determination of the number of significant components in liquid chromatography nuclear magnetic...
TRANSCRIPT

www.elsevier.com/locate/chemolab
Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151
Determination of the number of significant components in liquid
chromatography nuclear magnetic resonance spectroscopy
Mohammad Wasim, Richard G. Brereton*
School of Chemistry, University of Bristol, Cantock’s Close, Bristol BS8 1TS, UK
Received 19 October 2003; received in revised form 13 January 2004; accepted 14 January 2004
Available online 30 April 2004
Abstract
In this paper, the effectiveness of methods for determining the number of significant components is evaluated in four simulated and four
experimental liquid chromatography nuclear magnetic resonance (LC–NMR) spectrometric datasets. The following methods are tested:
eigenvalues, log eigenvalues and eigenvalue ratios from principal component analysis (PCA) of the overall data; error indicator functions
[residual sum of squares (rssq), residual standard deviation (RSD), ratio of successive residual standard deviations (RSDRatio), root mean
square error (RMS), imbedded error (IE), factor indicator functions, scree test and Exner function], together with their ratio of derivatives
(ROD); F-test (Malinowski, Faber–Kowalski and modified FK); cross-validation; morphological score (MS); purity-based approaches
including orthogonal projection approach (OPA) and SIMPLISMA; correlation and derivative plots; evolving PCA (EPCA) and evolving PC
innovation analysis (EPCIA); subspace comparison. Five sets of methods are selected as best, including several error indicator functions,
their ratio of derivatives, the residual standard deviation ratio, orthogonal projection approach (OPA) concentration profiles and evolving
PCA using an expanding window (EW). Omitting the dataset with the highest noise level, RSS, Malinowski’s F-test, concentration profiles
using SIMPLISMA and subspace comparison with PCA score also perform well.
D 2004 Published by Elsevier B.V.
Keywords: Liquid chromatography; Nuclear magnetic resonance; Principal component analysis; Noise level
1. Introduction introduced in the literature to find out the number of
Coupled chromatographic methods such as liquid chro-
matography coupled with diode array detector (LC–DAD),
liquid chromatography with infrared spectroscopy (LC–IR),
and gas chromatography with IR (GC–IR), gas chromatog-
raphy with mass spectrometry (GC–MS) are increasingly
common in the modern analytical laboratory and result in
large quantities of multivariate data, which often have to be
interpreted using chemometrics methods. For a successful
data analysis of this kind of data, the first step involves the
estimation of the correct rank, or number of significant
components in a multivariate data matrix. Ideally, this
should equal the number of compounds in a chromatograph-
ic cluster, but in practice, real data often contains artefacts,
e.g., baseline problems, different types of noise, errors
generated by data preprocessing (baseline correction,
smoothing, etc.) and so on. A variety of tests have been
0169-7439/$ - see front matter D 2004 Published by Elsevier B.V.
doi:10.1016/j.chemolab.2004.01.008
* Corresponding author. Fax: +44-117925-1295.
E-mail address: [email protected] (R.G. Brereton).
components present in a mixture, and several intercompar-
isons have been made to check the applicability and
limitations of these methods on different datasets [1–4].
Most of these methods produce excellent results when noise
distribution is white, normal and homoscedastic, and reso-
lution is reasonable but may fail to yield easily interpretable
results when these conditions are not fulfilled.
Most classical studies in chemometrics have been per-
formed on LC–DAD data, for which the principles of
resolution have been well established over the past decade.
In contrast, on-flow high-performance liquid chromatogra-
phy coupled with nuclear magnetic resonance (LC–NMR)
produces a relatively high level of noise [5]. The lower
sensitivity of NMR relative to uv/vis requires higher con-
centrations in LC–NMR compared to LC–DAD, often
requiring chromatographic columns to be overloaded and
leading to overlapped peaks in chromatographic direction in
many situations. In addition, the main features of NMR
spectra, are quite different from those obtained using a
DAD. NMR data usually consists of several Lorentzian-

M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151134
shaped peaks between which there is primarily noise, and, in
most of the cases, the noise regions occupy a larger portion
of the spectrum to the signals. Recently, we have reported
studies on the resolution of such chromatograms using
multivariate methods [6–8]. The present paper reports a
comprehensive study of methods employed for the determi-
nation of the number of significant components in LC–
NMR chromatographic peak clusters.
In the present study, we explore the effect of noise level
and resolution on the application of various methods for the
determination of rank. The performance is evaluated on both
simulated and real LC–NMR datasets containing different
numbers of components. The aim of this work is to identify
those methods which are suitable for rank analysis of LC–
NMR data, their advantages and disadvantages, and to group
methods producing similar results. In the performance of all
these methods, no priori knowledge about the pure spectra of
the main compounds or the instrumental noise is employed.
2. Experimental
The two-way data produced by LC–NMR results in
NMR spectra at several chemical shifts in one dimension
(along rows) and concentration (or chromatographic) pro-
files in the other dimension (along columns). Two different
groups of datasets are reported in this paper: one is simulated
data of three components and the other contains experimen-
tal data. Notation used in this paper is presented in Table 1.
2.1. Simulated data
Four simulated datasets, each consisting of three com-
ponent mixtures, were created using previously measured
NMR spectra of the compounds, 2,6-dihydroxynaphthalene
(I), 1,3-dihydroxynaphthalene (II), and 2,3-dihydroxynaph-
thalene (III), and simulating the chromatographic direction.
In contrast to the NMR spectra, which are usually modelled
as a sum of Lorentzian peaks, the chromatograms are best
based on a Gaussian model.
2.1.1. Chromatography
Three chromatographic peaks were created as follows:
f ðmÞ ¼ exp � m� lr
� �2� �
ð1Þ
where r is related to the standard deviation of the peak; m is
the chromatographic elution time; and l is the mean or
centre of the chromatographic peak. The value of r was
calculated from peaks of real chromatograms of the three
isomers of dihydroxynaphthalene. An average value of 20 s
was used for r where points were sampled every s. Each
chromatogram consisted of 252 spectra, resulting in a C
matrix over 252 points in time. While the theoretical model
of a Gaussian peak approaches zero asymptotically, to make
the chromatographic profiles resemble real data more close-
ly, i.e., with zero intensity at the start and end, all the
profiles were set to zero at a height of 10% of the original
Gaussian peakshape, and an offset was added to all the data
points. Chromatographic points outside this window were
set at zero for each compound. Three chromatographic
profiles were created using equal heights.
2.1.2. Spectra
Real NMR spectra were recorded using on-flow LC–
NMR for the three isomers of dihydroxynaphthalene, 100
mM in concentration each. Three spectra with maximum
peak height were selected from two-way datasets of each
isomer. All the regions were replaced with zeros in all NMR
spectra where there was no resonance peak. This procedure
resulted in 989 chemical shift values over the aromatic
region. A matrix S of dimensions 3� 989 was created,
consisting of the spectra of the three compounds.
2.1.3. Bilinear data matrices
Bilinear data matrices X of dimensions 252� 989 was
created by multiplying C with S and adding normally
distributed random noise as
X ¼ CS þ E ð2Þwhere E is error matrix. Four simulated datasets were
created using different peak positions and different noise
levels as follows:
Dataset 1: Peak maxima at time 90 (I), 110 (II) and 130
(III); noise 1% of maximum signal height,
corresponding to a resolution of 0.250.
Dataset 2: Peak maxima at time 90 (I), 110 (II) and 130
(III); noise 5% of maximum signal height,
corresponding to a resolution of 0.250.
Dataset 3: Peak maxima at time 80 (I), 110 (II) and 140
(III); noise 1% of maximum signal height,
corresponding to a resolution of 0.374.
Dataset 4: Peak maxima at time 80 (I), 110 (II) and 140
(III); noise 5% of maximum signal height,
corresponding to a resolution of 0.374.
2.2. Experimental data
A list of compounds used for the experimental mixtures
is presented in Table 2. The following four mixtures were
created by combining different compounds.
Dataset 5: Three-component mixture—consists of A, B and
C, 125 mM each.
Dataset 6: Four-component mixture—consists of D, E, F,
and G, 100 mM each.
Dataset 7: Seven-component mixture—consists of B, C, D,
G, H, I and J, 50 mM each.
Dataset 8: Eight-component mixture—consists of B, D, G,
H, I, J, K and L, 50 mM each.

