development of new density functionals and new methods for
TRANSCRIPT

Development of new density functionals and new methods for
analysis of convergence of ab initio molecular dynamics simulations
• DOE Early Career DE-SC0003871 DOE: DE-FG02-09ER16052
Marivi Fernandez-Serra Michelle Fritz Jose M. Soler

INTRODUCTION SIMULATING WATER
Improving ab initio molecular
dynamics (AIMD) for liquid water
(liquid structure, diffusion coefficient,
eqm. density, etc.)
• Correction of self-interaction errors
- Include exact exchange (PBE0)
+
• Inclusion of vdW interactions (TS-
vdW)
+
• Inclusion of Nuclear Quantum
Effects (NQE)
- Path Integral Molecular
Dynamics (PIMD)
- Temperature increase of 30 K
Results shown are from DiStasio Jr., R. A. et al. The
J. Chem. Phys. 141, 084502 (2014)

Mean absolute deviation (MAD) from Quantum Monte Carlo (QMC)
calculations of liquid water configurations
INTRODUCTION MB-POL
Number of configurations
MB-pol performs better
than all other models
compared.
Medders, G. R. et al. J. Chem. Phys. 143, 104102 (2015).

MB-pol1,2 is the most accurate potential for water available today.
•Incorporates the Partridge and Schwenke water monomer potential3
to describe 1-body energies.
•Fitted to 2- and 3-body energies calculated with CCSD(T).
Many-body expansion of potential energy for N interacting molecules:
•Many-body effects included with polarization model employed by the TTM4-F potential.4
INTRODUCTION MB-POL
2-body energies 3-body energies
1Babin, V., Leforestier, C., and Paesani, F. J. Chem. Theory Comput. 9, 5395 (2013).2Babin, V., Medders, G. R., and Paesani, F. J. Chem. Theory Comput., 10, 1599 (2014).3Partridge, H. and Schwenke, D. W. J. Chem. Phys. 106, 4618 (1997).4Burnham, C. J. et al. J. Chem. Phys. 135, 144502 (2011).
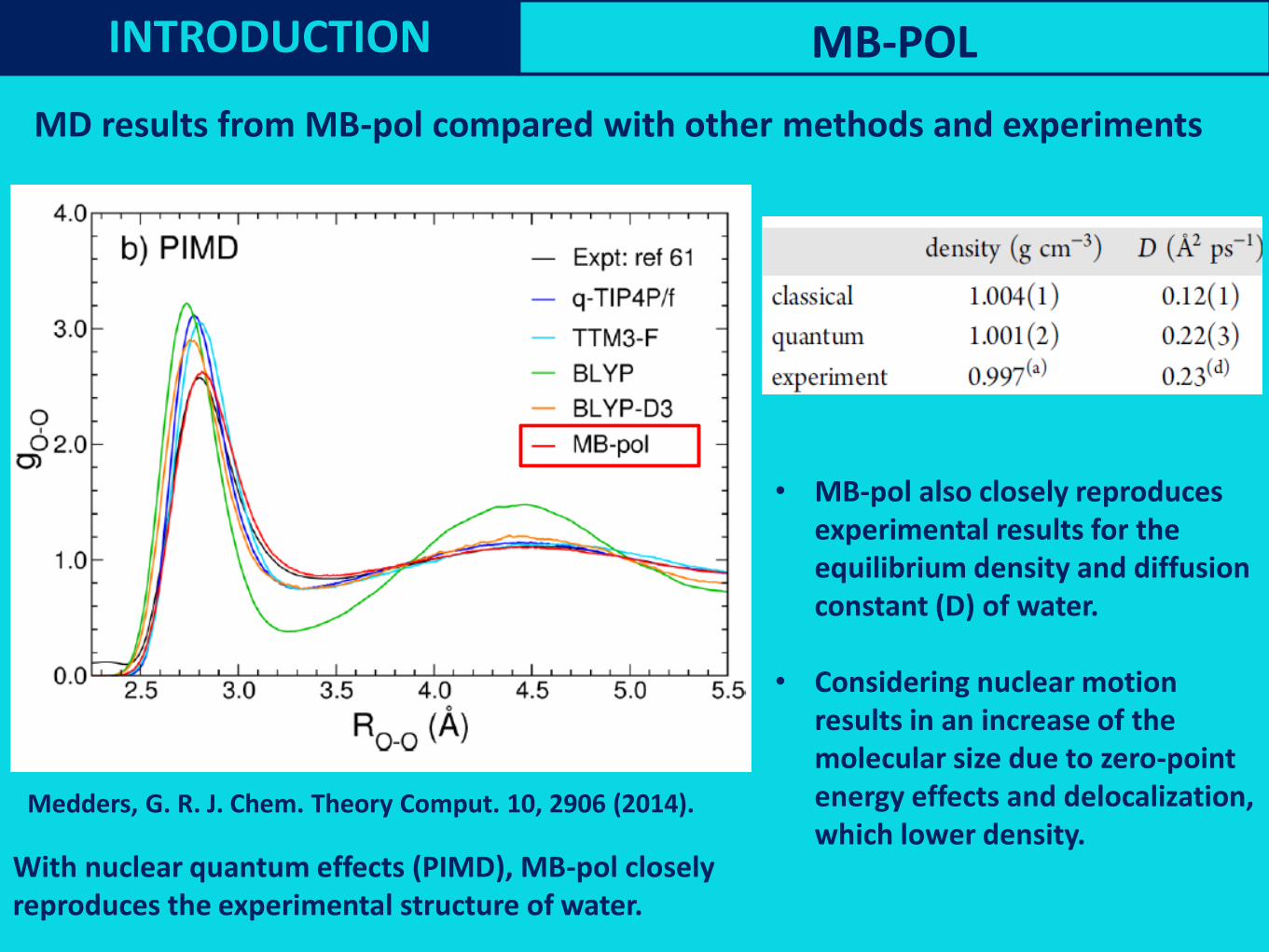
MD results from MB-pol compared with other methods and experiments
INTRODUCTION MB-POL
Medders, G. R. J. Chem. Theory Comput. 10, 2906 (2014).
With nuclear quantum effects (PIMD), MB-pol closely reproduces the experimental structure of water.
• MB-pol also closely reproduces experimental results for theequilibrium density and diffusion constant (D) of water.
• Considering nuclear motion results in an increase of the molecular size due to zero-point energy effects and delocalization, which lower density.

