dienstag 14. april 2009 9:15 - ams.med.uni-goettingen.de · tim beißbarth bioinformatik system...
TRANSCRIPT

Dienstag14. April 20099:15
UMG Georg-August-Universität GöttingenAbt. Medizinische Statistik - Biostatistik
Bioinformatik/System BiologieSommersemester 2009
Prof. Dr. Tim Beißbarth

Tim Beißbarth Bioinformatik
Biological Samples& Data Generation
Data Analysis& Statistics
Technology& Development
FunctionalInterpretation &Systems Biology
Gene expressionMicroarrays
Tissue fromPatients -Cancer
DifferentCell lines -Model Systems
Cellular AssaysRNAi Screens
Protein ArraysLysate Arrays
Differential GeneExpression -Statistical Testing
Classification -Machine Learning
Clustering -Visualization
CellularPertubation- Knockdown- overexpression
Find overrepresentedFunctional Groups.
Integration of differentdata.
Reconstruction ofbiological networks.
GO:GO:GO: p-value
p-value
p-value
p-value
p-value
p-value
p-valuep-value
p-valuep-value
p-valuep-value
p-valuep-value
p-valuep-value
p-valuep-value ??
S1 S3S2
EE
E
E E
S4
EEE
E
Likelihood model
Von Biologischen Problemen zu System-Biologischen Modellen

Tim Beißbarth Bioinformatik
System Biologie – verschiedene Herangehensweise
• Bottom-Up• Vernetzung weniger Proteine zu
Interaktionsmodellen (Pathways)• Ziel: Simulation von einfachen
biologischen Systemen und vorhersage von Zuständen.
• Modellierungsansätze:• Differentialgleichungen• Petri-Netze• Bayes-Netze• Boolsche Netze
• Top-Down• Genomische Ansätze zur Erhebung
von Daten und Modellierung der Proteinnetzwerke einer ganzen Zelle.
• Ziel: Besseres Verständnis der komplexen biologischen Zusammenhänge einer Zelle.
• Methodische Ansätze:• Protein-Protein-Interaktion• Gen-Expressionsnetze• Knock-Down Screens• Topologische Analyse von
Netzwerken

Tim Beißbarth Bioinformatik
Übersicht
• Di 14.4.: Vorbesprechung - Microarrays und statistische Datenanalyse
• Di 21.4.: Normalisierung/Differentielle Genanalyse
• Di 28.4.: Clustern
• Fr 8.5.: Klassifikation
• Di 19.5.: Gene-Ontologies
• Di 26.5.: Bayes-Netze

Tim Beißbarth Bioinformatik
Online
• Vorlesungsslides und R-Skripte:http://www.ams.med.uni-goettingen.de/biostatistics/sysbio09
• Lectures Terry Speed, Berkeley: http://www.stat.berkeley.edu/users/terry/Classes/
• Kurs NGFN „Practical DNA Microarray Analysis”: http://compdiag.molgen.mpg.de/lectures.shtml
• R/Bioconductor Dokumentation (Vignetten):http://www.bioconductor.org
• R Tutorial von Günther Sawitzkihttp://statlab.uni-hd.de/users/gs
• Google, Pubmed, Wikipedia

Tim Beißbarth Bioinformatik
1. Vorlesung
• Überblick über Methoden zur Genexpressionsanalyse und spezifische Probleme.
1. SAGE
2. Verschiedene Microarray Plattformen
1. cDNA
2. Affymetrix• Design von Microarray Experimenten• Auswertung von Microarray Experimenten (Überblick)
1. Verschiedene Level von Wiederholungen
2. Normalisierung
3. Clusterung
4. Klassifikation

Tim Beißbarth Bioinformatik
*****
GeneChip Affymetrix
cDNA microarray
Nylon membrane
Agilent: Long oligo Ink Jet
Illumina Bead Array
CGH
SAGE
VerschiedeneTechnologien

Tim Beißbarth Bioinformatik
SAGE
Normal Krank
isoliere mRNA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
Schneide bei CATG
Extrahiere tags(14-21bp)
Konkatenieretags
Ditag Ditag
Nla II
I
Nla II
I
Tag 1 Tag 2 Tag 3 Tag 5 Nla II
IDitag
Nla II
I
Tag 4 Tag 6
Sequenziere
Normal Krank
Quantifiziere tags

Tim Beißbarth Bioinformatik
cDNA und Affimetrix (kurze, 25 bp) Oligo Technologien.Lange Oligos (6075 bp) werden so ähnlich wie cDNA benutzt.

