department of chemistrychemistry.slu.edu/mo-inorganic/program/program_final.pdf · saint louis...
TRANSCRIPT

Saint Louis University
Department of Chemistry
Saturday, May 8th 2010

2
Program Chairs
Paul A. Jelliss, PhD
Saint Louis University
Chuck C. Kirkpatrick, PhD
Saint Louis University
Session Chairs
Janet Braddock‐Wilking, PhD
University of Missouri – St Louis
Nikolai Gerasimchuk, PhD
Missouri State University
Liviu Mirica, PhD
Washington University in St Louis
Paul Sharp, PhD
University of Missouri – Columbia
Acknowledgements
Steve Buckner – Chairman, Department of Chemistry, SLU
Shontae Williams – Department of Chemistry, SLU
Chuck Kirkpatrick ‐ website design
http://chemistry.slu.edu/MO‐Inorganic/

3
Location
Symposium, breakfast: Baer‐Fuller Lecture Hall (building #24) – LH 2
Lunch: Busch Student Center (building #7) – room 253
Poster session: Busch Student center (building #7) – room 251
Department of Chemistry (building # 34, Monsanto Hall)
Parking in the multi‐story parking garage @ Olive/Compton is recommended –
enter from Olive St (nr. building #48 on the map)
LH 2BSC 253 BSC 251
Parking

4
Schedule
8:00‐9:00am Continental breakfast (served outside Lecture Hall 2 (Baer‐Fuller))
9:00‐9:05 Opening remarks
9:05‐10:35am Oral Session 1 – Session Chair: Janet Braddock‐Wilking
Time Presenter Affiliation Title
9:05 Nikolai Gerasimchuk
Missouri State University
Pt‐cyanoximates: self‐assembled nano‐size electrical conductors
9:35 Liviu Mirica Washington University in St Louis
Stable mononuclear organometallic Pd(III) complexes and their C–C bond formation reactivity
10:05 Charles Chusuei Missouri University of Science & Technology
Charging nanowalls: adjusting the carbon nanotube isoelectric point via surface functionalization
10:35‐10:45am Break
10:45‐11:45am Oral Session 2 – Session Chair: Nikolai Gerasimchuk
10:45 Bryn Lutes Washington University in St Louis
Synthesis, structure and spectroscopy of heteropentadienyl‐cobalt complexes
11:15 Alice Karikachery
University of Missouri ‐ Columbia
Photochemistry studies on Pt(IV) naphthalenyl complexes
11:45am‐12:45pm Lunch (Busch Student Center, room 253)
12:45‐2:15pm Poster Session (Busch Student Center, room 251)
2:15‐3:45pm Oral Session 3 – Session Chair: Liviu Mirica
2:15 Doug Hammerstroem
Saint Louis University Aluminum nanoparticles capped using alkyl substituted epoxides
2:45 Stephen Chung Saint Louis University Size‐dependent Al nanoparticle oxidation enthalpy
3:15 Mark Conradi Washington University in St Louis
Discovery of a new species in the hydrogen chemistry of NaAlH4, by in situ NMR
3:45‐4:00am Break – cookies/coffee served outside LH 2
4:00‐5:00pm Oral Session 4 – Session Chair: Paul Sharp
4:00 Jim Bashkin University of Missouri – St Louis
An inorganic chemist ventures into drug discovery and bio‐organic chemistry
4:30 Bill Buhro Washington University in St Louis
The Pathway from a molecular precursor to silver nanoparticles: the prominent role of aggregative growth
5:00 Closing Remarks

5
Abstracts – Oral Session

6
Pt‐cyanoximates: self‐assembled nano‐size electrical conductors
Jessica Ratcliiff,a Vladimir Kolesnichenko,b Mikhail Berezin,c Vadim Pal’shin,d Carl Cheadlea and Nikolay Gerasimchuka,*
a Department of Chemistry, Temple Hall 431, Missouri State University, Springfield, MO 65897. b Department of Chemistry, Xavier University of Louisiana, New Orleans, LA. c Washington University Medical Center, St. Louis, MO. d CAMD, Baton Rouge, LA.
Recently several Pt‐based cyanoxime complexes synthesized and characterized in our research laboratory for the purpose of their in vitro/ in vivo cytotoxicity studies.1,2 However, two PtL2 complexes with the substituted cyanacetamide ligands shown below exist as dark blue‐green solids and appeared to be similar to the “Magnus green salt,” which possesses 1D structure.
N
O
NHO
NC
OH(MCO)
N
O
NHO
NC
H(PiPCO) Complexes exhibit strong intervalence charge transfer band at ~700 nm both in solid state and
in solutions, and have strong propensity to “poker chips” stacks of nanometers size. Strong donor solvents such as DMSO and Py disrupt 1D coordination polymer and promote formation of yellow solutions of monomeric complexes. Nevertheless, both compounds form dark‐green solutions in CH3CN, DMF and DMAA from which complexes can be deposited on a dielectric surface as thin films. Unfortunately, no single crystal data available for these 1D solids, but the EXAFS structure of powdery aggregated complexes revealed short Pt‐‐‐Pt bonds at ~3.15 Å.
Both PtL2 complexes demonstrated significant electrical conductivity in solid state at the level of 22 – 33 S/cm, which increases with partial chemical oxidation of Pt‐centers by Br2 or AgPF6.
1 Gerasimchuk , N.; Goeden, L.; Durham, P.; Barnes, C.; Cannon, J.F. Inorg. Chim. Acta, 2008, 361, pp.1983‐2001.
2 Eddings, D., Barnes, C., Durham, P., Gerasimchuk, N.N., Domasevich, K.V. Inorganic Chemistry, 2004, 43 (13), pp. 3894‐3909.
Talk
9:05

7
Stable mononuclear organometallic Pd(III) complexes and their C–C bond formation reactivity
Julia R. Khusnutdinova, Nigam P. Rath, and Liviu M. Mirica*
Department of Chemistry, Washington University, St. Louis, Missouri 63130‐4899
Both dinuclear and mononuclear organometallic Pd(III) complexes have been recently proposed as active intermediates in the oxidative functionalization of C–H bonds and oxidatively induced reductive elimination reactions, respectively. While a few dinuclear organometallic Pd(III) complexes have been characterized, no mononuclear organometallic Pd(III) complexes have been isolated to date. Reported herein is the synthesis and characterization of a series of Pd(III) complexes supported by the tetradentate ligand N,N’‐di‐tert‐butyl‐2,11‐diaza[3.3](2,6)pyridinophane (N4). Chemical or electrochemical oxidation of the Pd(II) complexes (N4)PdII(R)(X) (R = Me, X = Cl: 1; R = Ph, X = Cl: 2; R = X = Me: 3) generates [(N4)PdIIIMeCl]+ (1+), [(N4)PdIIIPhCl]+ (2+), and [(N4)PdIIIMe2]
+ (3+), respectively. These stable Pd(III) complexes were isolated and characterized by X‐ray diffraction, cyclic voltammetry, UV‐vis, EPR, magnetic moment measurements, and DFT calculations to confirm the presence of paramagnetic d7 Pd(III) centers. Interestingly, these Pd(III) complexes undergo light‐induced C–C bond formation reactions to give the corresponding homocoupled products ethane or biphenyl. Particularly remarkable is the observation for the first time of ethane formation from a monomethyl Pd complex, 3+. This transformation has direct implications in the development of catalysts for oxidative oligomerization of methane in particular, and oxidatively‐induced Pd‐catalyzed C–C bond formation reactions in general.
Talk
9:35

