% Nafion solution to produce Hb/
Nafion/MCM-41 suspension. 6 lL of Hb/Nafion/MCM-41
suspension was dropped on the surface of the modified GC
electrode. After the electrode was dried at room tempera-
ture and rinsed with water, the Hb/Nafion/MCM-41/Au–
PtNP/GC electrode was obtained. The electrode was stored
in refrigerator at 4 �C before use.
3 Results and Discussion
3.1 Characterization of the Modified Electrode by SEM
To investigate the surface structure and morphology of the
modified electrode, we performed SEM. Figure 1 shows
the SEM images of Au–PtNPs/NFs/GCE (Fig. 1a) MCM-
41/Au–PtNPs/NFs/GCE (Fig. 1b) and Hb/MCM-41/Au–
PtNP/GC electrodes (Fig. 1c). It can be seen that uniform
Au–PtNPs aligned along the surface of NFs. Generated
NPs homogenously distributed in the matrix of the inter-
laced NFs (Fig. 1a). After the subsequent deposition pro-
cess, one can see that uniform Au–PtNPs with an average
diameter around 40 nm aligned along the surface of those
NFs (Fig. 1a). The generated NPs were homogenously
distributed in the matrix of interlaced NFs. Figure 1b
shows the SEM image of MCM-41/Au–PtNPs/NFs/GCE.
As can be seen from the Fig. 1b, some small flakes were
well dispersed on the surface of the Au–PtNPs/NFs/GCE,
which indicated that the mesoporous sieves MCM-41 were
successfully modified on the Au–PtNPs/NFs/GCE. Fig-
ure 1c shows the SEM image of Hb/MCM-41/Au–PtNP/
GC with loose and porous structure and morphology. The
porous structure of Hb film on MCM-41/Au–PtNP/GC
could allow the small molecule to go through the film very
easily, which was beneficial to the electron transfer of
proteins in the film with underlying electrodes.
3.2 FT-IR Spectroscopy
FT-IR spectroscopy is a useful method for monitoring the
secondary structure of proteins. The characteristic amide I
and amide II bands of proteins provide detailed information
on the secondary structure of polypeptide chain [28]. The
amide I band (1,700–1,600 cm-1) is attributed to C=O
stretching vibration of peptide linkages in the backbone of
protein, and the amide II band (1,620–1,500 cm-1) is
caused by a combination of N–H bending and C–N
stretching of the peptide group. The intensity of the amide I
and amide II bands will significantly weaken or even dis-
appear if Hb is denatured [28]. The amide I and amide II
bands (1,647.33 and 1,537.26 cm-1) of Hb in MCM-41
had similar shapes and positions to those obtained for pure
Hb (1,651.99 and 1,533.02 cm-1) as described in the lit-
erature [28]. However, MCM-41 did not show obvious
Fig. 1 SEM images of Au–PtNPs/NFs/GCE (a), MCM-41/Au–PtNPs/NFs/GCE (b) and Hb/MCM-41/Au–PtNP/GC electrodes (c)
J Inorg Organomet Polym
123
signals in this wave number range. Therefore, it could be
proposed that the Hb immobilized in the MCM-41 almost
retained its native secondary structures.
3.3 UV–Vis Spectroscopy
Figure 2 shows the UV–Vis spectra of Hb and Hb/MCM-
41 in PBS. The Soret band of Hb is located at 406 nm
(Fig. 2, curve a) while that of Hb/MCM-41 is 407 nm
(Fig. 2, curve b). The band position of Hb/MCM-41 is
slightly shifted in comparison with that of Hb, which is
attributed to the weak interactions of MCM-41 with Hb
molecule.