DPPS METHODOLOGY
Motivation: Optimization of a density functional for water
• Hybrid functionals are not yet as accurate as MB-pol and theircomputational cost is still too large.
• We take the MB-pol approach to fit a GGA+vdW-DF functional,using a new method: Data Projection on Parameter Space.
• In practice, there is not one ab initio DFT, but many.
• Trial and error: does a functional fit the data?
• DPPS: what functional fits best the data?
Data Projection onto Parameter Space (DPPS)

II. DATA PROJECTION ONTO PARAMETER SPACE (DPPS)
Optimization of an exchange-correlation density functional for water, Michelle Fritz, Marivi Fernandez-Serra and Jose M. Soler, J. Chem. Phys. 144, 224101 (2016)

Optimization of GGA Exchange-Correlation (XC)
• Change in the XC energy is needed to reproduce reference energies.
• Express density and energy density in terms of 𝒌𝑭 = 𝟑𝝅𝝆 𝟏 𝟑 and 𝒌𝑭 = 𝛁𝝆𝝆 .
DPPS METHODOLOGY
Non –XC energy
> @ > @ > @ > @ nn non XC n XC n XC n refE E E E EU U U U� � � '
Set of electron densities at a given, initial functional
Initial XC ΔEXC to reproduce reference energy
Reference energies
> @ 3( ) ( ( ) , ( ) )
( , ) ( , )
GGAXC XC
GGAF G XC F G F G
E d
k k k k dk dk
U U H U U
U H
' ' �
'
³³ ³
r r r r
Δεxc to reproduce reference energy
Electron density in kF, kG
G

Density histogram for a water monomer
Electron density in real space (e-/Bohr3)
DPPS METHODOLOGY: GGA XC
Electron density in kF ,kG.
Electron density in kF, kG:
xy
z
H2O ( )U r
3 2 1 3( , ) ( ) ( (3 ( )) ) ( ( ) ( ))F G F Gk k d k kU U G S U G U U � � �³ r r r r r
( )( )GkUU�
rr
12 3(3 ( )) ,Fk S U r

• Set interpolation points kFα, kGβ.
• The reference exchange energies arethe projections of the known densitiesonto the set of parameters:
DPPS METHODOLOGY
( , )XC F Gk kDE D EH H
XC energy density
n nref XCE � xH U
Set of parameters
^ `DEH H ^ `n nDEU U
Electron densities in parameter space
Optimization of GGA Exchange-Correlation (XC)
𝑘
..
.
..
𝜀(𝑘
,𝑘
.
..
..
𝜀
..

