differential effects of estrogen and progesterone on at...
TRANSCRIPT

Differential Effects of Estrogen and Progesterone on AT1Receptor Gene Expression in Vascular Smooth Muscle Cells
Georg Nickenig, MD; Kerstin Strehlow, MD; Sven Wassmann, MD; Anselm T. Ba¨umer, MD;Katja Albory, MS; Heinrich Sauer, MD; Michael Bo¨hm, MD
Background—The beneficial vasoprotective effects of a postmenopausal estrogen replacement therapy may be preventedby a concomitant administration of progestins. To investigate the differential effects of estrogens and progesterone, weexamined their influence on AT1 receptor gene expression in vascular smooth muscle cells (VSMCs).
Methods and Results—17b-Estradiol caused downregulation of AT1 receptor mRNA expression to 46614%, whereasprogesterone led to a significant upregulation to 201629%, as assessed by Northern analysis. Western blots revealedthat estrogen induced a downregulation and progesterone an upregulation of the AT1 receptor protein. Estrogen-induceddecrease of AT1 receptor expression was mediated through activation of estrogen receptors. Nuclear run-on assaysrevealed that 17b-estradiol did not alter AT1 receptor mRNA transcription rate, whereas progesterone caused anenhanced AT1 receptor mRNA transcription rate. 17b-Estradiol decreased the AT1 receptor mRNA half-life from 5 to2 hours, whereas progesterone induced a stabilization of AT1 receptor mRNA to a half-life of 10 hours. Preincubationof VSMCs with PD98059, SB203580, herbimycin, wortmannin, orNv-nitro-L-arginine suggested that 17b-estradiolcaused AT1 receptor downregulation through nitric oxide–dependent pathways. Progesterone caused AT1 receptoroverexpression via PI3-kinase activation. Angiotensin II–induced release of reactive oxygen species was inhibited byestrogens. Progesterone itself enhanced the production of reactive oxygen species.
Conclusions—Because AT1 receptor regulation plays a pivotal role in the pathogenesis of hypertension and atherosclerosis,the differential effects of estrogen and progesterone on the expression of this gene may in part explain the potentiallycounteracting effects of these reproductive hormones on the incidence of postmenopausal cardiovascular diseases.(Circulation. 2000;102:1828-1833.)
Key Words: receptorsn angiotensinn muscle, smoothn hypertensionn atherosclerosisn hormones
Several lines of epidemiological evidence indicate that estrogensplay an important role in the pathogenesis of hypertension and
atherosclerosis. First, at a young age, women suffer considerablyless cardiovascular disease than men. Second, after menopause, thenatural state of estrogen deficiency, the incidence of cardiovasculardisease rises steeply in women.1,2 Finally, hormone replacementtherapy potentially prevents the onset of cardiac events in post-menopausal women.3–5 The pathways by which estrogens interactwith the cardiovascular physiology are not completely understood.Estrogens lower plasma lipoproteins,3 influence the renin-angioten-sin system,6,7exert antioxidative properties,8 and may act as calciumblocking agents.9 In addition, estrogens exert direct effects on thevessel wall, such as an increase of vascular NO production andmodulation of expression of endothelial constitutive NO synthase(ecNOS).10–12
Numerous retrospective studies have shown that estrogenreplacement therapy may be useful in the primary preventionof cardiovascular diseases in postmenopausal women.3–5
However, the first prospective and randomized study on thatsubject, the HERS trial,13 showed no advantage of hormone
replacement therapy in terms of a lowered incidence ofcardiac events in postmenopausal women who suffer fromcoronary heart disease. Among other things, this surprisingoutcome has been attributed to the addition of progesterone tothe therapy regimen. Progesterone could display a number ofpotential adverse effects on the cardiovascular system thatmight overcome the beneficial influence of estrogens. In thiscontext, reduction of HDL levels, downregulation of estrogenreceptors, decreased carbohydrate tolerance, and reducedblood flow and vasodilatation have been reported.14–17In anyevent, the molecular mechanisms by which a concomitantprogesterone therapy could counteract the estrogen replace-ment treatment are not known in detail and are the subject ofcontroversy, because some study results could not support thedeleterious effects of progesterone.1,2
The AT1 receptor mediates many biological effects of therenin-angiotensin system (RAS), such as vasoconstriction, waterand sodium retention, free radical release, and cell growth.18
Therefore, activation of the AT1 receptor has been implicated inthe pathogenesis of cardiovascular disease. Recently, it was
Received March 10, 2000; revision received May 4, 2000; accepted May 8, 2000.From the Klinik III fur Innere Medizin and the Institut fur Physiologie (H.S.), Universitat zu Koln, Cologne, Germany.Correspondence to Dr Georg Nickenig, Klinik III fur Innere Medizin, Joseph-Stelzmann-Straße 9, 50924 Koln, Germany. E-mail georg.nickenig@
uni-koeln.de© 2000 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org
1828
by guest on June 22, 2018http://circ.ahajournals.org/
Dow
nloaded from

shown that estrogen deficiency causes AT1 receptor overexpres-sion in vivo, leading to enhanced biological effects of the RAS,which could in part serve as explanation for the increase incardiac events after menopause in women.19
We hypothesized that potentially counteracting effects ofestrogens and progestins on the cardiovascular system could takeplace at the level of AT1 receptor regulation. To further inves-tigate the direct effects of these reproductive hormones onvascular cells, we examined their influence on AT1 receptor geneexpression in vascular smooth muscle cells (VSMCs) and soughtto clarify the underlying molecular mechanisms.