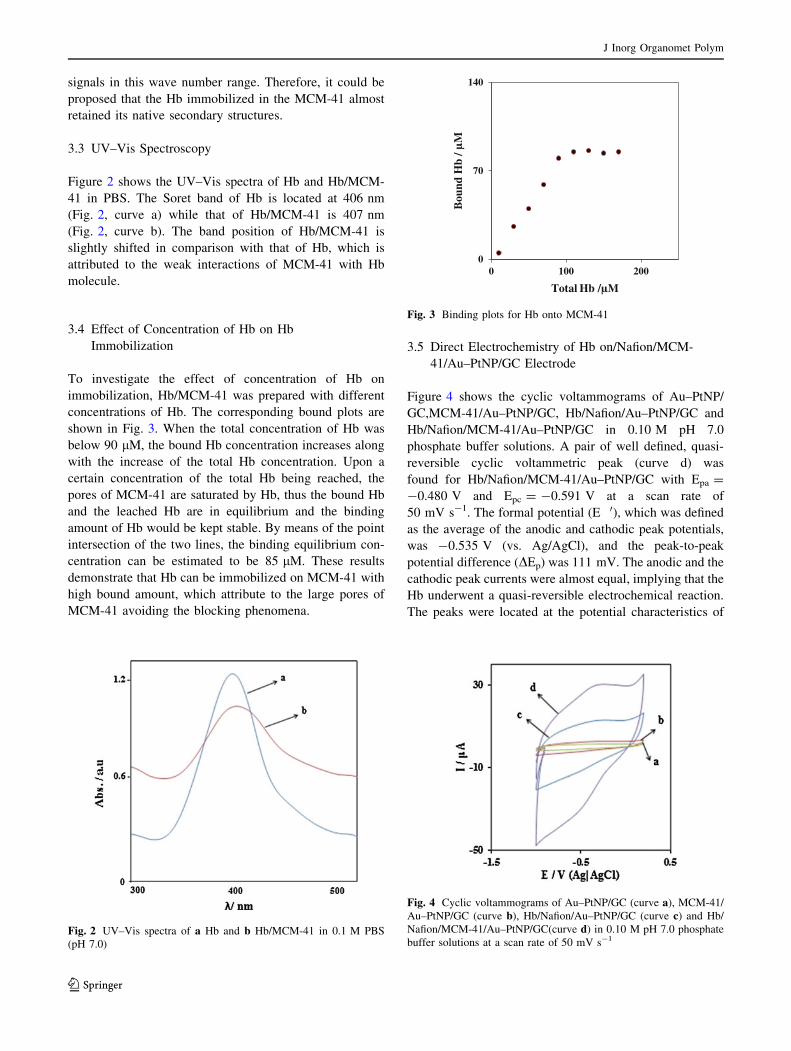
3.4 Effect of Concentration of Hb on Hb
Immobilization
To investigate the effect of concentration of Hb on
immobilization, Hb/MCM-41 was prepared with different
concentrations of Hb. The corresponding bound plots are
shown in Fig. 3. When the total concentration of Hb was
below 90 lM, the bound Hb concentration increases along
with the increase of the total Hb concentration. Upon a
certain concentration of the total Hb being reached, the
pores of MCM-41 are saturated by Hb, thus the bound Hb
and the leached Hb are in equilibrium and the binding
amount of Hb would be kept stable. By means of the point
intersection of the two lines, the binding equilibrium con-
centration can be estimated to be 85 lM. These results
demonstrate that Hb can be immobilized on MCM-41 with
high bound amount, which attribute to the large pores of
MCM-41 avoiding the blocking phenomena.
3.5 Direct Electrochemistry of Hb on/Nafion/MCM-
41/Au–PtNP/GC Electrode
Figure 4 shows the cyclic voltammograms of Au–PtNP/
GC,MCM-41/Au–PtNP/GC, Hb/Nafion/Au–PtNP/GC and
Hb/Nafion/MCM-41/Au–PtNP/GC in 0.10 M pH 7.0
phosphate buffer solutions. A pair of well defined, quasi-
reversible cyclic voltammetric peak (curve d) was
found for Hb/Nafion/MCM-41/Au–PtNP/GC with Epa =
-0.480 V and Epc = -0.591 V at a scan rate of
50 mV s-1. The formal potential (E Æ 0), which was defined
as the average of the anodic and cathodic peak potentials,
was -0.535 V (vs. Ag/AgCl), and the peak-to-peak
potential difference (DEp) was 111 mV. The anodic and the
cathodic peak currents were almost equal, implying that the
Hb underwent a quasi-reversible electrochemical reaction.
