continuous atom transfer radical polymerization in a tubular reactor
TRANSCRIPT
Full Paper
Continuous Atom Transfer RadicalPolymerization in a Tubular Reactor
Matthias Muller, Michael F. Cunningham,* Robin A. Hutchinson*
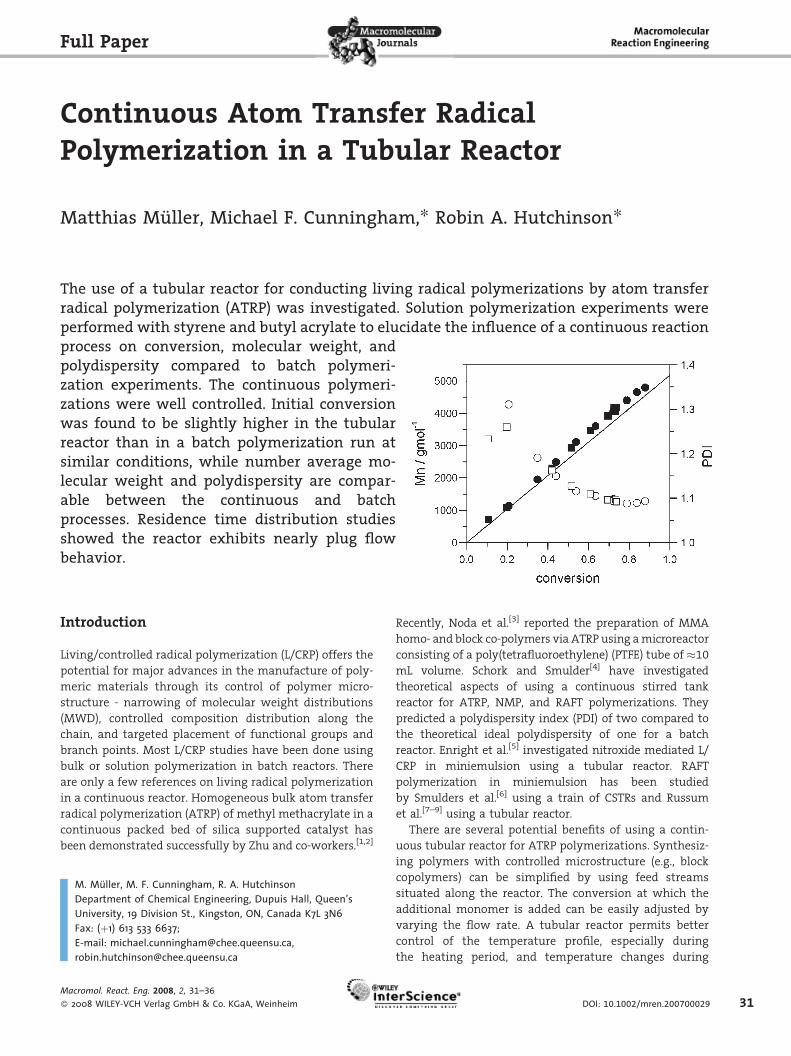
The use of a tubular reactor for conducting living radical polymerizations by atom transferradical polymerization (ATRP) was investigated. Solution polymerization experiments wereperformed with styrene and butyl acrylate to elucidate the influence of a continuous reactionprocess on conversion, molecular weight, andpolydispersity compared to batch polymeri-zation experiments. The continuous polymeri-zations were well controlled. Initial conversionwas found to be slightly higher in the tubularreactor than in a batch polymerization run atsimilar conditions, while number average mo-lecular weight and polydispersity are compar-able between the continuous and batchprocesses. Residence time distribution studiesshowed the reactor exhibits nearly plug flowbehavior.
Introduction
Living/controlled radical polymerization (L/CRP) offers the
potential for major advances in the manufacture of poly-
meric materials through its control of polymer micro-
structure - narrowing of molecular weight distributions
(MWD), controlled composition distribution along the
chain, and targeted placement of functional groups and
branch points. Most L/CRP studies have been done using
bulk or solution polymerization in batch reactors. There
are only a few references on living radical polymerization
in a continuous reactor. Homogeneous bulk atom transfer
radical polymerization (ATRP) of methyl methacrylate in a
continuous packed bed of silica supported catalyst has
been demonstrated successfully by Zhu and co-workers.[1,2]
M. Muller, M. F. Cunningham, R. A. HutchinsonDepartment of Chemical Engineering, Dupuis Hall, Queen’sUniversity, 19 Division St., Kingston, ON, Canada K7L 3N6Fax: (þ1) 613 533 6637;E-mail: [email protected],[email protected]
Macromol. React. Eng. 2008, 2, 31–36
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Recently, Noda et al.[3] reported the preparation of MMA
homo- and block co-polymers via ATRP using amicroreactor
consisting of a poly(tetrafluoroethylene) (PTFE) tube of�10
mL volume. Schork and Smulder[4] have investigated
theoretical aspects of using a continuous stirred tank
reactor for ATRP, NMP, and RAFT polymerizations. They
predicted a polydispersity index (PDI) of two compared to
the theoretical ideal polydispersity of one for a batch
reactor. Enright et al.[5] investigated nitroxide mediated L/
CRP in miniemulsion using a tubular reactor. RAFT
polymerization in miniemulsion has been studied
by Smulders et al.[6] using a train of CSTRs and Russum
et al.[7–9] using a tubular reactor.