Tim Beißbarth Bioinformatik
Microarray Hybridisierung
Gewebe 1 total RNA 1 Gewebe 2total RNA 2
reversen Strang synthetisieren,
labeln, mischen
Hybridisierung

Tim Beißbarth Bioinformatik
cDNA “A”Cy5 gelabelt
cDNA “B”Cy3 gelabelt
PROBE
TARGET
Definition von probe and target

Tim Beißbarth Bioinformatik
Microarrays Geschichte
• Basiert auf Southern BIot Technologie (Edward Southern, 1975, J. Mol. Biol.)
• 1990: erste high-density Nylonfilter Arrays (Lennon/Lehrach, 1991, Trends Genet., Review)
• 1995: cDNA-Microarrays beschrieben von Schena et al, Science
• 1996: Affymetrix Genechip Technologie beschrieben von Lockhart et al, Nat. Biotechnol.

Tim Beißbarth Bioinformatik
PCR Produkt Amplifikationpurification
cDNA Klone(probes)
printing
Microarray
0.1nl/spot
Hybridisiere Target mit Microarray
mRNA target)
excitation
laser 1laser 2
Emission
scanning
Analyse
Bilder überlagern
cDNA arrays zusammengefasst

Tim Beißbarth Bioinformatik
Zwei verschiedene Aspekte von Array Design
Design des Arrays Allokierung der mRNA Samples zu den Slides
Arrayed Library(96 oder 384well plates)
cDNAcDNA “A”Cy5 gelabelt
cDNA “B”Cy3 gelabelt
Hybridisierung
Spotte Microarrayauf Glas Slides
affy
MTWT

Tim Beißbarth Bioinformatik
cDNA Bibliotheken und Expressed Sequence Tags (ESTs)
Zelle mRNA-Pool
schneiden / reverse Transkription
cDNA -Bank
GAATTCGATATCTCA.....ATAGTCAGCATCAAGCTT GAATTCGATATCTCA.....ATAGTCAGCATCAAGCTT
5‘ Vector 5‘Read Vector 3‘ - 3‘ Vector 3‘Read Vector 5‘
cDNA - Klon
ESTs

Tim Beißbarth Bioinformatik
Beispiel EST Cluster visualisiert: http://genenest.molgen.mpg.de

Tim Beißbarth Bioinformatik
Alle Klone auf einem cDNA Array haben idealerweise …
• Gleiche Schmelztemperatur (ähnliche Länge und Basenzusammensetzung)
• Keine Sekundärstruktur (i.e. Selbstkomlementarität)
• Keine Homologen im Genom (i.e. Genspezifität)

Tim Beißbarth Bioinformatik
A Typen von Samples• Replikate – technische, biologische. • Poolen von Samples.• Amplifizieren von Samples.
B Verschiedene experimentelle Layouts• Ziel des Experiments.• Robustheit.• Erweiterbarkeit.• Effizienz:
• Anzahl der verbrauchten Slides.- Menge des benötigten Probenmaterials.
Verschiedene Aspekte von Design
2. Zuordnung der Samples zu den Slides

Tim Beißbarth Bioinformatik
Biologische Verifikation und Interpretation
Microarray Experiment
Experimentelles Design
Bildanalyse
Normalisierung
Biologische Fragestellung
TestenAuswerten KlassifizierungAnalyse
Clustern
Experimenteller Zyklus
Qualitäts Sicherung
fehlgeschlagen
In Ordnung
Vorverarbeitung

Tim Beißbarth Bioinformatik
mRNA Samples präparieren:
Maus ModelGewebe
präparieren
RNA Isolation
Amplification
Probelabelling
Hybridisierung

Tim Beißbarth Bioinformatik
mRNA Samples präparieren:
Maus ModelGewebe
präparieren
RNA Isolation
Amplification
Probelabelling
Hybridisierung
Biologische Replikate

Tim Beißbarth Bioinformatik
mRNA Samples präparieren :
Maus ModelGewebe
präparieren
RNA Isolation
Amplification
Probelabelling
Hybridisierung
Technische Replikate

