deregulated cyclin d1 expression is associated with...
TRANSCRIPT

Deregulated Cyclin D1 Expression Is Associated with DecreasedEfficacy of the Selective Epidermal Growth Factor ReceptorTyrosine Kinase Inhibitor Gefitinib in Head and NeckSquamous Cell Carcinoma Cell Lines
Larry H. Kalish,1 Rhonda A. Kwong,1
Ian E. Cole,2 Richard M. Gallagher,2
Robert L. Sutherland,1 andElizabeth A. Musgrove1
1Cancer Research Program, Garvan Institute of Medical Research,Darlinghurst, New South Wales, Australia, and 2Department ofOtorhinolaryngology Surgery, St. Vincent’s Hospital, Darlinghurst,New South Wales, Australia
ABSTRACTPurpose: Despite promising initial results, recent Phase
III trials of the selective epidermal growth factor receptor(EGFR) tyrosine kinase inhibitor gefitinib (“Iressa”; Astra-Zeneca, Wilmington, Delaware) in advanced head and necksquamous cell carcinoma (HNSCC) have been equivocal.Cyclin D1, an EGFR target gene, is frequently overex-pressed in HNSCC, has been implicated in its pathogenesis,and is strongly associated with poor prognosis in this dis-ease. Therefore, we examined the relationship between de-regulated cyclin D1 expression and sensitivity to gefitinib todetermine whether this frequently occurring oncogenicchange affected the cellular response to gefitinib.
Experimental Design: A panel of six EGFR-overex-pressing HNSCC cell lines was used to correlate CCND1gene copy number, cyclin D1 expression, and response togefitinib. The effect of constitutive overexpression of cyclinD1 was assessed by establishing stably transfected clonalSCC-9 cell lines.
Results: Three of six cell lines displayed cyclin D1 am-plification and/or overexpression, and these cell lines wereresistant to gefitinib. SCC 9 clones overexpressing cyclin D1continued to proliferate and maintained their S-phase frac-tion when treated with gefitinib, whereas empty vector con-trol clones and the parental SCC 9 cells were profoundlyinhibited and displayed marked reductions in S-phase. Theresistance of cyclin D1-overexpressing clones and cyclin D1-amplified cell lines was associated with maintenance of cy-clin D1 expression after gefitinib treatment.
Conclusions: These data suggest that deregulated cyclinD1 overexpression may be associated with resistance ofHNSCC to EGFR inhibitors. Therefore, the role of cyclin D1as a marker of therapeutic response and its utility as aprognostic marker in HNSCC warrant additional analysis.
INTRODUCTIONHead and neck cancers represent the sixth most common
malignancy worldwide. The vast majority (�90%) of thesecancers are squamous cell carcinomas. Despite advances in thetreatment of head and neck squamous cell carcinomas(HNSCC), the 5-year survival rate has remained �50% for thepast four decades, and the overall mortality rate is either stableor on the increase in Western countries (1).
The epidermal growth factor receptor (EGFR) has beenimplicated in cancer development and progression through over-expression (in the absence of gene amplification) in 36 to 100%of HNSCC and this is commonly associated with the activationof one of its ligands, transforming growth factor �, in anautocrine loop (reviewed in refs. 2, 3–6). These events occurearly in the pathogenesis of HNSCC (4) and are associated withreduced relapse-free survival and poor overall survival in manystudies of HNSCC patients (6, 7). Alterations in the function ofEGFR and other members of the tyrosine kinase receptor familyhave been linked with oncogenic transformation, autonomouscell growth, invasion, angiogenic potential, and development ofdistant metastases in a variety of cancers (8–10).
Advances in the understanding of the molecular biology ofvarious cancers have resulted in the development of noveltargeted therapeutics. Among these are therapies directed at theEGFR, which may have particular utility in HNSCC because ofthe frequent and early overexpression of EGFR in this disease.Potential therapeutic approaches include the use of monoclonalantibodies that compete with the binding of activating ligands tothe extracellular domain of the receptor, through to newer mo-dalities such as immunotoxins and antisense oligonucleotides.HNSCC patients have participated in clinical trials with most ofthe available classes of inhibitors and have seen some promisingresults (8, 11, 12). Gefitinib (“Iressa”; AstraZeneca, Wilming-
Received 1/12/04; revised 7/15/04; accepted 8/6/04.Grant support: This research was supported by the National Health andMedical Research Council of Australia, The Cancer Council New SouthWales, and the St. Vincent’s Clinic Foundation (Di Boyd Cancer Grant).L. Kalish is the recipient of a National Health and Medical ResearchCouncil Medical Postgraduate Research Scholarship and an AustralianPostgraduate Award. R. Kwong is the recipient of a Garnett Passe andRodney Williams Foundation Research Fellowship and a NationalHealth and Medical Research Council Medical Postgraduate ResearchScholarship.The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.Note: L. Kalish and R. Kwong contributed equally to this manuscript.Requests for reprints: Elizabeth A. Musgrove, Cancer Research Pro-gram, Garvan Institute of Medical Research, 384 Victoria Street, Dar-linghurst, New South Wales 2010, Australia. Phone: 612-9295-8328;Fax: 612-9295-8321; E-mail: [email protected].
©2004 American Association for Cancer Research.
7764 Vol. 10, 7764–7774, November 15, 2004 Clinical Cancer Research
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

ton, DE) is an orally active, low molecular weight (Mr 447),synthetic quinazalone derivative that acts as a selective revers-ible inhibitor of EGFR tyrosine kinase activity (13). It is alsoactive against other tyrosine kinases such as HER2, but onlywhen they are coexpressed with EGFR (14, 15). In one Phase IIclinical trial, 52 patients with advanced or metastatic HNSCCwere treated with gefitinib and showed an 8% complete re-sponse, 12% showed a partial response, and 35% of patients hadstable disease (16). Ongoing Phase III trials with gefitinib as amonotherapy in similar stage HNSCC patients have shownantineoplastic activity, although the results of these trials arestill premature (17) compared with the more established role ofgefitinib in non–small-cell lung carcinoma (18).
The underlying mechanism for the differential sensitivityof cancer cells to EGFR inhibitors is yet to be elucidated. Thelevel of EGFR expression alone is not sufficient to predict theresponse of cell lines to EGFR inhibitors in vitro (19, 20). EGFRmutations that correlate with sensitivity to gefitinib have re-cently been documented in small cell lung cancer (21, 22), butsimilar mutations were not found in any of the seven HNSCCcell lines examined (22). The cell cycle regulatory proteinscyclin D1 and p27KIP1 are targets for EGFR signaling (23–25)and are commonly deregulated in various cancers (26). CyclinD1 is well characterized as a marker of poor prognosis inHNSCC (27–35) and has been correlated with poor histologicaldifferentiation of HNSCC and local invasion (34–36). CyclinD1 associates with the cyclin-dependent kinases (CDK) CDK4and CDK6, activating them in mid-late G1 phase, and therebycontrolling progress through G1 phase (37). In addition, cyclinD1-CDK4/CDK6 complexes sequester the CDK inhibitorsp27KIP1 and p21WAF1/CIP1 (37). Because this titrates p27KIP1
and p21WAF1/CIP1 away from cyclin E-CDK2, another cyclin-CDK complex controlling progress through the G1-S–phasetransition, the balance between cyclin D1 and p27KIP1 levels canprofoundly affect the rate of cell cycle progression. EGFRinhibition induces arrest in G1 phase, and this is associated withdecreased cyclin D1 expression and increased p27KIP1 expres-sion (23, 38). Investigation of the role of these molecules in thesensitivity of HNSCC to gefitinib has largely focused on the roleof p27KIP1 and has shown that sensitivity is reduced in cellsexpressing antisense p27KIP1 constructs (39). However, the cen-tral roles of cyclin D1 and EGFR in the pathogenesis of HNSCCmake cyclin D1 an attractive downstream molecule of the EGFRpathway to explore for a potential association with clinicalsusceptibility to EGFR inhibitors. We hypothesized that EGFR-overexpressing HNSCC cell lines with coexisting amplificationof the cyclin D1 gene (CCND1) might show increased resistanceto gefitinib, and we have used six EGFR-overexpressingHNSCC cell lines and overexpression of cyclin D1 in SCC 9cells to test this idea.
