december 7 2013 - az9194.vo.msecnd.netaz9194.vo.msecnd.net/pdfs/131202/20302 petrou...
TRANSCRIPT

Cell, network and mouse modelling of genetic epilepsies for mechanism, diagnosis and therapy
December 7th 2013
Steven Petrou, PhD Deputy Director, The Florey Institute
Deputy Director, The Centre for Neural Engineering The University of Melbourne, Australia
American Epilepsy Society | Annual Meeting

Disclosure
Name of Commercial Interest
NONE
American Epilepsy Society | 2013 Annual Meeting
Type of Financial Relationship
NONE

Learning Objectives
• Understand how different models of genetic epilepsy can reveal “direct” and “emergent” pathologies
• Understand how this knowledge may inform therapeutic strategy
American Epilepsy Society | 2013 Annual Meeting

• Examined 264 trios (792 exomes)
• 329 variants discovered
Opportunity for translation provided by explosion in genetics knowledge

Models for revealing disease state biomarkers and pathology
1. Mouse
2. Single Cell
a. Biophysics
b. Structure-function
3. Neuronal Network a. Primary cultures
b. Human stem cell based

1. MOUSE MODELS

Mouse model reveals emergent pathologies
Novel epileptogenic mechanism in model of Early Onset Epileptic
Encephalopathy (EOEE) suggests targeted therapeutic intervention
Defined by frequent and severe epileptic seizures with progressive developmental regression, possible psychomotor deficits as seen in Dravet syndrome
Difficult to treat
Increasing recognition of underlying genetic defects
SCN1B gene, homozygous R125C (Patino, 2009)
Patient presented with Dravet Syndrome
We previously developed the het SCN1B(C121W) mouse
Does the homozygous mouse model Dravet?
Early onset epileptic encephalopathies (EOEE)
Modified from: Patino, G.A. (2009), A functional null mutation of SCN1B …

Characteristics of the homozygous C121W mouse
• Altered gait
• Premature death
• Spontaneous seizures
• More rapid progression to tonic-
clonic seizure following heating
Petrou, unpublished data 2013
Survival

Pharmacosensitivity of the homozygous C121W mouse similar to Dravet Syndrome
• Similar pharmacosensitivity to human Dravet patients
• Suggests shared pathological mechanism
Petrou, unpublished data 2013
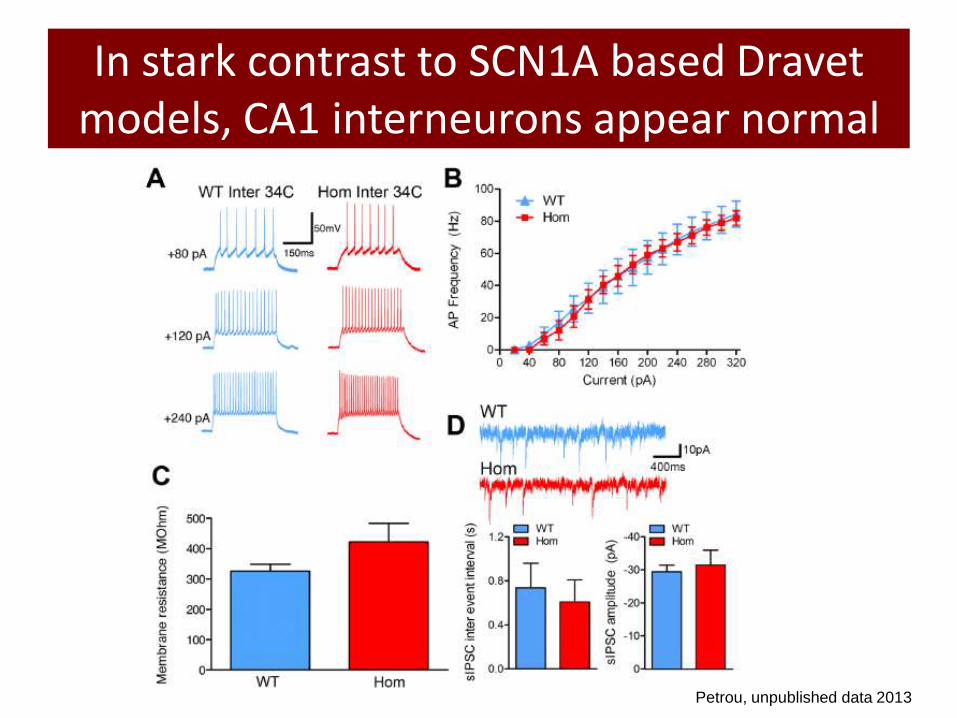
In stark contrast to SCN1A based Dravet models, CA1 interneurons appear normal
Petrou, unpublished data 2013