There are several potential benefits of using a contin-
uous tubular reactor for ATRP polymerizations. Synthesiz-
ing polymers with controlled microstructure (e.g., block
copolymers) can be simplified by using feed streams
situated along the reactor. The conversion at which the
additional monomer is added can be easily adjusted by
varying the flow rate. A tubular reactor permits better
control of the temperature profile, especially during
the heating period, and temperature changes during
DOI: 10.1002/mren.200700029 31

M. Muller, M. F. Cunningham, R. A. Hutchinson
32
polymerization, which in the end leads to better controlled
polymer properties. Furthermore, operating a tubular
reactor under pressure, which is needed close to or above
the boiling point of monomer or solvent, is much easier
than running a stirred tank reactor under the same
pressure, for both laboratory and industrial scale. This
paper describes initial results using a continuous tubular
reactor to produce homopolymers of butyl acrylate and
styrene via ATRP.
Experimental Part
The reactor setup is based on a similar setup used by Enright et al.[5]
to investigate nitroxide mediated polymerization inminiemulsion.
The tubular reactor setup consists of stainless steel tubing (150 m
length, 3.2 mm outer diameter, 2.1 mm inner diameter, reactor
volume �0.52 L) immersed in an oil bath. The flow was controlled
by aMasterflex peristaltic pump situated at the outlet of the reactor
tube. Between the pump and oil bath, a heat exchanger is used
to cool the reaction mixture down. The actual mass flow was
measured by a balance (Mettler Toledo PG 5002s) at the outlet. A
scheme of the reactor is shown in Figure 1. Two storage tanks were
used in order to be able to refill one storage tank while the other is
used for feeding the reactor. Therefore, operation time is not limited
by the volume of the storage tanks.
Batch reactions for comparison to the continuous reactor were
done utilizing a 1-L glass autoclave built into a Mettler-Toledo
LabMax system.
Materials
Styrene (99%, Aldrich), butyl acrylate (99%, Aldrich), toluene (99%,
Aldrich), benzonitrile (99%, Aldrich), acetonitrile (99%, Aldrich),
Cu(I)Br (98%, Aldrich), N,N,N0,N00,N0- pentamethyldiethylenetri-
amine (PMDETA, 99%, Aldrich), and methyl 2-bromopropionate
(MBrP, 98%, Aldrich) were used as received.
Figure 1. Continuous tubular reactor apparatus for ATRP polymerizat
Macromol. React. Eng. 2008, 2, 31–36
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Analyses
The molecular weight and PDI were measured by gel permeation
chromatography (GPC). The samples were first passed through a
column packed with basic alumina to remove the residual copper.
The GPC instrument was equipped with a Waters 2960 module
containing four Styragel columns with pore sizes of 100, 500, 103,
and 104 A, coupled with a Waters 410 differential refractive index
(RI) detector (480 nm) and a Wyatt Technology DAWN EOS
photometer multi-angle light scattering (LS) detector (690 nm,
30 mW Ga-As laser). THF was used as the eluent and the flow rate
was set to 1mL �min�1. The LSdetectorwas calibratedwith toluene,
normalizedwith 30000 g �mol�1 narrow polystyrene standard, and
the datawere processed using Astra (Version 4.90.08) software. The
system was calibrated with 11 narrow polystyrene standards.
Conversion was analyzed gravimetrically by drying small samples
at room temperature for 2 d and by gas chromatography using
a Varian CP 3800 system fitted with a 50 m� 0.25 mm inner
diameter fused-silica CP-Sil wall coated open tubular capillary
column (WCOT). Sample injectionwas controlled by a Varianmodel
CP-8410 injector. Analytes were detected using a flame ionization
detector. The GC was controlled by Varian Star software.
