die stationäre phase - papa-gey.depapa-gey.de/kolloquien/2010-12.pdf · oxidantien, analgetika,...
TRANSCRIPT

1
Die stationäre PhaseDie stationäre Phase
Sortimentsübersicht
Auswahlentscheidung
Sortimentsübersicht
Auswahlentscheidung

2
TrennmechanismenTrennmechanismen
Adsorptions-Chromatographie
Verteilungs-Chromatographie
Ionenaustausch-Chromatographie
Komplex-Chromatographie
Adsorptions-Chromatographie
Verteilungs-Chromatographie
Ionenaustausch-Chromatographie
Komplex-Chromatographie
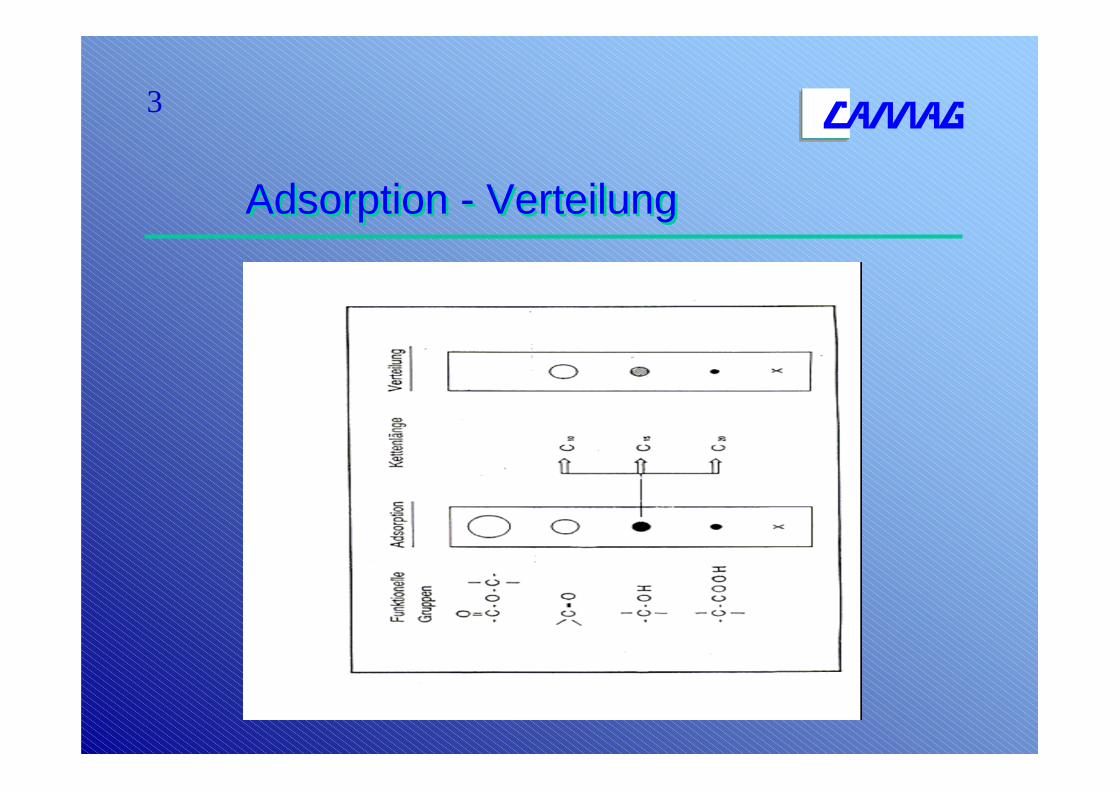
3
Adsorption - VerteilungAdsorption - Verteilung

4
Welche stationäre Phase ?Welche stationäre Phase ?AluminiumoxidKieselgelRP-SchichtenAmino-, Cyano und DiolphaseFlorisilPolyamidCelluloseKieselgurImprägnierte SchichtenIonenaustauscher-Schichten
AluminiumoxidKieselgelRP-SchichtenAmino-, Cyano und DiolphaseFlorisilPolyamidCelluloseKieselgurImprägnierte SchichtenIonenaustauscher-Schichten

5
Weiteres über stationäre PhasenWeiteres über stationäre Phasen
SchichtuntergrundBinderPhosphoreszenzindikatorHersteller-/ChargenabhängigkeitLabordämpfe- Vorwaschen der SchichtAktivität der SchichtDC-HPTLC
SchichtuntergrundBinderPhosphoreszenzindikatorHersteller-/ChargenabhängigkeitLabordämpfe- Vorwaschen der SchichtAktivität der SchichtDC-HPTLC

6
AbkürzungenAbkürzungen
(G Gips als Bindemittel
(H ohne artfremde Bindemittelzusätze
F Fluoreszenzindikator
245 Anregungswellenlänge des F
PSC präperative Schichten Schichtdicke > 0,25 mm
R hochgereinigtes Sorbens
RP 2,8,18 Reversed Phae/Umkehrphase mit
Kettenlänge/C-Atome 2,8,18
W wasserfeste,- benetzbare Schichten
S säurestabiler Indikator
60 mittlerer Porendurchmesser in Angström (=6 nm)

7
Terminologie & PolaritätTerminologie & Polarität
Normalphasen-Chromatographie(Straight Phase):polare SP + unpolare MP
Umkehrphasen-Chromatographie(Reversed Phase):unpolare SP + polare MPPolarität der Schichten
Si > NH2 > CN/Diol > RP-2 > RP-8 > RP-18
Normalphasen-Chromatographie(Straight Phase):polare SP + unpolare MP
Umkehrphasen-Chromatographie(Reversed Phase):unpolare SP + polare MPPolarität der Schichten
Si > NH2 > CN/Diol > RP-2 > RP-8 > RP-18

8
Stationäre Phasen auf Kieselgel-BasisStationäre Phasen auf Kieselgel-BasisCuCu++
polarepolare PhasePhase
88]]
OOSiSi
SiSiOO
OO
OO
SiSiOO
CHCH33
SiSi
SiSi
OO
OOOO
OO
SiSi
SiSi
SiSi
SiSi
OO
OO OOOO
[[
CHCH33
]][[ 55
]][[
CHCH33
33 CHCH33CHCH33
CC NN
OOOHOH
OHOH
NHNH22
SiSi
OOHH
]]88
NNOOOO OHOH
[[chiralechirale PhasenPhasen mittelpolaremittelpolare chemischchemischgebundenegebundene PhasenPhasen
unpolareunpolare chemischchemischgebundenegebundene PhasenPhasen

