cytokeratin dynamics in living cells - ukaachen.de · elvanol, prepared by dissolving 1 g mowiol...
TRANSCRIPT

INTRODUCTION
The polypeptide subunits of the 10 nm intermediate filaments(IFs) in epithelial cells can be grouped into two large multigenefamilies, termed type I and type II cytokeratins (CKs) whichare expressed in stoichiometrically equal amounts in a tissue-specific and cell type-restricted pattern (Fuchs and Weber,1994; Moll, 1998). CK filaments (CKFs) are considered to beparticularly important for epithelial resilience to mechanicalstress by forming a stable network which is attached to specificcell-cell contacts of the desmosome type (Schmidt et al., 1994;Bornslaeger et al., 1998; Fuchs and Cleveland, 1998).Disturbance of this supracellular network results in reducedtissue coherence and increased cell fragility as it occurs inseveral blister-forming genodermatoses which are caused bydominant negative-acting CK mutants (Fuchs and Weber,1994; McLean and Lane, 1995; Fuchs and Cleveland, 1998).The stability of CKFs is reflected by their remarkablebiochemical properties, i.e. their resistance to non-ionicdetergents, their insolubility in high salt buffers, and the stronginteractions of their subunits forming heterodimers (Fuchs andWeber, 1994; Herrmann and Aebi, 1998). Lateral exchange of
subunits enabling the continuous maintenance of the entirefilament network (Miller et al., 1991, 1993) and the scarceevidence for CK-specific motor proteins further underscore thestatic nature of CKFs and distinguishes them from the actin-based microfilaments and tubulin-based microtubules.
Given that the stabilising properties of CKFs fulfil importantfunctions in epithelial tissue integrity it is at the same timeimportant that they are dynamic structures to avoid interferencewith processes that are needed for tissue homeostasis andmaintenance such as mitosis, stratification, wound healing andmigration, or with other more specialised functions such assecretion. Exactly how these two opposing principles arerealised is not known although various levels of regulation maybe operating that affect filament dynamics by influencingfilament organisation, expression of distinct CK pairs withspecific dynamic properties, association with regulatorypolypeptides and protein modification such as phosphorylationand glycosylation (Eriksson et al., 1992; Skalli et al., 1992;Fuchs and Weber, 1994; McGowan and Coulombe, 1998;Omary et al., 1998).
Studies trying to elucidate in vivo dynamics of cellularelements often deduce sequential reaction patterns from results
4521Journal of Cell Science 112, 4521-4534 (1999)Printed in Great Britain © The Company of Biologists Limited 1999JCS0841
To monitor the desmosome-anchored cytokeratin networkin living cells fusion protein HK13-EGFP consisting ofhuman cytokeratin 13 and the enhanced green fluorescentprotein was stably expressed in vulvar carcinoma-derivedA-431 cells. It is shown for A-431 subclone AK13-1 thatHK13-EGFP emits strong fluorescence in fixed and livingcells, being part of an extended cytoplasmic intermediatefilament network that is indistinguishable from that ofparent A-431 cells. Biochemical, immunological andultrastructural analyses demonstrate that HK13-EGFPbehaves identically to the endogenous cytokeratin 13 and istherefore a reliable in vivo tag for this polypeptide andthe structures formed by it. Time-lapse fluorescencemicroscopy reveals that the cytokeratin 13-containingnetwork is in constant motion, resulting in continuousrestructuring occurring in single and migratory cells, aswell as in desmosome-anchored cells. Two major types ofmovement are distinguished: (i) oscillations of mostly long
filaments, and (ii) an inward-directed flow of fluorescenceoriginating as diffuse material at the cell periphery andmoving in the form of dots and thin filaments toward thedeeper cytoplasm where it coalesces with other filamentsand filament bundles. Both movements are energydependent and can be inhibited by nocodazole, but not bycytochalasin D. Finally, disassembly and reformation ofcytokeratin filament networks are documented in dividingcells revealing distinct and rapidly occurring stages ofcytokeratin organisation and distribution.
Movies available on-line:http://www.biologists.com/JCS/movies/jcs0841.htmlhttp://www.uni-mainz.de/FB/Medizin/Anatomie/Leube/
Key words: Cytoskeleton, Intermediate filament, Cytokeratin,Mitosis, GFP
SUMMARY
Detection of cytokeratin dynamics by time-lapse fluorescence microscopy in
living cells
Reinhard Windoffer and Rudolf E. Leube*
Department of Anatomy, Johannes Gutenberg-University Mainz, Becherweg 13, D-55099 Mainz, Germany*Author for correspondence (e-mail: [email protected])
Accepted 12 October; published on WWW 30 November 1999

4522
obtained at single time points from a large number of cells.This is particularly problematic when the examined cells donot undergo changes in a synchronised fashion and whenvariable phenotypic alterations are observed in different cellsof the same culture. These difficulties were realised early onin studies of mitotic CKF rearrangement which was shown todiffer between different cell types and even among individualcells in a given cell line (Franke et al., 1982, 1978a,b; Aubinet al., 1980; Horwitz et al., 1981; Lane et al., 1982; Celis etal., 1983; Jones et al., 1985; Turner and Ruane, 1985; Kitajimaet al., 1985; Tölle et al., 1987). With the methods at hand itwas at that time not possible to continuously follow CKFdynamics in single cells. Hence, it was not possible tounambiguously establish successive changes in IF organisationand to relate these changes to specific cellular conditions.Furthermore, it was difficult to determine whether CKFs areimmobile rods simply stabilising a rather rigid cytoskeletonor continuously flexible structures able to fine tuneinstantaneously according to specific cellular requirements bychanging shape, composition or spatial arrangement.
Monitoring of intracellular structures has become possibleby using fluorescent protein chimeras, allowing to tagspecifically the domain of interest. The green fluorescentprotein GFP, isolated originally from the jelly fish Aequoreavictoria, and several improved mutants have been particularlyuseful for the expression and detection of such fusion proteinsin living cells (Rizzuto et al., 1995). We now describe achimera consisting of human cytokeratin 13 and the GFP-variant enhanced green fluorescent protein (EGFP) thatintegrates into the IF cytoskeleton of stably transfected cells.We show that these labelled filaments can be visualised overextended time periods to characterise their dynamics ininterphase and during mitosis. The analyses of the obtainedimages demonstrate that CKFs are dynamic structuresdisplaying characteristic types of movement andrearrangement in interphase and mitosis.
MATERIALS AND METHODS
Construction of cDNA coding for CK 13-EGFP chimerasand expression in A-431 cellsTo prepare an expression construct for a CK 13-green fluorescentprotein (GFP) chimera a 1409 bp HindIIII/StuI-fragment coding forhuman CK 13 and lacking only the last carboxy-terminal codon forproline was first excised from plasmid CK133 (Kuruc et al., 1989;Hofmann and Franke, 1997). This fragment was subcloned intothe HindIII- and SmaI-sites of plasmid vector pBluescript KS+(Stratagene, La Jolla, CA) thereby generating plasmid pHK13-∆stop.The insert of this construct was subsequently excised with BamHI andHindIII and cloned into plasmid pEGFP-N3 (Clontech Laboratories,Palo Alto, CA). The resulting plasmid pHK13∆P.EGFP thereforecodes for almost the entire human CK 13 fused to the enhanced greenfluorescent protein (EGFP) at its carboxy terminus. Expression isdriven by the immediate early promoter of cytomegalovirus, and aneomycin-resistance cassette is present in the vector for selection.
Purified plasmid DNA was introduced into human vulvarcarcinoma-derived A-431 cells (line E3; cf. Moll et al., 1982; Leubeet al., 1988; ATCC CRL1555) using a modified calcium phosphateprecipitation method (cf. Leube et al., 1989). Stably transfected cellswere selected by addition of up to 1 mg/ml geneticin (Sigma, St Louis,MO). Individual colonies exhibiting filamentous fluorescence werepicked and subcloned for subsequent analyses.
Preparation and analysis of CK-enriched fractionsIF-containing fractions were prepared by standard methods (cf. Mollet al., 1982; Leube et al., 1988). Polypeptides contained in high salt-resistant pellets were separated by SDS-PAGE and stained withCoomassie Brilliant Blue (CBB; Serva, Heidelberg, FRG), orindividual polypeptides were identified after immunoblotting byspecific antibodies (see below), followed by horseradish peroxidase(HRP)-coupled secondary antibodies (Jackson ImmunoResearchLaboratories, West Grove, PA) and detection with anenhanced chemiluminescence system (Amersham Life Science,Buckinghamshire, UK).
Immunofluorescence microscopyFor indirect immunofluorescence microscopy cultured cells weregrown on coverslips, fixed with methanol (precooled to −20°C) for5 minutes and treated for 20 seconds with acetone (also precooled to−20°C). Cells were then dried, rehydrated in phosphate buffered saline(PBS) and incubated with primary antibodies for 30 minutes at roomtemperature, washed three times with PBS for 5 minutes each, andincubated with fluorochrome-labelled secondary antibodies for 30minutes. After several washes with PBS slides were mounted inElvanol, prepared by dissolving 1 g Mowiol 4-88 (CalbiochemGmbH, Frankfurt, Germany) in 4 ml distilled water and 2 ml glycerol,to which DAPI (Sigma) was added in some cases for nuclear staining.Fluorescence was viewed in an epifluorescence microscope(Axiophot, Carl Zeiss, Jena, FRG) using filtersets number 9 andnumber 2 from Zeiss and recorded with a digital camera (Hamamatsu4742-95, Hamamatsu, Herrsching, FRG).
