controlled ion-molecule reactions of ch3 and c3n · controlled ion-molecule reactions of ch 3 + and...
TRANSCRIPT

Poster : DivisionsChimie-Physique et Astrophysique Congrès général SFP 2017
Controlled ion-molecule reactions of CH3+ and C3N
-
A. Lopes1, C. Romanzin1, R. Thissen1, A. Cernuto2, D. Ascenzi2, B. Cunha de Miranda3, I. Zymak4,J. Žabka4, M. Polášek4, L. Koch4, F. Lindén5, W. D. Geppert5,
J.-C. Guillemin6, C. Alcaraz1 1LCP, UMR8000 CNRS, Univ. Paris Sud, Orsay – France 2Atomic & Molecular Physics Lab., Univ. di Trento, Povo – Italy 3 LIDYL, URA 2453CEA-CNRS, Gif sur Yvette– France 4J. Heyrovsky Institute of Physical Chemistry of the ASCR, Prague – Czech Republic 5 Stockholm University Astrobiology Centre, Stockholm Univ., Stockholm – Sweden 6ENSC Rennes, UMR6226CNRS, Rennes – France
In this contribution, we will present recent results concerning controlled ion-molecule reactions
of both cations and anions obtained with the CERISES set-up: a guided ion-beam experiment.The
originality of these experiments is to monitor the effect on the reactivityof either the parent-ion
excitation (vibrational, electronic) or the collision energy.
First, we will report on reactions of the CH3+ ion with several small hydrocarbon molecules
(CH4, C2H2, C2H4…). The control of the parent-ion excitation is achieved by producing the CH3+
ions via direct photoionization of a molecular beam of CH3∙ radicals and by tuning the energy of
the VUV photon (DESIRS beamline, synchrotron SOLEIL). Branching ratios and absolute reaction
cross-sections have been obtained for different reactive systems, their dependence on vibrational
and electronic excitation of the parent ion and collision energy will be discussed [1]. This will guide
us to identify the systems for which further state-to-state reactivity experiments are relevant. The
CERISES experiment indeed also allows for pure state selection of the parent ion thanks to
photoelectron-photoion coincidence techniques [2].
In addition to cations, it is now possible to study the reactivity of anions on CERISES. The first
results obtained concern the reaction of the C3N- ion with the acetylene molecule. In this latter
experiment, we focus on the control of collision energy. Three reactive channels leading
respectively to C2H-, CN- and C5N
- have been observed. Although, formation of the last two
products is exoergic, all reaction pathways exhibit large barriers as confirmed by the observation
of energetic reaction thresholds for all of them [3]. The contribution of this reaction to the growth of
larger anions is thus unlikely.
1.A. Lopes et al., in prep 2.B.Cunha de Miranda et al., J. Phys. Chem. A, 119, 6082 (2015) 3.C.F. Lindén et al., J. Phys. Chem. A, 120, 5337-5347 (2016)
1

O1
H.HADDAG, N. E. CHAKRI*, D. MESSADI
Laboratoire de Sécurité Environnementale et Alimentaire, Université BADJI Mokhtar, BP 12, 23000 Annaba, Algérie.
E-mails: [email protected], [email protected]
Abstract
The acentric factors of 18 hydroxy compounds (alcohols, phenols) were linearly correlated with 2
molecular descriptors of geometrical type selected by genetic algorithm, among more than 1600 derived
from the molecular modeling software DRAGON.
The different statistics calculated (multiple determination and prediction coefficients; roots of the
mean quadratic errors; Y-scrambling) show the quality, the robustness and the good internal predictive
capacity of the constructed model. No outliers or influential observation was found.
Key words: Alcohols and phenols – Numerical representation of chemical structure – Acentric factor – Hybrid SPR
model..
Corresponding Author : [email protected], [email protected]
RELATION STRUCTURE/ FACTEUR ACENTRIQUE D'ALCOOLS ET DE PHENOLS : Approche
Algorithme Génétique – Régression Linéaire Multiple.
.
Pour les posters : DCPhys Congrès général SFP 2017
Répliqua en PDMS de réseaux de surface d’azopolymère pour le dépôt de nanoparticules.
Fabien COUSSEAU1, Shahla GOLGAHASEMI SORKHABI1, 2, Matthieu LOUMAIGNE 21,Régis BARILLE1
1. Laboratoire MOLTECH-Anjou, CNRS(UMR6200), Universitéd’Angers, MOLTECH-Anjou, UFR
Sciences, 2 Bd Lavoisier, 49045 Angers France. 2. Research Institute for AppliedPhysics and Astronomy (RIAPA), University of Tabriz,Tabriz, (Iran).