BUT, hippocampal excitatory neurons from our homozygous C121W mice are more excitable
WT (n =8), Hom (n = 8)
Petrou, unpublished data 2013

Changes in input resistance explains neuronal excitability
WT (n=15), Hom (n=10); *P<0.05
Petrou, unpublished data 2013
• Increased input resistance (Rin) may be major contributor to increased excitability

Reduced neuronal arborisation might explain the changes in cellular properties
Petrou, unpublished data 2013

• Drugs that specifically target and reduce Rin may rescue the seizure phenotype
• Retigabine
– Activates KCNQ2/3 channels
– Reduces Rin
Can we use our information on cellular dysfunction to predict drug efficacy?
Modified from: Surti TS (2005), Identification by mass spectrometry and…

In vitro: Retigabine reverses the neuronal deficit
Reduces input resistance in homozygous neurons
Shifts homozygous input-output relationship
Petrou, unpublished data 2013

In vivo: Retigabine reduces seizure susceptibility
Petrou, unpublished data 2013

• Homozygous mice as a mouse model of Dravet – Phenotype corresponds to SCN1A mouse model of Dravet
• Thermogenic seizure susceptibility
• Premature death, unstable gait, severe seizures
• Altered excitatory neuron firing distinguishes this pathology from that of SCN1A based models – Increased Rin underlies changes in firing properties
– Mutant neurons display reduced dendritic arborisation as primary pathology • Emergent properties
– Retigabine reverses cellular and behavioral deficits
• Potential example of disease mechanism based therapy
Summary

2. SINGLE CELL MODELS

Genotype-phenotype correlation and pharmacological modulation of hKCNT1 channel
mutations causing epilepsy

KCNT1 based epilepsies
• In 2011 ADNFLE (Autosomal Dominant Nocturnal Frontal Lobe Epilepsy) was thought to be a disorder of nicotinic acetylcholine receptors
• In 2012 KCNT1 was discovered (Heron et al., 2012, Nature Genetics) as a gene for more severe ADNFLE with psychiatric features expanding the genetic architecture
• At the same time Barcia et al (2012, Nature Genetics) showed that KCNT1 was also associated with a very severe epileptic encephalopathy, Epilepsy of Infancy with Migrating Focal Seizures (EIMFS) characterised by drug-resistant seizures and developmental delay
• EIMFS and ADNFLE are allelic yet clinically distinct and the mechanism by which KCNT1 mutations produce this phenotypic spectrum is intriguing

KCNT1: (Slo2.2, KCa4.1, SLACK)
Na+ activated potassium channel
Thought to contribute to RMP and to slow
hyperpolarisation following repetitive firing
NH2
COOH
R398Q
Y796H
R928C
(R907C)
(R899C)
RCK domains NAD+ binding
domain [Na+
]
M896I
A934T
I760M R428Q
R474H
P924L
[Na+]
KCNT1 mutations in ADNFLE and EIMFS
M896I R398Q Y796H R928C R428Q A934T P924L
ADNFLE mutants EIMFS mutants
Most severe Least severe Disease severity