MATERIALS AND METHODSCell Culture and Conditions. Established human squa-
mous cell carcinoma lines, FaDu, Detroit 562, SCC 9, SCC 15,SCC 25, and CAL 27 were obtained from American TypeCulture Collection (Rockville, MD) and cultured in a humidifiedatmosphere of 5% CO2 and 95% air at 37°C. SCC 9, SCC 15,and SCC 25 cells were grown in 1:1 mixture of DMEM and
Ham’s F12 medium containing 1.2 g/L sodium bicarbonate, 2.5mmol/L L-glutamine, 15 mmol/L HEPES, and 0.5 mmol/L so-dium pyruvate (DMEM-F12) supplemented with 10% FCS, 400ng/mL hydrocortisone, and 80 �g/L Gentamicin. Detroit 562and FaDu were grown in EMEM supplemented with 2 mmol/LL-glutamine, 0.1 mmol/L nonessential amino acids, 1 mmol/Lsodium pyruvate, 10% FCS, and 80 �g/L Gentamicin. CAL 27cells were grown in DMEM containing 10% FCS, 2 mmol/LL-glutamine, 0.5 mmol/L sodium pyruvate, and 80 �g/L Gen-tamicin. All of the cell lines were derived from primary SCC ofthe tongue except for the FaDu cell line, which was derivedfrom a primary SCC from the hypopharynx, and Detroit 562,which is derived from a metastatic pharyngeal SCC. The controlcell lines MDA-MB-134 (American Type Culture Collection)and MCF-7 human breast cancer cells (Michigan Cancer Foun-dation, Detroit, MI) were cultured in RPMI 1640 supplementedwith 5% FCS, insulin (10 �g/mL), and gentamicin (10 �g/mL)at 37°C.
DNA Purification and Southern Blot Analysis. ForSouthern blot analysis, genomic DNA was prepared from thecell lines with the Qiagen DNeasy kit (Qiagen Pty Ltd.; CliftonHills, Victoria, Australia) according to the manufacturer’s in-structions. The cyclin D1 probe was a 1.3-kb HindIII restrictionfragment encompassing the coding region. Fifty nanograms ofthis DNA fragment were labeled by random priming with[�-32P]dCTP (110 Btq/mmol/L; Amersham, Castle Hill, Aus-tralia) and hybridized at a final probe concentration of 1 to 2ng/mL. The progesterone receptor (PR) probe used as a controlwas hPR1, a 1.2-kb human PR fragment encoding most of theDNA binding domain and part of the A/B domain.
Hybridization was carried out for 16 hours at 65°C in 0.5mol/L sodium phosphate (pH 6.9), 7% SDS, 0.5% Blotto instantskim milk, 0.2% sodium azide, and 1 mmol/L EDTA. Filterswere washed to a final stringency of 0.05 � SSC [20 � SSC: 3mol/L sodium chloride, 0.3 mol/L sodium citrate (pH 7.0)] �0.1% SDS for 30 minutes at 65°C. Autoradiography was done at-70°C with 2 intensifying screens (DuPont, Wilmington, DE).Images were also captured by PhosphoImager (Molecular Dy-namics 445 SI, Molecular Dynamics, Sunnyvale, CA).
Proliferation Assays. Cells (500 to 2,000) were plated in96-well microtiter plates (six replicates per plating density)according to predetermined growth characteristics and optimalplating densities for the respective cell lines. Inoculates wereincubated overnight at 37°C to allow cell attachment beforeaddition of gefitinib at t � 0. Control cells were treated with anequivalent concentration of DMSO (vehicle). Relative cell num-bers were estimated daily for 5 days, with the Cell Titer 96 assay(Promega, Madison, WI) in accordance with the manufacturer’sinstructions. The IC50 was defined as the drug concentrationrequired to reduce the cell number to 50% of the control.
Clonogenic Assays. These were established under con-ditions similar to the proliferation assays. Cells (6,000 to12,000) were plated overnight in 6-well plates before addition ofgefitinib or DMSO vehicle. Plated cells were observed twiceweekly, and the medium was replaced weekly with fresh me-dium containing either gefitinib (at 2 �mol/L or 10 �mol/Lconcentration) or DMSO. At the conclusion of the experiment,the cells were fixed and stained with a Diff Quick Stain (Lab
7765Clinical Cancer Research
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

Aids, Narrabeen, New South Wales, Australia) in accordancewith the manufacturer’s guidelines.
Expression of Cyclin D1. The entire open reading frameof cyclin D1 was amplified by PCR with PFU polymerase(Promega) with the pLib-D1 vector as template (40) and thencloned into the Gateway pDONR201 vector (Invitrogen, Rock-ville, MD). After confirmation by sequencing, recombinationinto the destination vector pcDNA-Dest 47 (Invitrogen) pro-duced a mammalian construct for expression.
SCC 9 cells were transfected for 24 hours with pcDNA-Dest 47–cyclin D1 and FuGENE (Roche) in a ratio of 1 �g to3 �L. Control SCC 9 cells were transfected with the emptypcDNA-Dest 47 vector with the same protocol. The cells wereselected for 21 days in 400 �g/mL Geneticin (Invitrogen), andclones isolated and expanded in the presence of 400 �g/mLGeneticin.
Cell Lysis. Cells were lysed as follows: cell monolayerswere washed twice with ice-cold PBS and then scraped intoice-cold “normal” lysis buffer [0.5% deoxycholate, 150 mmol/LNaCl, 1% sodium PPI, 50 mmol/L Tris (pH 8.0), 0.1% SDS,10% glycerol, 5 mmol/L EDTA, 20 mmol/L NaF, 10 �g/mLapoprotinin, 10 �g/mL leupeptin, 1 mmol/L phenylmethylsufo-nyl fluoride, and 200 �mol/L sodium orthovanadate] or Sherrlysis buffer [50 mmol/L HEPES (pH 7.5), 1 mmol/L dithiothre-itol, 150 mmol/L NaCl, 10% (v/v) glycerol, 0.1% Tween 20, 1mmol/L EDTA, 2.5 mmol/L EGTA, 10 mmol/L �-glycerophos-phate, 10 �g/mL aprotinin, 10 �g/mL leupeptin, 1 mmol/Lphenylmethylsulfonyl fluoride, 0.1 mmol/L sodium orthovana-date, and 1 mmol/L NaF]. Alternatively, cell monolayers weretrypsinized at the selected time points, and an aliquot wasresuspended in DMEM-F12 10% FCS then stained for later flowcytometric DNA analysis by addition of ethidium bromide (50�g/mL) and Triton X-100 (0.2%), with the remainder resus-pended in lysis buffer. Cell suspensions in lysis buffer wereincubated for 5 minutes on ice, and the cellular debris wascleared by centrifugation (13,000 rpm, 5 minutes, 4°C). Thecleared lysates were stored at 80°C. After normalization ofprotein concentration, SDS sample buffer was added, and thelysates were heated to 95°C for 3 minutes, separated by SDS-PAGE, and transferred to nitrocellulose membranes. Mem-branes were incubated (2 hours at room temperature or over-night at 4°C) with primary antibodies directed against thefollowing: cyclin D1 (DCS6; Novacastra Laboratories, New-castle-upon-Tyne, United Kingdom); p27KIP1 (K25020) andp21WAF1/CIP1 (C24420) from Transduction Laboratories (Lex-ington, KY); phospho-Rb (Ser-780) from Cell Signaling Tech-nology (Beverly, MA); and �-actin antibody (Sigma, St. Louis,MO). After incubation (1 hour at room temperature) with horse-radish peroxidase-conjugated antimouse or antirabbit secondaryantibody (Santa Cruz Biotechnology, Santa Cruz, CA), proteinswere visualized with the enhanced chemiluminescence detectionsystem (Amersham).