La génération de réseaux de surface par interaction lumière-matière offre de nombreuses perspectives dans des domaines variés
1. Nous nous intéressons à la déposition de nano-objets par une
réplique de réseaux de surfacetransparente et déformable pour l’étude spectroscopique d’objets unique. Nous réalisons des films minces d’un polymère azo-benzénique qui a la particularité de s’organiser
dans l’espace sous l’effet de la lumière2. Ainsi est produit un réseau de surface complexe (SRG pour
Surface-Relief Grating) qui n’est autre que la superposition de plusieursréseaux linéaires avec différentes orientations. Cette superposition génère de nombreuses cavités au sein du film mince.
Le SRG obtenu est utilisé comme moule. Après application d’un élastomère(PDMS) sur le SRG nous obtenons un réseau de surface transparent et étirable. Nous caractérisons l’original et sa réplique par l’AFM et l’étude de la figure de diffraction optique. La similarité des résultats nous dispense de l’analyse AFM et seul l’optique est utilisé pour caractérisé le SRG sous contraintes.L’étirement et la relaxation du SRG en PDMS déforment les cavités qui peuvent alors capturer des nano-objets (particules sphériques, nanotubes, …).
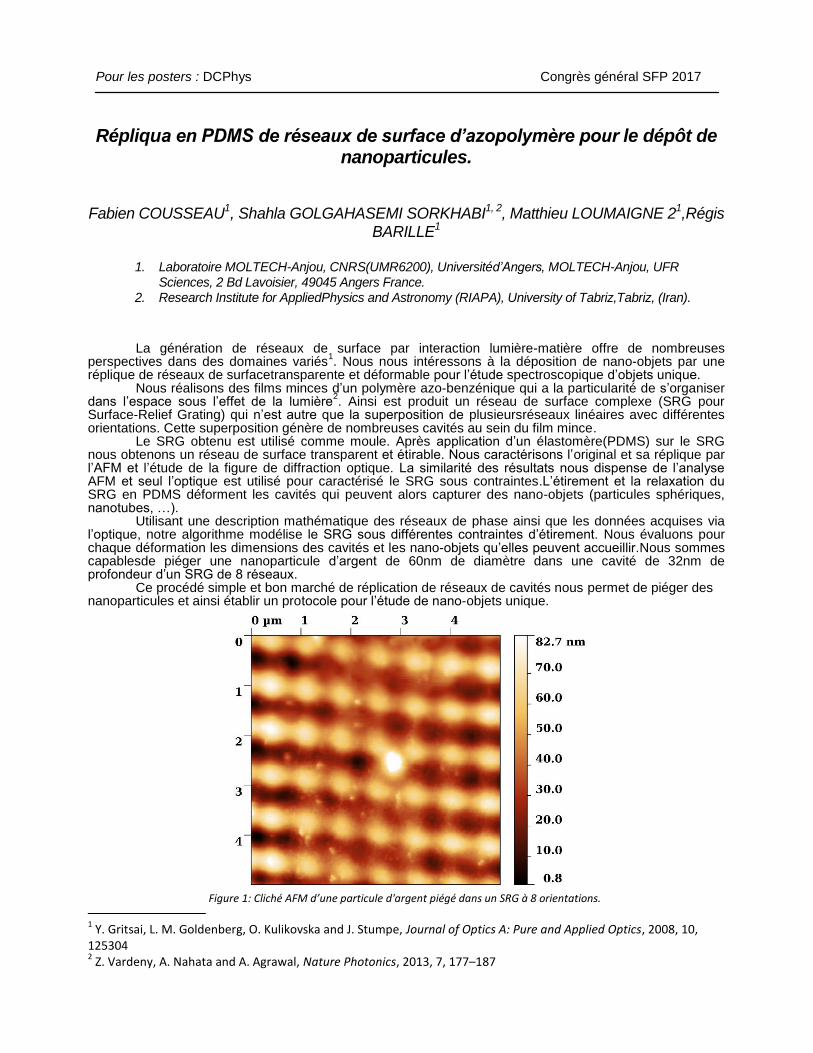
Utilisant une description mathématique des réseaux de phase ainsi que les données acquises via l’optique, notre algorithme modélise le SRG sous différentes contraintes d’étirement. Nous évaluons pour chaque déformation les dimensions des cavités et les nano-objets qu’elles peuvent accueillir.Nous sommes capablesde piéger une nanoparticule d’argent de 60nm de diamètre dans une cavité de 32nm de profondeur d’un SRG de 8 réseaux. Ce procédé simple et bon marché de réplication de réseaux de cavités nous permet de piéger des nanoparticules et ainsi établir un protocole pour l’étude de nano-objets unique.