Table 1
Notations used in the paper
Exner function
wsdm Standard deviation of mth row
r A parameter related to the standard deviation of a
Gaussian Peak
m Degrees of freedom
l Mean or centre of a chromatographic peak
zmn Normalized intensity at mth row and nth column
Ym A matrix in OPA containing reference spectra
and mth row of X
xm1 First reference spectrum in Ym
z Normalized average spectrum of X in OPA
xmn LC–NMR signal intensity at mth row and nth column
xm The mth row vector of X
X Data matrix containing LC–NMR signals
um Weight value in SIMPLISMA
Trace Mathematical Trace function
tmk PCA scores at mth row for kth componentT (superscript) Denoting transpose of a matrix
T Scores matrix obtained by PCA
tr Trace value of a matrix in subspace comparison
S Matrix of spectral profiles
rssq Residual sum of square calculated in SIMPLISMA
RSS Residual sum of square, an error indicator function
RSD Residual standard deviation
RPV Residual percent variance
ROD Ratio of derivatives of the error indicator function
RMS Root mean square error
Rm Matrix containing one or more reference spectra
in SIMPLISMA
RSDRatio Ratio of RSD function
h A positive multiplier in modified Faber–Kowalski
F-test
r Correlation coefficient
qm Ratio of standard deviation to corrected mean intensity
PRESS Predicted residuals error sum of squares
pum Purity value in SIMPLISMA
P Loadings matrix obtained by PCA
offset An adjustable parameter in SIMPLISMA
N Total number of variables (columns) in X
n Index for columns
MSnl Morphological score of noise level
MS Morphological score
M Total number of spectra (rows) in X
m Index for rows
K Total number of compounds present in mixture
k Index for number of compounds or factors
INF Error indicator function
IND Factor indicator function
IE Imbedded error
gk Eigenvalue of the kth component
A Matrix containing orthogonal spectra selected by a
variable selection method
f( ) Probability distribution function
F The value of F-test
B Matrix containing orthogonal spectra selected
by a variable selection method
emn Residual error in data point xmn after model fitting
EMC[ ] Expectation value calculated by Monte Carlo simulation
E Error matrix
E[ ] Expectation value
dw Durbin–Watson statistics
dm Dissimilarity value in OPA for row m
det Determinant of a matrix
Exner function
wD Subspace discrepancy function
C Matrix of chromatographic profiles
l Vector containing sum along variables of X matrix
j Overall average of the data matrix X
xmn Modelled value at m time and n variable for K PCs
x Vector of mean signal along elution time of matrix X
wm Mean signal height of mth row
w Vector containing all wm values
| | xm | | Magnitude of mth row
l Least-squares approximation of the total
intensity spectrum
dxmn/dm Derivative of xmn with respect to m
# Largest principal angle in subspace comparison
d Chemical shift
Table 1 (continued)
M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151 135
2.2.1. Chromatography
All the datasets, except dataset 6, were run on a Waters
(Milford, MA, USA) HPLC system, which consisted of a
717 Plus Autosampler, a 600S Controller, a model 996
Diode Array Detector with a model 616 pump. Dataset 6
was run on an HPLC system consisted of a Jasco pump PU-
980 and a UV detector UV-975.
2.2.2. Spectra
Acetonitrile (HPLC grade, Rathburn Chemicals, Walk-
burn, UK) and deuterated water (Goss Scientific Instruments
Great Baddow, Essex, UK) were used as solvent and mobile
phase in concentrations as presented in Table 3. A few drops
of tetramethylsilane (TMS; Goss Scientific) were added as a
chemical shift reference. A 4 m PEEK tube with width of
0.005 in. was used to connect the eluent from the Waters
HPLC instrument to a flow cell (300 Al) into NMR probe on
a 500 MHz NMR spectrometer (Jeol Alpha 500, Tokyo,
Table 2
The name of compounds, their code and supplier
Compound
Label
Compound Name Purity and Supplier
A Phenyl acetate 99%, Aldrich Chemical,
Milwaukee, WI, USA
B Methyl p-toulenesulphonate 98%, Lancaster,
Morecambe, UK
C Methyl benzoate 99%, Lancaster
D 2,6-dihydroxynaphthalene 98%, Avocado, Research
Chemicals, Heysham, UK
E 1,6-dihydroxynaphthalene 99%, Lancaster
F 1,3-dihydroxynaphthalene 99%, Aldrich, Steinheim,
Germany
G 2,3-dihydroxynaphthalene 98%, Acros Organics,
Geel, Belgium, UK
H 1,2-diethoxybenzene 98%, Lancaster
I 1,4-diethoxybenzene 98%, Lancaster
J 1,3-diethoxybenzene 95%, Lancaster
K Diethyl maleate 98%, Avocado
L Diethyl fumarate 97%, Avocado

Table 3
Experimental Conditions for real LC–NMR data acquisition
Dataset 5 Dataset 6 Dataset 7 Dataset 8
Column C18 Waters Symmetry
(150� 2.1 mm, 5.0 Am)
C18 Phenomenex
(150� 4.6 mm, 5.0 Am)
C18 Waters Symmetry
(100� 4.6 mm, 3.5 Am)
C18 Waters Symmetry
(100� 4.6 mm, 3.5 Am)
Composition of solvent
and mobile phase (v/v)
50:50 60:40 80:20 80:20
Flow rate (ml min� 1) 0.1 0.5 0.5 0.2
Injection volume (Al) 20 20 50 50
Acquisition time (s) 1.4850 1.1698 1.1698 1.1698
Pulse delay (s) 0.9 0.9 2 2
M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151136
Japan). For each spectrum, the spectral width was 7002.8
Hz and a pulse angle of 90j was employed.
2.3. Software
The data analysis was performed by computer pro-
grams written in Matlab by the authors of this paper.
Fourier transformation and preprocessing for LC–NMR
data was performed by in-house written software called
LCNMR, which runs under Matlab.
3. Methods
3.1. Data preprocessing
No data preprocessing was required for the datasets
which were simulated in the frequency or spectral domain.
The experimental data was acquired in the form of Free
Induction Decays (FIDs) during chromatographic elution,
then the following processing was performed on each FID.
Spectra were apodised and Fourier transformed. The first
spectrum in time was removed as it contained artefacts; the
spectra were aligned to the TMS reference and phase
corrected. In order to remove errors due to quadrature
detection, which results in regular oscillation of intensity,
a moving average of each frequency over every four points
in time was performed. This procedure has been described
elsewhere [9] in detail.
In LC–NMR, many parts of the spectrum correspond
primarily to noise channels, so it is important to select only
those variables where the signal to noise ratio is acceptable
(the noise level was defined by visual inspection of standard
deviation plots). In addition, there may only be certain
portions of the chromatographic (or time) direction where
compounds elute and other regions can be discarded.
Variable selection is important because, in multivariate
methods, if noise is dominant in the data, then the results
produced will be dominated by noise and become difficult
to interpret. For this purpose, the standard deviation was
calculated first over each variable (chemical shift), and only
those variables were retained where the standard deviation
was acceptable. The same procedure was repeated in the
time dimension.
3.2. Chemometrics data analysis
In literature, several methods have been proposed for the
determination of chemical rank and are described below.
3.2.1. Eigenvalue, logarithm of eigenvalues and eigenvalue
ratio
Principal component analysis (PCA) [10,11], when per-
formed on a data matrix X (M�N), with suitable K number
of components selected, decomposes the data matrix into a
scores matrix T, a loadings matrix P and an error matrix E of
dimensions M�K, K�N and M�N, respectively. Mathe-
matically, it can be defined as
X ¼ TP þ E ð3Þ
The size of each PC is measured by its eigenvalue [12],
which is calculated as
gk ¼XMm¼1
t2mk ð4Þ
where gk is the eigenvalue for kth component and tmk is the
score of mth row and kth component. Eigenvalues that
correspond to real factors should be considerably higher
than later eigenvalues, which correspond to noise. Thus, the
significant components can be identified easily in a plot of
eigenvalues against the number of components. The loga-
rithm of eigenvalues [13,14] and eigenvalue ratio [15] have
also been reported as useful methods for differentiating the
significant components from the remaining components. In
our study, the eigenvalues, logarithm of eigenvalues and
eigenvalue ratio were plotted against the number of compo-
nents, and the data rank was determined by finding a break
in the plot for the first two methods and locating a separate
group of data points in the eigenvalue ratio method.
In the methods described below, the speed of calculation
is influenced by the dimensions of the data matrix. There-
fore, in the calculation of eigenvalues, the data matrix was
formed in such a way that the number of columns became
less than the number of rows. If the original data matrix had
more rows than the columns, it was transposed. Thus, in all
of these methods the maximum number of eigenvalues was
equal to the number of columns. The data–matrix is defined

M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151 137
as X withM rows and N columns. No preprocessing, such as
centring, was performed on the data matrix.
3.2.2. Error indicator functions (INF)
In the following subsections, some methods have been
defined collectively as error indicator functions, as all of
these methods calculate some kind of noise present in the
data and will be denoted by INF.
3.2.2.1. Residual sum of squares plot (RSS). The RSS [16]
gives the sum of squares of error values after data repro-
duction using K eigenvalues. RSS is defined as
RSSðKÞ ¼XMm¼1
XNn¼1
x2mn �XKk¼1
gk ¼XMm¼1
XNn¼1
e2mn ð5Þ
where xmn is signal intensity at time m and chemical shift n;
gk is the kth eigenvalue; and emn is the error.
The RSS plot provides information about the size of
residuals after increasing number of eigenvalues is selected.
If data were noise free, the RSS value drops to zero after
selecting a suitable number of significant components, but
with real data, the RSS values plot show a change of slop.
The number of true components then becomes equal to the
K value after which this change is observed.
3.2.2.2. Residual standard deviation (RSD). The RSD [2]
represents the difference between raw data and pure data
with no experimental error, and it measures lack of fit of a
PC model. After selecting a suitable number of eigenvalues,
RSD should be equal or lower than the experimental error. It
is calculated as
RSDðKÞ ¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiXNk¼Kþ1
gk
MðN � KÞ
vuuuut: ð6Þ
RSD(K) is plotted against K; when it reaches the value of
instrumental error, it levels off. At that point, the number of
components become equal to K and all the points including
K, with a flatness in the plot that is observed visually.
3.2.2.3. Root mean square error (RMS). The RMS [2]
measures the difference between raw and regenerated data
after selecting suitable eigenvalues. Although the RMS is
closely related to the RSD, it produces lower values than the
RSD. It is defined as
RMSðKÞ ¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiXNk¼Kþ1
gk
MN
vuuuut¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiðN � KÞ
N
rRSDðKÞ: ð7Þ
RMS(K) is plotted against K, and the graph flattens when
the correct number of factors is determined. The criteria for
finding the number of components is the same as discussed
for RSD method.
3.2.2.4. Imbedded error (IE). The IE function [2,17] is
defined by
IEðKÞ ¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiK
XNk¼Kþ1
gk
NMðN � KÞ
vuuuut ¼ffiffiffiffiffiK
N
rRSDðKÞ: ð8Þ
It decreases until all the significant eigenvalues have
been calculated, and then starts to increase. Theoretically,
the number of significant factors is obtained when the
minimum is reached. In practice, a steady increase is rarely
observed because errors are not uniformly distributed and it
becomes difficult to find a true minimum.
In our study, we determined the number of components
by calculating the minimum of IE(K) function and finding a
change in slope in the IE vs. K plot.
3.2.2.5. Factor indicator function (IND). The IND, pro-
posed by Malinowski [2,17] is calculated as
INDðKÞ ¼ 1
ðN � KÞ2
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiXNk¼Kþ1
gk
MðN � KÞ
vuuuut ¼ 1
ðN � KÞ2RSDðKÞ:
ð9Þ
It reaches a minimum when the true number of compo-
nents are employed and provides a more pronounced
minimum than the IE function. IND is much more sensitive
than the IE in finding the true number of components [2].
The criteria for finding the rank of data using IND function
are the same as defined for IE function.
3.2.2.6. Residual percent variance (RPV; scree test). The
scree test reported by Cattell [18] suggests that the residual
variance should level off after a suitable number of factors
have been computed. It provides information on the relative
importance of each eigenvalue to the overall sum of squares
of the data and is defined by
RPVðKÞ ¼
XNk¼Kþ1
gk
XNk¼1
gk
0BBBB@
1CCCCA100: ð10Þ
This function is plotted against K. The plot levels off at
the point when the number of significant components has
been found. This point is taken as the true dimensionality of
the data.