9
Gebundene Phasen - DIOLGebundene Phasen - DIOL
Weniger beeinflusst durch Feuchtigkeit
Einsatz als Normal- oder Umkehrphase
Brauchbarste der gebundenen Phasen
Grosse Ähnlichkeit mit Kieselgel
Geringere Retention
Hormone, Steroide
Weniger beeinflusst durch Feuchtigkeit
Einsatz als Normal- oder Umkehrphase
Brauchbarste der gebundenen Phasen
Grosse Ähnlichkeit mit Kieselgel
Geringere Retention
Hormone, Steroide

10
Gebundene Phasen - AminopropylGebundene Phasen - Aminopropyl
Als Normalphase Trennung nach Art und Anzahl funktioneller Gruppen
Als NP: H+-Donor oder H+-Akzeptor
Als RP: schwacher Anionenaustauscher
Fluoreszierende Flecken mit Zuckern
Carbonsäuren, Phenole, Nucleotide, Vitamine, Xanthin-Derivative, Zucker
Als Normalphase Trennung nach Art und Anzahl funktioneller Gruppen
Als NP: H+-Donor oder H+-Akzeptor
Als RP: schwacher Anionenaustauscher
Fluoreszierende Flecken mit Zuckern
Carbonsäuren, Phenole, Nucleotide, Vitamine, Xanthin-Derivative, Zucker

11
Gebundene Phasen - CyanoGebundene Phasen - Cyano
Mittelpolar
Als Normalphase wie desaktiviertes Kieselgel
Hochselektiv für polare Heterocyclen
Als Umkehrphase zur Ionenpaar-Trennung
von Säuren
Pharmazeutische Konservierungstoffe
Mittelpolar
Als Normalphase wie desaktiviertes Kieselgel
Hochselektiv für polare Heterocyclen
Als Umkehrphase zur Ionenpaar-Trennung
von Säuren
Pharmazeutische Konservierungstoffe

12
Umkehrphasen (RP 2, RP 8, RP 18)Umkehrphasen (RP 2, RP 8, RP 18)
Unpolare Stoffe (Lipide)• Fettsäuren, Carotenoide, SteroidePolare Stoffe• Basische und saure WirkstoffeStoffe, die auf aktiven Schichten labil sind• Vitamine, Antibiotika, CannabinoideInsektizide, Alkaloide, Barbiturate, Anti-oxidantien, Analgetika, Lebensmittelfarben
Unpolare Stoffe (Lipide)• Fettsäuren, Carotenoide, SteroidePolare Stoffe• Basische und saure WirkstoffeStoffe, die auf aktiven Schichten labil sind• Vitamine, Antibiotika, CannabinoideInsektizide, Alkaloide, Barbiturate, Anti-oxidantien, Analgetika, Lebensmittelfarben
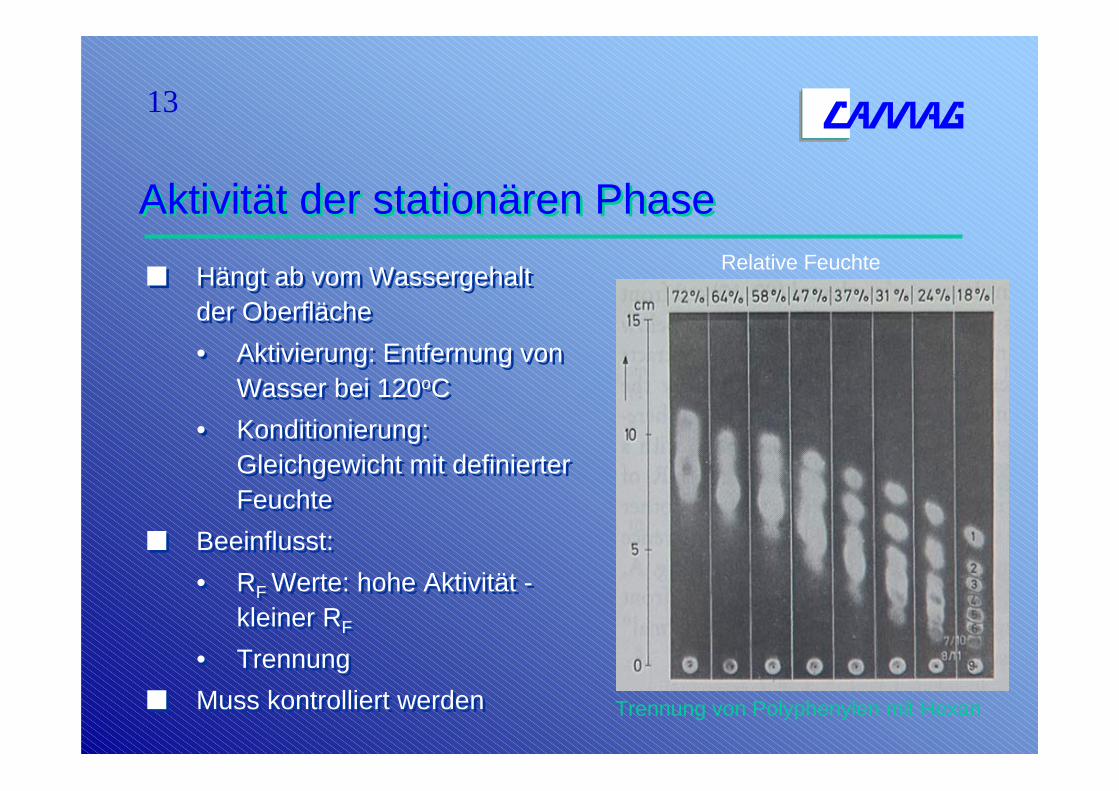
13
Trennung von Polyphenylen mit Hexan
Relative Feuchte
Aktivität der stationären PhaseAktivität der stationären Phase
Hängt ab vom Wassergehalt der Oberfläche• Aktivierung: Entfernung von
Wasser bei 120oC• Konditionierung:
Gleichgewicht mit definierter Feuchte
Beeinflusst: • RF Werte: hohe Aktivität -
kleiner RF
• TrennungMuss kontrolliert werden
Hängt ab vom Wassergehalt der Oberfläche• Aktivierung: Entfernung von
Wasser bei 120oC• Konditionierung:
Gleichgewicht mit definierter Feuchte
Beeinflusst: • RF Werte: hohe Aktivität -
kleiner RF
• TrennungMuss kontrolliert werden