DPPS METHODOLOGY
( , )XC F Gk kDE D EH H
XC energy density
n nref XCE � xH U
Set of parameters
^ `DEH H ^ `n nDEU U
Electron densities in parameter space
( , )XC F Gk kHDPPS
nU
nref XCE �
Optimization of GGA Exchange-Correlation (XC)
• Set interpolation points kFα, kGβ.
• The reference exchange energies arethe projections of the known densitiesonto the set of parameters:

Optimization of GGA Exchange
• Change in the exchange energy is needed to reproduce reference energies.
• Exchange energy density is the porduct of the LDA exchange energy densityand a functional of the reduced density gradient 𝒔 ≡ 𝒌𝑮 𝟐𝒌𝑭.
• Express density in terms of s.
DPPS METHODOLOGY
Initial exchange ΔEX to reproduce reference energy
Reference energies
( , ) ( ) ( )LDAX F G X F Xk k k F sH H
Exchange enhancement factor
> @ ( ) ( )LDAX X XE s F s dsU U H' '³
ΔFX to reproduce reference energyElectron density in s
> @ > @ > @ > @ nn non X n X n X n refE E E E EU U U U� � � '
Non –exchange energy

Density histogram for a water monomer
( ) ( , ) ( 2 )F G F G G Fs dk dk k k s k kU U G �³ ³Electron density in s:
Electron density in kF ,kG.
DPPS GGA EXCHANGE OPTIMIZATION FOR WATER
s=0.25
0.51.0
2.0
Reduced density gradient:
Electron density in s.
( , )F Gk kU ( )sU
> @ ( ) ( )LDAX X XE s F s dsU U H' '³

• Set interpolation points sα.
• The reference exchange energies arethe projections of 𝛈 = 𝝆𝜺𝑿𝑳𝑫𝑨 onto theset of parameters:
DPPS METHODOLOGY
( )XF F sD D
)𝑭 𝑿(𝒔
..
.
.
.
s
n nref XE � xF K
Set of parameters
^ `XF D F ^ `n nDK K
Known data (product of 𝝆 and 𝜺𝑿𝑳𝑫𝑨) in parameter space
Optimization of GGA Exchange
𝑭𝑿𝜶
𝜂 = 𝜌𝜀

Theoretical constraints: Bayesian
� �2
2
1
0
( | ) ( | ) ( )
( | ) exp2
1( ) exp ( ) ( , ') ( ') '
2
( ) ( ) ( ) , ( , ') ( ) ( ')
{ PBE, rev
n nref GGA
nn
x x
k k k kx x x x x
k
P theory facts N P facts theory P theory
E EP facts theory
E
P theory F s C s s F s ds ds
F s F s F s C s s F s F s
k
G G
G G G
�
½�° ° �® ¾'° °¯ ¿
½ �® ¾¯ ¿
�
¦
³³¦
PBE, PBEsol, BLYP, PW86, ... }
Probability that a theory is true given that some facts are true
Probability that some facts are true given that a theory is true
Probability that a theory is true
Error bars in the energies
Avg. of exchange functionals
DPPS METHODOLOGY: BAYESIAN CONSTRAINTS

Theoretical constraints: Bayesian DPPS METHODOLOGY: BAYESIAN CONSTRAINTS
Ab initio GGA exchange enhancement factors used as prior information in Bayesian optimizatons.

Optimization of GGA Exchange for Water
• Reference energies 𝑬𝒓𝒆𝒇 𝑿𝒏 are MB-pol energies of water
monomers in 300 geometries.+
• Input densities from initial functional: vdW-DF-cx1
vdW-DF XC general form:
• Apply DPPS to find optimal form of 𝑭𝑿(𝒔).
DPPS METHODOLOGY
> @ > @ > @ > @0 nlXC X C CE E E EU U U U � �
> @ 3 31 ' ( ) ( , ') ( ')2
nlCE d rd rU U I U ³³ r r r r
1K. Berland and P. Hyldgaard, Phys. Rev. B 89, 035412 (2014).

