department of clinical chemistry university of helsinki...
TRANSCRIPT

Department of Clinical ChemistryUniversity of Helsinki
MOLECULAR GENETICS
OF
AUTOSOMAL RECESSIVE CONGENITAL ICHTHYOSIS
Elina Virolainen, M.D
Academic Dissertation
To be publicly discussed, with the permission of the Faculty of Medicine, University of Helsinki,
in Auditorium 2 of the Meilahti Hospital, Haartmaninkatu 4, Helsinki,
on September 12th, 2000, at 12 noon
Helsinki 2000

2
Supervisor Professor Aarno Palotie, M.D.Departments of Clinical Chemistry, University of Helsinki andPathology and Laboratory Medicine, University of California, Los Angeles
Reviewers Professor Angela M. Christiano, Ph.D.Department of Dermatology and Genetics & Development, Columbia University,New York
Professor Leena Pulkkinen, Ph.D.Departments of Dermatology and Cutaneous Biology, Thomas Jefferson University,Philadelphia
Opponent Professor Leena Ala-Kokko, M.D., PhDDepartment of Medical Biochemistry, University of Oulu and Center for Gene Therapy,Tulane University Medical Center, New Orleans
ISBN 952 - 91 - 2463 - 5 (nid.)ISBN 952 - 91 - 2463 - 3 (PDF version)YliopistopainoHelsinki 2000

3
It seems as though I had not drunkfrom the cup of wisdom,but had fallen into it
Søren Kierkegaard
To Panu

4
CONTENTS
LIST OF ORIGINAL PUBLICATIONS .............................................................................................. 7
ABBREVIATIONS ................................................................................................................................. 8
1 INTRODUCTION .......................................................................................................................... 11
2 REVIEW OF THE LITERATURE .............................................................................................. 12
2.1 EPIDERMAL DIFFERENTIATION..................................................................................................... 122.1.1 The program of terminal differentiation.............................................................................. 122.1.2 Regulation of epidermal differentiation .............................................................................. 132.1.3 Cornified cell envelope ....................................................................................................... 14
2.2 TRANSGLUTAMINASES ................................................................................................................ 152.2.1 Function of transglutaminases in keratinocytes .................................................................. 162.2.2 Transglutaminase 1.............................................................................................................. 18
2.2.2.1 Gene ................................................................................................................................. 182.2.2.2 Structure........................................................................................................................... 182.2.2.3 Expression and tissue distribution ................................................................................... 192.2.2.4 Protein function ............................................................................................................... 202.2.2.5 Substrates......................................................................................................................... 212.2.2.6 Animal models of the transglutaminase 1 and the cornified cell envelope..................... 22
2.3 ICHTHYOSES................................................................................................................................ 232.3.1 Classification of ichthyoses................................................................................................. 232.3.2 Major primary ichthyoses.................................................................................................... 23
2.3.2.1 X-linked ichthyosis ........................................................................................................... 232.3.2.2 Ichthyosis vulgaris ........................................................................................................... 232.3.2.3 Harlequin ichthyosis ........................................................................................................ 242.3.2.4 Bullous ichthyosiform erythroderma (epidermolytic hyperkeratosis) ............................. 242.3.2.5 Ichthyosis bullosa of Siemens .......................................................................................... 24
2.3.3 Major ichthyosiform syndromes.......................................................................................... 262.3.3.1 Netherton’s syndrome ..................................................................................................... 262.3.3.2 Sjögren-Larsson syndrome .............................................................................................. 262.3.3.3 Refsum disease ................................................................................................................. 262.3.3.4 Ichthyosis Brittle hair Impaired intelligence Decreased fertility and Short staturesyndrome (IBIDS).......................................................................................................................... 26
2.3.4 Acquired ichthyoses ............................................................................................................ 272.4 AUTOSOMAL RECESSIVE CONGENITAL ICHTHYOSIS (ARCI)....................................................... 27
2.4.1 Classification and clinical features...................................................................................... 272.4.1.1 Lamellar Ichthyosis (LI) and Congenital Ichthyosiform Erythroderma (CIE)................ 272.4.1.2 Ultrastructural classification........................................................................................... 28
2.4.2 Molecular genetics of ARCI............................................................................................... 282.4.3 Therapeutic strategies of ARCI........................................................................................... 29

5
2.5 SEARCH FOR DISEASE GENES FOR MONOGENIC DISORDERS.......................................................... 302.5.1 Methods for identifying genes for human monogenic disorders ......................................... 302.5.2 Genetic mapping.................................................................................................................. 30
2.5.2.1 Genetic maps and markers............................................................................................... 302.5.2.2 Statistical analysis in monogenic disorders –linkage analysis........................................ 322.5.2.3 The genetic isolate of Finland and an internal isolate of Larsmo archipelago.............. 332.5.2.4 Homozygosity mapping.................................................................................................... 34
3 AIMS OF THE PRESENT STUDY.............................................................................................. 35
4 MATERIALS AND METHODS................................................................................................... 36
4.1 CHARACTERIZATION OF TGM 1 MUTATIONS .............................................................................. 364.1.1 ARCI patients ...................................................................................................................... 364.1.2 Isolation of DNA and DNA-amplification .......................................................................... 364.1.3 Mutation screening and characterization............................................................................. 36
4.1.3.1 Sequence analysis ............................................................................................................ 364.1.3.2 Solid-phase minisequencing............................................................................................. 374.1.3.3 Haplotype analysis........................................................................................................... 374.1.3.4 Modeling .......................................................................................................................... 374.1.3.5 Creating the expression construct, mutagenesis, and transfection.................................. 374.1.3.6 Analysis of transfection efficiency and steady state mRNA levels ................................... 374.1.3.7 Western blot ..................................................................................................................... 384.1.3.8 Metabolic labeling and immunoprecipitation.................................................................. 384.1.3.9 Transglutaminase activity measurement by ammonia release assay............................... 38
4.2 SEARCH FOR A NEW GENE UNDERLYING ARCI............................................................................ 384.2.1 Genotyping .......................................................................................................................... 38
4.2.1.1 Family .............................................................................................................................. 384.2.1.2 Candidate genes............................................................................................................... 384.2.1.3 Genome scan.................................................................................................................... 39
4.2.2 Statistical analysis ............................................................................................................... 394.2.2.1 Linkage analysis............................................................................................................... 394.2.2.2 Haplotypes ....................................................................................................................... 39
5 RESULTS AND DISCUSSION..................................................................................................... 40
5.1 IDENTIFICATION OF MUTATIONS IN TGM 1 GENE ........................................................................ 405.2 CHARACTERIZATION OF TGM 1 MUTATIONS .............................................................................. 41
5.2.1 Mutation-prone region in TGM 1........................................................................................ 415.2.2 Mutational modeling of Arg-142-Cys and Arg-141-His..................................................... 425.2.3 In vitro studies of Arg-142-Cys........................................................................................... 425.2.4 Characterization of other mutants – Gly-217-Ser, Val-378-Leu, Arg-395-Leu, and 3339A-to-G 43
5.3 GENOTYPE-PHENOTYPE CORRELATION OF ARCI......................................................................... 445.3.1 Clinical phenotypes of ARCI patients with TGM 1 mutations .......................................... 445.3.2 Morphological findings in patients with TGM 1 mutations................................................ 455.3.3 TGM 1 mutations and phenotype....................................................................................... 455.3.4 Locus heterogeneity and the phenotype .............................................................................. 45

6
5.3.5 Challenges in phenotype evaluation.................................................................................... 465.3.6 Clinical and morphological findings in a subgroup of ARCI.............................................. 47
5.4 ASSIGNMENT OF THE NEW ARCI LOCUS ON CHROMOSOME 19P13.1.-13.2.................................. 475.4.1 Exclusion of candidate loci ................................................................................................. 485.4.2 Genome scan ....................................................................................................................... 485.4.3 The new ARCI locus on chromosome 19 ........................................................................... 50
6 SUMMARY AND CONCLUSIONS............................................................................................. 51
ACKNOWLEDGEMENTS.................................................................................................................. 52
REFERENCES...................................................................................................................................... 55
ORIGINAL ARTICLES I-IV .............................................................................................................. 65

7
LIST OF ORIGINAL PUBLICATIONS
This thesis is based on four publications which are referred to in the text by their Roman numbers(Studies I-IV). In addition, some unpublished data are present.
I Elina Laiho (Virolainen), Jaakko Ignatius, Hanna Mikkola, Vivien C.Yee, David C Teller,Kirsti-Maria Niemi, Ulpu Saarialho-Kere, Juha Kere, Aarno Palotie: Transglutaminase 1mutations in congenital ichthyosis: Private and recurrent mutations in an isolated population.American Journal of Human Genetics 1997, 61:529-538
II Elina Laiho (Virolainen), Kirsti-Maria Niemi, Jaakko Ignatius, Juha Kere, Aarno Palotie andUlpu Saarialho-Kere: Clinical and morphological correlations for transglutaminase 1 genemutations in autosomal recessive congenital ichthyosis. European Journal of Human Genetics1999, 7:625-632
III Elina Virolainen, Maija Wessman, Iiris Hovatta, Kirsti-Maria Niemi, Jaakko Ignatius, JuhaKere, Leena Peltonen and Aarno Palotie: Assignment of a novel locus for autosomal recessivecongenital ichthyosis to chromosome 19p13.1-p13.2. American Journal of Human Genetics2000, 66:1132-1137
IV Elina Virolainen, Kirsti-Maria Niemi, Agneta Gånemo, Juha Kere, Anders Vahlquist and UlpuSaarialho-Kere. Abnormal keratinosomes and keratohyaline as markers for a severe subgroup ofcongenital ichthyosis. Submitted

8
ABBREVIATIONS
AIDS acquired immunity deficiency syndromeAP 1/2 activator protein 1/2ARCI autosomal recessive congenital ichthyosisATP2A2 and 2C1 Ca2+ -transporting ATPase 2A2 and 2C1ATPase adenosine thiphosphataseBIE bullous ichthyosiform erythrodermabp base pair1. bc before ChristBPAG2 bullous pemphigoid antigen 2Ca2+ calciumCACNA1A voltage-dependent Ca2+ channelcDNA complementary DNACE cornified cell envelopeCHILD congenital hemidysplasia with ichthyosiform erythroderma or naevus and
unilateral limb defectscM centimorganCIE congenital ichthyosiform erythrodermaCoA coenzyme AdCTP deoxy cytidine triphosphatedGTP deoxy guanosine triphosphateDR 5 direct repeat 5DNA deoxyribonucleic aciddNTP deoxy nucleoside triphosphateEAAT4 exitatory amino acid transporter 4EB epidermolysis bullosaEDTA ethylene diamine tetraactetic acidED/SF srd ectodermal dysplasia/skin fragility syndromeEKV erythrokeratodermia variabilisEM electron microscopyERCC2 and 3 excision repair complementing defective in Chinese hamsterEST expressed sequence tagFXIII coagulation factor XIIIGJB2 and 3 gap junction protein beta 2 and 3HGP human genome projectHL hearing lossHMG CoA hydroxy-methylglutaryl coenzyme AIBIDS ichthyosis brittle hair impaired intelligence decreased fertility and short stature
syndromeIBS ichthyosis bullosa of SiemensIC ichthyosis congenitaIg immunoglobulinIOSCA infantile onset spinocerebellar ataxiakDa kilodalton

9
K1/5/10/14 keratin 1/5/10/14 (protein)KID keratitis ichthyosis and deafness syndromeKRT 1/2e/10 keratin 1/2e/10 (gene)LEKTI lympho-epithelial kazal-type related inhibitorLI lamellar ichthyosisLLNL Lawrence Livermore national laboratorymRNA messenger RNAmt DNA mitochondrial DNANaOH sodium hydroxideNEPPK non-epidermolytic palmoplantar keratodermaNER nucleotide excision repairNHK normal human keratinocytesNIH National Institutes of HealthOMIM online mendelian inheritance in manPAGE polyacrylamide gel electrophoresisPC 1 and 2 pachonychia congenitaPCR polymerase chain reactionPPK palmoplantar keratoderma;PPK+HL palmoplantar keratoderma + hearing loss;PSEK progressive symmetric erythrokeratodermaRAR retinoid acid receptorRARE retinoid acid response elementRFLP restriction length polymorphismRNA ribonucleic acidRT reverse transcriptaseRXR retinoid X receptorSDS sodium dodecyl sulphateSERCA2 sarcoendoplasmic reticulum ATPase 2SPPK striate palmoplantar keratodermaSLC1A6 solute carrier family 1 member 6SNP single nucleotide polymorphismSP 1 specificity protein 1SPINK5 serine protease inhibitor, Kazal-type 5SPR small proline-rich proteinsSSCP single-stranded conformation polymorphismSTR short tandem repeatsSTS sequence-tagged site/steroid sulphataseSV40poly expression vector with simian virus 40 early promoterTFIIH transcription factor II HTGase 1/2/3/4 Transglutaminase protein 1/2/3/4TGM 1/2/3/4 transglutaminase 1/2/3/4 geneUS United StatesVNTR variable number of tandem repeatµCi microcurieÅ Ångström

10

11
1 INTRODUCTION
The largest organ in the human body is the skin, which produces a vital barrier between theenvironment and human tissues and body fluids. The most important contribution to the barrierfunction comes from the uppermost layer of the epidermis, the cornified cell layer. This layer formsenvelope networks, a lipid envelope and a protein envelope, which are impermeable structures keepingharmful organisms outside and important agents inside the body. The importance of this barrierfunction is demonstrated by an inherited defect of barrier function observed in certain scaling diseasesof the skin, known as the ichthyoses. For newborns, these are life-threatening conditions involving theloss of body fluids and risk of sepsis.
Ichthyoses form a heterogeneous group of diseases or syndromes, connected by defective keratinizationand scaling of skin. Ichthyotic diseases have a long history; the earliest observation is described in 250BC in an Indian text as “Ekakushtha, skin disease like scales of fish” (Menon and Hoberman 1969). Itwas not until the 1900s when the classification of ichthyoses became clearer after separation of X-linked ichthyosis, ichthyosis vulgaris, lamellar ichthyosis, and syndromic variants in 1950-1970(Griffiths et al. 1998). The first ichthyosis described biochemically was X-linked ichthyosis, when thesteroid sulphatase deficiency was discovered in patients with X-linked ichthyosis (Shapiro et al. 1978).
The development of modern technologies in molecular genetics has revolutionized the study ofinherited diseases such as ichthyoses. Breakthroughs emerging from clinical studies in study of thepathogenesis of ichthyoses during the last decade have revealed involvement of at least eight differentknown genes and three different chromosomal loci in the pathogenesis of different forms of ichthyosesand ichthyosiform syndromes. Specifically, these studies have illuminated the importance of keratins,lipid metabolism, and the cornified cell envelope for normal function of the skin. The geneticheterogeneity underlying the ichthyoses has been surprisingly large: for example, in autosomalrecessive ichthyosis alone at least five (and probably more) different genes underlie the disorder.Despite considerable progress within the field, the pathogenesis of about 10 ichthyotic diseases orsyndromes remains unknown.

12
2 REVIEW OF THE LITERATURE
2.1 EPIDERMAL DIFFERENTIATION
2.1.1 The program of terminal differentiationThe epidermis is a continuously renewing tissue, comprised of four histologically distinct cellularlayers (the basal layer, the spinous layer, the granular layer, and the cornified layer), each with a distinctmaturation state of the keratinocyte (Figure 1). Keratinocytes arise from stem cells in the basal layer ofthe epidermis and in the bulge region of the hair bulb (reviewed by Hall and Watt 1989), and movethrough a series of differentiation events until they are finally sloughed into the environment(desquamation) (reviewed by Eckert et al. 1997). In the normal epidermis, a balance exist between theprocess of proliferation and desquamation that results in its complete renewal approximately every 28days. (Roop 1995).
The basal epidermal layer consists of a single layer of relatively undifferentiated cells that are anchoredto the basal lamina via hemidesmosomal junctions (Borradori and Sonnenberg 1996). Two types ofproliferative keratinocytes have been found in the basal layer: 1) The stem cell, which has a high self-renewal capacity and low probability of terminal differentiation and expresses a high amount of β1integrins, 2) The transit-amplifying cell, the daughter of a stem cell that is destined to differentiatewithin about three to five rounds of division (Zhu et al. 1999). A typical feature of basal cells is toexpress keratins 5 (K5) and 14 (K14) (Steinert 1993). Impelled by some as-yet-unidentified trigger(s) ofterminal differentiation basal cells will begin their journey to the skin surface. As basal cellsdifferentiate they downregulate the expression of K5/K14 and induce expression of keratins 1 and 10,and later also keratin 2. In palmar and plantar skin additional keratins, keratins 6a, 6b, 9, 16, and 17 areexpressed (Fuchs 1993).
In the later stages of terminal differentiation the expression of a number of proteins is activated. One ofthem is profilaggrin, a histidine-rich basic protein, which is proteolytically processed into filaggrinmonomers. Filaggrin functions as an intermediate filament-associated protein that aggregates keratinfilaments in cornified cells (Ishida-Yamamoto et al. 1999). Lamellar granules, produced earlier, fusewith the plasma membrane and release lipids into the intercellular spaces of granular and stratumcorneum cells. An important step in terminal differentiation is the formation of the cornified cellenvelope (CE), a structure which consists of a lipid and protein envelope and forms an impermeablebarrier in the cell. The earliest CE protein to be expressed is involucrin in the upper spinous layers,followed by expression of later differentiation proteins such as loricrin (Fuchs 1993; Rossi et al. 1998).
The end-product of terminal differentiation is the corneocyte, a flattened cell whose organelles havebroken down and which is metabolically inactive. A corneocyte consists simply of a keratin filamentmatrix embedded in a filaggrin matrix and encased by a cornified cell envelope. The extracellularmatrix is replaced by lipid lamellae, and together the CE and lipids form an impermeable barrier againstthe environment. Finally, the corneocytes peel off from the skin as dead debris (Eckert et al. 1997). Thepathway of epidermal differentiation is presented in Figure 1.

13
Figure 1. Morphological progression ofkeratinocytes during differentiation withepidermal layers on the left, as well ascell organelles and certain moleculesinvolved in non-syndromic geno-dermatoses. Corresponding geno-dermatoses are shown on the right.Abbreviations: ARCI, autosomalrecessive congenital ichthyosis; ATP2A2and ATP2C1, Ca2+ transporting ATPase2A2 and 2C1; BIE, bullousichthyosiform erythroderma; BPAG2,bullous pemphigoid antigen 2; EB,epidermolysis bullosa; ED/SF srd,ectodermal dysplasia/skin fragilitysyndrome; EKV, erythrokeratodermiavariabilis; GJB2 and 3, gap junctionprotein beta 2 and 3; HL, hearing loss;IBS, ichthyosis bullosa of Siemens; K,keratin; NEPPK, non-epidermolyticpalmoplantar keratoderma; PC 1 and 2,pachonychia congenita 1 and 2; PPK,palmoplantar keratoderma; PPK+HL,palmoplantar keratoderma + hearingloss; PSEK, progressive symmetricerythrokeratoderma; SPPK, striatepalmoplantar keratoderma; SPRs, smallproline rich proteins (Adapted andreproduced with permission from Roop,D. (1995), Science 267:474-475).
2.1.2 Regulation of epidermal differentiationFactors that trigger epidermal cell commitment and differentiation are still incompletely understood,although specific factors have been identified that regulate keratinocyte proliferation anddifferentiation.
Calcium belongs to a group of important regulatory factors of the skin. A calcium gradient is known tobe present in the epidermis, such that the calcium content is fourfold higher in the superficial than inthe basal epidermis (Eckert et al. 1997). Calcium-dependent processes include a number of cellularevents such as the activation of enzymes, for example transglutaminase 1 (TGase 1), the secretion oflamellar bodies, desmosome formation, and cell adhesion (Vicanová et al. 1998). Recently, twoinherited skin diseases, Darier-White disease (OMIM 124200) and Hailey-Hailey disease (Benignchronic pemphigus) (OMIM 169600, were shown to be associated with defects in calcium pumpsexpressed in the skin (Hu et al. 2000; Sakuntabhai et al. 1999). Sarcoendoplasmic reticulum Ca2+
ATPase ATP2A2 (SERCA2) was shown to be the cause of the Darier-White disease, whereasmutations in ATP2C1, which in yeast pumps Ca2+ from cytoplasm to the Golgi, were found in Hailey-
Hailey disease. These two diseases further provide evidence for the importance of the Ca2+ signalingpathway in the skin.

