detailed atomistic analysis of the hiv-1 protease interface
TRANSCRIPT

Published: May 05, 2011
r 2011 American Chemical Society 7045 dx.doi.org/10.1021/jp200075s | J. Phys. Chem. B 2011, 115, 7045–7057
ARTICLE
pubs.acs.org/JPCB
Detailed Atomistic Analysis of the HIV-1 Protease InterfaceS�ergio Filipe Sousa, Bruno Tamames, Pedro Alexandrino Fernandes, and Maria Jo~ao Ramos*
REQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciencias, Universidade do Porto,Rua do Campo Alegre, 687, 4169-007 Porto, Portugal
bS Supporting Information
’ INTRODUCTION
The Acquired Immune Deficiency Syndrome (AIDS) is adisease of the human immune system caused by the HumanImmunodeficiency Virus type-1 virus (HIV-1) that currentlyinfects something like 33 million people worldwide, with 25million fatal cases so far.1 The immense magnitude of thesenumbers, together with their terrible socioeconomic and demo-graphic implications, has led to unprecedental political attentionand to a massive financial response, making this virus one of themost important targets for the development of new drugs.
The several enzymes and structural proteins that constitute theHIV-1 virus are synthesized in the form of two large polyproteins:gag and gag-pol. These two proteins are inactive and need to becleaved into the individual proteins and enzymes for properbiological activity. This essential process of cleavage is performedexclusively by the enzyme HIV-1 protease (PR), in what may beregarded as one of the most critical parts of the HIV-1 virus lifecycle. For this reason, the enzyme HIV-1 PR has become a veryattractive target for the development of new anti-HIV drugs.2�10
Structurally, HIV-1 PR is composed by two identicalmonomers,11,12 each with 99 amino acid residues, interlockedinto one another to form a dimer.13,14 PR acts on gag and gag-polproteins by breaking these large inactive proteins through veryprecise hydrolysis reactions at specific sequence points. Inparticular, this enzyme recognizes sequences of eight amino acidresidues in the two large proteins discussed above and for each ofthese sequences breaks the peptidic bond between the aminoacid residues 4 and 5.15
A total of nine different sequences of eight amino acid residueshave shown to be recognized by HIV PR, with some of thesesequences differing in all the eight amino acid residues thatcharacterize them.15,16 In particular, in such sequences thisenzyme has been shown to catalyze the hydrolysis of thefollowing peptidic bonds: Tyr-Pro, Leu-Ala, Met-Met, Phe-Leu, Phe-Pro, Phe-Tyr, and Leu-Pro.15 Despite this interestingdiversity, a single amino acid change in one of the recognizedsequences is enough to transform a substrate of PR into anonsubstrate. These subtle requirements for PR recognitionand the relatively large diversity of recognized sequences forsuch a small enzyme make the PR active site a particularlychallenging target for the development of new drugs.6,17 Whileseveral PR inhibitors have reached the market or are in advancedstages of clinical testing,2�10 resistance to these drugs as a resultof an increased prevalence of new strains containing mutations atthe active site is an additional limitation to the success of suchinhibitors.4,18 New strategies for the inhibition of this importantenzyme would therefore be of great relevance and are currentlyunder active investigation.19�23
Early experimental denaturation studies have suggested thatfolding and HIV-1 dimerization occur simultaneously,24�26
proposing also that the folded dimer exists in equilibrium withthe unfolded monomers and that individual folded monomers do
Received: January 4, 2011Revised: April 6, 2011
ABSTRACT: HIV-1 protease is a very attractive target for thedevelopment of new anti-HIV drugs and has been extensivelystudied over the past decades. In this study, we present adetailed atomic level characterization of the dimer interface inthe enzyme HIV-1 protease through computational alaninescanning mutagenesis and molecular dynamics simulations. Inaddition to a full mapping of the amino acid residues present atthe subunit interface, in terms of the corresponding energeticcontribution for dimer formation and of their classification ashot spots, warm spots, and null spots, we trace a dynamicanalysis of the subunit interacting and solvent accessible surface areas and of the most important hydrogen bonds between subunits.The results presented illustrate the high energetic importance for dimer formation of a small set of five amino acid residue pairs at thesubunit interface—Leu5, Ile50, Arg87, Leu97, and Phe99—and provide important clues on the most important structural andenergetic determinants for dimer formation. In addition, the results presented suggest several key targets at the subunit interface forthe development of newmolecules that aim to inhibit HIV-1 protease (PR) activity through blocking the formation of the fully activePR homodimeric form, providing important clues for drug design.

7046 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
not exist at appreciable concentration, as they are intrinsicallyunstable. However, other more recent studies have shown thatmonomeric HIV-1 PR appears to be relatively stable27 and thatthe isolated monomer has secondary and tertiary structure that isvery similar to that of the bound monomer. In addition, severalstudies of HIV-1 protease variants have shown that mutations ator near the interface can shift the monomer�dimer equilibriumin favor of the folded but inactive monomeric form.24,25,28,29
Also, a foldedmonomeric HIV-1 PR for several mutants has beenreported in the literature.28,30,31 Together these results suggestan alternative route for the development of anti-HIV-1 drugs:inhibiting the formation of the active PR dimer, through thedevelopment of dimerization inhibitors.19,20,32�37
Alanine scanning mutagenesis (ASM) is a very powerful toolfor the study of protein�protein or subunit interfaces,38 whichcan be applied to identify the regions that are responsible formost of the binding energy between the two components thatmake up the interface. The residues at these regions are called hotspots and have been defined as those residues that upon alaninemutation result in a binding free energy difference of 4.0 kcal/mol or greater.39,40 Other relevant residues for the bindingprocess between subunits are the warm spots, which uponalanine mutation result in a binding free energy differencebetween 2.0 and 4.0 kcal/mol. Amino acid residues that uponalanine mutation result in a binding free energy difference belowthis 2.0 kcal/mol threshold are called null spots.39,40
Computational alanine scanning mutagenesis was developedas a faster alternative to the much slower experimental ASM,allowing large interfaces to be efficiently analyzed for thepresence of hot spots, warm spots, and null spots. Severaldifferent schemes, based on the application of molecular dy-namics simulations, have been described in the literature, atdifferent levels of sophistication and computational cost and withdifferent levels of success.41�46 In particular, a variation of thestandard computational ASM method46 has been developed inour group,47 based on the use of the well-establishedMM�PBSA(molecular mechanics/Poisson�Boltzmann surface area)approach,48,49 optimized for the use of a continuum solvationdescription considering different internal dielectric constantvalues for different types of amino acid residues, and has beenshown to yield particularly high accurate results in a variety ofdifferent biological systems,50�52 with an overall success of 82%in the identification of hot spots.47
In this study, we assess a relatively new and innovative PRinhibition paradigm, by targeting the subunit interface of thiscritical enzyme, to identify highly important points for PRdimerization inhibition. For this, we have conducted a systematicapplication of the computational ASM method to a total of 70residues present at the interface of the two subunits of thisenzyme, identifying the ones that have a higher contribution tothe formation of the dimer. While some previous pharmacolo-gical studies have focused on the inhibition of PRdimerization,19,20,32�37 showing it as a viable and very promisingstrategy for the development of new anti-HIV drugs, theexistence of specific spots for dimerization inhibition at thesubunit interface remains very poorly understood. Hence, theknowledge arising from the computational ASM analysis of theHIV PR interface described in this study provides importantclues about the association determinants for the two PR subunitsand could serve as a basis for the development of a new class ofHIV-1 PR inhibitors designed to block the association of the twosubunits to yield the fully active PR homodimeric form.