The peaks were located at the potential characteristics of
Fig. 2 UV–Vis spectra of a Hb and b Hb/MCM-41 in 0.1 M PBS
(pH 7.0)
0
70
140
0 100 200
Bou
nd H
b/ µ
M
Total Hb /µM
Fig. 3 Binding plots for Hb onto MCM-41
Fig. 4 Cyclic voltammograms of Au–PtNP/GC (curve a), MCM-41/
Au–PtNP/GC (curve b), Hb/Nafion/Au–PtNP/GC (curve c) and Hb/
Nafion/MCM-41/Au–PtNP/GC(curve d) in 0.10 M pH 7.0 phosphate
buffer solutions at a scan rate of 50 mV s-1
J Inorg Organomet Polym
123
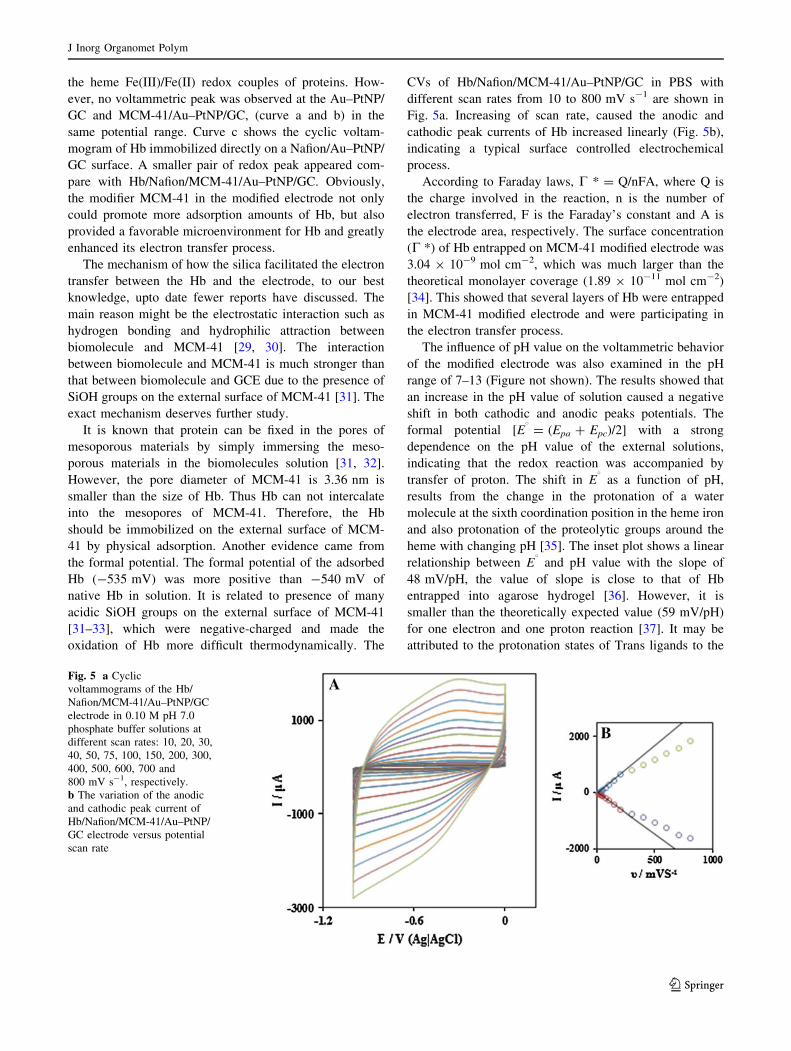
the heme Fe(III)/Fe(II) redox couples of proteins. How-
ever, no voltammetric peak was observed at the Au–PtNP/
GC and MCM-41/Au–PtNP/GC, (curve a and b) in the
same potential range. Curve c shows the cyclic voltam-
mogram of Hb immobilized directly on a Nafion/Au–PtNP/
GC surface. A smaller pair of redox peak appeared com-
pare with Hb/Nafion/MCM-41/Au–PtNP/GC. Obviously,
the modifier MCM-41 in the modified electrode not only
could promote more adsorption amounts of Hb, but also
provided a favorable microenvironment for Hb and greatly
enhanced its electron transfer process.
The mechanism of how the silica facilitated the electron
transfer between the Hb and the electrode, to our best
knowledge, upto date fewer reports have discussed. The
main reason might be the electrostatic interaction such as
hydrogen bonding and hydrophilic attraction between
biomolecule and MCM-41 [29, 30]. The interaction
between biomolecule and MCM-41 is much stronger than
that between biomolecule and GCE due to the presence of
SiOH groups on the external surface of MCM-41 [31]. The
exact mechanism deserves further study.
It is known that protein can be fixed in the pores of
mesoporous materials by simply immersing the meso-
porous materials in the biomolecules solution [31, 32].