14
Einstellen der relativen FeuchteEinstellen der relativen Feuchte
Masse %H2SO4
%relativeFeuchte
GesättigteSalzlösung
% relativeFeuchte
10 96 Pb(NO3)2 9820 88 KBr 8430 75 NaNO2 6640 56 NaHSO4
. H2O 5250 35 KF 3160 16 HCOOK 2170 3 ZnCl2. 1.5 H2O 10
Masse %H2SO4
%relativeFeuchte
GesättigteSalzlösung
% relativeFeuchte
10 96 Pb(NO3)2 9820 88 KBr 8430 75 NaNO2 6640 56 NaHSO4
. H2O 5250 35 KF 3160 16 HCOOK 2170 3 ZnCl2. 1.5 H2O 10

15
TLCTLC HPTLCHPTLCMittlere Korngrösse: 10 - 15 µm 5 - 7 µmKorngrössenverteilung: weit engSchichtdicke: 250 µm 100, 200 µmProbenzahl: max. 12 36 - 72Laufstrecke: 100 - 150 mm 30 - 50 mmEntwicklungsdauer: 30 - 200 min 3 - 20 minLM-Verbrauch: 50 ml 5 - 10 mlDetektionsgrenze, Abs.: 100 - 1000 ng 10 - 100 ng
Fluor.: 1 - 100 ng 0,1 - 10 ng
DC oder HPTLC ?DC oder HPTLC ?

16
HPTLC – DC - VergleichHPTLC – DC - Vergleich
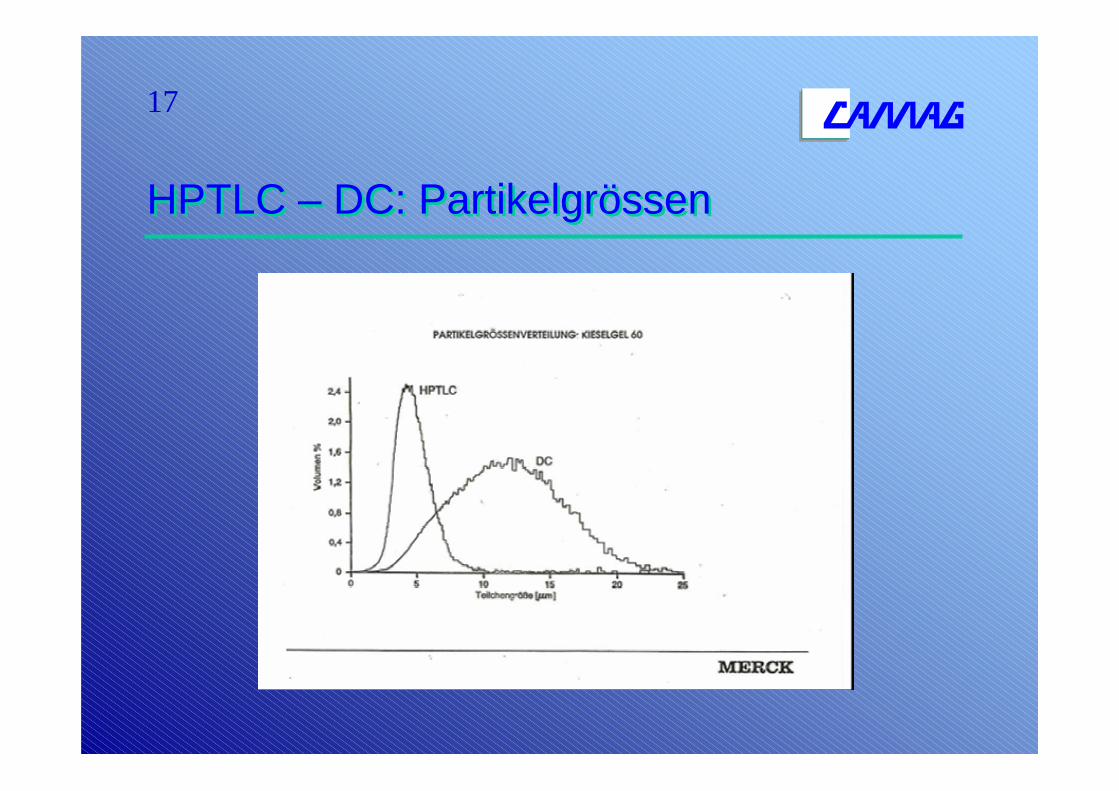
17
HPTLC – DC: PartikelgrössenHPTLC – DC: Partikelgrössen

18
Herstellung von Kieselgel und RPHerstellung von Kieselgel und RP

19
HPTLC-SorbenzienLichrospher - HPTLC klassisch
HPTLC-SorbenzienLichrospher - HPTLC klassisch

20
HPTLC - LichrospherHPTLC - Lichrospher

21

22

23
UTLCUTLC

24
HPTLC – DC: Optimale TrennstreckeHPTLC – DC: Optimale Trennstrecke

Fließmittelauswahl Methodenentwicklung in der DC

Anforderungen an das Fließmittel
Darf nicht mit der Probe reagieren
Ausreichende Selektivität und StärkeGeringe ToxizitätViskosität /sollte gering sein
Zusammen mit Oberflächenspannung bestimmt sie die Geschwindigkeit der Fließmittelfront und die Trennzeit
Dampfdruck/ nicht hoch nicht niedrigHoher Dampfdruck erhöht Risiken die mit Adsorption und Desorption des FM-Dampfes an bzw. von der Schicht.Selektive Verdampfung möglich
Genügende Reinheit und Stabilität

Anforderungen an das FließmittelReinheit
Trennung von 4 Polyhydroxybutyraten mit Chloroform
Stabilisator: Ethanol Amylen

0 20 40 60 80 100mm
0
20
40
60
80
100
120
140
160
180
200AE
A190
A200
A220
A240
A260
A280
A300
Analyse w:
Dateiname:0122.... Bahn:16Currenta SER-ANT, Chempark Dormagen, Geb. Da 5, 41538 Dormagen
3/FEB/2009
0 20 40 60 80 100mm
0
20
40
60
80
100
120
140
160
180
200AE
A190
A200
A220
A240
A260
A280
A300
Analyse w:
Dateiname:0160.... Bahn:6Currenta SER-ANT, Chempark Dormagen, Geb. Da 5, 41538 Dormagen
14/MAR/2009
Eindampfrückstand aus 4 mL t-BME (Fluka 20257 , LOT 1390414)Im AMD-Gradient, UV-Detektion
Eindampfrückstand aus 4 mL t-BME (SIAL 34875 , LOT 8323A)Im AMD-Gradient „PF-Absicherung“, UV-Detektion
Anforderungen an das FließmittelReinheit

Anforderungen an das FließmittelStabilität
• Gereinigte Lösemittel gut verschlossen und gegebenenfalls lichtgeschützt aufbewahren
• Fließmittel immer frisch bereiten Systeme wie n-Butanol/Eisessig/Wasser neigen zur Veresterung!Noch bedenklicher sind Verseifungen- es entstehen freie Säuren und Alkohol !

