conformational sampling with stochastic proximity embedding and self-organizing superimposition:...
TRANSCRIPT

Conformational Sampling with Stochastic Proximity Embedding and Self-OrganizingSuperimposition: Establishing Reasonable Parameters for Their Practical Use
Gary Tresadern*,† and Dimitris K. Agrafiotis‡
Johnson & Johnson, Pharmaceutical Research & Development, Janssen-Cilag S.A., Calle Jarama 75, PoligonoIndustrial, Toledo 45007, Spain, and Johnson & Johnson, Pharmaceutical Research & Development, L.L.C.,
665 Stockton Drive, Exton, Pennsylvania 19341
Received May 29, 2009
Stochastic proximity embedding (SPE) and self-organizing superimposition (SOS) are two recently introducedmethods for conformational sampling that have shown great promise in several application domains. Ourprevious validation studies aimed at exploring the limits of these methods and have involved rather exhaustiveconformational searches producing a large number of conformations. However, from a practical point ofview, such searches have become the exception rather than the norm. The increasing popularity of virtualscreening has created a need for 3D conformational search methods that produce meaningful answers in arelatively short period of time and work effectively on a large scale. In this work, we examine the performanceof these algorithms and the effects of different parameter settings at varying levels of sampling. Our goalis to identify search protocols that can produce a diverse set of chemically sensible conformations and havea reasonable probability of sampling biologically active space within a small number of trials. Our resultssuggest that both SPE and SOS are extremely competitive in this regard and produce very satisfactoryresults with as few as 500 conformations per molecule. The results improve even further when the rawconformations are minimized with a molecular mechanics force field to remove minor imperfections andany residual strain. These findings provide additional evidence that these methods are suitable for manyeveryday modeling tasks, both high- and low-throughput.
INTRODUCTION
Conformational sampling is an essential part of themolecular modeling process. Three-dimensional (3D) con-formations are used in molecular overlays, pharmacophorehypothesis generation, pharmacophore and shape searching,3D QSAR, field-based virtual screening, docking, and otherrelated applications. These analyses are often facilitatedthrough the construction of databases containing a finitenumber of precomputed conformations for each moleculeof interest. The underlying aim of conformational samplingis to generate 3D molecular structures, which represent thechemical compound as it exists in a biologically relevantcontext. A molecule can often adopt many energeticallyaccessible conformations in solution, but it generally assumesa distinct conformation, referred to as the bioactive confor-mation, when bound to a target protein. Clearly, to be usefulin drug design, a conformational search method must be ableto reproduce the bioactive conformation. Because thatconformation is, in turn, dependent on the structure anddynamics of the protein host and some ligands adopt differentbinding conformations with different proteins,1 it is importantthat the sampling of conformational space is as completeand unbiased as possible.
Conformation generation algorithms generally fall into twobroad categories: deterministic methods, which exhaustively
enumerate all possible torsions at certain discrete intervals,and stochastic methods, which use a random element toexplore the molecule’s conformational space.2 Althoughsystematic search can be very effective for molecules withlimited conformational flexibility, the exponential growth ofthe search space with the number of rotatable bonds, as wellas problems associated with ring closures, limit its utility asa general conformational sampling technique.3-6 For flexiblemolecules, stochastic methods designed to sample low energyconformations represent a viable alternative. In its simplestform, a stochastic method randomly perturbs the currentconformation of the molecule, minimizes it in energy, andrepeats the process to generate a sequence of minimizedconformations.7,8 Standard simulation techniques, such asmolecular dynamics and Monte Carlo methods, have beenused to generate an ensemble of conformations that lie inthe low energy regions on the potential energy surface.9-11
All of these methods generate conformations in a continuoustrajectory, in that each trial conformation is derived fromthe preceding one by a relatively small change. Because ofthis continuity, a large number of conformations are gener-ated between the important low energy ones, and a consider-able amount of computer time is spent on the calculationand minimization of potential energies for these transitionalconformations.
There have been numerous studies comparing computa-tionally and experimentally determined conformations as ameans of validating different search methods and protocols.Some of these studies focus on small molecule crystalstructures and others on bioactive conformations.12-14 In
* Corresponding author phone: +34-925-24-5782; e-mail: [email protected].
† Johnson & Johnson, Spain.‡ Johnson & Johnson, PA.
J. Chem. Inf. Model. 2009, 49, 2786–28002786
10.1021/ci9001926 CCC: $40.75 2009 American Chemical SocietyPublished on Web 11/17/2009

general, the conformation of a molecule in its crystal rarelymatches the protein-bound structure.15 Earlier work on asmall data set of 10 ligands cocrystallized with a targetprotein showed that the bioactive conformation is similar tothe lowest energy conformation in solution,16 but subsequentstudies on larger data sets suggest that it may be significantlyhigher in energy as compared to the global minimum.14,17
In fact, the relative conformational energies derived withcurrent molecular mechanics force fields introduce consider-able errors, which hinder the accurate calculation of freeenergies of binding.18 This emphasizes the need for cautionand good judgment when applying energy cutoffs to filterout irrelevant conformations. As for the differences ingeometry between free and bioactive conformations, Dillerand Merz have shown that bound conformations tend to bemore extended than the average random conformationsgenerated in vacuo.19 Together, these studies cast doubt overthe efficiency of conventional search methods in locatingbioactive conformations.
Two newer techniques that have shown considerablepromise in sampling the entire conformational space acces-sible to a given molecule are stochastic proximity embedding(SPE) and self-organizing superimposition (SOS). Bothmethods are based on a distance geometry formalism thatgenerates conformations that satisfy a set of geometricconstraints derived from the molecular connectivity table.20,21
There are two forms of constraints: (1) distance constraintsencoded in the form of upper (uij) and lower (lij) bounds forevery interatomic distance, dij (such that lij e dij e uij), and(2) volume constraints that prevent the signed volume Vijkl
formed by four atoms i, j, k, l from exceeding certain limits.The latter are used to enforce planarity of conjugated systemsand correct chirality of stereocenters. By generating coor-dinates that satisfy these constraints, one should in theorybe able to sample the entire conformational space. Theadvantage of distance geometry is that it generates chemicallysensible conformations without any direct energy calculation.The conformations can then be rapidly minimized with anysuitable force field to identify the corresponding energyminima.
SPE starts from a random initial conformation andgradually refines it by repeatedly selecting an individualconstraint at random and updating the respective atomiccoordinates toward satisfying that specific constraint. Thisprocedure is performed repeatedly until a reasonable con-formation is obtained.22-24 SOS utilizes a similar algorithm,but makes use of precomputed fragment geometries, whichcontributes to reduced CPU times and more physicallyrealistic geometries.25 A more detailed description of thesealgorithms can be found in the references and in the Methodssection.
Because of the nature of the embedding procedure andthe use of random initial atomic coordinates, both of thesemethods tend to produce conformations that are relativelycompact. To overcome this problem, we introduced a simpleboosting heuristic that can be used in conjunction with SOSand SPE to bias the search toward more extended (or morecompact) geometries.26 This is accomplished through a seriesof embeddings, each seeded on the result of the previousone. By probabilistically promoting extended geometries, theboosting heuristic biases the search toward the likelybioactive region of conformational space, but maintains
sufficient sampling power to ensure that important conforma-tions will not be missed.22-30
In the present work, we study in greater detail theperformance of SPE and for the first time the SOS algorithmin their ability to sample biologically relevant conformations.In a previous paper, we compared SPE to seven com-mercially available programs by generating 10 000 confor-mations for each of a set of 59 ligands, and showedexcellence performance for SPE and conformational boost-ing.29 The aim of the present study is not to repeat such anextensive comparison under idealized conditions, but ratherto establish a reasonable set of parameters that would allowreliable sampling of bioactive space using considerably fewertrials, and to provide practical guidelines on how to best usethese methods in everyday modeling problems. To have moreconfidence in our findings, we utilize a larger data set ofprotein-ligand crystal structures, which combines data fromthe work of Bostrom et al.,12 Diller and Merz,19 Perola andCharifson,14 and Chen and Foloppe (which, for the sake ofbrevity, will hereafter be identified by the name of their firstauthor).31 The performance of the search is assessed in fiveways: (1) the ability to retrieve the bioactive conformation,(2) the diversity of the generated conformations, (3) com-parison to a gold standard ensemble, (4) the strain energyof the conformations, and (5) the computational cost required.More specifically, we use as metrics the rmsd of thegenerated conformations as compared to their respectivebioactive structures, the number of unique 3D pharmacophorebits present in the resulting conformation families, theTanimoto similarity of the 3D pharmacophore bits to the goldstandard, the strain energy between each conformation andits nearest local minimum, and the CPU time required toperform the search. We show that the use of SPE and SOSwith subsequent energy refinement can lead to excellentperformance with respect to all of these criteria.
METHODS
Data Sets. The data set used in this study was assembledfrom several previous related works.12,14,19,31 The Bostromsubset contained 32 ligands from cocrystal structures withresolution e2.0 Å, most of which had temperature factorsbelow 30 Å2. The Diller subset contained 59 ligands withresolution e3.0 Å, of which 39 were e2.0 Å. This is thesame data set that we used in our previous comparison ofSPE to several widely used conformational analysis por-grams.29 The third subset consisted of the 100 publicallyavailable structures in the Perola data set, all of which hadresolution e3.0 Å and 48 of them e2.0 Å. Our validationsuite was completed with 130 ligands from the Chen dataset, all of which had resolution e2.5 Å and 54 of them e2.0Å. The Chen subset included a diverse selection of structuresfrom the PDB with resolution e2.5 Å, MW e 600, and less12 rotatable bonds, which were not already present inBostrom and Perola collections.
The entire data set was made up exclusively of nonco-valently bound ligands. The structures were downloaded andextracted from their respective Protein Data Bank (PDB)entries, protonation states were assigned using the MOE washfunction, tautomer states were manually assigned, and theresults were visually inspected. All ligands were convertedinto 2D and used in that form as input to the differentconformational search programs.