14
Retinoids (vitamin A and related agents) are potent modulators of keratinocyte differentiation (reviewed by Fisher and Voorhees 1996). In keratinocytes, retinoids bind to retinoid receptors (RARsand RXRs) which activate gene expression by interaction with DNA response elements located nearresponsive genes or by interacting with other transcription factors. The effects of retinoic acid in theepidermis in vivo and in vitro differ. In cultured keratinocytes, retinoids inhibit terminal differentiation(Jetten 1990), which is manifested by decreased expression of differentiation-associated keratins 1 and10 (Fuchs and Green 1981), filaggrin (Asselineau et al. 1990) and cell-envelope proteins (Griffiths et al.1992; Hohl et al. 1991; Magnaldo et al. 1992; Marvin et al. 1992; Yuspa and Harris 1974). In vivo,retinoids stimulate keratinocyte proliferation and growth, and no suppression of terminal differentiationis observed (Fisher et al. 1991). Differences of in vivo and in vitro responses to retinoids have beensuggested to depend on the absence of other signals, including the influence of the important dermalsignal, which is not present in culture (Aaronson et al. 1991; Tavakkol et al. 1992). The complexity ofretinoid metabolism in the skin is emphasized by the fact that pathogenic thickening in many skindiseases is ameliorated with retinoids.
Many other effectors such as transforming growth factor B, keratinocyte growth factor, interferon γ, andvitamin D are also known to affect epidermal differentiation (Eckert et al. 1997). However, each markerhas its own peculiar regulatory features and each marker form a complex network of signals.Elucidation of these networks and their role in epidermal differentiation remains to be determined(Mariniello et al. 1995).
2.1.3 Cornified cell envelopeThe cornified cell envelope (CE) is a 15 to 20-nm-thick structure formed on the inner surface of the cellmembrane by aggregation of a series of proteins that are cross-linked together both by disulfide bondsand by the Nε-(γ-glutamyl)lysine isopeptide crosslinks. Transglutaminases (Tgases) catalyze theformation of cross-links in CE (Figure 2) (Steinert 1995). The CE is a highly insoluble protein complexconstituting about 10% of the mass of a terminally differentiated corneocyte. CE consists of two parts:a protein envelope and a lipid envelope. The lipid envelope is located on the exterior of the proteinenvelope and consists of a monomolecular layer of ω-hydroxyceramides attached to the proteinenvelope by ester bonds. ω-hydroxyceramides provide a Teflon-like coating to the cell and alsointerdigitate with the intercellular lipid lamellae (Nemes and Steinert 1999).
The CE is a common feature of all stratified squamous epithelia of various species, although betweenepithelia its precise composition and structure vary widely. It is unclear when the assembly of the CEbegins in the epidermis, but evidence suggests that the assembly is a multi-stage process. Recentimmunogold-labeling and protein-sequencing data suggest that the most critical components of theearliest stages of CE formation are involucrin and perhaps also putative membrane-anchoring proteinssuch as envoplakin and periplakin (Steinert and Marekov 1999). When the local Ca2+ concentrationrises during the epidermal differentiation, TGase 1 starts cross-linking involucrin polymers andpossibly envoplakin and periplakin at the interdesmosomal region to form a scaffold for the CE (Nemeset al. 1999). Involucrin has also been shown to attach to ω-hydroxyceramides via ester-bond formationcatalyzed by TGase 1 (Nemes et al. 1999). This process is continued by the attachment of other CEproteins, such as cystatin A, to the involucrin network. This layer serves as a substrate for deposition ofloricrin together with other proteins. Loricrin is the major protein of nearly all CEs characterized to

15
date, accounting for approximately 60 to 80% of the protein mass of the CE. (Jarnik et al. 1998; Stevenand Steinert 1994). Small proline-rich proteins are suggested to function as stabilizers of CE-formingcross-links between loricrin and other CE substrates in reactions catalyzed by both TGase 1 and 3(Candi et al. 1999).
Figure 2. Model of the structure of the cornified cell envelope. The cytoplasmic surface of the protein envelope consistsmostly of loricrin admixed with many other proteins; the surface facing the lipid envelope consists of other structuralproteins such as envoplakin, involucrin, and desmoplakin. The keratin cytoskeleton is suggested to associate in two wayswith CE (Adapted and reproduced with permission from Steinert, P. (1998), J Biol Chem 273:11758-11769).
2.2 TRANSGLUTAMINASES
Transglutaminases (Tgases for protein, TGM for gene) represent a family of enzymes capable ofstabilizing protein assemblies via γ-glutamyl-ε-lysine cross-links (reviewed by Aeschlimann andPaulsson 1994; Greenberg et al. 1991). Enzymes of this family catalyze Ca2+ -dependent transferreactions between the γ-carboxamide group of a peptide-bound glutamine residue and various primaryamines, most commonly the ε-amino group of lysine residues (Figure 3). It appears that nature has notprovided an enzyme for the cleavage of the TGase cross-link. Therefore the action of these enzymesresults in the formation of irreversibly cross-linked, often insoluble molecular structures. Sevendifferent TGM gene products with amino acid homology of 20 to 40% have been characterized in thehuman genome thus far by determination of their primary structures (Table 1). Furthermore, secondarystructure predictions show even greater degrees of structural conservation (60-70%).

16
Figure 3. Simplified model of transglutaminase catalyzed formation of the isopeptide bond.A) TGase 1 (TG, open circle) is inactive in the absence of calcium and is anchored to theplasma membrane. Glutamine substrate is shown in the cytoplasm as solid rod. B) In thepresence of calcium, TGase 1 is activated (shaded circle) and forms an enzyme-substrateintermediate with the glutamine substrate with a concomitant release of ammonia. C) Theintermediate then reacts with the lysyl substrate (primary amin donator) to form a cross-linkD) The completed cross-link is shown (Adapted and reproduced with permission fromEckert, R. (1993), J Invest Dermatol 273:11758-11769).
TGases appear early in evolution, and enzymes with similar function to vertebrate TGases have beendetected in invertebrates, plants, and bacteria, actually in all organisms studied (Polakowska et al.1999). Considering the rates of divergence of the TGM genes between different species, it is likely thatvertebrate TGM were distinct from each other before reptiles separated from fish about 400 millionyears ago (Aeschlimann and Paulsson 1994). Two evolutionarily different lineages in human beingshave been suggested on the basis of homology: 1) TGase 2, 3 and band 4.2, and 2) TGase 1 and FXIII.In plants TGases have been found from pollen and chloroplast, suggesting the role in fertilization andlight capture (Del Duca et al. 1994) (Del Duca et al. 1997). TGases could also have an interestingindustrial applications in pharmaceutical and food industry as a biological glue and for cross-linkingfood proteins (Jurgensen et al. 1997; Matheis and Whitaker 1987).
2.2.1 Function of transglutaminases in keratinocytesDuring different stages of epidermal differentiation four TGases are expressed. TGase 2 is expressedprimarily in the basal cell layer early in epidermal differentiation. The function of TGase 2 inkeratinocytes is unclear, but it has been suggested to play a role in stabilization of the dermo-epidermaljunction. (Kim et al. 1995; Raghunath et al. 1996). TGase 1 and 3 are associated with terminaldifferentiation events of keratinocytes and cross-linking the structural proteins forming the cornifiedcell envelope, but they act in different ways. The major part of TGase activity in the suprabasal cellscomes from TGase 1, whereas the expression of TGase 3 in those cells is relatively low. Although thedetails of CE formation by TGases are still unclear, it has been speculated that TGase 1 forms the majorscaffold of the CE and that TGase 3 strengthens the structure (Kim et al. 1995). TGase X is a recentlydiscoverd, novel transglutaminase, which is expressed in terminally differentiating keratinocytes andwhose function in skin is still unknown (Aeschlimann et al. 1998). Based on their spatial and temporalexpression and functional significance, the family of TGases are strong candidates for disorders ofepidermal differentiation and cornification.

17
Table 1 Human transglutaminases
Name Synonym Symbol Tissueexpression
Physiological role Diseases Amino acidhomol. toTGM 1
Genesize
Chrom.locus
Protein size
Transglut-aminase 1
Keratinocytetransglutaminase
TGase 1,TgK
epidermis,epithelia,brain,
Formation of CE ARCIAD?
100% 14 kb 14q11.2-13 106 kDa67/33/10 kDa
Transglut-aminase 2
Tissuetransglutaminase
TGase 2,TgC
ubiquitous Apoptosis, GTP-bindingand signaling, stabilizingextracellular proteinassemblies
CD1,AD2?,PD3?,TED4?
36% unknown 20q11.2 77 kDa
Transglut-aminase 3
Epidermaltransglutaminase
TGase 3,TgE
epidermis, hairfollicle
Formation of CE, hairformation
noassociation
37% 43 kb 20q11.2 77 kDa
Transglut-aminase 4
Prostatetransglutaminase
TGase 4,TgP
prostate unknown noassociation
35% 35 kb 3p21.33-p22
77 kDa
Transglut-aminase 5
4.2 Band protein TGase 5 erythrocytes Cytoskeleton component noassociation
27% 20 kb 15q15
Transglut-aminase 6
Coagulation factorXIII a subunit
FXIII platelets,macrophages,monocytes,megakaryocyteuterus, placenta(dimeric),plasma(tetrameric)
Cross-linking of fibrinnetwork in bloodcoagulation
BleedingdisorderFXIIIdeficiency
43% 160 kb 6p24-25 83 kDa
Transglut-aminase 7
transglutaminase X TGase 7 keratinocytes unknown noassociation
35% unknown unknown 81 kDa
1 CD (Celiac disease)2 AD (Alzhermer disease)3 PD (Parkinson disease)4 TED (Trinucleotide expansion diseases)

18
2.2.2 Transglutaminase 1
2.2.2.1 Gene
The gene transglutaminase 1 (TGM 1), coding for TGase 1, has been localized to chromosome14q11.2-13. (Kim et al. 1992). It is the smallest gene in the TGM family, spanning about 14 kb ofgenomic DNA. The TGM 1 gene, which exists as a single copy in the human genome, consists of 15exons interrupted by 14 introns. Sequences of both exon and intron regions are known and twosequence variants for TGM 1 in the human population have been noted. The rare smaller variant, with afrequency of approximately 4%, contains a two-nucleotide deletion near the 5’ end, uses an alternateinitiation codon, and differs from the common larger variant only in its first 15 amino acids. Intron 14of the TGM 1 gene contains a polymorphic dinucleotide area useful for genetic linkage analysis.Rodent and human TGM 1 gene sequences are highly conserved: 84% sequence identity and 92%amino acid identity between human and rat exist. (Phillips et al. 1992).
The molecular mechanisms that control the transcription of the TGM 1 gene are poorly understood, butsome data reveal transcription regulatory elements of the TGM 1 gene. Two transcription controlelements are suggested to regulate the TGM 1 gene: a proximal promoter in the 5’region and atranscription regulatory element in intron 1. Functional mapping of deletion mutations in regulatoryelements of the TGM 1 5’upstream promoter region and intron 1 demonstrated that maximal activity ofreporter enzyme activity was achieved when both elements were present. The proximal 5’regioncontains four different types of regulatory elements: 1) three AP2-like response elements conservedalso in the rabbit TGM 1 promoter region, 2) two AP1 sites, 3) SP1 site, and 4) direct repeat 5 (DR5)type of retinoid acid response element (RARE) mediating retinoid adic-receptor (RAR) signaling. The5’region also contains two sub-elements silencing and enhancing Ca signaling. Intron 1 is suggested tocontain two major sites important to transcription of TGM 1: 1) the negative element containing areverse CCAAT box and a short RARE element and 2) a 194 bp element, which may function underunidentified conditions as an alternative promoter. In addition, intron 1 contains SP1, AP1, and AP2sites similar to those found in the 5’region. The precise regulatory role of all these elements is stillunclear (Polakowska et al. 1999).
2.2.2.2 Structure
The only TGase for which the three-dimensional structure is known is the A subunit of FXIII. Becauseof significant amino acid homology (43%), TGase 1 is suggested to share the same three-dimensionalstructure (Figure 4). This structure consists of four sequential domains; the β-sandwich, the catalyticcore domain, and the β-barrel domains 1 and 2. The secondary structure of the core domain is a mixtureof α-helix and β-sheet, whereas the remaining three domains consist predominantly of β-sheet (Yee etal. 1994). The highest degree of homology is exhibited in the catalytic core domain, which contains thethree conserved catalytic triad residues: Cys376, His432, and Asp456. Most of the structuraldifferences between FXIII and TGase 1 are in the C- and N-terminal regions. The N-terminus in TGase1 contains a membrane anchor region, whereas in FXIII this area contains the activation peptide, whichis cleaved by thrombin in the coagulation cascade leading to activation of FXIII. However, recent datasuggest that TGase 1 is also proteolytically processed at Arg 92, a region corresponding to the thrombincleavage site in FXIII (Candi et al. 1998). Accordingly, it has been suggested, although no structuraldata are available, that the membrane anchorage region of

19
Figure 4. Stereoviews showing amino acid residues conserved in human TGase 1and blood coagulation factor XIII A-subunit. The three-dimensional structure ofthe factor XIII A-subunit is shown as a coil structure, with the N- and C- terminiand four domains labeled. Thick dark lines connecting alpha carbon atoms are thethree regions which are sites of amino acid deletions in TGase 1 relative to factorXIII. Superimposed on the coil structure are the side chain moieties of amino acidresidues conserved in human TGM 1 and factor XIII sequences (299 residues,42%). The high level of sequence conservation, and the distribution of conservedresidues throughout indicate that the factor XIII A-subunit and TGase 1 possessthe same protein fold. (Courtesy from Dr. Vivien Yee, Cleveland ClinicFoundation, Ohio).
TGase 1 may fold onto the body of the TGase 1 enzyme, like activation peptide in FXIII, to partiallyobstruct access to the active site.
2.2.2.3 Expression and tissue distribution
In human epidermis, TGM 1 is first expressed in fetal periderm cells at the stage of the appearance of atwo-layered epidermis (appears at 7-10 weeks of gestation) and later in the entire epidermis, althoughthe expression is strongest in the upper epidermis (Akiyama et al. 1999; Lee et al. 1999). In maturehuman epidermis, TGM 1 expression is detected in minor quantities in the proliferative basal layer, inmodest amounts in the suprabasal cells committed to differentiation, and then in much larger amountsin the granular layer as terminal differentiation in the epidermis and cornified cell assembly proceeds(Steinert et al. 1996). There are controversial results surrounding the expression of TGM 1 in epidermalappendages, depending on the antibody used. The goat polyclonal anti-TGase 1 antibody reveals itsexpression in hair follicle, sebaceous, and sweat glands, whereas the mouse monoclonal B.C1 antibodydoes not stain sebaceous glands (Yoneda et al. 1998). TGase 1 expression is also detected in otherepithelial tissues that do not keratinize, such as oral-cavity, esophagus, trachea, lung, liver, and intestine(Bradway et al. 1992; Chang and Chung 1986; Hiiragi et al. 1999; Saunders et al. 1993). Recent dataalso suggests expression in brain and increased expression and cross-linking in Alzheimers disease(Kim et al. 1999).

20
Endogenous expression of TGM 1 can be regulated by physiological and pharmacological effectorssuch as calcium, retinoids, and glucocorticoids. Protein kinase C agonists produce a dramaticstimulation of expression in primary cultures of keratinocytes. Retinoid acid treatment suppresses itsexpression in culture; however, a hyperplastic response increasing expression appears to dominate invivo. (Mariniello et al. 1995)
2.2.2.4 Protein function
The TGase 1 enzyme is involved in the formation of a cornified cell envelope in terminallydifferentiating, stratified squamous epithelia by cross-linking a series of defined structural proteins. Asmentioned earlier, TGase 1 catalyzes two kinds of reactions in epidermis: formation of χ-glutamyl-ε-lysine cross-links (see Figure 3) and formation of ester bonds between ω-hydroxyceramides andinvolucrin (see Figure 5) (Nemes et al. 1999).
Figure 5. Model of the ester bond formation of TGase 1 and epidermal ceramides. The TGase 1 (solid circle)and involucrin (solid ovoid) are bound to cellular membrane in the presence of calcium (left). TGase 1 and
involucrin forms an acyl-enzyme intermediate which is exposed to the ω-hydroxyl group of ceramide (center).
Exposing of intermediate to ω-hydroxyl group results in high likelihood of the attack of the hydroxyl group tointermediatede. TGase 1 is released and ester bond is formed (right) (Adapted and reproduced with permissionfrom Nemes, Z. (1999), Proc Natl Acad Sci 96: 8402-8407).
TGase 1 exists in epidermal keratinocytes in several forms that are differentially partitioned betweenthe cytosol and membranes (Steinert et al. 1996). The cytosolic enzyme exists as a full-length form of106 kDa with low specific activity, and two proteolytically processed forms that have 5- to 10 foldhigher specific activities, one of 67 kDa and a second consisting of 33 kDa fragments held together bynon-covalent bonds (Kim et al. 1995). Together, these three forms constitute from between 5 and 35 %of the total TGase 1 enzyme activity in proliferating or terminally differentiating cells. The membrane-bound enzyme exists in two forms: a full-length essentially inactive zymogen; or a complex of67/33/10 kD chains that is highly active. The proteolytic cleavage at Arg 92 in terminally

21
differentiating keratinocytes leads to the formation of the 67/33/10 kDa highly active form showing a200-fold increase in specific activity (Candi et al. 1998). The membrane-bound zymogen formconstitutes the bulk of TGase 1 protein in keratinocytes. Most of the TGase 1 expressed during terminaldifferentiation remains as the full-length inactive membrane-bound form, but up to half of it may beproteolytically processed into the highly active 67/33/10 kDa form while still anchored to themembranes.
The TGase 1 enzyme activity is partitioned between the cellular membrane and cytosol compartmentsduring proliferation and differentiation by the addition of lipid acyl adducts (see Figure 6) (Steinert etal. 1996). All forms of TGase 1 are N-myristoylated at a cluster of five cystein residues located on theamino-terminal peptide, which is unique to the TGase 1 enzyme. However, in proliferating anddifferentiating cells, the TGase 1 is differentially S-myristoylated or S-palmitoylated, which appears toconfer differing degrees of attachment and provide a variable mechanism for cycling on or offmembranes.
Figure 6. Model of TGase 1 processing and cycling incultured keratinocytes. A) All membrane-bound or cytosolicforms of intact TGase 1 are constitutively labeled by an N-acyl myristate linkage on glycine residue 3, probably duringor immediately after translation. In proliferating cultures, theTGase 1 is also thio-esterified. This ”triple-barreled”labeling affords a more robust anchorage to the membranesand may serve as a more stable storage form of the zymogen.Because the half-life of this S-myristoylated label is 18h,less than for the protein itself, it may provide a novelmechanism by which TGase 1 can cycle off membranes. B)During terminal differentiation a much larger amount ofTGase 1 is expressed, all of which becomes N-myristoylated. After initiation of the differentiation signal,this newly synthesized protein is also palmitoylated. The”double-barreled” TGase 1 is less firmly attached to themembranes, since the half-life of S-palmitate is much lessthan the protein’s allowing for a much larger rate of cyclingoff membranes (Adapted and reproduced with permissionfrom Steinert, P. (1996), J Biol Chem 271:26242-26250).
2.2.2.5 Substrates
By in vitro cross-linking studies, TGase 1 has been found to cross-link a number of proteins involved inthe formation of the cornified cell envelope (Candi et al. 1995; Candi et al. 1999; Nemes et al. 1999;Simon and Green 1988). These proteins also show glutamyl-lysine isodipeptide bonds catalyzed byTGase when extracted from the CE in vivo. One of these proteins is involucrin (reviewed by Eckert etal. 1993), which has been assumed to be the major initial component of the CE, forming a scaffold ontowhich other proteins are incorporated (Nemes et al. 1999). Recent data also show that involucrin is themajor target for the attachment of ceramide lipids on the exterior surface of the CE (Marekov andSteinert 1998). Involucrin is a rod-shaped protein of 68 kD rich in glutamine/glutamic acid consistingof three domains, having an ideal shape for cross-bridging with a variety of molecules (Yaffe et al.1992). It is expressed by most, if not all, stratified squamous epithelia and is thought to be a ubiquitousmember of the CE (Eckert et al. 1993). In the epidermis, its expression is strongest in the cornified

22
layer, but suprabasal cells also show expression in the cell periphery (Rice and Green 1979). However,not all involucrin protein is incorporated into the CE, and it is possible that other biological functionsof involucrin may exist in the cytoplasm.
Loricrin is a glycine-, serine-, and cysteine-rich, highly insoluble protein expressed in the superficialgranular cells of the epidermis (Mehrel et al. 1990). Loricrin contains repeats of a unique, highlyflexible structure referred to as a ”glycine loop.” Three glycine-rich domains are interrupted withglutamine-rich domains (Hohl et al. 1991). Loricrin is the major component of the epidermal CE,constituting as much as 70% of the mass of CE (Steven and Steinert 1994). It is a substrate of TGase 1,2, and 3 in vitro (Candi et al. 1995). The predominant reaction of TGase 1 is in establishing inter-molecular cross-links to form very large loricrin oligomers, whereas most of the cross-links formed byTGase 3 involve intra-molecular cross-links. The preferential Gln/Lys residues used for cross-linkagediffer between TGase 1 and TGase 3, suggesting distinct and perhaps complementary functions of thetwo TGases (Candi et al. 1995). Recently, loricrin has been detected as a defective gene in certainsubtypes of two genodermatoses: Vohwinkel’s syndrome (OMIM 604117) and progressive symmetricerythrokeratoderma (OMIM 602036) (Ishida-Yamamoto et al. 1997; Korge et al. 1997; Maestrini et al.1996).
Small proline-rich proteins (SPRs) (cornifins, pancornulins) are rather small (10-30 kDa) and rich inproline, consisting of three domains (Kartasova and van de Putte 1988; Kartasova et al. 1988). Thecentral domain contains a variable number of repeat elements and is flanked by glutamine- and lysine-rich terminal domains homologous to the corresponding terminal domains in loricrin and involucrin.Three types of SPRs exist in humans, forming a complex family of proteins: SPRs 1 (two members),SPRs 2 (seven members) and SPRs 3 (one member) (Gibbs et al. 1993). Their composition in differentCEs varies widely. In normal human epidermis, SPR 1a is expressed mainly in skin appendages, SPR2is found in the granular layer, but SPR3 is absent (Hohl et al. 1995). Cross-linking of SPRs occurs atthe amino- and carboxyl-terminal regions, suggesting that SPRs may function as molecular cross-bridges to connect two proteins. SPRs are substrates for both TGase 1 and 3, but these enzymes cross-link SPRs in different ways. TGase 3 is suggested first to form small interchain polymers betweenloricrin and SPRs1, and further oligomerization then occurs via TGase 1. SPRs2 are mainly cross-linked by TGase 3 (Candi et al. 1999; Tarcsa et al. 1998).
2.2.2.6 Animal models of the transglutaminase 1 and the cornified cell envelope
Animal models, especially mouse models, have brought interesting insights into the field of TGase 1and the cornified cell envelope. For example, TGM 1 newborn knock-out mouse have erythematousskin with coarse wrinkles, and are smaller than their normal littermates. Their skin barrier function ismarkedly impaired, and these mice die within 4 to 5 h after birth (Matsuki et al. 1998). Overexpressionof human involucrin in transgenic mice results in scaling and abnormal hair (Crish et al. 1993).Unexpectedly, in mice, disturbances of the expression of the major structural gene of CE, loricrin, haverelatively minor effects on their clinical phenotype. Transgenic mice overexpressing the loricrin gene,and mice in whom the loricrin gene has been knocked out, have apparently normal phenotypes,although the latter exhibit transient neonatal erythroderma (Korge et al. 1997; Yoneda and Steinert1993). However, transgenic mice with 1 bp (C) insertion at 1190 in the loricrin gene ( Vohwinkelsyndrome mutation) show neonatal erythroderma, impaired barrier function, a marked parakeratosis,and a constricting band encircling the tail (Suga et al 1999).

