decomposition of furan on pd(111)
TRANSCRIPT

ORIGINAL PAPER
Decomposition of Furan on Pd(111)
Ye Xu
Published online: 24 April 2012
� Springer Science+Business Media, LLC 2012
Abstract Periodic density functional theory calculations
(GGA-PBE) have been performed to investigate the
mechanism for the decomposition of furan up to CO for-
mation on the Pd(111) surface. At 1/9 ML coverage, furan
adsorbs with its molecular plane parallel to the surface in
several states with nearly identical adsorption energies of
-1.0 eV. The decomposition of furan begins with the
opening of the ring at the C–O position with an activation
barrier of Ea = 0.82 eV, which yields a C4H4O aldehyde
species that rapidly loses the a H to form C4H3O
(Ea = 0.40 eV). C4H3O further dehydrogenates at the dposition to form C4H2O (Ea = 0.83 eV), before the a–b C–
C bond dissociates (Ea = 1.08 eV) to form CO. Each step
is the lowest-barrier dissociation step in the respective
species. A simple kinetic analysis suggests that furan
decomposition begins at 240–270 K and is mostly com-
plete by 320 K, in close agreement with previous experi-
ments. It is suggested that the C4H2O intermediate delays
the decarbonylation step up to 350 K.
Keywords Furan � Palladium � Decomposition �Kinetics � Hetoregenous catalysis �Density functional theory
1 Introduction
Furan compounds have gained significant attention in recent
years in the effort to convert biomass, a renewable resource,
into fuels and value-added chemicals. For instance,
5-hydroxymethylfurfural has been proposed as a versatile
platform that can be converted into many industrial com-
pounds including additional furan compounds and linear
oxygenates such as levulinic acid [1]. It has also been shown
that furan, furfural, and HMF can be converted to fuel
compounds including butanol [2], methyl-tetrahydrofuran
[3], and large alkanes [1, 4]. 2,5-dimethylfuran has been
proposed as a suitable fuel that has a number of desirable
characteristics compared to ethanol [5]. These furan com-
pounds can be synthesized from sugars, cellulose, hemi-
cellulose, and agricultural and forestry byproducts [5–8].
The conversions of furan compounds involve modifying
or removing side groups, saturating the furan ring, and
opening the furan ring for making linear compounds, all
of which can be accomplished through metal-catalyzed
hydrogenation and hydrogenolysis, among other catalytic
processes. Knowing what catalyst and reaction conditions
favor ring opening versus hydrogenation and vice versa will
contribute to enhancing the versatility of the furan platform,
and is also relevant to the removal of heteroatoms from
heterocyclic compounds, an important step in the processing
of heavy feed [9]. Finite pressure experiments have shown,
for instance, that at 373 K furan hydrogenation on Pd/zir-
conia produces little tetrahydrofuran (THF) at low H2:furan
ratio but solely THF at high H2:furan ratio [10]. Furan
hydrogenation on Pt(111) and (100) produces primarily tet-
rahydrofuran and butanol, and the selectivity for one comes
at the expense of the other [2]. The hydrogenation of furfural,
a singly substituted furan compound, can produce different
distributions of furfuryl alcohol, furan, and ring-opening
products on SiO2-supported Cu, Pd, and Ni catalysts, and the
product selectivity for hydrogenation decreases and that for
decarbonylation and ring opening increases, with increasing
temperature [11].
Y. Xu (&)
Center for Nanophase Materials Sciences, Oak Ridge National
Laboratory, 1 Bethel Valley Road, P.O. Box 2008, MS-6493,
Oak Ridge, TN 37831, USA
e-mail: [email protected]
123
Top Catal (2012) 55:290–299
DOI 10.1007/s11244-012-9797-z

To better understand the surface chemistry of furan
compounds on Pd surfaces, I begin by conducting a
density functional theory (DFT) study of the decomposi-
tion of furan, as the simplest of furan compounds, on
Pd(111) in the low coverage limit. Several surface science
studies of furan on single-crystal Pd(111) surfaces have
been reported in the literature. At sub-saturation coverage
furan lies flat on the surface with the ring above fcc or
hcp threefold hollow site, as determined by scanning
tunneling microscopy (STM) [12], near-edge X-ray
absorption fine structure (NEXAFS), and photoelectron
diffraction (PhD) [13]. The flat geometry is corroborated
by DFT calculations [14]. The molecular desorption of
furan is observed for dosages above 0.25 L and starts at
200–250 K and peaks at up to 360 K [15]. At lower
dosage furan decomposition starts at 230–270 K and is
essentially complete by 320 K as determined by X-ray
photoelectron spectroscopy (XPS), high-resolution elec-
tron energy loss spectroscopy (HREELS), ultraviolet
photoelectron spectroscopy (UPS) [15], PhD [13], and
laser-induced thermal desorption (LITD) [16]. Tempera-
ture programmed desorption (TPD) shows desorption-
limited H2 and CO evolution from the surface starting at
*300 and 450 K respectively, indicating that H and CO
form at these temperatures or lower [15]. LITD identifies
concomitant increase in CO coverage with the disap-
pearance of furan starting at 270 K and the formation of
a small amount of benzene at 350 K [16]. Based on the
formation of CO and the appearance of benzene, the
carbon fragment has been concluded to be C3H3 [13, 15,
16] and studied in detail using NEXAFS, PhD as well as
DFT calculations [17, 18].