Intelligent Laboratory Systems 72 (2004) 133–151
3.2.2.7. Exner function (w). The Exner function proposed
by Kindsvater et al. [19] is an empirical function, denoted
by the Greek letter w; it varies from zero to infinity. A wvalue of 1.0 is the upper limit of physical significance but
Exner [20] proposed 0.5 as the largest acceptable value. In
our study, we used w(K) < 0.1 for determining the number
of significant components. The function is defined as
follows:
wðKÞ ¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiMN
XMm¼1
XNn¼1
ðxmn � xmnÞ2
ðMN � KÞXMm¼1
XNn¼1
ðxmn � jÞ2
vuuuuuuut ð11Þ
where xmn is reproduced data point after K components
have been calculated, and j is the overall average of the
data matrix. The criterion for determining the number of
components is the same as defined in the scree test.
3.2.3. Ratio of derivatives of error indicator function (ROD)
All of the error indicator functions defined above can be
plotted against the number of components, and the estima-
tion of the true number of components is based on the
proper interpretation of the plots. Another way to find data
rank is to plot the derivative of any one of the error indicator
functions (defined in Sections 3.2.2.1–3.2.2.7) and find out
the minimum or maximum in the plot. Elbergali et al. [21]
suggested the second derivative, third derivative and ratio of
the second to third for finding a break-even point in the plot.
In our study, we selected only the ratio of the second to third
derivative defined as
RODðKÞ ¼ INFðKÞ � INFðK þ 1ÞINFðK þ 1Þ � INFðK þ 2Þ ð12Þ
where INF is an error indicator function.
The number of components present in a mixture can be
identified either by locating a minimum in the INF function
or maximum in ROD function. In Ref. [21], no further
explanation is given in the selection criteria when the num-
ber of components found is not the same by this criterion.
3.2.4. Ratio of RSD (RSDRatio)
The error indicator functions defined in Sections
3.2.2.1–3.2.2.7 can also be expressed as a ratio of the form
RSDRatioðKÞ ¼ INFðKÞINFðK � 1Þ : ð13Þ
The assumption is that the ratio changes smoothly over
the number of components that belongs to PCs that are not
significant. The ratio arising from the components
corresponding to noise will fall on a line and form a distinct
group of points as compared with those coming from
M. Wasim, R.G. Brereton / Chemometrics and138
significant components. We define the following ratio for
the RSD function as
RSDRatioðKÞ ¼ RSDðKÞRSDðK � 1Þ ¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiðN � K þ 1Þ
XNk¼Kþ1
gk
ðN � KÞXNk¼K
gk
vuuuuuuut :
ð14Þ
Although such a ratio could be computed for different
error indicator functions, we restrict to the RSD in this
paper, as similar results were obtained for the other indicator
functions.
3.2.5. F-test
Malinowski [22,23] developed a test based on Fisher
variance ratio called the F-test to determine the rank of data.
This calculates a ratio of two variances, which come from
independent pools of samples. The error in the data is
assumed to have normal distribution. In the application of
F-test, variances are eigenvalues, which derive from inde-
pendent sets of PCs. Mathematically, the F-test is defined by
FKðm1; m2Þ ¼gKXN
k¼Kþ1
gK
XNk¼Kþ1
ðM � k þ 1ÞðN � k þ 1Þ
ðM � K þ 1ÞðN � K þ 1Þ
ð15Þ
where m1 = 1 and m2=(N�K) are the degrees of freedom. The
calculated F-ratio is compared with F-inverse values at a
defined level of confidence (99% in our study). The number
of significant components is set equal to K when the F-value
exceeds F-inverse.
Faber and Kowalski [24,25] noticed that the degrees
of freedom used in Malinowski’s F-test were not consis-
tent with the definition of F-test. They defined an F-test
based on Mandel’s [26] degree of freedom (called FK F-
test) as
FKðm1; m2Þ ¼gKXN
k¼Kþ1
gK
m2m1
ð16Þ
where m1 and m2 are the degrees of freedom calculated by
m1 ¼ E½gK � ð17Þ
m2 ¼ ðM � K þ 1ÞðN � K þ 1Þ � m1 ð18Þ
and E[ gK] is the expectation value of the Kth eigenvalue,
which is based on the premise that the expected value of
an eigenvalue corresponding to noise divided by the

M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151 139
appropriate degree of freedom is an unbiased estimate of
the error variance r2 defined by
r2 ¼ E½gK �mK
: ð19Þ
The E[ gK] is measured by the average of Monte Carlo
simulations of several matrices of random values drawn
from a normal distribution with variance r2 = 1. The degrees
of freedom will then be defined by
mK ¼ EMC½gK � ð20Þ
where the subscript MC denotes values calculated by the
Monte Carlo simulations.
Following the Faber–Kowalski suggestions for F-test,
Malinowski [27] reported that the FK F-test is very sensitive
and overestimates the rank; he suggested a modification in
the calculation of degree of freedom as
mK ¼ hEMC½gK � ð21Þ
where h is a positive number, which will reduce the
sensitivity of the test.
In the present study, we used all of these variations and
reported their results with different significance levels.
3.2.6. Cross-validation
Cross-validation [28,29] is one of the most common
chemometrics method for determining the number of sig-
nificant components in a dataset. An implementation of
cross-validation known as leave-one-out is usually applied
to find out the number of factors. In the leave-one-out
method, a data matrix X is divided into submatrices of
dimension (M� 1,N) by taking one row out of the matrix X,
creating M submatrices. The deleted row is modelled by
varying the number of PCs. Each modelled row gives a
Predicted Residuals Error Sum of Squares (PRESS) for
varying number of PCs. PRESS is defined by
PRESSðKÞ ¼XMm¼1
XNn¼1
ðxmn � xmnÞ2 ð22Þ
where xmn is the modelled value at m time and n variable for
K PCs.
There are different ways to present the results, but a
common approach is to compare PRESS using (K + 1) PCs
to RSS using K PCs. If PRESS(K + 1)/RSS(K) is signifi-
cantly greater than 1, then K PCs gives the rank of the
data.
3.2.7. Morphological score (MS)
The morphological score was first presented in the
chemometrics literature by Shen et al. [30]. The method is
based on the fact that the ratio of the norm of a spectrum to
the norm of its first difference is higher for a profile of a
component than a profile generated only by noise. Mathe-
matically, it is defined by
MSðxÞ ¼ Nðx � xÞNNMOðx � xÞN ð23Þ
where MS(x) is the morphological score calculated for
vector x. MO(x) is called the morphological operator, and
it calculates the difference between sequential values of
the vector x. It has been shown that the score is scale
invariant and is not affected by the magnitude of the
noise; it is also independent of the baseline offset. The
procedure utilises the significant key spectra (normalized
to unit length) from a method suitable for selecting key
or pure variables in the form of matrix. Pure variables are
defined as those that best describe a single component in
the mixture. Then PCA is performed on that matrix and
the morphological score of each loadings vector, pk, is
calculated using Eq. (23). The morphological score of the
noise level is calculated using the formula given in Eq.
(24):
MSnl ¼ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiðN � 1ÞFðN � 1;N � 2Þ
N � 2
rð24Þ
where N is the number of elements in the vector, and F
is the F-test value at certain level of confidence at
(N� 1,N� 2) degrees of freedom. The rank of the data
becomes equal to the number of all vectors for which
MS>MSnl.
3.2.8. Orthogonal projection approach (OPA)
The orthogonal projection approach (OPA) [31] is a
stepwise method for finding the pure variables in the data
sequentially. Each step involves identifying a new compo-
nent in the data. The method can be applied in time as well
as in chemical shift direction. Here, the method is explained
only for chromatographic dimension.
The first step involves the creation of a matrix Ym for mth
spectrum
Ym ¼ ½zxm� ð25Þ
where z is the normalized average spectrum of X, and xm is
the mth spectrum in X. After the first component is
identified, the spectrum z is replaced by a reference
spectrum.
A dissimilarity value, dm, is calculated as
dm ¼ detðYmYTmÞ ð26Þ
where det is determinant, and YmT is transpose of matrix
Ym.
The value of dm is plotted against elution time. The time
corresponding to the maximum value of dm in the plot
indicates the most dissimilar spectrum in the data matrix as
compared with the average spectrum and is selected as the