1 Y. Gritsai, L. M. Goldenberg, O. Kulikovska and J. Stumpe, Journal of Optics A: Pure and Applied Optics, 2008, 10,
125304 2 Z. Vardeny, A. Nahata and A. Agrawal, Nature Photonics, 2013, 7, 177–187
Figure 1: Cliché AFM d’une particule d'argent piégé dans un SRG à 8 orientations.

PAMO/DCPhys Congrès général SFP 2017
Chlorination de molécules à liaison hydrogène intramoléculaire : effets contrastés sur la liaison H
A. Gutiérrez Quintanilla1,2, M. Chevalier1, R. Platakyte1,3, J. Ceponkus3, C. Crépin1 1 Institut des Science Moléculaires d'Orsay, CNRS Univ. Paris-Sud, Université Paris-Saclay, Bât.210, Campus d’Orsay, 91400 Orsay France 2 Instituto Superior de Tecnologías y Ciencias Aplicadas, La Habana, Cuba 3 Department of General Physics and Spectroscopy,Vilnius University,Sauletkio av. 9 bat. III, Vilnius, Lituanie
Le malonaldéhyde (MA) et l’acetylacétone (AcAc) sont deux molécules modèles pour l’étude
des liaisons hydrogène intramoléculaires (LHI).Toutes deux présentent une LHI fortedans leur
forme énol (voir figure), qui se traduit spectroscopiquement par de forts élargissementsdes
bandes vibrationnelles liées à la liaison H. La bande du mode d’élongation OH est trop large pour
être correctement mesurée en IR. Nous
avons étudié par spectroscopie IR en
matrice cryogénique les dérivés
monochlorés décrits sur la figure (MACl et
AcAcCl). Les bandes de AcAcCl restent
larges alors que celles de MACl s’affinent
par rapport à MA. Les expériences ont été
complétées par des calculs de chimie
quantique sur ces molécules et leurs
conformères. L’ensemble donne accès à
plusieurs indicateurs de la force de la liaison
H qui montrent tous que cette liaison
s’affaiblit en passant de MA à MACl alors
qu’elle se renforce en passant d’AcAc à
AcAcCl. Ces données seront discutées.
Les expériences de photoisomérisation en matrice ont également mis en lumière les
spécificités des dérivés chlorés avec la caractérisation de conformères « ouverts » (c’est-à-dire
sans LHI) stabilisés par une liaison non covalente Cl…H.
1
Figure

Congrès général SFP 2017
Poster:Division Chimie Physique (DCPhys)
Détermination des constantes élastiques K11 et K33 dans la phase nématique d’un composé possédant une phase nématique twist-bend.
G. Damême1, C. Meyer1, I. Dokli2, A. Lesac2, P. Davidson3, I. Dozov3 1 Physique des Systèmes Complexes, Université de Picardie Jules Verne, 80039 Amiens, France. 2Division of Organic Chemistry and Biochemistry,Institut Ruder Boskovic,10000 Zagreb, Croatie. 3 Laboratoire de Physique des Solides, CNRS, UniversitéParis-Saclay Bât.510, Campus d’Orsay, 91400 Orsay France.
En 2001, une nouvelle phase cristal-liquide, appelée phase nématique twist-bend NTB, a été
prédite théoriquement1 ; cette théorie prévoit que la constante élastique de ‘bend’ K33dans la
phase NTB devient négative.La phase NTBest spontanément distordue : elle présente à la fois de
latorsion (twist) et de laflexion (bend). Dans la phase NTB, le directeur nématique tourne sur un
cône avec un pas extrêmement court,de l’ordre de 10nm. En conséquence,la phase possède des
propriétés intéressantes : une chiralité macroscopique doublement dégénérée alors que les
molécules elles-mêmes sont achirales etdes temps de réponse extrêmement courts
soussollicitations électriques (<1µs). Récemment, la phase NTB a été mise en évidence
expérimentalement par un grand nombre de techniques différentes2.En particulier, des mesures
électro-optiques via l’effet électrocliniqueont permis de montrer que les domainesadjacents
visibles en microscopie optique dans la phase NTB possèdent des chiralités opposées3.