However, the pore diameter of MCM-41 is 3.36 nm is
smaller than the size of Hb. Thus Hb can not intercalate
into the mesopores of MCM-41. Therefore, the Hb
should be immobilized on the external surface of MCM-
41 by physical adsorption. Another evidence came from
the formal potential. The formal potential of the adsorbed
Hb (-535 mV) was more positive than -540 mV of
native Hb in solution. It is related to presence of many
acidic SiOH groups on the external surface of MCM-41
[31–33], which were negative-charged and made the
oxidation of Hb more difficult thermodynamically. The
CVs of Hb/Nafion/MCM-41/Au–PtNP/GC in PBS with
different scan rates from 10 to 800 mV s-1 are shown in
Fig. 5a. Increasing of scan rate, caused the anodic and
cathodic peak currents of Hb increased linearly (Fig. 5b),
indicating a typical surface controlled electrochemical
process.
According to Faraday laws, C * = Q/nFA, where Q is
the charge involved in the reaction, n is the number of
electron transferred, F is the Faraday’s constant and A is
the electrode area, respectively. The surface concentration
(C *) of Hb entrapped on MCM-41 modified electrode was
3.04 9 10-9 mol cm-2, which was much larger than the
theoretical monolayer coverage (1.89 9 10-11 mol cm-2)
[34]. This showed that several layers of Hb were entrapped
in MCM-41 modified electrode and were participating in
the electron transfer process.
The influence of pH value on the voltammetric behavior
of the modified electrode was also examined in the pH
range of 7–13 (Figure not shown). The results showed that
an increase in the pH value of solution caused a negative
shift in both cathodic and anodic peaks potentials. The
formal potential [E8 = (Epa ? Epc)/2] with a strong
dependence on the pH value of the external solutions,
indicating that the redox reaction was accompanied by
transfer of proton. The shift in E8 as a function of pH,
results from the change in the protonation of a water
molecule at the sixth coordination position in the heme iron
and also protonation of the proteolytic groups around the
heme with changing pH [35]. The inset plot shows a linear
relationship between E8 and pH value with the slope of
48 mV/pH, the value of slope is close to that of Hb
entrapped into agarose hydrogel [36]. However, it is
smaller than the theoretically expected value (59 mV/pH)
for one electron and one proton reaction [37]. It may be
attributed to the protonation states of Trans ligands to the
Fig. 5 a Cyclic
voltammograms of the Hb/
Nafion/MCM-41/Au–PtNP/GC
electrode in 0.10 M pH 7.0
phosphate buffer solutions at
different scan rates: 10, 20, 30,
40, 50, 75, 100, 150, 200, 300,
400, 500, 600, 700 and
800 mV s-1, respectively.
b The variation of the anodic
and cathodic peak current of
Hb/Nafion/MCM-41/Au–PtNP/
GC electrode versus potential
scan rate
J Inorg Organomet Polym
123

heme iron and amino acids around the heme, or the pro-
tonation of the water molecule coordinated to the central
iron [38].
3.6 Electrocatalytic Activity to Reduction of H2O2
Application of the modified electrode for reduction of
H2O2 was evaluated by CV. The cyclic voltammetric
responses of Au–PtNP/GC, MCM-41/Au–PtNP/GC, Hb/
Nafion/Au–PtNP/GCand Hb/Nafion/MCM-41/Au–PtNP/
GC electrodes (Fig. 6) in PBS (pH 7.0) in the absence and
presence of 9 mM of H2O2 were presented in Fig. 6. H2O2
did not undergo oxidation at AuNPs/GC and MCM-41/Au–
PtNP/GC electrodes in the potential window of -1–0.2 V
in PBS (curves b and d). However, presence of Hb film on
bimetallic Au–Pt inorganic–organic hybrid nanocomposite
had a catalytic effect for oxidation of H2O2. As shown in
Fig. 6, a pair of redox peak corresponding to the Fe(III)/
Fe(II) couple were observed at the surface of the electrodes
tested (curves e and g). Upon addition of H2O2, an
enhancement in the cathodic peak current is observed and
the anodic peak current tended to decrease (curves f and h).
The reason for this increase is that, along with the anodic
potential sweep, H2O2 oxides Fe(II) to Fe(III), while
simultaneous reduction of the regenerated Fe(III) causes an
increase in the cathodic current.