23
2.3 ICHTHYOSES
2.3.1 Classification of ichthyosesIchthyosis describes dry, rough skin with persistent, visible scaling over the body that may resemblefish scales (ichthys means fish in the Greek). Several different forms exist, and together they comprise aheterogeneous group of diseases. The clinical diversity of ichthyoses and the rarity of some of them haslead to a confusing array of classifications over the past century. According to the Textbook ofDermatology the major types of ichthyoses are congenital ichthyoses, ichthyosiform syndromes andacquired ichthyoses. Inherited forms of ichthyoses are presented in Table 1 (Griffiths et al. 1998).
Inherited or acquired ichthyoses lead to pathological conditions ranging from fairly mild disease tolethal conditions. Although inherited ichthyoses are quite rare as causes of human diseases, the study ofrare genodermatoses had provided a powerful tool for understanding the function of skin on a detailedmolecular level, thus opening a whole new set of possibilities for developing diagnostic and therapeutictools against ichthyotic disorders. The following two sections introduce the major primary ichthyosesand ichthyosiform syndromes with their known gene defects.
2.3.2 Major primary ichthyoses
2.3.2.1 X-linked ichthyosis
The first ichthyotic disorder with an established molecular basis was X-linked ichthyosis (OMIM308100). This was first recognized as a separate disorder in 1933 (Cockayne 1933) and wasdifferentiated from ichthyosis vulgaris in 1966 (Wells and Kerr 1966). Only males are affected, whilefemale carriers are asymptomatic. Scaling usually becomes evident at birth or during the first year oflife and is more prominent on the extremities. A variety of extracutaneous features affecting thegenitalia and eyes have been noted (Lykkesfeldt et al. 1985; Lykkesfeldt et al. 1983; Traupe and Happle1983). Steroid sulphatase deficiency was noted in association with the disease as early as 1978(Marinkovic-Ilsen et al. 1978; Webster et al. 1978). The first mutations in the steroid sulphatase gene(STS) were characterized in 1987 by (Yen et al. 1987). Over 90% of patients with STS defect have acomplete or partial deletion of the STS gene. The substrate of steroid sulphatase, cholesterol 3-sulphate,is elevated in many tissues including the epidermis, where it accounts for 12 to 30% of stratumcorneum lipids in X-linked ichthyosis instead of the 3% in normal skin (Elias et al. 1984). This disturbsthe normal lipid balance within the epidermis. Cholesterol 3-sulphate is also suggested to disturbTGase 1 function and therefore the normal assembly of the CE (Nemes et al. 2000).
2.3.2.2 Ichthyosis vulgaris
Ichthyosis vulgaris (OMIM 146700) is the most common form of the inherited ichthyosis with areported incidence of 1:250 (Wells and Kerr 1966). It is an autosomal dominant condition, and in themajority of affected individuals the phenotype is relatively mild. A close association between ichthyosisvulgaris and atopic diseases has been suggested, 37 to 50% of patients with ichthyosis vulgaris showingfeatures of atopic dermatitis (Fartasch et al. 1989; Kuokkanen 1969; Wells and Kerr 1966). Althoughthe molecular mechanism of ichthyosis vulgaris is unclear, abnormalities in filaggrin metabolism hasbeen suggested (Nirunsuksiri et al. 1998; Sybert et al. 1985). Recently, a preliminary linkage study ofone ichthyosis vulgaris family with five affected mapped a suggestive disease locus to the epidermal

24
differentiation complex on chromosome 1q21. However, sequencing of the profilaggrin gene located onthis region failed to reveal any mutation (Compton et al. 1998).
2.3.2.3 Harlequin ichthyosis
The most severe form of ichthyosis is harlequin ichthyosis (OMIM 242500), which is characterized bya severe erythrodermic ichthyosis with a distinctive and grotesque appearance at birth. It was invariablyassociated with stillbirth or early neonatal death until first surviving case was reported in 1985 (Lawlor1988; Lawlor and Peiris 1985). Since then, several other survivors have been reported, and the outcomeof these patients has been a severe erythrodermic ichthyosis (Haftek et al. 1996; Prasad et al. 1994;Roberts 1989). The inheritance pattern of harlequin ichthyosis is assumed to be autosomal recessive.However, although the molecular mechanism of the disease is unknown, a spontaneously mutatedharlequin ichthyosis mouse line has been reported (Sundberg et al. 1997). The disease locus of thesemice have been mapped to mouse chromosome 19. The corresponding homologous regions in thehuman genome are scattered over chromosomes 9, 10, and 11. The responsible gene has never beencloned neither in the mouse nor in the human, but calpain I, lying on chromosome 19 in mice and onchromosome 11q13 in human beings, is an interesting candidate gene for harlequin ichthyosis. CalpainI is a calcium-activated protease and is widely expressed in the epidermis. Recently, poor expression ofcalpain I was found in harlequin ichthyosis patients.
2.3.2.4 Bullous ichthyosiform erythroderma (epidermolytic hyperkeratosis)
Bullous ichthyosiform erythroderma (BIE) (OMIM number 113800) is an autosomal dominant disorderof keratinization which, in its early stages, is associated with blistering. Characteristic histologicalfindings with lysis and tonofilament clumping in suprabasal keratinocytes suggest an underlyinggenetic defect in keratin synthesis involving keratins 1 and 10 (K1, K10) (Ishida-Yamamoto et al.1992). Subsequently, several groups have described mutations in the highly conserved regions of K1and K10 (Cheng et al. 1992; Chipev et al. 1992; McLean et al. 1994). Mutations in these regions affectintermediate filament assembly and integrity and are predicted to disrupt higher filament assembly in adominant negative fashion, leading to the collapse of the network and to filament clumping around thenucleus.
2.3.2.5 Ichthyosis bullosa of Siemens
The clinical picture of ichthyosis bullosa of Siemens (IBS) (OMIM 146800) resembles that in BIE;however, it appears to be milder (Traupe et al. 1986). IBS and BIE are often clinically distinguishablefrom each other because of the similar clinical appearance and variability. However, they can bediscriminated at the molecular level since the type II keratin 2e, which is expressed in the upperkeratinocytes, has been recognized as the defective gene in ichthyosis bullosa of Siemens (Rothnagel etal. 1994).
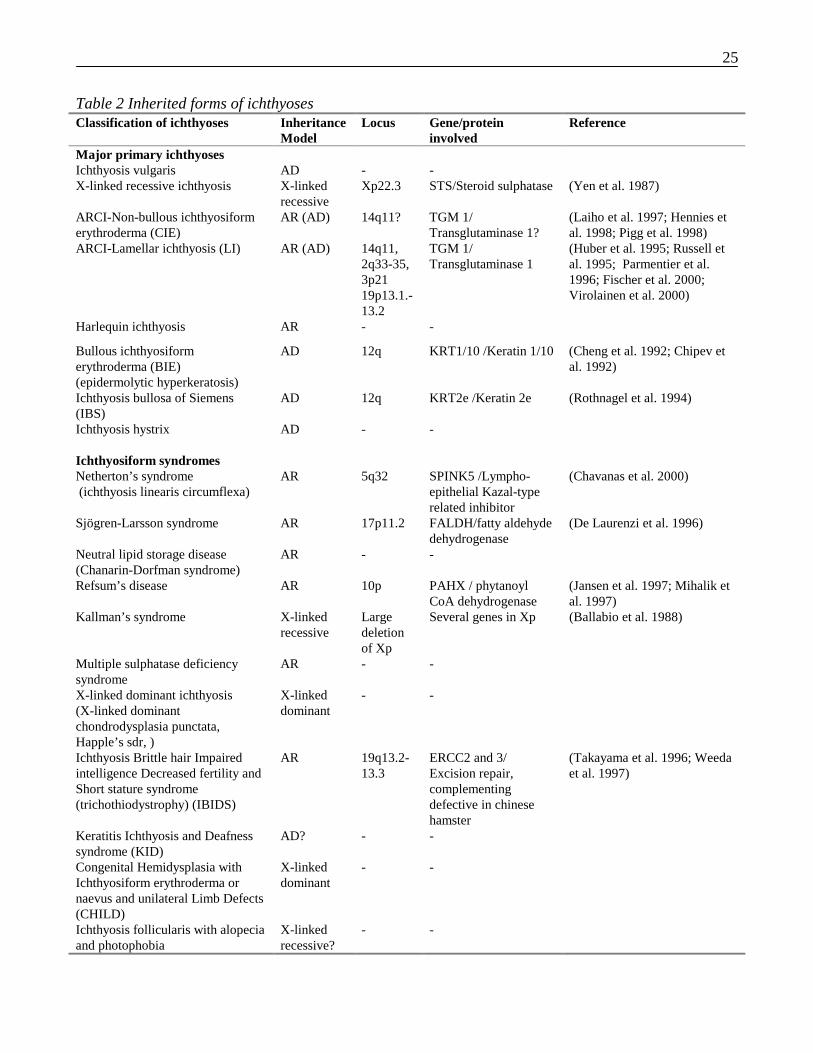
25
Table 2 Inherited forms of ichthyosesClassification of ichthyoses Inheritance
ModelLocus Gene/protein
involvedReference
Major primary ichthyosesIchthyosis vulgaris AD - -X-linked recessive ichthyosis X-linked
recessiveXp22.3 STS/Steroid sulphatase (Yen et al. 1987)
ARCI-Non-bullous ichthyosiformerythroderma (CIE)
AR (AD) 14q11? TGM 1/Transglutaminase 1?
(Laiho et al. 1997; Hennies etal. 1998; Pigg et al. 1998)
ARCI-Lamellar ichthyosis (LI) AR (AD) 14q11,2q33-35,3p2119p13.1.-13.2
TGM 1/Transglutaminase 1
(Huber et al. 1995; Russell etal. 1995; Parmentier et al.1996; Fischer et al. 2000;Virolainen et al. 2000)
Harlequin ichthyosis AR - -
Bullous ichthyosiformerythroderma (BIE)(epidermolytic hyperkeratosis)
AD 12q KRT1/10 /Keratin 1/10 (Cheng et al. 1992; Chipev etal. 1992)
Ichthyosis bullosa of Siemens(IBS)
AD 12q KRT2e /Keratin 2e (Rothnagel et al. 1994)
Ichthyosis hystrix AD - -
Ichthyosiform syndromesNetherton’s syndrome(ichthyosis linearis circumflexa)
AR 5q32 SPINK5 /Lympho-epithelial Kazal-typerelated inhibitor
(Chavanas et al. 2000)
Sjögren-Larsson syndrome AR 17p11.2 FALDH/fatty aldehydedehydrogenase
(De Laurenzi et al. 1996)
Neutral lipid storage disease(Chanarin-Dorfman syndrome)
AR - -
Refsum’s disease AR 10p PAHX / phytanoylCoA dehydrogenase
(Jansen et al. 1997; Mihalik etal. 1997)
Kallman’s syndrome X-linkedrecessive
Largedeletionof Xp
Several genes in Xp (Ballabio et al. 1988)
Multiple sulphatase deficiencysyndrome
AR - -
X-linked dominant ichthyosis(X-linked dominantchondrodysplasia punctata,Happle’s sdr, )
X-linkeddominant
- -
Ichthyosis Brittle hair Impairedintelligence Decreased fertility andShort stature syndrome(trichothiodystrophy) (IBIDS)
AR 19q13.2-13.3
ERCC2 and 3/Excision repair,complementingdefective in chinesehamster
(Takayama et al. 1996; Weedaet al. 1997)
Keratitis Ichthyosis and Deafnesssyndrome (KID)
AD? - -
Congenital Hemidysplasia withIchthyosiform erythroderma ornaevus and unilateral Limb Defects(CHILD)
X-linkeddominant
- -
Ichthyosis follicularis with alopeciaand photophobia
X-linkedrecessive?
- -

26
2.3.3 Major ichthyosiform syndromes
2.3.3.1 Netherton’s syndrome
Nethertons’s syndrome (OMIM number 256500) is the commonest of the ichthyotic syndromes, withan incidence of 1:100 000 (Netherton 1958). It is an autosomal recessive condition characterized bycongenital ichthyosis (hyperkeratosis is mild), a special hair defect (trichorrexis invaginata or “bamboohair”) and atopic manifestations (atopic dermatitis, hayfever, high serum IgE levels, andhypereosinophilia) (Griffiths et al. 1998). Recently, the disease gene SPINK 5 (serine proteaseinhibitor, Kazal type 5), encoding the serine protease inhibitor LEKTI (lympho-epithelial Kazal-typerelated inhibitor), was found from 13 families with Netherton’s syndrome (Chavanas et al. 2000). Themechanism by which LEKTI can cause the features of the disease is still unclear. However, as theLEKTI is expressed in thymus, it may cause abnormal maturation of T-lymphocytes, which may disturbnormal responsiveness to allergens and lead to atopic manifestations. LEKTI may also regulate yetunknown signaling pathways.
2.3.3.2 Sjögren-Larsson syndrome
Sjögren-Larsson syndrome (OMIM 270200) is an autosomal recessive neurocutanaous conditioncomprised of congenital ichthyosis, spastic diplegia, retinopathy, and mild to moderate mentalretardation (Jagell et al. 1981; Sjögren and Larsson 1957). A defect in essential fatty acid metabolismhas been detected, specifically in the enzyme fatty aldehyde dehydrogenase (De Laurenzi et al. 1996).Affected patients have impaired fatty alcohol oxidation and consequently accumulate long-chainalcohol in their tissues, which is assumed to be responsible for symptoms of the disease.
2.3.3.3 Refsum disease
Refsum disease (OMIM 266500) resembles in part Sjögren-Larsson disease: it is a neurocutaneousdisorder, and the causative agent is involved in fatty acid metabolism (Refsum 1946). However, thedisease is usually diagnosed only in early adult life, since clinical features like retinitis pigmentosa,cerebellar ataxia, peripheral polyneuropathy, ichthyosis, and increased cerebrospinal fluid proteinprogress slowly. This defective gene in Refsum disease is phytanoyl-CoA hydroxylase, which is aperoxisomal protein catalyzing the first step in the oxidation of phytanic acid (Jansen et al. 1997;Mihalik et al. 1997). This gene defect leads to accumulation of phytanic acid to tissues. Phytanic acid isderived from the diet and cannot be synthesized by human tissues. Effective dietary therapy is availableif the diagnosis is made in time (Djupesland et al. 1983; Gibberd et al. 1979).
2.3.3.4 Ichthyosis Brittle hair Impaired intelligence Decreased fertility and Short stature syndrome(IBIDS)
IBIDS (OMIM number 601675) is a rare and heterogeneous genodermatosis first described in 1968(Pollit et al. 1968). Major features are described in its name. The central feature of the disorder is areduced amount of sulphur-containing amino acids, cysteine and proline, in the hair. Clinicalphotosensitivity is present in approximately 50% of IBIDS patients but is not associated with anelevated frequency of cancers. Virtually all photosensitive IBIDS patients have a deficiency in thenucleotide excision repair (NER) of UV-induced DNA damage that is indistinguishable from that ofxeroderma pigmentosum (XP) complementation group D patients. Two genes encoding DNA repairgenes ERCC2 and 3 (Excision repair complementing defective in Chinese hamster) had been suggestedas disease causing gene in IBIDS. ERCC2 gene defect has been found altogether in 12 patients(Botta et

27
al. 1998; Coin et al. 1998; Takayama et al. 1997) and ERCC3 defect in two mildly affected patients(Weeda et al. 1997). ERCC2 and 3 has DNA helicase activity and they are subunits of thetranscription/repair factor TFIIH (Transcription factor II H) (Schaeffer et al. 1993). TFIIH is a basaltranscription factor with a second function in DNA repair. Thus, mutations in TFIIH components may,in addition of a repair defect, also cause transcriptional insufficiency, which may explain part of theother clinical features of IBIDS.
2.3.4 Acquired ichthyosesAcquired ichthyoses are generally associated with an underlying pathology such as a malignancy.Clinically, the ichthyosis of the patients is mild, resembling ichthyosis vulgaris. The most commonlyreported malignancy associated with acquired ichthyosis is Hodgkin’s disease (Cooper et al. 1980;Sneddon 1955). The production of transforming growth factor α, a potent enhancer of keratinocytemigration and proliferation, has been proposed as a causative factor (Lucker and Steijlen 1995).Acquired ichthyosis can also be detected with other malignancies, such as multiple myeloma, breast,lung, and cervix cancers (Bluefarb 1955; Flint et al. 1975). Acquired ichthyosis may occur with chronicmetabolic derangement such as malnutrition or malabsorbtion which may disturb lipid or possiblyvitamin absorption (Flint et al. 1975). Some endocrinological diseases, such as hypopituitarism, orgranulomatous diseases, such as sarcoidosis, are also associated with ichthyosis (Dykes and Marks1977; Feind-Koopmans et al. 1996). Several drugs which affect cholesterol or lipid metabolism in theskin, such as nicotinic acid or hydroxy-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors,can also sometimes predispose to ichthyosis (Williams et al. 1987).
2.4 AUTOSOMAL RECESSIVE CONGENITAL ICHTHYOSIS (ARCI)
2.4.1 Classification and clinical features
2.4.1.1 Lamellar Ichthyosis (LI) and Congenital Ichthyosiform Erythroderma (CIE)
In the Anglo-Saxon literature, two major types of autosomal recessive ichthyosis are distinguished:congenital ichthyosiform erythroderma (CIE) (OMIM 242100) and lamellar ichthyosis (LI) (OMIM242300) (Williams and Elias 1985). Terminology used in the description of ARCI is confusing; someauthors use the term LI for both LI and CIE patients, but most authors consider them separate entities.There is little data about the epidemiology, but estimated incidences are 1:300 000 for CIE and 1:500000 for LI (Griffiths et al. 1998; Wells and Kerr 1966). The mode of inheritance is usually autosomalrecessive, but dominant cases have been described in both disease groups (Traupe et al. 1984). Both LIand CIE patients are often born as collodion babies, encased in a plastic-like collodion membrane,which peels away within a few weeks. Collodion babies are in risk of sepsis and dehydration, butusually survive. Later in life, scaling in CIE patients is usually generalized and whitish, anderythrodermia is present. In contrast, in LI, the scales are thicker and brownish, and erythrodermia isvariable. Some patients have an intermediate phenotype between LI and CIE, and a clear-cut diagnosismay be difficult to make. In both patients groups, the severity of the phenotype varies. The rare subtypeof LI phenotype is an appearance of ichthyosis only on the trunk of the patients. In this type of LI, “ thebathsuit type,” the extremities of the affected individual have relatively normal skin, whereas the trunkis strongly affected.

28
2.4.1.2 Ultrastructural classification
An electron microscopic classification was presented by Anton-Lamprecht and other investigatorsbetween 1980 and 1994 (Anton-Lamprecht 1992; Arnold and Anton-Lamprecht 1988; Niemi et al.1991; Niemi et al. 1994; Niemi et al. 1992; Niemi et al. 1993). In this classification, ARCI patients aredivided into four different groups according to electron-microscopic findings. This classification is inuse in some European countries such as Finland, Sweden, and Germany.
IC type I: Clinically, type I patients correspond to CIE patients. They have generalized fine scaling withvariable erythroderma, but there is marked heterogeneity among affected individuals. Ultrastructurally,clear-cut criteria are lacking, although the presence of lipid droplets in cornified cells is detectable.However, lipid droplets are not a specific finding for ichthyoses (Niemi et al. 1994) (See Figure 1A inpublication II).IC type II: Of the IC groups, type II is the most clearly defined. Ultrastructurally, crystalloid structurescalled cholesterol clefts appear in the thickened corneal layer as a rather specific ultrastructural markerfor IC type II. Patients have a clinical picture consistent with classic lamellar ichthyosis with its largebrown scales (Niemi et al. 1991) (See Figure 1B in publication II).IC type III: In this group, elongated perinuclear membrane structures, abnormal keratinosomes, andvesicular complexes in the upper epidermal and cornified cells are detectable by electron microscopy.The clinical picture differs from the other types in that the onset of ichthyosis is variable. Ichthyosis aswell as erythroderma can be patchy or generalized, and in these patients flexures are typically ichthyotic(Niemi et al. 1992) (See Figure 1C in publication II).IC type IV: Only a few cases of type IV ichthyosis have been described. The clinical course innewborns may be severe, and infants are at risk of respiratory distress syndrome. Later, the clinicalpicture is milder, and follicular hyperkeratosis gives the skin a goose skin-like appearance. In electronmicroscopy, the packages of trilaminar membrane structures in upper epidermal and cornified cells area typical finding (Niemi et al. 1993) (See Figure 1 D in publication II).
2.4.2 Molecular genetics of ARCIThe first hints as to pathogenesis of ARCI were revealed in 1993 when a diminishedimmunohistochemical staining of TGase 1 was found in three LI patients (Hohl et al. 1993). A geneticstudy of inbred and outbred families demonstrated linkage to a region on chromosome 14q11, wherethe TGM 1 gene is located (Russell et al. 1994). Mutations in the TGM 1 gene resulting in reducedTGase 1 activity were identified soon after (Huber et al. 1995; Russell et al. 1995). To date over 30different mutations have been identified in the TGM 1 gene by several research groups (Study I and II)(Bichakjian et al. 1998; Candi et al. 1998; Hennies et al. 1998; Hennies et al. 1998; Huber et al. 1997;Parmentier et al. 1995; Petit et al. 1997; Pigg et al. 1998; Tok et al. 1999). Most of these are pointmutations or small deletions resulting in amino acid substitutions, stop codons, or splicing defects.However, not all ARCI patients carry the TGM 1 gene defect (Bale et al. 1996; Huber et al. 1995). Likemany other genetic diseases, ARCI is genetically heterogeneous, and in addition to the TGM 1 locus asecond locus for ARCI has been identified on chromosome 2q33-35 (Parmentier et al. 1996). However,the defective gene in that region remains still unknown (Parmentier et al. 1999). The most recentlinkage studies have revealed three new chromosomal loci for ARCI in chromosomes 3p21, 19p13.1.-13.2, and 19p12-q12, bringing the total number of ARCI loci to at least five (Fischer et al. 2000) (StudyIII). Unexpectedly, none of the other TGM genes are co-localized to the linkage intervals. Because a

29
great number of affected individuals show no linkage with any of these loci, there most probably areseveral other loci linked to ARCI.
2.4.3 Therapeutic strategies of ARCITreatment of ARCI involves the use of various topical emollients such as urea, propylene glycol, andalpha-hydroxy acids (AHA), and a combination of lactic acid and propylene glycol, as well as retinoids(oral vitamin A derivatives) (Gånemo and Vahlquist 1997; Steijlen et al. 1994). The mechanism of theeffectiveness of retinoid therapy in ARCI is unclear. However, therapeutic results are often incomplete,and side-effects are common. Recently, some interesting gene-therapeutic experiments have been madeto restore the TGase 1 activity (Choate et al. 1996) (Choate et al. 1996) (Choate and Khavari 1997).These studies used the human skin/immunodeficient mouse xenograft model. TGase 1-deficientprimary keratinocytes from LI patients were transfected with functional TGase 1. These keratinocyteswere then grafted onto immune deficient mice. Engineered LI epidermis on the immunodeficient mouseexpressed TGase 1 normally, whereas TGase 1 expression was absent from unengineered LI murineepidermis. Successful phenotypic correction in engineered LI epidermis was achieved, but transgeneexpression loss and loss of corrected phenotype appeared from week 6 after grafting (Choate et al.1996). Despite the limited results, gene replacement strategies in ARCI hold the promise of noveltherapeutic approaches.