Polymerization Procedure
Monomer, solvent, CuBr, and PMDETA were premixed for at least
30 min in an Erlenmeyer flask while purging the solution with
nitrogen. If there was no insoluble copper at the bottom after the
initialmixing period, the initiator was added and the solutionwas
poured into the two storage tanks of the tubular reactor setup. In
the case where insoluble copper was observed, the mixing period
was extended by 30 min, followed by 20 min to let the insoluble
parts settle to prevent transfer to the storage tanks. In most cases
therewas no insoluble catalyst. After purgingwith nitrogen for an
additional 10 min the storage tanks were closed and the nitrogen
valve was regulated to provide a pressure of 2 bar.
To begin the continuous reactions, the reactor tube was quickly
ion.
filled from storage tank #2 which took 5–6
min. The pump head of the peristaltic pump
was closed and the pump started with the
desired flow rate. Samples were taken at
intervals at the outlet of the reactor, which
results in samples with successively longer
reaction times and therefore higher conver-
sion until the full residence time of the tubular
reactor is reached. In principle it is possible to
take samples in the middle of the reactor
using T-junctions, but thiswould interrupt the
flow pattern and therefore was not done in
this study.
Residence Time Distributions (RTDs)
The RTD of the tubular reactor was deter-
mined using water. After applying a pulse of
NaCl solution, the conductivity was measured
using a conductivity meter placed between
DOI: 10.1002/mren.200700029
Continuous Atom Transfer Radical Polymerization in a Tubular Reactor
the heat exchanger and the peristaltic pump. Control experiments
without the reactor were done to determine the time the salt
solution needs to reach the reactor from the injection point and to
reach the conductivity meter after leaving the tubular reactor.
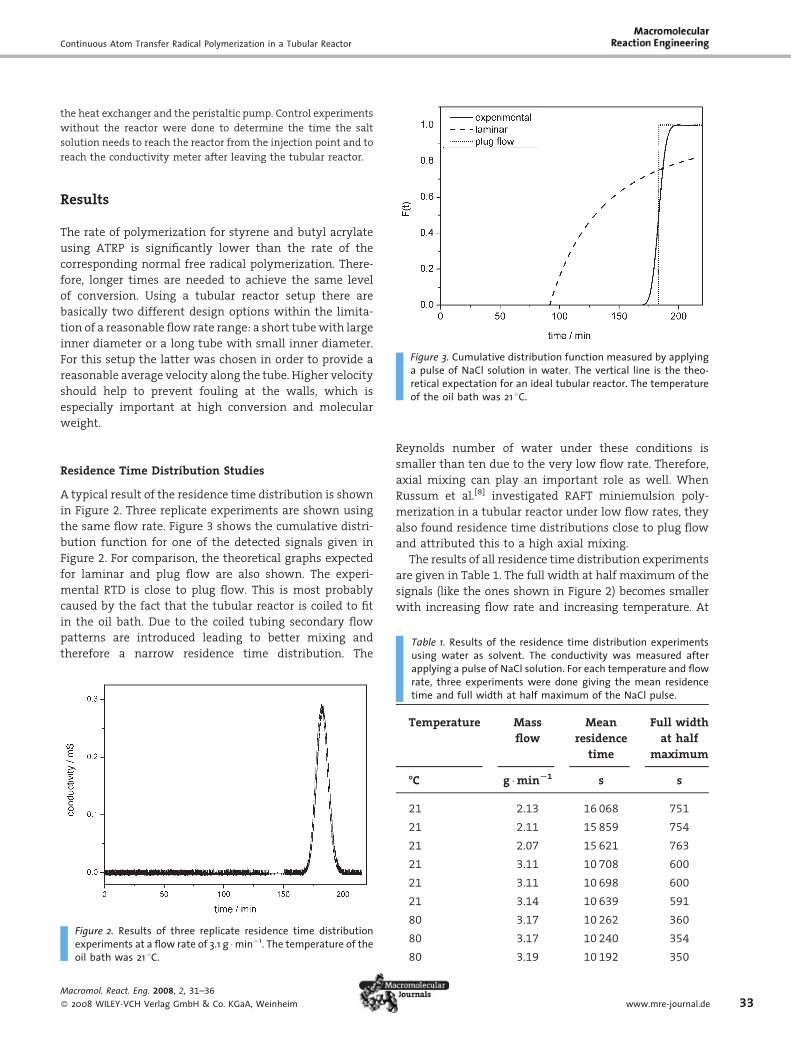
Figure 3. Cumulative distribution function measured by applyinga pulse of NaCl solution in water. The vertical line is the theo-retical expectation for an ideal tubular reactor. The temperatureof the oil bath was 21 8C.
