differences phenotypiques enfants adultes - sfeim · cftde-necker x.03 differences phenotypiques...
TRANSCRIPT

CFTDE-Necker X.03
Differences phenotypiques
enfants adultes
Jean-Marie Saudubray
SFEIM Grenoble Juin 2016

CFTDE-Necker X.03
De la chimie cosmique au DACU
(d’après Christian de Duve)
Chimie cosmique déterministe et
reproductible, non biosélective :C,H,O,N,
mais aussi Sucres,Acides aminés,Acides
gras ,Bases azotées
ARN (1ere molécule porteuse
d’informations réplicable)
Chimie +Sélection
Formation du Dernier Ancétre Commun
Universel (DACU)
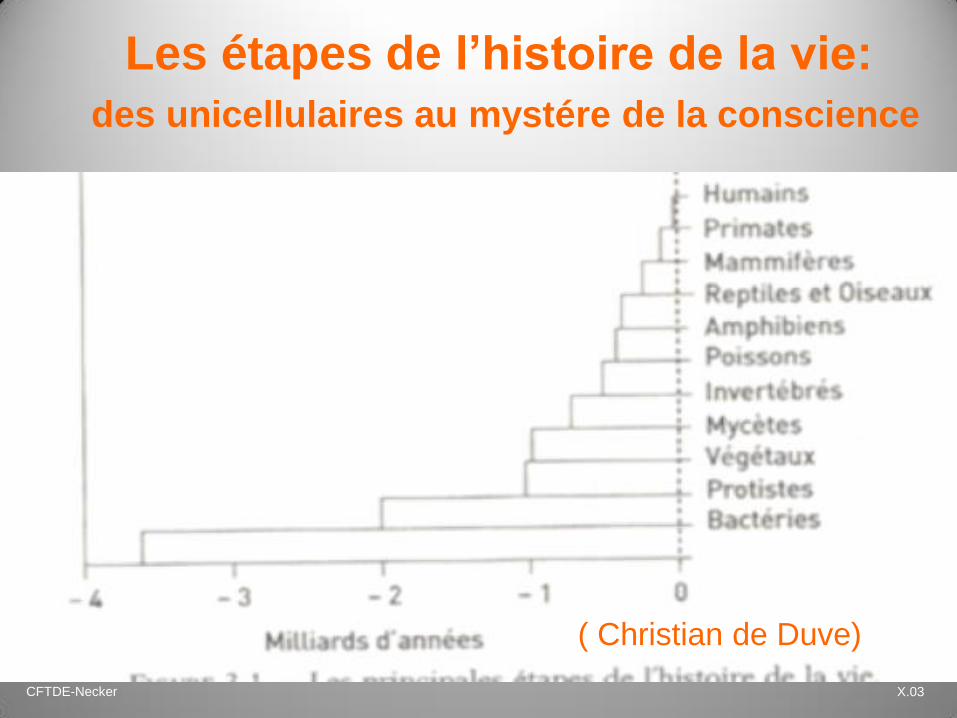
CFTDE-Necker X.03
Les étapes de l’histoire de la vie:
des unicellulaires au mystére de la conscience
( Christian de Duve)

CFTDE-Necker X.03
Le minimum vital du DACU
Une membrane
Une source d’énergie
Une molécule porteuse d’information
pouvant étre répliquée (ARN) et un
archivage des informations (ADN)
Des substances nutritives

CFTDE-Necker X.03
La leçon d’anatomie Rembrandt 1632

CFTDE-Necker X.03
La cellule humaine 2016
From Dr. Joe Clarke
Lysosome Localization of hydrolytic enzymes involved in
the degradation of large, complex substrates,
such as proteins, glycoconjugates, nucleic
acids, complex lipids
Mitochondria 1. Oxidative phosphorylation (respiratory chain)
2. Citric acid cycle
3. β-Oxidation of fatty acids, with production of acetyl-
CoA
4. Oxidative degradation of some amino acids
5. Proximal steps of urea biosynthesis
6. Biosynthesis of some amino acids
7. Proximal and distal steps of porphyrin biosynthesis
Plasma membrane 1. Limiting loss of intracellular components of the
cell,
ensuring concentration and preservation of
optimum local milieu
2, Regulating uptake of amino acids
3. Neurotransmitter binding and uptake (nervous
tissue)
4. Regulating uptake of metabolites, drugs,
chemicals, hormones, etc.
5. Uptake (endocytosis) and intracellular
trafficking of material suspended in extracellular
milieu
Peroxisomes 1. Synthesis of bile acids
2. Synthesis of some steroid hormones
3. Synthesis of plasmalogens
4. Transmission of glyoxylate to glycine
5. Catalase
6. Oxidation of D-amino acids
7. α-Oxidation of branched chain fatty acids, e.g.,
phytanic acid
8. β-Oxidation of long-chain and very long-chain fatty
acids
9. Oxidation of pipecolic acid
10. Spermine and spermidine oxidation
Golgi complex Post-translational modification of N-linked
oligosaccharides of nascent glycoproteins,
including glycosaminoglycan biosynthesis
Rough endoplasmic
reticulum (ER) Translation (mRNA-directed synthesis
of polypeptides)
Smooth endoplasmic
reticulum (ER)
1. Post-translational modification of nascent
polypeptides, including N- and O-glycosylation
2. Biosynthesis of cholesterol, phospholipids,
triglycerides, glycosphingolipids
3. Steroid hormone biosynthesis (endocrine tissues)
4. Detoxifications (P450, liver)
5. Glucose-6-phosphate (liver)
6. Calcium sequestration (sarcoplasmic reticulum,
muscle)
7. Porphyrin degradation