Afin de déterminer les propriétés élastiques de la phase nématique proche de la transition
N/NTB, nous étudions les propriétés diélectriques de la phase nématique d’un composé
nouvellement synthétisé qui possède la transition N/NTB4. Nous présentons des mesures de
capacitance en géométrie confinée en faisant varier la tension de mesure appliquée et ce pour
différentes valeurs de la température dans toute la gamme d’existence de la phase nématique. En
réalisant un ajustement précis de ces courbes expérimentales, nous remontonsdirectement à une
estimation de la tension seuil de Fréederickzs, à l’angle de prétilt de la cellule ainsi qu’à
l’évolution des constantes élastiques K11 et K33 en fonction de la température.La diminution
observée de la constante élastique de ‘bend’ lorsque la température diminue est tout à fait
compatible avec le changement de signe théoriquement attendu lors de la transition vers la phase
NTB.De plus, nous comparons l’évolutiondes constantes élastiques dans la phase nématique du
composé à celle d’un composé ne possédant pas de phase NTB en dessous de la phase
nématique mais une phase smectique A.
Les résultats sont en accord semi-quantitatif avec les prédictions théoriques1et la constante K33 est un indicateur efficace de l'existence d'une phase NTB, (réelle ou virtuelle) dans le diagramme de phase de la molécule mésogène fléchie.
1I. Dozov, Europhys. Lett., 56 (2), pp. 247–253 (2001).
2M. Cestari et al.Phys. Rev. E 84, 031704 (2011).
3 C. Meyer, G. R. Luckhurst, I. Dozov, Phys. Rev. Lett.111, 067801 (2013).
4T. Ivsic, U. Baumeister, I. Dokli, A. Mikleusevic& A. Lesac, Liq. Cryst.,44:1, 93-105 (2017).

Congrès général SFP 2017 Division de Chimie Physique
Indirect photodesorption processes in rare-gas solids on top of CO ices
R. Dupuy1, M. Bertin1, G. Féraud1, C. Romanzin2, L. Philippe1, M. Doronin1, P. Jeseck1, X. Michaut1 and J.-H. Fillion1
1 LERMA, Sorbonne Universités, UPMC Univ. Paris 06, Observatoire de Paris, PSL Research University, CNRS, F-75005, Paris, France 2 LCP, Université Paris Sud, CNRS UMR 8000, 91405 Orsay, France
UV Photodesorption, i.e. the ejection of molecules or atoms from a surface following a photon-
induced electronic transition, has been a topic of renewed interest in the last few years owing to
its relevance in astrophysical contexts. In cold regions of space, molecules are found in a solid icy
phase, for example forming the ice mantle covering the surface of interstellar dust grains. Thermal
desorption being negligible, the interaction between the gas phase and the solid phase in these
regions must be driven by non-thermal processes such as photodesorption induced by stellar UV
radiation, which laboratory experiments aim at quantitatively and qualitatively describing.
While progress has been made in the understanding of photodesorption from pure ices of a
single species (CO, CO2, H2O...), molecules in astrophysical ices are typically mixed together,
which can affect the physical and chemical processes occuring. In particular, we have shown that
the CO molecule, which is very abundant in space and has a very efficient photodesorption
mechanism, can induce desorption of other neighbouring molecules of a different species1,2
.
In order to better understand such indirect photodesorption mechanisms, rare-gas solids
(RGS) on top of CO is an ideal system to study. RGS are one of the few systems where
photodesorption effects3 and energy transfer mechanisms
4 are well understood. We present
experiments on RGS on top of CO ices performed with tunable synchrotron radiation in the VUV
range on the DESIRS beamline (SOLEIL, France). By selectively exciting one or the other species
(the RGS or CO), we observe simultaneous desorption of both species. Absolute photodesorption
yields are derived, which allows the efficiency of CO-induced desorption for the
Argon/Krypton/Xenon series to be compared. Experimental observations lead to the conclusion
that CO is mixed to some degree inside the RGS layers, and that RGS excitons localize at CO
sites, leading to strong CO desorption and enhanced total desorption yields.