While the basic feature of furan decomposition seems
plain enough, subtle questions remain, including how the
process is initiated, and whether the a C–H bond scission
precedes or follows ring opening. As Caldwell et al. have
reasoned, it is not safe even to rule out C–C bond scission
as the first step, and they have pointed out the lack of
information on the activation barrier for the breaking of the
C–O bond in an aromatic ring to be an obstacle for better
understanding the decomposition mechanism of furan on
Pd(111) [19]. The loss of the a H is common in five-
membered heterocycles, including on reactive surfaces
such as Ru(0001), to form a-furyl species [20]. Yet, anal-
ysis of kinetic isotope effects suggests that a C–H bond
scission cannot be the sole rate-determining step [19]. In
this study all the C–H, C–O, and C–C bond scission steps
up to the formation of CO are considered. C–O bond
scission is determined to be the first step in the decompo-
sition process, followed by the loss of up to two H atoms
before decarbonylation occurs to yield CO. Kinetic anal-
ysis based on the DFT results predicts that the decompo-
sition of furan on Pd(111) begins at 240–270 K and is
largely completely 320 K, in close agreement with avail-
able experimental evidence.
2 Methods
Periodic DFT calculations have been performed using the
Vienna Ab initio Simulation Package (VASP) [21, 22] in
the generalized gradient approximation (GGA-PBE [23]).
The core electrons are described by the projector aug-
mented wave (PAW) method [24], and the Kohn–Sham
valence states are expanded in plane wave basis sets up to
400 eV. A first-order Methfessel-Paxton scheme [25] is
used to smear the electronic states with a temperature of
0.1 eV. All total energies are extrapolated back to 0 K. The
equilibrium lattice constant of bulk fcc Pd is calculated to
be 3.953 A, in close agreement with the experimental value
(3.89 A) [26].
The Pd(111) surface is modeled by a four-layer slab
with a (3 9 3) surface unit cell, giving a coverage of 1/9
monolayer (ML) per adsorbate. The top two layers of the
slab are relaxed. Neighboring slabs are separated in the z
direction by a 20 A thick vacuum region. The surface
Brillouin zone is sampled with a 3 9 391 Monkhorst–
Pack k-point grid. Increasing the k-point grid density
to 7 9 791 changes the total energies of surfaces with
adsorbed furan by less than 0.1 eV and so the lower grid
density is used. A (3 9 4) surface unit cell is used to
achieve a high coverage of 1/6 ML, with all the other
parameters being identical to those mentioned above.
The adsorption energy (DE) for an atom or molecule
adsorbed on the Pd surface is calculated as:
DE ¼ EA=Pd þ ZPEA=Pd
� �� EA þ ZPEAð Þ � EPd
where EA/Pd is the total energy of a Pd(111) slab with an
adsorbate; ZPEA/Pd is the zero point energy (ZPE) of the
adsorbate; EA and ZPEA are the total energy and zero-point
energy of the isolated neutral adsorbate in the gas phase,
respectively; and EPd is the energy of the clean Pd(111)
slab. A more negative adsorption energy therefore corre-
sponds to stronger adsorption. Geometry optimization for
adsorbates and the top two layers of the Pd(111) slab is
converged to 0.03 eV/A for each relaxed atomic degree of
freedom.
The transition states (TSs) of elementary steps are cal-
culated using the dimer method [27], with geometry opti-
mization converged to 0.01 eV/A for each relaxed atomic
degree of freedom. Each TS is verified to possess a single
imaginary vibrational frequency that corresponds to the
reaction coordinate of the elementary step and is verified to
converge back to the initial state (IS) when perturbed in
the backward direction. The activation barrier for an ele-
mentary step is defined to be the difference between the
Top Catal (2012) 55:290–299 291
123

energies of the TS and IS: Ea = (ETS ? ZPETS) - (EIS ?
ZPEIS). The reaction energy is defined to be the difference
between the energies of the product or final state (FS) and the
IS: DErxn = (EFS ? ZPEFS) - (EIS ? ZPEIS). If the IS or the
FS involves more than one adsorbate, its energy is calculated
with each adsorbate in their most stable configurations at 1/9
ML coverage.
The ZPE values are listed in the Appendix.
3 Results
3.1 Adsorption of Furan
Furan prefers to adsorb with its molecular plane lying
parallel to the Pd(111) surface so that either the a–b or the
b–b C–C bond is located on the top of a Pd atom, and the
center of the ring is located either over a threefold site or a
bridge site. Thus there are several such adsorption states
available for furan on Pd(111) (see Fig. 1 for illustrations
of some of the adsorption states; their associated adsorption
energies; and the site and C atom labeling). Those states
with the ring located above the fcc or hcp threefold sites
are energetically degenerate, with an adsorption energy of
*-1.0 eV at the coverage of 1/9 ML. The states with the
ring located over the bridge site have adsorption energies
that are *0.4 eV more positive but are also stable local
minima. Furan with its ring located on top of a Pd atom or
perpendicular to the surface is not stable at this coverage.
The TPD study of Ormerod et al. [15] has found that while
furan dosed at\0.25 L on Pd(111) can desorb only as CO
and H2, exposures C0.25 L permit it to desorb molecularly.
The maximum molecular desorption temperature observed
is 360 K at 0.25 L, which suggests that the desorption
barrier for furan is *0.9 eV or greater [28]. LITD exper-
iments have enabled Caldwell et al. to calculate the
desorption barrier to be 0.98 eV [19]. The GGA-PBE
adsorption energy of *-1.0 eV at 1/9 ML is therefore
numerically in line with the experimental findings. Bradley
et al. [14] reported the adsorption energies of furan to be
-1.1 eV in GGA-PW91 and -0.45 eV in GGA-RPBE.