Flow Cytometry. Flow cytometric analysis was done ona FACSCalibur (Becton Dickinson Immunocytometry Systems,San Jose, CA) with CELLQuest 2.0 software (Becton DickinsonImmunocytometry Systems). The proportions of cells in G1, S,and G2�M phases of the cell cycle were calculated from theresulting DNA histograms with ModFit LT analysis software(Verity Software House, Inc., Topsham, ME).
Image and Data Analysis. Images captured by Phospho-Imager (Molecular Dynamics 445 SI; Molecular Dynamics) ordensitometer scanning (Molecular Dynamics PDSI) of X-rayfilm were quantitated with IP Lab Gel H analysis program(Signal Analytics, Vienna, VA). Quantification of protein levelsby this method was linear over the range of intensities measured.All statistical analysis was done with Statview 4.5 Software(Abacus Concepts, Berkeley, CA).
RESULTSCyclin D1 Overexpression and CCND1 Gene Amplifi-
cation in HNSCC Cell Lines. We hypothesized that cyclinD1 overexpression, as a consequence of gene amplification,may be sustained despite inhibition of EGFR signaling. To testthis premise, we characterized the response to gefitinib of sixEGFR-overexpressing HNSCC cell lines, FaDu, Detroit 562,SCC 9, SCC 15, SCC 25, and CAL 27, after first determiningtheir cyclin D1 protein expression levels and gene copy numbercompared with MCF-7 and MDA-MB-134, two well-character-ized breast cancer cell lines with normal and amplified CCND1(41). Western blots showed an 5-fold variation in cyclin D1protein expression within the HNSCC cell lines (Fig. 1A). Thelowest expression of cyclin D1 in the HNSCC cell lines wasobserved in SCC 9, SCC 15, and CAL27 (relative expression 1.0to 1.6), whereas SCC 25 and Detroit 562 had intermediateexpression (relative expression 2.5 to 3.5), and FaDu expressedthe highest levels of cyclin D1 (relative expression 5.3). Thus,we classified SCC25, Detroit 562, and FaDu as overexpressingcyclin D1 relative to the other three HNSCC cell lines. Bothbreast cancer cell lines expressed high levels of cyclin D1compared with the HNSCC cell lines (relative expression 4.8 to6.9; Fig. 1A), likely reflecting tissue-specific control of basalexpression of this gene. Because of the differences in proteinexpression levels and evidence for CCND1 amplification inHNSCC, we went on to examine gene copy number by Southernblot of HNSCC cell lines compared with MCF-7 and MDA-MB-134 (Fig. 1B). CCND1 is located at 11q13; therefore, tocontrol for chromosome copy number, the same filters werereprobed with PR, because the PR gene is also located atchromosome 11 (11q22), and PR amplification has not beenpreviously reported in HNSCC.
SCC 9, SCC 15, and CAL 27 had CCND1 gene copynumbers of 0.99 to 1.55 relative to the MCF-7 cell line and werenot considered to be amplified (Fig. 1B). Similarly, SCC 25cells, which overexpressed cyclin D1 but with a gene copynumber of 2.0, were not considered amplified. The Fadu andDetroit 562 cell lines had increased CCND1 copy numbers of24.3 and 3.3, respectively, after taking chromosome 11 numberinto account, and thus displayed CCND1 amplification (Fig.1B). These results are consistent with published data for thesecell lines (42). The large increase in CCND1 copy number in theFaDu cell line was similar to that of MDA-MB-134, and thesecell lines both expressed high levels of cyclin D1, although therelative increase in protein expression was smaller than theincrease in gene copy number. The two HNSCC cell lines withthe highest cyclin D1 expression (FaDu and Detroit 562) dis-played CCND1 amplification, whereas those with the lowest
7766 Cyclin D1 Expression and Gefitinib Resistance
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

cyclin D1 expression (SCC9, SCC15, and CAL 27) had noevidence for amplification at this locus.
EGFR-Overexpressing HNSCC Cell Lines that Over-express Cyclin D1 Are Less Sensitive to Gefitinib. BecauseCCND1 amplification and cyclin D1 protein overexpressionwere common in HNSCC cell lines, we examined whether thesensitivity of the six HNSCC cell lines to gefitinib was relatedto cyclin D1 overexpression. We treated cell lines that either did(FaDu, Detroit 562, and SCC 25) or did not (SCC 9, SCC 15,and CAL 27) overexpress cyclin D1, with 2 �mol/L gefitinib,10 �mol/L gefitinib, or DMSO (vehicle). After treatment, col-ony formation was assessed, and relative cell numbers weremeasured with colorimetric assays (Fig. 2). The cell lines thatoverexpressed cyclin D1 formed numerous colonies in the pres-
ence of 2 �mol/L or 10 �mol/L genitinib, although these weresmaller than for vehicle-treated cells (Fig. 2A). In contrast, theremaining cell lines formed very few colonies in the presence ofgefitinib (Fig. 2A). Similarly, the cell lines that overexpressedcyclin D1 continued to proliferate when treated with up to 10�mol/L gefitinib, but cell numbers remained static or decreasedafter gefitinib treatment in the cell lines that did not overexpresscyclin D1 (Fig. 2B). Thus, the clonogenic assays and growthcurves both indicated that the cell lines overexpressing cyclinD1 were resistant to gefitinib.
DNA analysis by flow cytometry indicated that 10 �mol/Lgefitinib induced arrest in G1 phase, with a resulting meandecrease in S phase in all of the cell lines (Table 1). Aftergefitinib treatment, the relative S-phase fraction varied from0.65 in Detroit 562 to 0.25 in SCC 9. As a group, the three celllines overexpressing cyclin D1 (FaDu, Detroit 562, and SCC 25)displayed a significantly smaller reduction in S-phase fractionthan the remaining three cell lines (P � 0.020).
In more detailed experiments, we examined the response ofthe panel of six cell lines to a range of concentrations ofgefitinib. Data from replicate experiments with colorimetricassays to measure relative cell number were pooled (Fig. 3) andused to estimate the concentration at which the relative cellnumber was reduced to 50% relative to control cells, after 5 daysof treatment (IC50). The IC50 ranged from 0.4 �mol/L for SCC9 to 14.4 �mol/L for FaDu (Table 1). Previous studies with arange of cancer cell lines have determined that resistant celllines have an IC50 of 3 to 16 �mol/L and sensitive cell lineshave an IC50 of 0.07 to 1.4 �mol/L (19). Consequently, weclassified Fadu, Detroit 562, and SCC 25 as resistant, and SCC9, SCC 15, and CAL 27 as sensitive to gefitinib. Interestingly,the most resistant cell line, FaDu, displayed the highest level ofCCND1 amplification and overexpression. Collectively, the datain Figs. 2 and 3 and Table 1 suggest that resistance to gefitinibis associated with cyclin D1 overexpression. These dataprompted us to additionally investigate the possibility that ge-fitinib resistance might result from the deregulation of cyclin D1expression.
Effects of Cyclin D1 Overexpression in SCC 9 Cells.To additionally explore the role of deregulated cyclin D1 ex-pression in sensitivity to gefitinib, we constitutively overex-pressed cyclin D1 in the SCC 9 cell line. This cell line isrepresentative of the majority of HNSCC, displaying overex-pression of EGFR and c-erb2 as well as mutation of p53. Thereis no evidence of amplification of the cyclin D1, D2, and D3genes or deletion or inactivation of p16INK4A, a CDK inhibitorspecifically targeting cyclin D-dependent kinases. We con-firmed the lack of CCND1 amplification (Fig. 1B) and noted lowcyclin D1 protein expression compared with other EGFR-over-expressing HNSCC cell lines (Fig. 1A).