For the same reason, the anodic current is smaller in
the presence of H2O2, indicating that Fe(II) is consumed
during a chemical step. Taking into account all these
observations, a possible mechanism of H2O2 electro
reduction Hb/Nafion/MCM-41/Au–PtNP/GC electrode
may be as follows:
HbFe IIIð Þ þ Hþ þ e�HbHFe IIð Þ ð1Þ
Fig. 6 a Cyclic voltammograms of Au–PtNP/GC (a and b), MCM-
41/Au–PtNP/GC (c and d), Hb/Nafion/Au–PtNP/GC (e and f) and Hb/
Nafion/MCM-41/Au–PtNP/GC electrodes (g and h) in 0.10 M pH 7.0
phosphate buffer solutions at scan rate of 50 mV s-1 in the absence
(a, c, e, g) and the presence (b, d, f, h) of 9 mM H2O2, respectively
Fig. 7 a Cyclic
voltammograms of the modified
electrode in the presence of
5 mM H2O2at various scan
rates: 10, 20,30, 40, 50, 75, 100,
150, 200, 300, 400 and
500 mV s-1 in 0.10 M pH 7.0
phosphate buffer solutions.
b Variation of anodic peak
currents versus square root of
potential scan rate c dependence
of the anodic peak potential
versus Log(v)
J Inorg Organomet Polym
123

2HbHFe IIð Þ þ H2O22HbFe IIIð Þ þ 2H2O ð2Þ
Moreover, the electrocatalytic reduction peak current of
H2O2 at Hb/Nafion/MCM-41/Au–PtNP/GC electrode was
-189 lA, which was 2.4 times larger than that at Hb/
Nafion/Au–PtNP/GC electrode (-7 lA). These results
indicate that presence of MCM-41 in the modified
electrode supplied a larger surface area to allow more
deposition of Hb for reduction of H2O2. Due to theirs
ordered uniform pore structure, huge surface areas, high
loading capacity, good biocompatibility and fine
environment for electron transfer, the use of MCM-41 as
modifier could greatly improve the behavior of Hb.
Figure 7a illustrates cyclic voltammograms of 5 mM
H2O2 using modified electrode that recorded at potential
sweep rates ranging from 10 to 500 mV s-1. The anodic
peak currents obtained were linear with respect to the
square root of the potential sweep rate (Fig. 7b), which
indicates the mass transfer controlled process. Also, the
results show that by increasing the scan rate, the cathodic
peak potential shifts toward negative potentials, suggesting
a kinetic limitation in the reaction between redox sites of
the Hb/Nafion/MCM-41/Au–PtNP/GC electrode and H2O2.
The a value of the electrode reaction can be evaluated from
the following equation [39]:
Ep ¼ b=2ð Þlog vð Þ þ constants ð3ÞOn the basis of Eq. (3), the slope of Ep versus log v plot
is b/2, where b indicates the Tafel slope. The plot of Ep
versus log v indicates a linear variation for scan rates
ranging 10–500 mV s-1 (Fig. 7c). The a value was
obtained using the recorded I–E curve of electrocatalytic
reduction of Hb/Nafion/MCM-41/Au–PtNP/GC (slope of
Log I vs. E plot). From the result obtained a transfer
coefficient of a was estimated as 0.49.
3.7 Amperometric Detection of H2O2 at Hb/Nafion/
MCM-41/Au–PtNP/GC Electrode
Since amperometry under stirred conditions has much
higher current sensitivity than CV, it was used to estimate
the lower limit of detection. Figure 8 displays a typical
steady-state catalytic current time response of the rotated
modified electrode (2,000 rpm) with successive injection
of 2.5 lM of H2O2 at an applied potential of -0.53 V
versus the reference electrode. As shown, during the suc-
cessive addition of H2O2, a well-defined response was
observed, demonstrating stable and efficient catalytic
ability of the Hb immobilized on the bimetallic Au–Pt
inorganic–organic hybrid nanocomposite and MCM-41
films. The response current is linear in the range of
2.5–40 lM of H2O2. The calibration plot over the con-
centration range of 2.5–40 lM has a slope with the cor-
relation coefficient of 0.998 and the detection limit of
0.25 lM at the signal to noise ratio of 3.
Detection limit and linear calibration range of the pro-
posed method were compared with those obtained in other
reports and the results are summarized in Table 1.
Although the linear range of the proposed modified elec-
trode is smaller than those reported in some previous
works, its detection limit is comparable or better than the
results reported for H2O2 determination at the surface of
recently fabricated modified electrodes [40–47].