Tim Beißbarth Bioinformatik
Gepoolte vs einzeln gemessene Samples
• Poolen kann als “biologisches Mitteln” gesehen werden.• Trade off zwischen
• Kosten für eine Hybridisierung.• Kosten für die mRNA Samples.
• Fall 1: Kosten für mRNA Sample << Kosten für HybridisierungPoolen kann helfen die Anzahl der Hybridisierungen zu reduzieren.
• Fall 2: Kosten für mRNA Sample >> Kosten für HybridisierungJedes Sample einzeln auf Array Auftragen um maximiale Information über biologische Varianz zu erhalten.
• Referenzen:• Han, E.-S., Wu, Y., Bolstad, B., and Speed, T. P. (2003). A study of the effects of pooling
on gene expression estimates using high density oligonucleotide array data. Department of Biological Science, University of Tulsa, February 2003.
• Kendziorski, C.M., Y. Zhang, H. Lan, and A.D. Attie. (2003). The efficiency of mRNA pooling in microarray experiments. Biostatistics 4, 465-477. 7/2003
• Xuejun Peng, Constance L Wood, Eric M Blalock, Kuey Chu Chen, Philip W Landfield, Arnold J Stromberg (2003). Statistical implications of pooling RNA samples for microarray experiments. BMC Bioinformatics 4:26. 6/2003

Tim Beißbarth Bioinformatik
Design eines Dye-Swap Experimentes
• Wiederholungen sind essentiell um die Qualität der Experimente beurteilen zu können.
• Ein Beispiel für Replikate ist der Dye-Swap, d.h. Replikate welche den Gleichen mRNA Pool benutzen, aber die Label vertauschen.
• Der Dye-Swap gibt Aufschluß darüber ob bei den Experimenten ein Farbbias eingeführt wird.

Tim Beißbarth Bioinformatik
Graphische Repräsentation
Knoten: mRNA samples;Kanten: Hybridisierungen;Richtung: dye Zuordnung.
Cy3 sample
Cy5 sample

Tim Beißbarth Bioinformatik
Eine einfache Design Frage:Direkte oder indirekt Vergleiche
Zwei Samples (A vs B)
e.g. KO vs. WT oder mutant vs. WT
A BA
BR
Direkt Indirekt
σ2 /2 2σ2
mittelwert (log (A/B)) log (A / R) – log (B / R )
Diese Berechnungen nehmen Unabhängigkeit der Samples an und sind in Wirklichkeit komplizierter.

Tim Beißbarth Bioinformatik
Experimentelle Resultate
• 5 Sets von Experimenten mit ähnlicher Struktur.
• Vergleiche Y-AxeA) SE für aveMmt
B) SE für aveMmt – aveMwt
SE

Tim Beißbarth Bioinformatik
4 Samples
C
A.BBA
B
C
A.B
A
B
C
A.B
A
B
C
A.B
A
Experimente für welche mehrere Designs in Frage kommen

Tim Beißbarth Bioinformatik
T2 T3 T4T1
RefT2 T3 T4T1
T2 T3 T4T1 T2 T3 T4T1
Experimente für welche mehrere Designs in Frage kommen
Zeitreihen

Tim Beißbarth Bioinformatik
Experimentelles Design
• Benutze hoch korrelierte Referenz-Samples um die Nachteile des Common Reference Designs auszugleichen.
• Benutze biologische Replikate an Stelle von technischen Replikaten soweit möglich.
• Effizienz kann mit verschieden Massen gemessen werden• Anzahl der Slides oder Hybridisierungen;• Menge des verbrauchten biologischen Materials.
• Vergleiche, an denen der Experimentator besonders interessiert ist, sollten nach Möglichkeit auf einem Slide direkt gemacht werden.
Referenzen• T. P. Speed and Y. H Yang (2002). Direct versus indirect designs for cDNA microarray experiments.
Sankhya : The Indian Journal of Statistics, Vol. 64, Series A, Pt. 3, pp 706-720• Y.H. Yang and T. P. Speed (2003). Design and analysis of comparative microarray Experiments In T. P
Speed (ed) Statistical analysis of gene expression microarray data, Chapman & Hall.• R. Simon, M. D. Radmacher and K. Dobbin (2002). Design of studies using DNA microarrays. Genetic
Epidemiology 23:21-36.• F. Bretz, J. Landgrebe and E. Brunner (2003). Efficient design and analysis of two color factorial
microarray experiments. Biostaistics.• G. Churchill (2003). Fundamentals of experimental design for cDNA microarrays. Nature genetics
review 32:490-495.• G. Smyth, J. Michaud and H. Scott (2003) Use of within-array replicate spots for assessing
differential experssion in microarray experiments. Technical Report In WEHI.• Glonek, G. F. V., and Solomon, P. J. (2002). Factorial and time course designs for cDNA microarray
experiments. Technical Report, Department of Applied Mathematics, University of Adelaide. 10/2002