CONFORMATIONAL SAMPLING WITH SPE AND SOS J. Chem. Inf. Model., Vol. 49, No. 12, 2009 2787
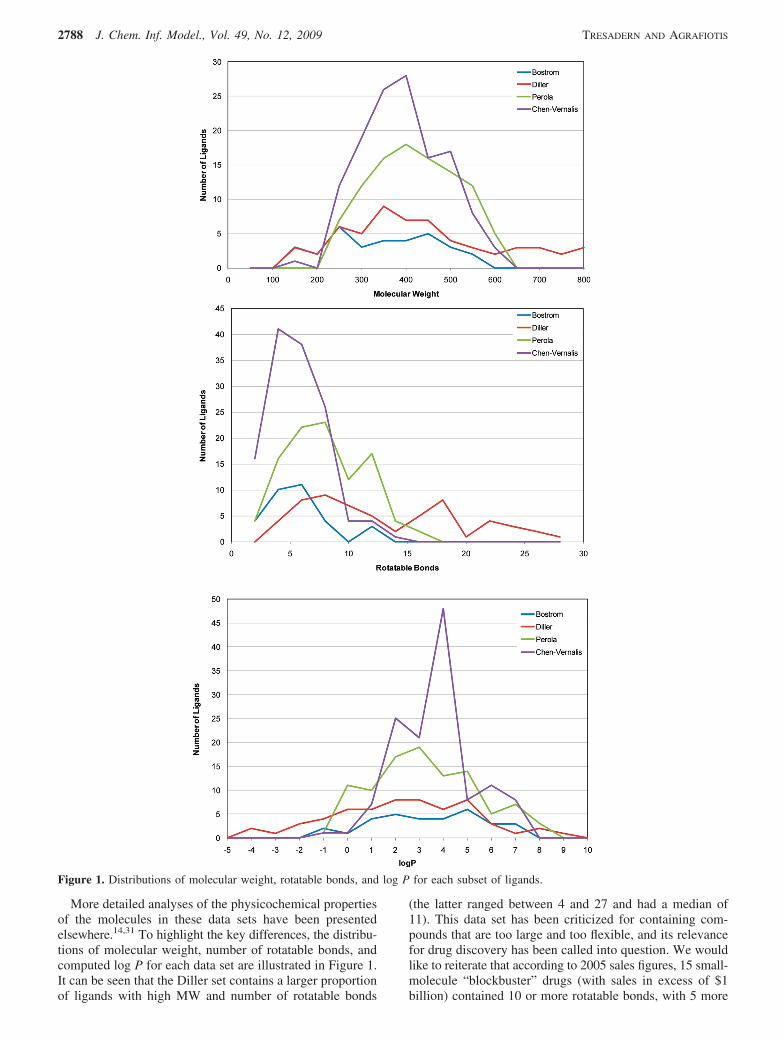
More detailed analyses of the physicochemical propertiesof the molecules in these data sets have been presentedelsewhere.14,31 To highlight the key differences, the distribu-tions of molecular weight, number of rotatable bonds, andcomputed log P for each data set are illustrated in Figure 1.It can be seen that the Diller set contains a larger proportionof ligands with high MW and number of rotatable bonds
(the latter ranged between 4 and 27 and had a median of11). This data set has been criticized for containing com-pounds that are too large and too flexible, and its relevancefor drug discovery has been called into question. We wouldlike to reiterate that according to 2005 sales figures, 15 small-molecule “blockbuster” drugs (with sales in excess of $1billion) contained 10 or more rotatable bonds, with 5 more
Figure 1. Distributions of molecular weight, rotatable bonds, and log P for each subset of ligands.
2788 J. Chem. Inf. Model., Vol. 49, No. 12, 2009 TRESADERN AND AGRAFIOTIS

of these molecules (like clarithromycin) having just as muchflexibility due to large rings.29 Also, a recent study of 147antibacterial compounds that are either on the market or inclinical development showed noticeably different MW andpolar surface area properties as compared to drugs in othertherapeutic areas.32 Therefore, in the antibacterial field, wherefinding initial hits is a known challenge,33 drug discoveryefforts may be forced toward higher molecular weights. Inaddition, larger and more flexible compounds present achallenge to many conformational search methods, and, aswill be shown in the sequel, allow for differences inperformance to be more clearly discerned. The Diller dataset is thus a useful complement to the compounds found inthe Bostrom, Perola, and Chen collections, the last two beingperhaps the most “drug-like”, with 88% of the compoundshaving MW between 200 and 500, and 88% of them havinge10 rotatable bonds.
The protein structures from which these ligands wereextracted span a range of different targets, including kinases,HIV reverse transcriptase, nuclear receptors, phosphotases,oxido-reductases, metalloproteases, isomerases, and lyases.In summary, our data set consists of drug-like ligands foundin multiple binding site environments, and contains a smallfraction of challenging, more flexible molecules that test thelimits and range of applicability of the conformational searchmethods.
Comparison Metrics. The main objective of the presentstudy was to assess how many conformations need to begenerated to have sufficient confidence that the biologicallyactive conformation (or some other conformation similar toit) is identified. Stated differently, we wanted to examinehow much does the performance of the various algorithmsdecline as fewer and fewer conformations are generated, andestablish sensible trade-offs between quality of sampling andcomputational speed. To address these questions, eachmethod was asked to produce 50, 100, 500, 1000, 5000, and10 000 conformations for each molecule, in six independentruns. Several variations of these methods were evaluated,
using different parameter settings as described in the sequel.A list of these parameter settings along with their abbreviatednames are shown in Table 1. Five basic methods werecompared: SPE with and without conformational boosting,SOS with and without conformational boosting, MOEconformational import, MOE bond rotation, and MOEstochastic search.
Stochastic Proximity Embedding. As mentioned in theIntroduction, SPE attempts to generate conformations thatsatisfy a set of distance and volume (chiral) constraints. Thefirst step of the process is to establish the lower and upperdistance bounds for every pair of atoms, as well as the desiredchiral volumes for every chiral center and planar system inthe molecule. These bounds are derived from the molecularconnectivity table and standard covalent geometry rules, ina manner similar to other distance geometry methods. Oncethe constraints are established, the program assigns randomcoordinates to all of the atoms, and uses a self-organizingscheme to rapidly refine the atomic positions so as to satisfyall of the input constraints.24,26,27 This refinement is ac-complished by randomly selecting one constraint at a time,adjusting the positions of the respective atoms so as to bettersatisfy that specific constraint, and repeating the processmany times until a sensible geometry is obtained. Themagnitude of the atomic adjustments is controlled by aparameter called the learning rate, which is gradually reducedduring the course of the refinement to improve convergenceand avoid oscillatory behavior. Distance and volume con-straints are selected with different probabilities, with theformer being sampled more frequently than the latter. Moredetails about the algorithm can be found in several previouspublications.24,26,27
Self-Organizing Superimposition. The self-organizingsuperimposition (SOS) algorithm employs an iterative schemesimilar to that SPE, but makes use of precomputed confor-mational templates of rigid fragments to enforce the desiredgeometry.25 In SPE, the embedding is carried out oneconstraint at a time, and the coordinate adjustments are
Table 1. Details of All Calculations Performed in This Worka
name maximum number of conformations description
SPE 50, 100, 500, 1000, 5000, and 10 000 SPE with bidirectional boosting toward both compact and extendedconformations in a 3:5 ratio
SPE_COM 50, 100, 500, 1000, 5000, and 10 000 SPE with three levels of boosting toward more compact geometriesSPE_EXT 50, 100, 500, 1000, 5000, and 10 000 SPE with five levels of boosting toward more extended geometriesSPE_MIN_0.1 50, 100, 500, 1000, 5000, and 10 000 same as “SPE” above, followed by MMFF94s minimization and
removal of duplicates within 0.1 Å rmsdSPE_COM_MIN_0.1 50, 100, 500, 1000, 5000, and 10 000 same as “SPE_COM” above, followed by MMFF94s minimization
and removal of duplicates within 0.1 Å rmsdSPE_EXT_MIN_0.1 50, 100, 500, 1000, 5000, and 10 000 same as “SPE_EXT” above, followed by MMFF94s minimization
and removal of duplicates within 0.1 Å rmsdSPE_0.1 50, 100, 500, 1000, 5000, and 10 000 same as “SPE” above, followed by removal of duplicates within 0.1
Å rmsdSPE_0.4 50, 100, 500, 1000, 5000, and 10 000 same as “SPE” above, followed by removal of duplicates within 0.4
Å rmsdSPE_0.8 50, 100, 500, 1000, 5000, and 10 000 same as “SPE” above, followed by removal of duplicates within 0.8
Å rmsdSOS 50, 100, 500, 1000, 5000, and 10 000 SOS without boostingSOS_EXT 50, 100, 500, 1000, 5000, and 10 000 SOS with five levels of boosting toward more extended geometriesMOE_CI 50, 100, 500, 1000, 5000, and 10 000 MOE conformational import with default settingsMOE_BR 50, 100, 500, 1000, 5000, and 10 000 MOE bond rotation with default settingsMOE_SS 10 000 MOE stochastic search with Chen settings
a See main text for details on parameter settings. Minimization performed for a maximum of 1000 cycles using the MMFF94s force fieldignoring the electrostatic term.
CONFORMATIONAL SAMPLING WITH SPE AND SOS J. Chem. Inf. Model., Vol. 49, No. 12, 2009 2789

applied only to the atoms involved in that specific constraint.However, most organic molecules contain locally rigidfragments connected through freely rotatable bonds. Forinstance, a phenyl ring can be thought of as a rigid fragmentthat assumes a regular hexagonal geometry regardless of itssurrounding environment. SOS takes advantage of this byusing precomputed conformational templates for each rigidpart of the molecule, and by adjusting the positions of all ofthe atoms in each fragment at once using a least-squaresfitting procedure.
More specifically, the SOS algorithm starts by identifyingall rotatable bonds in the molecule, and then removes thesebonds to generate a set of disconnected fragments. Bondsare considered rotatable if they are single, nonterminal, andthey are not part of a delocalized system or a ring of size 6or less. Each resulting fragment is encoded as a canonicalSMILES string, which is then used as a key to retrieve thecorresponding conformation from a precomputed 3D frag-ment library. If the requested fragment is not present in thelibrary, the program will generate a reasonable conformationusing standard SPE.
While the coordinates of core atoms in a template are takendirectly from the retrieved fragment conformation, thecoordinates of the attached atoms are not immediatelyavailable because they are not present in the fragmentmolecule. However, all hydrogen atoms of the fragmentconformations in the library are explicitly represented with3D coordinates. The coordinates of each attached atom aredetermined by replacing a corresponding hydrogen atom inthe fragment conformation and adjusting the bond lengthaccordingly. With the steps described above, the templatescan be quickly constructed for any input molecule duringthe initialization phase.
The actual embedding is carried out by an alternating seriesof template fitting operations and pairwise distance adjust-ments. Template fitting involves two basic steps. In the firststep, a rigid-body transformation (by translation and rotation)is performed on the reference template, which essentiallyplaces it in the closest position to the corresponding fragmentin the molecule. This can be done using the standard rigid-body rmsd superimposition technique or the improvedmethod described in ref 34. In the second step, thecoordinates of the fragment atoms in the molecule arereplaced by the new coordinates of the respective atoms inthe superimposed reference template. In essence, the fittingoperation applies the smallest possible adjustment to thefragment atoms in the molecule to achieve the geometry ofthe reference template. Each template fitting operation isfollowed by a number of pairwise distance adjustmentsbetween randomly chosen atoms from different fragmentsto remove steric clashes (similar to SPE). This process iscarried out for each fragment in the molecule, and the entireprocess is repeated until a predetermined convergencecriterion is met.