3.8 Effect of Interferences on H2O2 Oxidation
In order to reduce the interference of AA and UA, a Naf-
ion-coated Hb/MCM-41/Au–PtNP/GC electrode was pre-
pared. The effect of AA and UA on the reduction of H2O2
at the surface of Hb/Nafion/MCM-41/Au–PtNP/GC and
0
–70
–140
0 400 800
I / µ
A
E/ V (Ag|AgCl)
0
–70
–140
0 20 40 60
I / µ
A
C / µM
A
B
Fig. 8 a Amperometric
response of rotating sensor
during successive addition of
2.5 lM H2O2; conditions:
potential = -0.53 V,
pH = 7.0, and rotation speed of
2,000 rpm; b plots of current
versus H2O2 concentration
J Inorg Organomet Polym
123

Hb/MCM-41/Au–PtNP/GC were also investigated. Curve
a, b, c and d in Fig. 9a and b demonstrate a successive
additions of 15 lM H2O2, 15 lM uric acid, 15 lM AA and
15 lM H2O2 under the optimized experimental conditions
at the surface of Hb/MCM-41/Au–PtNP/GC (Fig. 9a) and
Hb/Nafion/MCM-41/Au–PtNP/GC (Fig. 9b) respectively.
Comparison of Fig. 9a and b shows that the current gen-
erated due to the interfering species at the surface of Hb/
Nafion/MCM-41/Au–PtNP/GC are negligible, indicating
high selectivity of the Hb/Nafion/MCM-41/Au–PtNP/GC
sensor. Therefore, a Nafion-coated Hb/MCM-41/Au–PtNP/
GC electrode may be used for the selective determination
of H2O2 in the presence of AA and UA. Nafion film is a
cation exchange polymer and repels AA and other nega-
tively charged species at optimized conditions. Nafion film
can provide a transport cannel only for the cations.
3.9 Stability and Reproducibility
Additional experiments were carried out to test the stability
and reproducibility. After several initial scans, no obvious
change of CV curves in pH 7.0 PBS could be observed for
about 100 continuous cyclic scans, suggesting that Hb
could tightly adsorb on the surface of MCM-41. The Hb/
Nafion/MCM-41/Au–PtNP/GC was stored in phosphate
buffer solution at pH 7.0 in the refrigerator at 4 �C for
2 weeks and no obvious change was found, indicating that
the modified electrode was quite stable. Repetitive mea-
surements were also carried out in buffer solution con-
taining 5 mM H2O2. Eight successive measurements
showed a relative standard deviation of 0.9 %, indicating
the modified electrode had excellent reproducibility.
3.10 Real Sample Analysis
The applicability of the proposed biosensor for H2O2
determination in serum samples investigated and results are
presented in Table 2. The standard addition method is
adopted for H2O2 detection in real sample and a calibration
curve obtained for each sample. The concentration of H2O2
in serum sample is found to be 200.5–205 lM, which is a
normal dosage for H2O2 in serum samples. To confirm the
validity of the results, the serum samples are spiked with
defined amount of H2O2 at levels similar to those found in
the samples. The obtained results in Table 2, demonstrate
satisfactory recoveries, varying between 97.5 and 101.5 %
for spiked H2O2. Therefore, the modified proposed sensor
can be used for H2O2 detection in real samples.