CFTDE-Necker X.03
Auscultation
Thoracique
par Laennec
en 1819
R A Thom’s
tableau
1954

CFTDE-Necker X.03
New Technologies 2016
La Metabolomique:
Mayo clinic’s biochemical genetics laboratory (Courtesy J Vockley)

CFTDE-Necker X.03
Le métabolisme intermédiaire
C ’ est l'ensemble des transformations
chimiques de dégradation (catabolisme) et de
fabrication (anabolisme) qui permettent un
cycle continu d'échanges entre les cellules et
les substances apportées par l'alimentation:
glucides, lipides et protides.
Immense réseau interconnecté

CFTDE-Necker X.03
Oxaloacetate
citrate
Pyruvate
Acetyl-CoA
NADH
NAD+
Isocitrate
PDH
IDH ATP
CTP
citrate
+
Glucose 1-P
Glucose 6-P
F 1,6-BP
PFK
GA3-P
6-P-
gluconolactone
Ribulose 5-P
NADPH
NADP+
G6PD -
Citrate lyase
Acetyl-CoA
Acetyl -CoA
carboxylase
Malonyl-CoA
Ceramide
Sphingolipids LCFA
VLCFA
Elongases
ELOV 1-6
NADP+
NADPH
Acyl-DHAP
Alkyl-DHAP
Plasmalogens
DHAPAT
ADHAPS
Fatty acyl-CoA
FAS NADP+
NADPH
Aceto-
acetyl-CoA
HMG-CoA
Mevalonate
Cholesterol
Ubiquinone
Dolichol
HMG-CoA
synthase
HMG-CoA
reductase
Co A
DHAP G3P DHase
Glycerol 3-P
TPI
phosphopantetheine
Vitamine B5
PANK2
CoA synthase
Serine
1,3-DPG
3-PG
Glyceraldehyde
3-P-DHase
P-Glycerate
kinase
Serine palmitoyl
transferase
Phospholipids
Diacylglycerol
Phosphatidate
SERAC-1
AGK
Cardiolipin
G3-P Acyltransferase
Triglycerides

CFTDE-Necker X.03
De novo PC/serine synthesis
From Lamari in Saudubray 2016
Serine

CFTDE-Necker X.03
Phospholipids from membrane remodeling
From Lamari et al in Saudubray 2016
Phosphatidyl choline and Eicosanoids Phosphatidyl-inositides

CFTDE-Necker X.03
Classification of IEMs >780 diseases
1. Intoxication: IEM of intermediary metabolism
(small molecules) >180 disorders
2. Energetic disorders: IEM of intermediary
metabolism >250 disorders (mostly mitochondrial)
3. Complex molecules: IEM of organelles metabolism >350 disorders (April 2016) (mostly complex lipids and glycosylation disorders)

CFTDE-Necker X.03 From Stephan Kolker

CFTDE-Necker X.03
From F Mochel and F Sedel Chapter 2 in Saudubray et al 2016
,Leigh syndrome

CFTDE-Necker X.03

CFTDE-Necker X.03
Acute crisis
,or nothing

CFTDE-Necker X.03
Phenotypic variations in IEM
1. Within a group of disorders :
Example : lysine, ornithine, arginine metabolism
pathophysiology of symptoms
2. Within a family with the same disorder
- With different residual enzyme activiy
genetic mechanisms
- With same residual enzyme activity
environmental factors
metabolic networks
development (ontogenic changes)

CFTDE-Necker X.03
Urea cycle interactions
12.Carbonic anhydrase VA (CAVA)
12
CO2 CO3H
Phenotypic variations within metabolic pathways
involved in hyperammonemia
After D Rabier

CFTDE-Necker X.03
Potential genetic mechanisms
- family bearing same mutation(s)-
1. Random X inactivation : OTC,PDH
2. Genetic imprinting : Niemann Pick A/B
3. Composite heterozygocity : MSUD
4. Mitochondrial segregation: NARP/LEIGH
5. Unfolding:TPI
6. Individual variations of cell quality control
7. Others …

CFTDE-Necker X.03
OTC deficiency: random X inactivation
v
1
1
1
III
II
I neonatal
death neonatal
death
2 3
I 1 :
I 2-3:
34
II 1:
as
III 1:
Permanent orotic aciduria
Brothers died in neonatal
period
Asymptomatic. No orotic
aciduria after protein loads.
Index case: hyperammonemic
coma at day 7; OTC activity =
12%
Molecular biology: Full deletion of OTC gene

CFTDE-Necker X.03
PDH : a family affair

CFTDE-Necker X.03
PDH activity and symptoms
Courtesy Dr. Brian Robinson

CFTDE-Necker X.03
Genetic imprinting in Sphingomyelinase deficiency
(Simonaro et al. Am J Human Genet 2006; 78:865-70)
The only allele expressed is that of the mother
Severe
Mild