30
2.5 SEARCH FOR DISEASE GENES FOR MONOGENIC DISORDERS
2.5.1 Methods for identifying genes for human monogenic disordersA number of approaches are possible for cloning disease genes of inherited disorders (Collins 1995).The main approaches include:1. Functional cloning. Traditionally, cloning was performed by the functional cloning approach in
which the identification of a defective gene was based on information about the basic biochemicaldefect without a reference to chromosomal map position. However, in most genetic diseases thebiochemical defect is unknown, a limitation which restricts the usefulness of this strategy
2. The candidate gene approach. In most genetic diseases, some knowledge of pathogenesis existswhich makes possible to make ”an educated guess” as to the possible defective genes. For example,in the genodermatoses some chromosomal regions include gene clusters of epidermaldifferentiation genes or keratins both of which have been shown to be defective in somegenodermatoses (Cheng et al. 1992; Maestrini et al. 1996; Mischke et al. 1996; Rosenberg et al.1988). Currently, an increasing amount of information from animal models provides ideas aboutpossible candidate genes through the use of comparative genomic strategies.
3. Positional cloning. In positional cloning, the search for the defective gene is based solely on itschromosomal localization. The power of positional cloning is that it can recognize disease geneswithout any knowledge of the disease mechanism. This can result in determination of a diseasegene which ”nobody would ever have guessed.” The positional candidate approach refers to theidentification of potential candidate genes from a restricted chromosomal region. A typical schemefor positional cloning is shown in Figure 7.
2.5.2 Genetic mappingIn the positional cloning strategy for a human disease gene, the second step after collecting suitablefamilies is to identify the disease locus by use of polymorphic markers and analysis of their segregationin pedigrees. In a simple monogenic disorder like ichthyosis, there should be only one disease locus inall affected individuals within a single pedigree. To localize disease loci, genetic maps of the humangenome are used.
2.5.2.1 Genetic maps and markers
Polymorphisms, simply defined as sequences that vary between individuals, exist in the humangenome. Different types of polymorphisms have been utilized in the creation of genetic maps. The firsthuman genetic map, containing approximately 400 loci, was based on restriction fragment lengthpolymorphisms (RFLPs) (Donis-Keller et al. 1987). The drawback of this restriction-enzyme basedmapping method is the labor intensiveness and low information content of biallelic markers. RFLPswere succeeded by minisatellite markers-variable number of tandem repeats (VNTRs)-which have ahigher information content (Jeffreys et al. 1985; Nakamura et al. 1987). The discovery ofmicrosatellites (STRs or short tandem repeats) and PCR techniques finally led to the modern techniqueof detecting microsatellites by PCR and polyacrylamide gel electrophoresis (PAGE) (Miesfeld et al.1981; Weber and May 1989). This progress started the spate of so-called second-generation maps of thehuman genome.

31
Figure 7. Principle of positional cloning approach. The first steps include a collection of suitable familiesand linkage studies of the genome (low resolution mapping). After identification of a disease locus, therefollows high resolution mapping; restriction of the critical region (e.g. by linkage disequilibrium methods). Ifthe sequence and transcripts of the region are mostly unknown, a physical mapping phase results. In thisphase, a contig, an arranged collection of sequences of the region of interest, is made. A visible cytogeneticrearrangement, if available, can greatly assist both the low and the high resolution mapping phases. Whenthe smallest possible region is achieved, all the transcripts in the area are identified, usually by utilizingESTs (partial cDNA sequences) mapped to the region. Finally, the disease-causing mutation is identified in acandidate gene or in the novel transcript by comparing sequences of affected individuals and healthycontrols. At present a growing amount of transcript data has changed the positional cloning approachtowards a positional candidate approach, which combines knowledge of map position with the increasinglydense human transcript map. Currently, when a human sequencing project is near its end, the laboriousphysical mapping phase is less often needed, whereas the use of different databases and computationalprediction programs in the interpreting of sequence data, “in silico” cloning, becomes increasingly important(Courtesy from Dr. Höglund).
Fluorescence-based, semi-automated typing of microsatellite markers is currently the method of choicefor genome-wide mapping projects, although SNP mapping is emerging. Advantages of microsatellitemarkers are their high informativeness and relatively dense distribution (once in every 30 kb)throughout the genome (Weber 1990). In genetic maps, the genetic distance is usually expressed incentimorgans (cM). One centimorgan corresponds to the average distance in which 1 recombination inevery 100 meiosis occurs. The human genetic map spans 3690 cM on average, but the male (2644 cM)and female (4481 cM) map differ considerably, indicating that recombination events are much morecommon in females (Gyapay et al. 1994). Today, several genetic microsatellite-based maps exist,comprised of thousands of markers. The most common type of microsatellites is dinucleotide repeats.Microsatellites still have disadvantages, such as manual gel loading, which makes completeautomatization difficult, and PCR amplification is costly and time-consuming. Completeautomatization would facilitate the laborious mapping projects of complex diseases, for whichhundreds of affected individuals are genotyped. Therefore, attention has recently turned to single-

32
nucleotide polymorphisms (SNPs), since they are the most suitable for automated genotyping, forexample by DNA microarrays, and a proportion of SNPs are thought to contribute to diseasesusceptibility (Brookes 1999; Collins et al. 1999). SNPs are the most frequently occurringpolymorphisms in humans, with an estimated frequency of 1/1000 bp. SNPs are distributed uniformlyon both the coding and non-coding regions of the genome. Large-scale SNP discovery efforts willprovide a dense, third-generation map of the human genome. Several organizations, such as the USNIH and the Genset company, have begun their own programs to detect tens of thousands SNPs, and ajoint academic/drug industry venture called the SNP consortium is committed to finding 300 000 SNPsand mapping half of them within a few years. Collectively, it is reasonable to expect many hundredthousand SNPs to enter the public databases in the near future (Brookes 1999).
Creating modern genetic and physical maps of the human genome has been a result of internationalcooperation within the Human Genome Project (HGP). It was originally initiated in the United States inthe late 1980s, but was soon extended to several other countries, and an International Human Genomeorganization was founded to coordinate the projects (Collins and Galas 1993). The results of the HGPare convincing. For example, a genetic map of under 2.0 cM density, and sequence-tagged site (STS)-anchored physical maps of 200-500 kb density have been created (Murray et al. 1994) (Dib et al. 1996;Hudson et al. 1995). The EST database contains 2 204 609 human ESTs (situation on 21th July 2000,dbEST release 072100, http://www.ncbi.nlm.nih.gov/dbEST/dbEST_summary. html), and many of themhave been positioned on the physical map. The main focus of the HGP is now on completing thesequencing of the human genome (Collins et al. 1998). Two alternative approaches to determine thehuman genome sequence have been proposed. The clone-by-clone approach constructs an organizedclone map which covers the human genome and which is then sequenced. In contrast, the shotgunsequencing strategy sequences unmapped clones, which are assembled afterwards ( The Sanger Centre1998) (Weber and Myers 1997). The public domain HGP follows a clone-by-clone approach and haspublished the sequences of two human chromosomes, chromosomes 22 and 21, and announced on 26th
June in a press release that the draft human sequence (sequencing finished, assembly not) is competed(Hattori et al. 2000). A private company, Celera Genomics, which utilizes the whole genome shotgunsequencing strategy, has already declared to have completed the draft sequence and the first assembly ofthe sequence. The high quality sequence (sequence and assembly are finished) is promised for 2003 inthe public HGP; the amount of high quality sequence in July 2000 was 21%: specifically 665 654 000base pairs are now sequenced and organized precisely within the human genome. The HGP also targetssequencing of other organisms. A number of genomes of microbial organisms such as Saccaromycescerevisiae have been sequenced, as well as two multicellular organisms, Caenorhabditis elegans andDrosophila melanogaster (Adams et al. 2000; The C. Elegans Sequencing Consortium 1998; Mewes etal. 1997).
2.5.2.2 Statistical analysis in monogenic disorders –linkage analysis
The most common approach to genetic mapping is linkage analysis, which has been successfullyapplied to hundreds of simple monogenic diseases. Two loci lying close together on the samechromosome are said to be “linked” if they have a tendency to co-segregate. Alleles at loci on the samechromosome should co-segregate at a rate related to the distance between them in the chromosome. Thespecific statistical figure, the recombination fraction (θ), describes this probability of a recombinationevent as occuring between the two loci. The recombination fraction ranges from θ = 0 for loci whichare tightly linked to θ = 0.5 for loci which are far apart (approaching random segregation). The

33
recombination fraction can be taken as a measure of the genetic distance between loci so that a mapdistance of 1cM corresponds approximately to a recombination fraction of 0.01 (Terwilliger and Ott1994).
In linkage analysis, the segregation of alleles at a disease locus and at a polymorphic marker locus isfollowed in the pedigree to determine whether the two loci tend to co-segregate more often than if theysegregated independently (for example, if they were not physically close together in the samechromosome). The object of the linkage analysis is to estimate the recombination fraction between thedisease and the marker loci, and, in case a recombination fraction is under 0.5, to determine whether thefinding is significant. The most commonly used statistical value to express the likelihood of linkage isthe LOD score (Morton 1955). LOD score is the logarithm to the base 10 of the likelihood ratio (thechance of observing the linkage of the trait and marker loci at a given recombination fraction to thechance given no linkage). A LOD score is considered as significant at a given recombination fraction ifit is greater than 3. A LOD score under –2 indicates significant evidence for exclusion of linkage.Computer methods have been developed to facilitate linkage calculations for several families andmarker loci (Ott 1974) (Lathrop and Lalouel 1984; Lathrop et al. 1984; Lathrop et al. 1986).
2.5.2.3 The genetic isolate of Finland and an internal isolate of Larsmo archipelago
Finland has been a representative example of the power of a genetic isolate in cloning of inheriteddisease genes. Analysis of mtDNA and Y chromosomal data in the Finns has revealed an exceptionallylow genetic diversity of Finns compared to that of other European populations (Sajantila et al. 1996);Kittles, 1998 #763]. This reflects the peculiar population history of Finland. Although Finland is likelyto have been inhabited without interruption since the glacial period, the majority of the genes of today`sFinnish population are thought to originate from recent small founder populations some 2000 years ago(Nevanlinna 1972). With time, the population has slowly grown, and although it experienced severalbottlenecks, during the last century the population has rapidly increased to its present 5.1 million.Religious and linguistic barriers, their geopolitical position, and most importantly geography haveensured that the Finns have remained in local and national isolation. This situation has resulted in thehigh prevalence of certain inherited diseases, grouped under the label "Finnish disease heritage,"including about 30 mostly recessive genetic traits which are exceptionally common in Finland, but arerare among other populations (de la Chapelle 1993; Norio et al. 1973; Peltonen et al. 1999).
Mapping of monogenic disease genes an isolated population like Finland offers advantages overmapping of heterogeneous populations. These include:1. The high incidence of some recessive disorders, which determines the sufficient number of families
for linkage studies.2. The high incidence of remotely consanguineous marriages, which enables utilization of efficient
mapping methods, such as homozygosity mapping.3. Monogenic disorders are likely to be caused by one or few founder mutations, making linkage
disequilibrium methods feasible.
Specific epidemiological data are lacking: no evidence exist that ARCI is enriched in Finland and is apart of the Finnish disease heritage. Despite this, internal isolates such as the archipelago of Larsmo(Luoto) in Osthrobotnia are highly consanguineous and provide families suitable for a random search ofnew disease loci (Figure 8). A good example of this is a collection of families and mapping of a disease

34
locus for tibial muscular dystrophy, “Larsmo disease”(Haravuori et al. 1998; Udd 1992) .
Figure 8. Location of the LarsmoArchipelago in the coast of Ostrobotnia nearthe town of Pietarsaari
Larsmo was probably not inhabited until the 13th century, and the origin of the founders is unknown.Because migration between the island and the mainland was very limited until this century,consanguineous marriages have thus been common as far back as family histories can be traced to the17th century (Udd 1992).
2.5.2.4 Homozygosity mapping
Rare recessive diseases in the offspring of consanguineous parents usually arise because the affectedindividuals have inherited two identical defective copies of the same gene from a common ancestor.Homozygosity mapping is a strategy to find the chromosomal localization of a recessive disease geneby identifying genomic regions of homozygosity in patients born from consanguineous marriages(Lander 1987). By definition, affected offspring of consanguineous marriages should also behomozygous for genetic markers near the responsible disease locus. The homozygosity mappingapproach was first applied in the localization of the genes causing ataxia with selective vitamin Edeficiency and alkaptonuria (Ben Hamida et al. 1993; Pollak et al. 1993). Isolated populations providean opportunity to apply this strategy even if only a few affected individuals are available for the primarygenome search. A good example is the mapping of the infantile-onset spinocerebellar ataxia (IOSCA)locus on chromosome 10q23.3-24.1 by use of only four affected individuals (Nikali et al. 1995).

35
3 AIMS OF THE PRESENT STUDY
Prior to this study, the TGM 1 gene was found to be the disease-causing gene in ARCI. Nothing wasknown about the molecular background of Finnish ARCI patients. Thus the main goal of the projectwas to characterize the molecular genetics of ARCI in Finland. The specific aims of this study were:
1. To detect and characterize the TGM 1 mutations in ARCI in Finnish patients2. To compare the clinical phenotype and histopathological findings of the patients with their
genotype3. To search for a new gene locus underlying the ARCI phenotype by genome-wide screening

36
4 MATERIALS AND METHODS
4.1 CHARACTERIZATION OF TGM 1 MUTATIONS
4.1.1 ARCI patients
We studied 49 patients from 38 Finnish families were studied. Patients were collected among ARCIpatients diagnosed with ichthyosis congenita types I-IV by electron microscopy at the Department ofDermatology of Helsinki University Central Hospital prior to this study. There were 24 type I, 10 typeII, 6 type III and 2 type IV ARCI patients, and 8 patients did not belong to any of these groups; 117unaffected family members also took part in the study. The diagnosis was based both on clinicalfindings and on electron microscopy. None of the individuals studied had neurological symptomssuggesting syndromic ichthyosis. The study was approved by the Ethics Committee of the Departmentof Dermatology of the Helsinki University Central Hospital.
4.1.2 Isolation of DNA and DNA-amplificationGenomic DNA was extracted from leukocytes by standard procedures (Sambrook et al. 1987). Tosearch for mutations, 15 exons and exon intron boundaries of the TGM 1 gene were amplified by PCR(Saiki et al. 1988). The reaction conditions were optimized for each individual region. RadioactivePCR for single-strand conformation polymorphism (SSCP) was performed by adding 2.5 µCi of α-32³²P dCTP or dGTP (Amersham, Aylesbury, UK) to each PCR assay.
4.1.3 Mutation screening and characterizationSSCP analysisThe radioactive PCR products were analyzed by SSCP (Orita et al. 1989) (Study I). The PCR productswere diluted 1:5 with 0.1% sodium dodecyl sulfate (SDS) and 10 mM EDTA and mixed with an equalvolume of stop solution (Sequenase kit; US Biochemicals, Cleveland, OH, USA). The samples weredenatured at 96°C for 5 min, cooled briefly on ice, and loaded on an 8 % polyacrylamide gel (60:1acrylamide/bisacrylamide; BioRad, Richmond, CA, USA) with 5% glycerol. Bands were visualized byautoradiography.
4.1.3.1 Sequence analysis
A solid-phase sequencing strategy was used for the regions where mobility shifts were detected inSSCP (Study I). PCR was performed using primer pairs in which one was biotinylated at its 5´ end. ThePCR product was purified with streptavidin-coated microbeads (Fluoricon; Baxter, Mundelein, IL,USA) (Syvänen et al. 1989). A total of 25 µl of the PCR product was incubated with microbeads inTENT buffer (0.01% Tween 20, 1 mM EDTA, 50 mM NaCl, and 40 mM Tris-HCL, pH 8.0 to 8.8) for0.5 hours at room temperature. The sample was washed with TENT buffer, and the bound PCR productwas denatured by incubation for 5 minutes in 50 mM NaOH. The unbiotinylated strand was removedby washing the product twice with TENT buffer. The single-strand template was sequenced directly bythe dideoxy chain termination method of Sanger (Sequenase version; US Biochemicals) (Sanger et al.1977).

37
4.1.3.2 Solid-phase minisequencing
After detection of the nucleotide alterations, the samples from all patients, family members, and healthycontrols were screened for each mutation by solid-phase minisequencing, which unequivocallyidentifies the genotype (Syvänen et al. 1990) (Study I). PCR for the minisequencing reaction wasperformed asymmetrically with one biotinylated primer and one unbiotinylated primer. The PCRproduct and phosphate-buffered saline/0.1% Tween in bovine serum albumin were added tostreptavidin-coated microtiter wells (Streptavidin-coated combiplate; Labsystems, Helsinki, Finland).The samples were incubated and washed afterwards five times with TENT buffer. The unbiotinylatedstrand was removed by denaturing, and a reaction mixture containing a detection primer, 3H dNTP(Amersham) Taq DNA polymerase (Promega, Madison, WI, USA), was added to each well, and thesamples were incubated and washed as above. The hybridized primer was released by denaturing, andthe radioactivity resulting from the incorporated 3H -labeled nucleotides was measured from each wellin a liquid scintillation counter (Microbeta; Wallac, Turku, Finland). The genotype was defined by theratios of incorporated mutant versus normal nucleotides (r value). To evaluate the frequency of theidentified point mutations in the general population, DNA samples of about 400 blood donors werescreened. The screening method was identical to that described above.
4.1.3.3 Haplotype analysis
Haplotypes of the affected individuals with TGM 1 mutations were detected as described in study I.Five polymorphic markers flanking TGM 1 gene were amplified by PCR, separated by electrophoresisand detected either by 5´end-labeled with ³²P-γATP (Amersham. Little Chalfont, UK) or by silverstaining of the gels.
4.1.3.4 Modeling
The structural consequences of the four missense mutations were predicted by utilizing the three-dimensional structure of the FXIII A-subunit zymogen dimer, determined by single-crystal X-raydiffraction (Yee et al. 1994), as a template for creating a homology model of human Tgase 1 (study I).Coordinates for the factor XIII structure were obtained by use of the program X-PLOR (Brunger 1992).Models of the TGase 1 mutant structures were generated with the computer program O (Jones et al.1991), and figures were drawn with the program MOLSCRIPT (Kraulis 1991).
4.1.3.5 Creating the expression construct, mutagenesis, and transfection
To explore the consequences of the Arg-142-Cys mutation, we performed in vitro expression studies inCOS cells utilizing the homology of the FXIII A subunit to TGM 1 (Study I). FXIII A-subunit cDNAwas inserted into a SV40poly expression vector, and the specific Arg-78-Cys mutation in factor XIIIcorresponding to the mutation site Arg-142-Cys in the TGM1 gene was created by oligonucleotide(Stacey and Schnieke 1990). COS cells were transfected with the mutant SV40poly cDNA construct bythe DEAE-dextran-chloroquine method (Sussman and Milman 1984). Cells were harvested after 48 to94 hours, and the consequences of the mutations were analyzed.
4.1.3.6 Analysis of transfection efficiency and steady state mRNA levels
The transfection efficiency and the steady state mRNA levels of the mutant FXIII in transfected cellswere studied by double transfecting the cells with an equal amount of wild type and mutant clones(Study I). The cells were harvested after 48 hours and the nuclear and cytoplasmic fractions wereseparated by lysis buffer and centrifugation (Jalanko et al. 1995). The ratio of the two clones in DNA

38
(nuclear fraction) and RNA (cytoplasmic fraction) were analyzed after PCR by solid-phaseminisequencing.
4.1.3.7 Western blot
The amount of mutant FXIII polypeptides accumulating inside the cells was studied by immunoblottingof the individually transfected cell homogenates (Study I). The cells were harvested 72 hours aftertransfection and analyzed by 10% SDS-polyacrylamide gel electrophoresis and polyclonal,monospecific anti- FXIII A-subunit antibody (Clotimmun, Behringwerke AG, Marburg, Germany)(Laemmli 1970). Visualization was performed by the enhanced chemiluminescence (Paunio et al.1994).
4.1.3.8 Metabolic labeling and immunoprecipitation
The intracellular stability of the mutant proteins was studied by pulse-chase analysis (Study I). The
transfected cells were metabolically labeled during a one-hour radioactive pulse with35
S-methionine(Amersham) Aylesbury, U.K), whereafter the cells were monitored after variable chase periods (4, 8,and 24 hours). FXIII polypeptides in the transfected cell lysates and chase media were analyzed byimmunoprecipitation with fixed FXIII A-subunit and Staphylococcus Aureus antibody(Immunoprecipitin, Gibco BRL, Gaithesburg, MD) (Proia et al. 1984). The precipitated proteins wereanalyzed in 10% SDS-PAGE gels and visualized by autoradiography.
4.1.3.9 Transglutaminase activity measurement by ammonia release assay
The enzymatic activity in the transfected cells was studied by the Berichrom activity assay(Behrichrom, Behringwerke AG, Marburg, Germany ) (Lorand et al. 1972) (Study I).
4.2 SEARCH FOR A NEW GENE UNDERLYING ARCI
4.2.1 Genotyping
4.2.1.1 Family
Only one family was selected for the genome scan due to the expected genetic heterogeneity of theichthyoses (Study III). This family originated from a known genetic subisolate in Finland, thearchipelago of Larsmo off the coast of Ostrobothnia. X-linked ichthyosis was excluded by measuringthe steroid sulphatase activity from one patient in the family, and the clinical picture and the pedigreeprofile were also not suggestive of X-linked ichthyosis. In histologic examination, the granular layerwas present in all patients, excluding ichthyosis vulgaris. All affected individuals displayed a non-lamellar, non-erythrodermic phenotype which resembled neither classic LI nor classic CIE (study III).
4.2.1.2 Candidate genes
The candidate gene approach served as the first strategy (Study III). We selected five possiblechromosomal loci, the TGM 2 and 3 locus in 20q11.2, Keratin loci in 17q and 12q11-q13, the secondlocus for ARCI in 2q33-q35 and the epidermal differentiation complex in 1q (Parmentier et al. 1996;Wang et al. 1994; Yoon et al. 1994). DNA was amplified by the touchdown-PCR procedure (Don et al.1991). PCR products were then pooled so that at most four PCR products of different sizes were pooledand gel electrophoresis was run on an ALF express automated sequencer (Pharmacia Biotech).Genotypes were analyzed by AlleleLinks™ 1.0 software package (Pharmacia Biotech).

39
4.2.1.3 Genome scan
After excluding the candidate genes, a genome-wide scan was performed using 370 highly polymorphicmicrosatellite markers of the Cooperative Human Linkage Center with fluorescent label (Dubovsky etal. 1995) (Study III). The average intermarker distance was 9.3 cM, the largest gaps being 24 cM onchromosomes 1 and 20.
4.2.2 Statistical analysis
4.2.2.1 Linkage analysis
Linkage analysis was performed assuming an autosomal recessive mode of inheritance for the ARCI
gene, with an estimated gene frequency of 10-3 in the Finnish population, and equal recombinationrates for males and females (Study III). Because of the early onset of the disease, complete penetrancecould be assumed. Data simulation analysis was performed by the SLINK option of the LINKAGEpackage of software (Ott 1989; Weeks et al. 1990).The two-point LOD score was calculated by theMLINK option of the LINKAGE package (Cottingham Jr et al. 1993; Lathrop and Lalouel 1984) andmultipoint linkage analysis were performed with the Simwalk2 program (Sobel and Lange 1996).Allele frequencies were ascertained from both the pedigree and from the unaffected individuals fromLarsmo island.
4.2.2.2 Haplotypes
Microsatellite markers at the ichthyosis locus on chromosome 19 were obtained from Genome DataBase (http://gdbwww.gdb.org) with the order of the markers and distances between them based on themetric physical map of chromosome 19 created by the Human Genome Center of Lawrence LivermoreNational Laboratory (http://www-bio.llnl.gov/brp/gebnome/genome.html) (Ashworth et al. 1995)(Study III). The most likely haplotypes were determined by the Simwalk2 program (Sobel and Lange1996).