The saturation surface coverage of furan has been esti-
mated at 15 % of a ML [19]. It can be seen in Fig. 1 that
furan, whether adsorbed in the more stable fcc/hcp states or
the less stable br states, occupies 6 Pd atoms. The maxi-
mum coverage in UHV conditions is therefore limited to
*1/6 ML without physical overlap or too small distances
between neighboring furan molecules. I have calculated the
differential adsorption energy of furan in the hcp/bbPd
state as the coverage increases from 1/9 ML to 1/6 ML to
be -0.11 eV, which suggests that the saturation coverage
is indeed around 1/6 ML.
In the gas phase, the two a–b C–C bonds in furan are
1.36 A, the b–b C–C bond is 1.43 A, and the two C–O bonds
are 1.37 A. In comparison to gas-phase ethanol, acetalde-
hyde, benzene, and ethylene molecules (Table 1), it can be
seen that C–C bonds in furan are intermediate between
single and double bonds, and the C–O bonds are
αα α
β β
f
h br
hcp/ββPd-1.00 eV
fcc/ββPd-0.99 eV
br/ββPd-0.61 eV
hcp/αβPd-0.98 eV fcc/αβPd
-0.98 eV
α1β2
β3α4 t
α α
β β
f
h br
hcp/ββPd-1.00 eV
fcc/ββPd-0.99 eV
br/ββPd-0.61 eV
hcp/αβPd-0.98 eV fcc/αβPd
-0.98 eV
α1β2
β3α4 t
Fig. 1 Several stable high-symmetry states for furan adsorbed on
Pd(111). The adsorption energy for each state is as labeled. The
labeling of the high-symmetry surface sites on Pd(111) (t top, brbridge, h hcp, f fcc) and the different C atoms in furan (a1, b2, b3, a4)
is indicated. Small black, red, white; and large white spheresrepresent C, O, H, and Pd atoms, respectively. Molecules are placed
close to each other for illustration purposes
Table 1 Comparison of C–C and C–O bond lengths (in A) in several
gas-phase molecules with furan in the gas phase and on Pd(111)
C–C C–O C–Pd
Ethanol 1.51 1.43 –
Acetaldehyde 1.50 1.22 –
Benzene 1.40 – –
Ethylene 1.33 – –
Furan (this work)
Gas-phase 1.36 (a–b) 1.37 –
1.43 (b–b)
hcp/bbPd 1.46 (a–b) 1.41 2.11 (a-Pd)
1.43 (b–b) 2.22 (b-Pd)
hcp/abPd 1.42, 1.47 (a–b) 1.39, 1.43 2.18, 2.10 (a-Pd)
1.46 (b–b) 2.27, 2.14 (b-Pd)
Furan (Bradley et al. from Ref. [14])
Gas-phase 1.35 (a–b) 1.36 –
1.42 (b–b)
Hollow-h 1.44 (a–b) 1.40 2.11 (a-Pd)
1.41 (b–b) 2.23 (b-Pd)
Off-hollow-h 1.40, 1.46 (a–b) 1.37, 1.41 2.21, 2.11 (a-Pd)
1.44 (b–b) 2.25, 2.13 (b-Pd)
292 Top Catal (2012) 55:290–299
123

intermediate between C–O and C=O, consistent with the
aromaticity of the molecule. On Pd(111), the C–O and a–bC–C bonds are both lengthened compared to the gas-phase
furan molecule, which suggests that the interaction with the
surface reduces the net bonding character of the p orbitals.
The calculated bond lengths of the adsorbed furan agree with
those calculated by Bradley et al. [14] in GGA-RPBE to
within 0.02 A (see Table 1 for comparison). The molecular
plane of furan is located at *2.1 A above the relaxed top
layer of Pd atoms. For comparison, the adsorption of thi-
opehene, the sulfur analog of furan, on MoS2 is completely
dominated by van der Waals (vdW) contributions at *-0.4 eV with the adsorption height being *3.5 A [29]. The
adsorption height of furan on Pd(111) as determined
by GGA functionals is much smaller than the likely vdW
equilibrium distance for furan, which suggests that vdW
contributions are not significant in furan adsorption on
Pd(111).
3.2 Dissociation of Furan
To investigate the decomposition of furan, I begin by
calculating C–H, C–O, and C–C bond scission in furan.
This is done for two adsorbed states, hcp/bbPd and hcp/
abPd. The hcp/bbPd state has five unique such steps: (1)
C–H bond scission at the a position; (2) C–H bond scission
at the b position; (3) C–O bond scission; (4) C–C bond
scission at the a–b position; (5) C–C bond scission at the
b–b position. The structure of the TS of each of the five
steps and the associated activation energies (Ea) relative to
the hcp/bbPd state are shown in Fig. 2.
As can be seen in Fig. 2, the TSs for the scission of the
C–H bonds always involve the dissociating H atom, the C
atom, and an underlying Pd atom forming a three-centered
bonding structure, a characteristic common of C–H bond
dissociation steps. The C–O and C–C bonds dissociate over
a Pd atom with the C atoms located in either a near-top or
bridge position at the TSs. The Ea for dissociating the
bC–H bond is 1.16 eV, whereas the Ea for dissociating the
aC–H bond is higher at 1.53 eV because the a H is closer to
the electron-withdrawing O atom. The C–O bond is the
weakest bond in furan, with Ea = 0.82 eV for its dissoci-
ation. At the TS, the dissociating C–O bond lengthens to
1.93 A over a Pd atom, and the C atom is stabilized in part
by two Pd atoms, while the remaining C–O bond contracts
to 1.33 A. The two different C–C bonds are also quite
difficult to break: The Ea for dissociating the a–b and the
b–b C–C bonds are calculated to be 1.62 and 1.49 eV,
respectively. Again I attribute the difference to the prox-
imity of the a–b C–C bonds to the O atom.