’METHODS
Model Setup and Molecular Dynamics Simulations. Twomodels were considered for this study: a substrate-free PRenzyme and a substrate-bound PR enzyme (substrate Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser). Even though some struc-tures available in the RCSB Protein Data Bank53 refer to thesubstrate-free PR form (2PC0, 2HB4, 2HB2, 1HHP, 3HVP, etc.)they typically contain only one of the two PR subunits and haveonly a moderate resolution. To obtain a representative high-resolution structure of the PR in the absence of substrate thatcontains full PR interface in atomic detail, a typical choice hasbeen to start from a high-resolution structure containing a smallinhibitor that does not disrupt the interface symmetry. Thisapproach typically guarantees that the full PR interface maintainsthe characteristics expected for the substrate-free form, in termsof range of interaction and symmetric nature. Following this idea,we used as a starting structure the 1T3R structure (resolution1.20 Å),54 after removing the small inhibitor present. For thesubstrate-bound PR enzyme, the model was prepared from the1F7A crystallographic structure (resolution 2.0 Å).55 This struc-ture contains a natural PR substrate (KARVLAEAM) and aD25N mutation to prevent catalytic activity. In preparing ourmodel, we have modeled an O� group at the position of the AsnNH2 group to have the wild-type Asp residue at position 25.Several mechanistic studies have demonstrated that the Asp25
amino acid residues of both subunits have different protonationstates and act together with a conserved water molecule tohydrolase the peptidic bond.56�59 In particular, the protonatedAsp25 residue establishes an important hydrogen bond with thepeptidic carbonyl oxygen of the substrate in the substrate-boundPR enzyme,60 while in the free enzyme, at optimal pH bothAsp25 residues interact with each other through a symmetric lowbarrier hydrogen bond (LBHB), that shifts from Asp25 to Asp25residues.61,62 According to this information, the Asp25 residuewith the proper orientation for such an interaction in thesubstrate-bound enzyme was protonated (subunit R in structure1F7A), while in the free enzyme an approximation had to bemade due to the intrinsic limitations of classic moleculardynamics, assigning the proton to one of the two Asp25 residues(subunit β in structure 1T3R). In both cases, the choice on theoxygen atom to be protonated was carefully done to maximizethe resulting hydrogen bonds. Hence, in the PR substrate-freeform, it was the “lower” oxygen atom that was protonated,maximizing the H-bond interaction with the “lower” oxygenatom of the Asp25 residue in the other subunit. In the substrate-bound form, it was the upper oxygen that was protonated,maximizing the hydrogen-bond interaction with the peptidesubstrate carbonyl group.Even though the optimal pH for HIV-1 PR activity is known to
be 4.7,56 conventional protonation states for all the amino acids(with the exception of Asp25) at pH 7 were considered toreproduce in more detail the more typical human physiologicalconditions. All the hydrogen atoms were added using the LEAPprogram in the AMBER 9.0 Software package.63 Cl� counterionswere used to neutralize the excess of positive charges in bothmodels, and a minimum distance of 12 Å of water (TIP3Pmodel)64 was considered to solvate the systems, allowing theapplication of periodic boundary conditions.TheCornell et al. force field was used in all calculations.65 Both
models were subjected to a four-stage refinement protocol usingthe SANDERmodule of AMBER 9.0, in which the constraints on

7047 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
the enzyme were gradually removed. In the first stage (10 000steps), 50 kcal mol�1 �2 harmonic forces were used to restrainthe positions of all atoms in the systems except the ones from thewater molecules. In the second stage (10 000 steps), theseconstraints were applied only to the heavy atoms and in thethird stage (30 000 steps) were limited to the CA and N atom-type atoms (backbone alfa carbons and nitrogens). This processended in a full energy minimization (fourth stage, maximum80 000 steps) until the rms gradient was smaller than 0.02 kcal/mol. Following an initial warming period during 40 ps (in anNVT ensemble), 10 ns of simulation (NPT) was performed foreachmodel, yielding a total simulation time of 20 ns. Only the last6 ns of each simulation was considered for the analysis presented.In the simulations performed, bond lengths involving hydrogenatoms were constrained using the SHAKE algorithm,66 and theequations of motion were integrated with a 2 fs time step. Anonbond-interaction cutoff radius of 10 Šwas adopted for usewith a Particle-Mesh Ewald scheme,67 whereas the Langevinthermostat68�70 was applied to maintain the temperature of thesystem at 310 K.Computational Alanine Scanning Mutagenesis. Our ala-
nine scanning mutagenesis protocol uses a single MD trajectoryof the wild-type system to calculate the binding free energies.From this trajectory, each interfacial residue of the HIV-1 PRsubunits is mutated to alanine, allowing the differences in thebinding free energy (ΔΔGbinding) for eachmutation in relation tothe wild-type to be calculated with the MM-PBSA approach,optimized by considering different internal dielectric constantvalues for different types of amino acid residues.The use of a single trajectory in this process to calculate
ΔΔGbinding has been shown to give a better agreement with theexperimental data than the use of multiple trajectories.47 In fact,for the use of a single trajectory, error cancelation has beenassumed to overcome the reduced sampling of the conforma-tional space.71,72
Following these general principles, the MM-PBSA scriptimplemented in AMBER was used to perform a postprocessingtreatment of the PR R/β dimer by using the structure of the fullenzyme and calculating its respective energy and those of theinteracting monomers (R and β). To generate the structure ofthe alanine mutants, a simple truncation of the mutated sidechain was made, replacing Cγ with a hydrogen atom and settingthe alanine Cβ�H bond direction to that of the former Cγ�Cβ.For the binding free energy calculations, 300 snapshots wereextracted from the last 6000 ps of the MD run.The binding free energy in solution [ΔGBinding(aq)] was
calculated using the thermodynamic cycle shown in Scheme 1in which ΔGBinding(g) is the interaction free energy between theR-subunit and β-subunit in the gas phase and ΔGR-subunit
Solv,ΔGβ-subunit
Solv, and ΔGEnzyme
Solv are the solvation free energy of
the R-subunit, β-subunit, and the dimer, respectively. Thebinding free energy difference between an alanine mutant andthe wild-type enzyme (ΔΔGBinding) in water is defined as
ΔΔGBinding ¼ ΔGBindingmutant �ΔGBindingwildtype
The binding free energy of the two subunits in water can bewritten as the difference between the free energy of the enzymeand that of the corresponding subunits.
ΔGBinding ¼ Gcomplex � ðGR-subunit þGβ-subunitÞThe free energy of the dimer and respective monomers in
water or in solvent can be calculated48 by summing the internalenergy (bond, angle, and dihedral), the electrostatic and the vander Waals interactions, the free energy of polar solvation, the freeenergy of nonpolar solvation, and the entropic contribution toyield the free energy of each component
Gcomponent ¼ Einternal þ Eelectrostatic þ EvdWþ GPolar Solv þ GNonpolarSolv � TS
In this equation, the first three terms were calculated using theforce field considered in the MD simulation, but with no cutoff.The electrostatic solvation free energy was calculated by solvingthe Poisson�Boltzmann equation with the software Delphiversion 4.73,74
According to this method, the protein is modeled as a di-electric continuum of low polarizability embedded in a dielectricmedium of high polarizability.74 The solvent is assumed to be ahomogeneous medium characterized by a single dielectric con-stant with a value that is usually near 80, which is taken to beequal to the bulk value for pure solvent. Separated by an abruptinterface, the solvent is in contact with the solute that isrepresented as a dielectric body whose shape is defined by atomiccoordinates and radii.74
For the energy calculations, three internal dielectric constantvalues, exclusively characteristic of themutated amino acids, wereused: 2 for the nonpolar amino acids, 3 for the polar residues, and4 for the charged amino acids.47 The different internal dielectricconstants considered have been shown to mimic the differentdegree of relaxation of the subunit interface when different typesof amino acids are mutated for alanine.47
The nonpolar contribution to solvation free energy due to vander Waals interactions between the solute and the solvent andcavity formation was modeled as a term that is dependent on thesolvent-accessible surface area of the molecule, estimatedthrough the use of the following empirical relation
ΔGnonpolar ¼ RAþ β
where A is the solvent-accessible surface area that was estimatedusing the molsurf program (based on the idea primarily devel-oped by Connolly)75 and R and β are empirical constants forwhich the values of 0.00542 kcal Å�2 mol�2 and 0.92 kcal mol�1
were used. Finally, the entropy term, obtained from the sum ofthe sum of translational, rotational, and vibrational components,was not calculated because it was assumed, based on previouswork, to have a neglectable contribution to ΔΔGbinding.