8
Charging nanowalls: adjusting the carbon nanotube isoelectric point via surface functionalization
Martin R. McPhail, Jacob A. Sells, Zhen He, and Charles C. Chusuei*
Chemistry Department, Missouri University of Science and Technology, Rolla, MO, USA
Controlling the point of zero charge (PZC) of carbon nanotubes is important for depositing finely dispersed metal nanoparticles from precursors in soln. for fabricating chemical and biological sensor and catalyst surfaces. Hi pressure carbon monoxide (HiPco) single‐walled carbon nanotubes (p‐SWNTs) were functionalized with carboxylic acid (COOH‐SWNT), nitroso (NO‐SWNT), and maleic anhydride (MA‐SWNT) groups. The MA‐SWNTs were synthesized using Diels‐Alder chemistry with graphene sheets serving as the diene. NO‐SWNTs were created via electrochemical oxidation, using HiPco sheets as a working electrode. The presence of attached moieties on the carbon nanotube surface was verified using X‐ray photoelectron (XPS) and attenuated total reflection IR (ATR‐IR) spectroscopies. PZC measurements (in parentheses) were in the descending order: NO‐SWNTs (7.5) > p‐SWNTs (3.5) > MA‐SWNTs (2.0) > COOH‐SWNTs (1.2). The trend in measured PZC values correlated with the electron
withdrawing character of the attached moieties, consistent with Hammett constants. Variations in the electron withdrawing character of the moieties led to SWNTs with differing semiconducting character, as observed in the UV‐vis‐NIR E11 semiconducting region and Raman D‐to‐G band ratios denoting changes in the semiconducting‐to‐metallic character of the functionalized SWNTs. These results suggest a tunability of the SWNT PZC via sidewall functionalization, a factor to consider for practical SWNT nanomaterial fabrication.
free-standing NTsheet as working
electrode
referenceelectrode,Ag/AgCl
counterelectrode(Pt wire)
free-standingsinglewalled
NT sheet
free-standing NTsheet as working
electrode
referenceelectrode,Ag/AgCl
counterelectrode(Pt wire)
free-standingsinglewalled
NT sheet
anthracene-likeportion of carbonnanotube (diene)
maleic anhydride(dienophile)
O
O
O
O
O
O
+
adduct
anthracene-likeportion of carbonnanotube (diene)
maleic anhydride(dienophile)
O
O
O
O
O
O
+
adduct
Talk
10:05

9
Synthesis, structure and spectroscopy of heteropentadienyl‐cobalt complexes
Bryn L. Lutes,a Donastas Sakellariou‐Thompson,a Michael Lipschutz,a John Seonghyun Lee,a John R. Bleeke,a,* and Nigam P. Rathb
a Department of Chemistry, Washington University, One Brookings Drive, St. Louis, Missouri 63130. b Department of Chemistry and Biochemistry, University of Missouri St. Louis, One University Boulevard, St. Louis, Missouri 63121
A heteropentadienyl ligand is one in which the terminal carbon of a pentadienyl ligand has been replaced with a heteroatom (O, P, N, S). The study of heteropentadienyl‐transition metal complexes has been a busy area of research in the last decade. These molecules can now be synthesized by generalized methods, allowing their interesting reactivity to be the focus of current research. These
molecules have shown the ability to adopt and shift between a variety of bonding modes (5 3
1), opening coordination sites at the metal center. The ultimate goal for heteropentadienyl‐transition metal complexes is their use as homogeneous catalysts.
This presentation will focus on the synthesis of a class of heteropentadienyl‐cobalt‐phosphine complexes, and their similarities to and differences from the existing heteropentadienyl‐iridium and –rhodium systems. Specifically, the synthesis of oxapentadienyl‐cobalt‐phosphine complexes from the reactions of potassium oxapentadienide and potassium 2,4‐dimethyl‐5‐oxapentadienide with ClCo(PMe3)3 and the characterization of these complexes by 1H, 31P and 13C nuclear magnetic resonance (NMR) and X‐Ray crystallography will be discussed. The oxapentadienyl‐cobalt‐phosphine complexes
have shown a remarkable stability of the all‐carbon 3 oxapentadienyl bonding mode. These complexes undergo ligand substitution reactions when exposed to carbon monoxide, losing phosphine without altering the oxapentadienyl bonding mode. The reaction of the oxapentadienyl‐cobalt‐phosphine complexes and their carbon monoxide ligand substitution products with small electrophiles, HOTf and MeOTf, was also investigated.
Talk
10:45

10
Photochemistry studies on Pt(IV) naphthalenyl complexes
Alice Raphael Karikachery, Joseph M. Clarkson, Han Baek Lee, Paul R. Sharp*
Dept. of Chemistry, University of Missouri, Columbia, MO 65211‐7600, USA
Energy storage in chemical bonds paves way for renewable and greener fuel technology. Hence there has been a huge interest in water splitting for the production of hydrogen, but this four electron process is difficult and not very economical. Alternatively hydrogen halide splitting, a more facile two electron process is also being explored in this regard. Photolytic halide elimination, from high valent late transition metal centers like Pt, Au and Rh, is a key step in designing HX splitting cycles. Further a higher quantum efficiency for the reductive elimination process will facilitate this. Previously in our group, photoelimination of bromine at Pt(IV) centers, coordinated to corannulene, phenanthrene and naphthalene ring systems have been investigated. The quantum yield for the Pt(IV) naphthaleny (1) complex was measured in the presence of an alkene trap and was close to 6%. Photochemistry of analogues 2 and 3 were studied to understand the factors promoting the halogen elimination. The proximity of the peri‐hydrogen to the metal center and effects of ring substitution will be explored.
Pt Pt
Pt
Br
Br
Et3P Br
PEt3
Br
Br
Et3P
PEt3Br
Br
PEt3
PEt3
Br
Br
Br
1 2 3
Talk
11:15