The following primary antibodies were used: rabbit polyclonalantibodies raised against GFP but also reacting with EGFP (MolecularProbes, Eugene, OR), and monoclonal antibodies against human CK4 (6B10; Organon Teknika GmbH, Eppelheim, Germany), human CK8 (Ks8-17.2; kindly provided by Werner W. Franke, German CancerResearch Center, Heidelberg, FRG), human CK 13 (Ks13.1; ProgenBiotechnics, Heidelberg, FRG), human CK 18 (Ks18.174; ProgenBiotechnics), desmoplakin (DPI/II; Progen Biotechnics), and α-tubulin (Amersham Life Science). Secondary antibodies were TexasRed-conjugated goat anti-mouse IgG, and Texas Red-conjugated goatanti-rabbit IgG (both from Jackson ImmunoResearch Laboratories).In addition, Texas Red-coupled phalloidin (Molecular Probes) wasused to label actin filaments.
Immunoelectron microscopyFor immunoelectron microscopy cells were grown to high density onglass coverslips. Cells were briefly washed in PBS (37°C) and fixedfor 10 minutes at room temperature in 2% formaldehyde freshlyprepared in PBS. After washing (two times 5 minutes) in PBS cellswere permeabilised with 0.1% saponin (Sigma) in PBS for 5 minutesat room temperature and washed again twice with PBS for 5 minutes.To block unspecific antibody binding sites cells were incubated for 15minutes in 5% normal goat serum (Sigma) in PBS followed by a 5minute wash step with PBS. Rabbit anti-GFP antibodies (1:8000 inPBS) were applied for 2 hours (this step was omitted in the negativecontrol). After washing three times for 5 minutes in PBS cells wereincubated with 1 nm gold-conjugated goat anti rabbit-IgG (1:50 inPBS; Nanoprobes, Stony Brook, NY) overnight at 4°C and thenwashed three times for 5 minutes in PBS. A second fixation stepensued using 2.5% glutaraldehyde in PBS for 15 minutes at roomtemperature which was washed out with distilled water. For silverenhancement cells were washed twice for 10 minutes in Hepes buffer(50 mM Hepes, 200 mM sucrose, pH 5.8), and were then treated withHQ Silver (Nanoprobes) for 6 minutes followed by two 5 minutewashings in a solution containing 50 mM Hepes and 250 mM sodiumthiosulfate (pH 7.5) and two 5 minute washings in PBS. For finalfixation 0.2% osmium tetroxide was applied for 30 minutes at roomtemperature which was washed out with distilled water (two times 5minutes). Cells were dehydrated in a graded ethanol series. Propylene
R. Windoffer and R. E. Leube

4523Cytokeratin dynamics in living cells
oxide was used as intermedium and cells were embedded in Epon 812and polymerized. Ultrathin sections were cut with a Reichert-JungUltramicrotome (Leica, Bensheim, FRG) after which sections werestained with 8% uranylacetate (in water) for 10 minutes. Grids wereviewed and documented in an EM10 electron microscope (Carl Zeiss).
Time-lapse microscopyTo view living cells for several hours a culture chamber wasconstructed that was mounted directly on the stage of anepifluorescence microscope (Axiophot, Carl Zeiss). The circularchamber (15 mm diameter; 1.5 mm height) was embedded in a steelframe with three holes (1 mm diameter) for in- and eflux of culturemedium and a temperature sensor. The bottom of the chamber waslocated on top of another larger chamber (20 mm diameter; 8 mmheight) filled with water that was continuously replaced by preheatedwater to maintain the temperature in the culture chamber precisely at37°C as determined by a sensor that was placed next to the observationfield. A coverslip was placed on top of the culture chamber withadhering cells now growing in an inverted position, and the chamberwas shut tightly by screwing a steel plate on top of the entire assembly.During recording cells where maintained in phenol red-free Hanks’medium (containing Hanks’ salt solution, 25 mM Hepes, MEM non-essential amino acid solution and MEM amino acid solution, 100U/ml penicillin, 100 µg/ml streptomycin, 5% FCS (all from LifeTechnologies Rockville, USA), 4.8 mM N-acetyl-L-cysteine (Sigma),pH 7.4). The culture medium was exchanged either continuously orin single steps using a pump with regulatable flow rates. Cellsremained viable for several days in the culture chamber whileproliferating and adhering to the coverslip. In some set-ups Hanks’medium was replaced with Hanks’ medium containing themicrotubule-disrupting agent nocodazole (Sigma; 10 µg/ml stocksolution in DMSO) at concentrations varying between 10 and 100 nM,or Hanks’ medium containing the actin filament-disrupting agentcytochalasin D (Sigma; 10 µM stock solution in Hanks’ medium) atfinal concentrations of between 0.1 µM and 2.5 µM.
EGFP epifluorescence was recorded using filterset no.10 from Zeissand a Hamamatsu digital camera (see above). HPD-CPx software(Hamamatsu) was used to grab the images and to control a shutter(Zeiss) which was opened only during image acquisition. Therecorded images where edited with Image-Pro Plus software(Media Cybernetics, Silver Spring, CA) and converted into movies(QuickTime 3.0). They are available at http://www.uni-mainz.de/FB/Medizin/Anatomie/Leube/ and at http://www.biologists.com/JCS/movies/jcs0841.html. Photoshop software (Adobe Photoshop 5.0) wasused to edit pictures.
Enrichment of mitotic cellsMitotic cells were enriched by synchronization employing twosequential thymidine blocks (Stein et al., 1994). To this end,exponentially growing cells were incubated in serum-free mediumwith 2 mM thymidine (Sigma) for 12-16 hours. Afterwards, cells werewashed two times with serum-free medium and incubated for 9 hoursin normal serum-containing medium that was supplemented with 24µM deoxycytidine (Sigma). The cycle was repeated once morestarting with thymidine treatment, and mitotic cells were imaged afterthe second incubation period in Hanks’ medium.
RESULTS
HK13-EGFP fusion proteins integrate into bona fidedesmosome-anchored IF networks in transfectedcellsA cDNA was constructed coding for chimera HK13-EGFPconsisting of human CK 13 lacking only the carboxy-terminalproline and the optimised GFP variant EGFP at the carboxy
terminus (Fig. 1A). The chimerical cDNA was transfectedinto the human vulvar carcinoma-derived epithelial cell lineA-431 (clone E3) known for its abundance of CKs includingCK 13 (cf. Leube et al., 1988; Kuruc et al., 1989). Thesecells express no or low amounts of other cytoplasmic IFproteins (Lai et al., 1993; own observations). Fluorescencemicroscopy of transiently transfected A-431 cells, either priorto or after fixation showed variable patterns of HK13-EGFPdistribution ranging from extensive networks to cytoplasmicaggregates of differing shapes and sizes as exemplified in Fig.1B.
Stable cell clones were established by selecting homogenouscell populations that express HK13-EGFP in normal-appearingfilaments. Only clone AK13-1 is described in thiscommunication because comparable results were obtained inother stable clones prepared in parallel. Clone AK13-1 exhibitsa strong and homogenous filamentous EGFP-fluorescence inpractically all cells. The survey micrograph in Fig. 2B depictsthe presence of an intricate cytoplasmic network that isintegrated into a mesh encompassing the entire group of cells.This pattern is indistinguishable from that seen for CK 13 inthe parent A-431 clone (not shown). The strong fluorescence
Fig. 1. Schematic representation of chimera HK13-EGFP (A) andfluorescence microscopy detecting the chimera in transientlytransfected vulvar carcinoma-derived A-431 cells (B). (A) Schemeshowing elements of chimera HK13-EGFP encompassing (i) humanCK 13 with its entire N-terminal head domain (H), the central coiled-coil rod region (ROD) consisting of subdomains 1a, 1b and 2 that areseparated by short spacer sequences (black boxes), and the C-terminal tail (T) lacking only a single proline from its carboxyterminus, and (ii) enhanced green fluorescent protein (EGFP) inframe at the carboxy-terminal end. The short linker sequenceGGSIAT between both modules is not shown. (B) Fluorescencemicrograph depicting differences in distribution of HK13-EGFP 48hours after transfection with an appropriate expression vector. Notethe different fluorescence patterns in different cells expressing thesame polypeptides. Bar, 10 µm.

4524
was equally well detectable in fixed cells (Fig. 2B) and in livingcells (see below).
IF-enriched high salt pellet fractions were prepared fromAK13-1 and wild-type A-431 cells to compare theircomposition by SDS-PAGE and immunoblotting. AK13-1
cells differed from A-431 cells by the presence of an additionalmajor CBB-stained polypeptide band of approximately 75 kDawhich corresponds closely to the molecular mass of 77 kDacalculated for the chimerical HK13-EGFP fusion protein (Fig.2A). In addition, this band reacted with antibodies directedagainst CK 13 and EGFP (Fig. 2A).