71
The ASM protocol used here,47 based in the well-knownMM-PBSA approach,46,48,49 has been used with success in the study ofseveral biological systems, including the IgG1 streptococcalprotein G (C2 fragment) complex,50 the ZipA:FtsZ complex,the complex formed between the hen egg white lysozyme (HEL)
Scheme 1. Thermodynamic Cycle Employed toCalculate theAssociation Free Energy (ΔGBinding)

7048 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
and the antibody HyHEL-10,51 and theMDM2�P53 complex,52
and in previous benchmarking studies against experimental datahas been shown to have an overall success of 82% in identifyinghot spots and 80% in identifying warm spots and to yield a meanunsigned error of around 0.5 kcal/mol.Mutant Selection. Residues to mutate were selected from a
detailed analysis of the HIV PR interface based on the 1F7Acrystallographic structure (resolution 2.0 Å).55 A total of 70interface residues were identified (35 in each subunit). Alanine,glycine, and proline interface residues were not considered inthis study.Alanine scanning mutagenesis of alanine residues is a redun-
dant process. Glycine has a smaller side chain than alanine andhence cannot be effectively mutated to alanine in a consistentfashion within the protocol considered for the other amino acids.In fact, in glycine the absence of a side chain confers to thebackbone an additional conformational freedom that typicallyresults in relevant structural rearrangements of the backbone.Hence, the experimental measure of the ΔΔGbinding for suchmutation does not correspond to the subunit interaction differ-ence between glycine and alanine but rather to a more globalinteraction difference that includes also the backbone rearrange-ment effects. This is an obvious limitation of the experimentalASM method that could be eventually surpassed computation-ally, namely with the use of a single trajectory. The difficulty herelies in the lack of experimental reference values for glyci-ne�alanine mutations, essential to fully calibrate the computa-tional ASM method employed to treat all the other amino acidresidues in this study.A similar problem can be highlighted for proline. In fact, the
proline ring restricts the geometry of the backbone chain in theproteins where it is present, sometimes resulting in a verydifferent backbone conformation than the one adopted whenan alanine or any other residue is present. Experimental ASM ofproline is therefore particularly troublesome, as it can produceabnormal changes to the binding free energies as a result of thebackbone conformational differences, masking the results.47,76
For these reasons, proline residues were also not evaluated.ΔΔGbinding values were calculated for all the other interface
amino acid residues for both the free enzyme and the substrate-bound enzyme.SASA Analysis. Solvent accessible surface area values were
calculated for all interface amino acid residues (backboneþ sidechain) using the program Visual Molecular Dynamics (VMD)77
and considering the standard probe radius for water of 1.4 Å.From theMD simulations performed and for each residue, SASAvalues were calculated for a total of 3000 MD snapshots from thelast 6 ns of simulation, considering: (1) the full dimer—SASAdimer; (2) only the residues from the same subunit, neglect-ing the shielding effect of the other subunit—SASAmonomer—and yielding a value for the potential SASA in themonomer. Finalvalues were expressed as a percentage of the potential SASA forthe free residue.
’RESULTS AND DISCUSSION
1. General Analysis of the MD Simulation. Prior to the ASMstudy, a general analysis of the MD trajectories generated wasperformed to evaluate the convergence and stability of the mostrelevant properties within the 10 ns simulation times considered.Properties evaluated included the potential and kinetic energies,the temperature, the pressure, the root-mean-square deviation, etc.From these general properties, the root-mean-square deviation(RMSd) is normally the most difficult to converge and is usuallytaken as a reference to assess the stability of an MD simulation.Figure 1 illustrates the RMSd values for the backbone CR
atoms in the MD simulations of the free enzyme and of thesubstrate-bound enzyme. In addition, this figure also shows theRMSd values for the subset of backbone CR atoms of the 70interface amino acid residues that will be the subject of moredetail in this study. The results show that both models are wellequilibrated after the initial 4 ns of simulation and in particularthe interface amino acid residues. In agreement with thisobservation, the remaining 6 ns of simulation was taken intoconsideration for the ASM calculations and subsequent analysis.2. Analysis of the Interface in the Free PR Enzyme. The PR
interface is remarkably extensive for such a small enzyme. In fact,upon dimerization, each monomer buries more than 50% of itstotal surface area, with 64% and 62% of its nonpolar and polarsurfaces becoming shielded from the solvent.26 The PR subunitinterface is defined to a great extent by a group of eightinterdigiting N- and C-terminal residues (residues 1�4 and96�99) from each of the two monomers, arranged in a set offour interlocking antiparallel beta strands.11,19 This set of 16residues has also been shown to represent 45% of the buriedsurface area during dimer formation.78
In this study, we are interested in understanding how theseeffects can be translated in terms of an energetic contribution to
Figure 1. RMSd analysis of the backbone CR atoms in theMD simulations of the free enzyme and of the substrate-bound enzyme. Particular attention isdedicated to the behavior of the interface amino acid residues.
7049 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
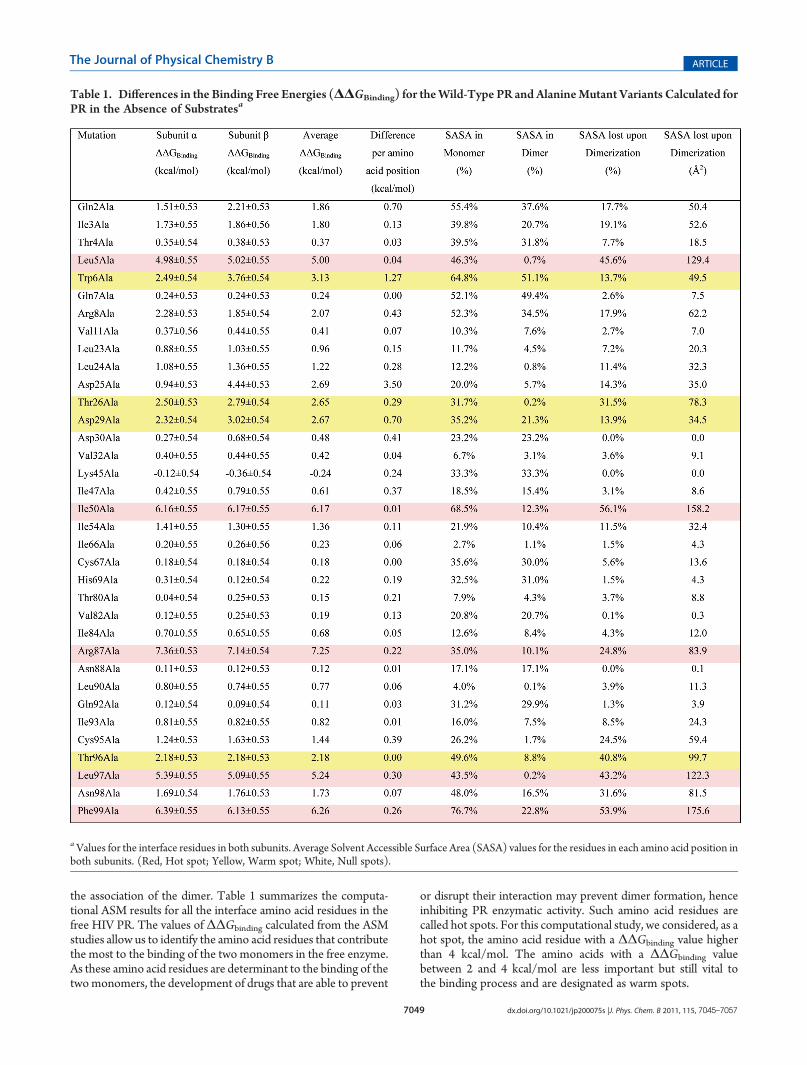
the association of the dimer. Table 1 summarizes the computa-tional ASM results for all the interface amino acid residues in thefree HIV PR. The values of ΔΔGbinding calculated from the ASMstudies allow us to identify the amino acid residues that contributethe most to the binding of the two monomers in the free enzyme.As these amino acid residues are determinant to the binding of thetwomonomers, the development of drugs that are able to prevent
or disrupt their interaction may prevent dimer formation, henceinhibiting PR enzymatic activity. Such amino acid residues arecalled hot spots. For this computational study, we considered, as ahot spot, the amino acid residue with a ΔΔGbinding value higherthan 4 kcal/mol. The amino acids with a ΔΔGbinding valuebetween 2 and 4 kcal/mol are less important but still vital tothe binding process and are designated as warm spots.
Table 1. Differences in the Binding Free Energies (ΔΔGBinding) for theWild-Type PR and AlanineMutant Variants Calculated forPR in the Absence of Substratesa
aValues for the interface residues in both subunits. Average Solvent Accessible Surface Area (SASA) values for the residues in each amino acid position inboth subunits. (Red, Hot spot; Yellow, Warm spot; White, Null spots).