Anforderungen an das FließmittelVerteilungsisotherme
Cs
cm
H.-P. Frey, K. Zieloff: Qualitative u. quantitative Dünnschichchromatographie, VCH (1993)
Verteilungsisotherme sollte linear sein
Zu trennende Substanzen sollen sich im System Fließmittel-Schicht gleichmäßig verteilen, damit der Fleck rund bleibt
Zusatz von Säuren, Basen , solvatisierenden Lösemitteln unterdrücken überlagernde Dissoziations- oder Assoziationsgleichgewichte- Substanzen werden in einheitliche Molekülform überführt

Charakterisierung von FließmittelnLösemittelstärke - εo
• Dimensionslose Größe zwischen 0 und 1
• Charakterisiert das Vermögen des Lösemittels zum Stofftransport
auf der Schicht
• Adsorptionsenergie des Lösemittels pro Flächeneinheit unabhängig
von Adsorptionsaktivität
• Pentan/Hexan ist als = 0.00 definiert

Geiss, Fundamentals of TLC, Huethig 1987
Eluotrope Reihe auf Kieselgel
Pentane / Hexane 0.00 Ethyl acetate 0.48
Cyclohexane 0.04 Acetone 0.50
CCl4 0.11 Acetonitrile 0.60
Toluene 0.27 Dioxane 0.60
Isopropyl ether 0.28 Pyridine 0.70
Dichloromethane 0.30 Propanol 0.82
Chloroform 0.36 Methanol 0.95
Diethylether 0.43 Acetic acid >>1

LösemittelstärkeZunehmende Fliessmittelstärke: Einfluss auf die Trennung eines Testfarbstoffes. MP: Hexan mit 0, 5, 10, 15, 20, 30, 40, und 90% Ethylacetat (von links nach recht)
CAMAG Applikationslabor

Lösemittelstärke binärer Mischungen
Die Lösemittelstärke einerMischung ist nicht additivBei Zugabe eines “starken” Lösemittels zu einem schwachen, ändert sich die Lösemittelstärke erstdrastisch, dann immer weniger
F. Geiß, Die Parameter der Dünnschichtchromatographie, Fr.Vieweg+Sohn, Braunschweig 1972

Lösemittelstärke und Selektivität
Vergleichende Entwicklung mitLösemittel εo
A,B = 0.18 auf Aluminiumoxid bei 58% r.F.
a) 7.1% Ethylacetat in Hexan
b) 9.53% Aceton in Hexan
c) 29.9% Toluen in Hexan
H.-P. Frey, K. Zieloff: Qualitative u. quantitative Dünnschichchromatographie, VCH (1993)

Polaritätsindex (P`) nach Snyder
• Maß für die Fähigkeit eines Fließmittels mit verschiedenen Lösungsmitteln (z.B. Ethanol, Dioxan, Nitromethan) in Wechselwirkung zu treten
• Zur Berücksichtigung der Protonenakzeptor-Protonendonator- und Dipoleigenschaften wurden die Verteilungskoeffizienten der Fließmittel gelöst in den oben genannten Lösemitteln hinzugezogen
• Aus diesen Daten berechnete Snyder log (K“)-Werte, die sich additiv zum P`-Wert zusammenstzen
P`= log (K“) Ethanol + log (K“) Dioxan + log (K“) Niromethan

Polaritätsindex (P`) nach Snyder
P`= log (K“) Ethanol + log (K“) Dioxan + log (K“) Niromethan
Es lassen sich daraus die Wechselwirkungsanteile der Fließmittel mit den Lösemitteln Etahanol, Dioxan, Nitromethan berechnen.
Zum Beispiel:
χe = log (K“)Ethanol / P´
P´= χeP´ + χdP´ + + χnP´
(χe + χd + + χn) = 1 P` Polaritätsindex
Χe Anteil Fließmittelwechselwirkung mit Ethanol (Protonenakzeptoreigensch.)
Χd Anteil Fließmittelwechselwirkung mit Dioxan (Protonendonatoreigensch.)
Χn Anteil Fließmittelwechselwirkung mit Nitromethan (Dipoleigensch.)

Einteilung der Lösemittel nachSelektivität (Snyder)
I Aliphatische Ether, Trialkylamine, TrialkylphosphateII Aliphatische AlkoholeIII Pyridin, THF, DMSO, DMF, DiethylenglycolIV Benzylalkohol, Ethylenglycol, Essigsäure, FormamidV Dichlormethan, 1,2-DichlorethanVI Ketone , Ester, Dioxan, NitrileVII Aromatische (halogenierte) Kohlenwasserstoffe,
NitroverbindungenVIII Chloroform, Wasser, Fluoroalkohole, m- Kresol

Polaritätsindex, Selektivitätsparameter, Selektivitätsgruppe
Xe- gemessene Protonenakzeptorstärke
Xd- gemessene Protonendonatorstärke
Xn- gemessene Dipolwirkung
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

Lösemittelstärke und Selektivität
A: Hexan /15% Ethylacetat
B: Hexan /20% Aceton
C: Hexan /10% Isopropanol
D: Chloroform
Aceton und Ethylacetat- gleiche Selektivitätsgruppe IV
Isopropanol II, Chloroform VIII
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

Zusammenfassung
• Selektivität- entscheidender Faktor zur Trennung von Substanzen gleicher Polarität
• Fließmittelstärke Faktor zur Trennung von Substanzen unterschiedlicher Polarität
• Isoelutrope Fließmittel- gleiche Stärke unterschiedliche Selektivität-unterschiedlicheTrennungen

Vorbetrachtungen zur MethodenentwicklungDefinition der analytischen Zielstellung
• Soll die Methode zur Routine oder für eine Entwicklung erarbeitet werden?
• Soll die Methode zur Untersuchung von Rohmaterial, Extrakten, Endprodukten oder Mischproben dienen?
• Möchten Sie die Identität einer Komponente oder der Komponente in einer Mischung analysieren?
• Möchten Sie quantifizieren?
• Welche Matrices erwarten Sie, welche NWG?
• Können Sie bereits auf eine bestehende Methode zurückgreifen?
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