MethodsCell CultureVSMCs were isolated from rat thoracic aorta (female Wistar-Kyoto,6 to 10 weeks old, Charles River GmbH, Sulzfeld, Germany) byenzymatic dispersion and cultured over several passages. Cells weregrown in a 5% CO2 atmosphere at 37°C in DMEM without phenoland 10% FBS (free of steroid hormones, S-15-M, c.c.pro GmbH).
mRNA Isolation and Northern AnalysisCulture medium was aspirated, and RNA was isolated with 1 mLRNA-Clean. Ten-microgram aliquots were electrophoresed, trans-ferred onto Hybond N membranes, and UV–cross-linked. Northernblots were prehybridized for 2 hours at 42°C in a buffer containing50% deionized formamide, 0.5% SDS, 63SSC, 10mg/mL dena-tured salmon sperm DNA (Sigma Aldrich Chemicals), and 53Denhardt’s solution and were then hybridized for 15 hours at 42°Cwith a random-primed, [32P]dCTP-labeled rat AT1 receptor cDNAprobe in the same buffer but without Denhardt’s solution.
Nuclear Run-On AssayAfter treatment, VSMCs were dispersed with trypsin and washed.The cell pellet was lysed for 10 minutes on ice, and the nuclei wereisolated by centrifugation. The nuclear pellet was resuspended in abuffer containing 40% glycerol, 50 mmol/L Tris, 5 mmol/L MgCl2,and 0.1 mmol/L EDTA. Nuclei ('53108 to 203108 nuclei perreaction) were used to carry out the transcription in a reactionmixture containing 40% glycerol, 50 mmol/L Tris, 5 mmol/L MgCl2,0.1 mmol/L EDTA, 0.5 mmol/L of CTP, GTP, and ATP, and 0.2 to0.3 mmol/L [32P]UTP (.3000mCi/mmol) at 30°C for 30 minutes.Reactions were terminated, and radioactive RNA was isolated andpurified. [32P]UTP-labeled RNA (53106 to 13107 cpm) was dis-solved in hybridization solution. Membranes were prehybridized for2 hours at 42°C and hybridized at 42°C for 16 hours.
Western BlottingVSMC samples were homogenized. Equal volumes of 23 SDS gelloading buffer (100 mmol/L Tris-HCl [pH 6.8], 200 mmol/L dithio-threitol, 4% SDS, 0.2% bromphenol blue, and 20% glycerol) wereadded, and samples were heated to 95°C for 10 minutes. The sampleswere then sonicated and spun at 10 000g for 10 minutes at roomtemperature. Twenty-five micrograms of protein of the supernatantwas run through a 10% polyacrylamide gel. Western blotting ofproteins was performed in a semidry blotting chamber (PharmaciaBiotech). Membranes were then incubated in 5% nonfat dry milk inPBS for 1 hour and washed with PBS-Tween (0.1%). The firstantibody (AT1 [N-10]: sc 1173, rabbit polyclonal IgG, Santa Cruz)was diluted 1:500 and incubated with the membrane for 1 hour atroom temperature. The second, horseradish peroxidase–labeled an-tibody (anti-rabbit, Sigma), was diluted to 1:5000 and incubated withthe membrane for 1 hour at room temperature. After the last washingstep, enzyme-linked chemiluminescence detection was carried outaccording to the manufacturer’s instructions (Amersham).
Measurement of Intracellular ReactiveOxygen SpeciesIntracellular reactive oxygen species (ROS) production was mea-sured by 29,79-dichlorofluorescein (DCF) fluorescence with confocallaser scanning microscopy techniques. Dishes of subconfluent cellswere washed and incubated in the dark for 30 minutes in the presenceof 10 mmol/L 29,79-dichlorodihydrofluorescein diacetate (H2DCF-DA, Molecular Probes). Culture dishes were transferred to a ZeissAxiovert 135 inverted microscope (Carl Zeiss), equipped with a325, numerical aperture 0.8, oil-immersion objective (Plan-Neofluar, Carl Zeiss) and Zeiss LSM 410 confocal attachment, andROS generation was detected as a result of the oxidation of H2DCF(excitation, 488 nm; emission long-pass LP515-nm filter set). Pixelimages (5123512) were collected by single rapid scans. In 4separate experiments, 5 groups of 25 cells each were randomlyselected from the image, and fluorescence intensity was taken. Therelative fluorescence intensities are average values of all experi-ments, and each value reflects measurements performed on aminimum of 100 cells for each sample.