SCC 9 cells were transfected with a vector expressingcyclin D1 or the corresponding empty vector, and clonal celllines were selected. The SCC 9 cells tolerated transfection withthe cyclin D1 expression vector and selection with no grossevidence of changes in cell morphology or cyclin D1-inducedcell death. At least two different clones were used for all of theadditional experiments, and these gave consistent results. Thelevels of cyclin D1 expression achieved, as measured by den-sitometry, were at least 3-fold greater in the two cyclin D1-
Fig. 1 Cyclin D1 expression and CCND1 amplification in HNSCC celllines. A, Western blot analysis of cyclin D1 in selected HNSCC celllines and control breast cancer cell lines MCF-7 and MDA-MB-134(MB-134). The same filter was reprobed with �-actin as a loadingcontrol. Relative intensities were normalized with �-actin, and thesignals expressed relative to SCC 9. B, Southern blot analysis of theCCND1 gene in HNSCC cell lines and control breast cancer cell lineswith known amplification status. Southern blots were sequentially hy-bridized with cyclin D1 and PR cDNA probes and autoradiographed for24 hours and 72 hours, respectively. The CCND1 signal was normalizedfor chromosome 11 copy number with the PR control. The gene copynumber is expressed relative to the MCF-7 breast cancer cells, which donot display amplification of CCND1.
7767Clinical Cancer Research
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
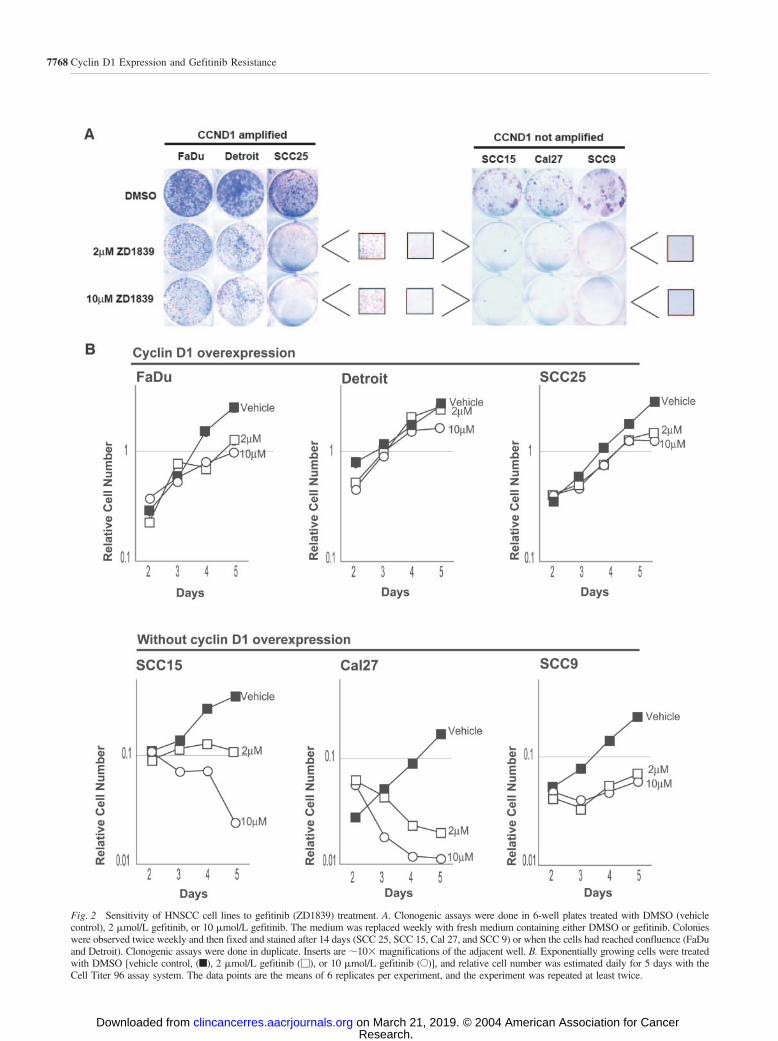
Fig. 2 Sensitivity of HNSCC cell lines to gefitinib (ZD1839) treatment. A. Clonogenic assays were done in 6-well plates treated with DMSO (vehiclecontrol), 2 �mol/L gefitinib, or 10 �mol/L gefitinib. The medium was replaced weekly with fresh medium containing either DMSO or gefitinib. Colonieswere observed twice weekly and then fixed and stained after 14 days (SCC 25, SCC 15, Cal 27, and SCC 9) or when the cells had reached confluence (FaDuand Detroit). Clonogenic assays were done in duplicate. Inserts are 10� magnifications of the adjacent well. B. Exponentially growing cells were treatedwith DMSO [vehicle control, (f), 2 �mol/L gefitinib (�), or 10 �mol/L gefitinib (E)], and relative cell number was estimated daily for 5 days with theCell Titer 96 assay system. The data points are the means of 6 replicates per experiment, and the experiment was repeated at least twice.
7768 Cyclin D1 Expression and Gefitinib Resistance
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
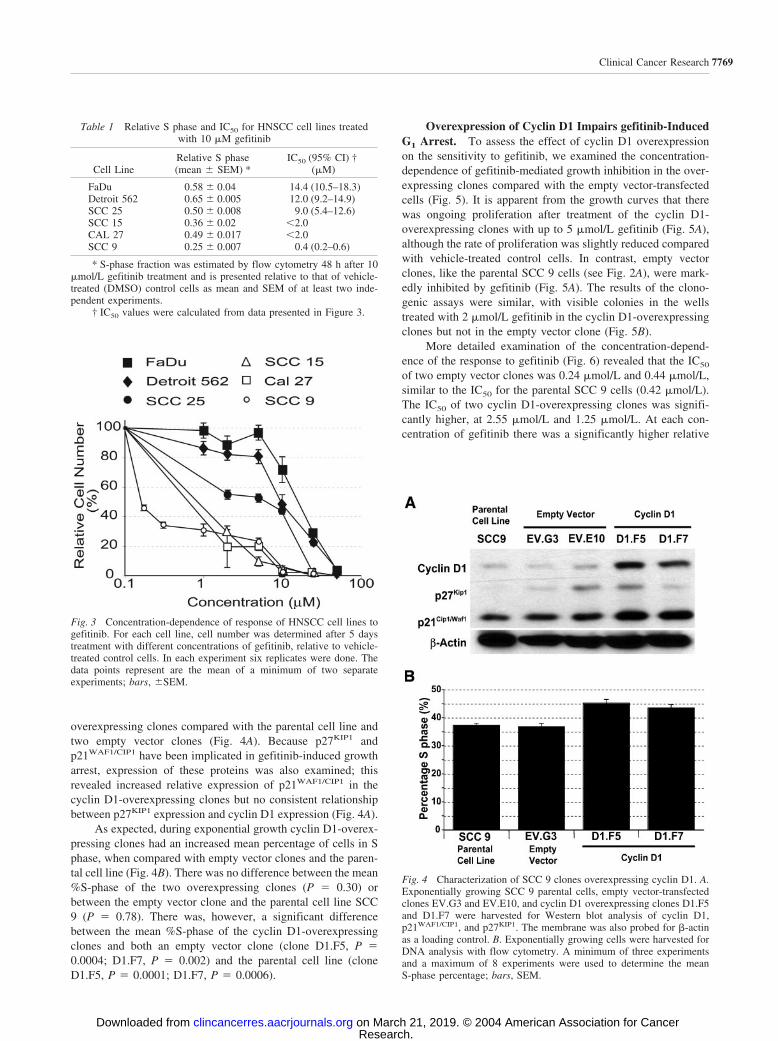
overexpressing clones compared with the parental cell line andtwo empty vector clones (Fig. 4A). Because p27KIP1 andp21WAF1/CIP1 have been implicated in gefitinib-induced growtharrest, expression of these proteins was also examined; thisrevealed increased relative expression of p21WAF1/CIP1 in thecyclin D1-overexpressing clones but no consistent relationshipbetween p27KIP1 expression and cyclin D1 expression (Fig. 4A).