The hcp/abPd state has lower symmetry than the hcp/
bbPd state, so all the bonds are nonequivalent. The struc-
ture and associated Ea for the TS of each of the steps for
the hcp/bbPd state are shown in Fig. 3. Despite the non-
Fig. 2 Structure of the TSs of
the bond dissociation steps in
furan in the hcp/bbPd state on
Pd(111): C–H bond scission at
the (a) a and (b) b positions;
(c) C–O bond scission; C–C
bond scission at the (d) a–b and
(e) b–b positions. In each panel,
top view is shown on the left and
a bonding scheme with bond
lengths indicated is shown on
the right (length of a C–H bond
is labeled only if it is
dissociating). The value
corresponding to the
dissociating bond is
underscored. The activation
energy (Ea) associated with
each TS is also indicated. Smallblack, red, white; and largewhite spheres represent C, O, H,
and Pd atoms, respectively
Top Catal (2012) 55:290–299 293
123

equivalence, it remains more difficult to dissociate the aC–H
bonds than the bC–H bonds, and more difficult to dissociate
the a–b C–C bonds than the b–b C–C bond. This suggests
that the relative strengths of the bonds in furan on Pd(111) is
mainly influenced by the atomic arrangement in the mole-
cule. C–O bond scission has Ea = 0.74 eV relative to the
hcp/abPd state, but in terms of the ZPE-corrected total
energy, this TS is only 0.06 eV more stable than the corre-
sponding TS for the hcp/bbPd state (Fig. 2c).
3.3 Dissociation of Ring-Opening Products
Since the opening of the furan ring preferentially occurs at
the C–O position with nearly identical TS energies for the
two different initial states, I use the higher-symmetry hcp/
bbPd state in the subsequent narrative for brevity. The
opening of the furan ring at the C–O position produces
what may be considered a dehydrogenated C4 aldehyde
species, C4H4O, the optimized structure for which is shown
in Fig. 4a. The dissociation of this linear C4H4O interme-
diate via four C–H bond scission steps at the a–d positions
and three C–C bond scission steps at the a–b, b–c, c–dpositions are calculated (see Fig. 4a for the labeling of the
C atoms). The structure and associated Ea for the TS of
each of these steps are depicted in Figs. 4b, h. I find that
C4H4O most readily undergoes C–H bond scission at the aposition, with Ea = 0.40 eV. C–H bond scission at the dand c positions is also facile, with Ea = 0.85 and 0.55 eV.
The dissociation of all three C–C bonds is difficult, with
each Ea exceeding 1 eV. The dissociation of the carbonyl
C=O bond is presumed to have a much higher barrier and
ignored in the subsequent calculations. This is in accord
with the fact that no oxygen-containing species is observed
to desorb in the TPD experiment besides CO [15].
Next I investigate the decomposition of the C4H3O
species that results from the loss of the a H in C4H4O
(Fig. 5a), and the C4H2O species that results from the C–H
bond dissociation at the d position in C4H3O (Fig. 6a).
Fig. 3 Structure of the TSs of
the bond dissociation steps in
furan in the hcp/abPd state on
Pd(111): C–H bond scission at
the (a) a1; (b) b2; (c) b3, and
(d) a4 positions; (e) C–O bond
scission at a4 position; C–C
bond scission at the (f) a1–b2;
(g) b2–b3; (h) b3–a4 positions.
In each panel, top view is shown
on the left and a bonding
scheme with bond lengths
indicated is shown on the right(length of a C–H bond is labeled
only if it is dissociating). The
value corresponding to the
dissociating bond is
underscored. The activation
energy (Ea) associated with
each TS is also indicated. Smallblack, red, white; and largewhite spheres represent C, O, H,
and Pd atoms, respectively
294 Top Catal (2012) 55:290–299
123

Three C–H bond scission steps, at the b, c, and d positions,
and three C–C bond scission steps at the a–b, b–c, c–dpositions are calculated for C4H3O (Figs. 5b, g). The most
facile dissociation step is C–H bond scission, occurring at
the d position, with Ea = 0.83 eV. C–C bond scission
at the a–-b position follows closely behind, with
Ea = 0.93 eV. For C4H2O, two C–H bond scission steps, at
the b and c positions, and three C–C bond scission steps at
the a–b, b–c, c–d positions are calculated (Figs. 6b, f).
Here C–C bond scission at the a–b position has the lowest
activation barrier (Ea = 1.08 eV), yielding CO.
3.4 Adsorption of Dissociated Products
Dissociated products including H, CO, CHO, C3H2, C3H3,
ring-opened benzene, and benzene have also been
calculated (Fig. 7). H, CO, and benzene are calculated in
previously reported preferred sites: fcc for H and CO
[30–32], and bridge prpr for benzene [33]. The adsorption
energies are calculated to be -2.81, -2.13, and -1.28 eV,
in close agreement with the previous DFT results. It should
be noted that the GGA-PBE predicted adsorption of CO is
known to be too strong [30]. CHO prefers to bind through
the C atom in a bridge site with the carbonyl oxygen located
in a near-top position. The C–O bond length is 1.27 A.
C3H2 prefers to be located with its C end in a threefold site
and the CH end bonded to the top of an adjacent Pd atom. Its
CH–CH bond is 1.36 A and CH–C bond is 1.45 A. C3H3
adsorbs with both ends in a near-top position and the
molecular plane tilted toward an adjacent Pd atom. Both of
the C–C bonds in C3H3 are 1.44 A, which therefore still has
a conjugated character.