Statistical AnalysisData are presented as mean6SEM. Statistical analysis was per-formed with the ANOVA test.
ResultsThe influence of estrogen and progestin on AT1 receptormRNA in VSMCs was measured. Cells were incubated for 0to 24 hours with 1 mmol/L of either 17b-estradiol orprogesterone, and AT1 receptor mRNA was measured byNorthern blotting. Figure 1, A and B, shows that 17b-estradiol caused a significant decrease of AT1 receptormRNA after 2 hours, reaching the maximum after 4 hours of46614% of control levels (P,0.05). In contrast, progester-one led to an upregulation of AT1 receptor mRNA, with amaximal effect of 201629% reached after 12 hours (Figure 1,A and B). Control experiments in which VSMCs wereincubated with vehicle showed that AT1 receptor andGAPDH mRNA levels remained stable over the experimentalperiod of 24 hours (data not shown). Both 17b-estradiol andprogesterone led to concentration-dependent modulation ofAT1 receptor mRNA in VSMCs (Figure 1C). After a 4-hourtreatment, the maximal effect was measured at 1mmol/L17b-estradiol, which led to a reduction of AT1 receptormRNA to 4166% of control cells (P,0.05). The maximalprogesterone effect was reached at a concentration of1 mmol/L after a 12-hour incubation (21567%). In allexperimental setups, GAPDH mRNA and 18S mRNA levelswere not altered (data not shown).
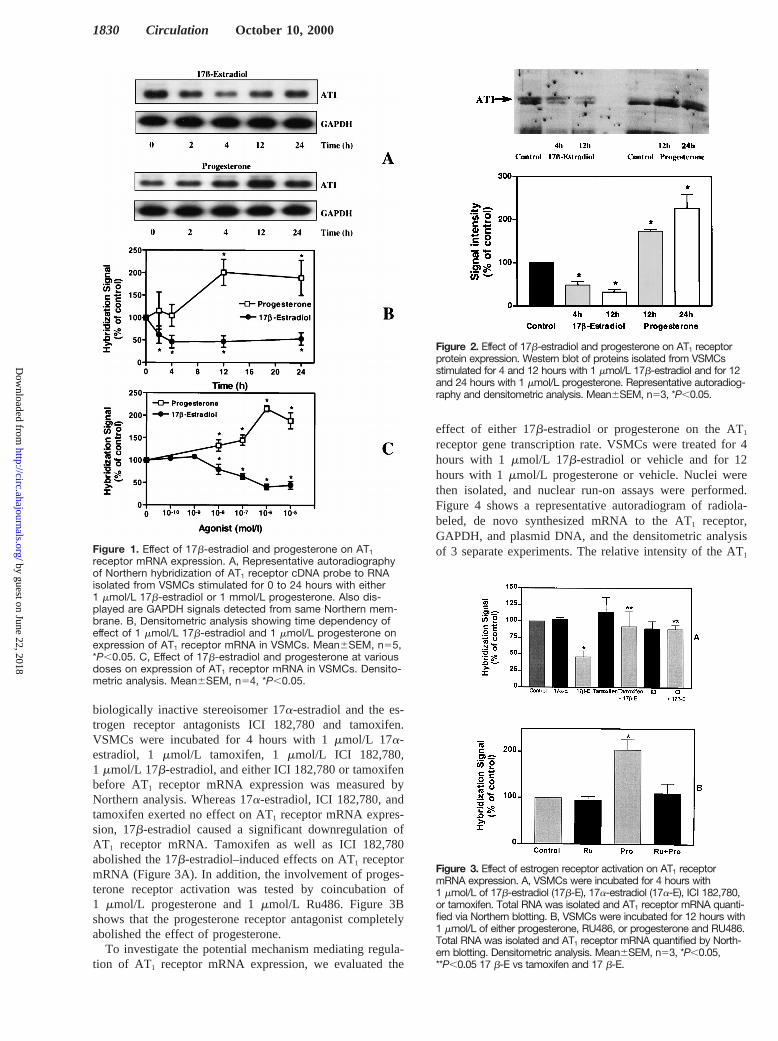
To assess whether this modulation of AT1 receptor mRNAexpression translates into comparable changes in AT1 receptormRNA protein levels, VSMCs were incubated for 4 and 12hours with 1mmol/L 17b-estradiol and for 12 and 24 hours with1 mmol/L progesterone. Total protein was isolated, and Westernanalysis was used to quantify AT1 receptor protein. Figure 2illustrates that 17b-estradiol caused a significant downregulation(4867% and 3265% of control levels), whereas progesteroneinduced an upregulation (17364% and 226632% of controllevels), of AT1 receptor protein expression. Control experimentshad shown that a 0- to 24-hour incubation with vehicle did notmodulate AT1 receptor expression (data not shown).