Vorbetrachtungen zur MethodenentwicklungAspekte
• Eine qualitative Methode dient als Fingerprint und sollte so viel wie möglich Komponenten auftrennen
• Steht Ihnen exklusives Referenzmaterial aus kontrolliertem Anbauzur Verfügung ist eine Identitätsprüfung weniger schwierig
• Ist Ihre Leitdroge Bestandteil einer Mischung kann ein komplexerFingerprint der Mischung entwickelt werden. Die spezifischen Banden der Leitdroge müssen herausgearbeitet werden, die übrigen werden als Matrix angesehen.
• Quantitative Methode setzt Basislinientrennung voraus. Das Chromatogramm kann so einfach wie möglich gestaltet werden. Extensive Probenvorbereitung kann erforderlich sein.
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

Vorbetrachtungen zur MethodenentwicklungAusgangspunkt
• Auftragen der Proben (Lösemittel, Probevolumen, Auftragegeschwindigkeit, Größe und Schärfe der Auftragezone)
• Modifikation der stationären Phase (Aktivität, Imprägnierung)
• Chromatographiekammer (Typ, Sättigungsgrad)
• Entwicklungsmodus (Einfach, Mehrfach, Gradient)
• Detektion und Derivatisierung (UV-Absorption, Fluoreszenz, Reagentien, Konditionierrung)
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

Was ist bekannt ?
• Anzahl der Komponenten• Chemische Struktur• Molekulargewicht• Löslichkeit• Matrix• Konzentrationsbereich• pKa ; UV Spektrum, etc. • Nichts
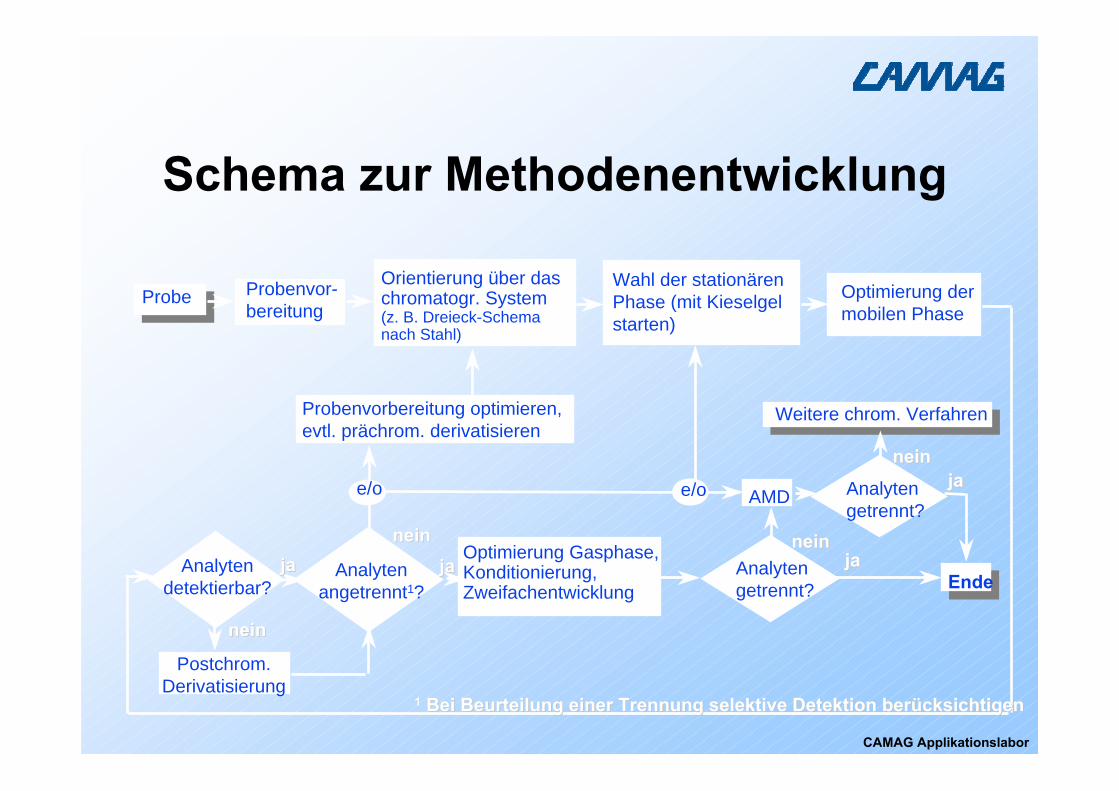
Probe Probenvor-bereitung
Analytengetrennt?
Orientierung über das chromatogr. System (z. B. Dreieck-Schema nach Stahl)
Wahl der stationären Phase (mit Kieselgel starten)
Analytenangetrennt1?
jajaneinnein
Optimierung Gasphase,Konditionierung, Zweifachentwicklung
Optimierung der mobilen Phase
jajaneinnein
AMD
Probenvorbereitung optimieren, evtl. prächrom. derivatisieren
Analytengetrennt?
jajaneinnein
Weitere chrom. Verfahren
Postchrom. Derivatisierung
jaja
neinnein
e/o e/o
Ende
11 Bei Beurteilung einer Trennung selektive Detektion berücksichtiBei Beurteilung einer Trennung selektive Detektion berücksichtigen gen
Analytendetektierbar?
Schema zur Methodenentwicklung
CAMAG Applikationslabor

Auswahl des FließmittelsPraktische Lösung
• Standardsysteme
• Fleckentest
• Prisma-Modell
• CAMAG - Optimierungsschema

Auswahl des FließmittelsStandardsysteme
Lösemittelgemische:1.Toluen/Ethylacetat 95+5 für unpolare Komponenten2. Chloroform/Methanol/Wasser
70+30+4 für Saponine und Lignane3. Ethylacetat/Essigsre/Ameisensre/Wasser
100+11+11+27 für Flavonoide4. Acetonitril/Wasser/Ameisensäure
30+8+2 für Aminosäuren5. Butanol/Essigsäure/Wasser
7+1+2 für polare Komponenten
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

1.Toluen/Ethylacetat 95+5 für unpolare Komponenten
2. Chloroform/Methanol/Wasser 70+30+4 für Saponine und Lignane
3.Ethylacetat/Essigsre/Ameisensre/Wasser 100+11+11+27 für Flavonoide
4. Acetonitril/Wasser/Ameisensäure 30+8+2 für Aminosäuren
5. Butanol/Essigsäure/Wasser 7+1+2 für polare Komponenten
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

FleckentestVersuch und Irrtum
• Lösen der Probe in unpolarem Lösemittel
• Applikation auf die DC-Platte
• Applikation ~ 20 µL unterschiedlicher Lösemittel
auf die Probenflecken
• Es erfolgt eine Zirkularentwicklung
• Trocknen der Lösemittel

Fleckentest
Goldenseal CAMAG Applikationslabor

Fleckentest
CAMAG Applikationslabor

Fleckentest
Lavender CAMAG Applikationslabor

Vor- und Nachteile Fleckentest
• Schnell und automatisiert• Lösmittelanzahl und Typ sind flexibel• Information über Lösemittelstärke und
Selektivität
• Ungesättigtes offenes System• Unsystematisch