40
5 RESULTS AND DISCUSSION
5.1 IDENTIFICATION OF MUTATIONS IN TGM 1 GENE
In the early 1990s, Finnish ARCI patients were collected to be classified into four different categoriesby electron microscopy according to criteria created by Anton-Lamprecht (Anton-Lamprecht et al 1992;Niemi et al. 1991; Niemi et al. 1994; Niemi et al. 1992; Niemi et al. 1993). In 1995, it was found that asubclass of ARCI patients carried mutations in the TGM 1 gene (Huber et al. 1995; Russell et al. 1995).Prior to that, nothing was known about the molecular defects underlying the Finnish patients’scondition. Since these patients were histologically and clinically well-characterized, we decided tobegin investigating molecular defects of this clinically and genetically heterogeneous disease group.
First, 49 affected individuals from 38 families were screened for TGM 1 mutations by SSCP (Study I).Those patients with positive findings in SSCP were sequenced. Detected mutations were screened forin affected individuals, family members, and healthy controls by minisequencing. 17 patients from 13families carried six different mutations: Arg-141-His, Arg-142-Cys, Gly-217-Ser, 3339A to G, Val-378-Leu, and Arg-395-Leu (Studies I-II). Four of the mutations, Arg-142-Cys, Gly-217-Ser, Val-378-Leu, and Arg-395-Leu, were novel (Figure 9). All of the mutations except 3339A to G were missensemutations resulting in single amino acid substitution; 3339A to G, situated in an exon-intron boundary,leads to a splicing error and subsequent premature termination codon. Most of the patients werecompound heterozygotes. Screening of 400 healthy controls for these mutations showed that thenucleotide changes observed were unlikely to represent common polymorphic variants in the generalpopulation.
Thus far, we and others have reported over 30 mutations in the TGM 1 gene (studies I-II), (Bichakjianet al. 1998; Candi et al. 1998; Hennies et al. 1998; Hennies et al. 1998; Huber et al. 1995; Huber et al.1997; Parmentier et al. 1995; Petit et al. 1997; Pigg et al. 1998; Russell et al. 1995; Tok et al. 1999).The six mutations detected in this study reflect the general pattern of mutations observed in the TGM 1gene. Most of the mutations are point mutations resulting in single amino acid substitutions, stopcodons or splicing errors. A few minor deletions or insertions resulting in a frameshift have beendetected, as well as one mutation in the Sp1 binding site in the promoter region, resulting in a reductionof TGM 1 transcript levels. Reduced TGM 1 activity is a consequence of most mutations. Although themutations are generally spread throughout the coding region, there seems to be a moderate clustering ofmutations in the catalytic core domain. We also identified a mutation-prone region in B-sandwichcoding for amino acid residues 141-144 (Study I). The mutation profile of the TGM 1 gene, except themutation-prone region mentioned above, is highly reminiscent of the mutation profile of FXIII in thebleeding disorder coagulation FXIII deficiency (Mikkola and Palotie 1996).
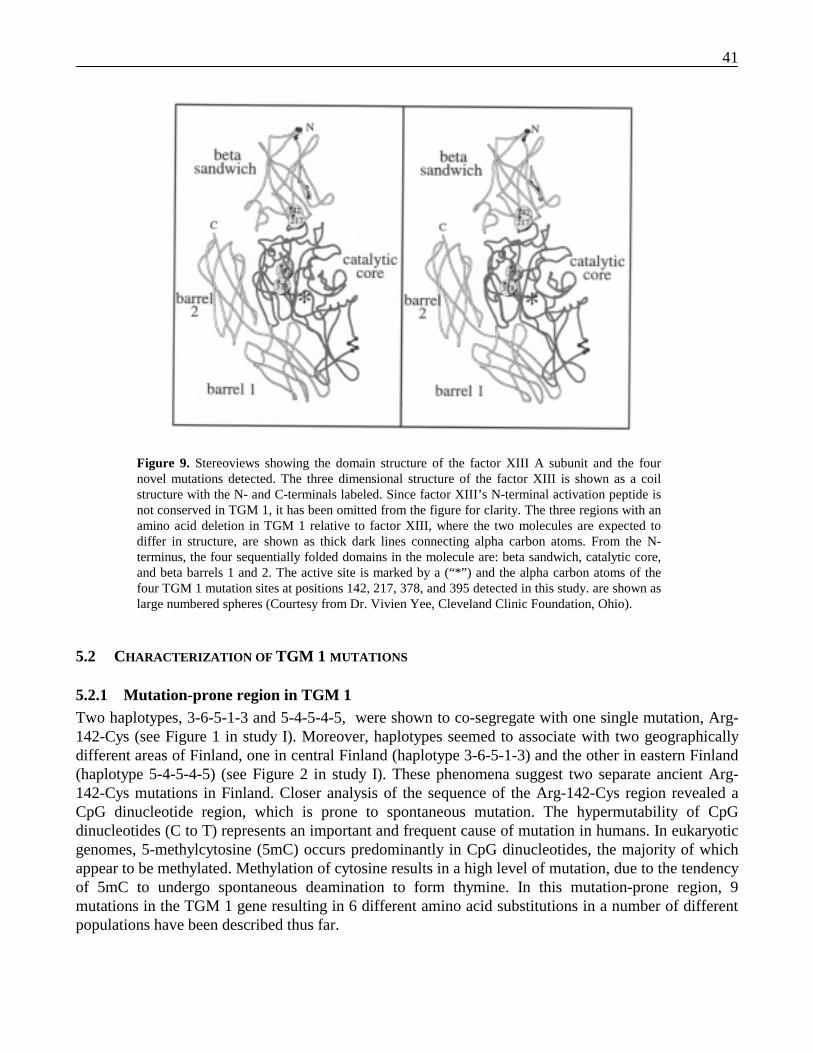
41
Figure 9. Stereoviews showing the domain structure of the factor XIII A subunit and the fournovel mutations detected. The three dimensional structure of the factor XIII is shown as a coilstructure with the N- and C-terminals labeled. Since factor XIII’s N-terminal activation peptide isnot conserved in TGM 1, it has been omitted from the figure for clarity. The three regions with anamino acid deletion in TGM 1 relative to factor XIII, where the two molecules are expected todiffer in structure, are shown as thick dark lines connecting alpha carbon atoms. From the N-terminus, the four sequentially folded domains in the molecule are: beta sandwich, catalytic core,and beta barrels 1 and 2. The active site is marked by a (“*”) and the alpha carbon atoms of thefour TGM 1 mutation sites at positions 142, 217, 378, and 395 detected in this study. are shown aslarge numbered spheres (Courtesy from Dr. Vivien Yee, Cleveland Clinic Foundation, Ohio).
5.2 CHARACTERIZATION OF TGM 1 MUTATIONS
5.2.1 Mutation-prone region in TGM 1Two haplotypes, 3-6-5-1-3 and 5-4-5-4-5, were shown to co-segregate with one single mutation, Arg-142-Cys (see Figure 1 in study I). Moreover, haplotypes seemed to associate with two geographicallydifferent areas of Finland, one in central Finland (haplotype 3-6-5-1-3) and the other in eastern Finland(haplotype 5-4-5-4-5) (see Figure 2 in study I). These phenomena suggest two separate ancient Arg-142-Cys mutations in Finland. Closer analysis of the sequence of the Arg-142-Cys region revealed aCpG dinucleotide region, which is prone to spontaneous mutation. The hypermutability of CpGdinucleotides (C to T) represents an important and frequent cause of mutation in humans. In eukaryoticgenomes, 5-methylcytosine (5mC) occurs predominantly in CpG dinucleotides, the majority of whichappear to be methylated. Methylation of cytosine results in a high level of mutation, due to the tendencyof 5mC to undergo spontaneous deamination to form thymine. In this mutation-prone region, 9mutations in the TGM 1 gene resulting in 6 different amino acid substitutions in a number of differentpopulations have been described thus far.

42
5.2.2 Mutational modeling of Arg-142-Cys and Arg-141-HisBecause of the interesting features of the mutation prone region, we decided to study the Arg-142region in more detail (study I). We used FXIII as a template to create a homology model to study thepossible structural consequences of the Arg-142-Cys mutation, since the three-dimensional structure ofTGase 1 has not been determined, and there is considerable amino acid homology (299 residues, 42%)between FXIII and TGase 1.
From structure-guided alignment of 19 sequences of proteins within the TGase family, Arg 141 andArg 142 are absolutely conserved residues. Residues which surround Arg 141 and Arg 142 areconserved in TGase 1 and F XIII, and thus it is very likely that the local protein structure is conservedin these regions as well, suggesting the structural importance of these residues.
Both Arg 141 and Arg 142 are found at the interface with the catalytic core domain far from the active-site catalytic triad residues (>25 Ångströms). The two arginine side-chains are involved in extensivehydrogen-bonding interactions which serve to stabilize the conformation of β-sandwich loops and,more importantly, to attach the β-sandwich to the catalytic core domain. Replacement of either of thesearginine residues with other amino acids which disrupt the hydrogen-bonding interactions is predictedto disrupt or destabilize the interdomain interface. Specifically, the Arg 142 side-chain forms a saltbridge with the conserved catalytic core Asp 253 residue, and with several main chain carbonyl groups(Pro 212, Ala 214, and Gly 315) (Figure 10). The introduction of the much smaller cysteine side-chainin the Arg-142-Cys mutant will abolish all five of these hydrogen bonds. Possible results are (1) analtered quaternary structure which has a modified substrate binding and recognition surface or is moresusceptible to proteolytic cleavage, or (2) a misfolded structure which is more easily proteolyzed, or isno longer capable of binding to or acting on a substrate.
5.2.3 In vitro studies of Arg-142-CysTo find out whether the result of the Arg-142-Cys mutation is a rapid proteolytic cleavage or anotherwise altered structure, the homology between TGM1 and FXIII was again utilized. The Arg-142-Cys mutation was introduced into the corresponding site of the FXIII A-subunit cDNA, and theconsequences of the mutation on the structurally homologous factor XIII A-subunit were studied invitro in a COS cell expression system (Study I). The mutant protein accumulated in the cells in reducedquantities compared to the wild type protein, as analyzed by immunoblotting of the transfected cellhomogenates. In pulse chase experiments, the mutant protein demonstrated marked instability andalmost complete degradation within 24 hours. Thus, on the basis of structural homology of the FXIIIand TGase 1 enzymes and conservation of the polypeptide sequence around the mutation, it is highlylikely that instability and susceptibility to proteolytic degradation is at least one of the consequences ofthe Arg-142-Cys mutation. The corresponding mutation in FXIII was also devoid of TGase activity.However, although models of Arg-142-Cys in FXIII seem to result in rapid protein degradation, TGase1 and FXIII have some distinct characteristics, and in vivo totally different functions. Moreover, thebiology of TGase 1 has revealed more complexity than was predicted at the start of our study. Theseaspects restrict the conclusions that can be drawn from our data.

43
Figure 10. Stereoviews of the modeled Arg-142-Cys mutation site. The beta sandwich andcatalytic core domains are shown as light and dark colored coil structures, respectively. Theside-chain moieties of the two conserved Arg 141 and Arg 142 residues in the sandwichdomain are shown as ball-and-stick structures. These two residues participate in a number ofhydrogen-bonding interactions which stabilize sandwich loop conformations and, moreimportantly, tether the sandwich domain to the catalytic core. The Arg 142 side-chain forms asalt bridge with the conserved core residue Asp 253, and hydrogen-bonds with three main-chain carbonyl groups. All of these interactions will be abolished in the Arg-142-Cys mutant(side-chain shown in dark-colored ball-and-stick). The side-chain group of Asp 253, as well asthe carbonyl groups involved in hydrogen-bonding interactions with Arg 141 and Arg 142, areshown as labeled ball-and-stick (Courtesy from Dr. Vivien Yee, Cleveland Clinic Foundation,Ohio).
Other groups have also studied the Arg-142 region in vitro. Expression studies of the mutants Arg-141-His, Arg-141-Cys, and Arg-142-His in TGase 1 by baculovirus/insect cell systems revealed reducedenzymatic activities as expected (Candi et al. 1998). Expression levels and half-times of mutants werecomparable to those of the wild type in Western blot, but specific pulse chase experiments were notperformed. In addition, the baculovirus system is not able to process TGase 1 to its most active form.As it is not a mammalian expression system, the post-translational modifications differ frommammalian cells, therefore challenging the interpretation of the results. Expression of Arg-141-His innormal human keratinocytes in the presence of both low and high calcium also demonstrated reducedactivity, but its stability was not examined (Candi et al. 1998).
5.2.4 Characterization of other mutants – Gly-217-Ser, Val-378-Leu, Arg-395-Leu, and 3339A-to-G
We utilized the homology model of TGase 1 also in mutational modeling of other identified mutations(Study I). Gly217 residue is located far from the active site (>20 Å). In the FXIII A-subunit structure,

44
the conserved sandwich Gly217 residue is located at the end of the β-strand near the core domaininterface and has a main chain conformation which is unfavorable for other amino acids with theirlarger side-chain groups that introduce short steric contacts. Based on this model, the Gly-217-Sermutation results in altered protein folding which can affect protein stability or function.
The conserved core Val378 residue is located in the active site helix, two amino acids downstreamfrom the catalytic Cys376 residue. The Val378 side chain is buried in a pocket formed by conservedhydrophobic residues. Substitution of the larger leucine side-chain introduces unfavorable contactswhich are likely to result in a limited local conformational change in the structure. These changes arelikely to extend to the catalytic residues and can drastically affect the catalytic behavior of the mutantmolecule.
Arg-385-Leu affects a conserved core residue which is at one end of a conserved string of hydrogen-bonding interactions (Arg395-Asp439-Tyr543-Trp437 in TGM 1; Arg333-Glu377-Tyr481-Trp375 inthe FXIII A-subunit) which span the body of the core domain. The substitution of a hydrophobicleucine side-chain for the positively charged Arg395 disrupts the conserved hydrogen-bonding patternand creates electrostatic conflicts, yielding a mutant protein with altered structure which extends to theactive site, or which is more unstable or easily degraded.
We did not study any consequences of the 3339 A to G mutation in our one patient, but previousstudies exists on this mutation. In Norwegian patients, 3339A to G results in an insertion of oneintronic G into the mRNA because of the A to G point mutation in the acceptor splice site of intron 5(Pigg et al. 1998). This mutation leads to a frameshift and premature termination at codon 3345. InNorway, this mutation is present in 80% of disease alleles, suggesting a single founder mutation in themajority of patients. The mutation has also been found in Swiss patients, but with differentconsequences (Huber et al. 1995). The mutation leads to a retention of intron 5 sequence in mRNA,resulting in a premature stop codon. No detailed studies exist on the precise consequences of thismutation, but it could lead to 1) reduced levels of mRNA due to nonsense mediated mRNA decay, 2)several variants of differently spliced mRNA transcripts, which has been reported with splice sitemutations, or 3) a truncated protein, which is unable to function properly because it lacks the catalyticsite. It has been shown, however, that the result of this mutation is lowered TGase activity (Huber et al.1995).
5.3 GENOTYPE-PHENOTYPE CORRELATION OF ARCI
5.3.1 Clinical phenotypes of ARCI patients with TGM 1 mutationsAfter the detection of TGM 1 mutations, we compared the genotype data with clinical findings (studyII), showing that, the majority of 13 families (8, 62%) with TGM 1 mutations had a phenotype ofclassic LI. However, the milder phenotype of patients in 5 families (38%) was also associated withTGM 1 mutations. All of the patients with TGM 1 mutations were quite severely affected, and none ofthem had the minimal white scaling described in some ichthyosis patients. The clinical presentationbetween affected family members was relatively homogeneous. In addition, in some patients withsevere classic LI, mutations were not detected in screening. It should be noted that the sensitivity of ourscreening method SSCP is only 60-80%, meaning that some of the mutations may have remainedundetected.

45
In most mutation reports, the phenotype of affected individuals has been a severe form of LI with abrown, platelike scale (Hennies et al. 1998; Russell et al. 1994). In some studies, however, a CIEphenotype has also been reported. In a Norwegian sample of 34 ARCI patients with TGM 1 mutations,30 LI patients (88%) and 4 CIE patients (12%) were found (Pigg et al. 1998). Furthermore, in a Germanstudy of 9 families, 3 of 9 patients had clinical CIE, 1 patient was mildly affected with minimal scaling,and the rest of the patients presented with the classic LI phenotype (Hennies et al. 1998). Some authors,however, have been skeptical as to whether CIE can be caused by mutations in TGM 1 (Shevchenko etal. 2000). In one study, TGM 1 mutations had been excluded from 5 CIE patients, although anabnormal cytoplasmic TGase 1 expression pattern and intermediate enzyme activity were observed inthose patients (Choate et al. 1998). Although the defect in these patients was not due to alterations inthe TGM 1 gene, the abnormal focal expression of TGase 1 may result in a pathogenic disruption inepidermal differentiation as well.
Although TGM 1 is expressed in some extracutaneous stratified epithelia such as intestinal and trachealepithelia, ARCI is considered purely a disease of the epidermis and its appendages. Some authors havespeculated that this may be due to the suggested role of TGase 1 in assembling both the lipid envelopeby esterbond formation and the protein envelope by cross-linking CE proteins in the skin (Nemes et al.1999). Other epithelial tissues also forms envelopes, but the lipid envelope is a unique feature in theskin. Therefore, the defective TGase 1 can possibly cause severe manifestations specifically in skin.
5.3.2 Morphological findings in patients with TGM 1 mutationsUltrastructurally, all of our TGM 1 patients belonged to either IC type I or type II (Study II). All of theIC type II patients and a third of the IC type I patients harbored TGM 1 mutations. Ultrastructuraldiagnosis of ARCI has been controversial, but it does seem to detect ARCI patients with TGM 1mutations to some extent. Besides ours, only one other study has been published in which the affectedpatients were classified ultrastructurally (Pigg et al. 1998). In that Norwegian study, only two of 34patients with TGM 1 mutations were classified as IC type I, whereas 32 patients has the type II disease.None of the ARCI patients with TGM 1 mutations has belonged to ARCI types III and IV in any study.
5.3.3 TGM 1 mutations and phenotypeAmong 13 families with TGM 1 mutations, most of the affected individuals were compoundheterozygotes; only two patients were homozygotes. We found no correlation between the type of theTGM 1 mutation and phenotype of the ARCI patients (Study II). Five different mutation combinationswere shown to cause type II ichthyosis, which is a clinically homogeneous group. Interestingly, thesame mutations were also characterized in patients with type I ichthyosis. Furthermore, two patients,who both carried the mutation combination Arg-142-Cys/Val-378-Leu, had different clinical picturesand different ultrastructural findings. Similar observations were demonstrated in the German study inwhich patients homozygous for the splicesite mutation 3339 A to G showed differences in their clinicalpictures (Hennies et al. 1998). Furthermore, 3 Norwegian CIE patients and 30 LI patients were allhomozygous for the same 3339 A to G mutation (Pigg et al. 1998).
5.3.4 Locus heterogeneity and the phenotypeThe genetic heterogeneity of ARCI is well-established, and five different loci have been linked to thedisease. Variable figures have been achieved on the proportion of ARCI patients associated with the

46
TGM 1 gene defect. In this study, 13 of 38 ARCI families (34%) showed linkes with TGM 1 mutations(Study II). An almost similar distribution was achieved in a recent linkage study performed by a Frenchgroup, involving a collection of 51 consanguineous families originating from North Africa, Turkey, andFrance. In this sample, 11 families (22%) were linked to the chromosome 14 (TGM 1) locus (Fischer etal. 2000). Based on biochemical observations utilizing TGase 1 activity measurements andimmunofluorescence studies, 50% of ARCI patients have been associated with the TGM 1 gene defect(Hohl et al. 1997). In 43 Norwegian families, 34 (80%) were found to carry a single founder TGM 1mutation (Pigg et al. 1998). Variations in these figures may reflect differences in diagnostic criteriaand/or differences in genetic disease profiles between different ethnic backgrounds.
Only the French study described above includes investigation of loci other than TGM 1 (Fischer et al.2000). In that study of 51 ARCI families, 17 families (34%) were linked to other chromosomal loci onchromosome 2 (5 families, 10%), chromosome 19 (6 families, 12%), and chromosome 3 (6 families,12%). In addition, 23 families (45%) were excluded from these loci, indicating the presence of at leastone additional locus for ARCI, if not more. In that study, the patients linked to chromosome 3 clinicallyresembled CIE, whereas the patients linked to chromosome 19 have a classical LI phenotype. Thephenotype in these families was reported to be more homogeneous than that in a group with TGM 1mutations. Patients of the Larsmo family, linked to chromosome 19, had an clearly milder phenotypewith only minimal scaling compared with chromosome 19-linked patients in the French study (StudyIII). It remains, however, possible that the locus of Larsmo family represents a separate entity from thatof French families. The chromosome 19 region demonstrating linkage was somewhat more broad andmore proximal in the French study (19p12-q12) than in our study (19p13.1.-13.2.). In a German study,comparison of six patients excluded from TGM1 with those linked to TGM 1 did not reveal anyspecific clinical features which could separate these groups from each other (Hennies et al. 1998). Bothshowed an entire spectrum of clinical features of ARCI from erythrodermic fine scaling to non-erythrodermic, large, dark scales.
Among inherited diseases, locus heterogeneity seems to be a rather common phenomenon. It is oftenfound in syndromes that result from failure of rather general pathways involving many genes (Strachanand Read 1996). Such complex pathways are for example, vision and hearing. A defect in any of thegenes of the same pathway may lead to similar clinical results. A striking example is Usher syndrome,an autosomal recessive combination of hearing loss and retinitis pigmentosa, where at least 8 loci havebeen found on chromosomes 1, 3, 10, 11, and 14 in three different clinical forms (Chaib et al. 1997;Hmani et al. 1999; Kaplan et al. 1992; Kimberling et al. 1992; Kimberling et al. 1990; Sankila et al.1995; Smith et al. 1992; Wayne et al. 1996). Moreover, defects in genes coding for a different part of asame structural unit can cause similar clinical phenotypes. A good example is epidermolysis bullosa, inwhich defects in at least 10 genes coding for different basement membrane zone proteins result inbullous disease with a rather similar outcome. The locus heterogeneity of ARCI seems to be significant.Although only one gene in the ARCI is known so far, it may be that some of the other ARCI loci revealgenes involved in the formation or function of CE, which is quite a complex structure itself, or revealgenes that disturb the normal function of TGase 1.
5.3.5 Challenges in phenotype evaluationThe evaluation of genotype-phenotype correlations in ARCI is challenging for many reasons. Diagnosisof ARCI is based on its clinical presentation and histology, and unfortunately, no more objective