KCNT1 epilepsy mutations show increased current magnitude and altered kinetics
Petrou, unpublished data 2013
W T
0 1
0
1 0
M 8 9 6 I
0 1
0
1 0
R 3 9 8 Q
0 1
0
1 0
Y 7 9 6 H
0 1
0
1 0
R 9 2 8 C
0 1
0
1 0
R 4 2 8 Q
1 0 0 0
0
1 0
A 9 3 4 T
0 1 0 0 0
0
1 0
P 9 2 4 L
1 0 0 0
0
1 0
2 A
1 0 0 m s
W T
0 1
0
1 0
M 8 9 6 I
0 1
0
1 0
R 3 9 8 Q
0 1
0
1 0
Y 7 9 6 H
0 1
0
1 0
R 9 2 8 C
0 1
0
1 0
R 4 2 8 Q
1 0 0 0
0
1 0
A 9 3 4 T
0 1 0 0 0
0
1 0
P 9 2 4 L
1 0 0 0
0
1 0
2 A
1 0 0 m s
W T
0 1
0
1 0
M 8 9 6 I
0 1
0
1 0
R 3 9 8 Q
0 1
0
1 0
Y 7 9 6 H
0 1
0
1 0
R 9 2 8 C
0 1
0
1 0
R 4 2 8 Q
1 0 0 0
0
1 0
A 9 3 4 T
0 1 0 0 0
0
1 0
P 9 2 4 L
1 0 0 0
0
1 0
2 A
1 0 0 m s
W T
0 1
0
1 0
M 8 9 6 I
0 1
0
1 0
R 3 9 8 Q
0 1
0
1 0
Y 7 9 6 H
0 1
0
1 0
R 9 2 8 C
0 1
0
1 0
R 4 2 8 Q
1 0 0 0
0
1 0
A 9 3 4 T
0 1 0 0 0
0
1 0
P 9 2 4 L
1 0 0 0
0
1 0
2 A
1 0 0 m s
WT M8961 R928C R428Q
R398Q Y796H A934T P924L
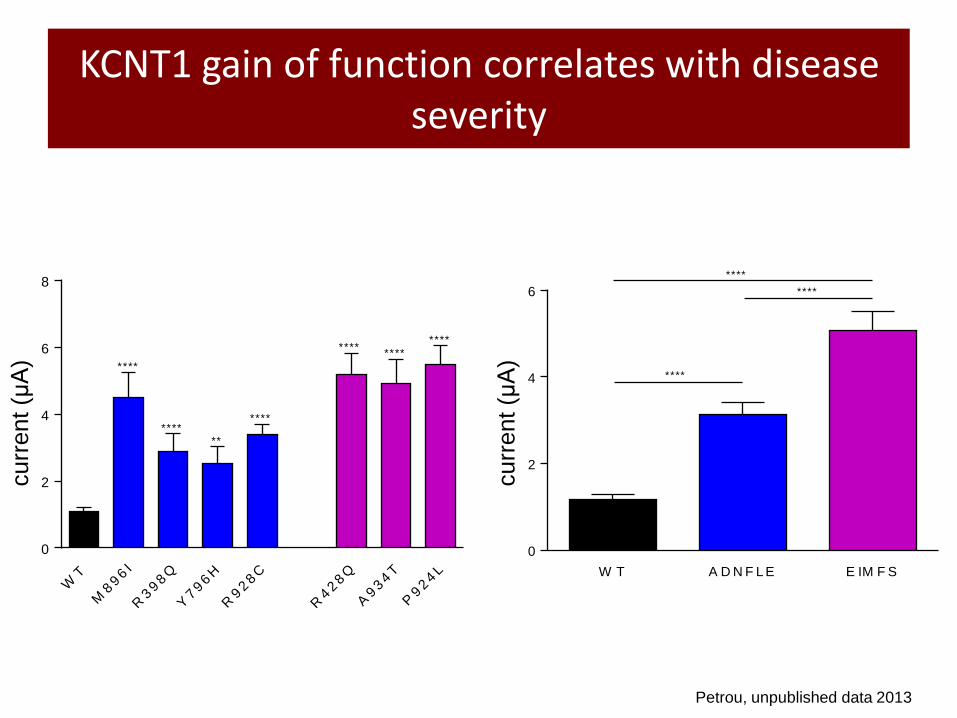
KCNT1 gain of function correlates with disease severity
Petrou, unpublished data 2013
WT
M896I
R398Q
Y796H
R928C
R428Q
A934T
P924L
0
2
4
6
8
cu
rre
nt
at
+1
0 m
V (
A)
****
******
****
********
****
cu
rre
nt
at
+1
0 m
V (
A)
W T A D N F L E E IM F S
0
2
4
6
****
****
****
curr
ent (μA
)
curr
ent (μA
)

Cinchona Tree Bark
quinine
quinidine
Yang et al., 2006
Quinidine as a potential therapy?
• The clinical severity of KCNT1 epilepsies
creates an urgent need for intervention
• Quinidine is an FDA approved antiarrhythmic
drug shown to inhibit rodent SLACK channels
• Does it act on hKCNT1 and how does it
interact with mutant channels?