1. Bertin et al., Physical Chemistry Chemical Physics, 14, 28,9929 (2012) 2. Bertin et al., The Astrophysical Journal, 779 (2), 120 (2013) 3. Zimmerer, Nuclear Instruments and Methods in Physics Research B, 91, 601-613 (1994) 4. Fugol’, Advances in Physics, 27, No 1, 1-87 (1978)
1

Chimie/Physique Congrès général SFP 2017
Caractérisation in situ et in operando des piles à combustibles à membranes échangeuses de protons
CF Cerqueira 1, PE Coulon1, G Rizza, MC Clochard1
1Laboratoire des Solides Irradiés, CEA DSM/IRAMIS, CNRS UMR 7642, Ecole Polytechnique, Université
Paris-Saclay, F-91128 Palaiseau, France
Une pile à combustible est un générateur
électrique qui, en présence de différents carburants
(hydrogène, alcool) et comburant, oxygène, réalise
la transformation directe de l'énergie chimique en
énergie électrique. Dans une pile à combustible à
membrane échangeuse de protons (PEMFC), la
membrane est un polyélectrolyte solide qui a, pour
double mission, d'opérer le transport des protons
de l'anode à la cathode, tout en assurant la
séparation des électrons et des gaz entre les
compartiments anodique et cathodique. Dans ce
cadre, I'optimisation de la formulation des
membranes et des électrodes en surface de la
membrane polyélectrolytique sont en cours de
développement et sont le sujet de recherches
actives. Néanmoins, la durée de vie des piles reste problématique. La dégradation des nano-
catalyseurs, composants essentiels des phases actives puisqu'ils permettent la dismutation de la
molécule d'hydrogène à I'anode et la recombinaison des protons avec l'oxygène à la cathode, est
un facteur limitant pour la commercialisation.
Bien que la dégradation des nano-catalyseurs ait été largement étudiée dans le passé,
essentiellement par des méthodes ex situ, l'utilisation d'une approche in situ et in operando
pourrait grandement améliorer notre compréhension des mécanismes intervenants lors du
cyclage électrochimique1. Ce travail a pour objectif le développement d'un nouvel outil de
caractérisation in situ et in operando en microscopie électronique à transmission pour étudier
l'évolution des propriétés structurales et électrochimiques des matériaux impliqués dans les
réactions de cyclage des piles à combustibles. Les différents mécanismes de dégradation des
matériaux constituant les piles sont en partie liés aux effets induits par les dynamiques
d'échanges à l'interface, tels que les réactions aux interfaces ionomère/catalyseurs/gaz
(hydrogène à l'anode ou oxygène à la cathode). Seront présentés les premiers résultats obtenus
avec aux interfaces Nafion+Carbon+acide/Platine/oxygène. Les expériences ont été réalisée à
I'aide du porte-échantillon TEM liquide/électrique Protochips Poséidon 510 sur un JEOL 2010F.
1. G.-Z. Zhu, S. Prabhudev, J. Yang, C. M. Gabardo, G. a. Botton, and L. Soleymani, “In Situ Liquid Cell TEM Study of Morphological Evolution and Degradation of Pt–Fe Nanocatalysts During Potential Cycling,” J. Phys. Chem. C, vol. 118, no. 38, pp. 22111–22119, (2014).
1
Figure 1: (a) C-Pt sec (b)C-Pt in liquide.
dfgdfgd

Congrès général SFP 2017
Ab-initio simulations on warm dense states of gold
A. Grolleau1, J.Gaudin2, B.Fabre2 1 Université de Bordeaux, 33405 Talence (France) 2 Centre Lasers Intenses et Applications, UMR 5107 CNRS - Université de Bordeaux - CEA, Université de Bordeaux, 33405 Talence (France)
An intense femtosecond laser pulse interacting with a solid leads to a highly excited
and non-equilibrium regime of matter called the Warm dense Matter (WDM). During the
few picoseconds following the interaction, electrons are heated to temperature of few eV
while atoms stay "cold". This non-equilibrium situation causes multiple specific processes
such as non-thermal melting (covalent materials), or bonds hardening (metals). In order to
understand the results of recent experiments conducted on gold at CELIA, simulations
are necessary. We used the ABINIT package to model the experimental conditions. The
gold structure has been simulated for different temperatures, and the dielectric function
has been computed for each system.