(a) (b) (c) (d)
(e) (f) (g) (h)
Ea = 0.40 eV Ea = 1.32 eV Ea = 0.85 eV
Ea = 0.55 eV Ea = 1.03 eV Ea = 1.48 eV Ea = 1.32 eV
αα
βγ
δ
Ea = 0.40 eV Ea = 1.32 eV Ea = 0.85 eV
Ea = 0.55 eV Ea = 1.03 eV Ea = 1.48 eV Ea = 1.32 eV
α
βγ
δ
Fig. 4 Top views of a the
C4H4O intermediate on Pd(111)
and b–h the TSs of the bond
dissociation steps: C–H bond
scission at the b a; c b; d c; and
e d positions; and C–C bond
scission at the f a–b; g b–c; and
h c–d positions. The activation
energy (Ea) associated with
each TS is indicated. Smallblack, red, white; and largewhite spheres represent C, O, H,
and Pd atoms, respectively. The
labeling of the four C atoms is
shown in (a)
(a) (b) (c) (d)
(e) (f) (g)
Ea = 1.40 eV Ea = 1.16 eV Ea = 0.83 eV
Ea = 0.93 eV Ea = 1.17 eV Ea = 1.68 eV
αα
βγ
δ
Ea = 1.40 eV Ea = 1.16 eV Ea = 0.83 eV
Ea = 0.93 eV Ea = 1.17 eV Ea = 1.68 eV
α
βγ
δ
Fig. 5 Top views of a the
C4H3O intermediate on Pd(111)
and b–g the TSs of the bond
dissociation steps: C–H bond
scission at the b b; c c; and d dpositions; and C–C bond
scission at the e a–b; f b-c; and
g c–d positions. The activation
energy (Ea) associated with
each TS is indicated. Smallblack, red, white; and largewhite spheres represent C, O, H,
and Pd atoms, respectively. The
labeling of the four C atoms is
shown in (a)
Top Catal (2012) 55:290–299 295
123

4 Discussion
Based on the TSs that have been located for the dissocia-
tion of the various bonds in furan and its dissociated
derivatives, a reaction energy profile for the decomposition
of furan leading up CO formation is constructed (Fig. 8).
The highest-energy TS on the reaction energy landscape is
that of C–O bond scission in furan and is located at 0.18 eV
below gas-phase furan, indicating that furan adsorbed on
Pd(111) prefers decomposition over desorption, which
agrees with the TPD results of Ormerod et al. [15]. No a-
furyl species is formed in contrast to Ru(0001) [20]. The
resulting C4H4O species undergoes rapid C–H bond dis-
sociation to produce C4H3O, whereas C–C bond scission in
C4H4O yielding CHO has a much higher activation barrier
(Path C in Fig. 8). C4H3O can dehydrogenate to produce
C4H2O with an activation barrier that is nearly identical to
the initial C–O bond scission (Path A in Fig. 8), or it can
undergo a–b C–C scission by overcoming an activation
barrier that is 0.1 eV higher to directly produce CO and
C3H3 surface species (Path B in Fig. 8). The ratio of
the dehydrogenation rate versus the decarbonylation rate,
calculated as
rA
rB¼ mA
mBe�EA
a �EAa
jT ;
with Ea= 0.83 eV; Ea= 0.93 eV (see Fig. 5); mA = 1.6 9
1014 and mB = 8.2 9 1013 s-1 (see Appendix), indicates that
only 1–2 % of C4H3O undergoes decarbonylation to yield CO
and C3H3 between 300 and 350 K. The majority of C4H3O
dehydrogenates further to C4H2O particularly since the rec-
ombinative desorption of H2 commences around 300 K to
eliminate H from the Pd(111) surface [34]. The loss of up to two
H atoms occurs after ring opening, which accounts for the
Fig. 6 Top views of a the C4H2O intermediate on Pd(111) and
b–f the TSs of the bond dissociation steps: C–H bond scission at the
b b; and c c positions; and C–C bond scission at the d a–b; e b–c; and
f c–d positions. The activation energy (Ea) associated with each TS is
indicated. Small black, red, white; and large white spheres represent
C, O, H, and Pd atoms, respectively. The labeling of the four C atoms
is shown in (a)
Fig. 7 Top views of a H; b CO; c CHO; d C3H2; e C3H3; f ring-
opened benzene; and g benzene in their respective preferred
adsorption states; h TS of 2C3H3 ? ring-opened benzene; i TS of
ring-opened benzene ? benzene. The activation energy (Ea) associ-
ated with each TS is indicated. Small black, red, white; and largewhite spheres represent C, O, H, and Pd atoms, respectively
Fig. 8 Reaction energy profile for furan decomposition on Pd(111).
All species are surface-adsorbed except for gas-phase furan (‘‘(g)’’).
The balance of H atoms is not shown for brevity. Points marked by
circles are TSs. The difference in energy between a pair of points
separated by a TS is the DErxn for that step as defined in ‘‘Methods’’.
The three alternate reaction paths (A, B, and C) are referenced to in
the text
296 Top Catal (2012) 55:290–299
123

desorption-limited H2 evolution observed by Ormerod et al.
[15], before decarbonylation. Therefore I conclude that on a
clean Pd(111) surface, the majority of furan undergoes ring
opening, dehydrogenation, and decarbonylation in that order.
Since C–C bond scission at the b–c and c–d positions consis-
tently has higher activation barriers than at the a–b position
(decarbonylation), CO2 is far less likely to form than CO on
Pd(111) [15, 35].