The distribution of HK13-EGFP was further examined byfluorescent co-localisation with other cytoskeletal proteins.No significant co-localisation was noted with phalloidin-stained actin filaments (not shown) and α-tubulin-positivemicrotubules (Fig. 2B,B′). On the other hand, there was a nearperfect match between EGFP-fluorescence and CK 13-immunofluorescence (Fig. 3A,A′). Furthermore, the stainingpatterns for HK13-EGFP and its type II partner CK 4 werevirtually identical (Fig. 3B,B′). Codistribution was also evidentfor CK 8, another potential type II polymerisation partner ofCK 13 (Fig. 3C,C′). In addition, HK13-EGFP filaments co-localised in most parts with type I CK 18-containing filaments(Fig. 3D,D′). Fig. 3E,E′ shows that HK13-EGFP-filamentsmeet desmoplakin-positive desmosomes at cell borders.
Finally, electron microscopy was performed to examine theCKF system that had incorporated HK13-EGFP. Normal-appearing IFs were seen in all cells (not shown). Byimmunoelectron microscopy specific label for EGFP wasfound on single 10 nm filaments as well as on larger filamentbundles (Fig. 4A). Labelled filaments were also detected nextto desmosomes and seen to traverse the outer plaque region(Fig. 4B). Taken together, it is concluded that HK13-EGFPpermits to faithfully monitor the CKF system in AK13-1 cells.
CKFs are mobile in interphase cells To find out whether CKFs are static or motile structures singleAK13-1 cells were viewed in a small culture chamber placeddirectly on the stage of an epifluorescence microscope (seeMaterials and Methods). Cells proliferated and remained viablefor several days under these conditions as determined by time-lapse recordings of interference contrast micrographs showingcontinuing membrane ruffling, organelle movement, cellmigration and division (not shown). In this set-up fluorescencemicroscopy pictures were then recorded every 60 seconds.Comparison of pictures of a region of a single cell taken 23
R. Windoffer and R. E. Leube
Fig. 2. Characterization of A-431 subclone AK13-1 stablyexpressing chimera HK13-EGFP by CBB-staining and immunoblotanalyses of SDS-PAGE-separated polypeptides present in high saltpellets (A) and by double fluorescence microscopy (B,B′).(A) Cytoskeletal proteins were enriched from wild-type A-431 cells(WT) and from AK13-1 cells (AK13-1). Equal amounts wereseparated in 12% SDS-polyacrylamide gels and polypeptides wereeither stained with CBB (Coomassie) or transferred ontonitrocellulose, subsequently reacted with mAb Ks13.1 (anti-CK13),or rabbit antibodies recognizing EGFP (anti-EGFP). Antibodybinding was detected with HRP-labelled secondary antibodies and anenhanced chemiluminescence system. Chimera HK13-EGFP is seenas an additional polypeptide band of apparent molecular mass~75 kDa (arrow) in the CBB-stained gel reacting with bothantibodies in immunoblots. Position and molecular mass ofcoelectrophoresed marker proteins in kDa are given on left margin.(B,B′) Note the filamentous EGFP-fluorescence in all methanol-acetone fixed cells (B; HK13-EGFP). It differs from the patternobtained by indirect immunofluorescence microscopy using primaryantibodies against α-tubulin (B′; anti-tubulin). Bar, 5 µm.
kDa

4525Cytokeratin dynamics in living cells
minutes apart shows that the extended cytoplasmic filamentnetwork differs profoundly between individual time pointsindicating that major rearrangements must have taken place(Fig. 5). Obviously, only very few filaments stayed in placeduring the entire observation period (arrowheads in Fig. 5). Totrace filament fates in detail it was necessary to examine theentire set of photomicrographs which are provided as movie 1(accessible together with all other movies at http://www.uni-mainz.de/FB/Medizin/Anatomie/Leube/ and at http://www.biologists.com/JCS/movies/jcs0841.html). Viewing of thismovie and many other similar recordings revealed two majortypes of CK movement. First, undulations were seen wherebyfilaments appear to move without major overall alterations inorientation. This was mostly seen for long filaments some ofwhich attached to desmosomes in dense cultures, and filamentsthat were located in the deeper cytoplasm, i.e. in a region thatwas often encircled by CKFs and CKF bundles. The secondtype of movement was most prominent in isolated cells butoccurred also in closely attached cells. It was detected as acontinuous inward movement of fluorescence from the cellperiphery toward the inner filament ring. In some instances,brightfield micrographs were recorded in parallel that showedthat the movements observed were not induced by cellmovement and did not simply reflect changes in cell shape. Inaddition, no correlation between lamellipodial extensions andCK movements were noted.
To examine the latter inward-directed movement in moredetail cells were seeded at low density and subjected to time-lapse recording as the movement could best be documented insingle cells. A representative sequence is shown in Fig. 6 andmovie 2. The movement appeared to originate from diffusefluorescence at the cell cortex just beneath the plasmamembrane subsequently accumulating in small dots and shortfilaments. These structures were seen to migrate into the deepercytoplasm toward the inner ring of CKFs. The fluorescent dotswere used as reference points to follow the movement as shownparadigmatically in Fig. 6 and movie 2. The speed of these dotsvaried considerably between different cells and ranged fromless than 100 nm/minute to more than 500 nm/minute. The dotsand attached filaments finally coalesced with the innerfilaments after which their fate could not be followed any more.
By time-lapse microscopy we were able to identify andmonitor migrating cells in large fields of sparsely-seeded cells.Some of these cells migrated over considerable distances (onewas filmed for 8.5 hours moving at an average speed of 13.5µm/hour) forming occasionally transient or stable contactswith other cells. Although it was difficult to keep filaments infocus in these rounded cells, the CK network and CK dynamicsappeared to be maintained during all phases (not shown). Theresolution, however, was not sufficient to pinpoint localchanges that seemed to occur at newly formed cell contactsites.
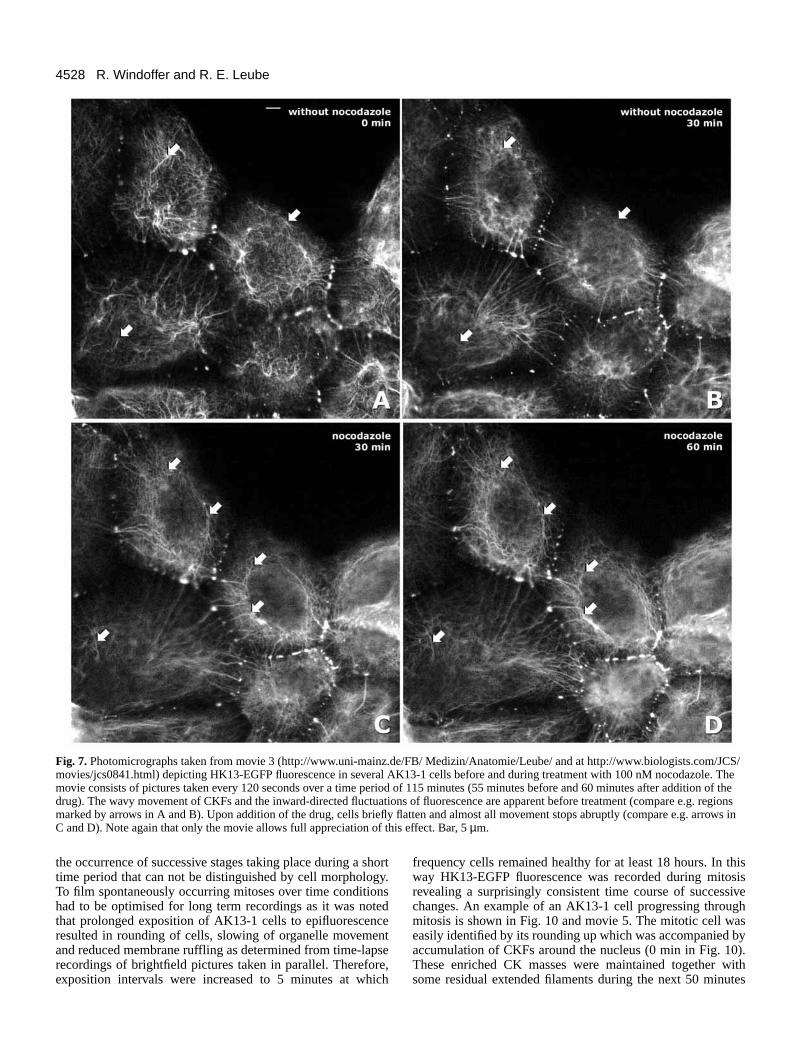
CK movements depend on microtubulesTo find out whether the above described CK dynamics rely onother major cytoskeletal networks the effect of microtubule-and microfilament-disrupting drugs was examined. First,nocodazole was applied at concentrations sufficient to disruptmicrotubules in AK13-1 cells. Disruption was confirmed byantibody staining of α-tubulin (not shown). The pictures inFig. 7 are taken from movie 3 showing the HK13-EGFP
Fig. 3. Fluorescent microscopical co-localization of chimera HK13-EGFP (A-E; HK13-EGFP) stably expressed in A-431 clone AK13-1together with endogenous CKs and desmosomal proteins. Cells werefixed with methanol/acetone and indirect immunofluorescencemicroscopy using Texas Red-coupled secondary antibodies wasperformed to detect CK 13 (A′; anti-ck13), CK 4 (B′; anti-ck4),cytokeratin 8 (C′; anti-ck8), CK 18 (D′; anti-ck18) and thedesmosomal plaque component desmoplakin (E′; anti-DP). Comparethe corresponding micrographs and note the almost complete co-distribution of HK13-EGFP with endogenous CKs and co-localization at desmoplakin-positive cell-cell contacts. Bars, 5 µm.

