confidential - amazon s3 · 2018-11-07 · confidential 115649 (mmr-161) protocol amendment 2...
TRANSCRIPT

CONFIDENTIAL
19-MAY-2015 1
Clinical Study ProtocolSponsor:
GlaxoSmithKline Biologicals
89, Rue de l’Institut, 1330 Rixensart, Belgium
Primary Study vaccine and number
GlaxoSmithKline (GSK) Biologicals’ live attenuated measles, mumps, rubella vaccine (MMR) Vaccine number 209762
Other Study vaccines Measles, mumps and rubella virus vaccine live(M-M-RII, Merck & Co., Inc., orM-M®RVaxPro®, Sanofi Pasteur/Merck Sharp and Dohme [SPMSD])
Varicella virus vaccine live (Varivax®, Merck & Co., Inc.)
Hepatitis A vaccine, inactivated (Havrix®, GSK Biologicals)
Pneumococcal 13-valent conjugate vaccine (diphtheria CRM197 protein) (Prevnar 13®, Pfizer Inc.)
eTrack study number and Abbreviated Title
115649 (MMR-161)
Investigational New Drug (IND) number
BB IND 7229
EudraCT number 2011-004905-26
Date of protocol Final: 22 June 2012
Date of protocol amendment/administrative change
Amendment 1 Final: 17 February 2014
Administrative Change 1 Final: 28 May 2014
Amendment 2 Final: 19 May 2015
Title Immunogenicity and safety study of GSK Biologicals’ Priorix® vaccine (209762) at an end of shelf-life potency compared to Merck & Co., Inc.’s MMR vaccine when both are given on a 2-dose schedule to healthy children in their 2nd year of life.
Detailed Title A phase IIIA, randomized, observer-blind, controlled, multinational study to evaluate the immunogenicity and safety of GSK Biologicals' MMR vaccine (209762) (Priorix®) at an end of shelf-life potency compared to Merck & Co., Inc.’s MMR vaccine(M-M-R®II), when both are co-administered with Varivax, Havrix and Prevnar 13 (subset of children), and given on a two-dose schedule to healthy children in their second year of life.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a3119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 2
eTrack study number and Abbreviated Title
115649 (MMR-161)
Investigational New Drug (IND) number
BB IND 7229
EudraCT number 2011-004905-26
Date of protocol amendment
Amendment 2 Final: 19 May 2015
Detailed Title A phase IIIA, randomized, observer-blind, controlled, multinational study to evaluate the immunogenicity and safety of GSK Biologicals' MMR vaccine (209762) (Priorix®) at an end of shelf-life potency compared to Merck & Co., Inc.’s MMR vaccine (M-M-R®II), when both are co-administered with Varivax, Havrix and Prevnar 13 (subset of children), and given on a two-dose schedule to healthy children in their second year of life.
Co-ordinating author , Scientific writer, ZeroChaos for GSK Biologicals
Contributing authors(Amended 19 May 2015)
, Project Statistician , Director, Statistical Manager (SynteractHCR for GSK Biologicals),
Study Delivery Lead , Study Delivery Lead , Study Manager , Study Manager (Keyrus Biopharma for GSK
Biologicals), Study Data Manager Study Data Manager , Clinical Safety and
Pharmacovigilance , , Clinical Safety
and Pharmacovigilance , GVCL Clinical Readout Lead , Clinical Immunology , Global Regulatory Affairs , GVCL Study Manager , Director, Clinical Research and
Development Leader, Vaccines for MMR and Varicella
, Vice President, Global Vaccine Development, GSK Biologicals
GSK Biologicals’ Protocol DS v 13.2Copyright 2012-2015 the GlaxoSmithKline group of companies. All rights reserved. Unauthorized copying or use of this information is prohibited.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a3219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 3
Protocol Amendment 2 Sponsor Signatory Approval
eTrack study number and Abbreviated Title
115649 (MMR-161)
IND number BB IND 7229
EudraCT number 2011-004905-26
Date of protocol amendment Amendment 2 Final: 19 May 2015
Detailed Title A phase IIIA, randomized, observer-blind, controlled, multinational study to evaluate the immunogenicity and safety of GSK Biologicals' MMR vaccine (209762) (Priorix®) at an end of shelf-life potency compared to Merck & Co., Inc.’s MMR vaccine(M-M-R®II), when both are co-administered with Varivax, Havrix and Prevnar 13 (subset of children), and given on a two-dose schedule to healthy children in their second year of life.
Sponsor signatoryVice President, Global Vaccine Development, GSK Biologicals
Signature
Date
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a3319-MAY-2015
- ----------------Checksum----------------!Ver.!Created On - -5212d944cbc02d700704d1740e5a9757538b86a3 2.0 5/22/2015 11:05:59 AM - ----------------------------------------------------------------------------- -
For internal use only

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 4
Protocol Amendment 2 Rationale
Amendment number: Amendment 2
Rationale/background for changes:
Serological assays for the determination of antibodies against measles, rubella and varicella viruses will now be performed by a new 3rd party Contract Research Organization (CRO) named NEOMED-LABS Inc. Initially the testing was planned to be performed by GSK Biologicals’ laboratory in Rixensart. The assays have been transferred to GSK Biologicals’ laboratory in Laval. As of April 1st 2015, the GSK Biologicals’ laboratory in Laval became part of Neomed. The only change between GSK Biologicals’ laboratory in Laval and NEOMED-LABS Inc. is the name of the laboratory: assays and facilities remain the same.