The specificity of the 17b-estradiol effect on the expres-sion of the AT1 receptor mRNA was tested by use of the
Nickenig et al AT1R Regulation by Estrogen and Progesterone 1829
by guest on June 22, 2018http://circ.ahajournals.org/
Dow
nloaded from
biologically inactive stereoisomer 17a-estradiol and the es-trogen receptor antagonists ICI 182,780 and tamoxifen.VSMCs were incubated for 4 hours with 1mmol/L 17a-estradiol, 1 mmol/L tamoxifen, 1 mmol/L ICI 182,780,1 mmol/L 17b-estradiol, and either ICI 182,780 or tamoxifenbefore AT1 receptor mRNA expression was measured byNorthern analysis. Whereas 17a-estradiol, ICI 182,780, andtamoxifen exerted no effect on AT1 receptor mRNA expres-sion, 17b-estradiol caused a significant downregulation ofAT1 receptor mRNA. Tamoxifen as well as ICI 182,780abolished the 17b-estradiol–induced effects on AT1 receptormRNA (Figure 3A). In addition, the involvement of proges-terone receptor activation was tested by coincubation of1 mmol/L progesterone and 1mmol/L Ru486. Figure 3Bshows that the progesterone receptor antagonist completelyabolished the effect of progesterone.
To investigate the potential mechanism mediating regula-tion of AT1 receptor mRNA expression, we evaluated the
effect of either 17b-estradiol or progesterone on the AT1
receptor gene transcription rate. VSMCs were treated for 4hours with 1 mmol/L 17b-estradiol or vehicle and for 12hours with 1mmol/L progesterone or vehicle. Nuclei werethen isolated, and nuclear run-on assays were performed.Figure 4 shows a representative autoradiogram of radiola-beled, de novo synthesized mRNA to the AT1 receptor,GAPDH, and plasmid DNA, and the densitometric analysisof 3 separate experiments. The relative intensity of the AT1
Figure 1. Effect of 17b-estradiol and progesterone on AT1
receptor mRNA expression. A, Representative autoradiographyof Northern hybridization of AT1 receptor cDNA probe to RNAisolated from VSMCs stimulated for 0 to 24 hours with either1 mmol/L 17b-estradiol or 1 mmol/L progesterone. Also dis-played are GAPDH signals detected from same Northern mem-brane. B, Densitometric analysis showing time dependency ofeffect of 1 mmol/L 17b-estradiol and 1 mmol/L progesterone onexpression of AT1 receptor mRNA in VSMCs. Mean6SEM, n55,*P,0.05. C, Effect of 17b-estradiol and progesterone at variousdoses on expression of AT1 receptor mRNA in VSMCs. Densito-metric analysis. Mean6SEM, n54, *P,0.05.
Figure 2. Effect of 17b-estradiol and progesterone on AT1 receptorprotein expression. Western blot of proteins isolated from VSMCsstimulated for 4 and 12 hours with 1 mmol/L 17b-estradiol and for 12and 24 hours with 1 mmol/L progesterone. Representative autoradiog-raphy and densitometric analysis. Mean6SEM, n53, *P,0.05.
Figure 3. Effect of estrogen receptor activation on AT1 receptormRNA expression. A, VSMCs were incubated for 4 hours with1 mmol/L of 17b-estradiol (17b-E), 17a-estradiol (17a-E), ICI 182,780,or tamoxifen. Total RNA was isolated and AT1 receptor mRNA quanti-fied via Northern blotting. B, VSMCs were incubated for 12 hours with1 mmol/L of either progesterone, RU486, or progesterone and RU486.Total RNA was isolated and AT1 receptor mRNA quantified by North-ern blotting. Densitometric analysis. Mean6SEM, n53, *P,0.05,**P,0.05 17 b-E vs tamoxifen and 17 b-E.
1830 Circulation October 10, 2000
by guest on June 22, 2018http://circ.ahajournals.org/
Dow
nloaded from

receptor signal is compared with GAPDH mRNA signalintensity. 17b-Estradiol had no effect on the rate of the denovo synthesis of AT1 receptor mRNA. Progesterone in-creased the AT1 receptor mRNA transcription rate from11762% to 15265% (AT1/GAPDH ratio). The hybridizationsignals elicited by the control vector were comparable be-tween the treatment groups.