7050 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
From the 35 pairs of residues evaluated at the interface of thesubunit of PR, only 9 (i.e., ca. 25%) were shown to have anenergetic contribution higher than 2 kcal/mol to the formation ofthe dimer. In this regard, the most important residues are Arg87,Phe99, Ile50, Leu97, and Leu5. All these 5 pairs of residues haveΔΔGbinding of more than 4 kcal/mol and hence are consideredhot spots for dimer formation. The remaining 4 pairs of residues,with ΔΔG binding values between 2 and 4 kcal/mol, are Trp6,Asp29, Thr26, and Thr96 and are warm spots. The remaining 26pairs of residues at the interface are null spots or cold spots,designations that refer, respectively, to amino acid residueswhose side chain has little effect on the stabilization of the dimer(between 0 and 2 kcal/mol) or whose contribution is detrimentalto dimer formation (negative contribution).A systematic study79 analyzing a total of 2325 alanine mutants
for which the change in the free energy of binding uponmutationto alanine had been experimentally measured has identified
tryptophan, tyrosine, arginine, isoleucine, and aspartate as themost common residues present among those contributing morethan 2 kcal/mol for the ΔΔGbinding.These general trends are in agreement with the identification of
Arg87, Ile50, Trp6, andAsp29 as hot spots andwarm spots inHIVPR. Leu5 and Leu97, however, are rather uncommon among hotspots and warm spots, with the above-mentioned study havingidentified only two leucine residues contributing more than 2kcal/mol to ΔΔG binding among a total of 242 leucine�alaninemutants considered. According to the same study,79 phenylala-nines and threonines are also rather uncommon as hot spots orwarm spots, with only five Phe and two Thr identified in a total of166 phenyalanine�alanine mutants and 131 threonine�alaninemutants. The unusually high energetic contribution calculated inthis study for Leu5, Thr26, Thr96, Leu97, and Phe99 could be adistinctive characteristic of HIV-1 protease and will be the subjectof particular detail over the subsequent sections.
Figure 2. Schematic representation of themost important interactions between residues from the different subunits, with indication of the hot spots andwarm spots identified.

7051 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
The values presented in Table 1 also allow one to have anenergetic view of the symmetric nature of the interface formed, byenabling a direct comparison of the ΔΔGbinding calculated uponalanine mutation for the same residue in each subunit. For mostinterface amino acid residues in the free PR study, there is anenergetic difference of less than 0.5 kcal/mol between the energeticcontribution of a given amino acid residue in subunit R and that ofthe same residue at subunit β. In addition, for 33 of the 35 pairs ofresidues there is a perfect qualitative agreement between theclassification of the residue as hot spot, warm spot, or null spotin both subunits. The only exceptions are Asp25 (for whichdifferent protonation states were assigned in the MD simulationsperformed as an approximation, even though experimentally a lowbarrier hydrogen bond—LBHB—shifting from one Asp25 to theother has been shown to take place)62,80 and the amino acidresidue Gln2. In line with the LBHD theory,62,80 for Asp25 the realvalue should be an average of the two extreme values calculated forthe protonated (0.94 kcal/mol) and deprotonated alternatives(4.44 kcal/mol). The importance of Asp25 for dimer formationhas been previously discussed in the literature.28,30,81 In fact,mutating Asp25 to Asn has been shown to destabilize dimerformation, however, to a lesser extent than the T26A mutation.28
Figure 2 illustrates the position of the hot spots and warmspots identified in this study in the structure of the HIV PRdimer, while Figure 3 gives a more detailed view of the twoindividual monomers.
The energetic agreement between the results calculated foramino acid position in both subunits is in line with previousstructural studies, which highlighted the symmetric nature of thePR enzyme in the absence of substrate,55 a characteristic featurethat is lost upon substrate binding. In fact, the average differencebetween the calculated ΔΔGbinding for all 35 pairs of residues ofthe two subunits is of only 0.31 kcal/mol in the free enzyme (0.17kcal/mol discarding the outliers Gln2, Pro6, and Asp25). Due tothis symmetric nature of the PR interface, we considered ascriteria for hot/warm/null spot for each amino acid position inthe absence of substrate the average ΔΔG binding (kcal/mol)obtained for each of the two subunits.A computational structure-based thermodynamic analysis of
the PR dimer had already suggested Leu5, Thr96, Leu97, andPhe99 as very important residues for dimerization.26 Deletion ofthe C-terminal β-strand of HIV-1 protease,30 which includes thehot spots Leu97 and Phe99 and the warm spot Thr96, has beenpreviously shown to produce a folded monomer in solution,instead of the normally more stable dimer. In addition, thestability of the PR dimer in a I50V mutant has been previouslyevaluated experimentally against that of the wild-type PR byassessing the PR activity with increasing concentrations ofdenaturing urea.82 In particular, the results showed that amutation at this position led to a 60% decrease in the dimerstability, a value which is in agreement with the high energeticcontribution of this residue envisioned from the present
Figure 3. Schematic representation of the two individual monomers that constitute the active HIV PR dimer, illustrating the relative position of all thehot spots and warm spots identified.

7052 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
computational study and with its role as a hot spot. Mutations atthis position have also been addressed in several other studiesanalyzing inhibitor binding in the PR dimer.83,84
Interestingly, this same study82 examined the importance ofmutations at position 54 (I54M and I54V), showing that muta-tions at this residue do not affect dimer stability, a result which isalso in agreement with the suggested role of this residue as a nullspot in the present analysis. Several other studies28,30,31 have alsodemonstrated that the introduction of mutations at the PR hotspot Arg87 (R87K), and into the warm spots Thr26 and Asp29(T26A, D29N, and D29A), disrupts the dimer interface contactsand destabilizes the protease dimer, causing the inhibition ofprotease dimerization.Several studies have suggestedDarunavir, a second generation,
recently accepted PR inhibitor, to block dimerization.20 Thebinding affinity of this inhibitor for the wild-type enzyme is about2 orders of magnitude stronger than that of first generation PRinhibitors,85 with structural studies showing that Darunavir canbind to two distinct sites: (1) the active site cavity, interactingwith the active-site amino acids Asp29 and Asp30 and (2) thesurface of one of the flexible flaps in the PR dimer, interactingwith residues Glu35, Trp42, Pro44-Met46, Lys55-Arg57, andVal77-Pro79.84,86,87 Darunavir and two other related experimen-tal PR inhibitors have been shown to block PR dimerization inconcentrations as low as 0.01 μM and to block HIV-PR replica-tion in vitro with IC50 values of 0.0002�0.48 μM.20 However,once the PRmonomers dimerize to yield a mature PR dimer, thedimer is not dissociated by these dimerization inhibitors, sug-gesting that these agents block dimerization at the nascent stageof protease maturation.20 Further studies analyzing the interac-tion of these inhibitors with the free monomer are required tofully understand their mechanism of interaction.3. Energetic Contribution to Hot Spots.Table 2 presents the
energetic contribution of all the individual components to thedifferences in the binding free energies (ΔΔGBinding) between thewild-type PR and alanine mutant variants for hot spots, warmspots, and null spots identified. The components presentedinclude the electrostatic energy term (ΔΔEelectrostatic), the vander Waals energy term (ΔΔEvdW), and the polar (ΔΔGPolarSolv)and nonpolar (ΔΔGNonPolarSolv) contributions to the solvationfree energy. The last two columns—hydrophilic and hydrophobic
contributions—represent, respectively, the sum of the electro-static energy term and of polar contribution to the solvation freeenergy (hydrophilic contribution) and of the van der Waals andnonpolar contribution to the solvation free energy (hydrophobiccontribution).Table 2 shows that the five hot spots identified have an average
ΔΔGBinding of 5.98 kcal/mol, while the five warm spots have anaverage value of 2.66 kcal/mol and the remaining interface aminoacids residues (null spots) of only 0.80 kcal/mol.Analyzing the several contributions to the ΔΔGBinding, it
can be observed that the hot spots identified have a lowerΔΔEelectrostatic and a dramatically higherΔΔEvdW in comparisonwith the other amino acid residues studied. In terms of thesolvation free energy terms, the hot spots identified have a higher(less negative) contribution to the ΔΔGPolarSolv and an alsohigher (more positive) contribution to the ΔΔGNonPolarSolv.
Combining these energy terms into hydrophilic and hydropho-bic contributions to the binding free energy (Table 2) highlightsthe importance of the hydrophobic interactions to the bindingfree energies in the hot spots identified (average contribution7.51 kcal/mol for hot spots, against 1.89 in the warm spots and0.89 in the null spots). The contribution of the hydrophilicenergy term is much smaller, and in this case the differences inthe average values calculated for hot spots, warm spots, and nullspots are also less dramatic.4. SASA Analysis. Bogan and Thorn79 in their seminal work
“Anatomy of Hot-Spots in Protein Interfaces” have found theexistence of very little correlation between the buried surface areaand the free energy of binding, and have concluded that theocclusion from solvent is a necessary although not sufficientrequirement for the occurrence of hot spots.In this section, we analyze this issue for the case of HIV PR but
from a different point of view. Table 1 presents also the solventaccessible surface area (SASA) values calculated for each residue,considering the full dimer (SASAdimer) and considering only theresidues in the same subunit (SASAmonomer). Given the largedifference in size that characterizes the different amino acidresidues present at the interface, the values are presented as apercentage of the potential SASA for the free residue.In the convention used in this study, a high SASAmonomer
indicates that a given residue is poorly shielded by residues from
Table 2. Energetic Contribution of All the Individual Components to the Differences in the Binding Free Energies (ΔΔGBinding)between the Wild-Type PR and Alanine Mutant Variants for the Hot Spots, Warm Spots, and Null Spots Identified in the Absenceof Substratea
hot spots warm spots null spots total
amino acid pairs identified 5 4 26 35
average ΔΔGBinding (kcal/mol) 5.98 2.66 0.80 1.75
average ΔΔEelectrostatic (kcal/mol) �0.78 6.84 1.10 1.49
average ΔΔEvdW (kcal/mol) 7.19 1.75 0.87 1.87
average ΔΔGPolarSolv (kcal/mol) �0.75 �6.07 �1.18 �1.68
average ΔΔGNonPolarSolv(kcal/mol) 0.32 0.14 0.02 0.08
average hydrophilic contribution (kcal/mol) �1.53 0.77 �0.09 �0.19
average hydrophobic contribution (kcal/mol) 7.51 1.89 0.89 1.95
average SASA in monomer (%) 54.0% 45.3% 25.1% 31.5%
average SASA in dimer (%) 9.2% 20.3% 17.2% 16.4%
average SASA lost upon dimerization (%) 44.7% 25.0% 7.9% 15.1%
average SASA lost upon dimerization (Å2) 133.9 65.5 21.5 42.6aAverage Solvent Accessible Surface Area (SASA) values.