Results
The rate of polymerization for styrene and butyl acrylate
using ATRP is significantly lower than the rate of the
corresponding normal free radical polymerization. There-
fore, longer times are needed to achieve the same level
of conversion. Using a tubular reactor setup there are
basically two different design options within the limita-
tion of a reasonable flow rate range: a short tubewith large
inner diameter or a long tube with small inner diameter.
For this setup the latter was chosen in order to provide a
reasonable average velocity along the tube. Higher velocity
should help to prevent fouling at the walls, which is
especially important at high conversion and molecular
weight.
Table 1. Results of the residence time distribution experimentsusing water as solvent. The conductivity was measured after
Residence Time Distribution Studies
A typical result of the residence time distribution is shown
in Figure 2. Three replicate experiments are shown using
the same flow rate. Figure 3 shows the cumulative distri-
bution function for one of the detected signals given in
Figure 2. For comparison, the theoretical graphs expected
for laminar and plug flow are also shown. The experi-
mental RTD is close to plug flow. This is most probably
caused by the fact that the tubular reactor is coiled to fit
in the oil bath. Due to the coiled tubing secondary flow
patterns are introduced leading to better mixing and
therefore a narrow residence time distribution. The
Figure 2. Results of three replicate residence time distributionexperiments at a flow rate of 3.1 g �min�1. The temperature of theoil bath was 21 8C.
Macromol. React. Eng. 2008, 2, 31–36
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Reynolds number of water under these conditions is
smaller than ten due to the very low flow rate. Therefore,
axial mixing can play an important role as well. When
Russum et al.[8] investigated RAFT miniemulsion poly-
merization in a tubular reactor under low flow rates, they
also found residence time distributions close to plug flow
and attributed this to a high axial mixing.
The results of all residence time distribution experiments
are given in Table 1. The full width at half maximum of the
signals (like the ones shown in Figure 2) becomes smaller
with increasing flow rate and increasing temperature. At
applying a pulse of NaCl solution. For each temperature and flowrate, three experiments were done giving the mean residencetime and full width at half maximum of the NaCl pulse.
Temperature Mass
flow
Mean
residence
time
Full width
at half
maximum
-C g �minS1 s s
21 2.13 16 068 751
21 2.11 15 859 754
21 2.07 15 621 763
21 3.11 10 708 600
21 3.11 10 698 600
21 3.14 10 639 591
80 3.17 10 262 360
80 3.17 10 240 354
80 3.19 10 192 350
www.mre-journal.de 33
M. Muller, M. F. Cunningham, R. A. Hutchinson
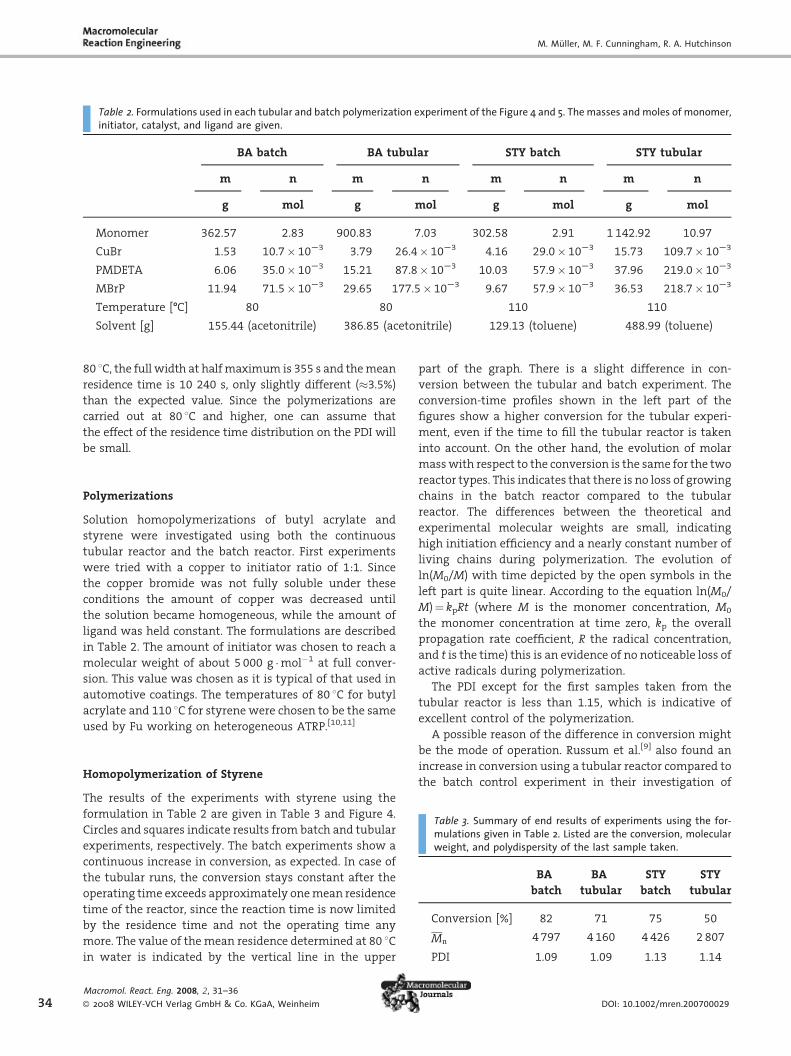
Table 2. Formulations used in each tubular and batch polymerization experiment of the Figure 4 and 5. The masses and moles of monomer,initiator, catalyst, and ligand are given.