4526
fluorescence in cells before and after addition of nocodazole.Both, undulations and inward-directed movements stoppedabruptly, i.e. within minutes after addition of the drug. Theentire network seemed to be immobilised and very little CK-motion was detected (compare structures labelled by arrows inFig. 7C,D). Brightfield pictures showed that lamellipodialmovements still continued in the presence of the drug whilethe motility of cytoplasmic organelles was considerably sloweddown. CK movements recovered after different lag periodsfollowing removal of the drug. The inhibitory effect ofnocodazole on CK movements was also observed at muchlower concentrations when no alterations in microtubuleorganisation were evident (not shown).
Similarly, cells were treated with cytochalasin D atconcentrations at which disruption of the microfilament systemoccurred in AK13-1 cells as assessed by phalloidin-labellingof actin fibres. Fig. 8 and the corresponding movie 4 show asequence of fluorescence pictures depicting two cells beforeand after addition of 0.1 µM cytochalasin D. The cellscontracted immediately (within minutes) after addition of thedrug. Concurrently, CKFs retracted from the periphery
R. Windoffer and R. E. Leube
Fig. 4. Immunogold electron microscopy of A-431 clone AK13-1 detecting chimera HK13-EGFP using rabbit antibodies recognizing EGFP.Note the abundant immunolabel (silver amplification technique) of single 8-10 nm filaments (small arrows) and filament bundles (large arrows)in the cytoplasm (A) and of filaments inserting into a large desmosome in B. N, nucleus. Bars, 0.5 µm.
Fig. 5. Three representative fluorescence micrographs taken from atime-lapse recording (movie 1; http://www.uni-mainz.de/FB/Medizin/Anatomie/Leube/ and at http://www.biologists.com/JCS/movies/jcs0841.html) showing alterations in distribution of HK13-EGFP in aliving AK13-1 cell. The movie covers 46 minutes and is composed ofpictures taken every 60 seconds showing that the HK13-EGFP-containing filaments are highly dynamic structures with only fewpresent during the entire observation period (arrowheads) but mostdisappearing (small arrows denoting filament gone by 23 minutes;large arrows delineating filament gone after 46 minutes). Two types ofmovement are discernible in the movie but can not be visualised instatic micrographs: (i) slow filament undulations predominantlyoccurring at the central parts of the cell, and (ii) inward-directed flowof fluorescence from the cell periphery toward the inner cytoplasm.Bar, 5 µm.

4527Cytokeratin dynamics in living cells
and accumulated in dense massesasymmetrically around the nucleus.Remarkably and in contrast to nocodazole-treated cells, the inward-directedmovement and wavy movements did notstop. The cells shown in Fig. 8 did notcontract very much but were chosen fordocumentation because the inward flow ofCK-reactivity could still be resolved in themovie (see also arrows in Fig. 8). Removalof cytochalasin D resulted in spreading ofcells within 10 minutes with continuedmovement of CKs (not shown).
Cells were treated with 50 mM 2-deoxy-D-glucose and 0.05% sodium azide(cf. Yoon et al., 1998) to determinewhether the observed microtubule-dependent movements require ATP. Withinless than 10 minutes almost all motionstopped in a manner comparable to thatseen in nocodazole-treated cells (notshown). Furthermore, CK movementsregained within 10 minutes after removalof the agents.
CKs are completely reorganizedduring mitosisProbably the most confusing and at thesame time most fascinating aspects of CK dynamics are theirfundamental rearrangements during mitosis. There have beena number of contradicting reports, even in the case of A-431cells where most observations agree that filaments are almostcompletely disassembled (Horwitz et al., 1981; Lane et al.,1982) although one communication claimed that CKFs wheremaintained (Tölle et al., 1987). Examination of spontaneouslyoccurring cell divisions of AK13-1 cells also revealed acomplex and heterogenous pattern of HK13-EGFPorganisation during mitotic stages (Fig. 9). The three cells, all
at pro-metaphase, exhibit three different phenotypes of the CKsystem. In one case a somewhat reduced but extended filamentnetwork was discernible (Fig. 9A), while in another cellfilaments were concentrated in the pericentriolar region (Fig.9B). In yet another cell no CKFs were seen, but only spheroidalgranular aggregates (also referred to as speckles in theliterature) were found distributed throughout the cytoplasm(Fig. 9C). In all instances, diffuse staining was noticeable todifferent degrees. These observations could be explained eitherby different patterns of CK-distribution in different cells, or by
Fig. 6. Images taken from a series offluorescence recordings of a single AK13-1cell illustrating inward movement of HK13-EGFP from the cell periphery toward innercircularly-arranged filament bundles. Imagesare shown inversely to optimize views. Tomeasure the inward-directed peripheralmovement, fluorescent ‘dots’ were tracedthrough several frames. Circles mark four suchdots whose position was followed. The pathtaken is shown in the first enlarged andduplicated micrograph by black lines withmeasuring points indicated by white dots. Thecomplexity and dynamics of the movementscan only be appreciated from the movie (movie2; http://www.uni-mainz.de/FB/Medizin/Anatomie/Leube/ and at http://www.biologists.com/JCS/movies/jcs0841.html) that was madeof pictures taken at 120 second intervals andshows a 240 minutes observation periodencompassing also the frames depicted in thisFig. 6 (arrows denoting the encircled dots).Bars, 5 µm (same magnification in lower sixpictures).
4528
the occurrence of successive stages taking place during a shorttime period that can not be distinguished by cell morphology.To film spontaneously occurring mitoses over time conditionshad to be optimised for long term recordings as it was notedthat prolonged exposition of AK13-1 cells to epifluorescenceresulted in rounding of cells, slowing of organelle movementand reduced membrane ruffling as determined from time-lapserecordings of brightfield pictures taken in parallel. Therefore,exposition intervals were increased to 5 minutes at which
frequency cells remained healthy for at least 18 hours. In thisway HK13-EGFP fluorescence was recorded during mitosisrevealing a surprisingly consistent time course of successivechanges. An example of an AK13-1 cell progressing throughmitosis is shown in Fig. 10 and movie 5. The mitotic cell waseasily identified by its rounding up which was accompanied byaccumulation of CKFs around the nucleus (0 min in Fig. 10).These enriched CK masses were maintained together withsome residual extended filaments during the next 50 minutes
R. Windoffer and R. E. Leube
Fig. 7. Photomicrographs taken from movie 3 (http://www.uni-mainz.de/FB/ Medizin/Anatomie/Leube/ and at http://www.biologists.com/JCS/movies/jcs0841.html) depicting HK13-EGFP fluorescence in several AK13-1 cells before and during treatment with 100 nM nocodazole. Themovie consists of pictures taken every 120 seconds over a time period of 115 minutes (55 minutes before and 60 minutes after addition of thedrug). The wavy movement of CKFs and the inward-directed fluctuations of fluorescence are apparent before treatment (compare e.g. regionsmarked by arrows in A and B). Upon addition of the drug, cells briefly flatten and almost all movement stops abruptly (compare e.g. arrows inC and D). Note again that only the movie allows full appreciation of this effect. Bar, 5 µm.

4529Cytokeratin dynamics in living cells
when the cell reached the final stages of prophase. Around thistime point almost all filamentous material disappeared andonly dots were visible together with diffuse cytoplasmicstaining. This transition was always very rapid and usually tookless than 10 minutes, i.e. two frames recorded (compare 50 minand 60 min in Fig. 10). The dotted material was very mobilemoving around the cytoplasm in various directions at highspeed. As the cells progressed through anaphase and telophasethe diffuse and dotted fluorescence remained. Enrichment ofaggregated material that sometimes coalesced was seenoccasionally at opposite cell poles (see 120 min in Fig. 10),and reformation of filamentous material was often noticeablebefore cytokinesis was completed. Remarkably, an enhanced
diffuse fluorescence was consistently seen below the plasmamembrane after cell division (130 min in Fig. 10) which wassubsequently further enriched in focal areas, mostly in regionswere cells had separated resulting in mirror images of labellingin both abutting cell poles (140 min in Fig. 10). From thisregion and other accumulations filamentous material extended(150 min in Fig. 10), and a new network was established. Inthe rounded cells shown in Fig. 10 some filaments remained indense accumulations around the nucleus (275 min) but in cellsconnected circumferentially to neighbouring cells bydesmosomes an extended network was seen soon aftercompletion of mitosis (see also below).