As expected, during exponential growth cyclin D1-overex-pressing clones had an increased mean percentage of cells in Sphase, when compared with empty vector clones and the paren-tal cell line (Fig. 4B). There was no difference between the mean%S-phase of the two overexpressing clones (P � 0.30) orbetween the empty vector clone and the parental cell line SCC9 (P � 0.78). There was, however, a significant differencebetween the mean %S-phase of the cyclin D1-overexpressingclones and both an empty vector clone (clone D1.F5, P �0.0004; D1.F7, P � 0.002) and the parental cell line (cloneD1.F5, P � 0.0001; D1.F7, P � 0.0006).
Overexpression of Cyclin D1 Impairs gefitinib-InducedG1 Arrest. To assess the effect of cyclin D1 overexpressionon the sensitivity to gefitinib, we examined the concentration-dependence of gefitinib-mediated growth inhibition in the over-expressing clones compared with the empty vector-transfectedcells (Fig. 5). It is apparent from the growth curves that therewas ongoing proliferation after treatment of the cyclin D1-overexpressing clones with up to 5 �mol/L gefitinib (Fig. 5A),although the rate of proliferation was slightly reduced comparedwith vehicle-treated control cells. In contrast, empty vectorclones, like the parental SCC 9 cells (see Fig. 2A), were mark-edly inhibited by gefitinib (Fig. 5A). The results of the clono-genic assays were similar, with visible colonies in the wellstreated with 2 �mol/L gefitinib in the cyclin D1-overexpressingclones but not in the empty vector clone (Fig. 5B).
More detailed examination of the concentration-depend-ence of the response to gefitinib (Fig. 6) revealed that the IC50
of two empty vector clones was 0.24 �mol/L and 0.44 �mol/L,similar to the IC50 for the parental SCC 9 cells (0.42 �mol/L).The IC50 of two cyclin D1-overexpressing clones was signifi-cantly higher, at 2.55 �mol/L and 1.25 �mol/L. At each con-centration of gefitinib there was a significantly higher relative
Table 1 Relative S phase and IC50 for HNSCC cell lines treatedwith 10 �M gefitinib
Cell LineRelative S phase(mean � SEM) *
IC50 (95% CI) †(�M)
FaDu 0.58 � 0.04 14.4 (10.5–18.3)Detroit 562 0.65 � 0.005 12.0 (9.2–14.9)SCC 25 0.50 � 0.008 9.0 (5.4–12.6)SCC 15 0.36 � 0.02 �2.0CAL 27 0.49 � 0.017 �2.0SCC 9 0.25 � 0.007 0.4 (0.2–0.6)
* S-phase fraction was estimated by flow cytometry 48 h after 10�mol/L gefitinib treatment and is presented relative to that of vehicle-treated (DMSO) control cells as mean and SEM of at least two inde-pendent experiments.
† IC50 values were calculated from data presented in Figure 3.
Fig. 3 Concentration-dependence of response of HNSCC cell lines togefitinib. For each cell line, cell number was determined after 5 daystreatment with different concentrations of gefitinib, relative to vehicle-treated control cells. In each experiment six replicates were done. Thedata points represent are the mean of a minimum of two separateexperiments; bars, �SEM.
Fig. 4 Characterization of SCC 9 clones overexpressing cyclin D1. A.Exponentially growing SCC 9 parental cells, empty vector-transfectedclones EV.G3 and EV.E10, and cyclin D1 overexpressing clones D1.F5and D1.F7 were harvested for Western blot analysis of cyclin D1,p21WAF1/CIP1, and p27KIP1. The membrane was also probed for �-actinas a loading control. B. Exponentially growing cells were harvested forDNA analysis with flow cytometry. A minimum of three experimentsand a maximum of 8 experiments were used to determine the meanS-phase percentage; bars, SEM.
7769Clinical Cancer Research
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

number of viable cells in the overexpressing clones than in theempty vector clones.
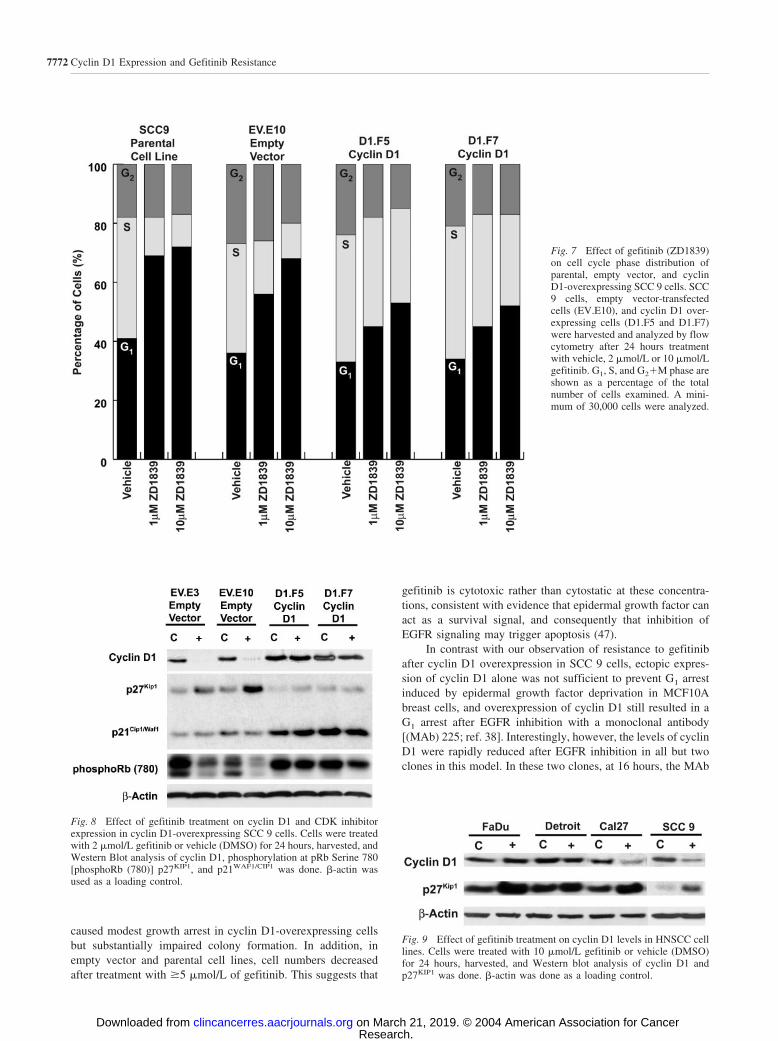
We also measured the cell cycle phase distribution of theclones after EGFR inhibition with gefitinib. After 24 hours oftreatment with 1 �mol/L or 10 �mol/L gefitinib (Fig. 7), it wasapparent that a greater proportion of empty vector and parentalcells were arrested in G1 phase after gefitinib treatment, com-pared with the cyclin D1-overexpressing clones. In the SCC 9parental cell line and empty vector clone after treatment withboth concentrations of gefitinib, the proportion of cells in Sphase was significantly reduced and that in G1 phase increasedrelative to vehicle-treated control cells (Fig. 7). In comparison,the relative S-phase fraction was reduced �20% in both of the
cyclin D1 overexpressing clones after 1 �mol/L of treatmentwith gefitinib and �30% after treatment with 10 �mol/L ge-fitinib. The continued proliferation of the cyclin D1-overex-pressing clones at these concentrations of gefitinib is thus asso-ciated with maintenance of a substantial proportion of cells in Sphase. Taken together, the 2- to 6-fold increase in IC50 com-bined with the continued exponential growth and high S-phasefraction of cyclin D1-overexpressing clones at concentrations ofgefitinib that resulted in growth arrest or cell death in parentalcells and empty vector clones is consistent with a role for cyclinD1 overexpression in resistance to gefitinib treatment.