The CUSTOM Fx improves 1-body energies from the initialfunctional vdW-DF-cx.
DPPS EXCHANGE OPTIMIZATION FOR WATER

DPPS EXCHANGE OPTIMIZATION FOR WATER

Density histogram for a water monomer
• As O-H bonds shorten, density moves towards oxygen and density gradient increases.
• Most changes in ρ(kF ,kG) needed are along a constant value of s.
DPPS GGA EXCHANGE OPTIMIZATION FOR WATER
xy
z
Change in H2O density (e-/Bohr3)
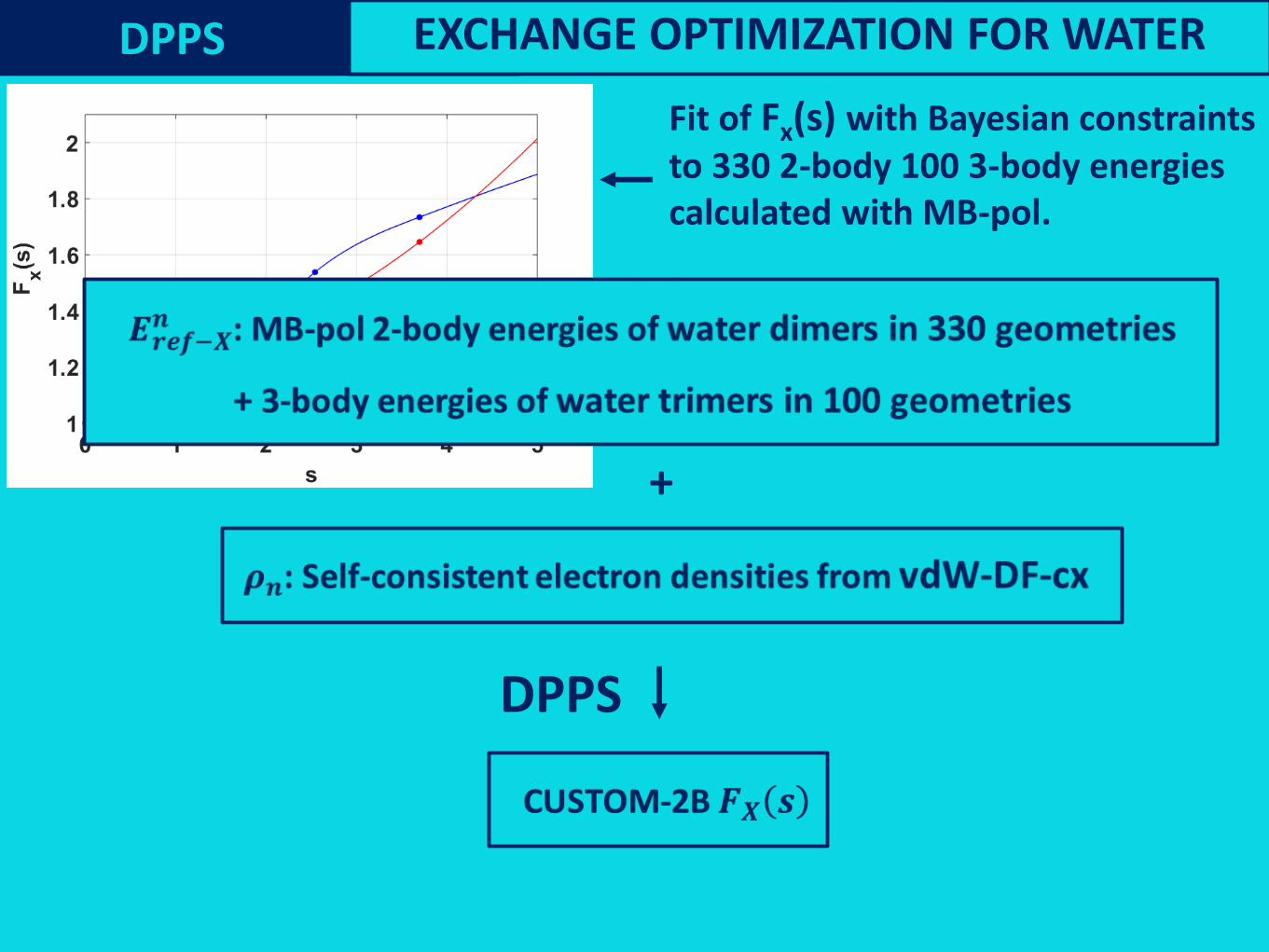
Fit of Fx(s) with Bayesian constraintsto 330 2-body 100 3-body energies calculated with MB-pol.
DPPS EXCHANGE OPTIMIZATION FOR WATER
+
DPPS