To further investigate the reproducibility of the observed
Fig. 8. HK13-EGFP fluorescence of two AK13-1 cells before and during cytochalasin D incubation (0.1 µM). Images are from movie 4(pictures taken every 60 seconds for 115 minutes) which is available at http://www.uni-mainz.de/FB/Medizin/Anatomie/Leube/ and athttp://www.biologists.com/JCS/movies/jcs0841.html. The cells show an extended cytoplasmic filament network at the beginning of therecording period (−55 minutes). Cells were first observed for 55 minutes in medium without cytochalasin D showing the characteristic inwardflow of fluorescence from the cell periphery and the wavy movement of inner filaments (see movie). After addition of cytochalasin D to theculture medium cells contract and CKFs condense around the nucleus (arrowheads in image taken at 41 minutes). Despite this condensation thewavy movement and inward-directed movement are still maintained. The latter is illustrated in images taken at 41 minutes and 51 minutes (seealso insets at two times higher magnification) by arrow that marks a fluorescent dot that moved appr. 1.6 µm toward the cell centre during thistime (bracket), i.e. appr. 266 µm/minute roughly corresponding to the mean speed of inward-movement determined from untreated cells. Notethat the complexity of dynamic movements can only be seen in the movie in which presentation was optimised to follow the comparativelyweak fluorescence in the cell periphery. Bar, 5 µm.
Fig. 9. Triple fluorescence microscopy of cells at comparable stages of mitosis (pro-metaphase) depicting different patterns of HK13-EGFPorganisation and distribution in AK13-1 cells. Cells were fixed with methanol/acetone and HK13-EGFP was visualised by direct excitation(green), tubulin was detected by indirect immunofluorescence microscopy with anti α-tubulin antibodies (red), and DNA was stained withDAPI (blue). The micrographs show that HK13-EGFP is either present in a residual extended network with diffuse cytoplasmic staining (A), orforms a highly aggregated filament system in the pericentriolar region with increased diffuse cytoplasmic staining (B), or is seen as‘speckles’/spheroidal granular aggregates throughout the cytoplasm together with diffuse fluorescence (C). Bar, 5 µm.

4530
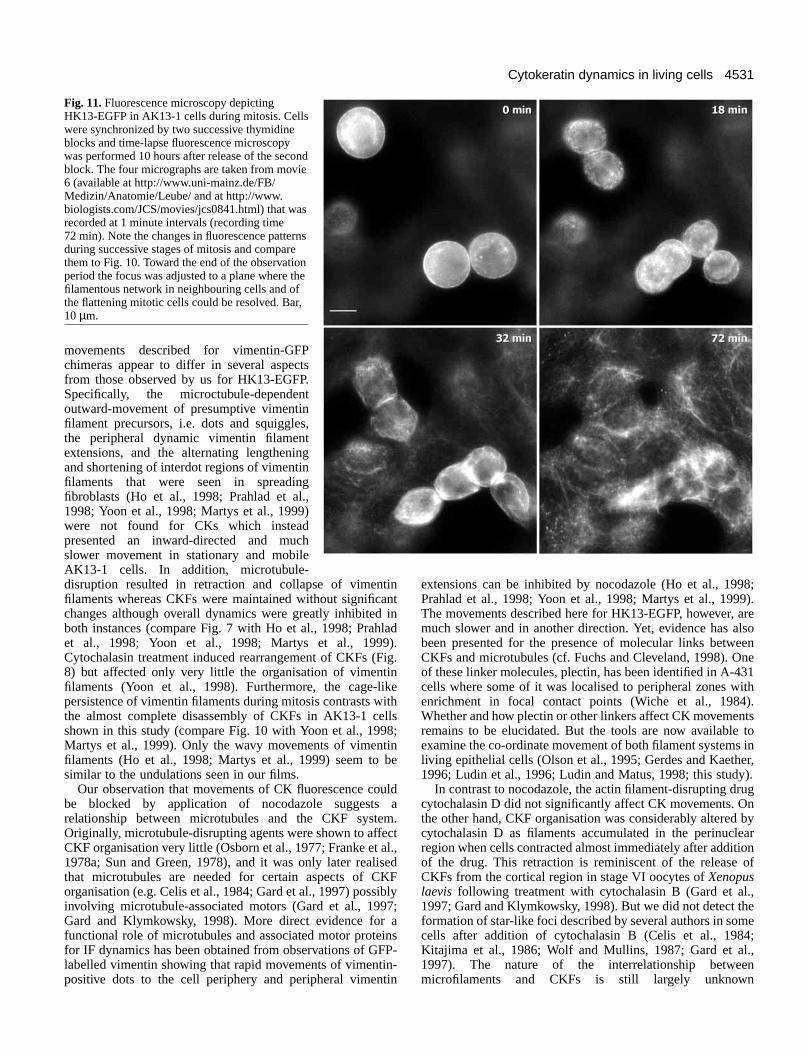
phenomena mitotic cells were enriched by subjecting AK13-1cells to successive cycles of thymidine treatment.Approximately 9 hours after the second thymidine block,multiple dividing cells could be captured in single frames. Oneexample is depicted in Fig. 11 and the corresponding movie 6in which three mitotic cells are seen that had already roundedup and had disassembled almost all of their CK filaments (0min in Fig. 11). Soon afterwards, cytokinesis started (18 minin Fig. 11). The local concentration of HK13-EGFPfluorescence below the plasma membrane with localaccumulations in areas where cell separation had taken placewas also apparent (e.g. lower cells at 32 min in Fig. 11), anda normal-appearing filamentous network was formed whilecells re-integrated into the coherent monolayer (upper cells at72 min in Fig. 11).
DISCUSSION
We report the generation of cell line AK13-1 synthesisingfluorescent CKFs for over 80 passages which wasaccomplished by stable integration of an expression constructcoding for a chimerical polypeptide consisting of human CK13 and EGFP fused to its carboxy terminus. We have shownthat the chimera was integrated into the endogenous filamentnetwork of stably transfected epithelial A-431 cells withoutnoticeable disturbance and can thus be used as a reliable tagfor the abundant CKFs present in this cell line. Extendedfluorescent microscopical observation of AK13-1 cells did not
result in significant fading of fluorescence althoughphototoxicity was noted after long exposure times at highfrequency. The cell system offers the opportunity to examinethe dynamic behaviour of CKFs and their subunit polypeptidesunder controlled in vitro conditions and enabled us toinvestigate changes of CK organisation and distribution thatoccur physiologically under standard culture conditionsspecifically those that take place in single cells and cell groupsduring interphase and mitosis. By this method it was possible,for the first time, to directly monitor and document the mobilityof CKFs, demonstrating that they form a dynamic and flexiblenetwork. In addition, novel types of microtubule-dependentCK movements, i.e. slow undulations of central filaments andperipheral inward-directed fluctuations of fluorescence, werediscerned. Several observations support the conclusion thatthese movements are not simply due to cell shape changes: (i)Brightfield pictures that were recorded in parallel showedlamellipodial movements and cell shape changes that were notlinked to CK movements. (ii) CK movements were the samewhether single cells were imaged or cells contained in densemonolayers (compare e.g. movies 1, 2 with movie 3). (iii)Lamellipodial extensions continued while CK movementsstopped in the presence of nocodazole (movie 3). (iv) Cellshape changes induced by cytochalasin D did not affect CKmovements significantly (movie 4).
It is important to note that the different IF types differsignificantly in many functional and dynamic aspects and thatresults obtained with one type can not necessarily beextrapolated to another. Therefore it is not surprising that
R. Windoffer and R. E. Leube
Fig. 10. HK13-EGFPfluorescence microscopy duringmitotic stages of a single AK13-1cell. The whole sequence ofpictures taken at 5 minuteintervals (recording time 280minutes) is available as movie 5 athttp://www.uni-mainz.de/FB/Medizin/Anatomie/Leube/ and athttp://www.biologists.com/JCS/movies/jcs0841.html (each framedisplayed in quadruplicate forbetter performance). Time point0 min shows a cell that hasalready rounded up and whoseCKFs have mostly aggregatedaround the nucleus. Late atprophase (50 min) a similarpattern is still prevalent, butaggregates and remainingfilaments are disassembledabruptly within less than 10minutes (60 min). Note that at thistime point only diffuse stainingand speckles/granular spheroidalaggregates are seen. Thisfluorescence pattern is maintainedduring the next hour with someenrichment at opposite cell poles(120 min). After cytokinesis (130min) a submembraneousenrichment of diffuse staining isvisible which further accumulates symmetrically to the plane of division at 140 min. Filaments emanating from the strongly fluorescentmaterial are seen 10 minutes later (150 min). A CK network with perinuclearly located CKFs are seen 125 minutes later (275 min). Bar, 5 µm.
4531Cytokeratin dynamics in living cells
movements described for vimentin-GFPchimeras appear to differ in several aspectsfrom those observed by us for HK13-EGFP.Specifically, the microctubule-dependentoutward-movement of presumptive vimentinfilament precursors, i.e. dots and squiggles,the peripheral dynamic vimentin filamentextensions, and the alternating lengtheningand shortening of interdot regions of vimentinfilaments that were seen in spreadingfibroblasts (Ho et al., 1998; Prahlad et al.,1998; Yoon et al., 1998; Martys et al., 1999)were not found for CKs which insteadpresented an inward-directed and muchslower movement in stationary and mobileAK13-1 cells. In addition, microtubule-disruption resulted in retraction and collapse of vimentinfilaments whereas CKFs were maintained without significantchanges although overall dynamics were greatly inhibited inboth instances (compare Fig. 7 with Ho et al., 1998; Prahladet al., 1998; Yoon et al., 1998; Martys et al., 1999).Cytochalasin treatment induced rearrangement of CKFs (Fig.8) but affected only very little the organisation of vimentinfilaments (Yoon et al., 1998). Furthermore, the cage-likepersistence of vimentin filaments during mitosis contrasts withthe almost complete disassembly of CKFs in AK13-1 cellsshown in this study (compare Fig. 10 with Yoon et al., 1998;Martys et al., 1999). Only the wavy movements of vimentinfilaments (Ho et al., 1998; Martys et al., 1999) seem to besimilar to the undulations seen in our films.