1
Figure 1 : LEFT – 32 gold atoms FCC lattice || RIGHT - 32 gold atoms melted box simulated using Molecular Dynamics at 5000 K

Colloque Jeunes chercheurs (colloque+ session poster) Congrès général SFP 2017
Imagerie AFM non-linéaire en détection de phase : application à des nanoparticules inorganiques enterrées dans des ligands organiques
M. Hurier, B. Donnio, J. L. Gallani, M. V. Rastei Institut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), CNRS, Université de Strasbourg
Nous présentons une technique de microscope à force atomique (AFM) basée sur les changements linéaires et non-linéaires de la phase d’oscillation d’une sonde AFM. La technique est développée pour observer des échantillons avec des zones à rigidité variable. Ici nous étudions le cas de nanoparticules d'or et de boites quantiques inorganiques rigides qui sont enfouies dans une matrice de ligands organiques plus souple. L'AFM en mode topographique ne suffit pas à détecter et imager efficacement des nanostructures inorganiques enterrées dans des ligands organiques. Les images topographiques se résument souvent à des infimes excroissances traduisant la présence de nanoparticules. En utilisant le mode dynamique d'un AFM, où la sonde est mise en vibration à l’aide d’un élément piézoélectrique, la phase d’oscillation de la sonde change en fonction des propriétés viscoélastiques de la zone sondée. La dissipation de l’énergie de la sonde est transmisse de manière irréversible sous forme de travail à l’échantillon. Ces phénomènes observables même avant le contact discontinu engendrent des réponses linéaires et non-linéaires de la sonde se traduisant finalement en images. Pour des mesures réalisées à basses pressions (P < 10
-5mbar) le régime non-linéaire domine la réponse de la sonde.
L’amplitude d’oscillation et la valeur moyenne de la distance pointe-échantillon sont des paramètres clés pour la détection, permettant d’ajuster le contraste. La technique permet l’observation des nanoparticules ayant des diamètres et des distances inter-particulaire de seulement quelques nanomètres. L’impact des différentes matrices organiques est aussi détaillé.
Figure 1 : Nanoparticules d’or enterrées dans une matrice organique

Screened potential constraint in a Reverse Monte Carlo (RMC)
modeling
M. KOTBI1, M. HABCHI2, M. Benkhaled3, S. M. MESLI4
1 Laboratoire de Physique Théorique (LPT), Université de Tlemcen (Algérie)
To explore a certain number of structural features of an aqueous electrolyte LiCl-6H2O
type, a Reverse Monte Carlo (RMC) modeling is applied [1, 2]. This is based essentially on
neutron scattering data [3, 4] consisting of four partial distribution functions issue from the
technique of the isotopic substitution. Instead of introducing the interaction potential as in the
classical methods (MD, MC), one computes a parameter 2 representing the difference
between the calculated structure function and that are of the experiment within standard
deviation.
One examines the system at glassy (120K) and liquid (300K) state compared to pure
water at room temperature. The chlorine and lithium ions charged -1 and +1, respectively,
the water molecule is represented by a flexible model [8] charged as -0.8476 for the oxygen
and +0.4238 for each hydrogen atom [7, 8]. The results one obtains could include some
artifacts [5, 6]. To remedy for this, we could make a propose choice of screened potential
model.
In conclusion, we could suggest that the choice of the interaction model as a function of
atomic or molecular properties forming the system could bring a meaningful improvement to
the results. An improvement in the coordination of this function is noticed. RMC is generally
limited to explore structural property of a system with or without interaction model.
Introducing potential as constraint in RMC simulation suggests a useful test of an interaction
potential model for classical methods as Monte Carlo (MC) and Molecular Dynamic (MD) with
which one can compute thermodynamic
_____________________________
1. R.L. Mc Greevy, M.A. Howe, J.D. Wicks, RMCA Version 3, A General Purpose Reverse Monte Carlo Code, October 1993.
2. R.L. Mc Greevy and Pusztai, Mol. Simul. 1 (1988) 359.
3. J.F. Jal, K. Soper, P. Carmona, J. Dupuy, J. Phys. Cond. Matter (3) (1991) 551.
4. B. Prével, J.F. Jal, J. Dupuy Philon, A.K. Soper, J. Chem. Phys. 103 (1995) 1886.
5. M. Kotbi, Hong Xu, M. Habchi, Z. Dembahri, Phy. Lett. A 315 (2003) 463.
6. H. Xu, M. Kotbi, Chem. Phys. Lett., 248 (1996) 89.
7. M.-C. Bellissent-Funel and G.W. Neilson, series C: Math. and Phys. Sciences Vol.205, ISBN 90-277-253469.
8. P.A. Bopp, I. Okada¸ H. Ohtaki and K. Heinsinger, 1985, Z. Naturforsch, 40a, 116.

DMC, DCPhys Congrès général SFP 2017
A correlation-hole approach to the electric double layer with counter-ions only
Ivan Palaia1, Ladislav Šamaj2, Emmanuel Trizac1
1 LPTMS, CNRS, Université Paris-Sud, Université Paris-Saclay, Orsay, France2 Institute of Physics, Slovak Academy of Science, Bratislava, Slovakia
In nature and in experiments colloidal particles – of artificial, mineral or biological origin – areoften electrically charged and surrounded by a solution of small oppositely charged ions, thatensure global neutrality and are called counter-ions. The electric potential and the counter-ionsdensity around the colloid are important observables in order to understand prominentphenomena, such as like-charge attraction, with applications ranging from cement solidification tothe mechanical properties of biological macromolecules. This motivates our interest in theproblem of the electric double layer, or how counter-ions distribute around a charged surface.