Further experiments measured the effect of either 17b-estradiol or progesterone on AT1 receptor mRNA stability.VSMCs were preincubated with either vehicle, 1mmol/L17b-estradiol (4 hours), or 1mmol/L progesterone (12 hours);transcription was blocked with 50mg/mL 5,6-dichloroben-zimidazole (DRB), and total RNA was isolated 0 to 8 hoursafter the addition of DRB. Figure 5 shows the quantificationof AT1 receptor mRNA. Under control conditions, the AT1
receptor mRNA half-life was measured at 5 hours. 17b-Estradiol destabilized AT1 receptor mRNA, resulting in anAT1 receptor mRNA half-life of 2 hours; progesterone stabi-
lized AT1 receptor mRNA to a half-life of'10 hours.GAPDH mRNA levels remained stable over the experimentalperiod (data not shown).
By means of various pharmacological inhibitors, the in-volved signal transduction pathways were characterized.VSMCs were preincubated with 1mmol/L PD98059 (p42/44MAP kinase inhibitor), 1mmol/L SB203580 (p38 MAPkinase inhibitor), 1mmol/L of the tyrosine kinase inhibitorherbimycin, 1 mmol/L of the phosphatidylinositol (PI3)-kinase inhibitor wortmannin, or the NO inhibitorNv-nitro-L-arginine (L-NNA, 10mmol/L) followed by a coincubationwith 1 mmol/L 17b-estradiol (4 hours) or 1mmol/L proges-terone (12 hours). The densitometric analysis of the Northernblots quantifying AT1 receptor mRNA is depicted in Figure 6.The estrogen-mediated downregulation of AT1 receptormRNA was inhibited only by L-NNA, suggesting an NO-dependent downregulation of the AT1 receptor. In contrast,the progesterone-caused AT1 receptor upregulation was abol-ished on coincubation with wortmannin, which supports theconcept that PI3-kinase is involved in this induced modulationof AT1 receptor gene expression. All inhibitors were also usedin concentrations from 10 nmol/L to 10mmol/L. PD98059,SB203580, and genistein did not blunt the effects of 17b-estradiol or progesterone on AT1 receptor expression, even ifapplied in high concentrations (data not shown).
AT1 receptor activation causes free radical release inVSMCs. To evaluate the functional relevance of the AT1
receptor regulation, cells were preincubated for various timepoints with either 1mmol/L 17b-estradiol or 1 mmol/L
Figure 4. Effect of 17b-estradiol and progesterone on AT1
receptor mRNA transcription rate. VSMCs were incubated withvehicle (4 and 12 hours), 1 mmol/L 17b-estradiol (4 hours), or1 mmol/L progesterone (12 hours). Nuclei were isolated andnuclear run-on assays performed. A representative autoradio-gram and densitometric analysis are demonstrated. Signal inten-sities of AT1 receptor and GAPDH mRNA were calculated. AT1
receptor/GAPDH ratio is shown. Mean6SEM, n53.
Figure 5. Effect of 17b-estradiol and progesterone on AT1 receptormRNA stability. Cells were preincubated for 4 hours with 17b-estradiol or 12 hours with 1 mmol/L progesterone before 50 mg/mLDRB was added to block transcription. Total RNA was isolated atindicated time points, and AT1 receptor mRNA was quantified byNorthern analysis. Mean6SEM, n53, *P,0.05.
Figure 6. Signal transduction pathways involved in AT1 receptorregulation induced by 17b-estradiol and progesterone. VSMCswere coincubated with either 1 mmol/L 17b-estradiol (4 hours) or1 mmol/L progesterone (12 hours) with the following compounds:PD, PD98059 (1 mmol/L); SB, SB203580 (1 mmol/L); H, herbimycin(1 mmol/L); W, wortmannin (1 mmol/L); or NA, L-NNA (10 mmol/L).AT1 receptor mRNA was assessed after isolation of total RNA andNorthern blot. Densitometric analysis of Northern blots quantifyingAT1 receptor mRNA is depicted for 17b-estradiol (E) in A and pro-gesterone (P) in B. C indicates control. Mean6SEM, n53,*P,0.05, **P,0.05 E vs NA1E or P vs W1P.
Nickenig et al AT1R Regulation by Estrogen and Progesterone 1831
by guest on June 22, 2018http://circ.ahajournals.org/
Dow
nloaded from

progesterone, followed by a 4-hour incubation with 1mmol/Langiotensin II. Production of free radicals was assessed viaDCF fluorescence. Figure 7 shows that angiotensin II led to aprofound increase of ROS, which was inhibited by a 12-hourpreincubation with 17b-estradiol. 17b-Estradiol itself had noeffect on the basal release of free radicals. In addition, only along-term preincubation with estrogens (12 to 24 hours)lowered angiotensin II–caused production of ROS (data notshown). Progesterone itself significantly enhanced the releaseof free radicals. No further increase of angiotensin II–inducedproduction of ROS was detected.