Due to a delay in the availability of serologic data for the mumps Plaque Reduction Neutralization Test (PRNT) data analysis for this study will be conducted as follows: Part 1 will include a summary of measles, mumps and rubella Enzyme-Linked Immunosorbent Assay (ELISA) results post dose 1 (Day 42) and post dose 2 (Day 84). Part 2 will include a full immunogenicity analysis for post dose 1 (Day 42) and post dose 2 (Day 84) including mumps PRNT results post dose 1, and all safety data.
ELISA details were added in Appendix A.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a3419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 5
Protocol Amendment 2 Investigator Agreement
I agree:
To conduct the study in compliance with this protocol, any mutually agreed future protocol amendments or protocol administrative changes, and with any other study conduct procedures and/or study conduct documents provided by GlaxoSmithKline Biologicals (GSK Biologicals).
To assume responsibility for the proper conduct of the study at this site.
That I am aware of, and will comply with, ‘Good Clinical Practice’ (GCP) and all applicable regulatory requirements.
To ensure that all persons assisting me with the study are adequately informed about the GSK Biologicals’ investigational product(s) and other study-related duties and functions as described in the protocol.
To acquire the reference ranges for laboratory tests performed locally and, if required by local regulations, obtain the laboratory’s current certification or Quality Assurance procedure manual.
To ensure that no clinical samples (including serum samples) are retained onsite or elsewhere without the approval of GSK Biologicals and the express written informed consent of the child’s parent(s)/legally authorized representative(LAR(s)).
To perform no other biological assays on the clinical samples except those described in the protocol or its amendment(s).
To cooperate with a representative of GSK Biologicals in the monitoring process of the study and in resolution of queries about the data.
That I have been informed that certain regulatory authorities require the sponsor to obtain and supply, as necessary, details about the investigator’s ownership interest in the sponsor or the investigational product, and more generally about his/her financial ties with the sponsor. GSK Biologicals will use and disclose the information solely for the purpose of complying with regulatory requirements.
Hence I:
Agree to supply GSK Biologicals with any necessary information regarding ownership interest and financial ties (including those of my spouse and dependent children).
Agree to promptly update this information if any relevant changes occur during the course of the study and for one year following completion of the study.
Agree that GSK Biologicals may disclose any information it has about such ownership interests and financial ties to regulatory authorities.
Agree to provide GSK Biologicals with an updated Curriculum Vitae and other documents required by regulatory agencies for this study.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a3519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 6
eTrack study number and Abbreviated Titles
115649 (MMR-161)
IND number BB IND 7229
EudraCT number 2011-004905-26
Date of protocol amendment Amendment 2 Final: 19 May 2015
Detailed Title A phase IIIA, randomized, observer-blind, controlled, multinational study to evaluate the immunogenicity and safety of GSK Biologicals' MMR vaccine (209762) (Priorix®) at an end of shelf-life potency compared to Merck & Co., Inc.’s MMR vaccine(M-M-R®II), when both are co-administered with Varivax, Havrix and Prevnar 13 (subset of children), and given on a two-dose schedule to healthy children in their second year of life.
Investigator name
Signature
Date
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a3619-MAY-2015
- ----------------Checksum----------------!Ver.!Created On - -5212d944cbc02d700704d1740e5a9757538b86a3 2.0 5/22/2015 11:05:59 AM - ----------------------------------------------------------------------------- -
For internal use only

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 7
Sponsor Information
1. Sponsor
GlaxoSmithKline Biologicals89, Rue de l’Institut, 1330 Rixensart, Belgium
2. Sponsor Medical Expert for the Study
Refer to the local study contact information document.
3. Sponsor Study Monitor
Refer to the local study contact information document.
4. Sponsor Study Contact for Reporting of a Serious Adverse Event
GSK Biologicals Central Back-up Study Contact for Reporting SAEs: refer toprotocol Section 8.3.2.
5. GSK Biologicals’ Central Safety Physician On-Call Contact information forEmergency Unblinding
GSK Biologicals Central Safety Physician and Back-up Phone contact: refer toprotocol Section 8.7.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a3719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 8
SYNOPSIS
Detailed Title A phase IIIA, randomized, observer-blind, controlled, multinational study to evaluate the immunogenicity and safety of GSK Biologicals' MMR vaccine (209762) (Priorix®) at an end of shelf-life potency compared to Merck & Co., Inc.’s MMR vaccine (M-M-R®II), when both are co-administered with Varivax, Havrix and Prevnar 13 (subset of children), and given on a two-dose schedule to healthy children in their second year of life.
Indication GSK's candidate combined measles, mumps and rubella investigational vaccine (referred to as Inv_MMR vaccine throughout this document) will be used as a 2-dose series for active immunization of healthy children against measles, mumps and rubella diseases. The primary dose will be given at 12 to 15 months of age followed by a second dose 6 weekslater.