Given a charged plane immersed in a solution with its counter-ions, the entire statisticalmechanics of the system is determined by one dimensionless coupling parameter Ξ, that dependson the surface charge of the plane, on the ionic valence and on temperature, through the Bjerrumlength. The exact counter-ions density profile is known only for the two limit cases of Ξ→0,corresponding to the Poisson-Boltzmann mean-field theory, and Ξ→∞, where instead counter-ions have high planar correlation and are tightly bound to the charged plane.
We propose a new method for computing the electric potential and the counter-ions density atarbitrary Ξ, based on the observation that, especially at strong coupling, each counter-ion builds acylindrical depletion region around itself, inaccessible to other ions1,2. We derive approximateequations, whose solutions can be proven surprisingly to still satisfy many known exactproperties: the contact theorem holds, the correct behavior for zero or infinite coupling is fullyrecovered, at long distance mean-field is retrieved with the correct coefficient and the correct first-order finite-Ξ correction3. Moreover these equations are easy to solve numerically by a self-consistent iterative scheme, for any value of the coupling parameter and up to a very far distancefrom the plane: compared to available Monte Carlo simulations1, this algorithm performs better ormuch better than previously developed methods2, which are additionally unable to capture manyof the mentioned exact features.
Far away from the charged plane, the expected transition to mean field and its dependence on Ξemerge clearly from our numerical profiles. At very strong coupling it is even possible to defineunambiguously an effective Gouy-Chapman length, whose existence had already beenconjectured with good reason2.
1. A. G. Moreira and R. R. Netz, Simulations of counter-ions at charged plates, Eur. Phys. J. E 8, 33 (2002)2. Y. Burak, D. Andelman and H. Orland, Test-charge theory for the electric double layer, Phys. Rev. E 70, 016102 (2004)3. R. R. Netz and H. Orland, Beyond Poisson-Boltzmann: Fluctuation effects and correlation functions, Eur. Phys. J. E 1, 203 (2000)

Colloque : Nanostructures hybrides Congrès général SFP 2017
Co-Ni nano-dumbbells
G. Magnifouet1, T. Bhatnagar1, Y.Shin1, S. Mercone2, F. Schoenstein2, C. Meny1,
C. Ulhaq-Bouillet1, V. Pierron-Bohnes1
1 Institut de Physique et Chimie des Matériaux de StrasbourgUMR7540- Dép. Magnétisme et Objets
Nanostructurés, 23 rue du Loess BP 43, 67034 Strasbourg Cedex 2 France
2 Laboratoire des Sciences des Procédés et Matériaux - CNRS UPR 3407, Université Paris XIII, Sorbonne
Paris Cité,99 Ave Jean-Baptiste Clément, 93430 Villetaneuse, France
Analysis of EELS and EDX concentration maps and profiles showed concentration gradients that
can be correlated with the differences in morphology and crystal structure observed between the
center and the ends in the dumbbells. These observations are in agreement with the high
resolution electron microscopy images which show that the structure is hexagonal in the center of
the dumbbells, with increasing number of stacking faults when approaching the head where face-
centered cubic zones can be evidenced. Electron holography images have given access to the
local configuration of the magnetization in these dumbbells. The obtained magnetic phase images
are compared to simulations of a simple model. The result is consistent with all the other
observations. Preliminary results of NMR spectrum on Co-Ni alloys reflect the probabilities of
occurrence of various chemical/topological configurations and help to identify different crystal
structures around the probed nuclei.
1. A. Gaul. et al., Role of Morphology on the Large Coercive Behavior in Co80Ni20 Nanowires, MRS Bull. 01/2014; 1708:mrss14-1708-
vv06-08. DOI: 10.1557/opl.2014.555 (2014).
2. M. Pousthomis et al., Localized magnetization reversal processes in cobalt nanorods with different aspect ratios, Nano Res. 8, 2231
(2015).
3. S. Mercone et al., Morphology control of the magnetization reversal mechanism in Co80Ni20 nanomagnets, J. Applied Phys. 117,
203905 (2015).