Our observation that movements of CK fluorescence couldbe blocked by application of nocodazole suggests arelationship between microtubules and the CKF system.Originally, microtubule-disrupting agents were shown to affectCKF organisation very little (Osborn et al., 1977; Franke et al.,1978a; Sun and Green, 1978), and it was only later realisedthat microtubules are needed for certain aspects of CKForganisation (e.g. Celis et al., 1984; Gard et al., 1997) possiblyinvolving microtubule-associated motors (Gard et al., 1997;Gard and Klymkowsky, 1998). More direct evidence for afunctional role of microtubules and associated motor proteinsfor IF dynamics has been obtained from observations of GFP-labelled vimentin showing that rapid movements of vimentin-positive dots to the cell periphery and peripheral vimentin
extensions can be inhibited by nocodazole (Ho et al., 1998;Prahlad et al., 1998; Yoon et al., 1998; Martys et al., 1999).The movements described here for HK13-EGFP, however, aremuch slower and in another direction. Yet, evidence has alsobeen presented for the presence of molecular links betweenCKFs and microtubules (cf. Fuchs and Cleveland, 1998). Oneof these linker molecules, plectin, has been identified in A-431cells where some of it was localised to peripheral zones withenrichment in focal contact points (Wiche et al., 1984).Whether and how plectin or other linkers affect CK movementsremains to be elucidated. But the tools are now available toexamine the co-ordinate movement of both filament systems inliving epithelial cells (Olson et al., 1995; Gerdes and Kaether,1996; Ludin et al., 1996; Ludin and Matus, 1998; this study).
In contrast to nocodazole, the actin filament-disrupting drugcytochalasin D did not significantly affect CK movements. Onthe other hand, CKF organisation was considerably altered bycytochalasin D as filaments accumulated in the perinuclearregion when cells contracted almost immediately after additionof the drug. This retraction is reminiscent of the release ofCKFs from the cortical region in stage VI oocytes of Xenopuslaevis following treatment with cytochalasin B (Gard et al.,1997; Gard and Klymkowsky, 1998). But we did not detect theformation of star-like foci described by several authors in somecells after addition of cytochalasin B (Celis et al., 1984;Kitajima et al., 1986; Wolf and Mullins, 1987; Gard et al.,1997). The nature of the interrelationship betweenmicrofilaments and CKFs is still largely unknown
Fig. 11. Fluorescence microscopy depictingHK13-EGFP in AK13-1 cells during mitosis. Cellswere synchronized by two successive thymidineblocks and time-lapse fluorescence microscopywas performed 10 hours after release of the secondblock. The four micrographs are taken from movie6 (available at http://www.uni-mainz.de/FB/Medizin/Anatomie/Leube/ and at http://www.biologists.com/JCS/movies/jcs0841.html) that wasrecorded at 1 minute intervals (recording time72 min). Note the changes in fluorescence patternsduring successive stages of mitosis and comparethem to Fig. 10. Toward the end of the observationperiod the focus was adjusted to a plane where thefilamentous network in neighbouring cells and ofthe flattening mitotic cells could be resolved. Bar,10 µm.

4532
although observations have been presented demonstratingmorphological and functional linkage between both (Hirokawaet al., 1982; Green et al., 1987; Yang et al., 1996; Toivala etal., 1997).
Close inspection of HK13-EGFP inward-directedmovements showed that waves of diffuse, i.e. non-filamentous,fluorescence were generated at the most peripheral parts ofcells that subsequently seemed to accumulate in dots and/orfilamentous structures which then moved further toward innercell regions where they appeared to fuse with other moredensely packed filaments. To maintain homeostasis within acell this net inward-movement must be counterbalanced. Butwe were not able to detect an outward-directed movement offilaments in AK13-1 cells. Therefore, HK13-EGFP must beprovided locally in another form. Two mechanisms couldaccount for this: either HK13-EGFP is translated selectively inthe cell periphery and/or there is an outward flow of ‘invisible’HK13-EGFP. Indeed, compartmentalised translation has beendescribed for another IF protein, vimentin, whose mRNA wasshown to be preferentially distributed near the nucleus incultured cells (Lawrence and Singer, 1986). It is noteworthy,however, that fluorophore formation involves time-consumingposttranslational modifications (e.g. Heim et al., 1994). As anexplanation for the second mechanism, it is important to knowthat a certain percentage of CK subunits is present in a solublepool consisting of tetramers and/or short oligomers (Franke etal., 1987; Klymkowsky et al., 1991; Chou et al., 1993; Bachantand Klymkowsky, 1996) that is difficult to visualise byfluorescence microscopy. Local conditions could create agradient of these soluble CK forms. Factors such as therecently identified solubilisation co-factor 14-3-3 whichpreferentially associates with hyperphosphorylated CKsubunits (Liao and Omary, 1996; Ku et al., 1998) could favourthe topologically restricted availability of CKs. In support,phosphorylated CKs were shown to be enriched in thebasolateral and apical domain of hepatocytes and exocrinepancreatic acinar cells (Liao et al., 1995). Furthermore, ademand for soluble CK subunits may be created by theabundant presence of intermediate filament associated proteins(IFAPs) in cortical cell regions such as plectin (Wiche et al.,1984; Foisner et al., 1988; Eger et al., 1997), the desmosomalcomponents desmoplakin and plakophilins, and thehemidesmosome proteins plectin/HD1/IFAP300, BPAG1e andβ4 integrin (for review Schmidt et al., 1994; Bornslaeger et al.,1997; Hatzfeld, 1997; Fuchs et al., 1997).
If there is indeed a continuous replenishment of soluble andhyperphosphorylated CK subunits in the cell peripheryregional filament growth and assembly could be initiated fromthis area. The increased diffuse staining in the most peripheralparts supports this notion. This appears to be in contrast toother studies in which organising centres for local CKFassembly were thought to be located either at desmosomes(Bologna et al., 1986), the nucleus (Eckert et al., 1982; Albersand Fuchs, 1989) or throughout the cytoplasm (Kreis et al.,1983; Franke et al., 1984; Miller et al., 1991, 1993; Paramioand Jorcano, 1994; Paramio, 1999). It should be kept in mind,however, that cells were flooded with CKs in those experimentswhile we looked at steady state distributions of CKpolypeptides, and future experiments are needed to resolvethese discrepancies.
Recent studies had indicated that CKFs are broken down to
variable degrees during mitosis in cultured cells depending onthe cell type, even the specific subclone and the particularculture conditions (for ref. see Introduction). In native tissuesCKFs appear to be disrupted during mitosis in most instances(Brown et al., 1983). Initially, we noted different types offilament organisation during mitosis in AK13-1 cells (e.g. Fig.9) but all mitotic cells that we filmed so far exhibited an almostcomplete filament breakdown and a comparable time course ofCK rearrangements. This suggests that at least some of thedifferent phenotypes observed by us and also by othersrepresent successive stages that could not be resolved in thestill pictures of seemingly identical mitotic stages. Anotheradvantage of the methodology used in this study is that it is notinfluenced by epitope masking which may have alsocontributed to the confusion in the analysis of mitotic CKrearrangements. In fact, selective CK epitope availability hasbeen documented during mitosis in PtK2 cells (Franke et al.,1983) and was exploited for the specific detection ofphosphorylated CK isoforms (Liao et al., 1995). We showedthat ordered CK rearrangements take place during pro-metaphase. First, CKFs accumulate in perinuclear aggregates.Some filaments are usually retained until late during this stagewhen within less than 10 minutes almost all filamentousmaterial disappears. It will be interesting to find out whetherthis process is under the influence of specific kinases known toaffect CKF stability in vivo (e.g. Deery, 1993; Toivala et al.,1997; Paramio, 1999) and/or severing enzymes (for such amechanism during oocyte maturation see Klymkowsky et al.,1991). Disassembly is followed by reformation of new CKFswhich is initiated from cytoplasmic aggregates and fromamorphous fluorescence that is enriched underneath theplasma membrane in late telophase. This hitherto unknownsubmembraneous accumulation of non-filamentous CKsduring mitosis is another indication of the importance of thecortical region for CKF formation. It gives direct evidence forthe existence of a soluble and assembly-competent CK pool inthe cell periphery. Furthermore, the enrichment of this materialin regions where cell separation had taken place andsubsequently also in other restricted areas show that these CKsare subject to topological cues acting as regulators of subunitaggregation and filament formation.