DiscussionThe incidence of cardiovascular disease is low in premeno-pausal women, but it increases steadily in postmenopausalwomen. In addition, postmenopausal hormone replacementtherapy may reduce this rise of cardiovascular events, assuggested by retrospective studies.1–5 The vasoprotectivepotential of estrogens has been attributed primarily to itseffects on serum lipid concentrations.3 However, an increas-ing body of evidence indicates that direct effects of estrogenson blood vessels contribute significantly to their cardiopro-tective effects.20 Activation of estrogena- and b-receptorscauses long-term effects on cellular gene expression pro-grams, which are thought to be mediated by genomic effectsof the activated steroid receptors. Intriguingly, estrogens alsoinduce nongenomic, rapid effects in vascular cells thatobviously occur independently of modulation of gene expres-sion. One of the most prominent features seems to be a rapidvasodilatory effect of estrogen, which is elicited throughendothelium-dependent and endothelium-independent mech-anisms. Current data suggest that estrogens enhance thebioavailability of NO through stimulation of NOS and NOrelease and potentially also through the antioxidant propertiesof estrogens.10–12,20In addition to these short-term effects onthe vascular motility, the described rapid actions of estrogens,
such as stimulation of NO release, may also cause longer-lasting effects on vascular cells that ultimately contribute tothe atheroprotective effects of estrogens.
Progesterone, which is frequently administered concomi-tantly with estrogens in postmenopausal replacement therapy,is thought to counteract some of the atheroprotective featuresof estrogens. This has been attributed primarily to adverseeffects on lipid levels and carbohydrate metabolism.14–17
Although the cellular effects of progesterone on the vesselwall are currently unknown, it may be hypothesized that thepotentially harmful influences of progesterone on cardiovas-cular events are also mediated through a direct impact onvascular cells. The data presented demonstrate, for the firsttime, a direct effect of progesterone on vascular cells, whichmay relate to the epidemiological evidence of hazardouseffects of progesterone on the cardiovascular system. Somestudies, eg, the Nurses Health Study, have shown thatprogesterone did not blunt the beneficial effects of estro-gens.1,2 These controversial data display the remaining un-certainty with respect to the vascular effects of progesterone.
The AT1 receptor, which induces vasoconstriction, cellulargrowth, and free radical release in the vessel,18 is regulated byangiotensin II, lipoproteins, growth factors, and insulin, amongother things.21–24 It has recently been reported that estrogendeficiency leads to overexpression of the vascular AT1 receptor,which is preventable by estrogen replacement therapy.19 Mostlikely, the underlying mechanism for these changes is themodulation of AT1 receptor gene expression in VSMCs byestrogens, as shown in the present study. 17b-Estradiol leads toa dose- and time-dependent downregulation of AT1 receptors inVSMCs. This effect is mediated by estrogen receptors andinvolves the destabilization of the AT1 receptor mRNA. Thelatter seems to be the principal cellular mechanism by whichAT1 receptor expression is modulated.21 Although MAP kinaseactivation has been described on stimulation with estrogens,25
this signal transduction pathway is not participating in theestrogen-induced AT1 receptor regulation. Estrogen-caused re-lease of NO seems to be a prerequisite of the describedregulative pathways, as shown by the inhibition of estrogen-induced AT1 receptor downregulation by an NO inhibitor. Thisis in agreement with previous findings that NO is released onestrogen stimulation. Moreover, it has been shown that NO isinvolved in AT1 receptor downregulation by, for example,cytokines in VSMCs.26,27 The effect of estrogen seems to bemediated by estrogen receptors, because the estrogen receptorantagonists ICI 182,780 and tamoxifen abolished the AT1
receptor–downregulating effect of 17b-estradiol. 17a-Estradiol,which does not activate estrogena- or b-receptors, showed noeffect on AT1 receptor expression.
Surprisingly, progesterone causes a profound upregulationof AT1 receptor gene expression mediated through stabiliza-tion of the AT1 receptor mRNA. In striking contrast to themode of action of estrogen, this effect on AT1 receptor geneexpression is mediated through activation of PI3-kinase.
The current concept of heterologous AT1 receptor regula-tion ascribes a pivotal role to posttranscriptional mecha-nisms.21 Stabilization or destabilization of AT1 receptormRNA seems to participate decisively in the modulation ofAT1 receptor expression. The signal transduction pathways
Figure 7. Effect of progesterone and 17b-estradiol on angioten-sin II–induced intracellular production of ROS in VSMCs. Repre-sentative microscopic scan. VSMCs were preincubated for 12hours with 1 mmol/L progesterone or 17b-estradiol or vehicle,followed by a 4-hour incubation with 1 mmol/L angiotensin II.Free radical production is visualized through DCF fluorescence.