CFTDE-Necker X.03
Composite Heterozygocity in MSUD
(Frezal et al; Hum Genet. 1985;71:89-91

CFTDE-Necker X.03
Composite Heterozygocity in MSUD
(Frezal et al; Hum Genet. 1985;71:89-91

CFTDE-Necker X.03
Mitochondrial segregation
2.5-year-old proband
Weakness
Developmental delay
Hypotonia
Ptosis
Mild lactic acidemia
TCA cycle
intermediates
on urine organic acids
Courtesy Dr. Anita Beck

CFTDE-Necker X.03
Affected 12-month-old brother
Failure to thrive
Developmental
regression
Hypotonia
Seizures
Case 3
12
months 2.5
years
5
years

CFTDE-Necker X.03
Family history
Mother and younger
brother
(at age 12 months)
Courtesy Dr. Anita Beck Case 3

CFTDE-Necker X.03
Head MRI
Bilateral putamen and
globus pallidus
abnormalities:LEIGH
Case 3 Courtesy Dr. Anita Beck
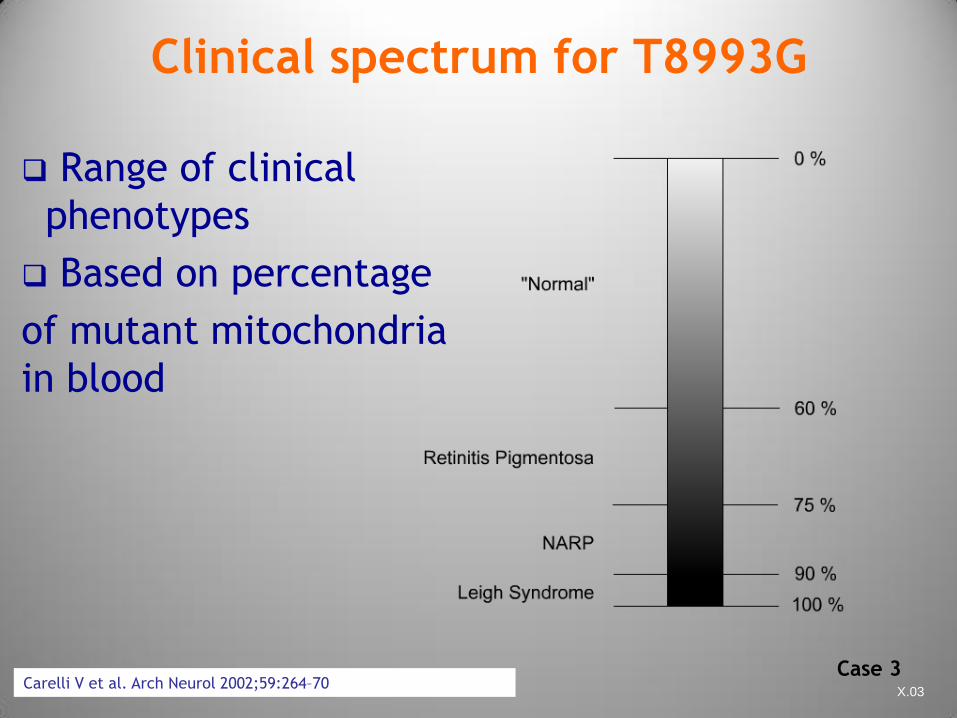
CFTDE-Necker X.03
Clinical spectrum for T8993G
Range of clinical
phenotypes
Based on percentage
of mutant mitochondria
in blood
Carelli V et al. Arch Neurol 2002;59:264–70 Case 3

CFTDE-Necker X.03
Case 3
80%
NARP
94%
Leigh
31%
carrier
?
Percentage blood
heteroplasmy

CFTDE-Necker X.03
Family matters
Phenotypically normal
sister
Case 3

CFTDE-Necker X.03
Tissue variation of 8993T>G
mtDNA
Case 3

CFTDE-Necker X.03
Conformational versus metabolic disease
Triose phosphate isomerase defect
F. Orosz et al. BBA 1792 (2009) 1168–1174
Null mutations:
Severe anemia
No neuro signs
Mild mutations:
No/mild anemia
Severe neuro signs