11
Aluminum nanoparticles capped using alkyl substituted epoxides
Douglas W. Hammerstroem,a Stephen W. Chung,a Elena A. Guliants,b Christopher E. Bunker,c Paul. A. Jelliss,a,* and Steven W. Bucknera,*
a Saint Louis University, Department of Chemistry 3501 Laclede Avenue, St. Louis, Missouri 63103. b Department of Electrical and Computer Engineering, University of Dayton Research Institute, Dayton, Ohio 45469. c Air Force Research Laboratory, Propulsion Directorate, Wright‐Patterson Air Force Base, Ohio 45433.
Aluminum has been a research interest of ours due to its many energy‐related applications, high abundance, and low cost. Micron‐sized pyrophoric metal particles have been used for thermite reactions, as propellants, for small‐scale hydrogen production, and in other applications in energy and fuels. Aluminum nanoparticles (Al NPs), in particular, have demonstrated great promise for these applications. Al NPs show faster burn rates, lower activation energy for H2 absorption and desorption, and have the potential for higher energy release. Al NPs are also suggested to have significant potential as dense hydrogen storage media. The synthesis and stabilization of Al NPs is a particularly challenging area in nanomaterial synthesis. Two of the main challenges with synthesis and stabilization of Al NPs is prevention of grain growth and inhibition of oxidation upon exposure to air. We have synthesized small (20‐30 nm) aluminum nanoparticles using a wet chemical protocol and passivated them using alkyl‐substituted epoxides as capping agents. These particles are capped using an epoxide reagent that subsequently polymerizes on the nanoparticle’s surface to produce a polyether. The FTIR and 13C NMR were used to help confirm polymerization of polyether chains. TEM and X‐ray powder diffraction were used to measure particle size. X‐ray powder diffraction and DSC/TGA were used to confirm fcc Al presence, absence of Al2O3, and particle degradation over time through air exposure. Capping and passivation Al NPs is effectively accomplished and stabilized against further growth, and oxidation is significantly inhibited by the organic capping layer. The organic shell that caps the nanoparticles is an oxygen‐rich polyether layer. While this oxygen‐rich layer is present, the product nanoparticles have very low aluminum oxide content.
Talk
2:15

12
Size‐dependent aluminum nanoparticle oxidation enthalpy
Stephen W. Chung,a Elena A. Guliants,b Christopher E. Bunker,c Paul A. Jelliss,a Steven W. Bucknera,*
a Saint Louis University, Department of Chemistry 3501 Laclede Avenue, St. Louis, Missouri 63103. b Department of Electrical and Computer Engineering, University of Dayton Research Institute, Dayton, Ohio 45469. c Air Force Research Laboratory, Propulsion Directorate, Wright‐Patterson Air Force Base, Ohio 45433.
Micron‐sized aluminum particles have been used for many high‐energy applications, including propellants.1 Currently, stabilized Al nanoparticles are being developed to enhance the reactivity, combustion rate, and possibly the enthalpy of reaction.2 The reactivity and combustion kinetics of aluminum nanoparticles (Al NPs) have been researched extensively.3 While there are some thermodynamic studies of Al NPs there are not detailed theoretical models available.
The enthalpy of reaction for bulk aluminum oxidizing with O2 is ‐1675.7 kJ/mol. However, the enthalpy of reaction for Al NPs reacting with oxygen may vary, based on the size of the nanoparticle. The origin of this variation in enthalpy of reaction from micron‐sized particle and bulk Al is due to surface effects in the Al NPs. The greater surface area means more surface Al atoms, which have fewer bonds with neighboring atoms than interior atoms. The nanoparticles are also assumed to have an oxide layer passivating the particle. This passivating layer causes an interfacial energy contribution to the enthalpy of reaction. Another aspect that must be considered is the nature of the oxidation product: Does it retain nanoscale dimensions or is there agglomeration? What is the crystal structure of the product? The product may be nano‐Al2O3, bulk Al2O3, bulk boehmite (AlO(OH)), bulk bayerite (Al(OH)3), or some combination of these. Here we describe a theoretical model that explicitly identifies the size dependence of oxide‐passivated Al nanoparticles on the enthalpy of the combustion reaction.
We use a thermodynamic cycle to formulate an equation relating to the reaction enthalpy of nano‐Al→nano‐Al2O3 (∆H°rxn,np,np) and nano‐Al→bulk Al2O3 (∆H°rxn,np,bk). We consider the size‐dependent cohesive energy and lattice energy (SDCE and SDLE) of Al and Al2O3, respectively. We use the Nanda model4 to describe the size dependencies of NPs.
1 Ramaswamy, A.; Kaste, P. Energ. Mater. 2005, 23, 1‐25. 2 Fernando, K. A. S.; Smith, M. J.; Harruff, B. A.; Lewis, W. K.; Guliants, E. A.; Bunker, C. E. J. Phys. Chem. C 2009, 113, 500‐503. 3 Galfetti, L.; De Luca, L. T.; Severini, F.; Meda, L.; Marra, G.; Marchetti, M.; Regi, M.; Bellucci, S. J. Phys.: Condens. Matter 2006, 18, S1991‐S2005. 4 Vanithakumari, S. C.; Nanda, K. K. J. Phys. Chem. B 2006, 110, 1033‐1037.
Talk
2:45

13
Discovery of a new species in the hydrogen chemistry of NaAlH4 by in situ NMR
Tim M. Ivancic and Mark S. Conradi*
Dept. of Physics, Washington University, St. Louis, MO 63130, USA
NaAlH4 has become the archetypal complex (ionic‐covalent) hydrogen storage solid, since the
discovery in 1997 that titanium and some other metals catalyze the reaction in both directions. Given
that spatially separated NaH and Al under excess H2 pressure can form NaAlH4, we have hypothesized a
mobile Al‐ or Na‐ bearing intermediate species in the reaction scheme.
27Al in situ NMR has been used to discover such a new species during dehydriding of NaAlH4.
Importantly, the new species can also be formed under excess H2 pressure without net evolution of Al
and H2; the new species can be returned to ambient for further study. The features of the new species
include: a very rapid 27Al T1, a sharp line (without MAS) at 300 K that broadens by ‐60oC, an
accompanying sharp hydrogen NMR line that is also motionally narrowed, CPMAS evidence of bonded
or at least nearby H, and a 27Al shift just above NaAlH4. The new species appears to be a highly defective
form of NaAlH4, where the large concentration of vacancies promotes rapid diffusion of Al and H.
Talk
3:15

14
An inorganic chemist ventures into drug discovery and bio‐organic chemistry: compounds designed to bind conserved regions of HPV DNA show broad‐spectrum activity against oncogenic (high‐risk) HPVs James K. Bashkin,a,* Terri G. Edwards,b Kevin J. Koeller,a Urszula Slomczynska,a and Chris Fisherb
a Dept. of Chemistry & Biochemistry, UM‐St. Louis, Saint Louis, MO 63121 USA b NanoVir, LLC, 4717 Campus Dr., Kalamazoo, MI 49008 USA
Cervical infections by the “high risk” human papillomaviruses (HPVs), including HPV16, 18 and 31 are usually not treated upon their discovery, but are flagged for later “follow‐up.” Traditional approaches to antiviral design for HPV have failed for a variety of reasons including the lack of traditional antiviral targets. Therefore, we designed novel antivirals to reduce viral DNA levels and persistence. We optimized several series of pyrrole‐imidazole polyamides based on lead structures designed to bind DNA sequences within the origin of replication (ori) of high‐risk HPV genotypes HPV16 and 18. Resulting compounds potently decreased cancer‐causing HPV DNA concentrations for HPV16, 18 and 31 in human cell and tissue cultures. We used human cells that maintain the viral DNA genomes as episomes (DNA circles, like plasmids, that aren’t part of human chromosomes): keratinocytes maintaining HPV16, 18 or 31 episomes were treated with increasing concentrations of polyamide or vehicle‐control for 48h in order to study dose response behavior. Loss of episomal DNA was measured by quantitative PCR (Q‐PCR). Of the 46 polyamides tested, including 16 control polyamides not derived from our core lead structures, 12 gave pseudo‐IC50s in the 30‐100 nM range against a least two genotypes, while several compounds decreased DNA levels of all three viral subtypes to undetectable levels. Treatment of cells with a lead polyamide, followed by removal of compound and passage of cells, resulted in a moderate rebound of viral DNA that did not return to control levels after 6 additional days in culture. Extension of the polyamide treatment period resulted in a remarkably‐effective delay and inhibition of episomal DNA rebound. These results illustrate that targeting of the HPV ori with polyamides has the potential for potent and long‐lasting effects on HPV DNA load. The structure of an active, lead compound is shown below. Isolated as its tris‐TFA salt, the compound has been prepared and purified on a 1.5 g scale; it is readily taken up by human keratinocytes in monolayer cell and 3‐D tissue cultures and is in preclinical development.
Talk
4:00