Density histogram for a water monomer
s=0.25
0.5
1.0
2.0
s=0.25
0.5
1.0
2.0
DPPS GGA EXCHANGE OPTIMIZATION FOR WATER
2-body interaction energy:
H-bonded configuration non-H-bonded configuration

Density histogram for a water monomer
s=0.25
0.51.0
2.0
DPPS GGA EXCHANGE OPTIMIZATION FOR WATER3-body interaction energy:

DPPS EXCHANGE OPTIMIZATION FOR WATER
Fit of Fx(s) with Bayesian constraints
to 330 2-body 100 3-body energies
calculated with MB-pol.
The CUSTOM-MB Fx improves 2-
body and 3-body energies, though
less than CUSTOM-2B and CUSTOM-
3B.
2-body energies 3-body energies

DPPS EXCHANGE OPTIMIZATION FOR WATER
Mean errors of the dataset MB-pol energies used in the optimizations.
• CUSTOM-1B results in significantly larger 2-body and 3-
body errors.
• If a 1-body correction term is added to the functional,
the best functional overall is CUSTOM-MB.

DPPS EXCHANGE OPTIMIZATION FOR WATER
The 1-body energy terms were corrected by the following approach:1
• vdW-DF-cx-Δ1 +1-body corrections
• CUSTOM-2B-Δ1 +2-body corrections
• CUSTOM-MB-Δ1 +3-body corrections
1Patridge, H. and Schwenke, D. W. J. Chem. Phys., 106, 4618 (1997).
1-body-corrected DFT energy
MB-pol 1-body energy
Uncorrected DFT energy
DFT 1-body energy (monomer potential energy surface1 fitted to DFT energies)

III. THERMODYNAMICINTEGRATION (TI) METHOD

Convergence of 𝒈𝑶𝑶(𝒓) withDM tolerance is unclear fromAIMD simulations.

Convergence of 𝒈𝑶𝑶(𝒓) withDM tolerance is unclear fromAIMD simulations.

Convergence of 𝒈𝑶𝑶(𝒓) withDM tolerance is unclear fromAIMD simulations.
𝒈 𝑶𝑶(𝒓)
Convergence of 𝑷 withBasis set is unclear fromAIMD simulations.

Thermodynamic Integration (TI)
• More efficient ab initio Molecular Dynamics (AIMD) convergence testing.– Only 1% of calculations in entire AIMD
simulations needs.
• Avoids masking by statistical fluctations.
• Can be used for convergence testing or to quickly test a new functional for a system.

• Consider the linear mix of two potentials:- 𝑽𝟎 (potential used in a full AIMD simulation)- 𝑽𝟐 (new potential for which properties will be estimated)
• Consider a number of snapshots described by potential 𝑽𝝀. The ensemble average of a property 𝜶 is
TI METHODOLOGY
𝛼 = ∫𝑑 𝑥 𝑒 𝛼 (𝑥)
𝛽 = 1𝑘𝑇
𝑍 = 𝑑 𝑥 𝑒
METHODOLOGY

• Consider the linear mix of two potentials:- 𝑽𝟎 (potential used in a full AIMD simulation)- 𝑽𝟐 (new potential for which properties will be estimated)
• Consider a number of snapshots described by potential 𝑽𝝀. The ensemble average of a property 𝜶 is
TI METHODOLOGY
𝛼 = ∫𝑑 𝑥 𝑒 𝛼 (𝑥)
𝛽 = 1𝑘𝑇
𝑍 = 𝑑 𝑥 𝑒

TI METHODOLOGY

• TI applied to convergence testing of parameters in AIMD simulations: – Basis Set– Mesh Cutoff– DM Tolerance
TI CONVERGENCE TESTING

• TI applied to convergence testing of parameters in AIMD simulations: – Basis Set– Mesh Cutoff– DM Tolerance
• Convergence of the following properties was tested: – Average pressure 𝑷– Average order parameter 𝜻 1
– 𝒈𝑶𝑶𝒎𝒊𝒏– 𝒈𝑶𝑶𝒎𝒂𝒙
TI CONVERGENCE TESTING
1Russo, J. and Tanaka, H. Nat. Commun. 5, 3556 (2014).