Rationale for the study and study design
Rationale for the study:
Measles, mumps and rubella are common viral illnesses of childhood. These highly infectious diseases and their complications are responsible for considerable morbidity and mortality throughout the world [WHO, 2000; WHO, 2001;WHO, 2004]. Measles can cause pneumonia and encephalitis. Mumps is associated with complications including aseptic meningitis, deafness and orchitis. Rubella symptoms in children include fever, swollen lymph nodes and rash, while rubella infection in pregnant women can cause congenital rubella syndrome. When immunization against these diseases is routinely practiced, significant reduction in disease incidence and associated complications has been reported [MMWR, 1998]. The trivalent Inv_MMR vaccine is licensed outside the United States (US) under the trade name of Priorix in over 100 countries, including Canada, Australia and all countries in the European Union.
This study is intended to support licensure of GSK Biologicals' trivalent combined measles, mumps and rubella vaccine (referred to as Inv_MMR vaccine in this document) in the US by generating immunogenicity and safety data in children 12 to 15 months of age. The US Centers for Disease Control (CDC) Advisory Committee on Immunization Practices (ACIP) recommends routine two-dose vaccination with MMR vaccine at 12 to 15 months and at 4 to 6 years of age. However, two doses of MMR vaccine may be given at a minimum of a one month interval (i.e., minimum of 28 days) if the first dose is
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a3819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 9
given on or after the first birthday [MMWR, 1998].
Rationale for the study design:
This study is designed to establish the end of shelf-life potency of Inv_MMR vaccine. In order to establish an end of shelf-life potency, the first dose of Inv_MMR vaccine will be given as one of two lots; one of a minimum potency, designated Inv_MMR_Min, and the other at a mid-range or medium potency designated Inv_MMR_Med. The primary endpoints of the study include: 1) evaluating the immunogenicity of a first dose of Inv_MMR_Min and/or the immunogenicity of a first dose of Inv_MMR_Med vaccine in contrast to a first dose of the US standard of care (MMR II vaccine referred to as Com_MMR within this document); and 2) demonstrating that the immune response for Inv_MMR_Min or Inv_MMR_Med meet a pre-specified acceptability criteria. Two lots of comparator vaccinewill be evaluated in this study, designated as Com_MMR_L1 and Com_MMR_L2. Throughout the study Com_MMR_L1 and Com_MMR_L2 will be analyzed as pooled lots. In the US, the primary dose of measles, mumps and rubella-containing vaccine is routinely co-administered with Varivax (VV), Havrix(HAV) and Prevnar 13 (PCV-13) at 12 to 15 months of age. Hence, in the US, the study will also evaluate the safety and immunogenicity of both Inv_MMR and Com_MMR vaccines when both are co-administered with these vaccines. Note: Given the heterogeneity in the use of pediatric pneumococcal vaccines globally, and administration of these vaccines generally begins in infancy, Prevnar 13 will be administered only to children enrolled in the US.
Six weeks later children randomized to receive either Inv_MMR_Min or Inv_MMR_Med will receive a second dose of the investigational vaccine, designated Inv_MMR_Release,which has a potency within the release range of the marketed vaccine. Children randomized to receive either Com_MMR_L1 or Com_MMR_L2 will receive a second dose of Com_MMR vaccine at the six week interval. Immune responses following a second dose of both Inv_MMR and Com_MMR will be evaluated in a sub-cohort of children enrolled in the US. Thus,the study will provide data on the immunogenicity (in a sub-cohort of children) and safety of Inv_MMR when administered on a two-dose schedule to children in the second year of life.
Objectives Primary
To control the risk of erroneous conclusions, a hierarchical procedure will be used for the multiple study objectives with the possibility to conclude on objectives 6-10 associated with Inv_MMR_Med even
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a3919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 10
though one or more of objectives 1-5 associated to Inv_MMR_Min are not met (see Section 10.3.1 for details).
Unless otherwise specified, all primary and secondary immunogenicity endpoints will be tested with an Enzyme-Linked Immunosorbent Assay (ELISA).
Minimum potency vaccine (Inv_MMR_Min):
1. To demonstrate non-inferiority of Inv_MMR_Min vaccine compared to pooled Com_MMR vaccine in terms of seroresponse rates (see Section 10.5 for definition) to measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% confidence interval (CI)
on the group difference (Inv_MMR_Min minus pooled Com_MMR) in seroresponse rate is ≥–5% for antibodies to measles, mumps and rubella viruses.
2. To demonstrate non-inferiority of Inv_MMR_Min vaccine compared to pooled Com_MMR vaccine in terms of GMCs for antibodies to measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group ratio of
GMCs (Inv_MMR_Min over pooled Com_MMR) is ≥0.67 for antibodies to measles, mumps and rubella viruses.
3. To demonstrate an acceptable immune response of Inv_MMR_Min vaccine in terms of seroresponse rates for measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI for the seroresponse
rate of Inv_MMR_Min is ≥90% for antibodies to measles, mumps and rubella viruses.
4. To demonstrate non-inferiority of the Inv_MMR_Min vaccine compared to pooled Com_MMR vaccine in terms of seroresponse rates (see Section 10.5 for definition) for mumps virus (by Plaque Reduction Neutralization Test (PRNT)) at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group
difference (Inv_MMR_Min minus pooled Com_MMR) in seroresponse rate is ≥-10% for antibodies to mumps virus.