15
The pathway from a molecular precursor to silver nanoparticles: the prominent role of aggregative growth
Vernal N. Richards,a Nigam P. Rath,b Shawn P. Shields,a and William E. Buhroa,*
a Department of Chemistry and Center for Materials Innovation, Washington University, St. Louis,
Missouri 63130‐4899 b Department of Chemistry and Biochemistry and Center for Nanoscience, University of Missouri – St. Louis, St. Louis, MO 63121
A mechanistic study of Ag‐nanoparticle growth by reaction of [(PPh3)2Ag(O2CC13H27)] and AIBN is reported. The half‐life for precursor disappearance at 130.0 ± 0.1 ºC under the reaction conditions is determined to be 3.65 ± 0.42 min, which defines the time scale for classical (LaMer) nucleation and growth to be within the first 15 min (4 half‐lives). The nanoparticle‐growth kinetics are separately determined by TEM monitoring and UV‐visible spectroscopy. Fits to the kinetic data establish that the active‐growth regime extends to 58 min, and that Ostwald ripening ensues shortly thereafter. Evidence for an aggregative nucleation and growth process is obtained. The quantitative data indicate that classical nucleation and growth, aggregative nucleation and growth, and Ostwald ripening occur in consecutive time regimes with little overlap, and that nanoparticle growth is dominated by the aggregative regime. Aggregative growth should be considered a potential contributing mechanism in all nanoparticle‐forming reactions.
Talk
4:30

16
Abstracts – Poster Session

17
Synthesis, characterization, and applications of 2,3,4,5‐tetraphenylgermoles containing coordinating substituents at the germanium center
J.B. Carroll, Janet Braddock‐Wilking,* Nigam P. Rath, and Teresa L. Bandrowsky
University of Missouri‐St. Louis, One University Boulevard, St. Louis, MO 63121
Several compounds based on 2,3,4,5‐tetraphenylgermole have been characterized containing various substituents at the germanium center. The compounds were synthesized via a salt‐metathesis coupling of 1,1‐dichloro‐2,3,4,5‐tetraphenylgermole1 and alkynyl lithium reagents terminated at one end with a coordinating moiety (an example is provided in Figure 1). These structures were characterized using multinuclear NMR, UV‐Vis and fluorescence spectroscopies, and X‐ray crystallography.
As the compounds presented herein have subsituents that can coordinate to metal centers, such as pyridyl or diphenylphosphino moieties, they can possibly be used for the chemodetection of metals. Detection can occur by altering the photoluminescent properties of these compounds through perturbation of the electronic or structural features of the germole ring (e.g., by affecting aggregation‐induced emission (AIE), a feature of related siloles2). Therefore, preliminary data describing reactions of 1,1‐bis[(pyridinyl)ethynyl]‐germoles synthesized with metal complexes will be presented.
Figure 1. Crystal structure of 1,1‐bis[(diphenylphosphino)ethynyl]‐2,3,4,5‐tetraphenylgermole
1. Curtis, M.D. “Synthesis and Reactions of Some Functionally Substituted Sila‐ and Germacyclopentadienes.” J. Am. Chem. Soc. 1969, 91, 6011‐6018. 2. (a) Lou, J.; Xie, Z.; Lam, J.W.Y.; Cheng, L.; Chen, H.; Qiu, C.; Kwok, H.S.; Zhan, X.; Liu, Y.; Zhu, D.; Tang, B.Z. “Aggregation‐induced emission of 1‐methyl‐1,2,3,4,5‐pentaphenylsilole.” Chem. Comm., 2001, 1740‐1741. (b) Chen, J.; Law, C.C.W.; Lam, J.W.Y.; Dong, Y.; Lo, S.M.F.; Williams, I.D.; Zhu, D.; Tang, B.Z. “Synthesis, Light Emission, Nanoaggregation, and Restricted Intramolecular Rotation of 1,1‐Substituted 2,3,4,5‐Tetraphenylsiloles.” Chem. Mater., 2003, 15, 1535‐1546.
Poster
1

18
Germoles designed to exhibit aggregation‐induced emission
Teresa Bandrowsky, Janet Braddock‐Wilking,* Nigam Rath, and J.B. Carroll
University of Missouri‐St. Louis, One University Boulevard, St. Louis, MO 63121
In recent years, much attention has been given to the development of silole derivatives which exhibit the unusual phenomenon referred to as aggregation‐induced emission [AIE]. AIE‐active molecules are non‐luminescent in solution, but emit strongly in the solid phase or as aggregated suspensions in solutions in which solubility is poor. AIE features are in sharp contrast to the well studied quenching of light emission referred to as aggregation‐caused quenching [ACQ]. With careful design, the unique propeller‐like geometry of AIE‐active molecules (an example is shown in Figure 1) inhibits the typical pathways that quench luminescence by impeding intramolecular motion of aryl substituents on the molecule and preventing conventional π‐π stacking interactions between adjacent molecules.1
While the extent of information concerning the synthesis and applications of AIE‐active siloles is vast, the development of the related germoles has remained largely undeveloped. As a result of the similarities in the energy levels of the HOMO and LUMO between the two analogs, germoles should also exhibit AIE behavior. Design of functionalized germoles, while more challenging synthetically than siloles, should yield a class of molecules with diverse, yet practical applications. AIE‐active germoles, like their predecessors, can be designed for real‐world applications as films in photoelectronic devices, chemical and explosive detection, pH sensing, cell imaging, immunoassay biolabels, and biological probes.1 In this work, a series of functionalized germoles have been successfully synthesized and their potential multifaceted applications are being investigated.
Figure 1. The figure of a functionalized germole and its crystal structure depicting the unique propeller‐like geometry necessary for AIE‐active molecules.
1 Lui, J.; Lam, J.W.Y.; Tang, B.Z J. Inorg. Organomet. Polym. 2009, 19, 249.
Poster
2