Because rigid fragments are precomputed, planarity andchirality constraints are automatically satisfied after thetemplate superimposition process, and local geometry isperfectly restored. Furthermore, because each embeddingstarts from completely random initial atomic coordinates,each new conformation is independent of those generatedin the previous runs, resulting in greater diversity and moreeffective sampling. As demonstrated in the sequel, because
the algorithm only involves pairwise distance adjustmentsand superimposition of relatively small fragments, it isimpressively efficient.
In this study, the required precomputed 3D library wasgenerated by breaking each molecule in an earlier versionof PubChem into fragments using the method outlined above,generating a conformation for each of these fragments usingSPE, and finally refining the resulting raw conformationsthrough MMFF94s energy minimization.
Conformational Boosting. Boosting is a simple heuristicthat can be used in conjunction with SPE and SOS togenerate increasingly extended or compact conformationsthrough iterations.26,28 In the first iteration, a normal SPEor SOS embedding is performed as described in the previoussections, generating a chemically sensible conformation c1.The lower bounds of all atom pairs lij are then replaced bythe actual interatomic distances dij in conformation c1 andused along with the unchanged upper bounds uij and volumeconstraints, Vl
ijkl and Vuijkl, to perform a second embedding
to generate another conformation, c2. This process is repeatedfor a prescribed number of iterations. The lower bounds arethen restored to their original default values, and a newsequence of embeddings is performed using a differentrandom number seed. Because the distance constraints in anyiteration are always equal to or greater than those in theprevious iterations, successively more extended conforma-tions should be generated. This process will never yield aset of distance constraints that are impossible to satisfy,because there exists at least one conformation (i.e., the onegenerated in the preceding iteration) that satisfies them.Therefore, the conformational space defined by the distanceconstraints will shrink but not vanish over the iterations, thuseffectively biasing the SPE sampling toward more extendedgeometries. An analogous procedure can be used to generateincreasingly compact conformations26 and has been em-ployed in this work as described in the following paragraphs.
SPE and SOS Parameters. The present study wasinitiated before the SOS algorithm was available; therefore,the majority of our experiments centered on SPE. Severalindependent conformational searches with different parametersettings were performed. For the SPE method, the adjustableparameters involved the level and direction of boosting, theuse of minimization, and the use of different rmsd thresholdsto eliminate duplicate conformations. The various parametersettings and the abbreviated names of the resulting searchprotocols are listed in Table 1.
Three different levels of boosting were evaluated for SPE.The first, referred to simply as SPE, used boosting in bothdirections to generate a combination of compact and extendedconformations in a ratio of 3:5, following the same protocolused previously.29 To generate the compact conformations,SPE was used with the options “-boost 3 -inVerse -keepall”.These options instructed the program to generate a series ofthree conformations for each trial, {ci,1, ci,2, ci,3}, where idenotes the ith trial. Conformation ci,1 was generated usingthe default distance bounds, ci,2 was generated using theinteratomic distances in ci,1 as upper bounds, and ci,3 wasgenerated using the interatomic distances in ci,2 as upperbounds. This process was repeated for N/3 trials, where Nwas the desired number of conformations. The first confor-mation in each trial was generated completely independentlyusing a different random number seed. The extended
2790 J. Chem. Inf. Model., Vol. 49, No. 12, 2009 TRESADERN AND AGRAFIOTIS

conformations were generated in a similar fashion using theoptions “-boost 5 -keepall”. The resulting conformations fromeach run were concatenated to form the final ensemble. Bydefault (see exceptions below), no duplicate check wasperformed, and all conformations were retained.
To further probe the impact of boosting, calculations werealso performed using only compact or only extendedconformations (denoted as SPE_COM and SPE_EXT, re-spectively). The parameter settings were virtually the sameas those described above, except that the number of trialswas increased accordingly to generate the desired numberof conformations.
The SPE and SOS algorithms generate conformationsbased on purely geometric considerations and do not requireany energy calculations. However, both programs have abuilt-in energy minimization option to allow conformationgeneration and subsequent energy refinement in the samecommand. To assess the impact of minimization, the SPE,SPE_COM, and SPE_EXT protocols were repeated usingidentical settings but with the additional -minimize flag. Thisapproach minimizes all generated conformations for amaximum of 1000 steps using the MMFF94s force field andthe BFGS variable metric minimization algorithm. Becauseminimization forces some raw conformations to convergeto the same local minima, we used an additional flag to re-move duplicate conformations from the final output, using anrmsd cutoff of 0.1 Å. The resulting ensembles are referred toas SPE_MIN_0.1, SPE_COM_MIN_0.1, and SPE_EXT_MIN_0.1, respectively.
The effect of applying a duplicate check to the raw SPEgeometries was also tested. Three different rmsd thresholdsfor removing duplicates were examined: 0.1, 0.4, and 0.8Å. The resulting ensembles are denoted by SPE_cutoff,SPE_COM_cutoff, and SPE_EXT_cutoff, where cutoff is thevalue of the rmsd threshold employed in the correspondingrun (i.e., 0.1, 0.4, and 0.8).
While SPE forms the majority of this work, we alsoincluded two SOS protocols to assess the general utility ofthe method. The first used the default parameters withoutany boosting (denoted as SOS), and the second used fivelevels of boosting toward more extended geometries, in amanner similar to the one described above (denoted asSOS_EXT). No inverse boosting was attempted, because atthat point there was increasing evidence that both SPE andSOS had a tendency to generate relatively compact geom-etries. In all cases, the number of trials was set such that thetotal number of output conformations was equal to 50, 100,500, 1000, 5000, and 10 000. The resulting conformationswere not subjected to rmsd duplicate filtering or energyminimization.
MOE Conformational Import. The MOE conformationalimport algorithm (MOE_CI) breaks the molecule intooverlapping fragments, retrieves precomputed conformationsof those fragments, and reassembles them by rigid bodysuperimposition. The first time a fragment conformation isencountered, should its conformation not exist in the frag-ment database, a stochastic approach is used to generatesuitable conformations. The coordinates for the fragmentsare then appended to the fragment database and used forsubsequent molecules containing the same structure. In thepresent study, fragment conformations with MMFF94x strainenergy greater than 5 kcal mol-1 were ignored. As with SOS
and SPE, six different conformational ensembles weregenerated for each molecule by setting the maximum numberof conformations to 50, 100, 500, 1000, 5000, and 10 000,respectively. All other parameters were left at their defaultvalues. Chen and Foloppe found that modification of otherparameters did not lead to significant improvements inperformance.31
MOE Bond Rotation. The MOE bond rotation (MOE_BR) sampling approach is based on the conformationgenerator used in the MOE docking and pharmacophoreelucidation routines. This relatively simple approach treatsall ring systems as rigid and generates new conformationsby bond rotation. The algorithm starts by converting the inputmolecule into 3D and minimizing its energy using theMMFF94x force field. From the resulting 3D configuration,new conformations are generated by modifying the torsionangles of the rotatable bonds based on a set of predefinedrules. There is no subsequent external refinement/minimiza-tion of the resulting conformations. The process generates auser-defined maximum number of conformations, which wasset to 50, 100, 500, 1,000, 5000, and 10 000, in accordancewith the protocol outlined above.
MOE Stochastic Search. MOE’s stochastic search(MOE_SS) is similar to Ferguson’s Random IncrementalPulse Search method8 in that new conformations are gener-ated via random perturbation of the parent conformation butdiffers in that the perturbation is of bonds rather thanCartesian coordinates. In this study, we have used stochasticsearch largely as a reference method, using the protocolsettings identified by Chen and Foloppe, which gave the bestresults in their comparative analysis of the MOE and Catalystconformational search algorithms. However, it was deemedto be a relatively low-throughput approach that was bestsuited for “detailed characterization of key compounds”.31
Our goal was to see if we could approach (or even improve)the performance of this protocol while reducing the com-putational cost. A 15 kcal mol-1 energy window was usedalong with the MMFF94x force field and the generalizedBorn (GB) solvation model, as implemented in MOE.35 Theinternal dielectric constant was set to 1 and the external to80. All other parameters were left at their default values. Amaximum of 10 000 conformations were requested for eachmolecule using an rmsd threshold of 0.1 Å to filter outduplicate structures (i.e., a new conformation was added tothe output set only if it differed by more than 0.1 Å rmsd toall previously identified conformations of the same molecule).
Analysis of Conformations. The first measure of searchperformance was to test if at least one of the generatedconformations was similar to the bioactive conformation ofthe molecule in question. Here, similarity was quantified bythe root mean squared distance (rmsd) between the generatedconformations and the corresponding bioactive structure.Each generated conformation was first aligned to the bio-active one using MOE’s least-squares superimpositionprocedure (SVL “superpose” function), and the rmsd betweenthe overlaid atoms was computed (heavy atoms only). (Wenote, parenthetically, that our group has recently developedan improved algorithm to determine the optimal rotation forleast-squares superposition using a Newton-Raphson quater-nion-based method and an adjoint matrix, that is at least anorder of magnitude faster than conventional inversion/decomposition methods.34) The total number of ligands with
CONFORMATIONAL SAMPLING WITH SPE AND SOS J. Chem. Inf. Model., Vol. 49, No. 12, 2009 2791

a conformation within an rmsd of 0.5, 1.0, 1.5, and 2.0 Å tothe bioactive conformation was determined and was used tomeasure the ability of each conformational search protocolto identify bioactive conformations.
Recently, the use of rmsd to assess docking poses to crystalstructures has been compared to the real space R-factor(RSR).36 That work was based on the observation thatexperimentally determined electron densities are sometimesincomplete and the exact coordinates of certain parts of thebound ligand can be equivocal. In such cases, comparingdocking poses to the electron density itself rather than exactatomic positions could help eliminate some of that ambiguity.These measures, however, are not entirely uncorrelated, inthat conformations with low rmsd also tend to have low RSR.Even when the electron density is inconclusive, the fittedstructure is still in the general vicinity of the true bioactiveconformation, and rmsd remains a relevant metric, and onethat is much easier to implement and faster to compute.
The diversity of the conformations produced by eachsearch protocol was quantified using the number of uniquepharmacophore triplets and quadruplets in the resultingensembles, as encoded by MOE’s 3D pharmacophorefingerprints. This approach is similar to that used in a recentstudy of library diversity.37 Both 3-point (piDAPH3) and4-point (piDAPH4) pharmacophores were computed, usingthe default definitions of hydrogen-bond donors, acceptors,and hydrophobic centers. These descriptors encode the 3Dpharmacophores of each conformation in a fixed-lengthbinary set, where each bit represents the presence or absenceof a particular pharmacophore feature. The union (inclusiveOR) of the binary sets of all of the conformations generatedfor a given molecule by a given method represents the entirepharmacophore space sampled by that method, with the totalcount of “on” bits serving as the ultimate measure ofpharmacophore diversity. To normalize for conformationalflexibility across different ligands, we report the averagenumber of pharmacophore bits per generated conformation,obtained by dividing the total number of pharmacophore bitsby the number of unique conformations identified by eachsearch protocol.