M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151140
first component. The spectrum at time m is selected as a first
reference vector, which replaces the average spectrum in Eq.
(25), so a new Ym matrix is obtained as
Ym ¼ ½xm1xm� ð27Þ
where xm1 is the first reference spectrum corresponding to
maximum dissimilarity value in the plot. Dissimilarity is
calculated again using Eq. (26) and presented graphically. If
a second reference spectrum, corresponding to maximum
dissimilarity, is present, then a second xm2 vector is added to
the Ym matrix.
In all subsequent steps, a new reference spectrum is
added every time to Ym and dissimilarity is calculated
again. This procedure is repeated until the dissimilarity plot
no longer contains an obvious peak or there is only random
noise left in the plot. The rank of the data becomes equal
to the number of reference spectra found in the whole
process.
3.2.9. SIMPLISMA
SIMPLISMA was first reported by Windig et al. [32]. It
determines the number of pure variables in the data and is
based on a stepwise process similar to OPA.
The first step is the identification of standard deviation
sdm and mean intensity wm of each spectrum, which become
the part of a ratio, defined as
qm ¼ sdm
wm þ ðoffset=100Þ �maxðwÞ ð28Þ
where max(w) is the maximum intensity of all the mean
intensities of the M spectra, and the offset is an adjustable
value. Each spectrum in the data matrix X is normalized to
unit length
zmn ¼xmn
NxmNð29Þ
and the matrix Rm is created as
Rm ¼ ½zm� ð30Þ
and the weight, denoted by um, of each spectrum is
calculated as
um ¼ detðRTmRmÞ ð31Þ
where Rm initially contains one normalized spectrum as
given in Eq. (30).
Eqs. (28) and (31) then provide a value for the purity
pum, defined as
pum ¼ um þ qm: ð32Þ
The value of pum is plotted as a function of time
(which is called a purity plot). The purest spectrum will
give the highest value of pum and the corresponding time
will be used to select the first reference spectrum. The
next steps are similar to OPA; a reference spectrum is
added to the Rm matrix and the process is repeated until
the purity plot does not show any peak or it exhibits only
noise. The rank becomes equal to the number of reference
spectra found.
The dissimilarity and purity data produced by OPA and
SIMPLISMA can also be explored by other methods for
determination of the rank of the data. In Sections 3.2.9.1–
3.2.9.3, we will describe three more procedures.
3.2.9.1. Residual sum of squares using SIMPLISMA.
SIMPLISMA provides many tools for the identification of
true number of components. Among these, residual sum of
squares (rssq) [1] gives good results. The calculation is as
follows. The total signal is calculated as l at all variables; for
example, it is possible to sum the chromatographic intensity
at every spectral datapoint, so l becomes a vector as a
function of chemical shift, each element corresponding to
the summed elution profile at the corresponding spectral
frequency. This is a linear combination of all the pure
spectra, which is then projected onto the space of the
spectra, as selected by SIMPLISMA, in a matrix S, and
the contribution of each selected spectrum on the total
spectrum is estimated. These values can then be used to
calculate the least-squares approximation of the total inten-
sity spectrum, as l.
l ¼ SðSTSÞ�1STl ð33Þ
When the proper number of spectra has been selected,
then l should be equal to l within the experimental error. The
difference between the two is calculated by residual sum of
squares (rssq).
rssq ¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiXNn¼1
ðln � lnÞ2
XNn¼1
l2n
vuuuuuuut : ð34Þ
Ideally, rssq should not change significantly after the
selection of true number of components. A graph of rssq
against K is plotted and the rank becomes equal to the first
rssq value after which graph does not show significant
change.
3.2.9.2. Durbin–Watson statistics using OPA and
SIMPLISMA. The Durbin–Watson (dw) [33–35] test
assumes that the observations, and so the residuals, have a
natural order. If residual is an estimate of error and all of
errors are independent, then the dw test checks for a
sequential dependence in which each error is correlated
with those before and after it in the sequence. In our study,
we used dissimilarity (dm) and purity ( pm) values as
residuals and the rank was estimated by locating a big
change in the dw values by plotting dw against the number

M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151 141
of components (or reference spectra). The test, for OPA
dissimilarity values, is defined as
dw ¼
XMm¼1
ðdm � dm�1Þ2
XMm¼1
d2m
ð35Þ
3.2.9.3. Concentration profiles using OPA and
SIMPLISMA. Dissimilarity plots obtained by OPA or
purity plots by SIMPLISMA on the spectral dimension
usually predict more components than the true rank, which
is due to the noise and to the fact that profiles are not well
aligned in the original data. This noise and drift generates
dispersion in the NMR peak positions, which causes the
incorrect estimation of a high rank. In this paper, we suggest
a slight variation in the interpretation of purity plots by
constructing the ‘‘concentration profiles’’. NMR spectra, in
contrast to UV, consist of a series of several resonances,
which result in quite distinct chromatographic profiles
compared to HPLC–DAD. This property of NMR signals
can be exploited in determining the true number of compo-
nents. When OPA or SIMPLISMA is performed on the
spectra, three types of concentration profiles can be ob-
served.
Single and pure profiles: these are retained.
Multiple profiles belonging to the same compound: the
repeated profiles are deleted.
Impure profiles starting from one component and extend-
ing over two or more than two pure concentration profiles:
some of these could be retained and some are deleted.
A visual inspection of these concentration profiles reveals
the correct number of components very quickly.
3.2.10. Correlation plots
The correlation coefficient [16] is a measure of similarity
between two vectors. It can be defined between two spectral
vectors at elution times m and m� 1 as
rðxm; xm�1Þ
¼
XNn¼1
ðxm;n � xmÞðxm�1;n � xm�1ÞffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiXNn¼1
ðxm;n � xmÞ2s ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiXN
n¼1
ðxm�1;n � xm�1Þ2s : ð36Þ
The correlation coefficient between two consecutive
spectra plotted against time provides useful information
about changes, which takes place in the data, which in turn
relates to the number of compounds in the mixture. The
correlation coefficient between two similar spectra will be
close to 1, while it will be close to 0 for totally dissimilar
spectra. The region in which the correlation coefficient is
close to 1 indicates a selective chromatographic region.
Normally, the logarithm of the correlation coefficient is
used for the vertical axis, providing correlation coefficients
are all positive, which is usual during regions of elution.
3.2.11. Derivative plots
Derivatives represent another approach for finding the
change in the characteristic spectra with time in the dataset.
However, derivatives also magnify noise, so it is important
that these are combined with smoothing functions. Savitsky
Golay (SG) filters [36–38] provide a facile combination of
smoothing and derivative functions in one step. One such
function of a first derivative with five-point quadratic
smoothing function is defined as
dxmn
dmcð�2xm�2;n � xm�1;n þ xmþ1;n þ 2xmþ2;nÞ=10 ð37Þ
In the process of derivative calculations, data is row
scaled (over wavelengths), and a Savitsky Golay first
derivative (absolute value) is calculated and presented as a
function of time. A full description of the method is reported
elsewhere [39]. The minima in the plot represent points of
highest purity corresponding to derivatives close to 0; thus,
the number of minima usually corresponds to the number of
compounds in the mixture.
3.2.12. Evolving principal component analysis (EPCA)
Two-way chromatographic data, corresponding to a se-
ries of unimodal peaks in elution time, have properties that
can be exploited for finding the row factors and concentra-
tion windows with the help of PCA. Evolving factor
analysis belongs to the family of self-modelling curve
resolution techniques [40], which can be implemented in
two steps. The first step, sometimes called as EPCA [41],
involves creating a window in the data matrix and applying
PCA on that window. The method can be implemented
either as expanding window (EPCA–EW) or fixed size
window evolving factor analysis (FSW–EFA). These meth-
ods will be described briefly below.
EPCA with expanding window is implemented in two
steps. The first step is forward window factor analysis,
which involves the formation of a submatrix from the data
matrix X, and then applying PCA to calculate K principal
components. This step will produce K eigenvalues. In the
next step, another spectrum (row) is added to the submatrix
formed in the last step, and again, PCA is performed for K
principal components. The procedure is repeated until all the
spectra (rows) in the matrix X are used. In the backward
window factor analysis (the second step), the exact proce-
dure is repeated but the rows are selected from the end of the
X matrix. Finally, the logarithm of eigenvalues is plotted
against corresponding number of rows in submatrices. In the
forward window, a rise in the eigenvalue curve indicates that
a new compound is detected in the submatrix, and in the
backward window, a fall in the eigenvalue curve indicates
that the compound has eluted completely. If the number of