The knowledge gained by time-lapse microscopy offluorescent CKs in living cells has helped us to begin to unravelthe complexity of their temporospatially-determined modesof dynamic behaviour which is of importance for theunderstanding of CK rearrangements that take place duringembryogenesis, migration and tissue proliferation.
The authors thank Antje Leibold, Sabine Thomas, Ursula Wilhelm,and Bernhard Beile for engaged and expert technical assistance. Wealso thank Dr Harald Herrmann (German Cancer Research Center,Heidelberg, FRG) for critical reading of the manuscript and helpfulcomments, Dr Werner W. Franke (German Cancer Research Center,Heidelberg, FRG) for gifts of antibodies, and Drs Andreas Hofer andRichard Funk (Institute of Anatomy, University Dresden, FRG) forhelpful ideas to optimise the culture chamber. The work wassupported by the Stiftung Rheinland-Pfalz für Innovation.
REFERENCES
Albers, K. and Fuchs, E. (1989). Expression of mutant keratin cDNAs in
R. Windoffer and R. E. Leube

4533Cytokeratin dynamics in living cells
epithelial cells reveals possible mechanisms for initiation and assembly ofintermediate filaments. J. Cell Biol. 108, 1477-1493.
Aubin, J. E., Osborn, M., Franke, W. W. and Weber, K. (1980).Intermediate filaments of the vimentin-type and the cytokeratin-type aredistributed differently during mitosis. Exp. Cell Res. 129, 149-165.
Bachant, J. B. and Klymkowsky, M. W. (1996). A nontetrameric species isthe major soluble form of keratin in Xenopus oocytes and rabbit reticulocytelysates. J. Cell Biol. 132, 153-165.
Bologna, M., Allen, R. and Dulbecco, R. (1986). Organization ofcytokeratin bundles by desmosomes in rat mammary cells. J. Cell Biol.102, 560-567.
Bornslaeger, E. A., Kowalczyk, A. P., Meng, J.-J., Ip, W. and Green, K. J.(1997). The role of the desmosomal plaque protein desmoplakin inintermediate filament anchorage and junction assembly. In Cytoskeletal-Membrane Interactions and Signal Transduction (ed. P. Cowin and M. W.Klymkowsky), pp. 181-198. Heidelberg: Springer.
Brown, D. T., Anderton, B. H. and Wylie, C. C. (1983). Alterations in theorganisation of cytokeratin filaments in normal and malignant humancolonic epithelial cells during mitosis. Cell Tissue Res. 233, 619-628.
Celis, J. E., Larsen, P. M., Fey, S. J. and Celis, A. (1983). Phosphorylationof keratin and vimentin polypeptides in normal and transformed mitotichuman epithelial amnion cells: behavior of keratin and vimentin filamentsduring mitosis. J. Cell Biol. 97, 1429-1434.
Celis, J. E., Small, J. V., Larsen, P. M., Fey, S. J., De Mey, J. and Celis, A.(1984). Intermediate filaments in monkey kidney TC7 cells: focal centersand interrelationship with other cytoskeletal systems. Proc. Nat. Acad. Sci.USA 81, 1117-1121.
Chou, C.-F., Riopel, C. L., Rott, L. S. and Omary, M. B. (1993). Asignificant soluble keratin fraction in ‘simple’ epithelial cells. Lack of anapparent phosphorylation and glycosylation role in keratin solubility. J. CellSci. 105, 433-444.
Deery, W. J. (1993). Role of phosphorylation in keratin and vimentin filamentintegrity in cultured thyroid epithelial cells. Cell Motil. Cytoskel. 26, 325-339.
Eckert, B. S., Daley, R. A. and Parysek, L. M. (1982). Assembly of keratinonto PtK1 cytoskeletons: evidence for an intermediate filament organizingcenter. J. Cell Biol. 92, 575-578.
Eger, A., Stockinger, A., Wiche, G. and Foisner R. (1997). Polarisation-dependent association of plectin with desmoplakin and the lateralsubmembrane skeleton in MDCK cells. J. Cell Sci. 110, 1307-1316.
Eriksson, J. E., Opal, P. and Goldman, R. D. (1992). Intermediate filamentdynamics. Curr. Opin. Cell Biol. 4, 99-104.
Foisner, R., Leichtfried, F. E., Herrmann, H., Small, J. V., Lawson, D. andWiche, G. (1988). Cytoskeleton-associated plectin: in situ localization, invitro reconstitution, and binding to immobilized intermediate filamentproteins. J. Cell Biol. 106, 723-733.
Franke, W. W., Schmid, E., Osborn, M. and Weber, K. (1978a). Differentintermediate-sized filaments distinguished by immunofluorescencemicroscopy. Proc. Nat. Acad. Sci. USA 75, 5034-5038.
Franke, W. W., Weber, K., Osborn, M., Schmid, E. and Freudenstein, C.(1978b). Antibody to prekeratin. Decoration of tonofilament-like arrays invarious cells of epithelial character. Exp. Cell Res. 116, 429-445.
Franke, W. W., Schmid, E., Grund, C. and Geiger, B. (1982). Intermediatefilament proteins in nonfilamentous structures: transient disintegration andinclusion of subunit proteins in granular aggregates. Cell 30, 103-113.
Franke, W. W., Schmid, E., Wellsteed, J., Grund, C., Gigi, O. and Geiger,B. (1983). Change of cytokeratin filament organization during the cell cycle:selective masking of an immmunologic determinant in interphase PtK2 cells.J. Cell Biol. 97, 1255-1260.
Franke, W. W., Schmid, E., Mittnacht, S., Grund, C. and Jorcano, J. L.(1984). Integration of different keratins into the same filament system aftermicroinjection of mRNA for epidermal keratins into kidney epithelial cells.Cell 36, 813-825.
Franke, W. W., Winter, S., Schmid, E., Söllner, P., Hämmerling, G. andAchtstätter, T. (1987). Monoclonal cytokeratin antibody recognizing aheterotypic complex: immunological probing of conformational states ofcytoskeletal proteins in filaments and in solution. Exp. Cell Res. 173, 17-37.
Fuchs, E. and Weber, K. (1994). Intermediate filaments: structure, dynamics,function and disease. Annu. Rev. Biochem. 63, 345-382.
Fuchs, E., Yang, Y., Dowling, J., Yu, Q.-C. and Guo, L. (1997). Intermediatefilament cytoarchitecture and BPAG1: a gene encoding two different typesof intermediate linker proteins. In Cytoskeletal-Membrane Interactions andSignal Transduction (ed. P. Cowin and M. W. Klymkowsky), pp. 167-180.Heidelberg: Springer.
Fuchs, E. and Cleveland, D. W. (1998). A structural scaffolding ofintermediate filaments in health and disease. Science 279, 514-519.
Gard, D. L., Cha, B. J. and King, E. (1997). The organization and animal-vegetal asymmetry of cytokeratin filaments in stage VI Xenopus oocytes isdependent upon f-actin and microtubules. Dev. Biol. 184, 95-114.
Gard, D. L. and Klymkowsky, M. W. (1998). Intermediate filamentorganization during oogenesis and early development in the clawed frog,Xenopus laevis. In Intermediate Filaments (ed. H. Herrmann and J. R.Harris), pp. 35-70. New York: Plenum Press.
Gerdes, H.-H. and Kaether, C. (1996). Green fluorescent protein:applications in cell biology. FEBS Lett. 389, 44-47.
Green, K. J., Geiger, B., Jones, J. C. R., Talian, J. C. and Goldman, R. D.(1987). The relationship between intermediate filaments and microfilamentsbefore and during the formation of desmosomes and adherens-type junctionsin mouse epidermal keratinocytes. J. Cell Biol. 104, 1389-1402.
Hatzfeld, M. (1997). Band 6 protein and cytoskeletal organization. InCytoskeletal-Membrane Interactions and Signal Transduction (ed. P. Cowinand M. W. Klymkowsky), pp. 49-60. Heidelberg: Springer.
Heim, R., Prasher, D. C. and Tsien, R. Y. (1994). Wavelength mutations andposttranslational autoxidation of green fluorescent protein. Proc. Nat. Acad.Sci. USA 91, 12501-12504.
Herrmann, H. and Aebi, U. (1998). Structure, assembly and dynamics ofintermediate filaments. In Intermediate Filaments (ed. H. Herrmann and J.R. Harris), pp. 319-362. New York: Plenum Press.
Hirokawa, N., Tilney, L. G., Fujiwara, K. and Heuser, J. E. (1982).Organization of actin, myosin, and intermediate filaments in the brushborder of intestinal epithelial cells. J. Cell Biol. 94, 425-443.
Ho, C.-L., Martys, J. L., Mikhailov, A., Gundersen, G. G. and Liem, R.K. H. (1998). Novel features of intermediate filament dynamics revealed bygreen fluorescent protein chimeras. J. Cell Sci. 111, 1767-1778.
Hofmann, I. and Franke, W. W. (1997). Heterotypic interactions and filamentassembly of type I and type II cytokeratins in vitro: viscometry anddeterminations of relative affinities. Eur. J. Cell Biol. 72, 122-132.
Horwitz, B., Kupfer, H., Eshhar, Z. and Geiger, B. (1981). Reorganizationof arrays of prekeratin filaments during mitosis. Exp. Cell Res. 134, 281-290.