TI CONVERGENCE TESTINGConvergence with Basis Set

IV. GAS AND LIQUID PHASE WATERRESULTS FROM OPTIMIZEDFUNCTIONALS
Optimization of an exchange-correlation density functional for water, Michelle Fritz, Marivi Fernandez-Serra and Jose M. Soler, J. Chem. Phys. 144, 224101 (2016)

Testing the CUSTOM functional: dimer binding energy curvesRESULTS WATER HEXAMERS
prism
cage
book
bag
cyclic
Relative binding energies of water isomers.
2-body and 3-body energy corrections improve the relative binding
energies of water hexamers.
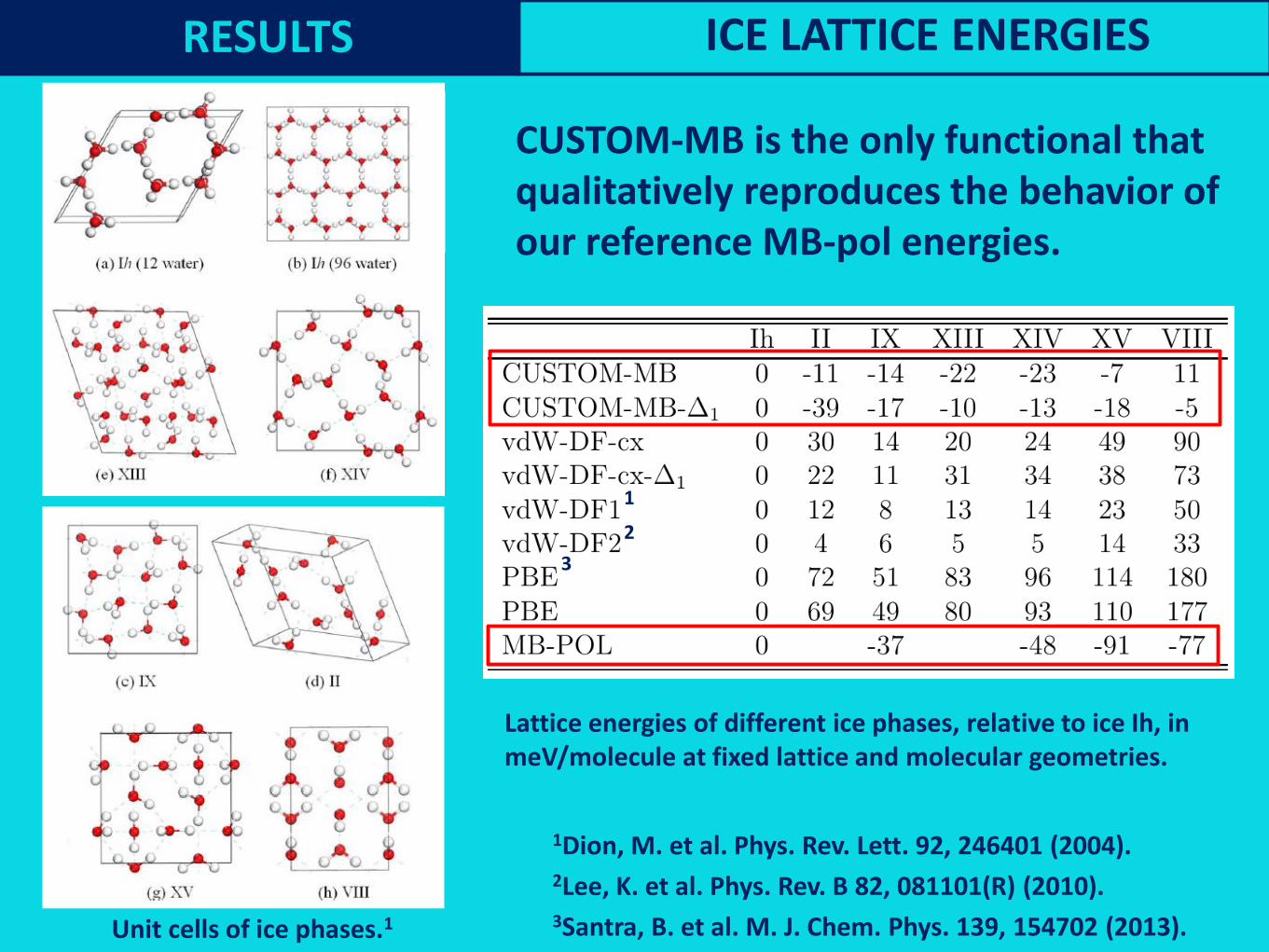
Testing the CUSTOM functional: dimer binding energy curvesRESULTS ICE LATTICE ENERGIES
Lattice energies of different ice phases, relative to ice Ih, in
meV/molecule at fixed lattice and molecular geometries.
1Dion, M. et al. Phys. Rev. Lett. 92, 246401 (2004).
2Lee, K. et al. Phys. Rev. B 82, 081101(R) (2010).
3Santra, B. et al. M. J. Chem. Phys. 139, 154702 (2013).
CUSTOM-MB is the only functional that
qualitatively reproduces the behavior of
our reference MB-pol energies.
Unit cells of ice phases.1
0
1
2
3