7053 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
its own subunit and exposed to the solvent and/or to residuesfrom the other subunit, while a low SASAmonomer indicates aresidue that is highly protected from other interactions by residuesof its own subunit. Similarly, a high SASAdimer illustrates that in thedimer a given residue is very much exposed to the solvent, while alow SASAdimer indicates a residue extremely protected by residuesfrom the two subunits. From the difference between SASAdimer
and SASAmonomer (described as SASA lost upon dimerization), it ispossible to get an idea of the contribution in terms of interactionsurface area of each residue to the subunit interface (valuesexpressed in percentage and in area).The results expressed in Table 1 show that all the hot spots
identified have a very low SASA in the dimer. Leu97 andLeu5 have average SASAdimer values of 0.2% and 0.7%, respec-tively, while Arg87 and Ile50 have average SASAdimer values of10.1% and 12.3%. Phe99 confirms this general tendency with aSASAdimer of 22.8%. These values contrast markedly with theestimated SASAmonomer values for these residues, for whichvalues between 35.0% (Arg87) and 76.7% (Phe99) weredetermined.Considering the SASAdimer and SASAmonomer values, it becomes
evident that dimerization leads to amajor decrease in the SASA forthese residues. In fact, for Leu5, Ile50, Leu97, and Phe99, thisdecrease is of more than 40% of their potential amino acid SASA.This difference is particularly noticeable in terms of absolute SASA(expressed in Å2) with the hot spots identified having typicallyaverage values of SASA lost upon dimerization higher than 100 Å2
(the only exception is Arg87, albeit with 83.9 Å2).Table 2 presents the average SASA values calculated for hot
spots, warm spots, and null spots, considering the categoriespresented above. Hot spots have clearly a higher SASA whenconsidering residues only from their own subunit(SASAmonomer), with 54% of their potential surface area freefor interaction with the other subunit and/or with solventmolecules, a value that decreases to 9.2% in the dimer (averagedecrease of 133.9 Å2 per residue).Warm spots are less exposed when considering only the
monomer (45%), and upon dimer formation the correspondingSASA decreases to 20.3%, which represents an average decreaseof 65.5 Å2 per amino acid residue. Null spots interact preferen-tially with their own subunit, exhibiting an average SASAmonomer
of only 25.1%, a value which decreases only slightly whenconsidering the dimer (to 17.4%, 21.4 Å2).Following the study of Bogan and Thorn,79 we have evaluated
the existence of a relationship between the solvent exposedsurface area of the interface amino acid residues and theircorresponding free energy of binding. For that, we have com-pared the free energy of binding with the SASA lost upondimerization for all the interfacial amino acid residue pairs.Results are presented in Figure 4, illustrating the behaviorobserved for the charged, polar, and nonpolar amino acidresidues. The results demonstrate the existence of an evidentcorrelation between the two quantities evaluated, particularly forthe nonpolar amino acid residues. In general, the higher thedecrease in the SASA when going from the monomer to thedimer in HIV PR (i.e., the SASA lost upon dimerization) thehigher the energetic contribution of that particular amino acid fordimer formation. This correlation is naturally less linear forcharged amino acid residues, for which other features (the chargefor example) came into play.Globally, these results confirm that for HIV protease the
contribution of the hot spots identified to the interfacial surfacearea is clearly higher than that of warm and null spots and that inthe dimer formed these residues have typically a much smallersolvent accessible surface area than warm spots and null spots.5. Hydrogen Bonding Analysis. One particular advantage
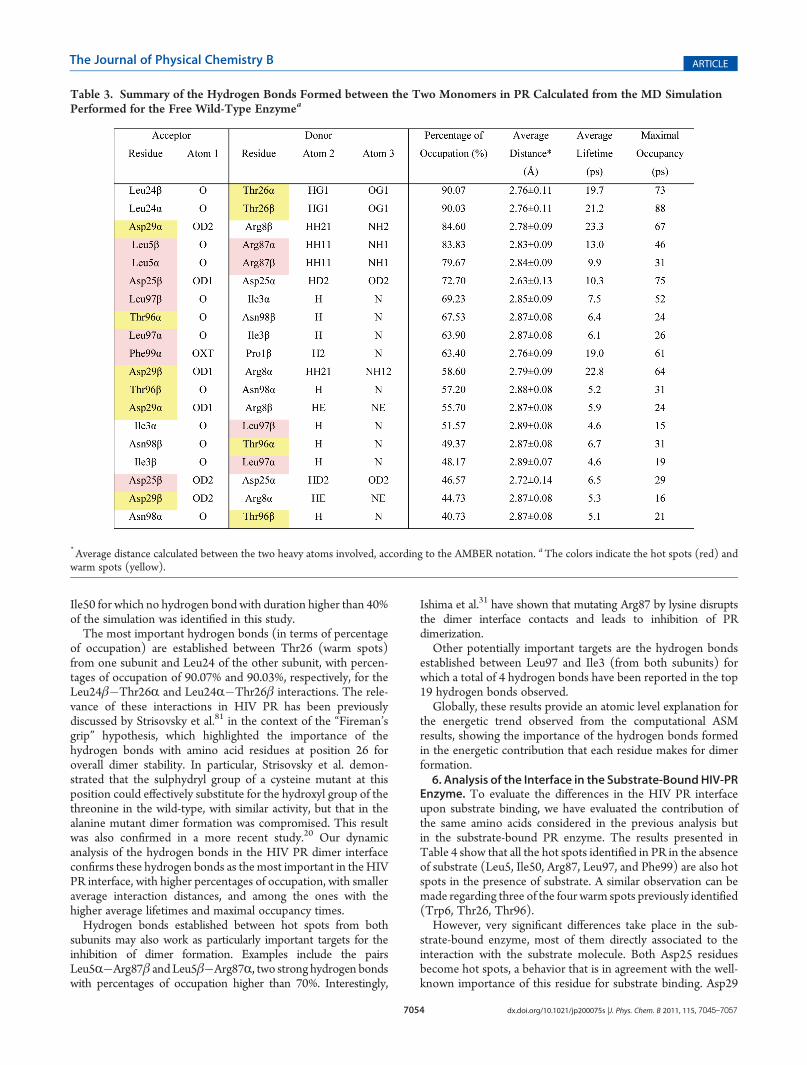
that MD simulations in general offer when studying biologicalsystems is the ability to determine a statistical picture on a varietyof atomic level dynamic events. This feature is particularlyimportant for events that are transient or at least short-lived,such as the formation and breaking of hydrogen bonds.To obtain a more detailed portrait of the PR subunit interface,
we have performed a hydrogen bonding analysis of all theinteractions between residues from the two subunits. Results arepresented in Table 3 and graphically illustrated also in Figure 2.The results show that there are 19 hydrogen bonds between
both subunits that are present during more than 40% of thesimulation. All of these interactions involve residues that havebeen identified as hot spots or warm spots. Nine of theseinteractions (47%) involve hot spots, with four out of the fivepairs of hot spots identified in this study participating in theseinteractions. The exception among the identified hot spots is
Figure 4. Representation of the SASA lost upon dimerization (Å2) as a function of the ΔΔGBinding (kcal/mol) for the interfacial charged, polar, andnonpolar amino acid residues.