Tim Beißbarth Bioinformatik
Gen-expressions Daten
Gene
mRNA Samples
geneexpressions level or ratio für Gen i in mRNA Sample j
M =Log2(rote Intensität / Grüne Intensität)
Vergleich jeweils zweier Bedingungen.
sample1 sample2 sample3 sample4 sample5 …1 0.46 0.30 0.80 1.51 0.90 ...2 0.10 0.49 0.24 0.06 0.46 ...3 0.15 0.74 0.04 0.10 0.20 ...4 0.45 1.03 0.79 0.56 0.32 ...5 0.06 1.06 1.35 1.09 1.09 ...
Gen-expressions Daten für G Gene und n Hybridisierungen. Gene x arrays Daten-matrix:
A =mittel: log2(rote Intensity), log2(Grüne Intensität)
Function (PM, MM) von MAS oder RMA

Tim Beißbarth Bioinformatik
Der Scatterplot - Aus Vorlesung von Rainer König vom Vorjahr
Daten Daten, logarithmisch

Tim Beißbarth Bioinformatik
MA Plot
A = 1/2 log2(RG)
M =
log 2
(R/G
)

Tim Beißbarth Bioinformatik
Vulcano Plot (B=Maß für die Reproduzierbarkeit, mehr dazu nächstes Mal)

Tim Beißbarth Bioinformatik
Rotierter Scatter Plot
M = log R/G = logR - logG A = ( logR + logG) /2
Positive Kontrollen
(in verschiedenen Konzentrationen gespottet) Negative Kontrollen
Leere Spots
Lowess Kurve

Dienstag14. April 20099:15
UMG Georg-August-Universität GöttingenAbt. Medizinische Statistik - Biostatistik
Analyse von Mikrorray Daten mit Hilfe von R/Bioconductor

Tim Beißbarth Bioinformatik
Was ist “R” ?
• Umgebung zur statistischen Datenanalyse
• Open source, weitgehend kompatibel mit Splus
• Sehr dynamisch durch einfache Einbindung neuer Funktionen („Packages“)
• In der wissenschaftlichen Gemeinschaft das (zusammen mit SAS) am weitesten verbreitete Statistik-Tool
• De facto - Standard bei Microarray-Analysen

Tim Beißbarth Bioinformatik
Installation von R/Bioconductor
http://cran.r-project.org

Tim Beißbarth Bioinformatik
Installation von R/Bioconductor

Tim Beißbarth Bioinformatik
Installation von R/Bioconductor

Tim Beißbarth Bioinformatik
Installation von R/Bioconductor
http://bioconductor.org

Tim Beißbarth Bioinformatik
Installation von R/Bioconductor

Tim Beißbarth Bioinformatik
Installation von R/Bioconductor

Tim Beißbarth Bioinformatik
Die R Syntax, elementare Rechenoperationen
=
20
1
0
x
=
==
20*20
1*1
0*0
20
1
0
*
20
1
0
*
xxy
> x = 0:20> y = x*x> plot(x,y)
0 5 10 15 20
010
020
030
040
0
xy

Tim Beißbarth Bioinformatik
Die R Syntax, elementare Rechenoperationen
=
4
3
2
1
x
> x = 1:4> y = x*2> z = x*c(0,1)
=
=
==
8
6
4
2
2
2
2
2
*
4
3
2
1
2*
4
3
2
1
2*xy
=
=
=
=
4
0
2
0
1
0
1
0
*
4
3
2
1
1
0*
4
3
2
1
1
0*xz