Previous experimental studies have identified the tem-
perature at which furan decomposition commences on
Pd(111) to be between 230 and 270 K as evidence by a
decrease of the furan vibrational signatures in HREELS
[15]; changes in the C signal in XPS [15] and PhD [13];
and a decrease in the furan desorption signal in LITD [16].
I perform a Redhead-like analysis to estimate the temper-
ature in TPD at which furan begins to dissociate on
Pd(111) using the DFT results. It is assumed that the ring-
opening step is a uni-molecular, first-order surface process,
and that the dissociation is not limited by the availability of
free sites, assumptions that are reasonable at the surface
coverage of 1/9 ML. As such, the classic Redhead equation
[28] applies:
r ¼ � dhdt¼ vhne�
EakT with T ¼ T0 þ bt:
For furan in the hcp/bbPd state undergoing C–O bond
dissociation, Ea is 0.82 eV (Fig. 2c); m = 5.6 9 1013 s-1
(see ‘‘Appendix’’); initial coverage h0 = 0.11; n = 1; and
the heating rate b is set to 10 K/s. The calculated coverage is
plotted as a function of temperature in Fig. 9, which shows
that the coverage of furan on Pd(111) begins to decrease
appreciably at 270 K and mostly reaches zero by 320 K. I
note briefly that a co-adsorbed H atom located in the 2nd
nearest-neighbor hcp site increases the Ea of the ring-
opening step slightly from 0.82 to 0.84 eV. For furan in the
hcp/abPd state, dissociation begins appreciably at 240 K.
The reaction pathway that I have constructed identifies
C4H2O as a stable surface intermediate prior to CO
formation. It is worth noting that in the HREELS experi-
ment by Ormerod et al. [15], a peak is initially seen at
1,750 cm-1 at 300 K and below, and it blue-shifts to
1,800 cm-1 and increases in intensity at 400 K. The
authors have interpreted this blue shift and increase in
intensity as due to increasing coverage of CO. Vibrational
spectroscopies of CO adsorbed on Pd(111) generally locate
the m(C=O) mode at 1,820–1,850 cm-1 for coverage up to
1/3 ML [36, 37]. On the other hand, C4H2O has a m(C=O)
mode calculated to be 1,700 cm-1 with a dipole compo-
nent perpendicular to the surface, and the m(C=O) fre-
quency of CO on Pd(111) is calculated to be 1,776 cm-1 at
1/9 ML. Therefore the observed C=O signature and its blue
shift may instead be due to the C4H2O intermediate and its
decarbonylation to yield CO.
The same kind of kinetic analysis as performed above
indicates that the decarbonylation of C4H2O becomes signif-
icant at 350 K and is mostly complete by 410 K (Fig. 9). This
does not directly agree with the LITD results of Caldwell et al.
[16], which show a simultaneous rise of the CO signal with the
decrease in the furan signal beginning at 270 K. I note that
after furan undergoes ring opening and subsequent dehydro-
genation, the surface C4 intermediate(s) has no ready desorp-
tion channel except undergoing further decomposition to
release CO as the first desorbing species. Laser-induced dis-
sociation of surface species is uncommon but can occur in
LITD experiments [38]. This may explain why CO formation
and furan disappearance appear to be correlated between 270
and 310 K and share similar kinetics in LITD experiments
[19]. I propose that the existing experimental evidence can
accommodate a ring-opened surface intermediate between 300
and 350 K, but whether this is true requires further experi-
mental investigation (e.g. using variable-temperature STM)
and kinetic analysis.
The exact fate of the C3 fragment is outside the scope of
this study. In the TPD experiments furan, CO, and H2 are
the only gaseous products observed [15]. No CxHy surface
species has been unambiguously characterized [18]. It
stands to reason that as furan cracks in the absence of
sufficient hydrogen in TPD, carbon residuals are left on the
surface that eventually become coke. In the LITD experi-
ments benzene is detected as an additional gaseous species