1832 Circulation October 10, 2000
by guest on June 22, 2018http://circ.ahajournals.org/
Dow
nloaded from

involved in this phenomenon are less clear. MAP kinaseactivation, cytosolic calcium release, cAMP accumulation,and NO have been implicated in transcriptional as well asposttranscriptional regulation of AT1 receptor gene expres-sion.21–24Nevertheless, the factors downstream of this signal-ing event are unknown. There is evidence that mRNA bindingproteins induce (de)stabilization of the AT1 receptor mRNA,but the detailed mechanisms have not yet been elucidated.21 Itmay be speculated that PI3-kinase activation as well as NOrelease interact directly or indirectly with these bindingproteins, leading to changes in AT1 receptor mRNA degra-dation. Interestingly, NO obviously plays a role in themodulation of ecNOS stability.28 According to recent find-ings, ecNOS stability is dependent on distinct mRNA bindingproteins. Therefore, NO could potentially play a role in theregulation of events involved in the AT1 receptor mRNAprocessing, as suggested by the presented data.
The AT1 receptor regulation by estrogens and progesteroneis accompanied by functional alterations in VSMCs, as shownby the altered angiotensin II–induced release of ROS. Ingeneral, activation of the AT1 receptor is a major source forROS in the vessel wall, and these ROS are closely involvedin the pathogenesis of atherosclerosis and hypertension. Ourdata indicate that the antioxidant properties of estrogenscould be mediated at least in part through the downregulationof the AT1 receptor. Furthermore, estrogen-induced AT1
receptor downregulation could be related to a decreasedvasoconstriction and cell growth. In contrast, progesterone-induced AT1 receptor upregulation would lead to the oppositeeffect. Progesterone itself enhances the release of free radi-cals, suggesting a direct interaction of progesterone withenzymatic systems such as NAD(P)H oxidase. The latter, andthe upregulation of the AT1 receptor, are possible mecha-nisms by which administration of progesterone causes adecreased blood flow and an increased vascular resistance inpostmenopausal women.29 The failure to show a reduction ofcardiovascular mortality by hormone replacement therapy inthe HERS trial has also been explained by the concomitantprogesterone treatment, which could be attributed at least inpart to the overexpression of AT1 receptors.
Our findings demonstrate a direct impact of estrogen andprogesterone on VSMCs, providing novel insights into themechanisms of action of reproductive hormones in thevasculature. Whereas the estrogen-induced, NO-dependentAT1 receptor downregulation may contribute to the athero-protective effects of estrogens, the progesterone-caused AT1
receptor overexpression may in part explain the adverseinfluences of progesterone on the cardiovascular system.
AcknowledgmentsThis work was supported by the Deutsche Forschungsgemeinschaft,the Koln Fortune Program/Faculty of Medicine, University ofCologne, and the Deutsche Herzstiftung.
References1. Wenger NK, Speroff L, Packard B. Cardiovascular health and disease in
women. N Engl J Med.1993;329:247–256.2. Colditz GA, Willett WC, Stampfer MJ, et al. Menopause and the risk of
coronary heart disease in women.N Engl J Med. 1987;316:1105–1110.3. Hong MK, Romm PA, Reagan K, et al. Effects of estrogen replacement
therapy on serum lipid values and angiographically defined coronary arterydisease in postmenopausal women.Am J Cardiol. 1992;69:176–178.
4. Heckbert SR, Weiss NS, Koepsell TD, et al. Duration of estrogenreplacement therapy in relation to the risk of incident myocardial infarctionin postmenopausal women.Arch Intern Med. 1997;157:1330–1336.
5. Nabulsi AA, Folsom AR, White A, et al. Association of hormone-replacement therapy with various cardiovascular risk factors in postmeno-pausal women: the Atherosclerosis Risk in Communities Study.N EnglJ Med.1993;328:1069–1075.
6. Erkkola R, Lammintausta R, Punnonen R, et al. The effect of estriolsuccinate therapy on plasma renin activity and urinary aldosterone inpostmenopausal women.Maturitas. 1978;1:9–14.
7. Schunkert H, Danser AH, Hense HW, et al. Effects of estrogenreplacement therapy on the renin-angiotensin system in postmenopausalwomen.Circulation. 1997;95:39–45.
8. Liehr JG. Antioxidant and prooxidant properties of estrogens.J Lab ClinMed. 1996;128:344–345.
9. Freay AD, Curtis SW, Korach KS, et al. Mechanism of vascular smoothmuscle relaxation by estrogen in depolarized rat and mouse aorta: role ofnuclear estrogen receptor and Ca21 uptake.Circ Res. 1997;81:242–248.