CFTDE-Necker X.03
Few potential ontogenic and
environmental mechanisms
Metabolic « switch » during development
Intercurrent events (fast,catabolism,diet,drugs)
Residual enzyme activity (+/-limitant step)
Acute versus slow progressive intoxication
Over energy expenditure
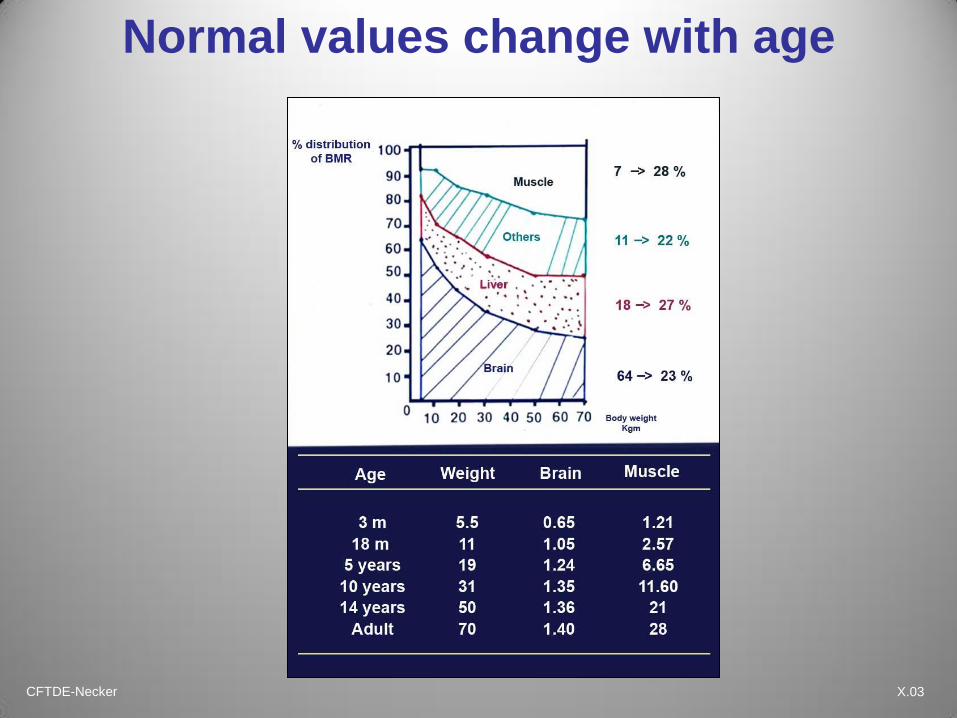
CFTDE-Necker X.03
Normal values change with age

CFTDE-Necker X.03
Nitrogen excretion related to age
Relationship between
urinary urea nitrogen
excretion and
body surface area

CFTDE-Necker X.03
Nitrogen production and outcome
100 g muscle contain about 20 g of proteins
Nitrogen protein content is 16%
20 g protein fully catabolized produces 3.2 g
nitrogen
3.2 g nitrogen = 228 mmol N (3200/14)
228 mmol nitrogen produces 114 mmol urea
(228/2) Urea:CO(NH2)2 114 mmol = 6.4 g
(93×60)
24h urea excretion allows to calculate the protein
catabolism

CFTDE-Necker X.03
1 g N = 6.25 g protein = 30 g muscle
Muscle amino acids
0
100
200
300
400
500
600
Leucine Valine Isoleucine Phenylalanine Methionine
mg AA/gN
(%/g N)
(8.5%) Leu MSUD: 500–700 mg/d
(6%) Val MSUD:300–400 mg/d
(4%) Phe PKU: 200–400 mg/d
(3.2%) Meth HSC: 120–250 mg/d
(5.9%) Ileu MSUD: 280–400 mg/d
Amino acid tolerance in IEM strictly reflects the muscle
amino acid composition

CFTDE-Necker X.03
Protein synthesis
Muscle protein content is 20g%
Nitrogen protein content is 16g%
1 g nitrogen = 6.25 g protein = 30 g muscle
Amino acid composition of natural proteins is
genetically determined (doesn’t depend on
the diet)
Amino acids from food are firstly and mostly
directed to protein synthesis
Essential amino acids are limiting factors

CFTDE-Necker X.03
Catabolism in control and PKU
Influence of diffusion space on blood
AA concentration
CONTROL
Muscle 100 g
Protein 20 g
20 AA 3.2 N
CO2+H2O UREA
(100 mmol)
PKU
Muscle 100 g
Protein 20 g
19 AA PHE (4%) 3.2 g N
(LEU, ILU…) 800 mg (186 mmol)
CO2+H2O UREA
(100 mmol)
LEU, ILU, PHE.. 186 mmol
Adult 60 kg 30 liters 800/30= 22 mg/l
Infant 4kg 3 liters 800/3 = 220 mg/l

CFTDE-Necker X.03
Anabolism in PKU is limited by
Phe intake
CLASSICAL PKU
Daily natural protein intake 5–8 g
(Phe 4% = 200–320 mg)
Phe free AA mixture 15g
8 essential AA Phe:300mg 3.2 g N
Maximum potentially
Synthesized protein 5–8 g
Muscle 25–35 g
CONTROL (theorical model)
Daily natural protein intake 20 g
9 essential AA 3.2 g N
No limitant factor
(Phe 4% about 800 mg)
Maximum potentially
synthesized protein 20 g
Muscle 100 g

CFTDE-Necker X.03
Coma
Tanyel Zubarioglu Indian J Pediatr 2016
DOI 10.1007/s12098-016-2077-3
OAT deficiency mimicking neonatal OTC

CFTDE-Necker X.03 From M Baumgartner et al chapter 21 in Saudubray et al 2016

CFTDE-Necker X.03

CFTDE-Necker X.03
20-50 years
• Pyramidal signs (77%)
• Cerebellar ataxia (64%)
• Cognitive decline (61%)
• Polyneuropathy (45%)
• Psychiatric signs (25%)
• Epilepsy (25%) • Learning difficulties (65%)
• Juvenile cataract (84%)
• Chronic diarrhea (40%)
• Tendon xanthomata (45%)
• Neonatal cholestasis
Childhood
Adulthood
Clinical spectrum of
Cerebrotendinous xanthomatosis
Estimated from a series of 135
published cases (Amador)
Courtesy F Sedel