VororientierungDreieck-Schema nach Stahl

Auswahl stationäre Phase
• Beginnen mit Kieselgel• Für spezielle Trennprobleme spezielle Schichten:
Bsp.:Eugenol, Methyleugenol, Isoeugenol
Kieselgel Amino- Cyano
MP: Toluen, D: Schwefelsäurereagenz,
1 Eugenol,
2 Methyleugenol,
3 Isoeugenol
OH
OMe
OMe
OMe
OH
OMe
1
2
3Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

Reine Fliessmittel
Herabsetzen / Erhöhen der Fliessmittelstärkem. Wasser oder stärkerenLSM gleiche Selektivitätsgruppe
Mischungen,Modifier-Zugabe(Säuren, Basen)
Umgebung der besten Mischung

Auswahl mobile Phase Stufe 1Beispiel Curcuma
A: TBME
B: Methanol
C: THF
D: Ethylacetat
E: DCM
F: Toluen
G: Chloroform
Gruppe A: die gewünschten Komponenten sind getrennt, die Rf-Werte sind im passenden Bereich, das Trennproblem ist gelöstGruppe B: Sie erhalten Rf-Werte über 0,8Gruppe C: Sie erhalten Rf-Werte kleiner 0,2
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

Auswahl mobile Phase Stufe 2Beispiel Curcuma
Lösemittel der Gruppe B werden mit n-Hexan „verdünnt“. Je höher die Lösemittelstärke, desto höher muss der Anteil an n-Hexan ausfallen. Sollte die Probe komplett in die Front laufen, sollte ein Verhältnis von mindestens 1:4 eingesetzt werden.
A-D 1:4 E und F 1:1
TBME Methanol THF Ethylacetat TBME Ethylacetat
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

Auswahl mobile Phase Stufe 2Beispiel Curcuma
Lösemitteln der Gruppe C wird ein polarer Modifier zugesetzt (z.B. Essigsäure, Ameisensäure oder Basen wie z.B. Diethylamin, Ammoniak). Ein erster guter Versuch sind ca. 10%. Auch der Zusatz von Wasser ist möglich, wenn die Mischbarkeit gegeben ist.
DCM/Essigsre Toluol/Essigsre Chloroform/Essigsre
9+1 8+2 9+1
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag

Auswahl mobile Phase Stufe 3Beispiel Curcuma
Kombination von 2 Lösemitteln aus den Gruppen B und C zunächst als Mischung 1:1, dann im Umkreis durch „pobieren“ beste Mischung findenEinstellung der Lösemittelstärke mit Modifiern um scharfe Trennzonen zu bekommen. Bei Fleckentailing ist z.B. eine Wasserzugabe von 0,5-1% nützlich. Modifizierung der Trennbedingungen (Kammersättigung, Feuchte)
Eike Reich, Anne Schibli „High-Performance Thin-Layer Chromatography for theAnalysis of Medical Plants“, Thieme Verlag
A: Isopropanol/Hexan/Wasser 1:6:0,2
B:TBME/Hexan/Ameisensre 5:5:0,1
C:Toluol/Methanol 4,5:0,5
D: Chloroform/Methanol/Ameisensäure 10:0,1:0,1

Feinabstimmung der Methode
Es wurde ein Fließmittel gefunden.
Variation des Auftragevolumens (qualitativ Maximum an Zonen, quantitativ linearer Arbeitsbereich)
Derivatisierung/Visualisierung ( spezifische und unspezifische Reagenzien, Optimierung der Derivatisierung für gute Reproduzierbarkeit)
Robustheit der Methode
Parameter der densitometrischen Messung (Wellenlänge, Spaltdimension, Geschwindigkeit)

Nun kann die Methode
validiert werden

Detektion/Derivatisierung in der DC

Detektion/DerivatisierungLokalisierung und Identifizierung
Lokalisierung (Ermittlung der Lage im Chromatogramm)
Physikalische VerfahrenChemische ReaktionenBiologische Reaktionen
Identifizierung (Verwendung von gruppenspezifischen Reagenzien)
Chemisches VerhaltenPhysikalische, chemische, biologische Eigenschaften

Physikalische Detektion

Chemische ReaktionenDerivatisierung
prächromatographisch
postchromatographisch
bei der Proben-vorbereitung am Start
chromatogr.Entwicklung
Chromatogramm
visuelle undphotom.
Auswertung

Prächromatographische in-situ-DerivatisierungChemische Reaktionen
Reagenzapplikation punkt- oderstrichförmig
Applikation Untersuchungslösung(in feuchte Reagenzzone)
eventuelle Wärmebehandlung(Startzone abdecken)
Chromatographische Entwicklung
Für Substanzen mit ähnlichen chromatographischen, aber unterschiedlichen chemischen Eigenschaften
Zur Erhöhung der Stabilität
Umsetzung flüchtiger Substanzen in Derivate
Erhöhung der Nachweisempfindlichkeit durch z.B.Erzeugung einer Fluoreszenz
Nachteil:Analyt wird mit Derivatisierungsreagenz verunreinigtund muss zusätzlich abgetrennt werden
Molekül wird größer und durch das Reagenz in der Struktur ähnlicher- dadurch schlechtere Trennung
Beispiel: DansylierungenDansylhydrazin für CarbonylgruppenDansylchlorid für NH und OH-GruppenDansylsemipiperazid für FettsäurenDansylazirin selektiv für SH-Gruppen

Prächromatographische in-situ-DerivatisierungChemische Reaktionen
Beispiel: DansylierungenDansylhydrazin für CarbonylgruppenDansylchlorid für NH und OH-GruppenDansylsemipiperazid für FettsäurenDansylazirin selektiv für SH-Gruppen
Gerad- und ungeradzahlige Fettsäuren nach in-situ-Derivatisierung mit Dansylsemicadaverid
1: C-24 bis C-16 2: C-24 bis C-6 3: C-20 bis C-12 4: C-20 bis C-11
5: C-19 bis C-11 6: C24 bis C-16

Derivatisierungstechniken
Tauchen
Chemische ReaktionenPostchromatographische Derivatisierung
Entwicklung des Chromatogramms
Erwärmen
Trocknen
SprühenBedampfen

Trocknen der Platte• Vertikale Lage der Platte, um Fließen des Restlösemittels
und damit Fleckendiffusion zu verhindern (so lange wie Feuchte offensichtlich).