10. Jiang C, Sarrel PM, Lindsay DC, et al. Endothelium-independentrelaxation of rabbit coronary artery by 17b-oestradiol in vitro.Br JPharmacol. 1991;104:1033–1037.
11. Lantin-Hermoso RL, Rosenfeld CR, Yuhanna IS, et al. Estrogen acutelystimulates nitric oxide synthase activity in fetal pulmonary artery endo-thelium. Am J Physiol. 1997;273:L119–L126.
12. Caullin-Glaser T, Garcia-Cardena G, Sarrel P, et al. 17b-Estradiol regu-lation of human endothelial cell basal nitric oxide release, independent ofcytosolic Ca21 mobilization.Circ Res. 1997;81:885–892.
13. Hulley S, Grady D, Bush T, et al, for the Heart and Estrogen/progestinReplacement Study (HERS) research group. Randomized trial of estrogenplus progestin for secondary prevention of coronary heart disease inpostmenopausal women.JAMA. 1998;280:605–613.
14. Sullivan JM, Shala BA, Miller LA, et al. Progestin enhances vasocon-strictor responses in post menopausal women receiving estrogenreplacement therapy.Menopause. 1995;4:193–197.
15. Miyagawa K, Rosch J, Stanczyk F, et al. Medroxiprogesterone interfereswith ovarian steroid protection against coronary vasospasm.Nat Med.1997;3:324–327.
16. Walsh, BW, Schiff I, Rosner B, et al. Effects of postmenopausal estrogenreplacement on the concentrations and metabolism of plasmalipoproteins.N Engl J Med. 1991;325:1196–1204.
17. Sarrel PM. How progestins compromise the cardioprotective effects ofestrogens.Menopause. 1995;2:187–190.
18. Griendling KK, Murphy TJ, Alexander RW. Molecular biology of therenin-angiotensin system.Circulation. 1993;87:1816–1828.
19. Nickenig G, Baumer AT, Grohe C, et al. Estrogen modulates AT1 receptorgene expression in vitro and in vivo.Circulation. 1998;97:2197–2201.
20. Mendelsohn ME, Karas RH. The protective effects of estrogen on thecardiovascular system.N Engl J Med. 1999;340:1801–1811.
21. Nickenig G, Murphy TJ. Enhanced AT1 receptor mRNA degradation andinduction of polyribosomal mRNA binding proteins by angiotensin II invascular smooth muscle cells.Mol Pharmacol. 1996;50:743–751.
22. Nickenig G, Murphy TJ. Down-regulation by growth factors of vascularsmooth muscle angiotensin receptor gene expression.Mol Pharmacol.1994;46:653–659.
23. Nickenig G, Sachinidis A, Michaelsen F, et al. Upregulation of vascularangiotensin II receptor gene expression by low-density lipoprotein invascular smooth muscle cells.Circulation. 1997;95:473–478.
24. Nickenig G, Roling J, Strehlow K, et al. Insulin induces upregulation ofvascular AT1 receptor gene expression by posttranscriptional mech-anisms.Circulation. 1998;98:2453–2460.
25. Migliaccio A, Di Domenico M, Castoria G. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7cells.EMBO J. 1996;15:1292–1300.
26. Sasmura H, Nakazato Y, Hayashida T, et al. Regulation of vascular type1 angiotensin receptors by cytokines.Hypertension. 1997;30:35–41.
27. Ichiki T, Usui M, Kato M, et al. Downregulation of angiotensin II type 1receptor gene transcription by nitric oxide.Hypertension. 1998;31:342–348.
28. Yuhanna IS, MacRitchie AN, Lantin-Hermoso RL, et al. Nitric oxide(NO) upregulates NO synthase expression in fetal intrapulmonary arteryendothelial cells.Am J Respir Cell Mol Biol. 1999;21:629–636.
29. Mercuro G, Pitzalis L, Podda A, et al. Effects of acute administration ofnatural progesterone on peripheral vascular responsiveness in healthypostmenopausal women.Am J Cardiol. 1999;84:214–218.
Nickenig et al AT1R Regulation by Estrogen and Progesterone 1833
by guest on June 22, 2018http://circ.ahajournals.org/
Dow
nloaded from

Sauer and Michael BöhmGeorg Nickenig, Kerstin Strehlow, Sven Wassmann, Anselm T. Bäumer, Katja Albory, Heinrich
Vascular Smooth Muscle Cells Receptor Gene Expression in1Differential Effects of Estrogen and Progesterone on AT
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2000 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/01.CIR.102.15.1828
2000;102:1828-1833Circulation.
http://circ.ahajournals.org/content/102/15/1828World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 22, 2018http://circ.ahajournals.org/
Dow
nloaded from