M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151142
PCs calculated is more than the true number of compounds
in the data matrix, the extra PCs will not show any
significant rise or fall in their eigenvalues.
FSW–EFA [42–45] is also implemented in two steps
similar to EPCA–EW. In the forward step, a submatrix of a
fixed size is created and its eigenvalues are calculated. Next,
the centre of matrix is shift by one row forward and PCA is
performed again. This procedure is repeated until the matrix
includes the last row of X. In the backward step, the
submatrix includes a fixed number of rows starting from
the end of data and moves backward. Finally, the logarithms
of the first few eigenvalues (typically 4 or 5) are plotted
against time, and number of components is estimated from
the plots.
3.2.13. Evolving principal components innovation analysis
(EPCIA)
The EPCIA [46–49] approach is based on curve fitting
using Kalman filters. The method assumes that two-dimen-
sional chromatographic data can be modelled as
X ¼ CS þ E ð38Þ
EPCIA is normally implemented in two stages: the first is
the calculation of pure variables, which provides chromato-
graphic profiles, and second is the modelling of data using
these chromatograms. In the procedure, each chromatogram
projects onto the (N� 1) two-dimensional subspaces and is
fitted to linear model recursively. Initially, a linear model
consisting of one component is obtained and the prediction
error is calculated. The predicted errors for the chromato-
gram at each spectral frequency are combined to give a root
mean square error (RMS). A plot of RMS against time gives
an indication of the number of components. If RMS is high
and shows peak-like structures, it suggests that another
component is required in the modelling. The procedure is
repeated until RMS becomes low and shows only random
structure. The algorithm to implement Kalman filters is
described in more detail in Chapter 3 of Ref. [16].
3.2.14. Subspace comparison
Subspace comparison [50] compares two subspaces, each
of which is described by a set of orthonormal vectors
selected by a method suitable for variable selection such
as OPA or SIMPLISMA. Suppose two subspaces are
defined as A={a1, a2, . . .aK} and B={b1, b2, . . .bK}. Forexample, A may be a matrix with 5 columns each consisting
of a profile obtained using SIMPLISMA for 5 pure compo-
nents; B may be the corresponding matrix obtained from
OPA. However, the components may be slightly different or
in a different order, so these matrices will not necessary be
identical, although ideally, they should contain similar
information. Note that K must be the same for both matrices;
the aim is to see whether, for example, the OPA construction
using 3 components is similar to that obtained from SIM-
PLISMA; if not, this is likely to be the wrong number of
components, so K is changed.
The vectors in A and B are orthogonalized by the
Schmidt procedure [35]. The next step is to calculate tr(K)
as
trðKÞ ¼ TraceðATBBTAÞ ð39Þ
where tr(K) varies between 0 and K; D(K), called subspace
discrepancy function, is calculated as follows:
DðKÞ ¼ K � trðKÞ ð40Þ
D(K) is the measure of that part of the subspace which is
in the orthogonal complement of the other. This becomes
zero when two subspaces are identical. Eigenvalues are
calculated on (ATB) matrix and are utilised as
sin2ð#KÞ ¼ 1� gK ð41Þ
where sin2(#K) is the largest principal angle. Ref. [50]
compared D(K) and sin2(#K) as a measure of disagreement
between the subspaces by plotting both values for K com-
ponents. The true number of factors becomes equal to the
largest value of K with D(K), or sin2(#K) close to zero.
4. Results and discussion
The results of different methods are discussed in separate
sections.
4.1. Eigenvalue, logarithm of eigenvalues and eigenvalue
ratio
The methods using logarithms and eigenvalue ratios
produced better results compared to the method using raw
eigenvalues (see Section 3.2.1). The eigenvalue plots deter-
mined the correct rank only in three datasets (see Table 4).
The plot of the logarithm of eigenvalues gave correct rank
for all datasets except the datasets 5, 6 and 8 where it was
difficult to find a true break among the values originating
from the significant and nonsignificant factors. The eigen-
value ratio yielded similar results as produced by the
logarithm of eigenvalue method except for dataset 5 where
it determined the correct rank. Fig. 1 is a plot of the
logarithm of eigenvalues for dataset 5 where there is no
indication about the data rank. For the same dataset, the
results of the eigenvalue ratio method are produced in Fig.
2. The results for these three methods have been compiled in
Table 4.
4.2. Error indicator functions (INF)
It has been explained in Section 3.2.2 that the rank was
determined by plotting error indicator functions against the
number of components and then locating a break in the
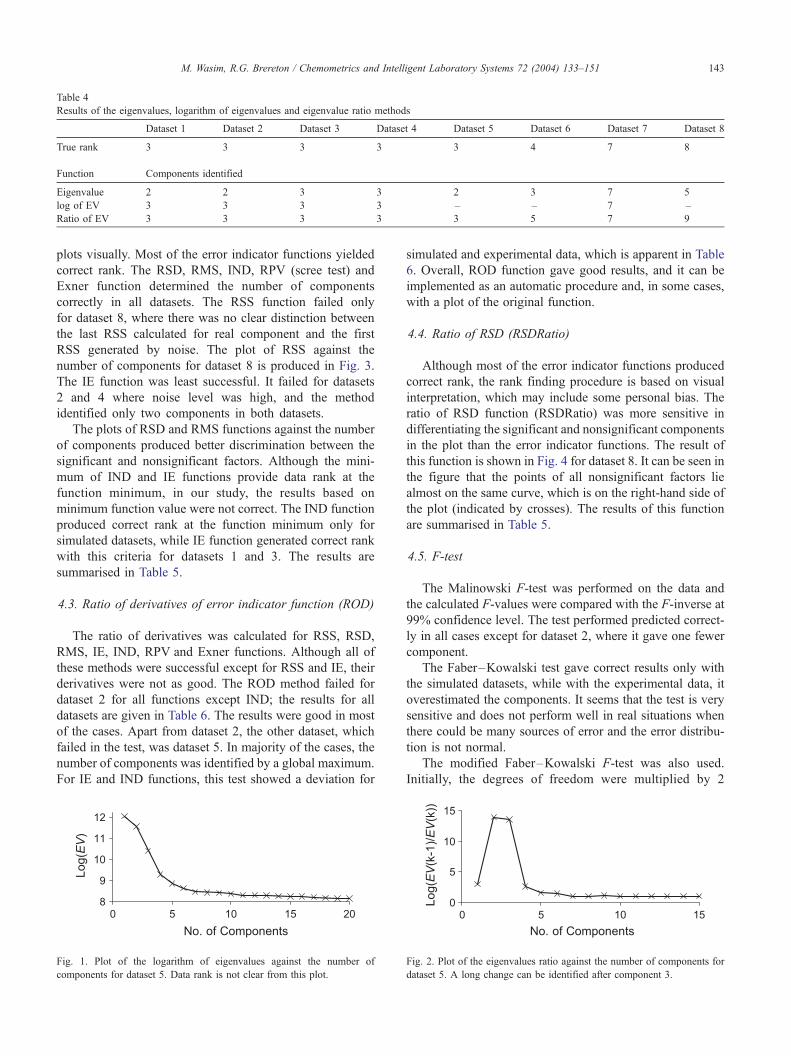
Table 4
Results of the eigenvalues, logarithm of eigenvalues and eigenvalue ratio methods
Dataset 1 Dataset 2 Dataset 3 Dataset 4 Dataset 5 Dataset 6 Dataset 7 Dataset 8
True rank 3 3 3 3 3 4 7 8
Function Components identified
Eigenvalue 2 2 3 3 2 3 7 5
log of EV 3 3 3 3 – – 7 –
Ratio of EV 3 3 3 3 3 5 7 9
M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151 143
plots visually. Most of the error indicator functions yielded
correct rank. The RSD, RMS, IND, RPV (scree test) and
Exner function determined the number of components
correctly in all datasets. The RSS function failed only
for dataset 8, where there was no clear distinction between
the last RSS calculated for real component and the first
RSS generated by noise. The plot of RSS against the
number of components for dataset 8 is produced in Fig. 3.
The IE function was least successful. It failed for datasets
2 and 4 where noise level was high, and the method
identified only two components in both datasets.
The plots of RSD and RMS functions against the number
of components produced better discrimination between the
significant and nonsignificant factors. Although the mini-
mum of IND and IE functions provide data rank at the
function minimum, in our study, the results based on
minimum function value were not correct. The IND function
produced correct rank at the function minimum only for
simulated datasets, while IE function generated correct rank
with this criteria for datasets 1 and 3. The results are
summarised in Table 5.
4.3. Ratio of derivatives of error indicator function (ROD)
The ratio of derivatives was calculated for RSS, RSD,
RMS, IE, IND, RPV and Exner functions. Although all of
these methods were successful except for RSS and IE, their
derivatives were not as good. The ROD method failed for
dataset 2 for all functions except IND; the results for all
datasets are given in Table 6. The results were good in most
of the cases. Apart from dataset 2, the other dataset, which
failed in the test, was dataset 5. In majority of the cases, the
number of components was identified by a global maximum.
For IE and IND functions, this test showed a deviation for
Fig. 1. Plot of the logarithm of eigenvalues against the number of
components for dataset 5. Data rank is not clear from this plot.
simulated and experimental data, which is apparent in Table
6. Overall, ROD function gave good results, and it can be
implemented as an automatic procedure and, in some cases,
with a plot of the original function.
4.4. Ratio of RSD (RSDRatio)
Although most of the error indicator functions produced
correct rank, the rank finding procedure is based on visual
interpretation, which may include some personal bias. The
ratio of RSD function (RSDRatio) was more sensitive in
differentiating the significant and nonsignificant components
in the plot than the error indicator functions. The result of
this function is shown in Fig. 4 for dataset 8. It can be seen in
the figure that the points of all nonsignificant factors lie
almost on the same curve, which is on the right-hand side of
the plot (indicated by crosses). The results of this function
are summarised in Table 5.
4.5. F-test
The Malinowski F-test was performed on the data and
the calculated F-values were compared with the F-inverse at
99% confidence level. The test performed predicted correct-
ly in all cases except for dataset 2, where it gave one fewer
component.
The Faber–Kowalski test gave correct results only with
the simulated datasets, while with the experimental data, it
overestimated the components. It seems that the test is very
sensitive and does not perform well in real situations when
there could be many sources of error and the error distribu-
tion is not normal.
The modified Faber–Kowalski F-test was also used.
Initially, the degrees of freedom were multiplied by 2
Fig. 2. Plot of the eigenvalues ratio against the number of components for
dataset 5. A long change can be identified after component 3.