5. To demonstrate non-inferiority of the Inv_MMR_Min vaccine compared to pooled Com_MMR vaccine in terms of geometric mean titer (GMT) for antibodies to mumps virus (by PRNT) at Day 42.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 11
Criteria: The lower limit of the two-sided 97.5% CI on the GMT ratio
(Inv_MMR_Min over pooled Com_MMR) is ≥0.67 forantibodies to mumps virus.
Medium potency vaccine (Inv_MMR_Med) [To be statistically analyzed only if one or more of the objectives 1-5 related to the minimum potency vaccine are not met]:
6. To demonstrate non-inferiority of Inv_MMR_Med vaccine compared to pooled Com_MMR vaccine in terms of seroresponse rates to measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group
difference (Inv_MMR_Med minus pooled Com_MMR) in seroresponse rate is ≥–5% for antibodies to measles, mumps and rubella viruses.
7. To demonstrate non-inferiority of Inv_MMR_Med vaccine compared to pooled Com_MMR vaccine in terms of GMCs for antibodies to measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group ratio of
GMCs (Inv_MMR_Med over pooled Com_MMR) is ≥0.67 for antibodies to measles, mumps and rubella viruses.
8. To demonstrate an acceptable immune response of Inv_MMR_Med vaccine in terms of seroresponse rates for measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI for the seroresponse
rate of Inv_MMR_Med is ≥90% for antibodies to measles, mumps, and rubella viruses.
9. To demonstrate non-inferiority of the Inv_MMR_Med vaccine compared to pooled Com_MMR vaccine in terms of seroresponse rates for mumps virus (by PRNT) at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group
difference (Inv_MMR_Med minus pooled Com_MMR) in seroresponse rate is ≥-10% for antibodies to mumps virus.
10. To demonstrate non-inferiority of the Inv_MMR_Med vaccine compared to pooled Com_MMR vaccine in terms of GMT for antibodies to mumps virus (by PRNT) at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the GMT ratio
(Inv_MMR_Med over pooled Com_MMR) is ≥0.67 for
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 12
antibodies to mumps virus.
Secondary
1. To assess the immunogenicity of Inv_MMR_Min followed by Inv_MMR_Release and pooled Com_MMR vaccine in terms of seroresponse rates and GMCs for antibodies to measles, mumps and rubella viruses at Day 84 (post Dose 2) (in a sub-cohort of children enrolled in the US).
2. To assess the immunogenicity of Inv_MMR_Med followed by Inv_MMR_Release and pooled Com_MMR vaccine in terms of seroresponse rate and GMCs for antibodies to measles, mumps and rubella viruses at Day 84 (post Dose 2) (in a sub-cohort of children enrolled in the US).
3. To assess the safety and reactogenicity of Inv_MMR_Min, Inv_MMR_Med, and Com_MMR when co-administered with Varivax, Havrix (to all children), and Prevnar 13 (only to children enrolled in the US).
Study design
Experimental design: Phase IIIA, observer-blind, randomized,controlled, multicenter, multi-country, end of shelf-life study with four parallel groups.
Duration of the study:
Primary Epoch: The study period is approximately 7.5 months starting at Visit 1 (Day 0) and ending at Visit 4 (Day 222).
Synopsis Table 1 Study groups and epochs foreseen in the study
Study groups Number of subjects Age (Min/Max) Primary Epoch
Inv_MMR_Min 150012 months – 15
months●
Inv_MMR_Med 150012 months – 15
months●
Com_MMR_L1 75012 months – 15
months●
Com_MMR_L2 75012 months – 15
months●
L = Lot
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 13
Control: active control
Com_MMR_L1 and Com_MMR_L2 vaccine.
Co-Administered vaccines:
All children will receive Havrix and Varivax as study vaccines, concomitantly with MMR vaccine at 12 to 15 months of age at Visit 1.
Prevnar 13 will only be administered to children recruited in the US at Visit 1.
At the end of the study, GSK will provide a second dose of Havrix and/or varicella vaccine to selected non-US countries if local health departments do not routinely provide hepatitis A and varicella vaccination. Note that the second dose of Havrixand varicella vaccine is not part of the study procedures. For information on administration of the 2nd dose of varicella vaccine in Malaysia, see Section 12.
Vaccination schedule: two-dose schedule
At Visit 1 children 12 to 15 months of age will be administered a dose from either one of two Inv_MMR vaccine lots (Inv_MMR_Min or Inv_MMR_Med), or from one of two active control Com_MMR vaccine lots (Com_MMR_L1 and Com_MMR_L2), depending on the study group. In addition, all children will be given two co-administered vaccines Varivax, and Havrix at Visit 1. All US children will also be given Prevnar 13.