In addition, a “gold standard” conformational ensemblewas generated by combining the output of all conformationalsearches and minimizing with the MMFF94x force field inMOE. As before, an rmsd threshold of 0.1 Å was used tofilter out duplicate structures. The same 3- and 4-pointpharmacophore fingerprints were computed, and the outputfrom each protocol was compared to the gold standard usingthe Tanimoto coefficient. This approach penalizes methodswith missing pharmacophores from energetically favorableconformations as well as excess pharmacophores fromstrained conformations.
The strain energy of the raw conformations produced bythe various methods was calculated using the E_straindescriptor in MOE. The strain energy is defined as the energyof the raw conformation minus the energy of the nearest localminimum, obtained by molecular mechanics force fieldminimization using the raw conformation as a starting point.To prepare the conformations for energy minimization,implicit hydrogen atoms were added and charges werecalculated. Energies were computed using the MMFF94xforce field as implemented in MOE using default settings.Distributions and simple statistics such as the mean strain
energy were used to identify which methods produced morehighly strained and unrealistic conformations. Preliminaryanalysis showed that the differences in strain energy betweendifferent search protocols were not sensitive to the maximumnumber of requested conformations; therefore, the strainstatistics were computed only for the runs involving 500conformations per ligand.
Finally, the computational time required to execute thesearches was recorded for a random selection of 10 ligandsfrom the combined data set (1GWX, 1I7Z, 1PXI, 1QPE,1URW, 1UYD, 2BAL, 7EST, 1LAH, and 3ERK; see Table1). CPU cost was evaluated in a separate postprocessing step,by repeating the calculations for the aforementioned ligandsusing the same settings that were used for the productionruns. All timings were performed on a Windows 2000 PCequipped with a single core 32-bit 2.0 GHz Intel PentiumM processor and 2 GB of RAM. The SPE and SOS programswere executed from the DOS shell (DOS command prompt)and were timed using the timethis.exe utility availablethrough the Windows 2000 resource kit. The MOE searcheswere spawned from within the MOE GUI using custom SVLscripts, and the CPU times were extracted from the SVLcommand window.
RESULTS AND DISCUSSION
Retrieval of Bioactive Conformations/DifferencesAcross Data Sets. As discussed in the Methods section andillustrated in Figure 1, the Diller collection contains com-pounds with generally higher molecular weight and morerotatable bonds than the Chen data set. One obvious questionis whether these differences have any significant impact onthe ability of the various conformational search methods tosample bioactive space. The ability to retrieve the bioactiveconformation at different levels of sampling is graphicallyillustrated in Figure 2 for the Diller and Chen data sets andthe SOS and MOE conformational import methods, respec-tively. Each of these plots shows the percentage of ligands(y-axis) for which the generated ensemble contained at leastone conformation close to the bioactive structure as deter-mined by different rmsd cutoff values (x-axis). Clearly, thegreater is the rmsd cutoff, the greater is the probability offinding at least one conformation close to the crystalstructure. For the Diller data set, both SPE and MOEconformational import improved as the number of generatedconformations increased regardless of the rmsd cutoffemployed (Figure 2a and b). More importantly, both methodswere able to outperform MOE stochastic search with theChen settings, which served as the reference methodthroughout this study. Strikingly, even with only 100 trials,SPE was able to identify the bioactive conformation for moreligands than MOE stochastic search with 10 000 trials. Thisis despite the fact that the latter method employed anelaborate solvation model and consumed vastly more com-putational resources than the former (see computational timesin Table 2 and detailed discussion in the following sections).MOE conformational import with 500 trials also showedimproved retrieval of bioactive conformations over thestochastic search method. This is in contrast to the findingsof Chen and Foloppe, who found that increasing themaximum number of conformations requested from confor-mational import made little appreciable difference in per-
2792 J. Chem. Inf. Model., Vol. 49, No. 12, 2009 TRESADERN AND AGRAFIOTIS
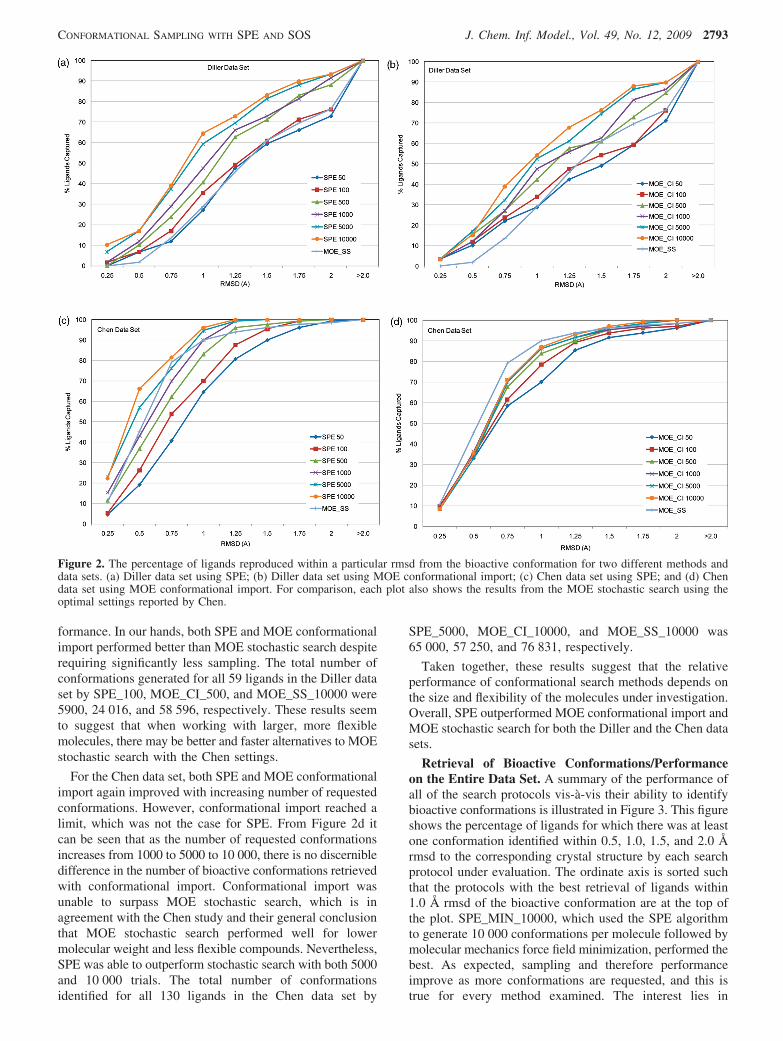
formance. In our hands, both SPE and MOE conformationalimport performed better than MOE stochastic search despiterequiring significantly less sampling. The total number ofconformations generated for all 59 ligands in the Diller dataset by SPE_100, MOE_CI_500, and MOE_SS_10000 were5900, 24 016, and 58 596, respectively. These results seemto suggest that when working with larger, more flexiblemolecules, there may be better and faster alternatives to MOEstochastic search with the Chen settings.
For the Chen data set, both SPE and MOE conformationalimport again improved with increasing number of requestedconformations. However, conformational import reached alimit, which was not the case for SPE. From Figure 2d itcan be seen that as the number of requested conformationsincreases from 1000 to 5000 to 10 000, there is no discernibledifference in the number of bioactive conformations retrievedwith conformational import. Conformational import wasunable to surpass MOE stochastic search, which is inagreement with the Chen study and their general conclusionthat MOE stochastic search performed well for lowermolecular weight and less flexible compounds. Nevertheless,SPE was able to outperform stochastic search with both 5000and 10 000 trials. The total number of conformationsidentified for all 130 ligands in the Chen data set by
SPE_5000, MOE_CI_10000, and MOE_SS_10000 was65 000, 57 250, and 76 831, respectively.
Taken together, these results suggest that the relativeperformance of conformational search methods depends onthe size and flexibility of the molecules under investigation.Overall, SPE outperformed MOE conformational import andMOE stochastic search for both the Diller and the Chen datasets.
Retrieval of Bioactive Conformations/Performanceon the Entire Data Set. A summary of the performance ofall of the search protocols vis-a-vis their ability to identifybioactive conformations is illustrated in Figure 3. This figureshows the percentage of ligands for which there was at leastone conformation identified within 0.5, 1.0, 1.5, and 2.0 Årmsd to the corresponding crystal structure by each searchprotocol under evaluation. The ordinate axis is sorted suchthat the protocols with the best retrieval of ligands within1.0 Å rmsd of the bioactive conformation are at the top ofthe plot. SPE_MIN_10000, which used the SPE algorithmto generate 10 000 conformations per molecule followed bymolecular mechanics force field minimization, performed thebest. As expected, sampling and therefore performanceimprove as more conformations are requested, and this istrue for every method examined. The interest lies in
Figure 2. The percentage of ligands reproduced within a particular rmsd from the bioactive conformation for two different methods anddata sets. (a) Diller data set using SPE; (b) Diller data set using MOE conformational import; (c) Chen data set using SPE; and (d) Chendata set using MOE conformational import. For comparison, each plot also shows the results from the MOE stochastic search using theoptimal settings reported by Chen.