Fig. 3. RSS plot of dataset 8 (real dataset). The data rank is not clear in the
plot.
Fig. 4. RSD(K)/RSD(K� 1) ratio of dataset 8 (real dataset). Significant
factors are indicated by w .
M. Wasim, R.G. Brereton / Chemometrics and Intell144
[h = 2 in Eq. (21)] and later with 10; in both cases, the
results were not correct (Table 7).
4.6. Cross-validation
As with other methods, cross-validation can also be
applied first by the inspection of plots, and then by
comparison based on PRESS and RSS values. Visual
inspection of all PRESS and RSS plots show true number
of components except in dataset 8, but the results based on
the ratio of PRESS and RSS gave the correct number of
components only in the case of simulated datasets and
predicted too many significant components in the experi-
mental datasets. Therefore, cross-validation based on the
visual inspection of PRESS and RSS plots leads to better
results.
4.7. Morphological score (MS)
The morphological score was, initially, applied to the
spectra obtained by OPA; the results were not correct
because OPA dissimilarity plots did not give true number
of components. It was also applied to the whole dataset,
which excluded the OPA calculations. The results showed
that the method worked only for simulated data, although it
provided good results when the number of components was
estimated by visual inspection of the plot of morphological
score against number of components, as shown in Figs. 5
and 6. Dataset 5 gave a correct result; the plot of dataset 6
showed the last two points, corresponding to real compo-
nents, slightly above the MS values of noise. Dataset 7 gave
six components, the first five MS values were well above
MS of noise but the sixth MS was below the noise level.
The seventh MS was higher than the noise level, thus
estimating six components in total.
4.8. Orthogonal projection approach (OPA)
OPAwas applied on object (time) and variable (chemical
shift) dimensions. As discussed in Section 3.2.8, OPA
produces dissimilarity plots, which can be utilised in differ-
ent ways to identify the number of components. We discuss
the results of these methods in Sections 4.8.1–4.8.3.
4.8.1. Dissimilarity plots
4.8.1.1. Object selection. All the simulated datasets pro-
vided correct results except dataset 1, which showed one
more component. In real datasets, only datasets 5 and 6
produced the correct results while datasets 7 and 8 gave
higher components.
4.8.1.2. Variable selection. All the simulated datasets
produced correct results except for dataset 2, in which
one less component was predicted than the true rank. In
all the real datasets, the number of significant components
was overestimated. Most of the results using dissimilarity
plots, on the time dimension, were not correct. Using the
variable dimension, the results were totally wrong be-
cause, in majority of cases, more components than
expected were found due to the reasons explained in
Section 3.2.9.3.
4.8.2. Durbin–Watson statistics (dw)
4.8.2.1. dw on object selection. The dw statistics produced
correct results only with datasets 1 and 3, which are
simulations. It found two components in datasets 2 and 4,
which contained high-noise levels. There was no clear
indication about the number of components in experimental
datasets. Figs. 7 and 8 show the dw plots for dataset 8 using
OPA data on two different dimensions.
4.8.2.2. dw on variable selection. The results calculated
by Durbin–Watson statistics were plotted and the number
of components calculated by determining where there was a
sharp increase in value. In most of the cases, less compo-
nents were found than the true rank. The success of the dw
test was high when it was used with dissimilarity values
obtained using variable dimension. It was successful in
most of the cases, as shown in Table 7. There was no
difference in simulated or real datasets from the success
point of view.
4.8.3. Concentration profiles
Concentration plots provided good results in all cases. As
OPA does not yield pure variables, therefore, some profiles
igent Laboratory Systems 72 (2004) 133–151

Table 5
Results of error indicator functions and RSD ratio compiled together
Dataset 1 Dataset 2 Dataset 3 Dataset 4 Dataset 5 Dataset 6 Dataset 7 Dataset 8
True rank 3 3 3 3 3 4 7 8
Function Components identified
RSS 3 3 3 3 3 4 7 7–8
RSD 3 3 3 3 3 4 7 8
RMS 3 3 3 3 3 4 7 8
IE 3 2 3 2 3 4 7 8
IND 3 3 3 3 3 4 7 8
RPV 3 3 3 3 3 4 7 8
w 3 3 3 3 3 4 7 8
RSDRatio 3 3 3 3 3 4 7 8
M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151 145
appear which extend over two or more than two compo-
nents. Fig. 9 shows concentration profiles of dataset 1. The
profiles for chemical shift, d, of 7.112 and 7.109 ppm
belong to the fastest eluting compound, and similarly those
for 6.722 and 6.726 ppm belong to the second eluting
compound. The profile for 7.196 ppm arises from the
slowest eluting compound, while the concentration plot
obtained by using 7.252 ppm starts with the middle eluting
compound and ends with the slowest eluting compound,
which can easily be identified as a combined profile that
originates from two close resonances (variables) from the
middle and slowest eluting compounds together. After
excluding the mixed profile, the presence of three compo-
nents is confirmed in dataset 1. Datasets 2 and 3 also show
similar profiles but at different chemical shifts. Dataset 4
does not show any common resonance, and the concentra-
tion plots are very clean for the three compounds.
In dataset 5, three components were correctly identified.
For dataset 6, initially, six concentration profiles were
generated. The second variable (7.112 ppm) is not a pure
variable and its concentration profile is indicative of two
components; it was not difficult to correctly identify the
true number of components. Dataset 7 resulted in interest-
ing plots; the first variable selected by OPA at 1.337 ppm
is not a pure variable and it belonged to compounds eluting
as sixth and seventh components in concentration plots
(see Fig. 10 which shows the first nine concentration
Fig. 5. Morphological score (MS) on dataset 1 (three components). Three
components can be easily discriminated (see horizontal line).
profiles). After deleting the first concentration profile
(which corresponds to a combination of the sixth and
seventh eluting compounds) and the last profile (which is
the same as the third component), it suggests that there is a
total of seven components in the mixture. Dataset 8 also
exhibited similar behaviour, and it also showed some
variables that correspond to a combination of two pure
components, but these were identified and the true dimen-
sionality of the data was determined correctly. Thus, using
the concentration profiles followed by OPA was a success-
ful approach in finding the true rank of the data in
simulated as well as in real datasets.
4.9. SIMPLISMA
SIMPLISMA is very sensitive to noise if the noise level
is high as it often selects components from noise. The effect
of noise could be reduced either by reducing the objects
dimension or by increasing the offset value. The latter
procedure is not recommended because, in that case, SIM-
PLISMAwill generate variables similar to OPA. The former
procedure was adopted, and chromatographic profiles were
discarded at the start and at the end of all datasets. It
significantly improved the results. SIMPLISMAwas applied
at an offset value of 15% on both dimensions.
Fig. 6. Morphological score (MS) on dataset 8 (eight components).
Discrimination is not good; only seven components can be found (see
horizontal line).

Fig. 7. Durbin –Watson statistics calculated on dissimilarity values
calculated by OPA on variable dimension using dataset 8 (eight
components).
Fig. 9. Concentration plots of the first six variables selected by OPA using
dataset 1 (simulated data).
M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151146
Like OPA, SIMPLISMA also makes use of several
methods in determining the data rank; in our study we used
four such methods. The results with these methods are
discussed in the Sections 4.9.1–4.9.4.
4.9.1. Purity plots
4.9.1.1. Object selection. Datasets 1, 3, 5 and 6 gave the
true rank, while datasets 2 and 4 gave one component less
than the true rank. Datasets 7 and 8 did not provide any
conclusive answers because the purity plots became very
complex after six components were detected, and so it was
difficult to decide the real number of components.
4.9.1.2. Variable selection. From datasets 1 and 3, three
components were predicted, and from datasets 2 and 4, two
components were predicted. Datasets 5 and 6 produced the
true rank. It was difficult to find out the true number of
components in datasets 7 and 8, where more components
appeared than the true rank.
Purity plots behaved in a similar manner to OPA dissim-
ilarity plots; that is, both of the methods yielded incorrect
rank in the majority of datasets. Like OPA, purity plots also
failed in the case of simulated datasets.
4.9.2. Residual sum of squares using SIMPLISMA
4.9.2.1. Object selection. Datasets 1, 3, 5 and 6 gave the
correct results. The remainder predicted a lower rank than
Fig. 8. Durbin –Watson statistics calculated on dissimilarity values
calculated by OPA on time dimension using dataset 8 (eight components).
the true one. These results suggest that rssq provides good
results in most of cases. In our present study, rssq plots
based on the variable selection gave better results than the
plots based on object dimension.
4.9.2.2. Variable selection. The rssq gave wrong results
for dataset 2, and dataset 6 did not produce good evidence
for the three components; there was some indication of
the existence of a fourth component. Similarly for dataset
8, either seven or nine components are suggested; there
was a slight decrease in the rssq value when the ninth
component was selected. The rssq plot of dataset 8 can be
seen in Fig. 11.
4.9.3. Durbin–Watson statistics (dw)
4.9.3.1. Object selection. Durbin–Watson gave correct
results only in dataset 3, and, in the rest of datasets, the
results were incorrect.
Fig. 10. Concentration plots of the first nine variables selected by OPA
using dataset 7 (real data).