Approximately 6 weeks later at Visit 2, children in either the Inv_MMR_Min orInv_MMR_Med groups will be administered a dose from a separate lot of the Inv_MMR vaccine at the release potency range (Inv_MMR_Release) for the second dose. Children who received Com_MMR_L1 or Com_MMR_L2 for the first dose will be administered a dose from one of two lots of Com_MMR vaccine for the second dose.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 14
Treatment groups:
Synopsis Table 2 Treatment groups
Treatment nameVaccine / Product
name
Treatment groupsInv_MMR_Min
(active)Inv_MMR_Med
(active)Com_MMR_L1
(control)Com_MMR_L2 (control)
Inv_MMR_Min*Inv_MMR_Min XInv_MMR Diluent X
Inv_MMR_Med*Inv_MMR_Med XInv_MMR Diluent X
Inv_MMR_Release†
Inv_MMR_Release X XInv_MMR_ReleaseDiluent§
X X
Com_MMR_L1*†
Com_MMR_L1 XCom_MMR_L1 Diluent¥
X
Com_MMR_L2*†
Com_MMR_L2 XCom_MMR_L2 Diluent¥
X
Varivax*Varivax X X X XVarivax Diluent X X X X
Havrix* Havrix X X X XPrevnar 13*‡ Prevnar 13 X X X XL= Lot* Administered during Visit 1 for Dose 1† Administered during Visit 2 for Dose 2§ The Inv_MMR_Release Diluent for Dose 2 is distinct from the Inv_MMR Diluent for Dose 1¥ The Com_MMR_L1 Diluent may be distinct from the Com_MMR_L2 Diluent‡ Prevnar 13 will only be given to children recruited in the US
Treatment allocation: Approximately 4500 healthy children 12 to 15 months of age will be randomized in a 2:2:1:1 ratio.
- For the first dose, children will be given a vaccination with a dose from either one of two Inv_MMR vaccine lots designated Inv_MMR_Min or Inv_MMR_Med (1500 children each); or from one of two Com_MMR vaccine lots designated Com_MMR_L1 or Com_MMR_L2 (750 children each).
- For the second dose, both the Inv_MMR_Min and Inv_MMR_Med groups will be given investigational vaccine from a separate lot designated Inv_MMR_Release, and both the Com_MMR_L1 and Com_MMR_L2 groups will be administered oneof two lots of Com_MMR vaccine.
- Overall, the randomization is 2:2:1:1 (Inv_MMR_Min:Inv_MMR_Med:Com_MMR_L1:Com_MMR_L2) and the treatment allocation at the investigator site will be performed using a central randomization system on the Internet (SBIR).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 15
Blinding: observer-blind.
- The study will be conducted in a double-blind fashion with regard to the twoInv_MMR vaccine lots (Inv_MMR_Min and Inv_MMR_Med) and in an observer-blind fashion for the lots of Inv_MMR vaccine versus the pooled Com_MMR vaccine lots.
Synopsis Table 3 Blinding of study epochs
Study Epoch BlindingPrimary Epoch observer-blind
Visits: Children from each treatment group will participate in 4 study visits (Day 0, Day 42, Day 84 and Day 222).
Sampling:
- Blood samples will be collected from each child at Day 0 and Day 42. In addition, a third blood sample will be collected from all US children at Day 84 (42 days post-dose 2).
Type of study: self-contained.
Data collection: Electronic Case Report Form (eCRF) using remote data entry (RDE).
Number of subjects
4500 children.
Endpoints Primary
Immunogenicity of the MMR vaccines at Day 42.
- Seroresponse to measles, mumps and rubella viruses (by ELISA) and to mumps virus (by PRNT) (see Section 10.5 for definition).
- Measles, mumps and rubella virus antibody concentrations (by ELISA) and mumps virus antibody titers (by PRNT).
Secondary
Immunogenicity of the MMR vaccines at Day 84 post-dose 2 (US sub-cohort).
- Seroresponse to measles, mumps and rubella viruses (by ELISA).
- Measles, mumps and rubella virus antibody concentrations (by ELISA).
Solicited local and general symptoms.
- Occurrence of solicited local symptoms in terms of injection site redness, pain and swelling from Day 0 to Day 3 after each vaccination.
- Occurrence of solicited general symptoms in terms of drowsiness,
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 16
loss of appetite and irritability from Day 0 to Day 14 after first vaccination.
- Occurrence of solicited general symptoms in terms of fever (temperature ≥38.0°C / 100.4°F), rash, parotid/salivary gland swelling, any sign of meningism (including febrile convulsions) from Day 0 to Day 42 after each vaccination.
Unsolicited adverse events.
- Occurrence of unsolicited symptoms according to the Medical Dictionary for Regulatory Activities (MedDRA) classification from Day 0 to Day 42 after each vaccination.
Adverse events of specific interest.
- Occurrence of new onset chronic disease (NOCD) (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies) and AEs prompting emergency room (ER) visits from Day 0 through the end of study.
Serious adverse events.