To better appreciate the molecular basis of the G1 arrest,we studied the expression of the molecules involved in the G1-S
Fig. 5 Response of cyclin D1-overexpressing SCC 9 cells togefitinib (ZD1839) treatment. A,Exponentially proliferating cy-clin D1-overexpressing (D1.F5and D1.F7) or empty vector-transfected clonal SCC 9 cells(EV.E10) were treated with 2�mol/L gefitinib (�), 5 �mol/Lgefitinib (E), or vehicle (C, f).Relative cell number was esti-mated daily with the Cell Titer96 assay kit. B, Clonogenic as-says were done in 6-well platestreated with DMSO (vehiclecontrol), 2 �mol/L gefitinib, or10 �mol/L gefitinib in the cyclinD1-overexpressing (D1.F5 andD1.F7) and empty vector-trans-fected clonal SCC 9 cells(EV.E10). The medium was re-placed weekly with fresh me-dium containing either DMSOor gefitinib. Colonies were ob-served twice weekly and thenfixed and stained after the cellshad reached confluence (D1.F5and D1.F7) or 40 days (EV.E10)after initial treatment. Clono-genic assays were done in atleast triplicate. Bars, �SEM
7770 Cyclin D1 Expression and Gefitinib Resistance
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

transition after gefitinib treatment of the cyclin D1-overexpress-ing clones. Initial examination of whether cyclin D1 overex-pression was maintained on gefitinib treatment of the cyclinD1-overexpressing clones revealed that cyclin D1 protein ex-pression was substantially reduced in the empty vector clonesbut maintained in the cyclin D1-overexpressing clones after 24hours treatment (Fig. 8). To determine whether this cyclin D1overexpression led to increased CDK4 activity, we used anantibody recognizing phosphorylation at serine 780, a residueof retinoblastoma protein (pRb) specifically phosphorylatedby cyclin D1-CDK4. This revealed that after gefitinib admin-istration, pRb serine 780 phosphorylation was sustained inthe overexpressing clones but reduced in the empty vector-transfected cells. Because both the CDK inhibitors p27KIP1
and p21WAF1/CIP1 have been implicated in the antiprolifera-tive effect induced by gefitinib in HNSCC (39), we alsoexamined their expression. None of the cell lines examineddisplayed increased p21WAF1/CIP1 expression after gefitinibtreatment. However, treatment of the empty vector-transfectedcells with 2 �mol/L gefitinib for 24 hours resulted in an up-regulation of p27KIP1 protein that was not apparent in either ofthe cyclin D1-overexpressing clones (Fig. 8).
These data suggested that failure to down-regulate cyclinD1 and up-regulate p27KIP1 was a central component of theresistance of the cyclin D1-overexpressing clones. To determinewhether this was also the case in the gefitinib-resistant cell linesdisplaying cyclin D1 overexpression and amplification at theCCND1 locus, we examined cyclin D1 and p27KIP1 expressionafter gefitinib treatment in the resistant FaDu and Detroit 562cell lines, compared with the gefitinib-sensitive CAL 27 andSCC 9 cell lines. The expression of p27KIP1 was increased aftergefitinib treatment of all of the cell lines, although the increasewas greater in the sensitive cell lines (Fig. 9). However, after
gefitinib treatment, the cyclin D1 levels were reduced by 50%in both CAL 27 and SCC 9 (Fig. 9). Thus, in both the cyclinD1-overexpressing cell lines and cyclin D1-transfected SCC 9cells, resistance to gefitinib was associated with maintenance ofcyclin D1 levels.
DISCUSSIONClinical studies have shown amplification of the cyclin D1
gene (CCND1) in up to 58% of HNSCC and overexpression ofcyclin D1 in up to 68% of HNSCC; overexpression in theabsence of gene amplification occurs in 4 to 55% of cases(27–34, 43). Both cyclin D1 overexpression as measured byimmunohistochemistry and gene amplification measured by dif-ferential PCR or FISH are consistent and strong markers of poorprognosis. Comparative studies have either shown no prognosticadvantage of protein expression over gene amplification or havefavored cyclin D1 gene amplification as a better marker of poorprognosis (33, 34, 44–46). The consequences of cyclin D1overexpression for response to therapy are not well understoodand may differ between cell types. We hypothesized that cyclinD1 overexpression may affect the efficacy of therapies thatreduce cyclin D1 expression, including those targeted at theEGFR. We have tested this hypothesis using six EGFR-overex-pressing HNSCC cell lines and SCC 9 tongue cancer cellsconstitutively overexpressing cyclin D1.
CCND1 gene amplification was present in two of sixEGFR-overexpressing HNSCC cell lines displaying differentialsensitivity to gefitinib. The FaDu cell line showed 24-foldCCND1 gene amplification, associated with high cyclin D1protein expression and resistance to gefitinib treatment (IC50
14.4 �mol/L). Similarly, the resistant Detroit 562 cell line (IC50
12 �mol/L) has evidence of increased gene copy number andoverexpression of cyclin D1 protein. In contrast, the sensitiveSCC 9, CAL 27, and SCC15 cell lines (IC50 0.4 to 1.4 �mol/L)had no evidence of increased gene copy number and relativelylow levels of cyclin D1 protein expression. The SCC 25 cell linehad an IC50 of 9 �mol/L, similar to the other two resistant celllines. However, at concentrations of 2 to 5 �mol/L gefitinib, theresponse of SCC 25 was intermediate between the sensitive celllines, which were inhibited by 80%, and the other resistantcell lines, which were inhibited by �20% (Fig. 3A). This mayreflect the modest degree of cyclin D1 overexpression and lackof CCND1 amplification of this cell line. Consistent with ourhypothesis, overall there seemed to be a relationship betweenthe degree of cyclin D1 overexpression and resistance to ge-fitinib, with the most resistant cell lines displaying CCND1amplification.
The SCC 9 cyclin D1 overexpression model showed thatderegulated expression of cyclin D1 could dampen the cellulareffects of the EGFR inhibitor gefitinib. Cyclin D1 transfectionresulted in an increased number of cells in S phase in thisalready transformed and fast-growing malignant cell line. Thecyclin D1-overexpressing clones were more resistant to ge-fitinib, resulting in a higher IC50, ongoing exponential growthafter treatment with up to 5 �mol/L gefitinib, and a significantlyreduced, although not totally ablated, G1 arrest. Colony forma-tion was impaired after treatment with 2 �mol/L of gefitinib.The highest concentrations of gefitinib tested (5 to 10 �mol/L)
Fig. 6 Concentration-dependence of response of cyclin D1-overex-pressing SCC9 cells to gefitinib treatment. For each cell line, cellnumber was determined after 5 days treatment with different concen-trations of gefitinib, relative to vehicle-treated control cells. In eachexperiment six replicates were done. The data points represent 12replicates from two experiments; bars, �SEM. Cyclin D1-overexpress-ing cells: D1.F5 (�); and D1.F7 (‚); empty vector-transfected cells:EV.G3 (E); and EV.E10 (�).
7771Clinical Cancer Research
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
caused modest growth arrest in cyclin D1-overexpressing cellsbut substantially impaired colony formation. In addition, inempty vector and parental cell lines, cell numbers decreasedafter treatment with �5 �mol/L of gefitinib. This suggests that
gefitinib is cytotoxic rather than cytostatic at these concentra-tions, consistent with evidence that epidermal growth factor canact as a survival signal, and consequently that inhibition ofEGFR signaling may trigger apoptosis (47).