47
diagnostic methods, such as laboratory tests, are available. This makes the diagnosis dependent on thesubjective judgments of the dermatologist and the pathologist. Furthermore, local medical practice maycreate differences in diagnostics. Clinical differentiation of LI and CIE is not straightforward and thereare intermediate forms of ARCI that are difficult to place in either of these entities. Some of the clinicalfeatures, such as erythrodermia, appers and disappears. Retinoid therapy and the patient’s own activityin treating the disease with emollients affects clinical features. This further complicates the evaluationof the phenotype. In addition, different criteria used for ARCI classifications confuse the correlation ofdifferent genotype-phenotype studies with each other. The final evaluation of phenotype-genotypecorrelations of ARCI will be accomplished only when more of the underlying genes have beencharacterized, and meaningful genotype-phenotype correlations are possible.
5.3.6 Clinical and morphological findings in a subgroup of ARCIDuring evaluation of genotype-phenotype correlations among patients with TGM 1 mutations, wefound a subgroup of ARCI patients with rather uniform electron-microscopic findings (Study IV). Inthat subgroup the morphological findings, abnormal keratohyalin and keratinosomes, resemble thosedescribed earlier only in harlequin ichthyosis patients. We demonstrated that similar ultrastructuralfindings can be detected in ARCI patients lacking the harlequin ichthyosis phenotype. The clinicalpicture of these patients was more variable than were their findings in electron microscopy, althoughsome clinical features, such as accentuation of ichthyosis in the scalp, large and thick scales,erythrodermia, ectropion, and alopecia were common features of nearly all patients. A peculiarmalformation of digits and toes exists in some of the patients. In this subgroup of ARCI patients TGM1 mutations were excluded.
5.4 ASSIGNMENT OF THE NEW ARCI LOCUS ON CHROMOSOME 19P13.1.-13.2.As TGM 1 was revealed to be the defective gene in only a third of the ARCI families, we decided tosearch for other defective genes in ARCI (Study III). Due to the genetic and clinical heterogeneity ofARCI, we decided to focus on consanguineous families in order to identify new genes for ARCI.Among the families not associated with a TGM 1 gene defect, one large IC type I family originatingfrom the genetically isolated island of Larsmo existed (Figure 11A). Close consanguinity could beobserved in the Larsmo pedigree. The parents of patient 5 were second cousins, and the mother ofpatient 5, the father of patients 6 and 7 and the mother of patient 9 shared a common grandparent, asdid the mother of patients 6 to 8 and the father of patient 9. Due to this consanguinity, a homozygositymapping strategy was applied to search for a new gene locus. In homozygosity mapping, affectedmembers of the family are expected to be homozygous for the disease-causing allele, and the wholesegment of the chromosome flanking the disease locus is predicted to be identical by descent. Aconservative prediction was that patient 5 should be homozygous for one mutant allele and this samemutant allele should be at least one of the disease alleles in patients 6 to 9. Patients 6 to 9 should alsoshare another mutant allele. Based on the hypothesis of one ancestral founder mutation in a geneticallyisolated population, we assumed that only one mutant allele should be found in this pedigree.
Prior to initiating the genotyping, data simulation analysis was carried out to determine whetherconclusive results could be achieved from linkage analysis of the chosen family. This simulationanalysis revealed a maximum expected LOD score of 3.57 and mean expected LOD score of 1.36 atrecombination fraction (θ) = 0.010. Therefore, an evenly spread set of informative markers could beexpected to yield a conclusive LOD score in the family material.

48
5.4.1 Exclusion of candidate lociFirst we selected 5 possible chromosomal loci, the TGM 2 and 3 loci on 20q11.2, Keratin (KRT) loci in17q and 12q11-q13, a second locus for ARCI on 2q33-q35, and the epidermal differentiation complexon 1q. TGase 2 and 3 are expressed in keratinocytes, and TGase 3, as well as genes in the epidermaldifferentiation complex, take part in CE formation, which make these loci interesting candidate regions.Keratins are shown to be defective in many genodermatoses and were included in the candidate locianalysis. Linkage for the ARCI gene to these candidate loci could be excluded since LOD scores below-2.0 were detected with every marker flanking these loci.
5.4.2 Genome scanAfter the candidate loci analysis, we performed a genome-wide scan using 370 polymorphic markersspaced at about 10 cM distances and analyzing five affected individuals as well as their parents, in totalnine samples. None of the initial markers revealed homozygosity between the affected individuals, butthe two-point linkage analysis revealed two interesting regions, one on chromosome 9 (D9S158 LODscore 2.05) and another on chromosome 19 (D19S432 LOD score 2.28 ). The chromosome 9 area wasexcluded as a disease locus by analysis of the haplotypes formed by four additional microsatellitemarkers around the area of interest (data not shown). In contrast, when 11 additional microsatellitemarkers were analyzed from the chromosomal region 19p13.1-p13.2, they revealed a region of about6.0 cM where all markers produced strong positive LOD scores (Figure 11B). In the final calculations,the allele frequencies from Larsmo population were used.
Haplotypes consisting of 14 microsatellite markers flanking the chromosome 19 region restricted thedisease locus spanning about 6 cM between markers D19S411 and D19S914 on 19p13.1-13.2 (Figure11A). Two alleles were identified in the disease chromosomes, with most of the markers revealing twocore haplotypes (Figure 11 A). Two adjacent loci in the middle of the haplotype, D19S415 andD19S179, revealed homozygosity in all affected individuals. However, these alleles were found to bequite common among the Larsmo population, and the heterozygosity of the markers was low. Thesignificance of this finding of homozygosity thus remains to be studied further and cannot yet beconsidered conclusive evidence for a shared chromosomal region among all affected individuals.
A French group has also published linkage on chromosome 19p12-q12 based a genome scan of nineconsanguineous, severely affected LI families (Fischer et al. 2000). However, according to the LLNLphysical map and the Genethon genetic map, these regions do not overlap, but have 2 cM betweenthem. The possibility exists that these regions represent two separate ARCI loci, which is supported bythe fact that phenotypes of our family and the nine French families differ from each other. Yet, errorsstill exist in published maps, and heterogeneity of ARCI patients is common, making it equally possiblethat these regions do overlap and represent only one ARCI locus on 19p.

49
2 31 13 11 62 12 13 23 12 22 32 22 21 35 4
2 33 12 11 62 12 11 23 11 22 31 21 22 32 4
2 33 12 11 62 12 11 23 11 22 31 22 22 32 4
2 31 11 16 61 11 12 21 12 23 32 22 23 33 4
3 11 24 22 31 12 12 13 22 32 32 33 23 21 3
3 41 13 11 63 12 11 22 13 21 33 23 22 36 4
3 11 23 21 33 12 11 12 23 31 33 33 22 26 3
3 11 23 21 33 12 11 12 23 31 33 33 22 26 3
3 11 14 12 61 12 12 23 12 22 32 23 23 31 4
4 11 21 26 31 11 12 11 22 33 32 32 23 24 3
4 11 21 26 31 11 12 11 22 33 32 32 23 24 3
4 11 21 26 31 11 12 11 22 33 32 32 23 24 3
3 31 13 41 23 12 21 22 33 21 23 23 32 36 1
3 31 13 41 23 12 21 22 33 21 23 23 32 36 1
2 31 13 15 61 11 11 22 13 22 33 22 22 22 1
5 22 12 34 52 12 14 12 24 31 24 33 23 21 2
2 31 13 15 61 11 11 22 13 22 33 22 22 22 1
1 32 12 13 61 11 11 22 13 23 33 22 22 23 1
5 32 12 14 61 12 14 22 14 21 34 23 23 21 1
D19S221D19S914D19S923D19S226D19S179D19S415D19S929D19S432D19S588D19S252D19S253D19S411D19S885D19S199
Ancestors
1 2
3 4
5
6 7 8
A
B
Figure 11A) Pedigree and haplotypes of the ARCI family on chromosome 19. Persons marked with * wereincluded in the genome scan. Recombinations in patient 9 restricted the area between D19S914and D19S411.B) Linkage of the ARCI family to chromosome 19p13.1.-13.2. Cloned genes and some of the locion the linkage region are shown in the middle. LOD scores to the loci are presented on the right.GCDH=Glutaryl CoA dehydrogenase, CALR=calreticulin, RAD23A=?, NFIX =nuclear factor I,X-type, LYL 1=lymphoid leukemia 1, CACNA 1A4= voltage-dependent Ca2+ channel,RFX 1=regulatory factor 1, OLFR =olfactory receptor, CD97 =cluster of differentiation, 97,SLC1A6R=solute carrier family 1 A, member 6, CYP4E3=cytocrome P450 4F2

50
5.4.3 The new ARCI locus on chromosome 19The new ARCI locus is localized at a region where significant progress has been made in the humangenome sequencing project. According to the LLNL, which is the major sequencing center for ofchromosome 19, the new ARCI locus is located on a 3.0 Mb area of which 1.34 Mb has beensequenced. Chromosome 19 is one of the most gene-rich chromosomes. Thirteen known genes and 65ESTs have been localized to the ARCI critical interval (Ashworth et al. 1995). There are several
possible candidate genes, including CACNA1A, a voltage-dependent Ca2+ channel gene (Diriong et al.1995), calreticulin, a major calcium storage protein in the lumen of the endoplasmic reticulum(McCauliffe et al. 1990), and SLC1A6, solute carrier family 1 member 6, an amino acid transporterregulating the level of extracellular glutamate (Figure 11B) (Fairman et al. 1995). Calcium is animportant regulator of epidermal differentiation as has been described. Although CACNA1A has beenpreviously shown to associate with three neurological diseases (familial hemiplegic migraine, episodicataxia type 2, and spinocerebellar ataxia type 6) (Ophoff et al. 1996; Zhuchenko et al. 1997), and itsexpression in keratinocytes is ambiguous, it remains, along with calreticulin, an interesting candidategene for ichthyosis. The functions of SLC1A6 or EAAT4 (excitatory amino acid transporter) associatedwith the glutamate signaling pathway has mainly been studied in neurons, but recent progress showsthat the glutamate signaling pathway is also present in the skin, making SLC1A6 an interestingcandidate gene (Maxfield et al. 1999).
One efficient way to search for candidate genes is to compare the human transcript map to maps ofother species, for example the mouse. Mouse homologies of genes located on 19p are widely dispersedin the mouse genome. At least 5 distinct homology segments exist in mouse chromosomes 8, 9, 10, and17. However, although most of our new ARCI locus on chromosome 19p13.1-13.2 seems to map onmouse chromosome 8 (MMU 8), the man-mouse homology relationship in this region is suggested tobe complex, and it is difficult to predict whether homologies of mouse genes in that region should mapto 19p in human beings (Mohrenweiser et al. 1996).

51
6 SUMMARY AND CONCLUSIONSAutosomal recessive congenital ichthyosis (ARCI) is a clinically and genetically heterogeneous groupof genodermatoses which typically result in a severe scaling disease of the skin. Substantial progresshas been made in the study of the pathogenesis of ARCI in recent years. In 1995, the transglutaminase 1gene (TGM 1) was revealed to have mutations in a subgroup of ARCI patients. The function of TGM 1is to catalyze the formation of the cornified cell envelope, a tightly cross-linked network of proteinsformed in the upper keratinocytes of the skin, in the later stages of epidermal differentiation.
The aim of this study was to further clarify the molecular mechanisms underlying ARCI. Fifty FinnishARCI patients exist, who prior to this study were ultrastructurally classified into four distinct groups. Inthe first stage, TGM 1 mutations were screened in these patients. Six different mutations were found inone third of the patients. Five of the mutations were point mutations causing amino acid substitutions.In addition, one splice site mutation was detected. Mutational modeling utilizing the three-dimensionalstructure of a homologous transglutaminase, coagulation factor XIII, showed that mutations werelocated in conserved regions of the protein and were likely to cause structural alterations and ultimatelythe disease. One of these mutations, Arg-142-Cys, was found to be mutated twice in Finnish patientsand to be located in a mutation-prone area. Due to these interesting features, we studied the mutationmore thoroughly by in vitro studies and demonstrated that the mutated protein is rapidly degraded incells.
The genotype-phenotype correlation studies implied that 62% of those patients with the TGM 1mutations were severely affected classical lamellar ichthyosis patients belonging to ultrastructuralgroup type II, and 32% were mildly affected and belonged to ultrastructural group type I. Surprisingly,the same mutations were shown to result in both ultrastructural types I and II, and furthermore, no clearcausative relationship between the phenotype and mutation type was observed. We also reported asubgroup of ARCI patients with ultrastructural findings resembling those of harlequin ichthyosis, butwithout the harlequin ichthyosis phenotype.
Since the TGM 1 gene defects explained only one third of ARCI cases, we decided to search for a newARCI gene. A primary random search of the human genome was performed in one largeconsanguineous family with five affected patients originating from a genetic subisolate, the archipelagoof Larsmo. This genome scan resulted in assignment of a new ARCI locus on chromosome 19p13.1.-13.2. Two core haplotypes were identified from a pedigree restricting the region to 6 cM. The ARCIgene remains to be identified within a region containing several possible candidate genes.
This study clarifies in part the molecular mechanisms behind autosomal recessive congenital ichthyosis.TGM 1 mutations and the genotype-phenotype correlation of ARCI and TGM 1 were characterized inFinnish patients. Due to the lack of a clear founder mutation for TGM 1 in Finland, DNA-baseddiagnostics is only partially feasible in Finland. In addition to detection of TGM 1 mutations, welocalized a new locus for an additional ARCI gene on chromosome 19p13.1.-13.2 and provided astepping stone for the cloning of a new ARCI gene. Further studies on the molecular mechanisms ofthis rare disease are needed to clarify its basic pathogenic mechanisms. These studies may offer newunderstanding of the molecules essential for the normal function of the skin.

52
ACKNOWLEDGEMENTS
This study was carried out during the years 1995 to 2000 in the Department of Clinical Chemistry,University of Helsinki, and the Laboratory of Molecular Genetics of the Helsinki University CentralHospital. I want to sincerely thank the Chairs of the Department of Clinical Chemistry: ProfessorsHerman Adlercreutz, Matti Härkönen, and Ulf-Håkan Stenman and the Chairs of the LaboratoryDepartment of the Helsinki University Central Hospital: Professors Raimo Tenhunen and LasseViinikka, for their supportive attitude towards young scientists and for creating facilities and aninfrastructure making this study possible.
I wish to express my deepest gratitude to the ARCI patients and families for participating in this study.
I am most indebted to my supervisor Professor Aarno Palotie for guiding me in my first steps into theworld of science. I was easily persuaded to begin work in Aarno’s lab with the sparkling wine we allshared at my first laboratory meeting… Aarno’s positive attitude towards life and people has created aspecial warm atmosphere in our laboratory, where work has been as enjoyable as it can be. I want alsoto thank him for the opportunity to work quite independently. These years in Aarno’s lab has been avery educating and valuable experience for me.
This work would never have been possible without Docent Kirsti-Maria Niemi. Her enthusiasm aboutichthyotic diseases and electron microscopy is something to admire. Although retired, she was alwayswilling to help and find time to comment on our study and give advice.
I owe my deepest gratitude to our collaborators in the ichthyosis team, comprising of Professor JuhaKere, Docent Ulpu Saarialho-Kere, and Docent Jaakko Ignatius. Juha’s wide knowledge of moleculargenetics and sharp ideas have been of great value in our research. I really appreciate Ulpu’s exactguidance in our clinical and ultrastructural co-work. Jaakko is thanked for thorough genealogical workwith our ichthyosis families. I shall miss the innovative, but yet relaxed meetings of our team in Juha’soffice.
I am most grateful to Professor Leena Peltonen (Palotie) for the time and expertise she has given to thisproject. I also wish to express my gratitude to Dr. Iiris Hovatta for advice in linkage analysis and thefruitful and cheerful collaboration. All the staff of the National Public Health Institute receives thanksfor both help and pleasant company.
I owe special thanks to Professor Vivien Yee for collaboration with FXIII crystal structure andunforgettable moments on Cheju Island, Korea.
I wish to acknowledge Professor Leena Pulkkinen from Thomas Jefferson University and ProfessorAngela Christiano for thorough pre-examination of my thesis. Dr. Carol Norris is thanked for editingthe language of the thesis over parmesan-popcorns.
I have been privileged to work with such “associate supervisors” as Maija Wessman and Arto Orpana.Maija’s positive energy for searching for migraine genes has been reflected in our ichthyosis work aswell, and I have enjoyed the fruits of her labor quite often, remaining indebted to her. Arto’s

53
methodological knowledge was more than welcome to our laboratory and touchdown PCR methodsaved me many working hours, for which I am grateful.
I am most grateful to all those wonderful characters and professionals in the lab with whome I have hadthe opportunity to share my everyday working life. Most of all I want to thank the “transglutaminasepersons,” Hanna Mikkola and Ulla Wartiovaara. Hanna was my first teacher in the laboratory, whichexplains why I never learned to wear a lab coat or to respect certain laboratory rules. She was theperson whose energy and ideas really started the ichthyosis project, and I am thankful for her of supportduring all times of the project. The friendship of Ulla Wartiovaara has been one of the most valuablethings during these years. What would the lunch hours be without your cheerful company! The three ofus really spent some unforgettable moments (mutation parties, fondue parties…) together. Jaakko“Jasu” Kaukonen is thanked for “free entertaining performances” during the working day. I also want tothank Jarkko Ihalainen, Mervi Heiskanen, Maris Laan, Nina Horelli-Kuitunen, Jakob Stenman, and PiaMarttila for collegiality and support during these years. Riitta Sallinen and Mari Kaunisto are thankedfor all kinds of help given to me and happy moments in the basement lab and in cafeteria during the lastyear. I would have love to spend a longer period with you. I also wish to thank Laura Oksanen, KirsiPiippo, Heidi Fodstad, and Päivi Laitinen and the other members of Kimmo Kontula’s group forcompany and advice during these years in the basement lab.
Good technical assistance is invaluable in research work. I want to express my gratitude to AgnetaBlomqvist-Kaksonen, Mervi Eeva, Elina Honkavaara, Sinikka Laine, Katariina Melasniemi, MarkoOts, Maritta Putkiranta, Tuula Salmivaara, and especially Eija Hämäläinen for all kinds of help in mywork and for taking care of those endless everyday routines in the lab.
I want to thank all my friends outside the lab for enjoyable moments together. The old friends fromhigh school and their partners are thanked for unforgettable parties in nearly every New Year, Vappu,and Midsummer. I want to thank our riding group in Inkoo, most importantly Hanna, and TiinaAsikainen for relaxing riding experiences. The “Finnish ABB community” in Switzerland deservesspecial thanks for taking care of so many practical things concerning our moving to Switzerland duringthe last winter. I also want to thank Panu’s family for all the numerous nice moments together.
I owe special thanks to my parents for both material and spiritual support during this project. It wouldhave been impossible for me to cope without the laptop…I also enjoyed of your generous hospitalityduring the last spring (my contribution to housekeeping was absolutely zero), which made our oddfamily arrangements possible. How can I ever thank you enough? I also want to thank my dear brotherPekka and sister Päivi, and I wish enjoyable moments with genetics for Päivi, who is starting her careeras a young scientist…
Finally, I owe my deepest thanks to my husband Panu, whose love and support have been essential forme in so many ways. Luckily, you are talented in housekeeping as well as with computers . . . . It hasbeen of a relief to relax with somebody with a common interest in sports, cooking, outdoor life, diving,and traveling. You have not left me much time to think of work during my leisure hours. I wish that thedays of “two homes” would finally be over, and we could be a “real family” again. I am truly lookingforward to the time with you during the next year in Switzerland.