- Occurrence of serious adverse events from Day 0 through the end of study.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 17
TABLE OF CONTENTS
PAGE
SPONSOR INFORMATION ............................................................................................7
SYNOPSIS......................................................................................................................8
LIST OF ABBREVIATIONS...........................................................................................25
GLOSSARY OF TERMS ...............................................................................................28
TRADEMARKS .............................................................................................................31
1. INTRODUCTION....................................................................................................321.1. Background ................................................................................................321.2. Rationale for the study and study design ....................................................32
1.2.1. Rationale for the study.................................................................321.2.2. Rationale for the study design......................................................32
2. OBJECTIVES.........................................................................................................332.1. Primary objectives ......................................................................................332.2. Secondary objectives..................................................................................35
3. STUDY DESIGN OVERVIEW ................................................................................36
4. STUDY COHORT...................................................................................................394.1. Number of study children/centers ...............................................................394.2. Inclusion criteria for enrollment ...................................................................404.3. Exclusion criteria for enrollment ..................................................................40
5. CONDUCT OF THE STUDY ..................................................................................415.1. Regulatory and ethical considerations, including the informed
consent process..........................................................................................415.2. Subject identification and randomization of treatment .................................42
5.2.1. Subject identification....................................................................435.2.2. Randomization of treatment.........................................................43
5.2.2.1. Randomization of supplies..........................................435.2.2.2. Treatment allocation to the study children ..................43
5.3. Method of blinding ......................................................................................445.4. General study aspects ................................................................................445.5. Outline of study procedures ........................................................................455.6. Detailed description of study procedures ....................................................46
5.6.1. Procedures prior to study participation.........................................465.6.1.1. Informed consent........................................................46
5.6.2. Procedures prior to the first vaccination .......................................475.6.2.1. Check inclusion and exclusion criteria ........................475.6.2.2. Collect demographic data...........................................475.6.2.3. Medical history ...........................................................475.6.2.4. Physical examination..................................................47
5.6.3. Procedures during the study/primary epoch.................................47
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 18
5.6.3.1. Check and record concomitant medication/vaccination and intercurrent medical conditions......................................................48
5.6.3.2. Check contraindications, warnings and precautions to vaccination ..........................................48
5.6.3.3. Assess pre-vaccination body temperature ..................485.6.3.4. Randomization ...........................................................485.6.3.5. Blood sampling for safety or immune response
assessments ..............................................................485.6.3.6. Treatment number assignment...................................485.6.3.7. Vaccination.................................................................495.6.3.8. Recording of AEs and SAEs.......................................495.6.3.9. Procedures during follow-up visits ..............................505.6.3.10. Study conclusion ........................................................50
5.7. Biological sample handling and analysis.....................................................505.7.1. Use of specified study materials ..................................................515.7.2. Biological samples .......................................................................515.7.3. Laboratory assays .......................................................................515.7.4. Biological samples evaluation ......................................................53
5.7.4.1. Immunological read-outs ............................................535.7.5. Immunological correlates of protection.........................................53
6. STUDY VACCINES AND ADMINISTRATION ........................................................546.1. Description of study vaccines......................................................................546.2. Storage and handling of study vaccines......................................................576.3. Dosage and administration of study vaccines .............................................586.4. Replacement of unusable vaccine doses....................................................596.5. Contraindications to subsequent vaccination ..............................................606.6. Warnings and precautions ..........................................................................616.7. Concomitant medication/vaccination...........................................................62
6.7.1. Medications/products that may lead to the elimination of a child from ATP analyses ..............................................................62
6.7.2. Time window for recording concomitant medication/vaccination in the eCRF.............................................63
6.8. Intercurrent medical conditions that may lead to elimination from an ATP cohort..................................................................................................64
7. HEALTH ECONOMICS ..........................................................................................64
8. ADVERSE EVENTS AND SERIOUS ADVERSE EVENTS.....................................648.1. Safety definitions ........................................................................................64
8.1.1. Definition of an adverse event......................................................648.1.2. Definition of a serious adverse event ...........................................658.1.3. Solicited adverse events ..............................................................668.1.4. Clinical laboratory parameters and other abnormal
assessments qualifying as adverse events or serious adverse events ............................................................................67
8.1.5. Adverse events of specific interest...............................................678.2. Detecting and recording adverse events and serious adverse
events.........................................................................................................678.2.1. Time period for detecting and recording adverse events