19
Investigation of catalytic hydrosilylation reactions using a platinum‐PTA complex
Sitaram Acharya, Janet Braddock‐Wilking, and Nigam P. Rath
Department of Chemistry and Biochemistry, University of Missouri‐St. Louis, St. Louis, Missouri‐63121
The metal‐mediated hydrosilylation reaction utilizing the water‐soluble platinum complex bearing the 1,3,5‐triaza‐7‐phosphaadamantane (PTA) ligand, cis‐dimethylbis(1,3,5‐triaza‐7‐phosphaadamantane) platinum(II), Pt(CH3)2(PTA)2 (1) has been investigated for the first time. The catalytic hydrosilylation reaction with different unsaturated systems such as alkenes, alkynes, ketones, and bis(alkenes/alkynes) has been explored utilizing different tertiary hydrosilanes as well as siloles and silafluorenes. Mostly, regioselective products were obtained and the β‐trans isomers were selectively formed during the hydrosilylation of the terminal alkynes and bis(alkynes) (eq 1). The effect of steric and electronic factors was examined on the selectivity of the hydrosilylated products. The results from these studies will be described.
Poster
3

20
Synthesis, electrochemistry, and magnetic properties of a binuclear bis(‐OMe) Co(II)Co(III) mixed‐valent complex
Jia Luo and Liviu M. Mirica*
Dept. of Chemistry, Washington University in St. Louis, St. Louis, MO, 63130, USA
A novel binuclear Co(II)Co(III) mixed‐valent complex containing ‐methoxo bridging ligands has been prepared where each cobalt center is capped by the tridentate N‐methyl‐N,N‐bis(2‐
pyridylmethyl)amine (L, C13H15N3): [LCo(II)‐(‐carboxylato)‐bis(‐methoxo)Co(III)L](ClO4)2. The corresponding Co‐O(acetate) and Co‐O(methoxo) bond distances at two cobalt centers differ significantly, which implies that the valences of cobalt ions are localized. The measurements of magnetic susceptibility confirm that cobalt(II) center is high‐spin in a pseudo‐octahedral environment. Electrochemical and spectroscopic characterizations, including UV‐Vis and EPR, were carried out to have a better understanding of the mixed‐valent nature of the complex. The relevance of this complex to the recently reported Co‐based water oxidation catalysts will also be discussed.
Poster
4

21
Silver(I) cyanoximates: synthesis, crystal structures and physical properties
Jeffrey R. Morton, and Nikolay N. Gerasimchuk*
Department of Chemistry, Missouri State UniversitySpringfield, MO 65897, United States of America
Ten silver(I) complexes with cyanoxime ligands, compounds that have general formula NC‐C(=NOH)‐R where R is an electron‐withdrawing group, were synthesized and characterized using the IR (nujol mulls), UV‐visible spectroscopy and X‐ray analysis. All ten synthesized AgL complexes have bright and unusual for silver(I) compounds color ranging from yellow, orange, to a deep purple. Crystal structures revealed formation of coordination polymers with cyanoxime anions acting as bridging ligands using both N and O atoms of the >CNO fragment for binding metal ions. Bond lengths between Ag(I) centers and anions in these polymers are significantly shorter than the sum of ionic radii of involved elements and evidenced their covalent character. Silver(I) cyanoximates demonstrate remarkable visible light insensitivity, but get sensitized and readily darken after the exposure to several gases such as CO, SO2, C2H2. That interesting property of solid AgL is currently under investigation.
The effect of UV irradiation on the UV‐vis diffuse reflectance spectra:
350 400 450 500 550 600 6500
10
20
30
40
50
60
70
80
% R
efle
ctan
ce
Wavelength (nm)
0 min 30 min 60 min 90 min 120 min 150 min 180 min 210 min 240 min 270 min
350 400 450 500 550 6000
10
20
30
% R
efle
ctan
ce
Wavelength (nm)
0 min 30 min 60 min 90 min 120 min 150 min 180 min 210 min 240 min 270 min 300 min 330 min 360 min 390 min 605 min
Poster
5

22
Binucleating ligands: control of the M‐M distance in transition metal complexes
Fengzhi Tang, Fengrui Qu, and Liviu Mirica*
Dept. of Chemistry, Washington University in St. Louis, St. Louis, MO 63130
Binuclear transition metal complexes are involved in a number of important chemical and enzymetic transformations, such as dioxygen activation and water oxidation. Reactivity of these complexes can be tuned by controlling the distance between the two metal centers. An efficient synthetic route for a series of tetradentate binucleating ligands was developed. The distance between the two metal binding sites of these ligands can be controlled by rigid 4,5‐xanthen‐di‐yl and 4,6‐dibenzofuran‐di‐yl linkers. Based on these ligands, binuclear copper and nickel complexes were synthesized and characterized by spectroscopic methods and X‐ray diffraction. A comparative study between the newly synthesized binuclear metal complexes and similar metal complexes with mono‐ and binucleating ligands will be presented, with a particular focus on water oxidation catalysis.
Poster
6

23
New chemical agents for controlling amyloid‐ peptide oligomerization/ aggregation in Alzheimer’s disease
Anuj K. Sharma, Nicholas. J. Hawco, Darren Finkelstein, S. Tucker, Ying Zhang, Nigam P. Rath, and Liviu M. Mirica*
Dept. of Chemistry, Washington University, St Louis, MO 63130, USA
Studies illustrate that amyloid‐β (Aβ) plaques are largely associated with the neuropathogenesis of Alzheimer’s disease (AD). However, recent in vivo studies have shown that the soluble aggregates of
A (hexamers, nonamers, and dodecamers) are most neurotoxic and their formation being correlated with memory loss and neurodegeneracy. Interactions of metal ions, especially Cu, Zn, and Fe, with the Aβ peptide are hypothesized to play an important role in the etiology of AD. We are trying to study the
interaction of these physiologically relevant soluble A oligomers with transition metal ions and to develop new chemotherapeutics and imaging agents for the prevention, diagnosis, and treatment of AD.
We are employing a bifunctional strategy to develop metal‐binding compounds that will interact
with the A oligomers and inhibit the formation of neurotoxic higher‐mass oligomers or amyloid plaques. The proposed bifunctional compounds contain molecular fragments such as 2‐phenyl‐
benzothiazole and o‐vanillin, which were shown to bind to soluble A oligomers and inhibit their aggregation. Synthesis and characterization of these bifuntional chelators and their interaction with metal ions by characterizing their metal complexes and the studies of their interaction with synthetic Aβ peptides will be highlighted.
Poster
7