CONFORMATIONAL SAMPLING WITH SPE AND SOS J. Chem. Inf. Model., Vol. 49, No. 12, 2009 2793
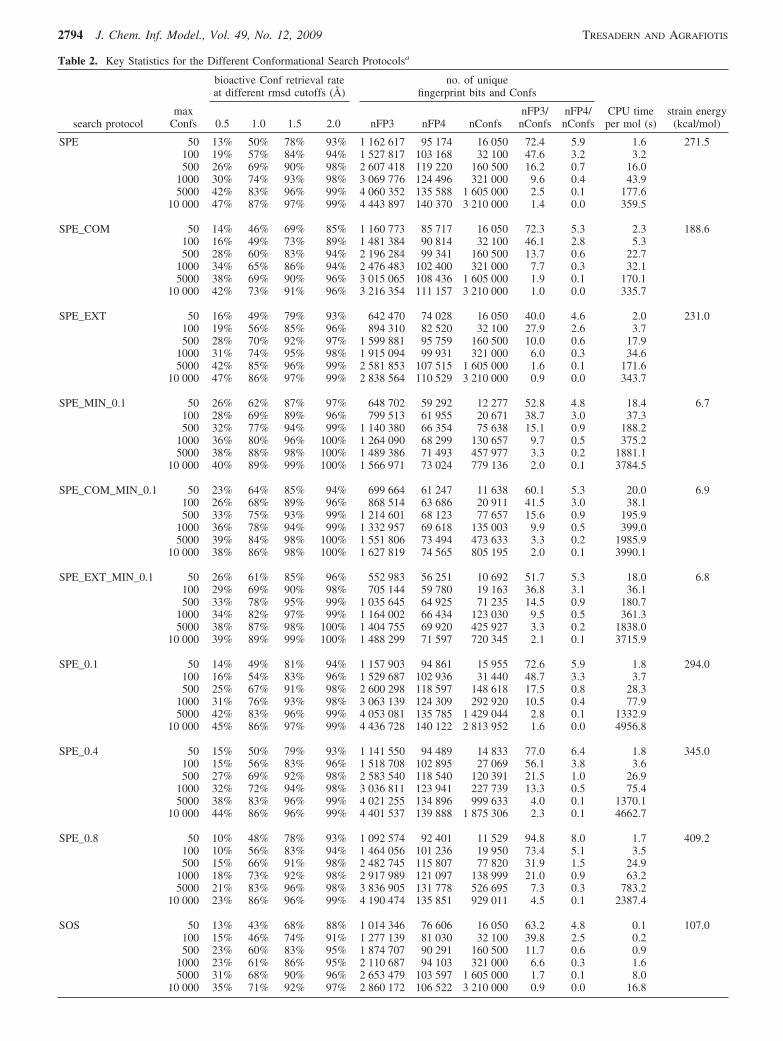
Table 2. Key Statistics for the Different Conformational Search Protocolsa
search protocolmax
Confs
bioactive Conf retrieval rateat different rmsd cutoffs (Å)
no. of uniquefingerprint bits and Confs
0.5 1.0 1.5 2.0 nFP3 nFP4 nConfsnFP3/nConfs
nFP4/nConfs
CPU timeper mol (s)
strain energy(kcal/mol)
SPE 50 13% 50% 78% 93% 1 162 617 95 174 16 050 72.4 5.9 1.6 271.5100 19% 57% 84% 94% 1 527 817 103 168 32 100 47.6 3.2 3.2500 26% 69% 90% 98% 2 607 418 119 220 160 500 16.2 0.7 16.0
1000 30% 74% 93% 98% 3 069 776 124 496 321 000 9.6 0.4 43.95000 42% 83% 96% 99% 4 060 352 135 588 1 605 000 2.5 0.1 177.6
10 000 47% 87% 97% 99% 4 443 897 140 370 3 210 000 1.4 0.0 359.5
SPE_COM 50 14% 46% 69% 85% 1 160 773 85 717 16 050 72.3 5.3 2.3 188.6100 16% 49% 73% 89% 1 481 384 90 814 32 100 46.1 2.8 5.3500 28% 60% 83% 94% 2 196 284 99 341 160 500 13.7 0.6 22.7
1000 34% 65% 86% 94% 2 476 483 102 400 321 000 7.7 0.3 32.15000 38% 69% 90% 96% 3 015 065 108 436 1 605 000 1.9 0.1 170.1
10 000 42% 73% 91% 96% 3 216 354 111 157 3 210 000 1.0 0.0 335.7
SPE_EXT 50 16% 49% 79% 93% 642 470 74 028 16 050 40.0 4.6 2.0 231.0100 19% 56% 85% 96% 894 310 82 520 32 100 27.9 2.6 3.7500 28% 70% 92% 97% 1 599 881 95 759 160 500 10.0 0.6 17.9
1000 31% 74% 95% 98% 1 915 094 99 931 321 000 6.0 0.3 34.65000 42% 85% 96% 99% 2 581 853 107 515 1 605 000 1.6 0.1 171.6
10 000 47% 86% 97% 99% 2 838 564 110 529 3 210 000 0.9 0.0 343.7
SPE_MIN_0.1 50 26% 62% 87% 97% 648 702 59 292 12 277 52.8 4.8 18.4 6.7100 28% 69% 89% 96% 799 513 61 955 20 671 38.7 3.0 37.3500 32% 77% 94% 99% 1 140 380 66 354 75 638 15.1 0.9 188.2
1000 36% 80% 96% 100% 1 264 090 68 299 130 657 9.7 0.5 375.25000 38% 88% 98% 100% 1 489 386 71 493 457 977 3.3 0.2 1881.1
10 000 40% 89% 99% 100% 1 566 971 73 024 779 136 2.0 0.1 3784.5
SPE_COM_MIN_0.1 50 23% 64% 85% 94% 699 664 61 247 11 638 60.1 5.3 20.0 6.9100 26% 68% 89% 96% 868 514 63 686 20 911 41.5 3.0 38.1500 33% 75% 93% 99% 1 214 601 68 123 77 657 15.6 0.9 195.9
1000 36% 78% 94% 99% 1 332 957 69 618 135 003 9.9 0.5 399.05000 39% 84% 98% 100% 1 551 806 73 494 473 633 3.3 0.2 1985.9
10 000 38% 86% 98% 100% 1 627 819 74 565 805 195 2.0 0.1 3990.1
SPE_EXT_MIN_0.1 50 26% 61% 85% 96% 552 983 56 251 10 692 51.7 5.3 18.0 6.8100 29% 69% 90% 98% 705 144 59 780 19 163 36.8 3.1 36.1500 33% 78% 95% 99% 1 035 645 64 925 71 235 14.5 0.9 180.7
1000 34% 82% 97% 99% 1 164 002 66 434 123 030 9.5 0.5 361.35000 38% 87% 98% 100% 1 404 755 69 920 425 927 3.3 0.2 1838.0
10 000 39% 89% 99% 100% 1 488 299 71 597 720 345 2.1 0.1 3715.9
SPE_0.1 50 14% 49% 81% 94% 1 157 903 94 861 15 955 72.6 5.9 1.8 294.0100 16% 54% 83% 96% 1 529 687 102 936 31 440 48.7 3.3 3.7500 25% 67% 91% 98% 2 600 298 118 597 148 618 17.5 0.8 28.3
1000 31% 76% 93% 98% 3 063 139 124 309 292 920 10.5 0.4 77.95000 42% 83% 96% 99% 4 053 081 135 785 1 429 044 2.8 0.1 1332.9
10 000 45% 86% 97% 99% 4 436 728 140 122 2 813 952 1.6 0.0 4956.8
SPE_0.4 50 15% 50% 79% 93% 1 141 550 94 489 14 833 77.0 6.4 1.8 345.0100 15% 56% 83% 96% 1 518 708 102 895 27 069 56.1 3.8 3.6500 27% 69% 92% 98% 2 583 540 118 540 120 391 21.5 1.0 26.9
1000 32% 72% 94% 98% 3 036 811 123 941 227 739 13.3 0.5 75.45000 38% 83% 96% 99% 4 021 255 134 896 999 633 4.0 0.1 1370.1
10 000 44% 86% 96% 99% 4 401 537 139 888 1 875 306 2.3 0.1 4662.7
SPE_0.8 50 10% 48% 78% 93% 1 092 574 92 401 11 529 94.8 8.0 1.7 409.2100 10% 56% 83% 94% 1 464 056 101 236 19 950 73.4 5.1 3.5500 15% 66% 91% 98% 2 482 745 115 807 77 820 31.9 1.5 24.9
1000 18% 73% 92% 98% 2 917 989 121 097 138 999 21.0 0.9 63.25000 21% 83% 96% 98% 3 836 905 131 778 526 695 7.3 0.3 783.2
10 000 23% 86% 96% 99% 4 190 474 135 851 929 011 4.5 0.1 2387.4
SOS 50 13% 43% 68% 88% 1 014 346 76 606 16 050 63.2 4.8 0.1 107.0100 15% 46% 74% 91% 1 277 139 81 030 32 100 39.8 2.5 0.2500 23% 60% 83% 95% 1 874 707 90 291 160 500 11.7 0.6 0.9
1000 23% 61% 86% 95% 2 110 687 94 103 321 000 6.6 0.3 1.65000 31% 68% 90% 96% 2 653 479 103 597 1 605 000 1.7 0.1 8.0
10 000 35% 71% 92% 97% 2 860 172 106 522 3 210 000 0.9 0.0 16.8
2794 J. Chem. Inf. Model., Vol. 49, No. 12, 2009 TRESADERN AND AGRAFIOTIS

identifying the methods that, for the same number or fewertrials, perform better. Some of the methods are highlightedin Figure 3 with different colors. The MOE stochastic searchusing the Chen and Foloppe settings, which is highlightedin yellow, showed average performance at all rmsd cutoffs.
Looking again at Figure 3, it appears that for protocolswith 5000 and 10 000 conformations, SPE slightly outper-forms SOS with or without boosting. (In the followingdiscussion, we use the term “boosting” to refer to biasingtoward more extended geometries, “inverse boosting” to referto biasing toward more compact conformations, and “bidi-rectional boosting” to refer to biasing in both directions, asdescribed in the Methods section.) Moreover, both SPE andSOS with boosting (SPE_EXT and SOS_EXT) rank higherthan SOS without boosting (SOS). However, as shown inTable 2, these differences are rather marginal; for 5000 trials,the difference in the bioactive conformation retrieval ratewith SPE_EXT and SOS_EXT is only 4% (85 and 81%,respectively). In fact, SOS_EXT performs within 0-2% ofthe SPE method for protocols involving 50, 100, 500, 1000,and 5000 trials. These data suggest that the use of precom-puted template geometries in the SOS method has anegligible impact on the ability to retrieve bioactiveconformations.
The positive impact of boosting on sampling bioactivespace is evident at all levels. Comparing the SOS andSOS_EXT protocols, where all parameters were identicalexcept for the use of boosting, the latter method showed 9%improvement over the former for the protocols involving 500trials (60% for SOS_500, and 69% for SOS_EXT_500). Thesame is true for SPE, where we observed a 10% improvement
from SPE_COM to SPE_EXT at 1.0 Å rmsd for protocolsinvolving 500 trials (60% for SPE_COM_500, and 70% forSPE_EXT_500). The results with bidirectional boosting,which represents a 3:5 mix of compact and extendedconformations, were very similar to those obtained fromregular boosting regardless of the number of trials requested.These results are fully consistent with our previous observa-tion that SPE tends to generate relatively compact conforma-tions and that inverse boosting offers little practical benefit.It also reflects the fact that bioactive conformations tend tobe more extended than random ones, and therefore biasingthe search toward extended geometries increases the likeli-hood of identifying the biologically active ones.