and serious adverse events .........................................................67
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 19
8.2.2. Evaluation of adverse events and serious adverse events...........698.2.2.1. Active questioning to detect adverse events
and serious adverse events........................................698.2.2.2. Assessment of adverse events...................................69
8.2.2.2.1. Assessment of intensity ..........................698.2.2.2.2. Assessment of causality .........................74
8.2.2.3. Assessment of outcomes............................................758.2.2.4. Medically attended visits.............................................76
8.3. Reporting of serious adverse events...........................................................768.3.1. Prompt reporting of serious adverse events to GSK
Biologicals ...................................................................................768.3.2. Contact information for reporting serious adverse events
to GSK Biologicals .......................................................................768.3.3. Completion and transmission of SAE reports to GSK
Biologicals ...................................................................................768.3.3.1. Back-up system in case the electronic SAE
reporting system does not work ..................................778.3.3.2. Updating of SAE information after freezing of
the child’s eCRF .........................................................778.3.4. Regulatory reporting requirements for serious adverse
events..........................................................................................778.4. Follow-up of adverse events and serious adverse events ...........................77
8.4.1. Follow-up of adverse events and serious adverse events ............778.5. Treatment of adverse events ......................................................................788.6. Unblinding...................................................................................................788.7. Emergency unblinding ................................................................................798.8. Subject card................................................................................................80
9. SUBJECT COMPLETION AND WITHDRAWAL.....................................................809.1. Subject completion .....................................................................................809.2. Child withdrawal..........................................................................................80
9.2.1. Child withdrawal from the study ...................................................809.2.2. Child withdrawal from investigational vaccine ..............................81
10. DATA EVALUATION: CRITERIA FOR EVALUATION OF OBJECTIVES ...............8110.1. Primary endpoint.........................................................................................8110.2. Secondary endpoints ..................................................................................8210.3. Estimated sample size ................................................................................82
10.3.1. Control on type I error ..................................................................8210.3.2. Power for Non-inferiority of Inv_MMR_Min to Com_MMR............8510.3.3. Power for a minimal seroresponse rate of Inv_MMR_Min
lots...............................................................................................8610.3.4. Power for Non-inferiority of Inv_MMR_Med to Com_MMR...........8710.3.5. Power for a minimal seroresponse rate of Inv_MMR_Med
lots...............................................................................................8810.4. Study cohorts to be evaluated.....................................................................89
10.4.1. Total vaccinated cohort................................................................8910.4.2. According-to-protocol cohort for analysis of safety.......................8910.4.3. According-to-protocol cohort for analysis of
immunogenicity............................................................................8910.5. Derived and transformed data.....................................................................9010.6. Conduct of analyses ...................................................................................91
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a31919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 20
10.6.1. Sequence of analyses (Amended 19 May 2015)..........................9110.6.2. Statistical considerations for interim analyses..............................91
10.7. Statistical methods......................................................................................9210.7.1. Methodology for computing CI .....................................................9210.7.2. Analysis of demographics/baseline characteristics ......................9210.7.3. Analysis of immunogenicity..........................................................92
10.7.3.1. Within groups assessment..........................................9210.7.3.2. Between group assessment post-dose1.....................9310.7.3.3. Interpretation of analyses ...........................................93
10.7.4. Analysis of safety.........................................................................93
11. ADMINISTRATIVE MATTERS ...............................................................................9511.1. Case Report Form/Remote Data Entry instructions ....................................9511.2. Monitoring by GSK Biologicals....................................................................9511.3. Archiving of data at study sites ...................................................................9611.4. Audits .........................................................................................................9711.5. Posting of information on Clinicaltrials.gov..................................................9711.6. Ownership, confidentiality and publication ..................................................97
11.6.1. Ownership ...................................................................................9711.6.2. Confidentiality ..............................................................................9711.6.3. Publication ...................................................................................9811.6.4. Provision of study results to investigators, posting to the
clinical trials registers and publication ..........................................98
12. COUNTRY SPECIFIC REQUIREMENTS...............................................................99
13. REFERENCES.....................................................................................................100
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 21
LIST OF TABLES
PAGE
Table 1 Study groups and epochs foreseen in the study.....................................37
Table 2 Treatment groups...................................................................................38
Table 3 Blinding of study epochs ........................................................................39
Table 4 Sub-cohorts............................................................................................39
Table 5 List of study procedures .........................................................................45
Table 6 Intervals between study visits.................................................................46
Table 7 Biological samples .................................................................................51
Table 8 Humoral Immunity (Antibody determination) (Amended 19 May 2015)......................................................................................................52
Table 9 Immunological read-outs ........................................................................53