Quinidine blocks human WT and mutant channels
Petrou, unpublished data 2013

3. NEURONAL NETWORK MODELS

The perfect storm for rapidly understanding the effect of mutations and drugs
INPUTS a. Genetics discoveries in epilepsy
b. Stem cell technology improvements
c. Multi-electrode array readiness
OUTPUTS a. Diagnostic markers
b. Efficacy markers
Convergence of technologies to enable breakthroughs in seizure neurobiology
and drug discovery toward the promise of precision medicine

Modeling complex genetic interactions
• Role of genetic background in determining clinical heterogeneity
– Compare multiple mutations in isogenic cell lines to simplify mutant analysis
– Compare patient mutation in cell line versus patient iPS neurons
– “Rescue” patient mutant to understand residual phenotype
• Create “disease state” models to explore drug action

CRISPR/Cas (Clustered Regularly Interspaced Palindromic Repeats/CRISPR-associated)
• Genome editing technologies
– Zinc Finger Nucleases
– TALENs
– CRISPR/Cas

CRISPR/Cas Homology Directed Repair
• Addressing off target effects:
• Nickase version of Cas
• WES of cell lines with established bioinformatics filtering
• Scalable

Stem Cell Differentiation
• Goal is to create a functioning network of neurons that can display sufficiently complex behavior to respond to genetic changes and drug exposure
• Differentiate human stem cells into
– Inhibitory neurons
– Excitatory neurons
– Glia

Multi Electrode Array: “Epilepsy in a dish”
Spikes and Bursts
MEA dish
Active Area
Contact Pads
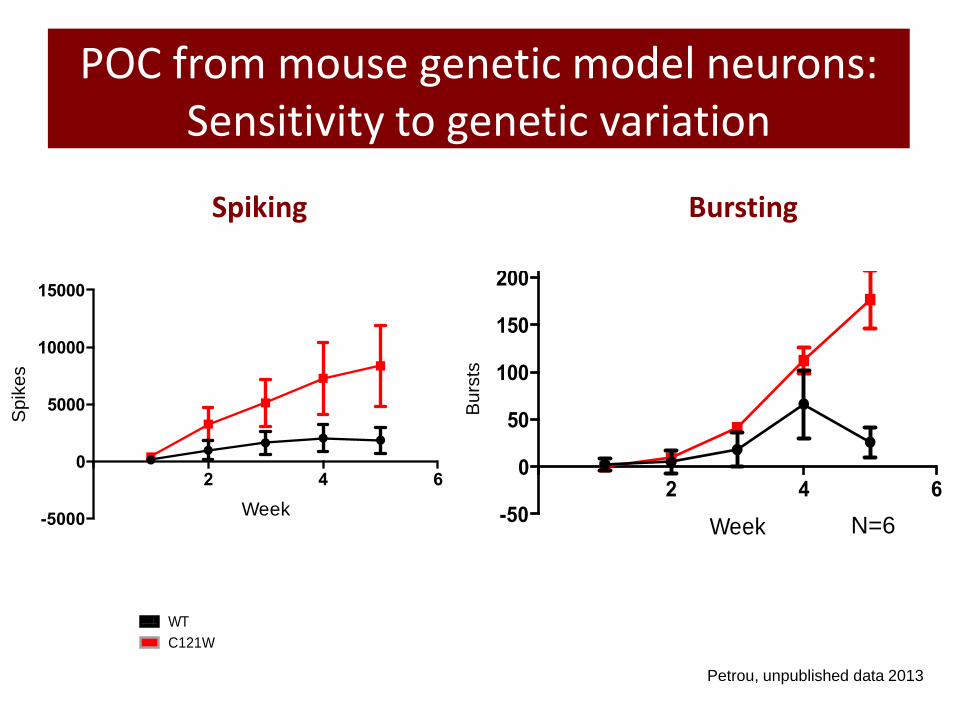
POC from mouse genetic model neurons: Sensitivity to genetic variation
2 4 6
-50
0
50
100
150
200
250
Week
Nu
mb
er
of U
nits
2 4 6
-5000
0
5000
10000
15000
Week
Un
its
Wild Type C121W
N=6
Sp
ike
s
Bu
rsts
Spiking
WT
C12
1W
0
20
40
60
80
ms
WT
C121W
Petrou, unpublished data 2013
Bursting