Kritische Trocknungim Luftstrom
Anethol in Anis- und FenchelölMP: Toluen/Ethylacetat 95+5 D: UV 254
Die etherischen Öle werden im Luftstrom von ihren Plätzen gerissen!
E.Reich,A. Schibli „HPTLC for the Analysis of Medicinal Plants“, Thieme 2006

DerivatisierungstechnikenThermochemische Reaktionen (Erwärmen)
Aminophase für die Trennung von ZuckernErhitzen der Platte 3-5 min auf 150°C

Bedampfen
• Doppeltrogkammer oder Konditionierkammer
• Nutzung von Joddämpfen , Ammoniak, Brom, Chlor,

Chemische ReaktionenPostchromatographische Derivatisierung
• Erzeugung einer Absorption im sichtbaren-oder UV-Bereich oder einer Fluoreszenz
• Verwendung von gruppenspezifischen Reagenzien zum stoffspezifischen Nachweis
• Steigerung der Selektivität• Steigerung der Nachweisempfindlichkeit

DerivatisierungstechnikenSprühen
Spraydosen
Laborsprüher
Sprühautomat (DESAGA)

DerivatisierungstechnikenSprühen
VorteileEinfach und schnellKeine besondere AusrüstungKleine Volumina
NachteileErzeugung von SprühnebelnSchwierige Gewährleistung der HomogenitätUndefiniertes VolumenRoutine zum exakten Sprühen muss erst erworben werdenEs könne partielle Feuchteinseln entstehen

DerivatisierungstechnikenTauchen
• Gleichmäßige Belegung mit Reagenz
• Reproduzierbare Ergebnisse durch standardisierte Bedingungen
• Keine Sprühnebel• Reinigen der Rückseite• Vertikale Lagerung um
Bandenverbreiterung auszuschließen
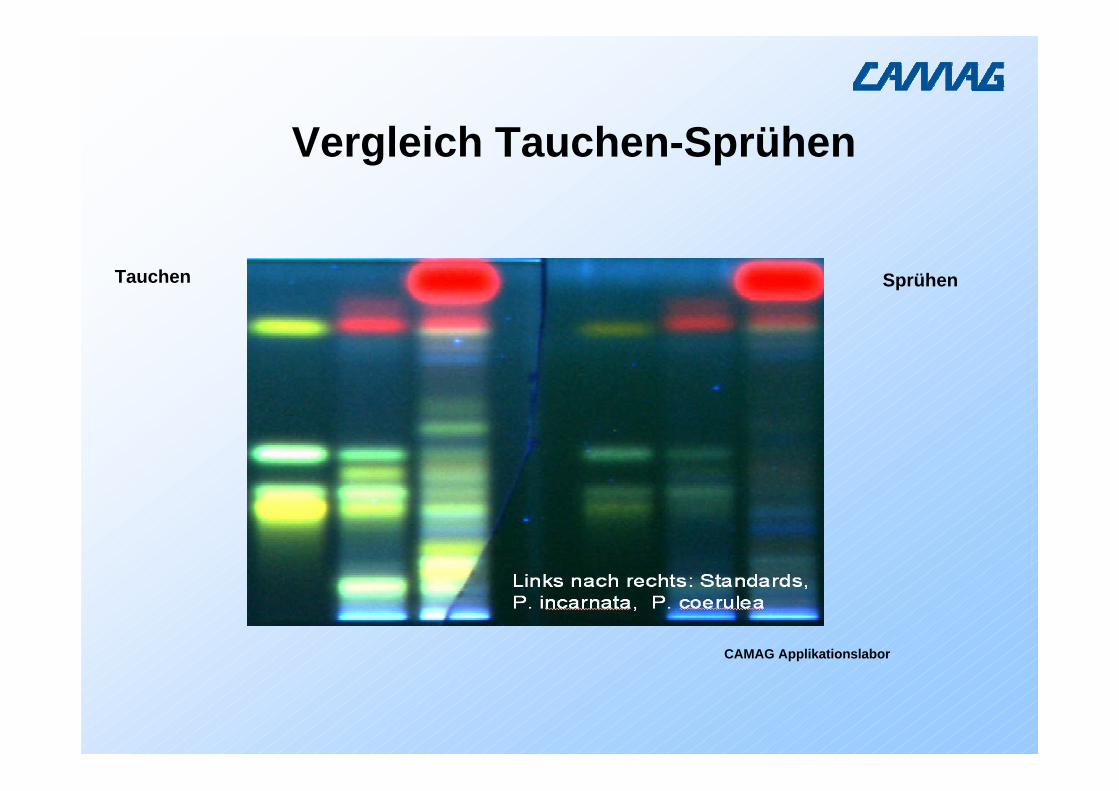
Vergleich Tauchen-Sprühen
Tauchen Sprühen
CAMAG Applikationslabor

Vergleich Tauchen-Sprühen
Diplomarbeit Katherine Gessler, Uni Basel

Vergleich Sprühen-Tauchen
Derivatisierung von Capsaicinmit Gibbs-Reagenz(Dichlorchinon chlorimidgefolgt von Base)
Sprühreagenz: 1%-ige Lsg. Gibbs-Reagen; 2%-ige Natriumcarbonat- Lösungen haben hohe Viskosität und lassen sich somit schlecht dosieren
Tauchreagenz: 0,025% GibbsReagenz und basische Bedampfung mit z.B. Ammoniak

ReagenzienUniversalreagenzien
Ansprühen mit Wasser macht lipophileSubstanzen kurzzeitig als helle Flecken aufgrauem Grund

ReagenzienUniversalreagenzien
5-10 min 120-160°CVersch. Gefärbte Zonen auf hellem Grund
Vanillin-Phosphorsäure
1-20 min 95-140°CFluoreszierende Zonen bei 366nm,oderverkohlte Zone
Schwefelsäure-Reagenz
10 min 150-180°CBlaue Flecken auf gelbem Grund
3% Phosphormolybdänsäurein Propan-2-ol
90-125°C 1-5 minUntersch. gef. Zonen, häufig bei 365nm Fluoreszenz
Anisaldehyd-Schwefelsäure-Reagenz

Anisaldehyd-SchwefelsäureWelche Rezeptur soll ich nehmen?
Kraus,Koch,Hoffstetter,Kuhn
100°C, bis zur Farbentw.
85ml10ml5ml0,5ml
85ml
Methanol
100-105°C
100°C, 2-5 min
1-15 min, 90-125°C
Bedingungen
Stahl100ml2ml1ml
Reich,Schibli10ml5ml0,5ml
Jork,Funk,FischerWimmer
97 ml2ml1ml
LiteraturEssigsäureSchwefelsäureAnisaldehyd