7054 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
Ile50 for which no hydrogen bond with duration higher than 40%of the simulation was identified in this study.The most important hydrogen bonds (in terms of percentage
of occupation) are established between Thr26 (warm spots)from one subunit and Leu24 of the other subunit, with percen-tages of occupation of 90.07% and 90.03%, respectively, for theLeu24β�Thr26R and Leu24R�Thr26β interactions. The rele-vance of these interactions in HIV PR has been previouslydiscussed by Strisovsky et al.81 in the context of the “Fireman’sgrip” hypothesis, which highlighted the importance of thehydrogen bonds with amino acid residues at position 26 foroverall dimer stability. In particular, Strisovsky et al. demon-strated that the sulphydryl group of a cysteine mutant at thisposition could effectively substitute for the hydroxyl group of thethreonine in the wild-type, with similar activity, but that in thealanine mutant dimer formation was compromised. This resultwas also confirmed in a more recent study.20 Our dynamicanalysis of the hydrogen bonds in the HIV PR dimer interfaceconfirms these hydrogen bonds as themost important in theHIVPR interface, with higher percentages of occupation, with smalleraverage interaction distances, and among the ones with thehigher average lifetimes and maximal occupancy times.Hydrogen bonds established between hot spots from both
subunits may also work as particularly important targets for theinhibition of dimer formation. Examples include the pairsLeu5R�Arg87β andLeu5β�Arg87R, two strong hydrogen bondswith percentages of occupation higher than 70%. Interestingly,
Ishima et al.31 have shown that mutating Arg87 by lysine disruptsthe dimer interface contacts and leads to inhibition of PRdimerization.Other potentially important targets are the hydrogen bonds
established between Leu97 and Ile3 (from both subunits) forwhich a total of 4 hydrogen bonds have been reported in the top19 hydrogen bonds observed.Globally, these results provide an atomic level explanation for
the energetic trend observed from the computational ASMresults, showing the importance of the hydrogen bonds formedin the energetic contribution that each residue makes for dimerformation.6. Analysis of the Interface in the Substrate-BoundHIV-PR
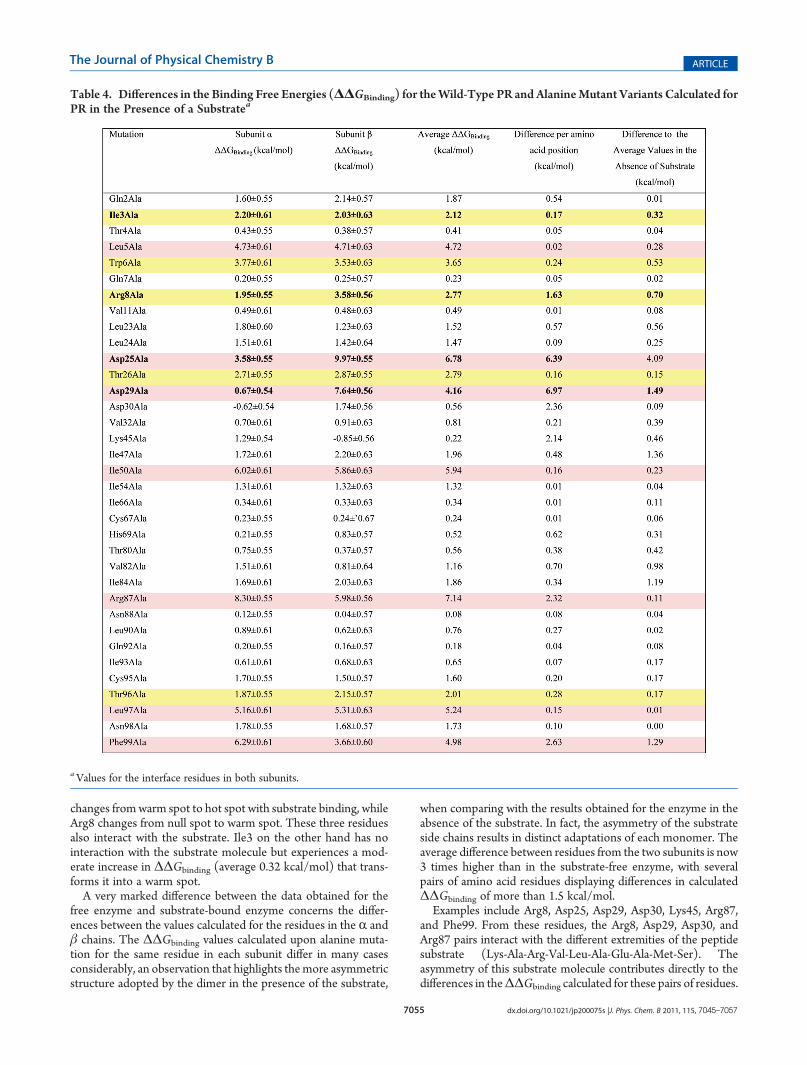
Enzyme. To evaluate the differences in the HIV PR interfaceupon substrate binding, we have evaluated the contribution ofthe same amino acids considered in the previous analysis butin the substrate-bound PR enzyme. The results presented inTable 4 show that all the hot spots identified in PR in the absenceof substrate (Leu5, Ile50, Arg87, Leu97, and Phe99) are also hotspots in the presence of substrate. A similar observation can bemade regarding three of the four warm spots previously identified(Trp6, Thr26, Thr96).However, very significant differences take place in the sub-
strate-bound enzyme, most of them directly associated to theinteraction with the substrate molecule. Both Asp25 residuesbecome hot spots, a behavior that is in agreement with the well-known importance of this residue for substrate binding. Asp29
Table 3. Summary of the Hydrogen Bonds Formed between the Two Monomers in PR Calculated from the MD SimulationPerformed for the Free Wild-Type Enzymea
*Average distance calculated between the two heavy atoms involved, according to the AMBER notation. aThe colors indicate the hot spots (red) andwarm spots (yellow).
7055 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
changes fromwarm spot to hot spot with substrate binding, whileArg8 changes from null spot to warm spot. These three residuesalso interact with the substrate. Ile3 on the other hand has nointeraction with the substrate molecule but experiences a mod-erate increase in ΔΔGbinding (average 0.32 kcal/mol) that trans-forms it into a warm spot.A very marked difference between the data obtained for the
free enzyme and substrate-bound enzyme concerns the differ-ences between the values calculated for the residues in the R andβ chains. The ΔΔGbinding values calculated upon alanine muta-tion for the same residue in each subunit differ in many casesconsiderably, an observation that highlights themore asymmetricstructure adopted by the dimer in the presence of the substrate,
when comparing with the results obtained for the enzyme in theabsence of the substrate. In fact, the asymmetry of the substrateside chains results in distinct adaptations of each monomer. Theaverage difference between residues from the two subunits is now3 times higher than in the substrate-free enzyme, with severalpairs of amino acid residues displaying differences in calculatedΔΔGbinding of more than 1.5 kcal/mol.Examples include Arg8, Asp25, Asp29, Asp30, Lys45, Arg87,
and Phe99. From these residues, the Arg8, Asp29, Asp30, andArg87 pairs interact with the different extremities of the peptidesubstrate (Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser). Theasymmetry of this substrate molecule contributes directly to thedifferences in theΔΔGbinding calculated for these pairs of residues.
Table 4. Differences in the Binding Free Energies (ΔΔGBinding) for theWild-Type PR and AlanineMutant Variants Calculated forPR in the Presence of a Substratea
aValues for the interface residues in both subunits.

7056 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
These observations are in agreement with previous X-ray studiesthat have analyzed how this symmetric enzyme adjusts to recognizeits asymmetric substrates.55 Although the specific alterations ob-served in this study are characteristic of the specific substrateconsidered, similar trends (albeit involving probably differentcombinations of residues) would also be observed for substrateshaving any of the other recognition sequences that HIV-1 proteasecleaves specifically, as all of them are structurally asymmetric.
’CONCLUSIONS
In this study, we presented a detailed atomistic analysis of thesubunit interface in the HIV-1 protease enzyme, an importanttarget in HIV therapy. In particular, we have evaluated theenergetic contribution of all the amino acid residues at thesubunit interface of this homodimeric enzyme, in an attempt toidentify the most important determinants for dimer formation.This analysis was further complemented with a dynamic analysisof the hydrogen bonds formed between residues from differentsubunits and of the solvent accessible surface areas of allinterfacial amino acid residues, providing relevant clues for thedevelopment of new inhibitors of HIV-1 protease designed toblock the association of the two subunits, thereby preventing theformation of the fully active PR homodimeric form.