gOO(r) from AIMD with vdW-DF-xc and CUSTOM functionals, comparedwith MB-pol and experimental results.
• CUSTOM-2B: increase in first peak and interstitial region, decrease insecond coordination shell.
• CUSTOM-MB decrease in coordination peaks, in the interstitial region.
• 1-body corrections shift the first coordination peak to the right.
RESULTS AIMD RESULTS

Testing the CUSTOM functional: dimer binding energy curvesRESULTS AIMD RESULTS
• vdW-DF-cx and CUSTOM-MB-Δ1 predict equilibrium densities that are too high.
• Density should improve with NQE and a better basis set.
• The compressibility predicted by CUSTOM-MB-Δ1 is in good agreement with experiments.1
1Wagner, W. and Pruss, A. J. Phys. Chem. Ref. Data 31, 387 (2002).
1
1

CONCLUSIONS
CONCLUSIONS:
•We have designed and implemented a general Bayesian method to optimize GGA exchange-correlation (DPPS).
•We have applied DPPS to the optimization of GGA exchange+vdWfunctional for 2-body and 3-body energies calculated with MB-pol.
•Optimization of 1-body energies is incompatible with that of interaction energies. A 1-body correction term was added to total energies.
•The TI method was introduced and applied to convergence testing for AIMD simulations.
•The functional optimized to 2-body and 3-body energies is shown to perform considerably better than vdW-DF-cx for clusters, ice, and water.
FUTURE WORK:
•Nuclear quantum effects should be taken into account to attempt better agreement with experiments.
•Application of DPPS to larger datasets and other functional forms.

CONCLUSIONS
CONCLUSIONS:
•We have designed and implemented a general Bayesian method to optimize GGA exchange-correlation (DPPS).
•We have applied DPPS to the optimization of GGA exchange+vdWfunctional for 2-body and 3-body energies calculated with MB-pol.
•Optimization of 1-body energies is incompatible with that of interaction energies. A 1-body correction term was added to total energies.
•The TI method was introduced and applied to convergence testing for AIMD simulations.
•The functional optimized to 2-body and 3-body energies is shown to perform considerably better than vdW-DF-cx for clusters, ice, and water.
FUTURE WORK:
•Nuclear quantum effects should be taken into account to attempt better agreement with experiments.
•Application of DPPS to larger datasets and other functional forms.
THANK YOU FOR YOUR ATTENTION!

TI METHODOLOGY
ESTIMATING CHANGES IN PRESSURE WITH FUNCTIONAL
1Vydrov, O. A. and Voorhis, T. V. J. Chem. Phys. 133, 244103 (2010).