CFTDE-Necker X.03
Mild intellectual
disability
Mild pyramidal signs
Chronic diarrhea
Mild intellectual
disability
Chronic diarrhea
Liver cholestasis
Chronic diarrhea CTX
Courtesy Dr Fanny Mochel

CFTDE-Necker X.03
Cerebrotendinous xanthomatosis
Su
rviv
al P
rob
ab
ility
0.0
0
.2
0.4
0
.6
0.8
1
.0
0 10 20 30 40 50 60
Time to Event (Year)
Psychiatric Symptoms
Walking Difficulties
Cataract
School Difficulties
Diarrhea
Courtesy Fanny Mochel
Cholestan
ol
Natural history of
cerebrotendinous
xanthomatosis:
a paediatric disease
diagnosed in adulthood
Degos et al. Orphanet
Journal of Rare Diseases
(2016) 11:41

CFTDE-Necker X.03
Neutral Lipid Storage Diseases
Steatosis,Myopathy Oxysterol
Spastic paraparesis
Cerebrotendinous
xanthomatosis
Cataract, Spasticity
Ataxia.Psychiatric signs
Niemann Pick C
Pyramidal sign, Ataxia
Cognitive decline
Neuropathy
PBD/IRD
RP. Deafness
Mental retardation
Transient neonatal to early infantile
Cholestasis or Liver failure
followed by neurodegeneration
MEGDEL
Hypotonia
Dystonia
Leigh like
Citrin
Late onset
neuropsychiatric signs
LCHAD
Myoglobinuria
RP.Neuropathy
Arginase
Spastic paraplegia

CFTDE-Necker X.03
Psychiatric signs,
dementia, ataxia,
dystonia
0 1 2 3 6 10 20 30 40 50
Vertical supranuclear gaze palsy
Early infantile
Late infantile
Juvenile
Adult
Systemic signs in NPC
Splenomegaly
Motor
delay Walking
problems,
clumsiness,
speech delay
Learning problems,
ataxia, seizures,
cataplexia
Age (years)
Neonatal
Fetal hydrops/neonatal cholestasis
Courtesy MT Vanier

CFTDE-Necker X.03
Few potential environmental
mechanisms
Metabolic « switch » during development
Intercurrent events (fast,catabolism,diet,drugs)
Residual enzyme activity (+/-limitant step)
Acute versus slow progressive intoxication
Over energy expenditure

CFTDE-Necker X.03
Glucose sources with fasting
4 8 12 16 20 24 28 32 2 8 16 24 0
10
20
30
40
Hours Days
Exogenous
Glucagon Catecholamines
Glucagon Catecholamines
Cortisol
GH
I II III IV
Insulin
Glycogen Gluconeogenesis
MEAL
Glucose used g/h
From JM Saudubray et al J.Inher.Metab.Dis 2000;23:197-214 . J..
Phase I: Post-prandial II: Short to
middle fast
III: Long fast IV: Very long fast
Glucose Source Exogenous Glycogen
Gluconeogenesis
Gluconeogenesis
(hepatic)
Glycogen
Gluconeogenesis
(hepatic and renal)
Consuming
tissues
All All but liver muscle Brain, Blood cell,
Adrenal medulla
Brain, Blood cell,
Adrenal medulla
Greatest brain
nutrient
Glucose Glucose Glucose Ketone bodies
Glucose
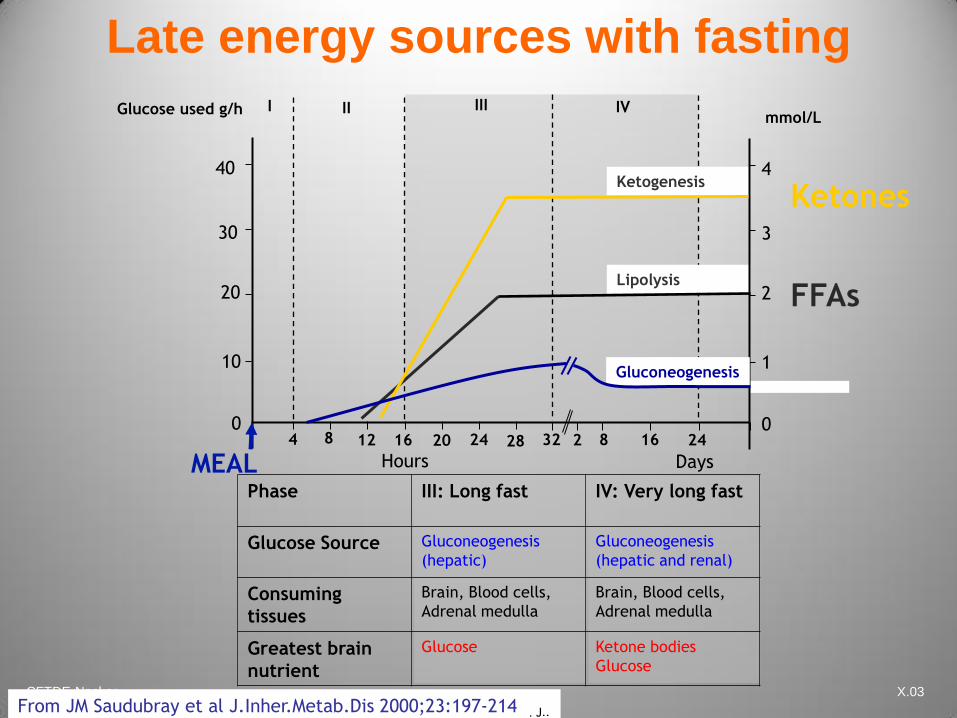
CFTDE-Necker X.03
Phase III: Long fast IV: Very long fast
Glucose Source Gluconeogenesis
(hepatic)
Gluconeogenesis
(hepatic and renal)
Consuming
tissues
Brain, Blood cells,
Adrenal medulla
Brain, Blood cells,
Adrenal medulla
Greatest brain
nutrient
Glucose Ketone bodies
Glucose
10
20
30
40
Late energy sources with fasting
4 8 12 16 20 24 28 32 2 8 16 24
Hours Days
Glucose used g/h I II III IV
MEAL
FFAs
Ketones Ketogenesis
Lipolysis
0
1
2
3
4
0
mmol/L
Gluconeogenesis
Courtesy of JM Saudubray, Paris Necker Hospital From JM Saudubray et al J.Inher.Metab.Dis 2000;23:197-214 . J..