BA batch BA tubular STY batch STY tubular
m n m n m n m n
g mol g mol g mol g mol
Monomer 362.57 2.83 900.83 7.03 302.58 2.91 1 142.92 10.97
CuBr 1.53 10.7� 10S3 3.79 26.4� 10S3 4.16 29.0� 10S3 15.73 109.7� 10S3
PMDETA 6.06 35.0� 10S3 15.21 87.8� 10S3 10.03 57.9� 10S3 37.96 219.0� 10S3
MBrP 11.94 71.5� 10S3 29.65 177.5� 10S3 9.67 57.9� 10S3 36.53 218.7� 10S3
Temperature [-C] 80 80 110 110
Solvent [g] 155.44 (acetonitrile) 386.85 (acetonitrile) 129.13 (toluene) 488.99 (toluene)
34
80 8C, the full width at halfmaximum is 355 s and themean
residence time is 10 240 s, only slightly different (�3.5%)
than the expected value. Since the polymerizations are
carried out at 80 8C and higher, one can assume that
the effect of the residence time distribution on the PDI will
be small.
Polymerizations
Solution homopolymerizations of butyl acrylate and
styrene were investigated using both the continuous
tubular reactor and the batch reactor. First experiments
were tried with a copper to initiator ratio of 1:1. Since
the copper bromide was not fully soluble under these
conditions the amount of copper was decreased until
the solution became homogeneous, while the amount of
ligand was held constant. The formulations are described
in Table 2. The amount of initiator was chosen to reach a
molecular weight of about 5 000 g �mol�1 at full conver-
sion. This value was chosen as it is typical of that used in
automotive coatings. The temperatures of 80 8C for butyl
acrylate and 110 8C for styrene were chosen to be the same
used by Fu working on heterogeneous ATRP.[10,11]
Table 3. Summary of end results of experiments using the for-mulations given in Table 2. Listed are the conversion, molecularweight, and polydispersity of the last sample taken.
BA
batch
BA
tubular
STY
batch
STY
tubular
Conversion [%] 82 71 75 50
Mn 4797 4 160 4 426 2807
PDI 1.09 1.09 1.13 1.14
Homopolymerization of Styrene
The results of the experiments with styrene using the
formulation in Table 2 are given in Table 3 and Figure 4.
Circles and squares indicate results from batch and tubular
experiments, respectively. The batch experiments show a
continuous increase in conversion, as expected. In case of
the tubular runs, the conversion stays constant after the
operating time exceeds approximately onemean residence
time of the reactor, since the reaction time is now limited
by the residence time and not the operating time any
more. The value of themean residence determined at 80 8Cin water is indicated by the vertical line in the upper
Macromol. React. Eng. 2008, 2, 31–36
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
part of the graph. There is a slight difference in con-
version between the tubular and batch experiment. The
conversion-time profiles shown in the left part of the
figures show a higher conversion for the tubular experi-
ment, even if the time to fill the tubular reactor is taken
into account. On the other hand, the evolution of molar
masswith respect to the conversion is the same for the two
reactor types. This indicates that there is no loss of growing
chains in the batch reactor compared to the tubular
reactor. The differences between the theoretical and
experimental molecular weights are small, indicating
high initiation efficiency and a nearly constant number of
living chains during polymerization. The evolution of
ln(M0/M) with time depicted by the open symbols in the
left part is quite linear. According to the equation ln(M0/
M)¼ kpRt (where M is the monomer concentration, M0
the monomer concentration at time zero, kp the overall
propagation rate coefficient, R the radical concentration,
and t is the time) this is an evidence of no noticeable loss of
active radicals during polymerization.