In contrast with our observation of resistance to gefitinibafter cyclin D1 overexpression in SCC 9 cells, ectopic expres-sion of cyclin D1 alone was not sufficient to prevent G1 arrestinduced by epidermal growth factor deprivation in MCF10Abreast cells, and overexpression of cyclin D1 still resulted in aG1 arrest after EGFR inhibition with a monoclonal antibody[(MAb) 225; ref. 38]. Interestingly, however, the levels of cyclinD1 were rapidly reduced after EGFR inhibition in all but twoclones in this model. In these two clones, at 16 hours, the MAb
Fig. 9 Effect of gefitinib treatment on cyclin D1 levels in HNSCC celllines. Cells were treated with 10 �mol/L gefitinib or vehicle (DMSO)for 24 hours, harvested, and Western blot analysis of cyclin D1 andp27KIP1 was done. �-actin was done as a loading control.
Fig. 7 Effect of gefitinib (ZD1839)on cell cycle phase distribution ofparental, empty vector, and cyclinD1-overexpressing SCC 9 cells. SCC9 cells, empty vector-transfectedcells (EV.E10), and cyclin D1 over-expressing cells (D1.F5 and D1.F7)were harvested and analyzed by flowcytometry after 24 hours treatmentwith vehicle, 2 �mol/L or 10 �mol/Lgefitinib. G1, S, and G2�M phase areshown as a percentage of the totalnumber of cells examined. A mini-mum of 30,000 cells were analyzed.
Fig. 8 Effect of gefitinib treatment on cyclin D1 and CDK inhibitorexpression in cyclin D1-overexpressing SCC 9 cells. Cells were treatedwith 2 �mol/L gefitinib or vehicle (DMSO) for 24 hours, harvested, andWestern Blot analysis of cyclin D1, phosphorylation at pRb Serine 780[phosphoRb (780)] p27KIP1, and p21WAF1/CIP1 was done. �-actin wasused as a loading control.
7772 Cyclin D1 Expression and Gefitinib Resistance
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

225-treated cells had more cyclin D1 bound to p27KIP1 thancontrol cells, and it was concluded that the remaining unboundcyclin D1 protein was at a level inadequate to maintain cellcycle progression. In contrast, in both SCC 9 cells expressingectopic cyclin D1 and two HNSCC cell lines with amplificationof the CCND1 gene, gefitinib insensitivity was associated withfailure to appreciably decrease cyclin D1 expression after treat-ment. These data are consistent with the conclusion that main-tenance of cyclin D1 levels is critical to the resistance these celllines display to gefitinib and confirm the central role of cyclinD1 down-regulation in the response to inhibition of EGFRsignaling. No clinical studies have addressed whether cyclin D1expression is maintained in CCND1-amplified cancers afterEGFR inhibition, and this is an issue warranting additionalinvestigation.
The CDK inhibitors p27KIP1 and p21WAF1/CIP1 are mem-bers of the CIP/KIP family, which interact with and inhibit thecyclin-CDK complexes essential for G1 to S-phase progression(37). Both p21WAF1/CIP1 and p27KIP1 have been implicated inthe growth arrest after disruption of EGFR kinase activity inEGFR- and HER2-overexpressing cells (23, 25, 39). Di Gennaroet al. (39) showed that in HNSCC-derived cell lines treated withgefitinib, there was an increase in p27KIP1 and p21WAF1/CIP1
protein expression. The resultant inhibition of cyclin E- andcyclin A-CDK2 complexes resulted in G1 phase arrest, growthinhibition, and apoptosis. In this study, p21WAF1/CIP1 expressionwas unchanged in all of the clones and parental cell lines aftergefitinib treatment. We observed an increase in p27KIP1 expres-sion as a response to gefitinib treatment in the SCC 9 parentalcell line and the empty vector clones, which was not apparent inthe cyclin D1-transfected cells. However, p27KIP1 expressionwas increased after gefitinib treatment of the resistant cell linesFaDu and Detroit 562, and thus there was no clear relationshipbetween p27KIP1 induction and sensitivity to gefitinib.
In summary, we have provided several lines of evidence insupport of the idea that deregulation of cyclin D1 confersresistance to the EGFR-targeted therapy with gefitinib: therelationship between CCND1 amplification, cyclin D1 overex-pression, and insensitivity to gefitinib; the ability of constitutiveoverexpression of cyclin D1 to confer resistance to gefitinib;and the correlation between the ability of gefitinib to down-regulate cyclin D1 and sensitivity to growth inhibition by thiscompound. CCND1 amplification is a frequent finding inHNSCC and thus warrants additional investigation as a potentialmarker of resistance. Identifying cyclin D1 amplification andtargeting it directly with antisense cyclin D1 gene therapy (42,48) or indirectly through CDK inhibitors (e.g., flavopiridol)together with EGFR inhibitors may be a useful therapeuticstrategy in a significant subset of HNSCC patients.
ACKNOWLEDGMENTSWe thank Christine Lee for advice and assistance with some
experiments.
REFERENCES1. Stewart BW, Kleihues P, editors. World Cancer Report. Lyon,France: IARC Press; 2003.2. O-Charoenrat P, Rhys-Evans PH, Archer DJ, Eccles SA. C-erbBreceptors in squamous cell carcinomas of the head and neck: clinical
significance and correlation with matrix metalloproteinases and vascularendothelial growth factors. Oral Oncol 2002;38:73–80.
3. Quon H, Liu FF, Cummings BJ. Potential molecular prognosticmarkers in head and neck squamous cell carcinomas. Head Neck 2001;23:147–59.
4. Grandis JR, Tweardy DJ. Elevated levels of transforming growthfactor alpha and epidermal growth factor receptor messenger RNA areearly markers of carcinogenesis in head and neck cancer. Cancer Res1993;53:3579–84.
5. Grandis JR, Melhem MF, Gooding WE, et al. Levels of TGF-alphaand EGFR protein in head and neck squamous cell carcinoma andpatient survival. J Natl Cancer Inst (Bethesda) 1998;90:824–32.
6. Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factorreceptor expression on survival and pattern of relapse in patients withadvanced head and neck carcinoma. Cancer Res 2002;62:7350–6.
7. Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. EurJ Cancer 2001;37(Suppl 4):S9–15.
8. Grunwald V, Hidalgo M. Developing inhibitors of the epidermalgrowth factor receptor for cancer treatment. J Natl Cancer Inst (Be-thesda) 2003;95:851–67.
9. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell2000;103:211–25.
10. Yarden Y. The EGFR family and its ligands in human cancer:signalling mechanisms and therapeutic opportunities. Eur J Cancer2001;37(Suppl 4):S3–8.
11. Mendelsohn J, Baselga J. Status of epidermal growth factor receptorantagonists in the biology and treatment of cancer. J Clin Oncol 2003;21:2787–99.
12. Ciardiello F, Tortora G. A novel approach in the treatment ofcancer: targeting the epidermal growth factor receptor. Clin Cancer Res2001;7:2958–70.
13. Woodburn JR. The epidermal growth factor receptor and its inhi-bition in cancer therapy. Pharmacol Ther 1999;82:241–50.
14. Moasser MM, Basso A, Averbuch SD, Rosen N. The tyrosinekinase inhibitor gefitinib (“Iressa”) inhibits HER2-driven signalling andsuppresses the growth of HER2-overexpressing tumor cells. Cancer Res2001;61:7184–8.
15. Moulder SL, Yakes FM, Muthuswamy SK, et al. Epidermal growthfactor receptor (HER1) tyrosine kinase inhibitor gefitinib (Iressa) inhib-its HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and invivo. Cancer Res 2001;61:8887–95.