because this technique favors high-entropy pathways such
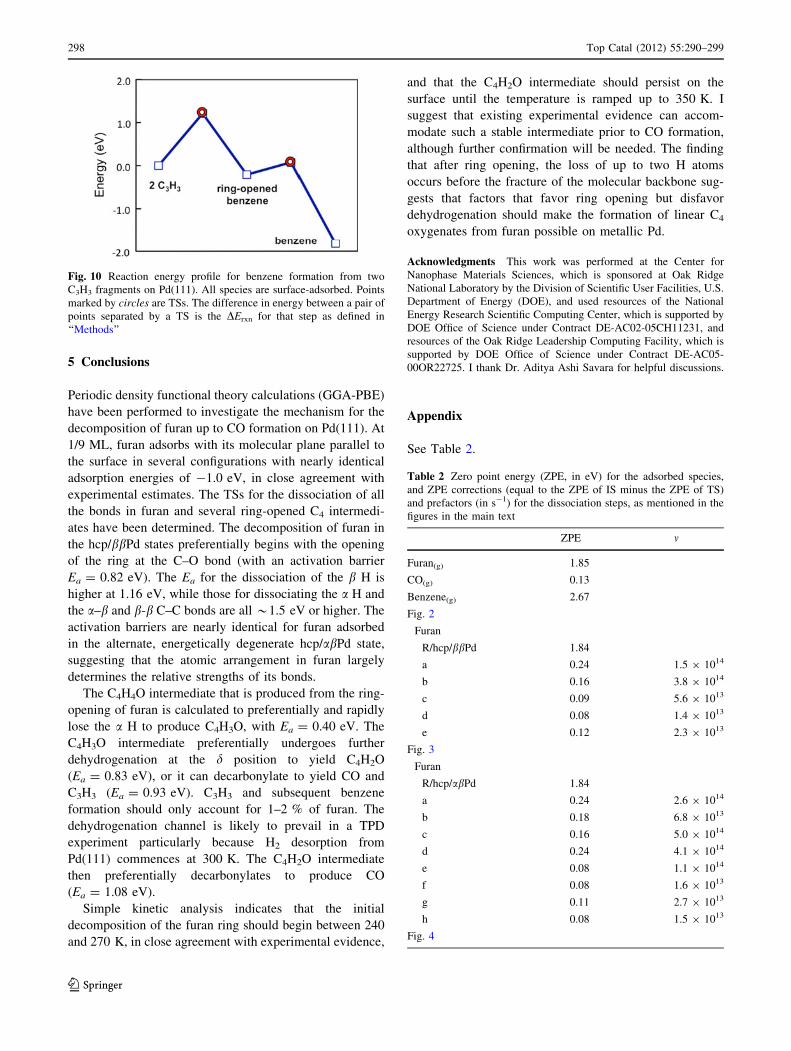
as desorption [16, 19]. I have calculated benzene on
Pd(111) [33] and its formation from two C3H3 fragments
(see Fig. 7f–i). It is a two-step process, with Ea = 1.21 and
0.29 eV for the 1st and 2nd C–C coupling steps, respec-
tively, and a large overall driving force of DErxn =
-1.82 eV (Fig. 10). This is not a major pathway even in
LITD experiments (2 % of furan is estimated to end up as
benzene [16]). This can be rationalized by our finding that
most of the C3 fragments that are produced in the
decarbonylation step are already hydrogen-poor (C3H2).
Fig. 9 Calculated coverages of furan (left solid curve) and the C4H2O
intermediate (right dashed curve) on Pd(111) both as functions of
temperature. See text for more detail
Top Catal (2012) 55:290–299 297
123
5 Conclusions
Periodic density functional theory calculations (GGA-PBE)
have been performed to investigate the mechanism for the
decomposition of furan up to CO formation on Pd(111). At
1/9 ML, furan adsorbs with its molecular plane parallel to
the surface in several configurations with nearly identical
adsorption energies of -1.0 eV, in close agreement with
experimental estimates. The TSs for the dissociation of all
the bonds in furan and several ring-opened C4 intermedi-
ates have been determined. The decomposition of furan in
the hcp/bbPd states preferentially begins with the opening
of the ring at the C–O bond (with an activation barrier
Ea = 0.82 eV). The Ea for the dissociation of the b H is
higher at 1.16 eV, while those for dissociating the a H and
the a–b and b-b C–C bonds are all *1.5 eV or higher. The
activation barriers are nearly identical for furan adsorbed
in the alternate, energetically degenerate hcp/abPd state,
suggesting that the atomic arrangement in furan largely
determines the relative strengths of its bonds.
The C4H4O intermediate that is produced from the ring-
opening of furan is calculated to preferentially and rapidly
lose the a H to produce C4H3O, with Ea = 0.40 eV. The
C4H3O intermediate preferentially undergoes further
dehydrogenation at the d position to yield C4H2O
(Ea = 0.83 eV), or it can decarbonylate to yield CO and
C3H3 (Ea = 0.93 eV). C3H3 and subsequent benzene
formation should only account for 1–2 % of furan. The
dehydrogenation channel is likely to prevail in a TPD
experiment particularly because H2 desorption from
Pd(111) commences at 300 K. The C4H2O intermediate
then preferentially decarbonylates to produce CO
(Ea = 1.08 eV).
Simple kinetic analysis indicates that the initial
decomposition of the furan ring should begin between 240
and 270 K, in close agreement with experimental evidence,
and that the C4H2O intermediate should persist on the
surface until the temperature is ramped up to 350 K. I
suggest that existing experimental evidence can accom-
modate such a stable intermediate prior to CO formation,
although further confirmation will be needed. The finding
that after ring opening, the loss of up to two H atoms
occurs before the fracture of the molecular backbone sug-
gests that factors that favor ring opening but disfavor
dehydrogenation should make the formation of linear C4
oxygenates from furan possible on metallic Pd.
Acknowledgments This work was performed at the Center for
Nanophase Materials Sciences, which is sponsored at Oak Ridge
National Laboratory by the Division of Scientific User Facilities, U.S.
Department of Energy (DOE), and used resources of the National
Energy Research Scientific Computing Center, which is supported by
DOE Office of Science under Contract DE-AC02-05CH11231, and
resources of the Oak Ridge Leadership Computing Facility, which is
supported by DOE Office of Science under Contract DE-AC05-
00OR22725. I thank Dr. Aditya Ashi Savara for helpful discussions.
Appendix
See Table 2.