24
Spectroscopic and computational studies of six‐membered bipyridine platinacycle complexes as solid‐state luminescent materials
Robert Robinson Jr. and Paul R. Sharp*
Department of Chemistry, University of Missouri‐Columbia, Columbia, MO 65211, USA
The six‐membered bipyridine platinacycles, Pt(C6H4XC6H4)(tBu2bpy) (X = NMe, O, CH2;
tBu2bpy = 4,4’‐bis‐t‐butyl‐2,2’‐bipyridine), have been synthesized and fully characterized by NMR spectroscopy and X‐ray crystallographic data. These platinacycle complexes are highly luminescent materials (solid‐state) and the luminescence originates from a “mixed‐metal/ligand‐to‐ligand charger transfer (MMLLCT) band.1 Spectroscopic (UV‐vis/fluorescence) studies have shown that both the absorption and emission properties are tunable by altering the bridging group (X) within the metallacyclic ring system. In addition, computational (DFT/TD‐DFT) studies indicate that the electronic properties of the bridging groups influence the mixed‐metal/ligand molecular orbital, which shifts the energy gap of the MMLLCT band.
1. De Crisci, A. G.; Lough, A. J.; Multani, K.; Fekl, U. Organometallics 2008, 27(8), 1765‐1779.
X = CH2 X = NMe X = O
Poster
8
25
trans cinnamic acid truxillic acid
h
trans cinnamic acid truxillic acid
h
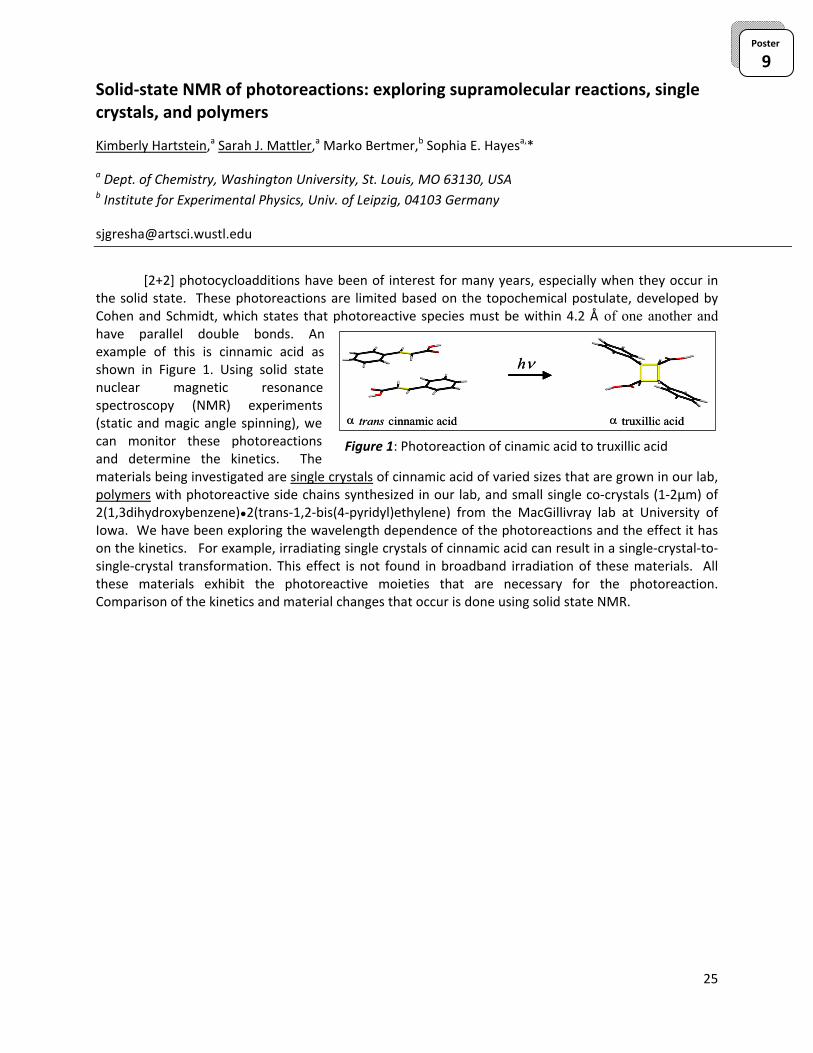
Figure 1: Photoreaction of cinamic acid to truxillic acid
Solid‐state NMR of photoreactions: exploring supramolecular reactions, single crystals, and polymers
Kimberly Hartstein,a Sarah J. Mattler,a Marko Bertmer,b Sophia E. Hayesa,*
a Dept. of Chemistry, Washington University, St. Louis, MO 63130, USA b Institute for Experimental Physics, Univ. of Leipzig, 04103 Germany
[2+2] photocycloadditions have been of interest for many years, especially when they occur in the solid state. These photoreactions are limited based on the topochemical postulate, developed by Cohen and Schmidt, which states that photoreactive species must be within 4.2 Å of one another and have parallel double bonds. An example of this is cinnamic acid as shown in Figure 1. Using solid state nuclear magnetic resonance spectroscopy (NMR) experiments (static and magic angle spinning), we can monitor these photoreactions and determine the kinetics. The materials being investigated are single crystals of cinnamic acid of varied sizes that are grown in our lab, polymers with photoreactive side chains synthesized in our lab, and small single co‐crystals (1‐2µm) of 2(1,3dihydroxybenzene)●2(trans‐1,2‐bis(4‐pyridyl)ethylene) from the MacGillivray lab at University of Iowa. We have been exploring the wavelength dependence of the photoreactions and the effect it has on the kinetics. For example, irradiating single crystals of cinnamic acid can result in a single‐crystal‐to‐single‐crystal transformation. This effect is not found in broadband irradiation of these materials. All these materials exhibit the photoreactive moieties that are necessary for the photoreaction. Comparison of the kinetics and material changes that occur is done using solid state NMR.
Poster
9

26
Optical pumping phenomena in si‐GaAs
Dustin Wheeler, Erika Sesti, Katherine M. Wentz, and Sophia E. Hayes*
Dept. of Chemistry, Washington University in St. Louis, St. Louis, MO 63130, USA
Electronic devices are becoming increasingly important to our society, and semiconductors make up the critical components for controlling these devices.Accurate characterization of semiconductor samples is therefore required as the demands on materials become more exacting. A newly emerging technique for probing the electronic band structure of (and hence the behavior of electrons in) semiconductors is optically‐pumped NMR (OPNMR).
Most recent OPNMRresearch has been focused on direct‐gap semiconductors, in particular III‐V semiconductors like GaAs.1,2 In the presence of a magnetic field, the relatively simple band structure splits into a series of quantized levels known as Landau levels. This quantization alters the optical excitation selection rules normally in effect for these structures, and in some cases it may change the overall polarization of electrons in the conduction band. Below the bulk semiconductor band edge, samples of a given semiconductor with different dopants or varied dopant levels can generate unique signal profiles with respect to photon energy. However, samples of a given semiconductor will have relatively constant profiles above the bulk band edge.
Using experimental and theoretical magnetoabsorption data, we have successfully modeled some of the fine structure features in OPNMR signal intensity for energies above the band edge of GaAs.3 The next step is to build on this success and attempt to use theoretical absorption data from the Stanton research group (Dept. of Physics, Univ. of Florida) as an input to our model to discern more fine structure in the high‐energy regime of the signal profile. Magnetoabsorption, seen as the sum of spin‐up and spin‐down electrons in the conduction band, is a valuable tool in capturing the major features in the profile, but data from the Stanton group suggests that the difference between conduction band electron spins may capture many of the finer features that the current version of the model misses. Our goal is to use these tools to create a generalized model that can be applied to many different semiconductor samples to predict the electronic structure and behavior of these samples.
1. Bowers. Microscopic interpretation of optically pumped NMR signals in GaAs. Solid State Nuclear Magnetic Resonance (1998) (11) pp. 11‐20 2. Mui et al. Physical insights from a penetration depth model of optically pumped NMR. J. Chem. Phys. (2008) 3. Mui et al. Manifestation of Landau level effects in optically‐pumped NMR of semi‐insulating GaAs. Phys. Chem. Chem. Phys. (2009) vol. 11 (32) pp. 7031.
600 400 200 0 -200
ppm1.48 1.50 1.52 1.54 1.56 1.58
-6
-4
-2
0
2
4
Photon Energy (eV)
OP
NM
R S
igna
l Int
ens
ity (
arb
. uni
ts)
+
–
Poster
10