CFTDE-Necker X.03
Few potential environmental
mechanisms
Metabolic « switch » during development
Intercurrent events (fast,catabolism,diet,drugs)
Residual enzyme activity (+/-limitant step)
Acute versus slow progressive intoxication
Over energy expenditure

CFTDE-Necker X.03
Late onset OTC
Born in 1951.
Died at 12 years old
from sudden, unexplained
coma. No previous history
JY 49
years

CFTDE-Necker X.03
K Died at 19 yr
Reye Syndrome
OTC deficiency — MALE to FEMALE transmission

CFTDE-Necker X.03
Few potential environmental
mechanisms
Metabolic « switch » during development
Intercurrent events (fast,catabolism,diet,drugs)
Residual enzyme activity (+/-limitant step)
Acute versus slow progressive intoxication
Over energy expenditure
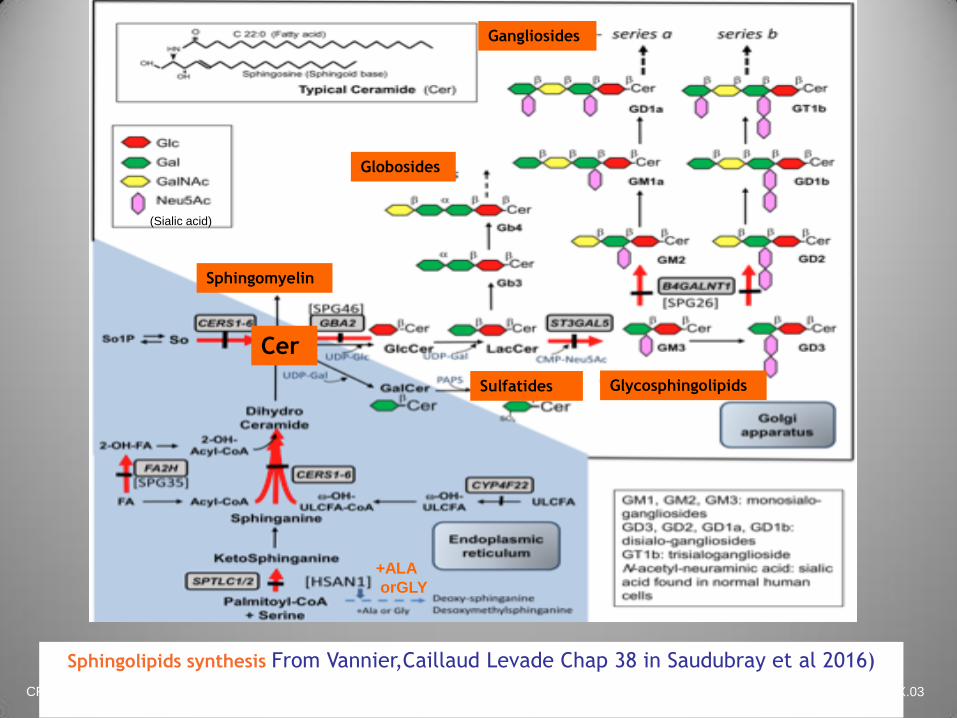
CFTDE-Necker X.03
Sphingolipids synthesis From Vannier,Caillaud Levade Chap 38 in Saudubray et al 2016)
(Sialic acid)
Cer
Globosides
Sulfatides
Sphingomyelin
Glycosphingolipids
Gangliosides
+ALA
orGLY