Fig. 10 Reaction energy profile for benzene formation from two
C3H3 fragments on Pd(111). All species are surface-adsorbed. Points
marked by circles are TSs. The difference in energy between a pair of
points separated by a TS is the DErxn for that step as defined in
‘‘Methods’’
Table 2 Zero point energy (ZPE, in eV) for the adsorbed species,
and ZPE corrections (equal to the ZPE of IS minus the ZPE of TS)
and prefactors (in s-1) for the dissociation steps, as mentioned in the
figures in the main text
ZPE m
Furan(g) 1.85
CO(g) 0.13
Benzene(g) 2.67
Fig. 2
Furan
R/hcp/bbPd 1.84
a 0.24 1.5 9 1014
b 0.16 3.8 9 1014
c 0.09 5.6 9 1013
d 0.08 1.4 9 1013
e 0.12 2.3 9 1013
Fig. 3
Furan
R/hcp/abPd 1.84
a 0.24 2.6 9 1014
b 0.18 6.8 9 1013
c 0.16 5.0 9 1014
d 0.24 4.1 9 1014
e 0.08 1.1 9 1014
f 0.08 1.6 9 1013
g 0.11 2.7 9 1013
h 0.08 1.5 9 1013
Fig. 4
298 Top Catal (2012) 55:290–299
123

References
1. Chheda JN, Huber GW, Dumesic JA (2007) Angew Chem Int Ed
46:7164
2. Kliewer CJ, Aliaga C, Bieri M, Huang WY, Tsung CK, Wood JB,
Komvopoulos K, Somorjai GA (2010) J Am Chem Soc
132:13088
3. Huber GW, Iborra S, Corma A (2006) Chem Rev 106:4044
4. Huber GW, Dumesic JA (2006) Catal Today 111:119
5. Roman-Leshkov Y, Barrett CJ, Liu ZY, Dumesic JA (2007)
Nature 447:982
6. Roman-Leshkov Y, Chheda JN, Dumesic JA (2006) Science
312:1933
7. Carlson TR, Vispute TP, Huber GW (2008) ChemSusChem 1:397
8. Yu S, Brown HM, Huang XW, Zhou XD, Amonette JE, Zhang
ZC (2009) App Catal A Gen 361:117
9. Furimsky E (2000) App Catal A Gen 199:147
10. Jackson SD, Canning AS, Vass EM, Watson SR (2003) Ind Eng
Chem Res 42:5489
11. Sitthisa S, Resasco DE (2011) Catal Lett 141:784
12. Loui A, Chiang S (2004) Appl Surf Sci 237:555
13. Knight MJ, Allegretti F, Kroger EA, Polcik M, Lamont CLA,
Woodruff DP (2008) Surf Sci 602:2524
14. Bradley MK, Robinson J, Woodruff DP (2010) Surf Sci 604:920
15. Ormerod RM, Baddeley CJ, Hardacre C, Lambert RM (1996)
Surf Sci 360:1
16. Caldwell TE, Abdelrehim IM, Land DP (1996) J Am Chem Soc
118:907
17. Knight MJ, Allegretti F, Kroger EA, Polcik M, Lamont CLA,
Woodruff DP (2008) Surf Sci 602:2743
18. Bradley MK, Duncan DA, Robinson J, Woodruff DP (2011) Phys
Chem Chem Phys 13:7975
19. Caldwell TE, Land DP (1999) J Phys Chem B 103:7869
20. Qiao MH, Yan FQ, Sim WS, Deng JF, Xu GQ (2000) Surf Sci
460:67
21. Kresse G, Furthmuller J (1996) Comput Mater Sci 6:15
22. Kresse G, Hafner J (1994) Phys Rev B 49:14251
23. Perdew JJ, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
24. Kresse G, Joubert D (1999) Phys Rev B 59:1758
25. Methfessel M, Paxton AT (1989) Phys Rev B 40:3616
26. Ashcroft NW, Mermin ND (1976) Solid state physics. Saunders
College, Orlando
27. Henkelman G, Jonsson H (1999) J Chem Phys 111:7010
28. Masel RI (1996) Principles of adsorption and reaction on solid
surfaces. Wiley, New York
29. Moses PG, Mortensen JJ, Lundqvist BI, Nørskov JK (2009) J
Chem Phys 130:104709
30. Gajdos M, Eichler A, Hafner J (2004) J Phys Condens Matter
16:1141
31. Lim KH, Chen ZX, Neyman KM, Rosch N (2006) J Phys Chem B
110:14890
32. Xu L, Xu Y (2011) Catal Today 165:96
33. Morin C, Simon D, Sautet P (2006) Surf Sci 600:1339
34. Conrad H, Ertl G, Latta EE (1974) Surf Sci 41:435
35. Medlin J, Horiuchi C, Rangan M (2010) Top Catal 53:1179
36. Bradshaw AM, Hoffmann FM (1978) Surf Sci 72:513
37. Surnev S, Sock M, Ramsey MG, Netzer FP, Wiklund M, Borg M,
Andersen JN (2000) Surf Sci 470:171
38. Hall RB (1987) J Phys Chem 91:1007
Table 2 continued
ZPE m
C4H4O
a 1.76
b 0.16 1.2 9 1014
c 0.20 6.6 9 1014
d 0.16 1.4 9 1014
e 0.12 4.2 9 1013
f 0.09 7.0 9 1013
g 0.09 3.2 9 1013
h 0.09 3.5 9 1013
Fig. 5
C4H3O
a 1.47
b 0.22 2.8 9 1014
c 0.24 2.6 9 1014
d 0.15 1.6 9 1014
e 0.07 8.2 9 1013
f 0.09 3.1 9 1013
g 0.08 3.8 9 1013
Fig. 6
C4H2O
a 1.19
b 0.19 7.5 9 1013
c 0.23 1.3 9 1014
d 0.09 6.4 9 1013
e 0.12 3.4 9 1013
f 0.05 7.2 9 1012
Fig. 7
H
a 0.17
CO
b 0.19
CHO
c 0.47
C3H2
d 0.92
C3H3
e 1.19
C6H6 half-ring
f 2.54
Benzene
g 2.63
h -0.06 2.9 9 1011
i 0.01 1.4 9 1013
Top Catal (2012) 55:290–299 299
123