27
An integrated synthetic methodology for surface passivation and kinetic stabilization of aluminum nanoparticles with polymerizable capping agents
Brandon Thomas,a Steven W. Buckner,a,* Chistopher E. Bunker,b Mark A. Burgers,a Steven W. Chung,a Elena A. Guliants,c Douglas W. Hammerstroem,a Sophia E. Hayes,d Paul A. Jelliss,a,* Carl D. Oberle,a and Katherine Wentzd
a Saint Louis University, Department of Chemistry 3501 Laclede Avenue, St. Louis, Missouri 63103. b Air Force Research Laboratory, Propulsion Directorate, Wright‐Patterson Air Force Base, Ohio 45433. c Department of Electrical and Computer Engineering, University of Dayton Research Institute, Dayton, Ohio 45469. d Dept. of Chemistry, Washington University in St Louis, St Louis, Missouri 63130.
We have developed a new synthetic methodology for chemical passivation of reactive aluminum nanoparticles (AlNPs), which are potential power sources for portable hydrogen fuel cellsused by military personnel in the field. Based on catalytic decomposition of alane‐adducts in toluene solution, our synthetic protocols have been refined to produce AlNPs with a reasonably narrow range of sizes, typically ~30 nm on average. Recent work has focused on utilizing the reactivity of surface aluminum atoms to initiate ring‐opening polymerization upon treatment with alkyl‐epoxide species. A solvent‐based thermal approach also promotes additional polymerization of an alkene functionality in the epoxide capping agent, designed to embed to the resulting kinetically stabilized AlNPs in a hydrophobic polymer matrix. We present results from FTIR, solid state NMR, and PXRD, along with titrimetric measurements to determine active aluminum content of the AlNPs.
Poster
11

28
Alkene oxidation by [(COD)Pt(OH)]nn+ : mechanistic studies
Nandita M.Weliange and Paul R.Sharp*
Department of Chemistry, University of Missouri‐Columbia, Columbia, MO 65211, USA
Pt‐hydroxo complex, [(COD)Pt(OH)]nn+ reacts with both acyclic and cyclic alkenes to yield
oxygenated products. Oxygenation of 2‐norbornene yields the protonated platinaoxetane,1 [(COD)Pt(OH)(C7H10)]
+ while ethene is oxidized to acetaldehyde. Both reactions are proton catalyzed. The ethene oxidation yields acetaldehyde and (COD)Pt(CH2CH3)(OTf) as the only products. The reaction proceeds via a Pt‐ vinyl hydroxo (π‐bonded) intermediate that was characterized by 1H and 13C NMR techniques. An analogous situation is seen in the case of propene oxidation, where acetone is produced. Details and proposed mechanisms for these alkene oxidation reactions will be presented.
1. Weliange N.M.; Szuromi E.; Sharp P.R. J. Am. Chem. Soc. 2009, 131, 8736-8737.
Poster
12

29
Ferracarboranes as electron transfers mediator for glucose oxidase
Scott S. Graham, Paul A. Jelliss,* Adela Josipovic
Dept. of Chemistry, Saint Louis University, St Louis, MO 63103‐2010
We present the synthesis of various ferracarborane derivatives for use as electron transfer mediators for glucose oxidase immobilized in a chitosan film cast upon a glassy carbon electrode as a bioanode for a fuel cell. The primary objective is to retain the ferracarborane electron transfer mediator at the electrode interface with glucose oxidase in the chitosan film, exploiting the reversible FeIII/II redox chemistry of ferracarborane sandwhich complexes. To prevent leaching, the ferracarborane mediators must be covalently tethered at the electrode by some means. The ferracarborane has been incorporated into the chitosan film in two ways. One method involves adding a pendant aldehyde to the mediator, allowing it to be covalently tethered to the chitosan backbone via reductive amination. The other method involves covalently attaching the mediator to activated carbon nanotubes functionalized with carboxylic acid groups which are added to the chitosan matrix using EDC (1‐ethyl‐3‐(3‐dimethylaminopropyl) carbodiimide) coupling . In both cases the thin film electrodes were examined using cyclic voltammetry. Upon the addition of glucose to the cell solution, a rise in peak current signifies proper mass transport of glucose through the film in addition to the retained activity of the enzyme. These ferracarborane mediator systems may also provide application for use as glucose sensors.
Poster
13

30
Bipyridyl nitrosyl rhenacarborane complexes for drug‐delivery vehicle applications
Hongjian Mai,a William Banks,b Trent Cameron,a Patrick Hawkins,b Paul A. Jelliss,a,* Xiaoming Shia
a Dept. of Chemistry, Saint Louis University, St Louis, MO 63103‐2010 b Saint Louis University School of Medicine, Division of Geriatrics, Department of Internal Medicine
Our preliminary work with rhenacarborane complexes has shown that they are especially promising as drug‐delivery candidates with regard to crossing the blood‐brain barrier (BBB), in particular. Our work is centered on the synthesis and adaptation of bipyridyl nitrosyl rhenacarborane complexes with a view to producing a connectable functionality for physiologically active cargo binding. We are pursuing two basic synthetic pathways to produce pendant hydroxyl groups, which can be exploited to bind to amino acid and oligopeptide molecules via ester linkages. This methodology is sought on the basis that the central nervous system (CNS) is replete with esterase enzymes, which can potentially jettison the delivery vehicle from the pharmacophore following BBB traversal. The first route involves the synthesis of complex 1, which bears a hydroxymethyl group on one of the bipyridyl rings. Spectroscopic data (FTIR, 1H and 11B NMR) support the proposed structure and we are planning experiments to use the DCC (dicyclohexylcarbodiimide) coupling methodology to bind this with the dual‐protected amino acid tyrosine, N‐fmoc‐O‐benzyl‐L‐tyrosine, via an ester linkage. We will then use established deprotection measures to expose a delivery‐vehicle‐bound tyrosine moiety. This work is intended to demonstrate proof‐of‐concept delivery‐vehicle‐cargo assembly and deprotection. Secondly, we are synthesizing asymmetric bipyridyl ligands for complexes such as 2, which bears a longer hydroxypentyl group and which we will use to couple to methionine enkaphalin (an endogenous opioid peptide neurotransmitter) based on protocols developed with our tyrosine work. Once successfully synthesized, attention will be focused on pharmacological studies.
N
Re
N
N
Me
OH
1
O
NO
Re
N
N
Me
2
On Ph
Poster
14