The PDI except for the first samples taken from the
tubular reactor is less than 1.15, which is indicative of
excellent control of the polymerization.
A possible reason of the difference in conversion might
be the mode of operation. Russum et al.[9] also found an
increase in conversion using a tubular reactor compared to
the batch control experiment in their investigation of
DOI: 10.1002/mren.200700029
Continuous Atom Transfer Radical Polymerization in a Tubular Reactor
Figure 4. Results of styrene homopolymerization. Tubular and batch experimental results aredepicted by squares and circles, respectively. Open symbols correspond to the right axis andclosed symbols to the left. The vertical line in the upper left part indicates the mean residencetime of the high temperature experiment in Table 1.
RAFT polymerization in miniemulsion. In case of the
batch experiments, the initiator is added after the reaction
mixture is heated up, to be as close as possible to the
tubular reactor, in which the reactionmixture is heated up
nearly instantaneously entering the reactor. Adding the
initiator in the batch experiments usually takes between
40 and 50 s and additional time to mix. Therefore, the
initiation period may be a little longer in the case of the
batch experiments.
After styrene polymerizations in the tubular reactor
were completed, at the beginning of the cleaning proce-
dure, it was observed that small particles were washed out
of the reactor. On one occasion the reactor became blocked
during the experiment. This is most probably due to
precipitation of the Cu(II) catalyst complex. The solubility
of the Cu(II) catalyst is lower than that of Cu(I).[10,11] During
polymerization there may be gradual accumulation of
Cu(II) due to a small but finite termination rate. While this
is not a serious problem in a stirred tank batch reactor, it
can cause serious operability issues with a tubular reactor.
There are several possible approaches to solve this
problem. The easiest would be to choose a different ligand
such as 4,4-di-(5-nonyl)-2,2-bipyridine (dNbpy) which
leads to a homogeneous reaction mixture.[12] However,
this ligand is not commercially available in large volumes
and is therefore too expensive for industrial applications.
Alternative approaches to make the reaction mixture
homogeneous are to reduce the amount of copper in the