The ASM results highlight the particular importance of a set offive amino acid pairs for dimer formation (hot spots): Arg87,Phe99, Ile50, Leu97, and Leu5 (Figure 2 and Figure 3). Mutatingone of these residues by alanine leads to a destabilization of theresulting dimer formed ofmore than 4 kcal/mol when comparingwith the wild-type PR. These residues have also been shown to bethe largest contributors, in terms of area, to the subunit interfaceof the dimer, with each of these residues losing on average 134 Å2
of solvent accessible surface area upon dimer formation. Fourother pairs of residues also make a significant energetic contribu-tion (albeit more modest than the latter) to dimer formation.These residues, identified as warm spots, are Trp6, Asp29, Thr26,and Thr96, and have also an intermediate contribution in termsof area to the subunit interface. All the remaining residuesevaluated have only a residual energetic contribution to dimerformation and a much more modest contribution to the inter-facial area between both subunits.
This study also shows that all the most important hydrogenbonds between residues from different subunits involve at leastone hot spot or one warm spot. In particular, the hydrogen bondsformed between Leu24 and Thr26 from the two subunits wereshown to be the most stable ones during the molecular dynamicssimulations performed, a result that is in line with previousexperimental studies that have shown the importance of thishydrogen bond to dimer formation.20,81 Other key hydrogenbonds involve the pairs Leu5�Arg87 and Ile3�Leu97.
Inhibitors designed to bind the residues identified in this studyas hot spots or warm spots, or to interfere or disrupt the morerelevant and persistent hydrogen bonds observed in the course oftheMD simulations, are much more likely to have a greater effecton blocking the formation of the fully active PR homodimericform. These features should be taken into account in future drugdesign and development studies targeting HIV-1 protease.
’ASSOCIATED CONTENT
bS Supporting Information. Detailed description and illus-tration on the protonation of the Asp 25 residues for both
subunits in the free and in the substrate-bound PR enzyme.Comparison of the ΔΔGBinding values calculated by ASM fora selected number of interfacial amino acid residues startingfrom a PR model prepared from an alternative X-ray structure(2PC0). This material is available free of charge via the Internetat http://pubs.acs.org.
’AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
’ACKNOWLEDGMENT
FCT (PTDC/QUI/68302/2006).
’REFERENCES
(1) Kallings, L. O. J. Int. Med. 2008, 263, 218–243.(2) Flexner, C. New Engl. J. Med. 1998, 338, 1281–1292.(3) Misson, J.; Clark, W.; Kendall, M. J. J. Clin. Pharm. Ther. 1997,
22, 109–117.(4) Deeks, S. G.; Smith, M.; Holodniy, M.; Kahn, J. O. J. Am. Med.
Assoc. 1997, 277, 145–153.(5) Wlodawer, A.; Vondrasek, J. Annu. Rev. Biophys. Biomol. Struct.
1998, 27, 249–284.(6) Mastrolorenzo, A.; Rusconi, S.; Scozzafava, A.; Supuran, C. T.
Exp. Opin. Ther. Pat. 2006, 16, 1067–1091.(7) Aruksakunwong, O.; Promsri, S.; Wittayanarakul, K.;
Nimmanpipug, P.; Lee, V. S.; Wijitkosoom, A.; Sompornpisut, P.;Hannongbua, S. Curr. Comput. Aided Drug Des. 2007, 3, 201–213.
(8) Martinez-Cajas, J. L.; Wainberg, M. A. Antiviral Res. 2007,76, 203–221.
(9) Fernandez-Montero, J. V.; Barreiro, P.; Soriano, V. Exp. Opin.Pharmacother. 2009, 10, 1615–1629.
(10) Tamames, B.; Fernandes, P. A.; Ramos, M. J. Curr. Bioact.Compd. 2006, 2, 43–62.
(11) Wlodawer, A.; Miller, M.; Jaskolski, M.; Sathyanarayana, B. K.;Baldwin, E.; Weber, I. T.; Selk, L. M.; Clawson, L.; Schneider, J.; Kent,S. B. H. Science 1989, 245, 616–621.
(12) Miller, M.; Schneider, J.; Sathyanarayana, B. K.; Toth, M. V.;Marshall, G. R.; Clawson, L.; Selk, L.; Kent, S. B. H.; Wlodawer, A.Science 1989, 246, 1149–1152.
(13) Copeland, T. D.; Oroszlan, S.Gene Anal. Tech. 1988, 5, 109–115.(14) Erickson, J. W.; Burt, S. K. Annu. Rev. Pharmacol. Toxicol. 1996,
36, 545–571.(15) Kurt, N.; Haliloglu, T.; Schiffer, C. A. Biophys. J. 2003, 85,
853–863.(16) Ozer, N.; Haliloglu, T.; Schiffer, C. A. Proteins 2006, 64, 444–456.(17) Moore, J. P.; Stevenson, M. Nat. Rev. Chem. Discovery 2000, 1,
40–49.(18) Condra, J. H.; Schleif, W. A.; Blahy, O. M.; Gabryelski, L. J.;
Graham, D. J.; Quintero, J. C.; Rhodes, A.; Robbins, H. L.; Roth, E.;Shivaprakash, M.; Titus, D.; Yang, T.; Teppler, H.; Squires, K. E.;Deutsch, P. J.; Emini, E. A. Nature 1995, 374, 569–571.
(19) Davis, D. A.; Brown, C. A.; Singer, K. E.; Wang, V.; Kaufman, J.;Stahl, S. J.; Wingfield, P.; Maeda, K.; Harada, S.; Yoshimura, K.;Kosalaraksa, P.; Mitsuya, H.; Yarchoan, R.Antiviral Res. 2006, 72, 89–99.
(20) Koh, Y.; Matsumi, S.; Das, D.; Amano, M.; Davis, D. A.; Li, J. F.;Leschenko, S.; Baldridge, A.; Shioda, T.; Yarchoan, R.; Ghosh, A. K.;Mitsuya, H. J. Biol. Chem. 2007, 282, 28709–28720.
(21) Spaltenstein, A.; Kamierski, W. M.; Miller, J. F.; Samano, V.Curr. Top. Med. Chem. 2005, 5, 1589–1607.
(22) Camarasa, M. J.; Velazquez, S.; San-Felix, A.; Perez-Perez, M. J.;Gago, F. Antiviral Res. 2006, 71, 260–267.
(23) Broglia, R. A.; Levy, Y.; Tiana, G. Curr. Opin. Struct. Biol. 2008,18, 60–66.

7057 dx.doi.org/10.1021/jp200075s |J. Phys. Chem. B 2011, 115, 7045–7057
The Journal of Physical Chemistry B ARTICLE
(24) Grant, S. K.; Deckman, I. C.; Culp, J. S.; Minnich, M. D.;Brooks, I. S.; Hensley, P.; Debouck, C.; Meek, T. D. Biochemistry 1992,31, 9491–9501.(25) Xie, D.; Gulnik, S.; Gustchina, E.; Yu, B.; Shao, W.; Qoronfleh,
W.; Nathan, A.; Erickson, J. W. Protein Sci. 1999, 8, 1702–1707.(26) Todd, M. J.; Semo, N.; Freire, E. J. Mol. Biol. 1998, 283,
475–488.(27) Levy, Y.; Caflisch, A. J. Phys. Chem. B 2003, 107, 3068–3079.(28) Louis, J. M.; Ishima, R.; Nesheiwat, I.; Pannell, L. K.; Lynch,
S. M.; Torchia, D. A.; Gronenborn, A. M. J. Biol. Chem. 2003,278, 6085–6092.(29) Levy, Y.; Caflisch, A.; Onuchic, J. N.; Wolynes, P. G. J. Mol. Biol.
2004, 340, 67–79.(30) Ishima, R.; Torchia, D. A.; Lynch, S. M.; Gronenborn, A. M.;
Louis, J. M. J. Biol. Chem. 2003, 278, 43311–43319.(31) Ishima, R.; Ghirlando, R.; Tozser, J.; Gronenborn, A. M.;
Torchia, D. A.; Louis, J. M. J. Biol. Chem. 2001, 276, 49110–49116.(32) Bowman, M. J.; Chmielewski, J. Bioorg. Med. Chem. 2009,
17, 967–976.(33) El Dine, R. S.; El Halawany, A.M.;Ma, C.M.; Hattori, M. J. Nat.
Prod. 2009, 72, 2019–2023.(34) Babe, L. M.; Rose, J.; Craik, C. S. Proc. Natl. Acad. Sci. U.S.A.