CFTDE-Necker X.03
Serine palmitoyltransferase (SPT)
deficiency
Causes adult hereditary sensory autonomic
neuropathy type I (autosomal dominant trait
HSAN I)
Progressive intoxication by deoxysphingoid bases
treatable by high doses of serine
Deoxysphingoid bases levels measured by tandem
MS in plasma are elevated (diagnostic test)
Diagnosis confirmed by molecular analysis of the
SPTLC1 and C2 genes
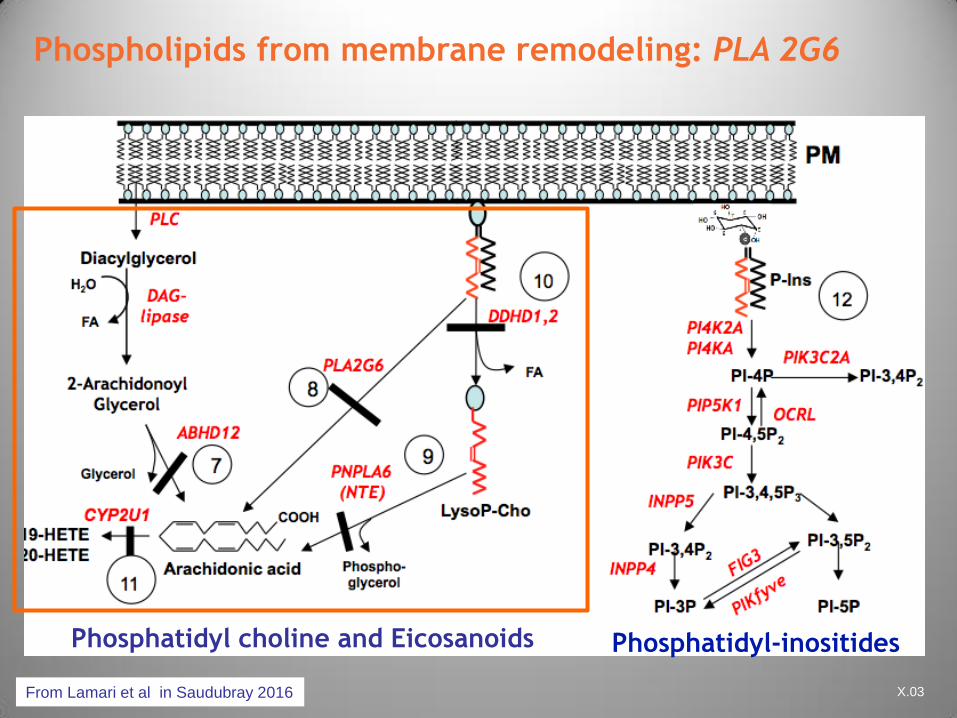
CFTDE-Necker X.03
Phospholipids from membrane remodeling: PLA 2G6
From Lamari et al in Saudubray 2016
Phosphatidyl choline and Eicosanoids Phosphatidyl-inositides

CFTDE-Necker X.03
Biochemistry
PLA2G6 encodes
– iPLA2b enzyme phospholipase group 4 (calcium
dependent)
– Alternative name phospholipase A2B
Releases FFA from phospholipids and
lysophospholipids membranes
Play a role in
– Intracellular signaling:
arachidonate,eicosanoids
– Inflammatory processes
– Apopotosis

CFTDE-Necker X.03
Phospholipase A2B deficiency
Three main clinical phenotypes:
Infantile neuroaxonal dystrophy (INAD)
Neurodegeneration with brain iron
accumulation (NBIA)
INAD and NBIA are caused by loss of
ability of PLA2 to catalyse FA release
which predicts accumulation of PL
(neuroaxonal spheroids):Intoxication ?
Dystonia-Parkinsonism in adults .
Mutations do not directly impair catalytic
function

CFTDE-Necker X.03
PLAN: gen PLA2G6
1- 6 years 4 – 40 years <3 years
PROTOCOL NBIA PLAN: gen PLA2G6
Dystonia))Parkinsonism)
INAD) Atypical)
AREP
Pure
Parkinsonism
L-dopa
responsive
Parkinsonism)
Courtesy Angela Garcia

CFTDE-Necker X.03
Infantile neuroaxonal dystrophy
Courtesy of Isabelle Desguerre, Paris
Cerebellar atrophy with signal hyperintensity in cerebellar cortex
occasional hypointensity in the pallida and substantia nigra ( iron deposits)

CFTDE-Necker X.03
Few potential environmental
mechanisms
Metabolic « switch » during development
Intercurrent events (fast,catabolism,diet,drugs)
Residual enzyme activity (+/-limitant step)
Acute versus slow progressive intoxication
Over energy expenditure

CFTDE-Necker X.03
Changing spectrum of PDH
Male, 25 years old, has normal intelligence
From age 15 months to adulthood:
– Recurrent episodes of ataxia, hypotonia, weakness,
lethargy when febrile (about once a year)
– Between episodes: progressive polyneuropathy,
scoliosis
– Diagnosis: Charcot-Marie-Tooth, type II
At age 19 years, developed left optic
neuropathy
At age 24 years: right optic neuropathy
Case F Sedel M Brivet