0
–30
–60
0 150 300
I / µ
A
t / s
0
–30
–60
0 150 300
I / µ
A
t / s
a
b
cd
a b cd
A BFig. 9 Successive additions of
15 lM H2O2 (a), 15 lM uric
acid (b), 15 lM ascorbic acid
(c) and15 lM H2O2 (d) under
the optimized experimental
conditions at the surface of Hb/
MCM-41/Au–PtNP/GC (a) and
Hb/Nafion/MCM-41/Au–PtNP/
GC (b) respectively
Table 2 Determination of H2O2 in human serum samples,
(%, ± R.S.D. calculated and based on five measurements)
Sample Added (lM) Found (lM) Recovery (%)
Serum sample 1 – 190.0 ± 4.0
30.0 224.0 ± 5.0 101.8 ± 2.0
70.0 259.6 ± 4.0 99.8 ± 1.5
Serum sample 2 – 195.5 ± 4.0
30.0 222.5 ± 4.0 98.6 ± 2.0
70.0 266.5 ± 4.5 100.3 ± 1.5
Table 1 Voltammetric response for H2O2 using various modified
electrodes
Electrode Linear range
(lM)
LoD
(lM)
References
Hb/nano–Au/ITO 10–70,000 45 [40]
Hb/silica sol–gel/CPE 10–100 3.9 [41]
Hb in silica sol–gel/CPE 1.00–280 0.86 [42]
Hb/CNT powder
microelectrodes electrodes
210–900 9.00 [43]
Hb/thiolated-viologen/ Au
electrode
10.0–125 0.25 [44]
Hb-Gel/GCE 50.0–1,200 3.40 [45]
Hb/CMC–TiO2-NTs/GCE 4.00–64.0 4.63 [46]
Hb/HNTs/ILs/GCE 7.50–97.5 2.40 [47]
This work 2.50–40.0 0.25
CPE carbon paste electrode, GCE glassy carbon electrode, HNTs
halloysite nanotubes, ILs ionic liquide
J Inorg Organomet Polym
123

4 Conclusions
In this paper, Hb could strongly adsorb onto the surface of
MCM-41 modified Au–PtNP/GCE to form a stable film
through immersion. MCM-41 can provide favorable
microenvironments for proteins due to the ordered uniform
pore structure, huge surface areas, high loading capacity
and good biocompatibility for electron transfer and they
provided a favorable microenvironment around Hb to
retain the enzymatic bioactivity and native structure of Hb.
So the proposed biosensor showed a stable, sensitive and
fast response to H2O2.
Also, Nafion-coated Hb/MCM-41/Au–PtNP/GC elec-
trode may be used for the selective determination of H2O2
in the presence of AA and UA. Nafion film is a cation
exchange polymer and repels AA and other negatively
charged species at optimized conditions.
Acknowledgments The authors gratefully acknowledge the finan-
cial support of this work by the Khorramabad Branch, Islamic Azad
University.
References
1. H.A.O. Hill, Coord. Chem. Rev. 151, 15 (1996)
2. S.F. Wang, T. Chen, Z.L. Zhang, X.C. Shen, Z.X. Lu, D.W. Pang,
K.Y. Wong, Langmuir 21, 9260 (2005)
3. L. Gorton, A. Lindgren, T. Larsson, F.D. Munteanu, T. Ruzgas, I.
Gazaryan, Anal. Chim. Acta 400, 91 (1999)
4. E. Ferapontova, L. Gorton, Bioelectrochemistry 66, 49 (2005)
5. D. Paolucci-Jeanjean, M.P. Belleville, G.M. Rios, Chem. Eng.
Res. Des. 83, 302 (2005)
6. P.L. He, N.F. Hu, G. Zhou, Biomacromolecules 3, 139 (2002)
7. H. Sun, N.F. Hu, H.Y. Ma, Electroanalysis 12, 1064 (2000)
8. C.C. Silva, H.H.B. Rocha, F.N.A. Freire, M.R.P. Santos, K.D.A.
Saboia, J.C. Goes, A.S.B. Sombra, Mater. Chem. Phys. 92, 260
(2005)
9. Y.D. Zhao, Y.H. Bi, W.D. Zhang, Q.M. Luo, Talanta 65, 489
(2005)
10. Z.H. Dai, S.Q. Liu, H.X. Ju, H.Y. Chen, Biosens. Bioelectron. 19,
861 (2004)
11. J.J. Yu, J.R. Ma, F.Q. Zhao, B.Z. Zeng, Electrochim. Acta 53,
1995 (2007)
12. H. Yamada, A.J. Bhattacharyya, J. Maier, Adv. Funct. Mater. 16,
525 (2006)
13. M. Hartmann, Chem. Mater. 17, 4577 (2005)
14. C. Lei, Y. Shin, J.K. Magnuson, G. Fryxell, L.L. Lasure, D.C.
Elliott, J. Liu, E.J. Ackerman, Nanotechnology 17, 5531 (2006)
15. H.S. Guo, N.Y. Hu, S.X. Ge, D. Yang, J.N. Zhang, Talanta 68, 61
(2005)
16. H. Ma, J. He, D.G. Evans, X. Duan, J. Mol. Catal. B-Enzym. 30,
209 (2004)
17. A.L. Briseno, S.C.B. Mannsfeld, E. Formo, Y.J. Xiong, X.M. Lu,
Z.N. Bao, S.A. Jenekhe, Y.N.J. Xia, J. Mater. Chem. 18, 5395
(2008)
18. T. Yoshida, J. Zhang, D. Komatsu, S. Sawatani, H. Minoura, T.
Pauporte, D. Lincot, T. Oekermann, D. Schlettwein, H. Tada, D.