One parameter that seems to have a significant impact isenergy minimization. Comparing SPE_COM_MIN, SPE_EXT_MIN, and SPE_MIN with the corresponding nonmini-mized ensembles shows a notable change in the rate ofretrieval of bioactive conformations. Even with just 100 trialsper molecule, these three methods outperformed all otherapproaches in this study and were within 1-2% of the MOEstochastic search using the best settings identified by Chenand Foloppe. This is particularly encouraging consideringthat these methods generated an order of magnitude fewerconformations than MOE stochastic search (∼20 000 ascompared to ∼240 000; see Table 2). Interestingly, mini-mization had the largest effect on the protocols with the fewertrials. For example, SPE with 50 trials retrieved 50% of thebioactive conformations within 1.0 Å rmsd, whereas withminimization this percentage increased to 62%. Generally,at lower rmsd cutoffs, minimization seems to work betterwhen the number of trials is small, but gets progressively
Table 2 Continued
search protocolmax
Confs
bioactive Conf retrieval rateat different rmsd cutoffs (Å)
no. of uniquefingerprint bits and Confs
0.5 1.0 1.5 2.0 nFP3 nFP4 nConfsnFP3/nConfs
nFP4/nConfs
CPU timeper mol (s)
strain energy(kcal/mol)
SOS_EXT 50 16% 50% 82% 94% 628 884 68 856 16 050 39.2 4.3 0.2 107.7100 19% 56% 87% 96% 856 681 75 793 32 100 26.7 2.4 0.3500 26% 69% 90% 97% 1 445 027 88 056 160 500 9.0 0.5 1.3
1000 31% 72% 91% 97% 1 720 402 92 721 321 000 5.4 0.3 2.05000 39% 81% 93% 98% 2 301 005 102 394 1 605 000 1.4 0.1 8.4
10 000 41% 82% 96% 99% 2 546 276 107 043 3 210 000 0.8 0.0 26.6
MOE_CI 50 24% 57% 78% 91% 259 866 39 266 11 856 21.9 3.3 11.2 39.7100 25% 62% 83% 93% 327 388 43 102 21 138 15.5 2.0 11.9500 28% 69% 88% 96% 480 491 49 292 68 831 7.0 0.7 13.3
1000 27% 72% 89% 97% 608 775 53 070 119 935 5.1 0.4 13.25000 28% 77% 93% 98% 823 604 57 822 381 249 2.2 0.2 28.9
10 000 29% 79% 94% 98% 884 862 58 817 644 565 1.4 0.1 28.9
MOE_BR 50 15% 54% 83% 95% 669 451 68 655 14 511 46.1 4.7 0.6 150.0100 20% 59% 83% 96% 887 337 73 759 28 017 31.7 2.6 0.7500 24% 67% 91% 98% 1 352 914 81 477 122 812 11.0 0.7 0.7
1000 27% 68% 94% 99% 1 542 185 83 648 225 446 6.8 0.4 0.85000 29% 74% 93% 99% 1 934 722 87 563 902 290 2.1 0.1 2.3
10 000 30% 75% 95% 99% 2 012 879 87 607 1 588 277 1.3 0.1 4.0
MOE_SS 10 000 30% 70% 88% 93% 1 097 628 70 979 240 770 4.6 0.3 5647.1 0.0
a nFP3 and nFP4 refer to the number of unique 3-point (piDAPH3) and 4-point (piDAPH4) pharmacophore bits as calculated in MOE.nConfs refers to the number of conformations identified by each protocol. FP3/nConfs and FP4/nConfs represent the average number of unique3- and 4-point pharmacophores per conformation. CPU time per molecule represents the amount of time required to complete the search for asingle molecule, averaged over 10 randomly selected molecules from the data set. Strain energy represents the median average local strainenergy in kcal/mol between each conformation and its nearest local minimum, calculated on the basis of the protocols involving 500 trials.
CONFORMATIONAL SAMPLING WITH SPE AND SOS J. Chem. Inf. Model., Vol. 49, No. 12, 2009 2795

worse as more and more conformations are generated. Athigher rmsd cutoffs, minimization almost always has apositive impact, although the effect becomes less pronouncedas the number of trials increases.
To further examine the effects of minimization, we alsosubjected the MOE conformational import (MOE_CI) andbond rotation (MOE_BR) methods to similar post processing.Without minimization, these two methods with 50 conforma-tions per molecule identified at least one conformation within1.0 Å rmsd from the bioactive structure for 57% and 54%of the ligands, respectively. Upon minimization, the retrievalrate was slightly reduced for conformational import (55%)and slightly increased for bond rotation (57%). Improvement,
if any, was much less pronounced than seen with SPE. Thissuggests that it is not sufficient to minimize the rawconformations from any method and expect such improve-ments. We attribute the improvement in performance to thesynergistic effects of minimizing more diverse conformationsas generated by SPE, as discussed in the following section.
The rmsd cutoff used to eliminate duplicate conformationshad minimal impact on the ability to retrieve the bioactivestructure. Duplicate filtering at 0.1 Å rmsd had almost noeffect, and at 0.4 and 0.8 Å the difference was no more than2-4% for virtually any level of sampling (number of trials).The only exception is that there were significantly fewerligands identified within 0.5 Å rmsd to the bioactive structure
Figure 3. The percentage of ligands reproduced within 0.5 (9), 1.0 ([), 1.5 (b), and 2.0 (-) Å rmsd from the bioactive conformation fordifferent methods and settings. The search protocols are sorted top to bottom in decreasing fraction of ligands reproduced within 1.0 Å,with the best performing methods placed at furthest left. Color-coding is used to identify selected methods: MOE_BR (purple), SPE (orange),SPE_MIN (red), SPE_EXT (green), SOS_EXT (blue), and MOE_SS (yellow).
2796 J. Chem. Inf. Model., Vol. 49, No. 12, 2009 TRESADERN AND AGRAFIOTIS

when filtering duplicates at 0.8 Å rmsd. This result, however,is expected because duplicate elimination is susceptible tothe order in which the conformations are generated. Indeed,a conformation within 0.5 Å from the crystal structure wouldhave been eliminated if it were within 0.8 Å to a previouslygenerated conformation that was itself more than 0.5 Å fromthe crystal structure. Therefore, we recommend the use of asensible rmsd cutoff that is large enough to eliminateredundant structures and minimize storage requirements, yetnot too large so as to preclude the sampling of importantregions of conformational space due to artifacts associatedwith the sampling sequence.
Considering the MOE conformational import and bondrotation methods, in general they performed well for fewerconformations but relatively worse as the number of trialsincreased. Indeed, MOE conformational import requesting50 conformations per molecule outperformed the standard,nonminimized SPE and SOS protocols. Performance con-tinued to improve with increasing number of conformations,but not to the same extent as with the other methods. Thedifference in percent retrieval between 50 and 10 000conformations was 22% for conformational import and 21%for bond rotation, but 37% for SPE. With 500 conformationsper molecule, SPE and conformational import showedcomparable performance, both identifying 69% of ligandswithin 1.0 Å rmsd of the crystal structure. On average, bondrotation was 2-4% worse than conformational import at alllevels of sampling within the same rmsd cutoff.
Conformation Diversity Captured with 3D Finger-prints. The extent of conformational coverage was thesecond important aspect considered. A good conformationalsearch method must cast a wide net over the potential energylandscape and sample as broad a range of moleculargeometries as possible. In some respects, the ability toretrieve the bioactive conformation is itself a measure of theextent of sampling. We have previously studied the perfor-mance of SPE using the radius of gyration with 10 000conformations per molecule.29 The boosting heuristic waspartly designed to promote the generation of more extendedgeometries, and we have shown that, when applied to SPEand SOS, they both performed very well in this regard. Inthis work, the extent of sampling was assessed using thenumber of unique 3-point and 4-point pharmacophorescombined with the overall number of conformations generated.
The numbers of unique 3- and 4-point pharmacophoresidentified with each protocol are listed in Table 2. The SPEmethod yields by far the greatest pharmacophore diversityof all approaches evaluated. With 10 000 trials per molecule,SPE generated ∼4.4 million 3-point and over 140 000 4-pointpharmacophores. In comparison, the MOE stochastic searchidentified ∼1.1 million and ∼71 000, respectively. However,upon force field minimization, the number of uniqueconformations and pharmacophores identified by SPE wasreduced by a factor of 2-4, due to the fact that many of theraw conformations were minimized to similar geometriescontaining equivalent pharmacophores. This suggests thatSPE produces some conformations and pharmacophores,which are physically unrealistic, and that additional refine-ment or minimization is necessary if this method is to beuseful in a practical setting. Eliminating duplicate conforma-tions prior to minimization reduced the number of conforma-tions significantly (particularly as the number of trials
increased), but the impact on the number of unique phar-macophores was minimal even at relatively high rmsd cutoffs(SPE vs SPE_0.1, SPE_0.4, and SPE_0.8 in Table 2).Interestingly, the average number of unique pharmacophoresper conformation (nFP3/nConfs and nFP4/nConfs in Table2) decreases upon minimization for searches involving veryfew trials, but increases as more and more conformationsare generated. The effect is more pronounced for extendedgeometries, and less pronounced for compact ones. Theseresults compare very favorably with MOE stochastic search;indeed, a mere 500 minimized conformations generated bySPE (SPE_MIN_0.1) captured just as much pharmacophorediversity as MOE_SS, whereas given the same number oftrials (10 000), SPE produced nearly 40% more 3-pointpharmacophores.
The SOS method did not yield as much pharmacophorediversity as raw SPE, but produced twice as many pharma-cophores as compared to SPE coupled with minimization.Furthermore, SOS was nearly 4 times as effective asMOE_CI, and almost 1.5-2 times as effective as MOE_BRat comparable levels of sampling. More impressively, even50 SOS conformations were enough to capture almost thesame amount of pharmacophore diversity as 10 000 confor-mations obtained with MOE stochastic search using the Chensettings.
Comparison to the Gold Standard Ensemble. A goodsearch method must produce conformations that are not onlydiverse but also energetically plausible. It was shown abovethat minimization reduced the number of pharmacophoresgenerated by SPE, suggesting the presence of physicallyunrealistic conformations in the output of the samplingprocedure. Although irrelevant high energy conformationscan be removed via energy minimization, this step impartsadditional computational cost. To assess the quality of thesampling protocols, we compared them to a “gold standard”ensemble constructed by combining the output from allprotocols, minimizing the conformations, and removingduplicates within an rmsd threshold of 0.1 Å. The resultingensemble comprised over six million conformations. The 3-and 4-point pharmacophores were calculated in the mannerdescribed above, and the output of each protocol wascompared to the gold standard via the Tanimoto similaritycoefficient. This metric effectively penalizes for missingconformations from incomplete sampling and high energyconformations from unproductive sampling. Similar trendswere observed for 3- and 4-point pharmacophores; therefore,details are given only for the 3-point pharmacophorefingerprints.