CFTDE-Necker X.03
Metabolic studies
Amino acids — elevated alanine (789 µM)
Fasting Postprandial
Lactate
(nl <1.8 mM)
1.6 mM 2.8 mM
Pyruvate
(nl <67 mM)
0.173 mM 0.250 mM
Lac/Pyr (nl<20) 9 11
Case 10
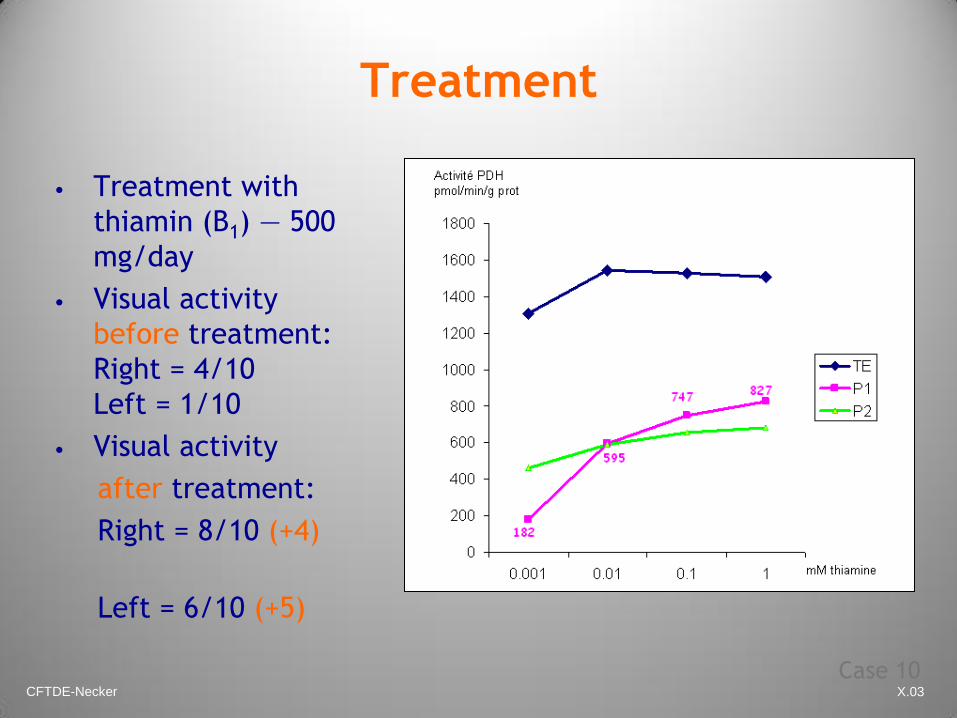
CFTDE-Necker X.03
Treatment
• Treatment with
thiamin (B1) — 500
mg/day
• Visual activity
before treatment:
Right = 4/10
Left = 1/10
• Visual activity
after treatment:
Right = 8/10 (+4)
Left = 6/10 (+5)
Case 10

CFTDE-Necker X.03
Late onset biotinidase deficiency
22-year-old man presented with a subacute
extensive myelopathy with bilateral optic
neuropathy
Mimicking seronegative neuromyelitis optica.
Metabolic evaluation revealed biotidinase
deficiency,
Treatment with high doses of biotin
progressively improved the clinical condition.
Multiple Sclerosis Journal 2015, Vol. 21(12) 1608–1609

CFTDE-Necker X.03
High dose biotin to the rescue of
demyelinating processes?
High doses of biotin (100–300 mg/day) in patients with
SPMS and PPMS have an impact on disease progression
in those with optic neuropathy,homonymous hemianopia,
and spinal cord involvement *
A phase III trial of high-dose biotin in progressive MS
shows improvement in MS-related disability outcomes (13
% of the exposed group vs 0 % of placebo group had
improvement of Expanded Disability Status Scale or timed
25-foot walk at month 9 and12)**.
*Sedel F et al. Mult Scler Relat Disord 2015;4:159–169.
**Tourbah A et al. The 67th Annual Meeting of the American Academy of Neurology, 2015
Tourbah A (2015) Biotin and demyelinating diseases – a new connection? Mult Scler J 21:1608-1609

CFTDE-Necker X.03
L P Saint Paul et al (2016) high-dose biotin in the treatment of progressive multiple sclerosis,
Expert Opinion on Drug Metabolism& Toxicology, 12:3, 327-344

CFTDE-Necker X.03
Clinical circumstances of care
in adult practice
1. IEM diagnosed in the pediatric age and
reaching adulthood :
treated / untreated natural history
2. IEM striking in adulthood :
- unknown presentations
- methods of investigations adapted to adults
3. IEM diagnosed in adulthood but starting at the
pediatric age

CFTDE-Necker X.03
Challenge of adult IEM patient care
1.Long term treatment maintenance is difficult
2. Natural history of treated patients is unknown
3. Pediatricians don’t know late complications
4.Late onset forms are missed by adult physicians
5.Pediatric forms undiagnosed by pediatricians
6.Diagnostic criteria adapted to adult mild forms
(control values)

CFTDE-Necker X.03
Conclusions
Cette différence phenotypique
« …qui est causée par l'âcreté des humeurs
engendrées dans la concavité du
diaphragme, il arrive que ces vapeurs...
Ossabandus, nequeyrs, nequer, potarimum,
potsa milus. Voilà justement pourquoi votre
fille est muette. »
From Sganarelle in Moliére
Le médecin malgré lui

CFTDE-Necker X.03
Conclusions:
Déjà Necker…!
Cette différence phenotypique
« …qui est causée par l'âcreté des humeurs
engendrées dans la concavité du
diaphragme, il arrive que ces vapeurs...
Ossabandus, nequeyrs, nequer, potarimum,
potsa milus. Voilà justement pourquoi votre
fille est muette. » From Sganarelle in Moliére
Le médecin malgré lui

CFTDE-Necker X.03
Chapters 1 and 2
6th edition

CFTDE-Necker X.03
THE END