54
This work has been financially supported by the Helsinki Biomedical Graduate School, the SigridJuselius Foundation, the Ulla Hjelt Fund of the Foundation for Pediatric Research, the FarmosResearch Foundation, the Finnish Medical Society Duodecim, the Instrumentarium ResearchFoundation, and the Research Institute of Helsinki University Central Hospital.
In the (for once silent) basement lab of Meilahti Hospital,
01:15 a.m., 7 August, 2000,

55
REFERENCES
Aaronson SA, Bottaro DP, Miki T, Ron D, Finch PW, Fleming TP, Ahn J, et al (1991) Keratinocyte growth factor. Afibroblast growth factor family member with unusual target cell specificity. Ann NY Acad Sci 638:62-77
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, et al (2000) The genome sequenceof Drosophila melanogaster [Review]. Science 287:2185-2195
Aeschlimann D, Koeller MK, Allen-Hoffmann BL, Mosher DF (1998) Isolation of a cDNA encoding a novel member of thetransglutaminase gene family from human keratinocytes. Detection and identification of transglutaminase geneproducts based on reverse transcription-polymerase chain reaction with degenerate primers. J Biol Chem 273:3452-60
Aeschlimann D, Paulsson M (1994) Transglutaminases: protein cross-linking enzymes in tissues and body fluids. ThrombHaemost 71:402-15
Akiyama M, Smith LT, Yoneda K, Holbrook KA, Hohl D, Shimizu H (1999) Periderm cells form cornified cell envelope intheir regression process during human epidermal development. J Invest Dermatol 112:903-909
Anton-Lamprecht (1992) The Skin. In: Papadimitriou JM, Henderson DW, Spagnolo DV (eds) Diagnostic ultrastucture ofnon-neoplastic diseases. 1th edn Churchill Livingstone, Edinburgh, pp 460-468
Arnold M-L, Anton-Lamprecht I (1988) Ichthyosis Congenita type III. Arch Dermatol Res 280:268-278Ashworth LK, Batzer MA, Brandriff B, Branscomb E, de Jong P, Garcia E, Garnes JA, et al (1995) An integrated metric
physical map of human chromosome 19. Nat Genet 11:422-7Asselineau D, Dale BA, Bernard BA (1990) Filaggrin production by cultured human epidermal keratinocytes and its
regulation by retinoic acid. Differentiation 45:221-9Bale SJ, Russell LJ, Lee ML, Compton JG, Digiovanna JJ (1996) Congenital recessive ichthyosis unlinked to loci for
epidermal transglutaminases. J Invest Dermatol 107:808-811Ballabio A, Parenti G, Carrozzo R, Coppa G, Felici L, Migliori V, Silengo M, et al (1988) X/Y translocation in a family
with X-linked ichthyosis, chondrodysplasia punctata, and mental retardation: DNA analysis reveals deletion of thesteroid sulphatase gene and translocation of its Y pseudogene. Clin Genet 34:31-7
Ben Hamida C, Doerflinger N, Belal S, Linder C, Reutenauer L, Dib C, Gyapay G, et al (1993) Localization of Friedreichataxia phenotype with selective vitamin E deficiency to chromosome 8q by homozygosity mapping [see comments].Nat Genet 5:195-200
Bichakjian CK, Nair RP, Wu WW, Goldberg S, Elder JT (1998) Prenatal exclusion of lamellar ichthyosis based onidentification of two new mutations in the transglutaminase 1 gene. J Invest Dermatol 110:179-82
Bluefarb S (1955) Cutaneous manifestations of multiple myeloma. Arch Dermatol Syphilol 72:506-22Borradori L, Sonnenberg A (1996) Hemidesmosomes: roles in adhesion, signaling and human diseases. Curr Op Cell Biol
8:647-56Botta E, Nardo T, Broughton BC, Marinoni S, Lehmann AR, Stefanini M (1998) Analysis of mutations in the XPD gene in
Italian patients with trichothiodystrophy: site of mutation correlates with repair deficiency, but gene dosage appearsto determine clinical severity. Am J Hum Genet 63:1036-1048
Bradway SD, Bergey EJ, Scannapieco FA, Ramasubbu N, Zawacki S, Levine MJ (1992) Formation of salivary-mucosalpellicle: the role of transglutaminase. Biochem J 284:557-64
Brookes AJ (1999) The essence of SNPs. Gene 234:177-86Brunger AT (1992) X-PLOR, A system for X-ray crystallography and NMR. Yale University, New HavenCandi E, Melino G, Lahm A, Ceci R, Rossi A, Kim IG, Ciani B, et al (1998) Transglutaminase 1 mutations in lamellar
ichthyosis. Loss of activity due to failure of activation by proteolytic processing. J Biol Chem 273:13693-702Candi E, Melino G, Mei G, Tarcsa E, Chung SI, Marekov LN, Steinert PM (1995) Biochemical, structural, and
transglutaminase substrate properties of human loricrin, the major epidermal cornified cell envelope protein. J BiolChem 270:26382-26390
Candi E, Tarcsa E, Idler WW, Kartasova T, Marekov LN, Steinert PM (1999) Transglutaminase cross-linking properties ofthe small proline-rich 1 family of cornified cell envelope proteins - Integration with loricrin. J Biol Chem274:7226-7237
The C.elegans sequencing consortium (1998) Genome Sequence of the Nematode C. elegans: A Platform for InvestigatingBiology. Science 282:2012-2018
Chaib H, Kaplan J, Gerber S, Vincent C, Ayadi H, Slim R, Munnich A, et al (1997) A newly identified locus for Ushersyndrome type I, USH1E, maps to chromosome 21q21. Hum Mol Genet 6:27-31

56
Chang SK, Chung SI (1986) Cellular transglutaminase. The particulate-associated transglutaminase from chondrosarcomaand liver: partial purification and characterization. J Biol Chem 261:8112-21
Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, Bonafe J-L, et al (2000) Mutations in SPINK5,encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet 25:141-142
Cheng J, Syder AJ, Yu QC, Letai A, Paller AS, Fuchs E (1992) The genetic basis of epidermolytic hyperkeratosis: adisorder of differentiation-specific epidermal keratin genes. Cell 70:811-9
Chipev CC, Korge BP, Markova N, Bale SJ, DiGiovanna JJ, Compton JG, Steinert PM (1992) A leucine----proline mutationin the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell 70:821-8
Choate KA, Khavari PA (1997) Sustainability of keratinocyte gene transfer and cell survival in vivo. Hum Gene Ther 8:895-901
Choate KA, Kinsella TM, Williams ML, Nolan GP, Khavari PA (1996) Transglutaminase 1 delivery to lamellar ichthyosiskeratinocytes. Hum Gene Ther 7:2247-53
Choate KA, Medalie DA, Morgan JR, Khavari PA (1996) Corrective gene transfer in the human skin disorder lamellarichthyosis. Nature Med 2:1263-7
Choate KA, Williams ML, Khavari PA (1998) Abnormal Transglutaminase 1 Expression Pattern in a Subset of Patients withErythrodermic Autosomal Recessive Ichthyosis. J Invest Dermatol 110:8-12
Cockayne E (1933) Inherited abnormalities of the skin and its appendages. Oxford University Press, LondonCoin F, Marinoni J-C, Rodolfo C, Fribourg S, Pedrini AM, Egly J-M (1998) Mutations in the XPD helicase gene result in
XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat Genet. 20:184-188
Collins A, Lonjou C, Morton NE (1999) Genetic epidemiology of single-nucleotide polymorphisms. Proc Natl Acad SciUSA 96:15173-7
Collins F, Galas D (1993) A new five-year plan for the U.S. Human Genome Project [see comments]. Science 262:43-6Collins FS (1995) Positional cloning moves from perditional to traditional [published erratum appears in Nat Genet 1995
Sep;11(1):104]. Nat Genet 9:347-50Collins FS, Patrinos A, Jordan E, Chakravarti A, Gesteland R, Walters L (1998) New goals for the U.S. Human Genome
Project: 1998-2003. Science 282:682-9Compton JG, DiGiovanna JJ, Bailey R, Austin F, Fleckman P, Bale SJ (1998) In pursuit of the molecular basis of ichthyosis
vulgaris. J Invest Dermato(Suppl) 110:619Cooper MF, Wilson PD, Hartop PJ, Shuster S (1980) Acquired ichthyosis and impaired dermal lipogenesis in Hodgkin's
disease. Brit J Dermatol 102:689-93Cottingham Jr RW, Idury RM, Schäffer AA (1993) Faster Sequential Genetic Linkage Computations. Am J Hum Genet
53:252-263Crish JF, Howard JM, Zaim TM, Murthy S, Eckert RL (1993) Tissue-specific and differentiation-appropriate expression of
the human involucrin gene in transgenic mice: an abnormal epidermal phenotype. Differentiation 53:191-200de la Chapelle A (1993) Disease gene mapping in isolated human populations: the example of Finland. J Med Genet 30:857-
865De Laurenzi V, Rogers GR, Hamrock DJ, Marekov LN, Steinert PM, Compton JG, Markova N, et al (1996) Sjögren-
Larsson syndrome is caused by mutations in the fatty aldehyde dehydrogenase gene. Nat Genet 12:52-7Del Duca S, Bregoli AM, Bergamini C, Serafini-Fracassini D (1997) Sex Plant Reprod 10:89-95Del Duca S, Tidu V, Bassi D, Serafini-Fracassini D, Esposito C (1994) Planta 193:283-289Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, et al (1996) A comprehensive genetic map of the
human genome based on 5264 microsatellites. Nature 380:152-154Diriong S, Lory P, Williams ME, Ellis SB, Harpold MM, Taviaux S (1995) Chromosomal localization of the Human Genes
for α1A, α1B,and α1C Voltage-Dependent Ca2+ Channel Subunits. Genomics 30:605-609
Djupesland G, Flottorp G, Refsum S (1983) Phytanic acid storage disease: hearing maintained after 15 years of dietarytreatment. Neurology 33:237-40
Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (1991) Touchdown PCR to circumvent spurious priming duringgene amplification. Nucl Acids Res 19:4008
Donis-Keller H, Green P, Helms C, Cartinhour S, Weiffenbach B, Stephens K, Keith TP, et al (1987) A genetic linkage mapof the human genome. Cell 51:319-37
Dubovsky J, Sheffield VC, Duyk GM, Weber JL (1995) Sets of short tandem repeat polymorphism for efficient linkagescreening of the human geneme. Hum Mol Genet 4:449-452

57
Dykes PJ, Marks R (1977) Acquired ichthyosis: multiple causes for an acquired generalized distrubance in desquamation. BrJ Dermatol 97:327-34
Eckert RL, Crish JF, Robinson NA (1997) The epidermal keratinocyte as a model for the study of gene regulation and celldifferentiation. Physiol Rev 77:397-424
Eckert RL, Yaffe MB, Crish JF, Murthy S, Rorke EA, Welter JF (1993) Involucrin--structure and role in envelope assembly.J Invest Dermatol 100:613-7
Elias PM, Williams ML, Maloney ME, Bonifas JA, Brown BE, Grayson S, Epstein EH, Jr. (1984) Stratum corneum lipids indisorders of cornification. Steroid sulfatase and cholesterol sulfate in normal desquamation and the pathogenesis ofrecessive X-linked ichthyosis. J Clin Invest 74:1414-21
Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995) An exitatory amino-acid transporter withproperties of a ligand-gated chloride channel. Nature 375:599-603
Fartasch M, Diepgen TL, Hornstein OP (1989) Atopic dermatitis--ichthyosis vulgaris--hyperlinear palms--an ultrastructuralstudy. Dermatologica 178:202-5
Feind-Koopmans AG, Lucker GP, van de Kerkhof PC (1996) Acquired ichthyosiform erythroderma and sarcoidosis. J AmAcad Dermatol 35:826-8
Fischer J, Faure A, Bouadjar B, Blanchet-Bardon C, Karaduman A, Thomas I, Emre S, et al (2000) Two new loci forAutosomal Recessive Ichthyosis on chromosomes 3p21 and 19p12-q12 and evidence for further geneticheterogeneity. Am J Hum Genet 66:904-913
Fisher GJ, Esmann J, Griffiths CE, Talwar HS, Duell EA, Hammerberg C, Elder JT, et al (1991) Cellular, immunologic andbiochemical characterization of topical retinoic acid-treated human skin [published erratum appears in J InvestDermatol 1991 Jun;96(6):814]. J Invest Dermatol 96:699-707
Fisher GJ, Voorhees JJ (1996) Molecular mechanisms of retinoid actions in skin. FASEB J 10:1002-13Flint GL, Flam M, Soter NA (1975) Acquired ichthyosis. A sign of nonlymphoproliferative malignant disorders. Arch
Dermatol 111:1446-7Fuchs E (1993) Epidermal differentiation and keratin gene expression. J Cell Sci (Suppl) 17:197-208Fuchs E, Green H (1981) Regulation of terminal differentiation of cultured human keratinocytes by vitamin A. Cell 25:617-
25Gånemo A, Vahlquist A (1997) Lamellar ichthyosis is markedly improved by a novel combination of emollients [letter]. Br
J Dermatol 137:1017-8Gibberd FB, Billimoria JD, Page NG, Retsas S (1979) Heredopathia atactica polyneuritiformis (refsum's disease) treated by
diet and plasma-exchange. Lancet 1:575-8Gibbs S, Fijneman R, Wiegant J, van Kessel AG, van De Putte P, Backendorf C (1993) Molecular characterization and
evolution of the SPRR family of keratinocyte differentiation markers encoding small proline-rich proteins.Genomics 16:630-7
Greenberg CS, Birckbichler PJ, Rice RH (1991) Transglutaminases: multifunctional cross-linking enzymes that stabilizetissues. FASEB J 5:3071-3077
Griffiths CE, Rosenthal DS, Reddy AP, Elder JT, Astrom A, Leach K, Wang TS, et al (1992) Short-term retinoic acidtreatment increases in vivo, but decreases in vitro, epidermal transglutaminase-K enzyme activity andimmunoreactivity. J Invest Dermatol 99:283-8
Griffiths WAD, Judge MR, Leigh IM (1998) Disorders of Keratinization. In: R.H. C, Burton JL, Burns DA, Breathnach SM(eds) Textbook of Dermatology. Blackwell Science, Oxford, pp 1501-04
Gyapay G, Morissette J, Vignal A, Dib C, Fizames C, Millasseau P, Marc S, et al (1994) The 1993-94 Genethon humangenetic linkage map [see comments]. Nat Genet 7:246-339
Haftek M, Cambazard F, Dhouailly D, Reano A, Simon M, Lachaux A, Serre G, et al (1996) A longitudinal study of aharlequin infant presenting clinically as non-bullous congenital ichthyosiform erythroderma. Br J Dermatol135:448-453
Hall PA, Watt FM (1989) Stem cells: the generation and maintenance of cellular diversity. Development 106:619-33Haravuori H, Mäkelä-Bengs P, Udd B, Partanen J, Pulkkinen L, Somer H, Peltonen L (1998) Assignment of the tibial
muscular dystrophy locus to chromosome 2q31. Am J Hum Genet 62:620-626Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, Toyoda A, et al (2000) The DNA sequence of human
chromosome 21. Nature 405:311-319Hennies HC, Küster W, Wiebe V, Krebsovà A, Reis A (1998) Genotype/Phenotype correlation in autosomal recessive
lamellar ichthyosis. Am J Hum Genet 62:1052-1061

58
Hennies HC, Raghunath M, Wiebe V, Vogel M, Velten F, Traupe H, Reis A (1998) Genetic and immunohistochemicaldetection of mutations inactivating the keratinocyte transglutaminase in patients with lamellar ichthyosis. HumGenet 102:314-8
Hiiragi T, Sasaki H, Nagafuchi A, Sabe H, Shen SC, Matsuki M, Yamanishi K, et al (1999) Transglutaminase type 1 and itscross-linking activity are concentrated at adherens junctions in simple epithelial cells. J Biol Chem 274:34148-54
Hmani M, Ghorbel A, Boulila-Elgaied A, Ben Zina Z, Kammoun W, Drira M, Chaabouni M, et al (1999) A novel locus forUsher syndrome type II, USH2B, maps to chromosome 3 at p23-24.2. Eur J Hum Genet 7:363-7
Hohl D, Aeschlimann D, Huber M (1997) In vitro and rapid in situ transglutaminase assays for concenital ichthyoses - acomparative study. J Invest Dermatol 110:268-271
Hohl D, de Viragh PA, Amiguet-Barras F, Gibbs S, Backendorf C, Huber M (1995) The Small Proline-Rich Proteinsconstitute a multigene family of differentially regulated cornified cell envelope precursor proteins. J InvestDermatol 104:902-909
Hohl D, Huber M, Frenk E (1993) Analysis of the cornified cell envelope in lamellar ichthyosis. Arch Dermatol 129:618-624
Hohl D, Lichti U, Breitkreutz D, Steinert PM, Roop DR (1991) Transcription of the human loricrin gene in vitro is inducedby calcium and cell density and suppressed by retinoic acid. J Invest Dermatol 96:414-8
Hohl D, Mehrel T, Lichti U, Turner ML, Roop DR, Steinert PM (1991) Characterization of human loricrin. Structure andfunction of a new class of epidermal cell envelope proteins. J Biol Chem 266:6626-36
Hu Z, Bonifas JM, Beech J, Bench G, Shigihara T, Ogawa H, Ikeda S, et al (2000) Mutations in ATP2C1, encoding acalcium pump, cause Hailey-Hailey disease. Nat Genet 24:61-5
Huber M, Rettler I, Bernasconi K, Frenk E, Lavrijsen SPM, Ponec M, Bon A, et al (1995) Mutations of keratinocytetransglutaminase in lamellar ichthyosis. Science 267:525-528
Huber M, Rettler I, Bernasconi K, Wyss M, Hohl D (1995) Lamellar ichthyosis is genetically heterogeneous - cases withnormal keratinocyte transglutaminase. J Invest Dermatol 105:653-654
Huber M, Yee VC, Burri N, Vikerfors E, Lavrijsen APM, Paller AS, Hohl D (1997) Consequences of seven novel mutationson the expression and structure of keratinocyte transglutaminase. J Biol Chem 272:21018-21026
Hudson TJ, Stein LD, Gerety SS, Ma J, Castle AB, Silva J, Slonim DK, et al (1995) An STS-based map of the humangenome [see comments]. Science 270:1945-54
Ishida-Yamamoto A, McGrath JA, Judge MR, Leigh IM, Lane EB, Eady RA (1992) Selective involvement of keratins K1and K10 in the cytoskeletal abnormality of epidermolytic hyperkeratosis (bullous congenital ichthyosiformerythroderma). J Invest Dermatol 99:19-26
Ishida-Yamamoto A, McGrath JA, Lam H, Iizuka H, Friedman RA, Christiano AM (1997) The molecular pathology ofprogressive symmetric erythrokeratoderma: a frameshift mutation in the loricrin gene and perturbations in thecornified cell envelope. Am J Hum Genet 61:581-9
Ishida-Yamamoto A, Tanaka H, Nakane H, Takahashi H, Hashimoto Y, Iizuka H (1999) Programmed cell death in normalepidermis and loricrin keratoderma. Multiple functions of profilaggrin in keratinization. J Invest Dermatol. SympProc 4:145-9
Jagell S, Gustavson KH, Holmgren G (1981) Sjögren-Larsson syndrome in Sweden. A clinical, genetic and epidemiologicalstudy. Clin Genet 19:233-56
Jalanko A, Manninen T, Peltonen L (1995) Deletion of the C-terminal end of aspartylglucosaminidase resulting in alysosomal accumulation disease: evidence for a unique genomic rearrangement. Hum Mol Genet :435-441
Jansen GA, Ofman R, Ferdinandusse S, Ijlst L, Muijsers AO, Skjeldal OH, Stokke O, et al (1997) Refsum disease is causedby mutations in the phytanoyl-CoA hydroxylase gene. Nat Genet 17:190-3
Jarnik M, Simon MN, Steven AC (1998) Cornified cell envelope assembly: a model based on electron microscopicdeterminations of thickness and projected density. J Cell Sci 111:1051-60
Jeffreys AJ, Wilson V, Thein SL (1985) Hypervariable 'minisatellite' regions in human DNA. Nature 314:67-73Jetten AM (1990) Multi-stage program of differentiation in human epidermal keratinocytes: regulation by retinoids. J Invest
Dermatol 95:44S-46SJones TA, Zou J-Y, Cowan SW, Kjelgaard M (1991) Improved methods for building models in electron density maps and
location of errors in these models. Acta Crystallogr A47:110-119Jurgensen K, Aeschlimann D, Cavin V, Genge M, Hunziker EB (1997) A new biological glue for cartilage-cartilage
interfaces: tissue transglutaminase. J Bone Joint Surg Am 79:185-193Kaplan J, Gerber S, Bonneau D, Rozet JM, Delrieu O, Briard ML, Dollfus H, et al (1992) A gene for Usher syndrome type I
(USH1A) maps to chromosome 14q. Genomics 14:979-87

59
Kartasova T, van de Putte P (1988) Isolation, characterization, and UV-stimulated expression of two families of genesencoding polypeptides of related structure in human epidermal keratinocytes. Mol Cell Biol 8:2195-203
Kartasova T, van Muijen GN, van Pelt-Heerschap H, van de Putte P (1988) Novel protein in human epidermalkeratinocytes: regulation of expression during differentiation. Mol Cell Biol 8:2204-10
Kim I-G, McBride W, Wang M, Kim S-Y, Idler WW, Steinert PM (1992) Structure and organization of the humantransglutaminase 1 gene. J Biol Chem 267:7710-7717
Kim SY, Grant P, Lee JH, Pant HC, Steinert PM (1999) Differential expression of multiple transglutaminases in humanbrain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer's disease. J Biol Chem274:30715-21
Kim S-Y, Chung S-I, Steinert PM (1995) Highly active soluble processed forms of the Transglutaminase 1 Enzyme inepidermal keratinocytes. J Biol Chem 270:18026-18035
Kimberling WJ, Moller CG, Davenport S, Priluck IA, Beighton PH, Greenberg J, Reardon W, et al (1992) Linkage of Ushersyndrome type I gene (USH1B) to the long arm of chromosome 11. Genomics 14:988-94
Kimberling WJ, Weston MD, Moller C, Davenport SL, Shugart YY, Priluck IA, Martini A, et al (1990) Localization ofUsher syndrome type II to chromosome 1q. Genomics 7:245-9
Korge BP, Ishida-Yamamoto A, Punter C, Dopping-Hepenstal PJ, Iizuka H, Stephenson A, Eady RA, et al (1997) Loricrinmutation in Vohwinkel's keratoderma is unique to the variant with ichthyosis. J Invest Dermatol 109:604-10
Kraulis PJ (1991) MOLSCRIPT, a program to produce both detailed and schematic plots of protein structures. J ApplCrystallogr 24:946-950
Kuokkanen K (1969) Ichthyosis vulgaris. A clinical and histopathological study of patients and their close relatives in theautosomal dominant and sex-linked forms of the disease. Acta Derm-Venereol. (Supp)l 62:1-72
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-5Laiho E, Ignatius J, Mikkola H, Yee VC, Teller DC, Niemi KM, Saarialho-Kere U, et al (1997) Transglutaminase 1
mutations in autosomal recessive congenital ichthyosis: private and recurrent mutations in an isolated population.Am J Hum Genet 61:529-538
Lander ES (1987) Homozygosity mapping: A Way to Map Human Recessive Traits with the DNA of Inbred Children.Science 236:1567-1570
Lathrop GM, Lalouel JM (1984) Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet36:460-5
Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad SciUSA 81:3443-3446
Lathrop GM, Lalouel JM, White RL (1986) Construction of human linkage maps: likelihood calculations for multilocuslinkage analysis. Genet Epidemiol 3:39-52
Lawlor F (1988) Progress of a Harlequin Fetus to Nonbullous Ichthyosiform Erythroderma. Pediatrics 82:870-873Lawlor F, Peiris S (1985) Harlequin fetus successfully treated with etretinate. Br J Dermatol 112:585-90Lee SC, Lee JB, Kook JP, Seo JJ, Nam KI, Park SS, Kim YP (1999) Expression of differentiation markers during fetal skin
development in humans: Immunohistochemical studies on the precursor proteins forming the cornified cellenvelope. J Invest Dermatol 112:882-886
Lorand L, Campbell-Wilkes LK, Cooperstein L (1972) A filter paper assay for transamidating enzymes using radioactiveamine substrates. Anal Biochem 50:623-631
Lucker GP, Steijlen PM (1995) Acrokeratosis paraneoplastica (Bazex syndrome) occurring with acquired ichthyosis inHodgkin's disease. Br J Dermatol 133:322-5
Lykkesfeldt G, Hoyer H, Ibsen HH, Brandrup F (1985) Steroid sulphatase deficiency disease. Clin Genet 28:231-7Lykkesfeldt G, Hoyer H, Lykkesfeldt AE, Skakkebaek NE (1983) Steroid sulphatase deficiency associated with testis
cancer. Lancet 2:1456Maestrini E, Monaco AP, McGrath JA, Ishida-Yamamoto A, Camisa C, Hovnanian A, Weeks DE, et al (1996) A molecular
defect in loricrin, the major component of the cornified cell envelope, underlies Vohwinkel's syndrome. Nat Genet13:70-7
Magnaldo T, Bernerd F, Asselineau D, Darmon M (1992) Expression of loricrin is negatively controlled by retinoic acid inhuman epidermis reconstructed in vitro. Differentiation 49:39-46
Marekov LN, Steinert PM (1998) Ceramides are bound to structural proteins of the human foreskin epidermal cornified cellenvelope. J Biol Chem 273:17763-70
Mariniello L, Qin Q, Jessen BA, Rice RH (1995) Keratinocyte transglutaminase promoter analysis - identification of afunctional response element. J Biol Chem 270:31358-31363