16. Cohen EE, Rosen F, Stadler WM, et al. Phase II trial of gefitinib inrecurrent or metastatic squamous cell carcinoma of the head and neck.J Clin Oncol 2003;21:1980–7.
17. Caponigro F. Rationale and clinical validation of epidermal growthfactor receptor as a target in the treatment of head and neck cancer.Anticancer Drugs 2004;15:311–20.
18. Ranson M, Wardell S. Gefitinib, a novel, orally administered agentfor the treatment of cancer. J Clin Pharm Ther 2004;29:95–103.
19. Bishop PC, Myers T, Robey R, et al. Differential sensitivity ofcancer cells to inhibitors of the epidermal growth factor receptor family.Oncogene 2002;21:119–27.
20. Ciardiello F, Caputo R, Bianco R, et al. Antitumor effect andpotentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosinekinase inhibitor. Clin Cancer Res 2000;6:2053–63.
21. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer:correlation with clinical response to gefitinib therapy. Science (WashDC) 2004;304:1497–500.
22. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in theepidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
23. Lenferink AE, Busse D, Flanagan WM, Yakes FM, Arteaga CL.ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 throughmultiple signalling pathways. Cancer Res 2001;61:6583–91.
7773Clinical Cancer Research
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

24. Hulit J, Lee RJ, Russell RG, Pestell RG. ErbB-2-induced mammarytumor growth: the role of cyclin D1 and p27Kip1. Biochem Pharmacol2002;64:827–36.25. Busse D, Yakes FM, Lenferink AE, Arteaga CL. Tyrosine kinaseinhibitors: rationale, mechanisms of action, and implications for drugresistance. Semin Oncol 2001;28:47–55.26. Bloom J, Pagano M. Deregulated degradation of the cdk inhibitorp27 and malignant transformation. Semin Cancer Biol 2003;13:41–7.27. Bova RJ, Quinn DI, Nankervis JS, et al. Cyclin D1 and p16INK4Aexpression predict reduced survival in carcinoma of the anterior tongue.Clin Cancer Res 1999;5:2810–9.28. Akervall JA, Michalides RJ, Mineta H, et al. Amplification ofcyclin D1 in squamous cell carcinoma of the head and neck and theprognostic value of chromosomal abnormalities and cyclin D1 overex-pression. Cancer (Phila) 1997;79:380–9.29. Okami K, Reed AL, Cairns P, et al. Cyclin D1 amplification isindependent of p16 inactivation in head and neck squamous cell carci-noma. Oncogene 1999;18:3541–5.30. Liu SC, Zhang SY, Babb JS, Ridge JA, Klein-Szanto AJ. Imagecytometry of cyclin D1: a prognostic marker for head and neck squa-mous cell carcinomas. Cancer Epidemiol Biomark Prev 2001;10:455–9.31. Kyomoto R, Kumazawa H, Toda Y, et al. Cyclin-D1-gene ampli-fication is a more potent prognostic factor than its protein over-expres-sion in human head-and-neck squamous-cell carcinoma. Int J Cancer1997;74:576–81.32. Muller D, Millon R, Velten M, et al. Amplification of 11q13 DNAmarkers in head and neck squamous cell carcinomas: correlation withclinical outcome. Eur J Cancer 1997;33:2203–10.33. Mineta H, Miura K, Takebayashi S, et al. Cyclin D1 overexpressioncorrelates with poor prognosis in patients with tongue squamous cellcarcinoma. Oral Oncol 2000;36:194–8.34. Michalides RJ, van Veelen NM, Kristel PM, et al. Overexpressionof cyclin D1 indicates a poor prognosis in squamous cell carcinomaof the head and neck. Arch Otolaryngol Head Neck Surg 1997;123:497–502.35. Jares P, Fernandez PL, Campo E, et al. PRAD-1/cyclin D1 geneamplification correlates with messenger RNA overexpression and tumorprogression in human laryngeal carcinomas. Cancer Res 1994;54:4813–7.36. Bellacosa A, Almadori G, Cavallo S, et al. Cyclin D1 gene ampli-fication in human laryngeal squamous cell carcinomas: prognostic sig-nificance and clinical implications. Clin Cancer Res 1996;2:175–80.
37. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative reg-ulators of G1-phase progression. Genes Dev 1999;13:1501–12.
38. Chou JL, Fan Z, DeBlasio T, et al. Constitutive overexpression ofcyclin D1 in human breast epithelial cells does not prevent G1 arrestinduced by deprivation of epidermal growth factor. Breast Cancer ResTreat 1999;55:267–83.
39. Di Gennaro E, Barbarino M, Bruzzese F, et al. Critical role of bothp27KIP1 and p21CIP1/WAF1 in the antiproliferative effect of gefitinib(‘Iressa’), an epidermal growth factor receptor tyrosine kinase inhibitor,in head and neck squamous carcinoma cells. J Cell Physiol 2003;195:139–50.
40. Musgrove EA, Hunter LJ, Lee CS, et al. Cyclin D1 overexpressioninduces progestin resistance in T-47D breast cancer cells despitep27(Kip1) association with cyclin E-Cdk2. J Biol Chem 2001;276:47675–83.
41. Buckley MF, Sweeney KJ, Hamilton JA, et al. Expression andamplification of cyclin genes in human breast cancer. Oncogene 1993;8:2127–33.
42. Sauter ER, Herlyn M, Liu SC, Litwin S, Ridge JA. Prolongedresponse to antisense cyclin D1 in a human squamous cancer xenograftmodel. Clin Cancer Res 2000;6:654–60.
43. Goto H, Kawano K, Kobayashi I, Sakai H, Yanagisawa S. Expres-sion of cyclin D1 and GSK-3beta and their predictive value of prognosisin squamous cell carcinomas of the tongue. Oral Oncol 2002;38:549–56.
44. Uhlman DL, Adams G, Knapp D, Aeppli DM, Niehans G. Immuno-histochemical staining for markers of future neoplastic progression inthe larynx. Cancer Res 1996;56:2199–205.
45. Capaccio P, Pruneri G, Carboni N, et al. Cyclin D1 protein expres-sion is related to clinical progression in laryngeal squamous cell carci-nomas. J Laryngol Otol 1997;111:622–6.
46. Miyamoto R, Uzawa N, Nagaoka S, Hirata Y, Amagasa T. Prog-nostic significance of cyclin D1 amplification and overexpression in oralsquamous cell carcinomas. Oral Oncol 2003;39:610–8.
47. Kari C, Chan TO, Rocha deq Uadros M, Rodeck U. Targeting theepidermal growth factor receptor in cancer: apoptosis takes center stage.Cancer Res 2003;63:1–5.
48. Sauter ER, Nesbit M, Litwin S, et al. Antisense cyclin D1 inducesapoptosis and tumor shrinkage in human squamous carcinomas. CancerRes 1999;59:4876–81.
7774 Cyclin D1 Expression and Gefitinib Resistance
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

2004;10:7764-7774. Clin Cancer Res Larry H. Kalish, Rhonda A. Kwong, Ian E. Cole, et al. Neck Squamous Cell Carcinoma Cell LinesReceptor Tyrosine Kinase Inhibitor Gefitinib in Head and
FactorDecreased Efficacy of the Selective Epidermal Growth Deregulated Cyclin D1 Expression Is Associated with
Updated version
http://clincancerres.aacrjournals.org/content/10/22/7764
Access the most recent version of this article at:
Cited articles
http://clincancerres.aacrjournals.org/content/10/22/7764.full#ref-list-1
This article cites 45 articles, 20 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/10/22/7764.full#related-urls
This article has been cited by 15 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://clincancerres.aacrjournals.org/content/10/22/7764To request permission to re-use all or part of this article, use this link
Research. on March 21, 2019. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from