Tim Beißbarth Bioinformatik
Die Datenstruktur exprSet
> getBioC(“vsn“)> library(vsn)> data(lymphoma)> class(lymphoma)[1] “exprSet“
lymphoma
lymphoma@exprs
lymphoma@phenodata
Gene
mRNA Samplessample1 sample2 sample3 sample4 …
1 0.46 0.30 0.80 1.51 ...2 0.10 0.49 0.24 0.06 ...3 0.15 0.74 0.04 0.10 ...4 0.45 1.03 0.79 0.56 ...5 0.06 1.06 1.35 1.09 ...
name colour typesample1 red CLL ...sample2 green CLL ...sample3 red DLCL ...sample4 green DLCL ...
(other slots)
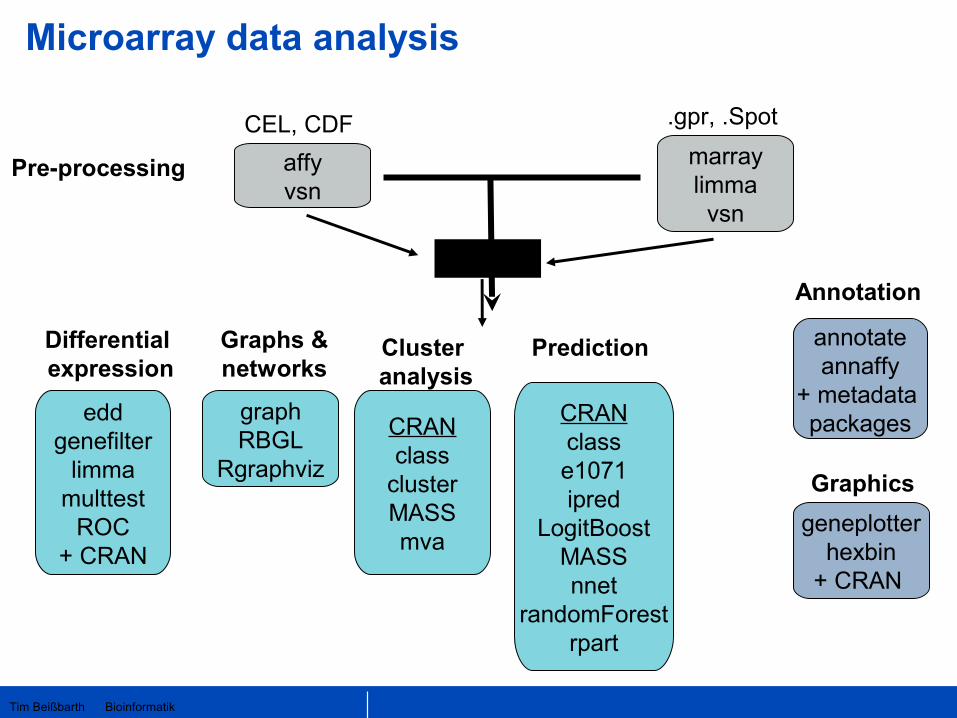
Tim Beißbarth Bioinformatik
CEL, CDF
affyvsn
.gpr, .Spot
Pre-processing
graphRBGL
Rgraphviz
eddgenefilter
limmamulttest
ROC+ CRAN
annotateannaffy
+ metadata packagesCRAN
classclusterMASSmva
geneplotterhexbin
+ CRAN
marraylimma
vsn
Differential expression
Graphs &networks
Cluster analysis
Annotation
CRANclasse1071ipred
LogitBoostMASSnnet
randomForestrpart
Prediction
Graphics
exprSet
Microarray data analysis

Tim Beißbarth Bioinformatik
Qualitätskontrolle II: Diagnostische Plots
• Scatterplot (absolute vs. logarithmische Skala)• M-A-Plot• Erkennen systematischer Fehler• QQ-Plot, Boxplot, • Ähnlichkeitsmatrix, Dendrogramm

Tim Beißbarth Bioinformatik
Visualisierung der Daten: Scatterplots
Vergleich zweier Microarraymessungen
Absolutskala logarithmische Skala
y = ½ x
y = ¼ x
y = 2x
y = 4x
y = ½ x
y = ¼ x
y = 2x
y = 4x
Vorteile der logarithmischen Transformation:
• Die Daten verteilen sich gleichmäßiger über den Plot• Linien konstanter Fold ratios bilden Parallelen zur Hauptdiagonalen
> x = exprs[,1]> y = exprs[,2]> plot(x,y)
> x = exprs[,1]> y = exprs[,2]> plot(x,y,log=“xy“)