Figure 4 shows the Tanimoto similarity for each protocolwith increasing number of requested conformations. Asexpected, the overall similarity is generally high as eachindividual method captures a reasonable fraction of the totalpharmacophore space. The worst protocol was MOE con-formational import (MOE_CI) with 50 requested conforma-tions per molecule, with a similarity of 0.86. In Figure 4,the similarity curves are separated into three different plotsaccording to the pattern of change in the similarity score asa function of the number of requested conformations. In thefirst plot, SPE_COM, SPE_EXT, SOS_EXT, and MOE_BRshow relatively little change, with the Tanimoto score slightlyincreasing for the first few hundred conformations, andslightly decreasing afterward. This is gratifying because it
CONFORMATIONAL SAMPLING WITH SPE AND SOS J. Chem. Inf. Model., Vol. 49, No. 12, 2009 2797
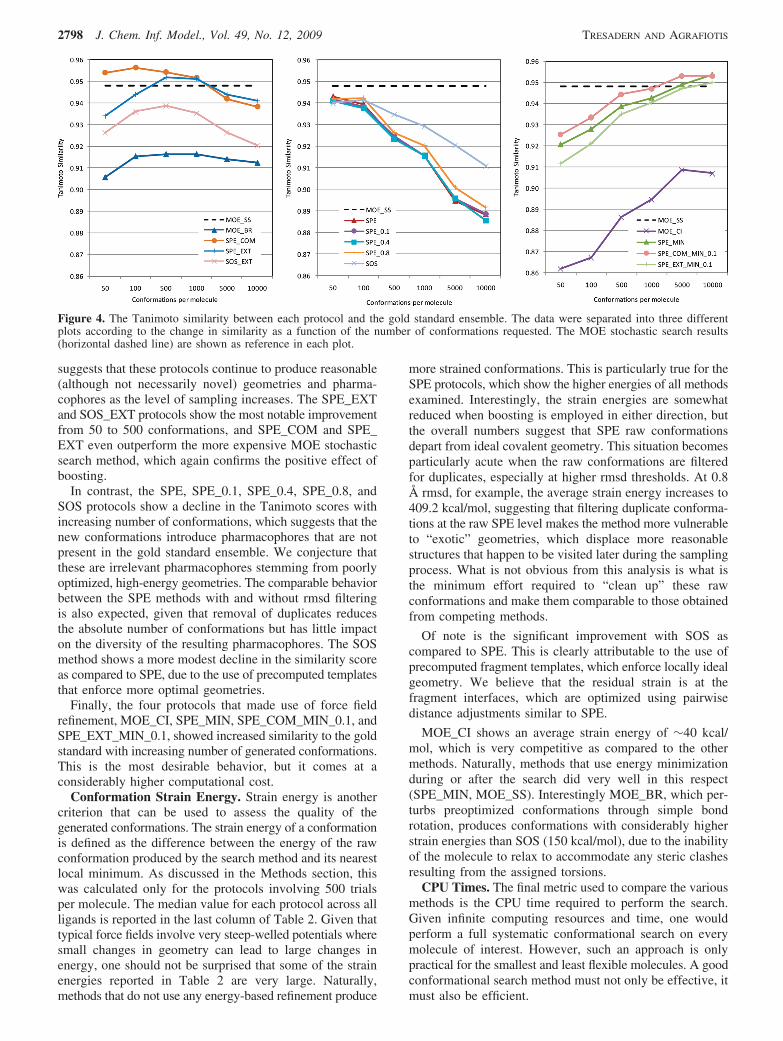
suggests that these protocols continue to produce reasonable(although not necessarily novel) geometries and pharma-cophores as the level of sampling increases. The SPE_EXTand SOS_EXT protocols show the most notable improvementfrom 50 to 500 conformations, and SPE_COM and SPE_EXT even outperform the more expensive MOE stochasticsearch method, which again confirms the positive effect ofboosting.
In contrast, the SPE, SPE_0.1, SPE_0.4, SPE_0.8, andSOS protocols show a decline in the Tanimoto scores withincreasing number of conformations, which suggests that thenew conformations introduce pharmacophores that are notpresent in the gold standard ensemble. We conjecture thatthese are irrelevant pharmacophores stemming from poorlyoptimized, high-energy geometries. The comparable behaviorbetween the SPE methods with and without rmsd filteringis also expected, given that removal of duplicates reducesthe absolute number of conformations but has little impacton the diversity of the resulting pharmacophores. The SOSmethod shows a more modest decline in the similarity scoreas compared to SPE, due to the use of precomputed templatesthat enforce more optimal geometries.
Finally, the four protocols that made use of force fieldrefinement, MOE_CI, SPE_MIN, SPE_COM_MIN_0.1, andSPE_EXT_MIN_0.1, showed increased similarity to the goldstandard with increasing number of generated conformations.This is the most desirable behavior, but it comes at aconsiderably higher computational cost.
Conformation Strain Energy. Strain energy is anothercriterion that can be used to assess the quality of thegenerated conformations. The strain energy of a conformationis defined as the difference between the energy of the rawconformation produced by the search method and its nearestlocal minimum. As discussed in the Methods section, thiswas calculated only for the protocols involving 500 trialsper molecule. The median value for each protocol across allligands is reported in the last column of Table 2. Given thattypical force fields involve very steep-welled potentials wheresmall changes in geometry can lead to large changes inenergy, one should not be surprised that some of the strainenergies reported in Table 2 are very large. Naturally,methods that do not use any energy-based refinement produce
more strained conformations. This is particularly true for theSPE protocols, which show the higher energies of all methodsexamined. Interestingly, the strain energies are somewhatreduced when boosting is employed in either direction, butthe overall numbers suggest that SPE raw conformationsdepart from ideal covalent geometry. This situation becomesparticularly acute when the raw conformations are filteredfor duplicates, especially at higher rmsd thresholds. At 0.8Å rmsd, for example, the average strain energy increases to409.2 kcal/mol, suggesting that filtering duplicate conforma-tions at the raw SPE level makes the method more vulnerableto “exotic” geometries, which displace more reasonablestructures that happen to be visited later during the samplingprocess. What is not obvious from this analysis is what isthe minimum effort required to “clean up” these rawconformations and make them comparable to those obtainedfrom competing methods.
Of note is the significant improvement with SOS ascompared to SPE. This is clearly attributable to the use ofprecomputed fragment templates, which enforce locally idealgeometry. We believe that the residual strain is at thefragment interfaces, which are optimized using pairwisedistance adjustments similar to SPE.
MOE_CI shows an average strain energy of ∼40 kcal/mol, which is very competitive as compared to the othermethods. Naturally, methods that use energy minimizationduring or after the search did very well in this respect(SPE_MIN, MOE_SS). Interestingly MOE_BR, which per-turbs preoptimized conformations through simple bondrotation, produces conformations with considerably higherstrain energies than SOS (150 kcal/mol), due to the inabilityof the molecule to relax to accommodate any steric clashesresulting from the assigned torsions.
CPU Times. The final metric used to compare the variousmethods is the CPU time required to perform the search.Given infinite computing resources and time, one wouldperform a full systematic conformational search on everymolecule of interest. However, such an approach is onlypractical for the smallest and least flexible molecules. A goodconformational search method must not only be effective, itmust also be efficient.
Figure 4. The Tanimoto similarity between each protocol and the gold standard ensemble. The data were separated into three differentplots according to the change in similarity as a function of the number of conformations requested. The MOE stochastic search results(horizontal dashed line) are shown as reference in each plot.
2798 J. Chem. Inf. Model., Vol. 49, No. 12, 2009 TRESADERN AND AGRAFIOTIS

The second-to-last column in Table 2 lists the mean CPUtime per molecule for each method and each level ofsampling, averaged over the 10 ligands as described in theMethods section. Dividing this by the maximum number ofrequested conformations yields an estimate of the time pergenerated (more accurately, retained) conformation. Thesetimings reveal several interesting trends. First, MOE sto-chastic search, which serves as our reference method, wasthe slowest of all methods, requiring an average of 0.565 sper conformation using the computer system described inthe Methods section. Clearly, this is a low-throughputtechnique that is best suited for detailed studies of relativelyfew compounds or small libraries.31
SPE was an order of magnitude faster, requiring 0.036 sper conformation. As expected, the computational time forthe SPE and SOS methods scales linearly with the numberof conformations, and this is true with or without boosting.Boosting introduces a negligible overhead to the overallprocess, which corresponds to the time required to measureall interatomic distances and update the bounds matrix. Therate-limiting step is the embedding itself, which is whatmakes boosting so appealing; it is simple, effective, andefficient. In contrast, minimization increases the CPU timeby a factor of 10, to an average of 0.36-0.40 s perconformation, which is comparable to MOE stochasticsearch. This time, however, also includes checking for andeliminating duplicate conformations, a process that isquadratic in nature and becomes the limiting factor as thenumber of conformations grows beyond a certain point. Thelower is the rmsd threshold, the more conformations areretained, and the longer it takes to detect duplicates.
SOS is remarkably fast, requiring an average of only 0.002s per conformation. MOE conformational import and MOEbond rotation are the methods that show the most unusualscaling, with the average cost per conformation becomingsmaller and smaller as the total number of generatedconformations increases. This reflects a relatively largeinitialization time for both methods, which becomes less ofa factor as sampling increases. At 10 000 conformations,MOE_CI is comparable to SOS with an average cost of 0.003s per conformation, but at 500 conformations (which is acommon practical limit for large multiconformation databaseconstruction), it is more than 10 times slower (0.027 s forMOE_CI versus 0.003 s for SOS). MOE_BR is the fastestof all methods examined, particularly at large samplinglevels, requiring less than 0.001 s per conformation at 10 000trials. However, as discussed above, the efficiency of bothMOE_CI and MOE_BR comes at the expense of conforma-tional diversity. For MOE_BR, it also comes at the expenseof the quality of conformations, which have relatively largestrain energies (although they are still significantly better thanthose generated by SPE without minimization).
CONCLUSIONS
The intent of the present study was to examine the utilityof the relatively newer SPE and SOS conformational searchalgorithms in everyday modeling tasks. Our aim was not toperform an exhaustive evaluation of all possible combinationsof options and parameter settings, but to assess whether thesettings recommended in the original publications would leadto sensible outcomes in a practical setting. The practicalapplication of any modeling tool always involves compromise.
Our analysis is reminiscent of the classic engineeringtriangle, which describes the interplay between the three coreelements of an engineering project, quality, time, and cost.These three attributes describe how well, how fast, and howcheaply a solution is obtained. In our context, the quality ofa conformational search method encompasses a number offactors such as the ability to sample energetically sensibleand biologically relevant space while maintaining sufficientdiversity to accommodate hitherto unobserved (but highlyprobable) binding modes. Time is equally important. Atimely result is far more valuable than one that is obtainedafter the fact and has no ability to influence experiments.On the surface, the third element, cost, may appear the leastrelevant, but it too has a subtle but profound impact.Licensing costs and prior familiarity are often the determiningfactors in what method is chosen in a particular environment.More importantly, licensing costs are often related to thedevelopment costs required to build and maintain thesolution, and ultimately reflect the complexity of the solutionitself. Indeed, simplicity is often an under-appreciated factor.