Fig. 11. Residual sum of square (rssq) calculated by SIMPLISMA on the
time dimension using dataset 8 (eight components).
Fig. 13. Concentration plots of the first five variables selected by
SIMPLISMA using dataset 5 (real data).
M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151 147
4.9.3.2. Variable selection. Durbin–Watson was totally
unable to provide answer about the number of components.
The plots were without minima and did not show any rise
after the true number of components.
4.9.4. Concentration profiles
When OPA and SIMPLISMA are applied to the variable
dimension, in most cases, they found more components than
the true number, as estimated by dissimilarity or purity
plots. In Section 4.8.3, it was discussed that concentration
profiles proved successful in OPA. The concentration pro-
files become complicated when the number of components
becomes large and requires a careful inspection of plots.
Fig. 12 shows the concentration plots of the first four
variables of dataset 1 obtained by SIMPLISMA. It is easy
to identify that the fourth component is the same as the
third component. Furthermore, this can be confirmed by
calculating the correlation coefficient between profiles 3
and 4, which is greater than 0.9, suggesting that both
profiles are very similar. Concentration profiles obtained
by SIMPLISMA on the time dimension gave correct
results in all cases except dataset 5 where it suggested
four components as shown in Fig. 13. It can be seen that
the fourth component is approximately similar to the third
Fig. 12. Concentration plots of the first four variables selected by
SIMPLISMA using dataset 1 (simulated data).
component but, due to the high level of noise present in
these profiles, it is not very clear, and the fifth component
is the same as the first component. Concentration profiles
of dataset 7 are given in Fig. 14. Among these profiles, the
profiles obtained using chemical shift d at 7.177 and 7.182
ppm belong to the same component; the profiles from the
chemical shifts at 2.450 and 2.464 ppm are also from a
single compound, while the profile at 1.323 ppm is a
combined profile of components 6 and 7. On deleting
these profiles, seven components remain, which is a
correct number for this dataset. Similarly, 8 components
are identified in dataset 8. Fig. 15 shows the concentration
profiles for dataset 8.
As concentration profiles selected by SIMPLISMA have
more noise than those selected by OPA, it makes the
identification process more difficult, and, sometimes, an
incorrect number of components is identified as in the case
of dataset 5.
Fig. 14. Concentration plots of the first ten variables selected by
SIMPLISMA using dataset 7 (real data).

Fig. 15. Concentration plots of the first eleven variables selected by
SIMPLISMA using dataset 8 (real data).
Fig. 16. EPCIA on dataset 6 (four components) fourth plot showing a peak-
like structure.
Fig. 17. EPCIA on dataset 6 (four components) after using five variables.
M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151148
4.10. Correlation plots
In our present study, correlation plots did not provide a
suitable approach in finding the true number of components,
and were successful only in one case, which was dataset 6.
It gave one component less than the true rank in the
simulated datasets and datasets 5 and 7. In dataset 8, this
method finds two components less than the true rank.
Therefore, it suggests that this technique is not very sensi-
tive and underestimates the true number of components.
Correlation plots work well when chromatographic resolu-
tion is high, although the data-preprocessing step removes
most of the noise but not completely. Noise is removed from
those frequencies where there appears no peak from all the
components under investigation.
4.11. Derivative plots
In the same way as correlation plots, derivatives yielded
incorrect results. Datasets 1, 3, 4 and 5 produced one
component less, and dataset 2 showed two components less
than the true rank. The derivatives in the present study were
successful only in the case of dataset 6. The method found
six components in datasets 7 and seven components in
dataset 8. The presence of noise and low resolution appears
to be the possible reason for the failure of the method.
4.12. Evolving principal component analysis (EPCA)
EPCA–EW was another successful method. This method
was not only successful in all cases but this method was the
easiest in interpretation of its plots. The results were correct
in all cases. The method of FSW–EFA was successful only
in two cases, datasets 6 and 7. In all other cases, it produced
one component less than the true rank, even with the
simulated datasets. This method did not perform well in
one of our previous study [5] of a three-component mixture,
which could be due to a high level of noise.
4.13. Evolving principal component innovation analysis
(EPCIA)
In EPCIA, the number of components can be found by
looking at the RMS plots, their shape and the changes in the
RMS plots after selecting different numbers of components.
In our study, we used OPA and SIMPLISMA data as an
input to EPCIA. Datasets 1, 3 and 4 gave similar OPA and
SIMPLISMA variables. Therefore, the results of EPCIA
were same and correct for these samples. We discuss results
of other datasets in Sections 4.13.1 and 4.13.2.
4.13.1. OPA selected variables
Dataset 2 suggests four significant components, and, in
dataset 5, there were three significant components. There
was a small peak in the third plot of dataset 5, which could
be assigned to a fourth component but this peak persisted
even after the addition of fourth profile. Therefore, this
method suggests that dataset 5 contains only three compo-
nents. Dataset 6 showed four components by both methods
(OPA and SIMPLISMA). It also behaved similarly to data-
set 5 and the last plot did not show complete randomness as
shown in Figs. 16 and 17. For dataset 7, 10 OPA variables
were used as input; among those, four appeared to have
small impact on the reduction of RMS values. If those
variables were regarded as impure or insignificant, this
suggests that dataset 7 consists of six significant compo-

Fig. 18. Subspace discrepancy function D(K) (diamonds) and sin2(#K)
(stars) for PCA scores and OPA variables using dataset 2.
M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151 149
nents. A similar observation was made for dataset 8,
yielding seven components when 10 OPA variables were
modelled.
4.13.2. SIMPLISMA selected variables
Datasets 2, 5 and 6 gave correct results. EPCIA proved to
be difficult when the number of components becomes large.
It has been found that, in the analysis, some specific
variables behave less effectively. One such variable can be
located in dataset 7, which is the fourth variable. If this is
removed and EPCIA is performed again with six variables,
the results are almost the same when seven components
were modelled. Thus, in this case, it can be concluded that
there are six true components in this mixture. Dataset
8 produces randomness in the RMS plot after eight variables
have been used. Again in this case, the fourth variable has
very low influence on the overall reduction of the RMS
value or the shape of RMS plot. If this is removed and
EPCIA is performed again with seven variables, the results
are the same. These observations lead to a conclusion that
some variables, which show a small change in the reduction
of RMS values, appear to be less effective and cannot be
taken as a new component. Finally, a small peak-like
structure was seen in the last RMS plot of all experimental
datasets, which indicates that either the variables are not
pure or the variables are not sufficient to model the data.
Table 6
Results of ROD on different error indicator functions
Components identified
Dataset 1 Dataset 2 Dataset 3 Dataset
True rank 3 3 3 3
Function Components identified
RSS 3 2 3 3
RSD 3 2 3 3
RMS 3 2 3 3
IE 3a 2b 3a 2b
IND 3b 3b 3b 3b
RPV 3 2 3 3
w 3 2 3 3
a First minimum in ROD plot and function minimum.b Global minimum in ROD plot and function minimum.
4.14. Subspace comparison
Subspace comparison produced promising results. PCA
scores and variables from OPA and SIMPLISMAwere used
as input for this test. The three methods produced three
combinations of variables, namely, PCA scores and SIM-
PLISMA variables, PCA scores and OPA variables, and
SIMPLISMA variables and OPA variables. It is difficult to
correctly interpret the results from the description given in
reference [50] because the criterion is a qualitative one.
Neither of the combinations could identify the correct
number of components in dataset 2; there is some indication
about the third component in the subspace discrepancy plot
using scores from PCA scores and OPA variables, as shown
in Fig. 18. A similar situation existed in other cases; for
example, dataset 6 appears to consist of either three or four
components (the third component has lower value than the
fourth component) using variables from SIMPLISMA and
OPA. Best results and better discrimination were obtained
using PCA scores and OPA variables, and PCA scores and
SIMPLISMA variables. Subspace comparison, although
relying on other methods for variable selection, is simple
to implement and fast to use.
5. Summary of results
All of the methods have been compiled in Tables 4–7.
Five methods could be selected as the best for providing true
rank. The first is a set of error indicator functions (RSD,
RMS, IND, RPVand Exner function), second is ROD on IND
function, third is RSD ratio, fourth is concentration plots
using OPA, and the last method is EPCA–EW. However, all
of these methods rely on visual interpretation of various plots,
except ROD on IND function. As the visual interpretation of
plots may vary from person to person, therefore, the final
results may be prone to some bias and are hard to automate.
Dataset 2 has the highest level of noise and lowest resolution.
4 Dataset 5 Dataset 6 Dataset 7 Dataset 8
3 4 7 8
2 4 7 8
3 4 7 8
3 4 7 8
3b 4 7 8
3 4 7 8
2 4 7 8
3 4 7 8