Tim Beißbarth Bioinformatik
Visualisierung der Daten: M(inus)-A(verage) Plot
Vorteile des M-A Plots: • Linien konstanter Fold ratios bilden Parallelen zur x-Achse• M-A Plot ermöglicht das Erkennen systematischer Änderungen des
Kanal1/Kanal2-Expressionsverhältnisses in Abhängigkeit von der mittleren Expressionsstärke eines Gens
1 10 100 1000 10000
11
01
00
10
00
10
00
0
x
y
0 2 4 6 8 10-4
-20
24
log(x * y)/2
log
(y/x
)
> x = log(exprs[,1])> y = log(exprs[,2])> plot(x,y)
…> xMA =(x+y)/2> yMA = y-x> plot(xMA,yMA)
Drehen um 45o
log
(fo
ld r
atio
vo
n y
und
x)
log (geometr. Mittel von x und y)

Tim Beißbarth Bioinformatik
Erkennen systematischer Fehler im M-A Plot
Keine (kaum) systematische Abhängigkeiten
Kanal 2 ist um einen konstanten Faktor stärker als Kanal 1
Multiplikativer Bias

Tim Beißbarth Bioinformatik
Erkennen systematischer Fehler im M-A Plot
Kanal 1 ist nur im niedrig exprimierten Bereich stärker als Kanal 2
Additiver Bias
Kombination aus additivem und multiplikativem Bias

Tim Beißbarth Bioinformatik
Quantile-Quantile plot (QQ-plot). Um zwei durch dieVektoren x und y gegebenen Verteilungen zu vergleichen, plotte für alle Werte q aus dem Intervall (0,1) das q-Quantil der x-Verteilung gegen das q-Quantil der y-Verteilung.
QQ-plot
Vergleich zweier Messungen: QuantileQuantileplots

Tim Beißbarth Bioinformatik
Interpretation:
Vergleich zweier Messungen: QQplots
Unähnliche Verteilungen:Der QQplot ist nicht linear, insbesondere nicht im Zentrum der QQ-Linie.
Ähnliche Verteilungen, die Enden der y-Verteiluing sind länger.
Ähnliche Verteilungen, die Enden der x-Verteilung sind länger.

Tim Beißbarth Bioinformatik
Five-point-Summary. Für einen Vektor x wird das 5-tuple der Quantile (xmin, x0.25, xmed, x0.75, xmax) das five-point-summary genannt.
Boxplot. Ein Boxplot ist die Visualisierung des (mehrerer) Five-point-summarys:
Boxplots sind zum schnellen Vergleich mehrerer Verteilungen besonders gut geeignet
x0.25
xmax
x0.75
xmed
xmin
Vergleich mehrerer Messungen: Boxplots
(1,1
)
(1,2
)
(1,3
)
(1,4
)
(2,1
)
(2,2
)
(2,3
)
(2,4
)
(3,1
)
(3,2
)
(3,3
)
(3,4
)
(4,1
)
(4,2
)
(4,3
)
(4,4
)
-2-1
01
2
Swirl array 93: pre-norm
PrintTip
M

Tim Beißbarth Bioinformatik
Vergleich mehrerer Messungen: Ähnlichkeitsmatrizen und Dendrogramme
> distanzen = dist(t(exprs))> dendrogramm = hclust(distanzen)> plot(dendrogramm)
CL
L-5
2
refe
ren
ce
refe
ren
ce
refe
ren
ce
CL
L-3
9
DL
CL
-00
24
DL
CL
-00
23
CL
L-1
3
CL
L-1
3
refe
ren
ce
refe
ren
ce
refe
ren
ce
refe
ren
ce
refe
ren
ce
DL
CL
-00
32
DL
CL
-00
29
05
00
00
10
00
00
15
00
00
Cluster Dendrogram
hclust (*, "complete")d
He
igh
t
Mit Dendrogrammen / Ähnlichkeitsmatrizen lassen sich oft Batcheffekte erkennen oder einzelne defekte Chips identifizieren.

Tim Beißbarth Bioinformatik
Acknowledgements – Slides geborgt von
• Achim Tresch
• Benedikt Brors
• Wolfgang Huber
• Terry Speed
• Jean Yang