Jones, J. C. R., Goldman, A., Yang, H.-Y. and Goldman, R. D. (1985). Theorganizational fate of intermediate filament networks in two epithelial celltypes during mitosis. J. Cell Biol. 100, 93-102.
Kitajima, Y., Inoue, S., Yoneda, K., Mori, S. and Yaoita, H. (1985).Alteration in the arrangement of the keratin-type intermediate filamentsduring mitosis in cultured human keratinocytes. Eur. J. Cell Biol. 38, 219-225.
Kitajima, Y., Inoue, S. and Yaoita, H. (1986). Reorganization of keratinintermediate filaments by the drug-induced disruption of microfilaments incultured human keratinocytes. J. InveSt Dermatol. 87, 565-569.
Klymkowsky, M. W., Maynell, L. A. and Nislow, C. (1991). Cytokeratinphosphorylation, cytokeratin filament severing and the solubilization of thematernal mRNA Vg1. J. Cell Biol. 114, 787-797.
Kreis, T. E., Geiger, B., Schmid, E., Jorcano, J. L. and Franke, W. W.(1983). De novo synthesis and specific assembly of keratin filaments innonepithelial cells after microinjection of mRNA for epidermal keratin. Cell32, 1125-1137.
Ku, N. O., Liao, J. and Omary, M. B. (1998). Phosphorylation of humankeratin 18 serine 33 regulates binding to 14-3-3 proteins. EMBO J. 17, 1892-1906.
Kuruc, N., Leube, R. E., Moll, I., Bader, B. L. and Franke, W. W. (1989).Synthesis of cytokeratin 13, a component characteristic of internal stratifiedepithelia, is not induced in human epidermal tumors. Differentiation 42,111-123.
Lai, Y.-K., Lee, W.-C. and Chen, K.-D. (1993). Vimentin serves as aphosphate sink during the apparent activation of protein kinases by okadaicacid in mammalian cells. J. Cell. Biochem. 53, 161-168.
Lane, E. B., Goodman, S. L. and Trejdosiewicz, L. K. (1982). Disruptionof the keratin filament network during epithelial cell division. EMBO J. 1,1365-1372.
Lawrence, J. B. and Singer, R. H. (1986). Intracellular localization ofmessenger RNAs for cytoskeletal proteins. Cell 45, 407-415.
Leube, R. E., Bader, B. L., Bosch, F. X., Zimbelmann, R., Achtstaetter, T.and Franke, W. W. (1988). Molecular characterization and expression ofthe stratification-related cytokeratins 4 and 15. J. Cell Biol. 106, 1249-1261.
Leube, R. E., Wiedenmann, B. and Franke, W. W. (1989). Topogenesis andsorting of synaptophysin: synthesis of a synaptic vesicle protein from a genetransfected into nonneuroendocrine cells. Cell 59, 433-446.

4534
Liao, J., Lowthert, L. A., Ku, N.-O., Fernandez, R. and Omary, M. B.(1995). Dynamics of human keratin 18 phosphorylation: polarizeddistribution of phosphorylated keratins in simple epithelial tissues. J. CellBiol. 131, 1291-1301.
Liao, J. and Omary, M. B. (1996). 14-3-3 proteins associate withphosphorylated simple epithelial keratins during cell cycle progression andact as a solubility cofactor. J. Cell Biol. 133, 345-357.
Ludin, B. and Matus, A. (1998). GFP illuminates the cytoskeleton. TrendsCell Biol. 8, 72-77.
Ludin, B., Doll, T., Meili, R., Kaech, S. and Matus, A. (1996). Applicationof novel vectors for GFP-tagging of proteins to study microtubule-associatedproteins. Gene 173, 107-111.
McGowan, K. and Coulombe, P. A. (1998). The wound repair-associatedkeratins 6, 16, and 17: insights into the role of intermediate filaments inspecifying keratinocyte cytoarchitecture. In Intermediate Filaments (ed. H.Herrmann and J. R. Harris), pp. 173-204. New York: Plenum Press.
McLean, W. H. I. and Lane, E. B. (1995). Intermediate filaments in disease.Curr. Opin. Cell Biol. 7, 118-125.
Martys, J. L., Ho, C.-L., Liem, R. K. H. and Gundersen, G. G. (1999).Intermediate filaments in motion: Observations of intermediate filaments incells using green fluorescent protein-vimentin. Mol. Biol. Cell 10, 1289-1295.
Miller, R. K., Vikstrom, K. and Goldman, R. D. (1991). Keratinincorporation into intermediate filament networks is a rapid process. J. CellBiol. 113, 843-855.
Miller, R. K., Khuon, S. and Goldman, R. D. (1993). Dynamics of keratinassembly: exogenous type I keratin rapidly associates with type II keratinin vivo. J. Cell Biol. 122, 123-135.
Moll, R., Franke, W. W., Schiller, D. L., Geiger, B. and Krepler, R. (1982).The catalog of human cytokeratin polypeptides: patterns of expression ofspecific cytokeratins in normal epithelia, tumors and cultured cells. Cell 31,11-24.
Moll, R. (1998). Cytokeratins as markers of differentiation in the diagnosis ofepithelial tumors. In Intermediate Filaments (ed. H. Herrmann and J. R.Harris), pp. 319-362. New York: Plenum Press.
Olson, K. R., McIntosh, J. R. and Olmsted, J. B. (1995). Analysis of MAP4 function in living cells using green fluorescent protein (GFP) chimeras. J.Cell Biol. 130, 639-650.
Omary, M. B., Ku, N.-O. Liao, J. and Price, D. (1998). Keratin modificationsand solubility properties in epithelial cells and in vitro. In IntermediateFilaments (ed. H. Herrmann and J. R. Harris), pp. 105-140. New York:Plenum Press.
Osborn, M., Franke, W. W. and Weber, K. (1977). Visualization of a systemof filaments 7-10 nm thick in cultured cells of an epitheloid line (PtK2) byimmunofluorescence microscopy. Proc. Nat. Acad. Sci. USA 74, 2490-2494.
Paramio, J. M. and Jorcano, J. L. (1994). Assembly dynamics of epidermalkeratins K1 and K10 in transfected cells. Exp. Cell Res. 215, 319-331.
Paramio, J. M. (1999). A role for phosphorylation in the dynamics of keratinintermediate filaments. Eur. J. Cell Biol. 78, 33-43.
Prahlad, V., Yoon, M. Moir, R. D., Vale, R. D. and Goldman, R. D.(1998). Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J. Cell Biol. 143,159-170.
Rizzuto, R., Brini, M., Pizzo, P., Murgia, M. and Pozzan, T. (1995).Chimeric green fluorescent protein as a tool for visualizing subcellularorganelles in living cells. Curr. Biol. 5, 635-642.
Schmidt, A., Heid, H. W., Schäfer, S., Nuber, U. A., Zimbelmann, R. andFranke, W. W. (1994). Desmosomes and cytoskeletal architecture inepithelial differentiation. Cell type-specific plaque components andintermediate filament anchorage. Eur. J. Cell Biol. 65, 229-245.
Skalli, O., Chou, Y.-H. and Goldman, R. D. (1992). Intermediate filaments:not so tough after all. Trends Cell Biol. 2, 308-312.
Stein, G. S., Stein, J. L., Lian, J. B., Last, T. J., Owen, T. and McCabe, L.(1994). Synchronization of normal diploid and transformed mammaliancells. In Cell Biology: A Laboratory Handbook (ed. J. E. Celis), pp. 282-287. New York: Academic Press.
Sun, T.-T. and Green, H. (1978). Immunofluorescent staining of keratin fibersin cultured cells. Cell 14, 469-476.
Toivala, D. M., Goldman, R. D., Garrod, D. R. and Eriksson, J. E. (1997).Protein phosphatases maintain the organization and structural interactionsof hepatic keratin intermediate filaments. J. Cell Sci. 110, 23-33.
Tölle, H.-G., Weber, K. and Osborn, M. (1987). Keratin filament disruptionin interphase and mitotic cells – how is it induced? Eur. J. Cell Biol. 43, 35-47.
Turner, B. M. and Ruane, M. (1985). Immunofluorescent localisation ofcytokeratin antigens in mitotic HeLa cells using monoclonal antibodies. Eur.J. Cell Biol. 36, 48-57.
Wiche, G., Krepler, R., Artlieb, U., Pytela, R. and Aberer, W. (1984).Identification of plectin in different human cell types andimmunolocalization at epithelial basal cell surface membranes. Exp. CellRes. 155, 43-49.
Wolf, K. M. and Mullins, J. M. (1987). Cytochalasin B-inducedredistribution of cytokeratin filaments in PtK1 cells. Cell Motil. Cytoskel.7, 347-360.
Yang, Y., Dowling, J., Yu, Q.-C., Kouklis, P., Cleveland, D. W. and Fuchs,E. (1996). An essential cytoskeletal linker protein connecting actinmicrofilaments to intermediate filaments. Cell 86, 655-665.
Yoon, M., Moir, R. D., Prahlad, V. and Goldman, R. D. (1998). Motileproperties of vimentin intermediate filament networks in living cells. J. CellBiol. 143, 147-157.
R. Windoffer and R. E. Leube