Table 7
Results of all methods except eigenvalue based methods
Dataset 1 Dataset 2 Dataset 3 Dataset 4 Dataset 5 Dataset 6 Dataset 7 Dataset 8
True components 3 3 3 3 3 4 7 8
Indicator Components identified
Malinowski’s F-test (a= 0.01) 3 2 3 3 3 4 7 8
Faber–Kowalski F-test 3 3 3 3 >3 >4 >7 >8
Modified FK F-test (h= 2) 3 3 3 3 >3 >4 >7 >8
Modified FK F-test (h= 10) 3 2 3 3 3 4 8 11
Morphological score 3 3 3 3 3 4 6 7
Cross-validation 3 3 3 3 6 5 8 10
OPA
Dissimilarity plots (time dim.) 4 3 3 3 4 4 9 10
Dissimilarity plots (var. dim.) 3 2 3 3 4 5 >7 >8
Durbin–Watson (time dim.) 3 2 3 2 3 2 – –
Durbin–Watson (var. dim.) 3 3 4 3 2 4 7 8
Concentration profiles 3 3 3 3 3 4 7 8
SIMPLISMA
Purity plots (time dim.) 3 2 3 2 3 4 9 >8
Purity plots (var. dim.) 3 2 3 2 3 4 7–8 >8
Durbin–Watson (time dim.) – – 3 – 2 2 7 10
Durbin–Watson (var. dim.) – – – – 2 3 7 6
RSS (time dim.) 3 2 3 2 3 4 6 6
RSS (var. dim.) 3 2 3 3 3 3 or 4 7 6
Concentration profiles 3 3 3 3 4 4 7 8
Correlation plot 2 2 2 2 2 4 6 6
Derivative plot 2 1 2 1 or 2 2 4 6–7 6
EPCA
Expanding window 3 3 3 3 3 4 7 8
Fixed size moving window 2 2 2 2 2 4 7 8
EPCIA
OPA variables 3 4 3 3 3 4 6 7
SIMPLISMA variables 3 3 3 3 3 4 6 7
Subspace comparison
SIMPLISMA and OPA 3 2 3 2 3 3 7 8
PCA scores and SIMPLISMA 3 2 3 3 3 4 7 8
PCA scores and OPA 3 2 3 3 3 4 7 8
M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151150
If the results of this dataset are omitted, then RSS, Malinow-
ski’s F-test, concentration profiles using SIMPLISMA and
subspace comparison with PCA score perform well. The
methods that least often predicted the correct rank were
Durbin–Watson statistics on SIMPLISMA variables, corre-
lation plots and derivative plots. The rank estimation in
dataset 3 was correctly determined by most of the methods;
this was primarily due to good resolution and a well-defined
noise distribution in the data. The relative effectiveness of
methods for LC–NMR data differ quite strongly to LC–
DAD because there are different characteristics in the spectra.
In NMR, there are often quite distinct and well-defined
spectral peaks, whereas in uv/vis spectroscopy, the spectral
peaks tend to be much broader and less well defined. In
addition, the noise levels in NMR are often much higher than
in uv/vis. In LC–NMR, it is not easy to eliminate noise by
preprocessing because there are always regions where there
are resonances in the spectrum of one compound and noise in
the spectrum of another, so that noise regions will be retained
even after variable selection. In addition, there can be slight
drifts in peak positions in LC–NMR peak positions during
recording which, because of the relatively sharp nature of
these peaks, has an influence on the effectiveness of certain
methods; this problem is not usually encountered in LC–
DAD. Hence, methods such as correlation coefficients and
derivatives that are very effective in LC–DAD because of the
smooth and relatively noise-free nature of the data are less
useful in LC–NMR which technique is a challenge for the
chemometrician.
Automation is a desirable feature for any algorithm.
Only two of the successful methods can readily be
automated, one is Malinowski’s F-test and the other is
ROD on IND function. The ROD was successful for all
datasets using IND function, while Malinowski’s F-test
failed for dataset 2 which had high noise and low
resolution.

M. Wasim, R.G. Brereton / Chemometrics and Intelligent Laboratory Systems 72 (2004) 133–151 151
As artefacts make it difficult to identify correct data rank,
all those methods which depend on eigenvalues, are strongly
influenced by the existence of artefacts. Moreover, the
interpretation becomes difficult when the noise level is high.
Successful methods, which do not rely on eigenvalues, are
OPA dissimilarity plots, SIMPLISMA purity plots and
concentration plots. Among these methods, concentration
plots using OPA variables was the most successful.
Most of the methods estimated less components than the
true rank except log of eigenvalues, eigenvalues ratio, FK F-
test, modified FK test, cross-validation, OPA and SIM-
PLISMA and subspace comparison (only by SIMPLISMA
and OPA data), which gave more components.
Acknowledgements
M.W. wishes to thank the Ministry of Science and
Technology, Government of Pakistan for their funding and
the support of PAEC, Pakistan. We thank Drs. C. Airiau and
M. Murray for help with the experimental work.
References
[1] F.C. Sanchez, B.V.D. Bogaert, S.C. Rutan, D.L. Massart, Chemom.
Intell. Lab. Syst. 34 (1996) 139–171.
[2] E.R. Malinowski, Factor Analysis in Chemistry, third ed., Wiley, New
York, 1991.
[3] M. Meloun, J. Capek, P. Miksik, R.G. Brereton, Anal. Chim. Acta
423 (2000) 51–68.
[4] J. Toft, Chemom. Intell. Lab. Syst. 29 (1995) 189–212.
[5] M. Wasim, M.S. Hassan, R.G. Brereton, Analyst 128 (2003)
1082–1090.
[6] H. Shen, C.Y. Airiau, R.G. Brereton, Chemom. Intell. Lab. Syst. 62
(2002) 61–78.
[7] H. Shen, C.Y. Airiau, R.G. Brereton, J. Chemom. 16 (2002) 165–175.
[8] H. Shen, C.Y. Airiau, R.G. Brereton, J. Chemom. 16 (2002) 469–481.
[9] J.C. Hoch, A.S. Stern, NMR Data Processing, Wiley-Liss, New York,
1996.
[10] S. Wold, K. Esbensen, P. Geladi, Chemom. Intell. Lab. Syst. 2 (1987)
37–52.
[11] H. Wold, in: J. Gani (Ed.), Soft Modelling by Latent Variables: the
Nonlinear Iterative Partial Least Squares (NIPALS) Algorithm, Per-
spectives in Probability and Statistics, Academic Press, London,
1975, pp. 117–142.
[12] T.M. Rossi, I.M. Warner, Anal. Chem. 54 (1986) 810–815.
[13] N. Ohta, Anal. Chem. 45 (1973) 553–557.
[14] I.T. Jollife, Principal Component Analysis, Springer, New York, 1986.
[15] R.F. Hirsch, G.L. Wu, Chemom. Intell. Lab. Syst. 1 (1987) 265–272.
[16] R.G. Brereton, Chemometrics: Data Analysis for the Laboratory and
Chemical Plant, Wiley, Chichester, 2003.
[17] E.R. Malinowski, Anal. Chem. 49 (1977) 612–617.
[18] R.B. Cattell, Multivariate Behav. Res. 1 (1966) 245–276.
[19] H.H. Kindsvater, P.H. Weiner, T.J. Klingen, Anal. Chem. 46 (1974)
982–988.
[20] O. Exner, Collection of Czech, Chem. Commun. 31 (1966)
3222–3251.
[21] A. Elbergali, J. Nygren, M. Kubista, Anal. Chim. Acta 379 (1999)
143–158.
[22] E.R. Malinowski, J. Chemom. 3 (1988) 49–60.
[23] E.R. Malinowski, J. Chemom. 4 (1990) 102.
[24] K. Faber, B.R. Kowalski, Anal. Chim. Acta 337 (1997) 57–71.
[25] N.K.M. Faber, Comput. Chem. 23 (1999) 565–570.
[26] J. Mandel, Technometrics 13 (1971) 1–18.
[27] E.R. Malinowski, J. Chemom. 13 (1999) 69–81.
[28] S. Wold, Technometrics 20 (1978) 397–405.
[29] D.W. Osten, J. Chemom. 2 (1988) 39–48.
[30] H. Shen, L. Stordrange, R. Manne, O.M. Kvalheim, Y. Liang, Che-
mom. Intell. Lab. Syst. 50 (2000) 37–47.
[31] F.C. Sanchez, J. Toft, B. Van den Bogaert, D.L. Massart, Anal. Chem.
68 (1996) 79–85.
[32] W. Windig, C.E. Heckler, F.A. Agblevor, R.J. Evans, Chemom. Intell.
Lab. Syst. 14 (1992) 195–207.
[33] N.R. Draper, H. Smith, Applied Regression Analysis, third ed., Wiley,
New York, 1998.
[34] S. Gourvenec, D.L. Massart, D.N. Rutledge, Chemom. Intell. Lab.
Syst. 61 (2002) 51–61.
[35] G. Strang, Linear Algebra and its Applications, third ed., Harcourt
Brace Jovanovich, Orlando, 1998.
[36] A. Savitzky, M.J.E. Golay, Anal. Chem. 36 (1964) 1627–1639.
[37] J. Steinier, Anal. Chem. 44 (1972) 1906–1909.
[38] K. Kavianpour, R.G. Brereton, Analyst 123 (1998) 2035–2042.
[39] S. Dunkerley, R.G. Brereton, J. Crosby, Chemom. Intell. Lab. Syst. 48
(1999) 99–119.
[40] M. Maeder, Anal. Chem. 59 (1987) 527–530.
[41] B.G.M. Vandeginste, D.L. Massart, L.M.C. Buydens, S. De Jong, P.J.
Lewi, J. Smeyers-Verbeke, Handbook of Chemometrics and Quali-
metrics, Part B, Elsevier, Amsterdam, 1998, pp. 274–278.
[42] H.R. Keller, D.L. Massart, Anal. Chim. Acta 246 (1991) 379–390.
[43] H.R. Keller, D.L. Massart, J.O. De Beer, Anal. Chem. 65 (1993)
471–475.
[44] J. Toft, O.M. Kvalheim, Chemom. Intell. Lab. Syst. 19 (1993) 65–73.
[45] F. Cuesta Sanchez, J. Toft, O.M. Kvalheim, D.L. Massart, Anal.
Chim. Acta 314 (1995) 131–139.
[46] S.C. Rutan, J. Chemom. 1 (1987) 7–18.
[47] G.H. Webster, T.L. Cecil, S.C. Rutan, J. Chemom. 3 (1988) 21–32.
[48] S.J. Vanslyke, P.D. Wentzell, Anal. Chem. 63 (1991) 2512–2519.
[49] P.D. Wentzell, S.G. Hughes, S.J. Vanslyke, Anal. Chim. Acta 307
(1995) 459–470.
[50] H. Shen, Y. Liang, O.M. Kvalheim, R. Manne, Chemom. Intell. Lab.
Syst. 51 (2000) 49–59.