POC: Sensitivity to drugs
Petrou, unpublished data 2013
Retigabine (10uM)
Wash
Spiking
Bath
Week 1 2 3 4
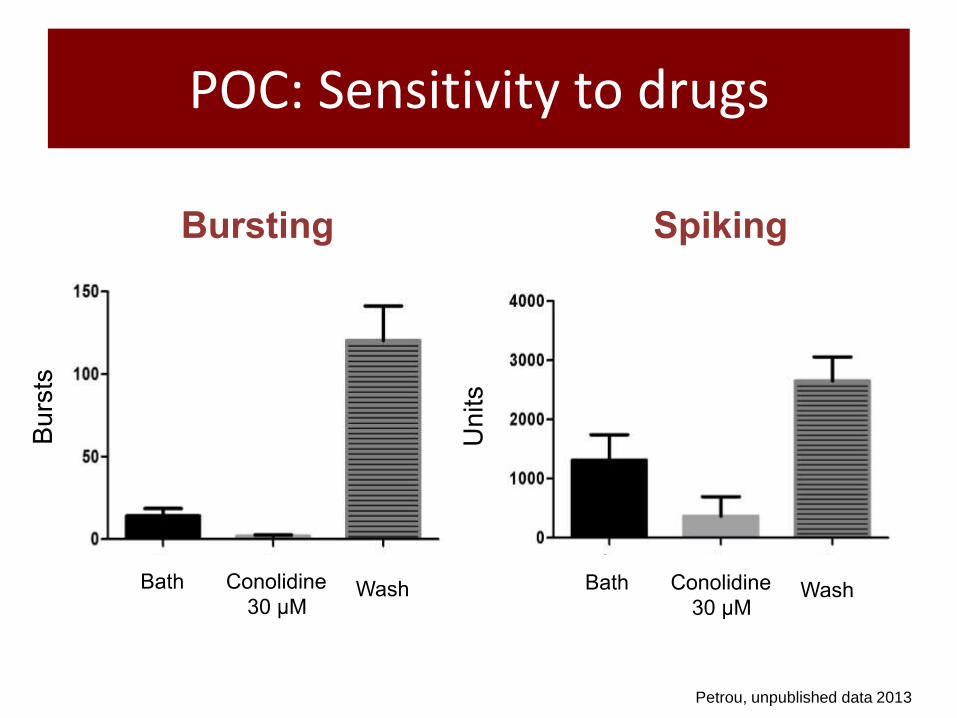
Petrou, unpublished data 2013
POC: Sensitivity to drugs
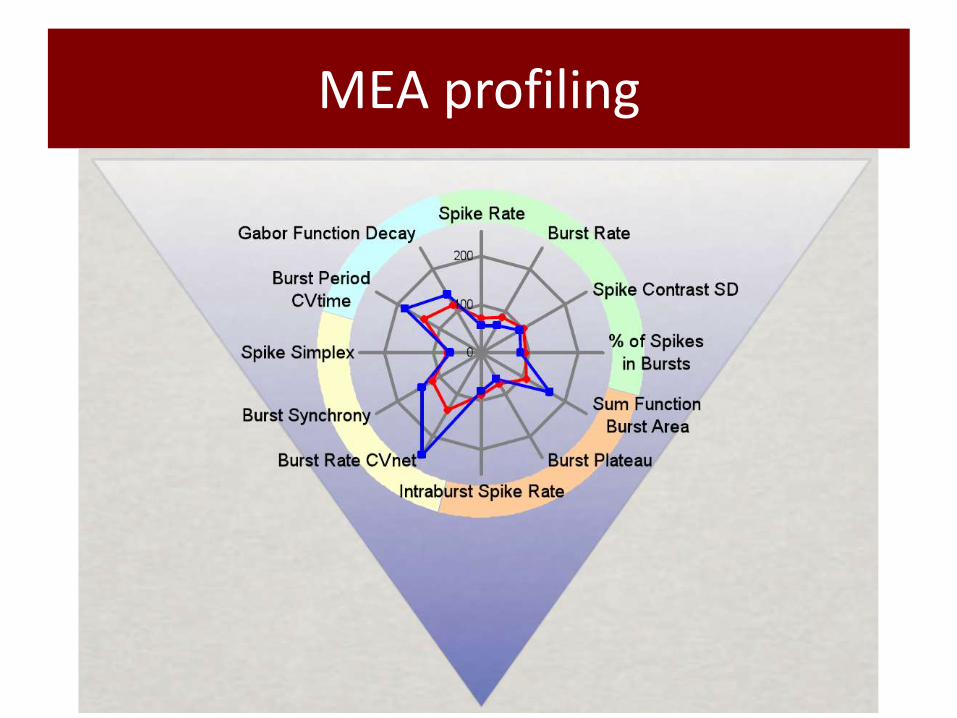
MEA profiling

Acknowledgements
• Chris Reid • Carol Milligan • Melody Li • Emma Morrisroe • Sophie Crux • Nuttawat “Pop”
Suphanatarida • Kay Richards • Elena Gazina • Verena Wimmer • Bryan Leaw
• Sam Berkovic • David Goldstein • Ingrid Scheffer • Leanne Dibbens • Sarah Heron