The results presented above give us hope that the SPEand SOS methods hold great promise for a variety ofmodeling applications. Comparing the bioactive conformationretrieval rates between SPE and MOE conformational importon the Diller and Chen data sets showed that SPE performedrelatively better for more flexible molecules. SPE outper-formed an elaborate variant of MOE stochastic search onthe Diller data set while generating significantly fewerconformations in a much shorter amount of time. In addition,SPE improved with increasing number of trials, whereasMOE conformational import reached a limit beyond whichadditional conformations did not help. Other work from ourgroup has shown that SPE and SOS perform excellently onmacrocyclic compounds, where most systematic searchmethods collapse.38 Taken together, these results suggest thatSPE should be considered among the best approaches forchallenging, flexible molecules.
While SOS and SPE performed similarly in terms ofretrieving bioactive conformations, SOS was an order ofmagnitude faster and generated much better geometries withlower strain energies. Confirming our previous studies, wealso found that the boosting heuristic had a positive impactin directing the search toward bioactive relevant space.
The question of most interest to us when we embarkedon this study was whether the SPE and SOS methods hadadequate performance at lower sampling levels, which woulddetermine their suitability for building multiconformationdatabases for pharmacophore and shape-based searching. Ourresults confirmed that SOS coupled with boosting and 500conformations per molecule had comparable performance toMOE stochastic search with 10 000 conformations, whilehaving vastly lower CPU demands.
Applying an rmsd filter to reduce the number of outputconformations did not adversely affect the bioactive retrievalrate, and the 3D pharmacophore diversity of the resultingensemble improved. However, the resulting conformationswere more dissimilar to the gold standard and had signifi-cantly higher strain energies, so we would not recommendrmsd filtering prior to minimization.
The high strain energies of the SPE conformations are ofsome concern. When minimized, the conformational en-sembles produced by SPE improved greatly, but this came
CONFORMATIONAL SAMPLING WITH SPE AND SOS J. Chem. Inf. Model., Vol. 49, No. 12, 2009 2799

at a significant cost in terms of speed. However, the keyresult of this study is that the sampling capacity of SPE issuch that fewer minimized conformations obtained by SPEcapture more biologically relevant conformational and phar-macophore diversity than much larger ensembles obtainedby other methods. Thus, generating fewer conformations withSPE and paying the additional price of minimization maystill be preferred over spending an equivalent amount of timesampling more extensively with alternative methods. Theseother methods, like MOE conformational import and MOEbond rotation, did not improve to the same extent withminimization, which suggests that they have significantlylower sampling potential.
The above also suggests a reasonable compromise. In thiswork, we used a relatively long minimization procedureinvolving up to 1000 steps. Yet, experience with gradientminimizers suggests that most of the strain is relieved in theearlier cycles and that the bulk of the effort is spent fine-tuning the geometry to perfection. We believe that mostpractical applications do not require “perfect” conformations,and therefore the number of minimization steps (andcomputational time) can be significantly reduced withoutaffecting the modeling outcome. This hypothesis is currentlyunder investigation.
REFERENCES AND NOTES
(1) Stockwell, G. R.; Thornton, J. M. Conformational diversity of ligandsbound to proteins. J. Mol. Biol. 2006, 356, 928–944.
(2) Leach, A. R. Survey of methods for searching the conformational spaceof small and medium-sized molecules. In ReViews in ComputationalChemistry; Lipkowitz, K. B., Boyd, D. B., Eds.; VCH: New York,1991.
(3) Lipton, M.; Still, W. C. The multiple minimum problem in molecularmodeling. Tree searching internal coordinate conformational space.J. Comput. Chem. 1988, 9, 343.
(4) Bruccoleri, R. E.; Karplus, M. Chain closure with bond anglevariations. Macromolecules 1985, 18, 2767.
(5) Bruccoleri, R. E.; Karplus, M. Prediction of the folding of shortpolypeptide segments by uniform conformational sampling. Biopoly-mers 1987, 26, 137.
(6) Go, N.; Scheraga, H. A. Ring closure and local conformationaldeformations of chain molecules. Macromolecules 1970, 3, 178.
(7) Saunders, M. Stochastic explorations of molecular mechanics energysurfaces. Hunting for the global minimum. J. Am. Chem. Soc. 1987,109, 3150.
(8) Ferguson, D. M.; Raber, D. J. A new approach to probing conforma-tional space with molecular mechanics: random incremental pulsesearch. J. Am. Chem. Soc. 1989, 111, 4371.
(9) Chang, G.; Guida, W. C.; Still, W. C. An internal-coordinate MonteCarlo method for searching conformational space. J. Am. Chem. Soc.1989, 111, 4379–4386.
(10) Li, Z.; Scheraga, H. A. Monte Carlo-minimization approach to themultiple-minima problem in protein folding. Proc. Natl. Acad. Sci.U.S.A. 1987, 84, 6611–6615.
(11) Jorgensen, W. L.; Tirado-Rives, J. Monte Carlo vs molecular dynamicsfor conformational sampling. J. Phys. Chem. 1996, 100, 14508–14513.
(12) Bostrom, J. Reproducing the conformations of protein-bound ligands:A critical evaluation of several popular conformational searching tools.J. Comput.-Aided Mol. Des. 2001, 15, 1137–1152.
(13) Bostrom, J.; Greenwood, J. R.; Gottfries, J. Assessing the performanceof OMEGA with respect to retrieving bioactive conformations. J. Mol.Graphics Modell. 2003, 21, 449–462.
(14) Perola, E.; Charifson, P. S. Conformational analysis of drug-likemolecules bound to proteins: An extensive study of ligand reorganiza-tion upon binding. J. Med. Chem. 2004, 47, 2499–2510.
(15) Nicklaus, M. C.; Wang, S.; Driscoll, J. S.; Milne, G. W. A.Conformational changes of small molecules binding to proteins.Bioorg. Med. Chem. 1995, 3, 411–428.
(16) Vieth, M.; Hirst, J. D.; Brooks, C. L. Do active site conformations ofsmall ligands correspond to low free-energy solution structures.J. Comput.-Aided Mol. Des. 1998, 12, 563–572.
(17) Kirchmair, J.; Laggner, C.; Wolber, G.; Langer, T. Comparativeanalysis of protein-bound ligand conformations with respect toCatalyst’s conformational space subsampling algorithms. J. Chem. Inf.Model. 2005, 45, 422–430.
(18) Tirado-Rives, J.; Jorgensen, W. L. Contribution of conformationfocusing to the uncertainty in predicting free energies for protein-ligand binding. J. Med. Chem. 2006, 49, 5880–5884.
(19) Diller, D. J.; Merz, K. M. Can we separate active from inactiveconformations. J. Comput.-Aided Mol. Des. 2002, 16, 105–112.
(20) Crippen, G. M. Rapid calculation of coordinates from distance matrices.J. Comput. Phys. 1978, 26, 449–452.
(21) Spellmeyer, D. C.; Wong, A. K.; Bower, M. J.; Blaney, J. M.Conformational analysis using distance geometry methods. J. Mol.Graphics Modell. 1997, 15, 18.
(22) Agrafiotis, D. K. Stochastic proximity embedding. J. Comput. Chem.2003, 24, 1215–1221.
(23) Rassokhin, D. N.; Agrafiotis, D. K. A modified update rule forstochastic proximity embedding. J. Mol. Graphics Modell. 2003, 22,133–140.
(24) Xu, H.; Izrailev, S.; Agrafiotis, D. K. Conformational sampling byself-organization. J. Chem. Inf. Comput. Sci. 2003, 43, 1186–1191.
(25) Zhu, F.; Agrafiotis, D. K. Self-organizing superimposition algorithmfor conformational sampling. J. Comput. Chem. 2007, 28, 1234–1239.
(26) Izrailev, S.; Zhu, F.; Agrafiotis, D. K. A distance geometry heuristicfor expanding the range of geometries sampled during conformationalsearch. J. Comput. Chem. 2006, 27, 1962–1969.
(27) Agrafiotis, D. K.; Xu, H. A self-organizing principle for learningnonlinear manifolds. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 15869–15872.
(28) Agrafiotis, D. K.; Gibbs, A.; Zhu, F.; Izrailev, S.; Martin, E.Conformational boosting. Aust. J. Chem. 2006, 59, 874–878.
(29) Agrafiotis, D. K.; Gibbs, A. C.; Zhu, F.; Izrailev, S.; Martin, E.Conformational sampling of bioactive molecules: A comparative study.J. Chem. Inf. Model. 2007, 47, 1067–1086.
(30) Agrafiotis, D. K.; Bandyopadhyay, D.; Carta, G.; Knox, A. J.; Lloyd,D. G. On the effects of permuted input on conformational samplingof drug-like molecules: an evaluation of stochastic proximity embed-ding. Chem. Biol. Drug Des. 2007, 70, 123–133.
(31) Chen, I. J.; Foloppe, N. Conformational sampling of druglike moleculeswith MOE and Catalyst: Implications for pharmacophore modelingand virtual screening. J. Chem. Inf. Model. 2008, 48, 1773–1791.
(32) O’Shea, R.; Moser, H. E. Physicochemical properties of antibacterialcompounds: Implications for drug discovery. J. Med. Chem. 2008,51, 2871–2878.
(33) Payne, D. J.; Gwynn, M. N.; Holmes, D. J.; Pompliano, D. L. Drugsfor bad bugs: confronting the challenges of antibacterial discovery.Nat. ReV. Drug DiscoVery 2007, 6, 29–40.
(34) Pu, L.; Agrafiotis, D. K.; Theobald, D. L. Fast determination of theoptimal rotational matrix for weighted superpositions. J. Comput.Chem., in press.
(35) Onufriev, A.; Bashford, D.; Case, D. A. Modification of the generalizedBorn model suitable for macromolecules. J. Phys. Chem. B 2000, 104,3712–3720.
(36) Yusuf, D.; Davis, A. M.; Kleywegt, G. J.; Schmitt, S. An alternativemethod for the evaluation of docking performance: RSR vs RMSD.J. Chem. Inf. Model. 2008, 48, 1411–1422.
(37) Martin, E. J.; Hoeffel, T. J. Oriented substituent pharmacophoreproperty space (OSPPREYS): A substituent-based calculation thatdescribes combinatorial library products better than the correspondingproduct-based calculation. J. Mol. Graphics Modell. 2000, 18, 383–403.
(38) Bonnet, P.; Agrafiotis, D. K.; Zhu, F.; Martin, E. Conformationalanalysis of macrocycles: Finding what common search methods miss.J. Chem. Inf. Model., Article ASAP.
CI9001926
2800 J. Chem. Inf. Model., Vol. 49, No. 12, 2009 TRESADERN AND AGRAFIOTIS