1Vydrov, O. A. and Voorhis, T. V. J. Chem. Phys. 133, 244103 (2010).
TI METHODOLOGY
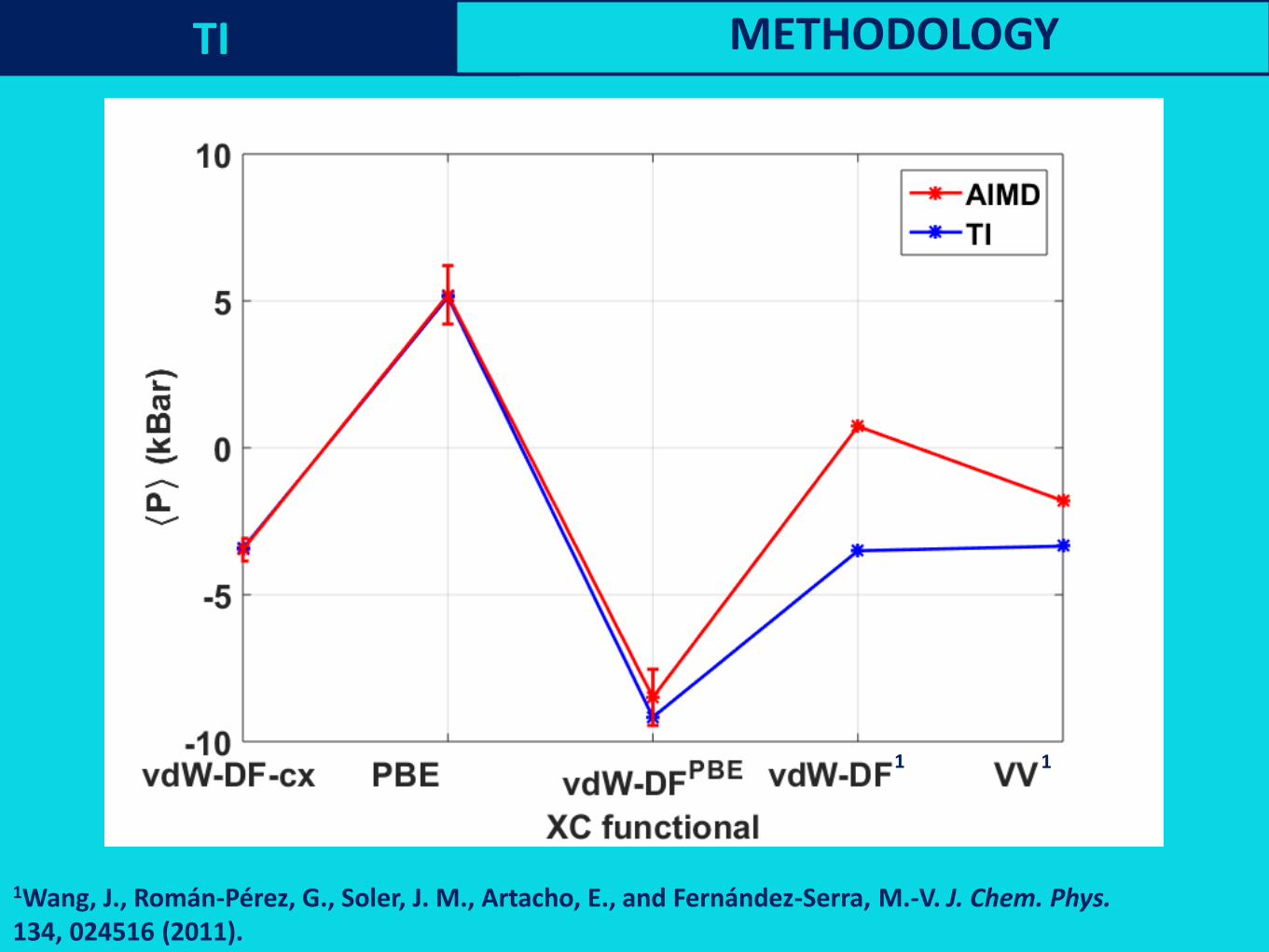
1Wang, J., Román-Pérez, G., Soler, J. M., Artacho, E., and Fernández-Serra, M.-V. J. Chem. Phys.134, 024516 (2011).
11
TI METHODOLOGY

TI METHODOLOGY

TI METHODOLOGY

TI METHODOLOGY