31
Solid‐State NMR of inorganic nanomaterials
Katherine M. Wentz,a Brandon Thomas,b Steven W. Buckner,b Mark A. Burgers,b Steven W. Chung,b Douglas W. Hammerstroem,b Paul A. Jelliss,b Carl D. Oberle,b Chistopher E. Bunker,c Elena A. Guliants,d Sophia E. Hayesa,*
a Department of Chemistry, Washington University in St. Louis, St. Louis, MO 63130, USA b Department of Chemistry, Saint Louis University, St. Louis, MO c Air Force Research Lab, Wright‐Patterson Air Force Base, Dayton, OH d University of Dayton Research Institute, Dayton, OH
Nuclear magnetic resonance (NMR) is a spectroscopic technique that can give insight into materials and their structure. Solution‐state NMR is widely accepted for many organic and inorganic materials but maintaining the solid phase has greater potential in discovering the structural information of tomorrow’s semiconductors and nanomaterials. By using techniques such as {1H}13C cross‐polarization magic angle spinning (CP‐MAS), both structural and spatial information can be obtained about the ligands on the surface of the nanoparticle. In addition to the surface structure, NMR can be done on the nanoparticle center itself. 27Al NMR has been performed on several samples of aluminum nanoparticles with different surface ligands as well as 13C CP‐MAS of the surface ligands themselves. 31P MAS NMR of InP nanowires was also evaluated.
Poster
15

32
Capping and passivation of aluminum nanoparticles and the environmental
effects on long‐term air stability
Brandon Thomas,a Steven W. Buckner,a,* Chistopher E. Bunker,b Mark A. Burgers,a Steven W. Chung,a Elena A. Guliants,c Douglas W. Hammerstroem,a Paul A. Jelliss,a,* and Carl D. Oberle,a
a Saint Louis University, Department of Chemistry 3501 Laclede Avenue, St. Louis, Missouri 63103. b Air Force Research Laboratory, Propulsion Directorate, Wright‐Patterson Air Force Base, Ohio 45433. c Department of Electrical and Computer Engineering, University of Dayton Research Institute, Dayton, Ohio 45469.
Unstable in their bare form, aluminum nanoparticles (Al NPs) require surface passivation to inhibit air oxidation. A variety of capping agents have been used to halt agglomeration and provide surface passivation to Al NPs. Most often, simple oxide passivation with alumina is used, while more recent caps include different organic compounds that bind strongly to the NP surface. Simple oxide passivation provides good stability but lowers energy density of the Al NPs. Organic capping agents have the potential to stabilize the particles while minimally reducing energy density. In this paper we discuss the long‐term stability of Al NPs capped with various organic compounds. Organic capping provides good protection from agglomeration but widely variable levels of oxidation protection. Our recently developed epoxide capping method provides the highest level of stability toward oxidation while traditional carboxylic acids afford minimal protection. We also observe that Al NP oxidation is strongly water vapor dependent.
Poster
16

33
Symposium Registrants
First Name Last Name Affiliation Email
Sitaram Acharya University of Missouri‐St. Louis [email protected]
Wipark Anutrasakda Washington University in St. Louis [email protected]
Teresa Bandrowsky University of Missouri‐St. Louis [email protected]
Lol Barton University of Missouri‐St. Louis [email protected]
Jim Bashkin UMSL and NanoVir, LLC [email protected]
John Bleeke Washington University in St. Louis [email protected]
Janet Braddock‐Wilking University of Missouri‐St. Louis [email protected]
Steven Buckner Saint Louis University [email protected]
J. B. Carroll University of Missouri‐St. Louis [email protected]
David Casto University of Missouri‐Columbia [email protected]
Stephen Chung Saint Louis University [email protected]
Joe Clarkson University of Missouri‐Columbia [email protected]
Mark Conradi Washington University in St. Louis [email protected]
Joyce Corey University of Missouri‐St. Louis [email protected]
Kevin Davis Southeast Missouri State University [email protected]
Emi Evangelio Washington University in St. Louis [email protected]
Nick Gerasimchuk Missouri State University [email protected]
Scott Graham Saint Louis University [email protected]
Doug Hammerstroem Saint Louis University [email protected]
Alexia Harris Southeast Missouri State University [email protected]
Michelle Harris Washington University in St. Louis [email protected]
Kimberly Hartstein Washington University in St. Louis [email protected]
Nicholas Hawco Washington University in St. Louis [email protected]
Sophia Hayes Washington University in St. Louis [email protected]
Kassondra Hess Southeast Missouri State University [email protected]
Greg A. Hogan University of Missouri‐St. Louis [email protected]
Benjamin Hooe Southeast Missouri State University [email protected]
Paul Jelliss Saint Louis University [email protected]
Alice Raphael Karikachery University of Missouri‐Columbia [email protected]
Julia Khusnutdinova Washington University in St. Louis [email protected]
Chuck Kirkpatrick Saint Louis University [email protected]
Aaron Klucker Southeast Missouri State University [email protected]
Yang Li University of Missouri‐Columbia [email protected]
Jia Luo Washington University in St. Louis [email protected]
Bryn Lutes Washington University in St. Louis [email protected]
Hongjian Mai Saint Louis University [email protected]
Sarah Mattler Washington University in St. Louis [email protected]
Dawn Mills Southeast Missouri State University [email protected]
Liviu Mirica Washington University in St. Louis [email protected]
Shawn Morningstar Southeast Missouri State University [email protected]
Jeffrey Morton Missouri State University [email protected]

34
Fengrui Qu Washington University in St. Louis [email protected]
Nigam P. Rath University of Missouri‐Columbia [email protected]
Robert Robinson Jr. University of Missouri‐Columbia [email protected]
Rebecca Sabo Southeast Missouri State University [email protected]
Erika Sesti Washington University in St. Louis [email protected]
Anuj Sharma Washington University in St. Louis [email protected]
Paul Sharp University of Missouri‐Columbia [email protected]
Susan Snyder Southeast Missouri State University [email protected]
Meghan Stouffer Washington University in St. Louis [email protected]
Meghan Stouffer Washington University in St. Louis [email protected]
Andy Surface Washington University in St. Louis [email protected]
Fengzhi Tang Washington University in St. Louis [email protected]
Brandon Thomas Saint Louis University [email protected]
Stephanie Tucker Washington University in St. Louis [email protected]
Alicia Webb University of Missouri‐Columbia [email protected]
Nandita Weliange University of Missouri‐Columbia [email protected]
Katherine Wentz Washington University in St. Louis [email protected]
Dustin Wheeler Washington University in St. Louis [email protected]
Cody Wicker Southeast Missouri State University