Anisaldehyd-Schwefelsäure-Reagenz
10 ml Schwefelsäure werden vorsichtig in eine Mischung aus
170 ml eisgekühltem Methanol und 20 ml Eisessig eingerührt. Zu dieser Mischung wird 1 ml Anisaldehyd zugefügt.
Das Reagenz ist farblos und sollte im Kühlschrank aufbewahrt werden. Sollte sich die Farbe ändern, muss es verworfen werden!
Die Platte wird in das Reagenz mit einer Verweilzeit von 1 s getaucht. Danach wird 2-5 min auf 100°C erhitzt.
Auswertung im Weißlicht und bei UV 366 nm

Ergebnis in Abhängigkeit vom zugeführten Reagenz
Diplomarbeit Katherine Gessler, Uni Basel

Stoffspezifische ReagenzienZucker und Glycoside
Schicht: HPTLC Kieselgel 60 (rel. Feuchte über 62% SchwefelsäureFließmittel: Chloroform: Essigsäure: Wasser 30+35+5 Derivatisierung: Diphenylamin, Anilin und Orthophosphorsäure gelöst in Aceton

Beeinflussung der Nachweisempfindlichkeitzugeführtes Reagenz
Funk, Fischer, Wimmer ,Dünnschichtchromatographie Bd.1

Nachbehandlung
Trocknen
Abzugvertikale Lagerung
ev. Ventilator oder FönSubstanzeigenschaften (Flüchtigkeit, Oxidationsempfindlichkeit
beachten)

Einwirkung von Wärme
TrockenschrankHeizplatte
Nicht auf das Gitternetz legen
Metallblock verwendenoder frei schwebend
Visuelle Verfolgunghomogene Umsetzung

Hinweise zur Benutzung von Wärmeplatten
• Legen Sie die Platte auf die noch kalte Heizplatte und heizen Sie Platte und Gerät gleichzeitig auf.
• Wollen Sie mehrere Platten derivatisieren kühlen Sie auf etwa 60°C ab.
• Haben Sie hohen Plattendurchsatz, dann legen Sie die Platten auf ca. 1mm dicke seitlich angebrachte Abstandhalter (das Luftpolster verhindert das Aufwölben der Platte. Wählen Sie eine 10°C höhere Temperatur um Wärmeverlust auszugleichen.

• Temperatur• Konzentration und Zusammensetzung
der Lösung• Stabilität der Lösung

DerivatisierungTemperatur
Flavonoids of St. John‘s Wort
1: 3 min Kaltluftstrom 2: 5 min 105°C3: 30 min 105°C
1 32
CAMAG Applikationslabor
366 nm, NP, NP/PEG
366 nm, NP, NP/PEG nach 30 min
366 nm, NP, NP/PEG

DerivatisierungTemperatur
Diplomarbeit Katherine Gessler, Uni Basel

Konzentration und Zusammensetzung der Lösung
Diplomarbeit Katherine Gessler, Uni Basel

DerivatisierungAlter des Reagenz
Diplomarbeit Katherine Gessler, Uni Basel

Lagerung der derivatisierten Platte
Diplomarbeit Katherine Gessler, Uni Basel

Einfluss des Plattenuntergrundes auf Derivatisierung

Reagenz im Sorbens
Etherische ÖleMolybdatophosphorsäure
DihydroxybenzoleSilbernitrat
LipideAmmoniumsulfat
PAHCoffein
Nachgewiesene SubstanzenReagenz
Reagenz wird in die Sorbensschicht eingearbeitet

Reagenz im FließmittelVoraussetzung: -gleichmäßige Verteilung auf der Schicht-Reagenz muss mit der Fließmittelfront laufen
Beispiele: Ninhydrin bei AminosäurenFluorescamin bei biogenen Aminen
Ninhydrin in der mobilen Phase vermindert das Rauschen um den Faktor 2. Die selektive Derivatisierungist bestens geeignet, da die nicht derivatisierte Matrix die Detektion nicht beeinflusst
Identifizierung und Quantifizierung von Aminosäuren und Peptiden (CBS 101,Hamon,Diancourt,Roy,Sbaffo-Poasevara)

Probleme beim Derivatisieren
Verdünnen Reagenz, Temperatur herabsetzen, Platte vorwaschen
Hohe Untergrundfärbung
Polarität Lösemittel ändern ( KG-Polarität erhöhen, RP-Polarität senken)
Benetzungsprobleme
Polarität Lösemittel ändern ( KG-Polarität senken, RP-Polarität erhöhen)
Fleckenelution bzw. Vergrößerung
Standardisierte Bedingungen einhalten
Schlechte Reproduzierbarkeit
ReaktionProblem

Wirkungsbezogene Detektiondirektes Biomonitoring
Direkte Messung der physiologischen Wirkung – Referenzsubstanzen müssen nicht vorhanden sein
-Enzymatische Reaktionen
Acetylcholinesterase- Hemmung durch Phosphorsäureester und Carbamate
Bestimmung von Schwermetallen mit Urease
-Antibiotische Wirkung
mit Bacillus subtilis
-Toxische Wirkung
mit Vibrio fisheriMarines BakteriumKontinuierlilches BiolumineszenzsignalNicht pathogenSeit 1979 für ökotoxische Tests im Einsatz(DIN 38412-431)

Wie funktioniert das ?
Züchtung der Bakterien
Tauchen der DC-Platte in die Bakteriensuspension
Beseitigung des Überschusses

Ergebnis
200
ng
150
ng
100
ng
A B C D E
100 μg
75 μ
g
50 μ
g
25 μ
g
10 μ
g
Biolumineszenz-löschung
Biolumineszenz-stimulation
A-E: Probenverdünnungennp: Nitrophenol (toxische Referenz)
np

BeispielePatulin in Apfelsaft

Das CAMAG TLC-MS Interface
Laser on/off
Stempel rauf/runter
Schalter Reinigung
Sicherheitschutzschild
on/off Schaltventil zu MS

Stempel

Prinzip TLC-MS Kopplung
CAMAG TLC-MS Interface
HPLC Pump fördert Lösungsmittel (MeOH)Flow rate: 100µl/min
MS-ESI (Electrospray Ionization Interface)

Funktionsprinzip TLC-MS Interface
Positionierung Platte mit Laserkreuz auf gewünschten Spot/Zone
Stempel senkt sich mit 4 bar ab und Extraktion beginnt

DC/HPTLC Platte mit Spots/Zonen
Stempel senkt sich mit kontrolliertem Druck
HPLC Pumpe fördertLösungsmittel Massenspektometer
analysiert Probe
Funktion Stempel und Extraktion
MS2