solution or switch to another solvent which is a better
solvent for the catalyst complex. The former choice would
lead to a lower conversion and likely negatively impact
Macromol. React. Eng. 2008, 2, 31–36
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
control of the polymerization, and
therefore we investigated the use
of a different solvent. Due to its
low boiling point of 82 8C, acet-onitrile was deemed unsuitable,
and benzonitrile (bp¼ 189 8C)was chosen instead.
Initial experiments with ben-
zonitrile showed no precipitation
in a batch experiment but when
used in the tubular reactor did
lead to a blocked tube after �2 h
of operation. This time the block-
age occurred in or following
the heat exchanger, when the
reaction mixture was cooling
down. Opening a three-way valve
attached between the reactor
and the heat exchanger for test-
ing purposes revealed that the
hot reaction mixture was homo-
geneous. Further tests are cur-
rently being done to optimize the
cooling unit to prevent blockage as a result of catalyst
precipitation during the cooling step.
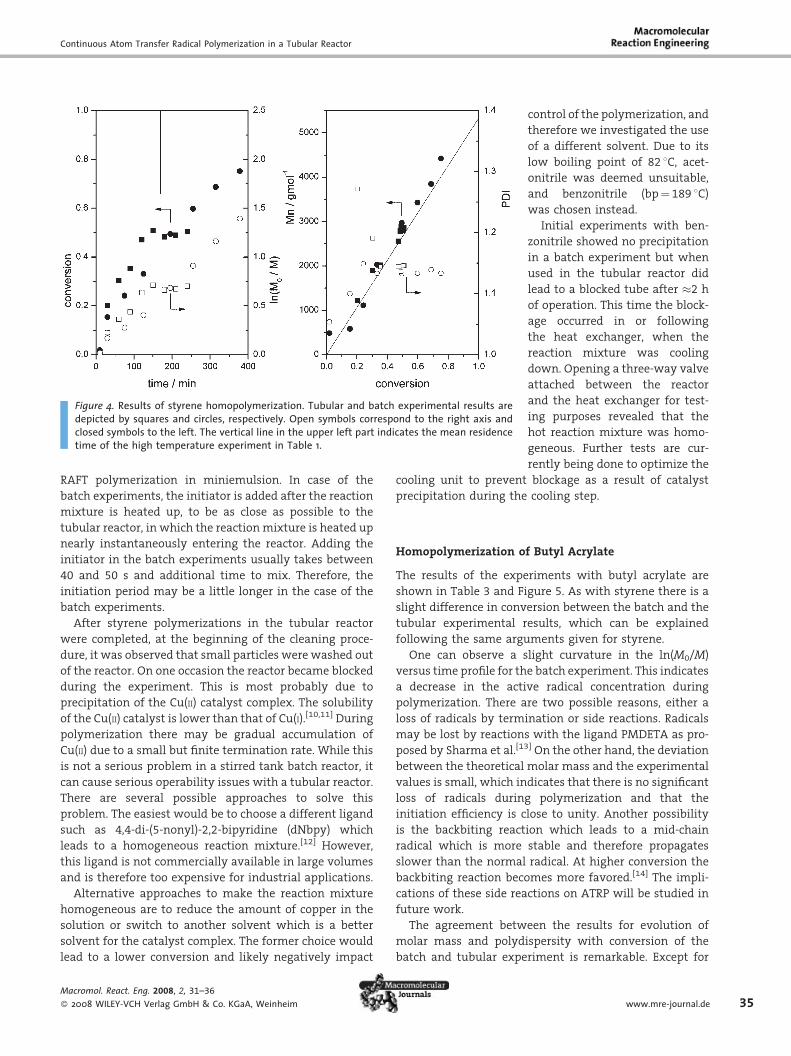
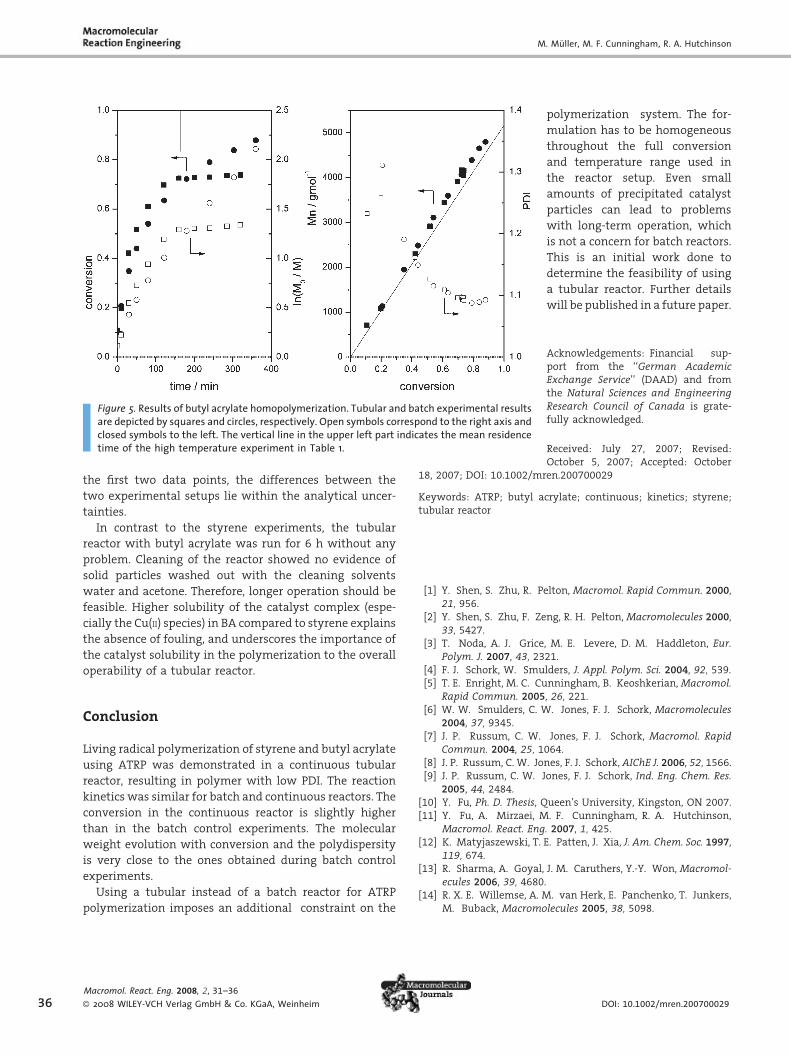
Homopolymerization of Butyl Acrylate
The results of the experiments with butyl acrylate are
shown in Table 3 and Figure 5. As with styrene there is a
slight difference in conversion between the batch and the
tubular experimental results, which can be explained
following the same arguments given for styrene.
One can observe a slight curvature in the ln(M0/M)
versus time profile for the batch experiment. This indicates
a decrease in the active radical concentration during
polymerization. There are two possible reasons, either a
loss of radicals by termination or side reactions. Radicals
may be lost by reactions with the ligand PMDETA as pro-
posed by Sharma et al.[13] On the other hand, the deviation
between the theoretical molar mass and the experimental
values is small, which indicates that there is no significant
loss of radicals during polymerization and that the
initiation efficiency is close to unity. Another possibility
is the backbiting reaction which leads to a mid-chain
radical which is more stable and therefore propagates
slower than the normal radical. At higher conversion the
backbiting reaction becomes more favored.[14] The impli-
cations of these side reactions on ATRP will be studied in
future work.