Co-Ni nanodumbells were synthesized by reduction of
Ni and Co acetates in polyol medium [1]. It has been
shown that the variation of the synthesis parameters
such as the basicity of the medium or the nature of the
nucleating agent allow to obtain nano-objects of
different morphology [2] which itself drives the
magnetization reversal mechanism. The measurements
of the global magnetic properties show a high coercivity,
strongly depends on both the shape anisotropy (related
to the shape details of the nanodumbbell head) and
magnetocrystalline anisotropy (required to maintain a
stable orientation of the magnetic moment inside the
nanoparticles) of the nano-object and are in good
agreement with the results of micromagnetic
simulations [3].
Electron Hologram of Co-Ni showed the shift of the fringe on the object (inset - Phase image)

Pour les colloques : Congrès général SFP 2017 Pour les posters : Chimie Physique
Dynamique du transfert de charge dans un switch intra-moléculaire chiral
Hatem Labidi1, Henry Pinto2, Jerzy Leszczynski2, Damien Riedel1
1 Institut des Sciences Moléculaires d’Orsay (ISMO), CNRS, Univ. Paris Sud, Université Paris-
Saclay, F-91405 Orsay, France 2 Interdisciplinary Center for Nanotoxicity, Department of Chemistry, Jackson State University, Jackson,
Mississippi 39217, USA
Les processus de transfert de charge
(CT) contôle une large gamme d’effets physico-
chimiques allant de l’émission de lumière dans
les OLEDs, à l’effet photoélectrique dans les
cellules photovoltaïques en passant par les
phases de l’attachement de l’oxygène dans
l’hémoglobine. Cependant, ces processus sont
souvent décrits macroscopiquement, en
moyennant les informations sur un ensemble
d’élements, alors que les CT trouvent souvent
leur origine dans la molécule individuelle ou à
l’échelle de l’atome. Alors que les méthodes optiques de type pompe-sonde sont souvent délicates
à mettre en œuvre à ces échelles, et pour mieux comprendre l’origine des CT au sein de la molécule
individuelle, il est nécessaire de developper de nouvelles méthodes et outils adaptées à cette
problèmatique.
Pour cela nous avons étudié la comutation de la molécule de Nickel-tetraphenylporphyrine
adsorbée sur la surface du Si(100) à basse température (9 K). Deux configurations chirales peuvent
être activées à tour de rôle par excitation électronique localisée via les électrons d’un microscope
à effet tunnel. Des simulations numériques utilisant la théorie de la fonctionnelle densité décrivent
la structure électronique des conformations moléculaires. En analysant les variations d’efficacité
quantique de la commutation en fonction de la position et de l’énergie de l’excitation, nous avons
pu mettre en évidence un lien étroit et quantitatif entre nos données expériementales et le modèle
de Marcus-Jordner décrivant des paramètres cruciaux du transfer de charge comme le couplage
électronique, l’énergie libre de réaction ou l’énergie de réorganisation. Cette méthode peut être
adaptée à différents systèmes moléculaires chimisorbées ou physisorbées.
Figure 1 : dynamique du transfert de charge
dans la molécule de NiTPP/Si(100).

Etude d’un liquide ionique confiné dans une silice nanoporeuse
Effets des interactions adsorbat - adsorbant
Maryam SAFARIAMIN*, Gilberte DOSSEH*, Frédéric VIDAL*;
*Laboratoire de Physicochimie des Polymères et des Interfaces, EA 2528
Université de Cergy-Pontoise
Les nanomatériaux hybrides associant des systèmes organiques et
inorganiquessuscitent un intérêt croissant à cause de leursapplications dans des domaines
aussi variés que la catalyse, les biocapteurs, le relargage des médicaments ou les
supercondensateurs. Pour les applications dans des dispositifs électrochimiques comme les
supercondensateurs, des liquides ioniques sont, soit incorporés dans des matrices
polymères, soit confinés dans des matrices poreuses. Dans ces matériaux, l’étude des
interactions aux interfaces organique - inorganique est indispensable pour comprendre les
propriétés structurales, thermodynamiques et dynamiques de l’adsorbat.
Nous avons étudié l’effet du confinement sur les propriétés thermiques et la
dynamique moléculaire d’un liquide ionique organique confiné dans des silices
nanoporeuses avec des tailles de pores de 15 et 30 nm. Les températures et enthalpies de
transitions de phase ont été déterminées par calorimétrie différentielle à balayage. Des
mesures de spectroscopie de résonance magnétique nucléaire du proton ont permis
d’étudier l’effet du confinement sur la dynamique des adsorbats. L’effet des interactions
entre l’adsorbat et l’adsorbant sur lespropriétés du liquide ionique confiné seradiscuté.