1995, 92, 10069–10073.(35) Babe, L. M.; Rose, J.; Craik, C. S. Protein Sci. 1992, 1, 1244–1253.(36) Zutshi, R.; Chmielewski, J. Bioorg. Med. Chem. Lett. 2000,
10, 1901–1903.(37) Shultz, M. D.; Bowman, M. J.; Ham, Y. W.; Zhao, X. M.; Tora,
G.; Chmielewski, J. Angew. Chem., Int. Ed. 2000, 39, 2710–2713.(38) Clackson, T.; Wells, J. A. Science 1995, 267, 383–386.(39) Pons, J.; Rajpal, A.; Kirsch, J. F. Protein Sci. 1999, 8, 958–968.(40) Keskin, O.; Ma, B. Y.; Nussinov, R. J. Mol. Biol. 2005,
345, 1281–1294.(41) Lopez, M. A.; Kollman, P. A. Protein Sci. 1993, 2, 1975–1986.(42) Kortemme, T.; Baker, D. Proc. Natl. Acad. Sci. U.S.A. 2002,
99, 14116–14121.(43) Schapira, M.; Totrov, M.; Abagyan, R. J. Mol. Recognit. 1999,
12, 177–190.(44) Aqvist, J.; Medina, C.; Samuelsson, J. E. Protein Eng. 1994, 7,
385–391.(45) Verkhivker, G. M.; Bouzida, D.; Gehlhaar, D. K.; Rejto, P. A.;
Freer, S. T.; Rose, P. W. Proteins 2002, 48, 539–557.(46) Massova, I.; Kollman, P. A. J. Am. Chem. Soc. 1999, 121,
8133–8143.(47) Moreira, I. S.; Fernandes, P. A.; Ramos, M. J. J. Comput. Chem.
2007, 28, 644–654.(48) Kollman, P.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong,
L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; Donini, O.; Cieplak, P.;Srinivasan, J.; Case, D. A.; Cheatham, T. E., III Acc. Chem. Res. 2000,33, 889–897.(49) Massova, I.; Kollman, P. A. Perspect. Drug Discovery Des. 2000,
18, 113–135.(50) Moreira, I. S.; Fernandes, P. A.; Ramos, M. J. J. Phys. Chem. B
2006, 110, 10962–10969.(51) Moreira, I. S.; Fernandes, P. A.; Ramos, M. J. Int. J. Quantum
Chem. 2007, 107, 299–310.(52) Moreira, I. S.; Fernandes, P. A.; Ramos, M. J. Theor. Chem. Acc.
2008, 120, 533–542.(53) Bernstein, F. C.; Koetzle, T. F.; Williams, G. J. B.; Meyer, E. F.;
Brice, M. D.; Rodgers, J. R.; Kennard, O.; Shimanouchi, T.; Tasumi, M.J. Mol. Biol. 1977, 112, 535–542.(54) Surleraux, D. L. N. G.; Tahri, A.; Verschueren, W. G.; Pille,
G. M. E.; de Kock, H. A.; Jonckers, T. H. M.; Peeters, A.; De Meyer, S.;Azijn, H.; Pauwels, R.; de Bethune,M. P.; King, N.M.; Prabu-Jeyabalan,M.;Schiffer, C. A.; Wigerinck, P. B. T. P. J. Med. Chem. 2005, 48, 1813–1822.(55) Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C. A. J. Mol. Biol.
2000, 301, 1207–1220.(56) Hyland, L. J.; Tomaszek, T. A.; Meek, T. D. Biochemistry 1991,
30, 8454–8463.
(57) Piana, S.; Bucher, D.; Carloni, P.; Rothlisberger, U. J. Phys.Chem. B 2004, 108, 11139–11149.
(58) Liu, H. Y.; MullerPlathe, F.; Vangunsteren, W. F. J. Mol. Biol.1996, 261, 454–469.
(59) Brik, A.; Wong, C. H. Org. Biomol. Chem. 2003, 1, 5–14.(60) Jaskolski, M.; Tomasselli, A. G.; Sawyer, T. K.; Staples, D. G.;
Heinrikson, R. L.; Schneider, J.; Kent, S. B. H.; Wlodawer, A. Biochem-istry 1991, 30, 1600–1609.
(61) Porter, M. A.; Molina, P. A. J. Chem. Theory Comput. 2006,2, 1675–1684.
(62) Piana, S.; Carloni, P. Proteins 2000, 39, 26–36.(63) Case, D. A.; Darden, T. A.; Cheatham, T. E., III; Simmerling,
C. L.; Wang, J.; Duke, R. E.; Luo, R.; Merz, K. M.; Pearlman, D. A.;Crowley, M.; Walker, R. C.; Zhang, W.; Wang, B.; Hayik, S.; Roitberg,A.; Seabra, G.; Wong, K. F.; Paesani, F.; Wu, X.; Brozell, S.; Tsui, V.;Gohlke, H.; Yang, L.; Tang, C.; Mongan, J.; Hornak, V.; Cui, G.; Beroza,P.; Mathews, D. H.; Schafmeister, C.; Ross, W. S.; Kollman, P. A.AMBER 9.0 Software package; University of California: San Francisco,2006.
(64) Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey,R. W.; Klein, M. L. J. Chem. Phys. 1983, 79, 926–935.
(65) Cornell, W. D.; Cieplak, P.; Bayly, C. I.; Gould, I. R.; Merz,K. M.; Fergunson, D. M.; Spellmeyer, D. C.; Fox, T.; Caldwell, J. W.;Kollman, P. A. J. Am. Chem. Soc. 1995, 117, 5179–5197.
(66) Ryckaert, J. P.; Ciccotti, G.; Berendsen, H. C. J. Comput. Phys.1977, 23, 327–341.
(67) Essman, V.; Perera, L.; Berkowitz, M. L.; Darden, T.; Lee, H.;Pedersen, L. G. J. Chem. Phys. 1995, 103, 8577–8593.
(68) Pastor, R. W.; Brooks, B. R.; Szabo, A. Mol. Phys. 1988,65, 1409–1419.
(69) Loncharich, R. J.; Brooks, B. R.; Pastor, R.W. Biopolymers 1992,32, 523–535.
(70) Izaguirre, J. A.; Catarello, D. P.; Wozniak, J. M.; Skeel, R. D.J. Chem. Phys. 2001, 114, 2090–2098.
(71) Huo, S.; Massova, I.; Kollman, P. A. J. Comput. Chem. 2002,23, 15–27.
(72) Gohlke, H.; Case, D. A. J. Comput. Chem. 2004, 25, 238–250.(73) Rocchia, W.; Alexov, E.; Honig, B. J. Phys. Chem. B 2001,
105, 6507–6514.(74) Rocchia, W.; Sridharan, S.; Nicholls, A.; Alexov, E.; Chiabrera,
A.; Honig, B. J. Comput. Chem. 2002, 23, 128–137.(75) Connolly, M. L. J. Appl. Crystallogr. 1983, 16, 548–558.(76) Moreira, I. S.; Fernandes, P. A.; Ramos, M. J. Proteins 2007,
68, 803–812.(77) Humphrey, W.; Dalke, A.; Schulten, K. J. Mol. Graphics 1996,
14, 33–&.(78) Weber, I. T. J. Biol. Chem. 1990, 265, 10492–10496.(79) Bogan, A. A.; Thorn, K. S. J. Mol. Biol. 1998, 280, 1–9.(80) Porter, M. A.; Molina, P. A. J. Chem. Theory Comput. 2006,
2, 1675–1684.(81) Strisovsky, K.; Tessmer, U.; Langner, J.; Konvalinka, J.;
Krausslich, H. G. Protein Sci. 2000, 9, 1631–1641.(82) Liu, F. L.; Kovalevsky, A. Y.; Tie, Y. F.; Ghosh, A. K.; Harrison,
R. W.; Weber, I. T. J. Mol. Biol. 2008, 381, 102–115.(83) Bottcher, J.; Blum, A.; Heine, A.; Diederich, W. E.; Klebe, G.
J. Mol. Biol. 2008, 383, 347–357.(84) Kovalevsky, A. Y.; Tie, Y. F.; Liu, F. L.; Boross, P. I.; Wang, Y. F.;
Leshchenko, S.; Ghosh, A. K.; Harrison, R.W.;Weber, I. T. J. Med. Chem.2006, 49, 1379–1387.
(85) King, N. M.; Prabu-Jeyabalan, M.; Nalivaika, E. A.; Wigerinck,P.; de Bethune, M. P.; Schiffer, C. A. J. Virol. 2004, 78, 12012–12021.
(86) Kovalevsky, A. Y.; Liu, F. L.; Leshchenko, S.; Ghosh, A. K.;Louis, J. M.; Harrison, R. W.; Weber, I. T. J. Mol. Biol. 2006, 363,161–173.
(87) Tie, Y. F.; Boross, P. I.; Wang, Y. F.; Gaddis, L.; Hussain, A. K.;Leshchenko, S.; Ghoshl, A. K.; Louis, J. M.; Harrison, R. W.;Weber, I. T.J. Mol. Biol. 2004, 338, 341–352.