Wohrle, K. Funabiki, M. Matsui, H. Miura, H. Yanagi, Adv.
Funct. Mater. 19, 17 (2009)
19. D.J. Milliron, I. Gur, A.P. Alivisatos, MRS Bull. 30, 41 (2005)
20. C. Sanchez, B. Julian, P. Belleville, M. Popall, J. Mater. Chem.
15, 3559 (2005)
21. J.W. Kriesel, T.D. Tilley, Adv. Mater. 11, 1645 (2001)
22. P. Gomez-Romero, Adv. Mater. 13, 163 (2001)
23. J. Gong, T. Zhou, D. Song, L. Zhang, X. Hu, Anal. Chem. 82, 567
(2010)
24. X. Dai, G.G. Wildgoose, C. Salter, A. Crossley, R.G. Compton,
Anal. Chem. 78, 6102 (2006)
25. V.R. Holland, B.C. Saunders, F.L. Rose, A.L. Walpole, Tetra-
hedron 30, 3299 (1974)
26. X.G. Zhao, J.L. Shi, B. Hu, L.X. Zhang, Z.L. Hua, Mater. Lett.
58, 2152 (2004)
27. J.H. Yang, H.S. Wang, L.H. Lu, Y.B. Wang, W.D. Shi, H.J.
Zhang, Synth. Met. 158, 572 (2008)
28. Y. Li, X. Zenga, X. Liua, X. Liub, W. Weia, S. Luo, Colloid
Surface B 79, 241 (2010)
29. W.B. Stockton, M.F. Rubner, Macromolecules 30, 2717 (1997)
30. P.L. He, N.F. Hu, J.F. Rusling, Langmuir 20, 722 (2004)
31. J.F. Diaz, K.J. Balkus, Mol. Catal. B 2, 115 (1996)
32. H. Takahashi, B. Li, T. Sasaki, C. Miyazaki, T. Kajino, S. Ina-
gaki, Micro. Meso. Mater. 45, 755 (2001)
33. I. Tinoco, K. Kauer, J.C. Wang, Physical chemistry principles
and applications in biological sciences (Prentice-Hall, Engle-
wood Cliffs, 1978), p. 606
34. S.F. Wang, T. Chen, Z.L. Zhang, D.W. Pang, K.Y. Wong,
Electrochem. Commun. 9, 1709 (2007)
35. J. Wyman, Rev. Biophys. 1, 35 (1968)
36. H.H. Liu, Z.Q. Tian, Z.X. Lu, Z.L. Zhang, M. Zhang, D.W. Pang,
Biosens. Bioelectron. 20, 294 (2004)
37. A.M. Bond, Modern polarographic methods in analytical chem-
istry (Marcel Dekker, New York, 1980), p. 27
38. I. Yamazaki, T. Araiso, Y. Hayashi, H. Yamada, R. Makino, Adv.
Biophys. 11, 249 (1978)
39. J.A. Harrison, Z.A. Khan, J. Electroanal. Chem. 28, 131 (1970)
40. J.D. Zhang, M. Oyama, Electrochim. Acta 50, 85 (2004)
41. L. Wang, G.X. Lu, B.J. Yang, Sens. Actuators. B. Chem. 99(50),
50 (2004)
42. Y.D. Zhao, Y.H. Bi, W.D. Zhang, Q.M. Luo, Talanta 65, 489
(2005)
43. A.K.M. Kafi, D.Y. Lee, S.H. Park, Y.S. Kwon, Microchem. J.
85(489), 308 (2007)
44. D. Shan, S.X. Wang, H.G. Xue, S. Cosnier, Electrochem. Com-
mun. 9, 529 (2007)
45. H. Yao, N. Li, J.Z. Xu, J.J. Zhu, Talanta 71, 550 (2007)
46. W. Zheng, Y.F. Zheng, K.W. Jin, N. Wang, Talanta 74, 1414
(2008)
47. Y. Zhang, H. Cao, W. Fei, D. Cui, N. Jia, Sens. Actuators.
B. Chem. 162, 143 (2012)
J Inorg Organomet Polym
123