The agreement between the results for evolution of
molar mass and polydispersity with conversion of the
batch and tubular experiment is remarkable. Except for
www.mre-journal.de 35
M. Muller, M. F. Cunningham, R. A. Hutchinson
Figure 5. Results of butyl acrylate homopolymerization. Tubular and batch experimental resultsare depicted by squares and circles, respectively. Open symbols correspond to the right axis andclosed symbols to the left. The vertical line in the upper left part indicates the mean residencetime of the high temperature experiment in Table 1.
36
the first two data points, the differences between the
two experimental setups lie within the analytical uncer-
tainties.
In contrast to the styrene experiments, the tubular
reactor with butyl acrylate was run for 6 h without any
problem. Cleaning of the reactor showed no evidence of
solid particles washed out with the cleaning solvents
water and acetone. Therefore, longer operation should be
feasible. Higher solubility of the catalyst complex (espe-
cially the Cu(II) species) in BA compared to styrene explains
the absence of fouling, and underscores the importance of
the catalyst solubility in the polymerization to the overall
operability of a tubular reactor.
Conclusion
Living radical polymerization of styrene and butyl acrylate
using ATRP was demonstrated in a continuous tubular
reactor, resulting in polymer with low PDI. The reaction
kinetics was similar for batch and continuous reactors. The
conversion in the continuous reactor is slightly higher
than in the batch control experiments. The molecular
weight evolution with conversion and the polydispersity
is very close to the ones obtained during batch control
experiments.
Using a tubular instead of a batch reactor for ATRP
polymerization imposes an additional constraint on the
Macromol. React. Eng. 2008, 2, 31–36
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
polymerization system. The for-
mulation has to be homogeneous
throughout the full conversion
and temperature range used in
the reactor setup. Even small
amounts of precipitated catalyst
particles can lead to problems
with long-term operation, which
is not a concern for batch reactors.
This is an initial work done to
determine the feasibility of using
a tubular reactor. Further details
will be published in a future paper.
Acknowledgements: Financial sup-port from the ‘‘German AcademicExchange Service’’ (DAAD) and fromthe Natural Sciences and EngineeringResearch Council of Canada is grate-fully acknowledged.
Received: July 27, 2007; Revised:October 5, 2007; Accepted: October
18, 2007; DOI: 10.1002/mren.200700029
Keywords: ATRP; butyl acrylate; continuous; kinetics; styrene;tubular reactor
[1] Y. Shen, S. Zhu, R. Pelton, Macromol. Rapid Commun. 2000,21, 956.
[2] Y. Shen, S. Zhu, F. Zeng, R. H. Pelton, Macromolecules 2000,33, 5427.
[3] T. Noda, A. J. Grice, M. E. Levere, D. M. Haddleton, Eur.Polym. J. 2007, 43, 2321.
[4] F. J. Schork, W. Smulders, J. Appl. Polym. Sci. 2004, 92, 539.[5] T. E. Enright, M. C. Cunningham, B. Keoshkerian, Macromol.
Rapid Commun. 2005, 26, 221.[6] W. W. Smulders, C. W. Jones, F. J. Schork, Macromolecules
2004, 37, 9345.[7] J. P. Russum, C. W. Jones, F. J. Schork, Macromol. Rapid
Commun. 2004, 25, 1064.[8] J. P. Russum, C. W. Jones, F. J. Schork, AIChE J. 2006, 52, 1566.[9] J. P. Russum, C. W. Jones, F. J. Schork, Ind. Eng. Chem. Res.
2005, 44, 2484.[10] Y. Fu, Ph. D. Thesis, Queen’s University, Kingston, ON 2007.[11] Y. Fu, A. Mirzaei, M. F. Cunningham, R. A. Hutchinson,
Macromol. React. Eng. 2007, 1, 425.[12] K. Matyjaszewski, T. E. Patten, J. Xia, J. Am. Chem. Soc. 1997,
119, 674.[13] R. Sharma, A. Goyal, J. M. Caruthers, Y.-Y. Won, Macromol-
ecules 2006, 39, 4680.[14] R. X. E. Willemse, A. M. van Herk, E. Panchenko, T. Junkers,
M. Buback, Macromolecules 2005, 38, 5098.
DOI: 10.1002/mren.200700029