60
Marinkovic-Ilsen A, Koppe JG, Jobsis AC, de Groot WP (1978) Enzymatic basis of typical X-linked ichthyosis [letter].Lancet 2:1097
Marvin KW, George MD, Fujimoto W, Saunders NA, Bernacki SH, Jetten AM (1992) Cornifin, a cross-linked envelopeprecursor in keratinocytes that is down-regulated by retinoids. Proc Natl Acad Sci USA 89:11026-30
Matheis G, Whitaker JR (1987) J Food Biochem 11:309-327Matsuki M, Yamashita F, Ishida-Yamamoto A, Yamada K, Kinoshita C, Fushiki S, Ueda E, et al (1998) Defective stratum
corneum and early neonatal death in mice lacking the gene for transglutaminase 1 (keratinocyte transglutaminase).Proc Natl Acad Sci USA 95:1044-9
Maxfield S, Maltman J, Kennovin G, Bowgen C, Raxworthy M, Wood E, Skerry T, et al (1999) Switching of keratinocytephenotype by a novel glutamate signalling pathway analogous to synaptic transmission in the central nervoussystem. J Invest Dermatol 112:563, A244
McCauliffe DP, Lux FA, Lieu T-S, Sanz I, Hanke J, Newkirk MM, Bachinski LL, et al (1990) Molecular cloning,expression, and chromosome 19 localization of a human Ro/SS-A autoantigen. J Clin Invest 85:1379-1391
McLean WH, Eady RA, Dopping-Hepenstal PJ, McMillan JR, Leigh IM, Navsaria HA, Higgins C, et al (1994) Mutations inthe rod 1A domain of keratins 1 and 10 in bullous congenital ichthyosiform erythroderma (BCIE). J InvestDermatol 102:24-30
Mehrel T, Hohl D, Rothnagel JA, Longley MA, Bundman D, Cheng C, Lichti U, et al (1990) Identification of a MajorKeratinocyte Cell Envelope Protein, Loricrin. Cell 61:1103-1112
Menon IA, Hoberman HF (1969) Dermatological writings of ancient India. Med His 13:387-392Mewes HW, Albermann K, Bahr M, Frishman D, Gleissner A, Hani J, Heumann K, et al (1997) Overview of the yeast
genome [published erratum appears in Nature 1997 Jun 12;387(6634):737]. Nature 387:7-65Miesfeld R, Krystal M, Arnheim N (1981) A member of a new repeated sequence family which is conserved throughout
eucaryotic evolution is found between the human delta and beta globin genes. Nucl Acids Res 9:5931-47Mihalik SJ, Morrell JC, Kim D, Sacksteder KA, Watkins PA, Gould SJ (1997) Identification of PAHX, a Refsum disease
gene. Nat Genet 17:185-9Mikkola H, Palotie A (1996) Gene defects in congenital factor XIII deficiency. Semin Thromb Hemost 22:393-8Mischke D, Korge BP, Marenholz I, Volz A, Ziegler A (1996) Genes encoding structural proteins of epidermal cornification
and S100 calcium-binding proteins form a gene complex ("epidermal differentiation complex") on humanchromosome 1q21. J Invest Dermatol 106:989-92
Mohrenweiser H, Olsen A, Archibald A, Beattie C, Burmeister M, Lamerdin J, Lennon G, et al (1996) Report of the thirdinternational workshop on human chromosome 19 mapping 1996. Cytogenet Cell Genet 74:161-186
Morton NE (1955) Sequential test for the detection of linkage. Am J Hum Genet 62:690-697Murray JC, Buetow KH, Weber JL, Ludwigsen S, Scherpbier-Heddema T, Manion F, Quillen J, et al (1994) A
comprehensive human linkage map with centimorgan density. Cooperative Human Linkage Center (CHLC).Science 265:2049-54
Nakamura Y, Leppert M, O'Connell P, Wolff R, Holm T, Culver M, Martin C, et al (1987) Variable number of tandemrepeat (VNTR) markers for human gene mapping. Science 235:1616-22
Nemes Z, Demeny M, Marekov LN, Fesus L, Steinert PM (2000) Cholesterol 3-sulfate interferes with cornified envelopeassembly by diverting transglutaminase 1 activity from the formation of cross-links and esters to the hydrolysis ofglutamine. J Biol Chem 275:2636-46
Nemes Z, Marekov LN, Fesus L, Steinert PM (1999) A novel function for transglutaminase 1: attachment of long-chainomega-hydroxyceramides to involucrin by ester bond formation. Proc Natl Acad Sci USA 96:8402-7
Nemes Z, Marekov LN, Steinert PM (1999) Involucrin cross-linking by transglutaminase 1 - Binding to membranes directsresidue specificity. J Biol Chem 274:11013-11021
Nemes Z, Steinert PM (1999) Bricks and mortar of the epidermal barrier. Experim Mol Med 31:5-19Netherton EW (1958) A unique case of trichorrexin nodosa, "Bamboo hairs". Arch Dermatol 78:483-487Nevanlinna HR (1972) The Finnish population structure: A genetic and genealogical study. Hereditas 71:195-236Niemi K-M, Kanerva L, Kuokkanen K (1991) Recessive ichthyosis congenita type II. Arch Dermatol Res 283:211-218Niemi K-M, Kanerva L, Kuokkanen K, Ignatius J (1994) Clinical, light and electron-microscopic features of recessive
congenital ichthyosis type I. Br J Dermatol 130:626-633Niemi K-M, Kanerva L, Wahlgren C-F, Ignatius J (1992) Clinical, light and electron-microscopic features of recessive
ichthyosis congenita type III. Arch Dermatol Res 284:259-265Niemi K-M, Kuokkanen K, Kanerva L, Ignatius J (1993) Recessive ichthyosis congenita type IV. Am J Dermatopathol
15:224-228

61
Nikali K, Suomalainen A, Terwilliger J, Koskinen T, Weissenbach J, Peltonen L (1995) Random search for sharedchromosomal regions in four affected individuals: the assignment of a new hereditary ataxia locus. Am J HumGenet 56:1088-1095
Nirunsuksiri W, Zhang SH, Fleckman P (1998) Reduced stability and bi-allelic, coequal expression of profilaggrin mRNAin keratinocytes cultured from subjects with ichthyosis vulgaris. J Invest Dermatol 110:854-61
Norio R, Nevanlinna HR, Perheentupa J (1973) Hereditary diseases in Finland; rare flora in rare soul. Ann Clin Res 5:109-41
Ophoff RA, Terwindt GM, Vergouve MN, van Eijk R, Oefner PJ, Hoffman SMG, Lamerdin JE, et al (1996) FamilialHemiplegic migrane and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNLA1A4.Cell 87:543-552
Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T (1989) Detection of polymorphisms of human DNA by gelelectrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA 86:2766-70
Ott J (1974) Estimation of the recombination fraction in human pedigrees: efficient computation of the likelihood for humanlinkage studies. Am J Hum Genet 26:588-97
Ott J (1989) Computer-simulation methods in human linkage analysis. Proc Natl Acad Sci USA 86:4175-4178Parmentier L, Blanchet-Bardon C, Nguyen S, Prud'homme J-F, Dubertret, Weissenbach J (1995) Autosomal recessive
lamellar ichthyosis: identification of a new mutation in transglutaminase 1 and evidence for genetic heterogeneity.Hum Mol Genet 4:1391-1395
Parmentier L, Clepet C, BoughdeneStambouli O, Lakhdar H, BlanchetBardon C, Dubertret L, Wunderle E, et al (1999)Lamellar ichthyosis: Further narrowing, physical and expression mapping of the chromosome 2 candidate locus.Eur J Hum Genet 7:77-87
Parmentier L, Lakhdar H, Blanchet-Bardon C, Marchand S, Dubertret L, Weissenbach J (1996) Mapping of a second locusfor lamellar ichthyosis to chromosome 2q33-35. Hum Mol Genet 5:555-559
Paunio T, Kangas H, Kalkkinen N, Haltia M, Palo J, Peltonen L (1994) Towards understanding the pathogenic mechanismin gelsolin-related amyloidosis: in vitro expression reveals an abnormal gelsolin fragment. Hum Mol Genet 3:2223-2229
Peltonen L, Jalanko A, Varilo T (1999) Molecular genetics of the Finnish disease heritage. Hum Mol Genet 8:1913-1923Petit E, Huber M, Rochat A, Bodemer C, Teillac-Hamel D, Muh JP, Revuz J, et al (1997) Three novel point mutations in the
keratinocyte transglutaminase (TGK) gene in lamellar ichthyosis: significance for mutant transcript level, TGKimmunodetection and activity. Eur J Hum Genet 5:218-28
Phillips MA, Stewart BE, Rice RH (1992) Genomic structure of keratinocyte transglutaminase. Recruitment of new exon formodified function. J Biol Chem 267:2282-6
Pigg M, GeddeDahl T, Cox D, Hausser I, AntonLamprecht I, Dahl N (1998) Strong founder effect for a transglutaminase 1gene mutation in lamellar ichthyosis and congenital ichthyosiform erythroderma from Norway. Eur J Human Genet6:589-596
Polakowska RR, Graf BA, Falciano V, LaCelle P (1999) Transcription regulatory elements of the first intron control humantransglutaminase type I gene expression in epidermal keratinocytes. J Cell Biochem 73:355-369
Pollak MR, Chou YH, Cerda JJ, Steinmann B, La Du BN, Seidman JG, Seidman CE (1993) Homozygosity mapping of thegene for alkaptonuria to chromosome 3q2 [see comments]. Nat Genet 5:201-4
Pollit RJ, Jenner FA, Davies M (1968) Sibs with mental and physical retardation and trichorrhexis nodosa with abnormalamino acid composition in the hair. Arch Dis Child 43:211-216
Prasad RS, Pejaver RK, Hassan A, al Dusari S, Wooldridge MA (1994) Management and follow-up of harlequin siblings. BrJ Dermatol 130:650-3
Proia RL, D'Azzo A, Neufield EF (1984) Association of alfa- and beta-subunits during biosythesis of beta-hexosaminidasein cultured fibroblasts. J Biol Chem 259:3350-3354
Raghunath M, Hopfner B, Aeschlimann D, Luthi U, Meuli M, Altermatt S, Gobet R, et al (1996) Cross-linking of thedermo-epidermal junction of skin regenerating from keratinocyte autografts - anchoring fibrils are a target for tissuetransglutaminase. J Clin Invest 98:1174-1184
Refsum S (1946) Heredopathia atactica polyneuritiformis. Acta Psychiatr Scand (Suppl) 38:1-303Rice RH, Green H (1979) Precence in human epidermal cells of a soluble protein precursor of the cross-linked protein. Cell
18:681-694Roberts LJ (1989) Long-term survival of a harlequin fetus. J Am Acad Dermatol 21:335-9Roop D (1995) Defects in the Barrier. Science 267:474-475Rosenberg M, RayChaudhury A, Shows TB, Le Beau MM, Fuchs E (1988) A group of type I keratin genes on human
chromosome 17: characterization and expression. Mol Cell Biol 8:722-36

62
Rossi A, Jang SI, Ceci R, Steinert PM, Markova NG (1998) Effect of AP1 transcription factors on the regulation oftranscription in normal human epidermal keratinocytes. J Invest Dermatol 110:34-40
Rothnagel JA, Traupe H, Wojcik S, Huber M, Hohl D, Pittelkow MR, Saeki H, et al (1994) Mutations in the rod domain ofkeratin 2e in patients with ichthyosis bullosa of Siemens. Nat Genet 7:485-90
Russell LJ, DiGiovanna JJ, Hashem N, Compton JG, Bale SJ (1994) Linkage of Autosomal Recessive Lamellar Ichthyosisto Chromosome 14q. Am J Hum Genet 55:1146-1152
Russell LJ, DiGiovanna JJ, Rogers GR, Steinert PM, Hashem N, Compton JG, Bale SJ (1995) Mutations in the gene fortransglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat Genet 9:279-283
The Sanger Centre and The Genome Sequencing Centre (1998) Toward a complete human genome sequence. Genome Res8:1097-108
Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, et al (1988) Primer-directed enzymaticamplification of DNA with a thermostable DNA polymerase. Science 239:487-91
Sajantila A, Salem A-H, Savolainen P, Bauer K, Gierig C, Pääbo S (1996) Paternal and maternal DNA lineages reveal abottleneck in the founding of the Finnish population. Proc Natl Acad Sci USA 93:12035-12039
Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, Monk S, Smith M, et al (1999) Mutations in ATP2A2,
encoding a Ca2+ pump, cause Darier disease. Nat Genet 21:271-277Sambrook J, Fritsch EF, Maniatis T (1987) Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory, New
YorkSanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA
74:5463-7Sankila EM, Pakarinen L, Kaariainen H, Aittomaki K, Karjalainen S, Sistonen P, de la Chapelle A (1995) Assignment of an
Usher syndrome type III (USH3) gene to chromosome 3q. Hum Mol Genet 4:93-8Saunders NA, Bernacki SH, Vollberg TM, Jetten AM (1993) Regulation of transglutaminase type I expression in squamous
differentiating rabbit tracheal epithelial cells and human epidermal keratinocytes: effects of retinoic acid andphorbol esters. Mol Endocr 7:387-98
Schaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers JHJ, Chambon P, et al (1993) DNA repairhelicase: a component of BTF2 (THFIIH) basic transcription factor. Science 260:58-63
Shapiro LJ, Weiss R, Webster D, France JT (1978) X-linked ichthyosis due to steroid sulphatase deficiency. Lancet :70-72Shevchenko YO, Compton JG, Toro JR, DiGiovanna JJ, Bale SJ (2000) Splice-site mutation in TGM 1 in congenital
recessive ichthyosis in American families: molecular, genetic, genealogic, and clinical studies. Hum Mol GenetSimon M, Green H (1988) The glutamine residues reactive in transglutaminase-catalyzed cross-linking of involucrin. J Biol
Chem 263:18093-8Sjögren T, Larsson T (1957) Oligophrenia in association with congenital ichthyosis and spastic disorders. Acta Psychiatr
Scand 32 (Suppl 113):1-112Smith RJ, Lee EC, Kimberling WJ, Daiger SP, Pelias MZ, Keats BJ, Jay M, et al (1992) Localization of two genes for
Usher syndrome type I to chromosome 11. Genomics 14:995-1002Sneddon I (1955) Aquired ichthyosis in Hodgkin's disease. Br Med J 1:763-4Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker
sharing statistics. Am J Hum Genet 58:1323-1337Stacey A, Schnieke A (1990) SVpoly: a versatile mammalian expression vector. Nucl Acid Res 18:2829Steijlen PM, Van Dooren-Greebe RJ, Van de Kerkhof PC (1994) Acitretin in the treatment of lamellar ichthyosis. Br J
Dermatol 130:211-4Steinert PM (1993) Structure, function, and dynamics of keratin intermediate filaments. J Invest Dermatol 100:729-34Steinert PM (1995) A model for the hierarchial structure of the human epidermal cornified cell envelope. Cell Death
Different 2:33-40Steinert PM, Chung SI, Kim SY (1996) Inactive zymogen and highly active proteolytically processed membrane-bound
forms of the transglutaminase 1 enzyme in human epidermal keratinocytes. Biochem Biophys Res Commun221:101-106
Steinert PM, Kim S-Y, Chung S-Y, Marekov LN (1996) The transglutaminase 1 enzyme is variably acylated by myristateand palmitate during differentiation in epidermal keratinocytes. J Biol Chem 271:26242-26250
Steinert PM, Marekov LN (1999) Initiation of assembly of the cell envelope barrier structure of stratified squamousepithelia. Mol Biol Cell 10:4247-61
Steven AC, Steinert PM (1994) Protein composition of cornified cell envelopes of epidermal keratinocytes. J Cell Sci107:693-700

63
Strachan T, Read AP (1996) Molecular pathology Human Molecular Genetics. BIOS Scientific Publishers, Oxford, UK, pp419
Suga Y, Ogawa H, Bundman D, Roop D (1999) Transgenic mice expressing a mutant form of loricrin exhibit anerythroderma similar to Vohwinkel syndrome. J Invest Dermatol (Suppl) 112:551
Sundberg JP, Boggess D, Hogan ME, Sundberg BA, Rourk MH, Harris B, Johnson K, et al (1997) Harlequin ichthyosis(ichq): a juvenile lethal mouse mutation with ichthyosiform dermatitis. Am J Pathol 151:293-310
Sussman DJ, Milman G (1984) Short-term, high-efficiency expression of transfected DNA. Mol Cell Biol 4:1641-3Sybert VP, Dale BA, Holbrook KA (1985) Ichthyosis vulgaris: identification of a defect in synthesis of filaggrin correlated
with an absence of keratohyaline granules. J Invest Dermatol 84:191-4Syvänen AC, Aalto-Setälä K, Harju L, Kontula K, Söderlund H (1990) A primer-guided nucleotide incorporation assay in
the genotyping of apolipoprotein E. Genomics 8:684-92Syvänen AC, Aalto-Setälä K, Kontula K, Söderlund H (1989) Direct sequencing of affinity-captured amplified human DNA
application to the detection of apolipoprotein E polymorphism. FEBS Lett 258:71-4Takayama K, Danks DM, Salazar EP, Cleaver JE, Weber CA (1997) DNA repair characteristics and mutations in the
ERCC2 DNA repair and transcription gene in a trichothiodystrophy patient. Hum Mutat 9:519-525Takayama K, Salazar EP, Broughton BC (1996) Defects in the DNA repair and transcription gene ERCC2(XPD) in
trichothiodystrophy. Am J Hum Genet 58:263-70Tarcsa E, Candi E, Kartasova T, Idler WW, Marekov LN, Steinert PM (1998) Structural and transglutaminase substrate
properties of the small proline-rich 2 family of cornified cell envelope proteins. J Biol Chem 273:23297-23303Tavakkol A, Elder JT, Griffiths CE, Cooper KD, Talwar H, Fisher GJ, Keane KM, et al (1992) Expression of growth
hormone receptor, insulin-like growth factor 1 (IGF-1) and IGF-1 receptor mRNA and proteins in human skin. JInvest Dermatol 99:343-9
Terwilliger JD, Ott J (1994) Handbook of human genetic linkage. The John Hopkins University Press, BaltimoreTok J, Garzon MC, Cserhalmi-Friedman P, Lam HM, Spitz JL, Christiano AM (1999) Identification of mutations in the
transglutaminase 1 gene in lamellar ichthyosis. Exp Dermatol 8:128-33Traupe H, Happle R (1983) Clinical spectrum of steroid sulfatase deficiency: X-linked recessive ichthyosis, birth
complications and cryptorchidism. Eur J Pediatr 140:19-21Traupe H, Kolde G, Hamm H, Happle R (1986) Ichthyosis bullosa of Siemens: a unique type of epidermolytic
hyperkeratosis. J Am Acad Dermatol 14:1000-5Traupe H, Kolde G, Happle R (1984) Autosomal dominant lamellar ichthyosis: a new skin disorder. Clin Genet 26:457-461Udd B (1992) Limb-girdle type muscular dystrophy in a large family with distal myopathy: homozygous manifestation of a
dominant gene? J Med Genet 29:383-389Vicanová J, Boelsma E, Mommaas AM, Kempenaar JA, Forslind B, Pallon J, Egelrud T, et al (1998) Normalization of
epidermal calcium distribution profile in reconstructed human epidermis is related to improvement of terminaldifferentiation and stratum corneum barrier formation. J Invest Dermatol 111:97-106
Wang M, Kim IG, Steinert PM, McBride OW (1994) Assignment of the human transglutaminase 2 (TGM2) andtransglutaminase 3 (TGM3) genes to chromosome 20q11.2. Genomics 23:721-2
Wayne S, Der Kaloustian VM, Schloss M, Polomeno R, Scott DA, Hejtmancik JF, Sheffield VC, et al (1996) Localizationof the Usher syndrome type ID gene (Ush1D) to chromosome 10. Hum Mol Genet 5:1689-92
Weber JL (1990) Informativeness of human (dC-dA)n.(dG-dT)n polymorphisms. Genomics 7:524-30Weber JL, May PE (1989) Abundant class of human DNA polymorphisms which can be typed using the polymerase chain
reaction. Am J Hum Genet 44:388-96Weber JL, Myers EW (1997) Human whole-genome shotgun sequencing. Genome Res 7:401-9Webster D, France JT, Shapiro LJ, Weiss R (1978) X-linked ichthyosis due to steroid-sulphatase deficiency. Lancet 1:70-2Weeda G, Eveno E, Donker I, Vermeulen W, Chevallier-Lagente O, Taieb A, Stary A, et al (1997) A mutation in the
XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet 60:320-329Weeks DE, Ott J, Lathrop GM (1990) SLINK: a general simulation program for linkage analysis. Am J Hum Genet 62:620-
626Wells RS, Kerr CB (1966) Clinical features of autosomal dominant and sex-linked ichthyosis in an english population. Br
Med J 1:947-950Williams ML, Elias PM (1985) Heterogeneity in autosomal recessive ichthyosis: clinical and biochemical differentiation of
lamellar ichthyosis and non-bullous congenital ichthyosiform erythroderma. Arch Dermatol 121:477-488Williams ML, Feingold KR, Grubauer G, Elias PM (1987) Ichthyosis induced by cholesterol-lowering drugs. Implications
for epidermal cholesterol homeostasis. Arch Dermatol 123:1535-8

64
Yaffe MB, Beegen H, Eckert RL (1992) Biophysical characterization of involucrin reveals a molecule ideally suited tofunction as an intermolecular cross-bridge of the keratinocyte cornified envelope. J Biol Chem 267:12233-8
Yee VC, Pedersen LC, le Trong I, Bishop PD, Stenkamp RE, Teller DC (1994) Three-dimensional structure of atransglutaminase: human blood coagulation factor XIII. Proc Natl Acad Sci USA 91:7296-300
Yen PH, Allen E, Marsh B, Mohandas T, Wang N, Taggart RT, Shapiro LJ (1987) Cloning and expression of steroidsulfatase cDNA and the frequent occurrence of deletions in STS deficiency: implications for X-Y interchange. Cell49:443-54
Yoneda K, Akiyama M, Morita K, Shimizu H, Imamura S, Kim SY (1998) Expression of transglutaminase 1 in human hairfollicles, sebaceous glands and sweat glands. Br J Dermatol 138:37-44
Yoneda K, Steinert PM (1993) Overexpression of human loricrin in transgenic mice produces a normal phenotype. ProcNatl Acad Sci USA 90:10754-8
Yoon SJ, LeBlanc-Straceski J, Ward D, Krauter K, Kucherlapati R (1994) Organization of the human keratin type II genecluster at 12q13. Genomics 24:502-8
Yuspa SH, Harris CC (1974) Altered differentiation of mouse epidermal cells treated with retinyl acetate in vitro. Exp CellRes 86:95-105
Zhu AJ, Haase I, Watt FM (1999) Signaling via beta1 integrins and mitogen-activated protein kinase determines humanepidermal stem cell fate in vitro. Proc Natl Acad Sci USA 96:6728-33
Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, et al (1997) Autosomal dominantcerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium
channel. Nat Genet 15:62-69