Table 10 Study vaccines.......................................................................................55
Table 11 Dosage and administration.....................................................................58
Table 12 Solicited local adverse events at Inv_MMR and Com_MMR injection sites .........................................................................................66
Table 13 Solicited general adverse events............................................................66
Table 14 Reporting periods and methods for adverse events and serious adverse events.......................................................................................68
Table 15 Intensity scales for solicited symptoms...................................................70
Table 16 Brighton Collaboration levels of diagnostic certainty...............................72
Table 17 Timeframes for submitting serious adverse event reports to GSK Biologicals..............................................................................................76
Table 18 Probability to meet the non-inferiority criteria with respect to seroresponse rate for antibodies to measles, mumps and rubella viruses for Inv_MMR_Min compared to Com_MMR ...............................85
Table 19 Probability to meet the non-inferiority criteria with respect to GMCs of antibodies to measles, mumps and rubella viruses for Inv_MMR_Min compared to Com_MMR ................................................86
Table 20 Probability to meet the minimum level of seroresponse rate for antibodies to measles, mumps and rubella for Inv_MMR_Min................86
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 22
Table 21 Probability to meet the non-inferiority criteria with respect to seroresponse rate for antibodies to measles, mumps and rubella viruses for Inv_MMR_Med compared to Com_MMR ..............................87
Table 22 Probability to meet the non-inferiority criteria with respect to GMCs of antibodies to measles, mumps and rubella viruses for Inv_MMR_Med compared to Com_MMR ...............................................88
Table 23 Probability to meet the minimum level of seroresponse rate for antibodies to measles, mumps and rubella for Inv_MMR .......................88
Table 24 GSK Biologicals’ laboratories (Amended 19 May 2015) .......................102
Table 25 Outsourced laboratories (Amended 19 May 2015) ...............................102
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 23
LIST OF FIGURES
PAGE
Figure 1 Study design ..........................................................................................36
Figure 2 Sequence for evaluating the study objectives in order to control the overall type I error below 2.5% .........................................................84
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 24
LIST OF APPENDICES
PAGE
APPENDIX A LABORATORY ASSAYS (Amended 19 May 2015)..............................101
APPENDIX B CLINICAL LABORATORIES ................................................................102
APPENDIX C AMENDMENTS AND ADMINISTRATIVE CHANGES TO THE PROTOCOL.........................................................................................103
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 25
LIST OF ABBREVIATIONS
Ab Antibody
ACIP Advisory Committee on Immunization Practices
AE Adverse event
ANOVA Analysis of variance
Anti-measles Antibody to measles virus
Anti-mumps Antibody to mumps virus
Anti-rubella Antibody to rubella virus
ATP According-To-Protocol
BB IND Biological Investigational New Drug
°C Degree Celsius
CCID Cell Culture Infectious Dose
CDC Centers for Disease Control
CD-ROM Compact Disc Read-Only Memory
CI Confidence Interval
cm Centimeter
Com_MMR_L1 Comparator MMR Vaccine Lot 1
Com_MMR_L2 Comparator MMR Vaccine Lot 2
CPE Cytopathic Effect
CRM197 Cross-Reactive Material (mutant diphtheria toxin)
CRO Contract Research Organization (Amended 19 May 2015)
DC Diary Card
DNA Deoxyribonucleic Acid
eCRF electronic Case Report Form
ED50 End-point Dilution 50%
EDTA Ethylenediamine-Tetraacetic Acid
ELISA Enzyme-Linked Immunosorbent Assay
ER Emergency Room
EU ELISA Units
°F Degree Fahrenheit
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 26
Flu Influenza
GCP Good Clinical Practice
GMC Geometric Mean Concentration
GMT Geometric Mean Titer
GSK GlaxoSmithKline
HAV Hepatitis A Vaccine Havrix
Hib Haemophilus influenzae type b conjugate vaccine
HIV Human Immunodeficiency Virus
ICF Informed Consent Form
ICH International Conference on Harmonization
IEC Independent Ethics Committee
IgG Immunoglobulin class G
IM Intramuscular
IND Investigational New Drug
Inv_MMR_Med Investigational MMR Medium Potency Vaccine
Inv_MMR_Min Investigational MMR Minimum Potency Vaccine
Inv_MMR_Release Investigational MMR Release Potency Vaccine
IRB Institutional Review Board
IU International Unit
L Lot
LAR(s) Legally Acceptable Representative(s)
LOD Limit of Detection
LOQ Limit of Quantitation
LSLV Last Subject’s Last Visit
mcg / g Microgram
MedDRA Medical Dictionary for Regulatory Activities
Mg Milligram
mIU Milli International Unit
mL Milliliter
Mm Millimeter
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 27
MMR Measles, Mumps and Rubella vaccine
MMWR Morbidity and Mortality Weekly Report
MRC Medical Research Council
NB Nota Bene
NIBSC National Institute for Biological Standards and Control
NOCD New Onset Chronic Disease
PASS Power Analysis and Sample Size (software)
PCV-13 Pneumococcal Conjugate Vaccine Prevnar 13
PFU Plaque Forming Unit
PPD Pharmaceutical Product Development, Inc.
PPM Parts Per Million
PRNT Plaque Reduction Neutralization Test
RDE Remote Data Entry
SAE Serious Adverse Event
SAS® Statistical Analysis System
SBIR Randomization System on Internet
SC Subcutaneous
SD Standard Deviation
SDV Source Document Verification
SOP Standard Operating Procedures
SPM Study Procedures Manual
SPMSD Sanofi Pasteur/Merck Sharp and Dohme
TCID Tissue Culture Infectious Dose
US United States (of America)
Vacc Vaccination
vs. Versus
VV Varicella Vaccine Varivax
VZV Varicella Zoster Virus
WHO World Health Organization
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 28
GLOSSARY OF TERMS
Adverse event: Any untoward medical occurrence in a patient or clinical investigation child, temporally associated with the use of a medicinal product, whether or not considered related to the medicinal product.
An AE can therefore be any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease (new or exacerbated) temporally associated with the use of a medicinal product. For marketed medicinal products, this also includes failure to produce expected benefits (i.e., lack of efficacy), abuse or misuse.
Blinding: A procedure in which one or more parties to the trial are kept unaware of the treatment assignment in order to reduce the risk of biased study outcomes. The level of blinding is maintained throughout the conduct of the trial, and only when the data are cleaned to an acceptable level of quality will appropriate personnel be unblinded or when required in case of a serious adverse event. In an observer-blind study, the child/parent(s)/Legally Acceptable Representative(s) (LAR(s)) and the site and sponsor personnel involved in the clinical evaluation of the children are blinded while other study personnel may be aware of the treatment assignment. In a double blind study, the child/parent(s)/LAR(s), the investigator and sponsor staff who are involved in the treatment or clinical evaluation of the children and the review or analysis of data are all unaware of the treatment assignments. Partially-blind is to be used for study designs with different blinding levels between different groups, e.g., double-blinded consistency lots which are open with respect to the control group.
Child: Term used throughout the protocol to denote an individual who has been contacted in order to participate or participates in the clinical study, either as a recipient of the investigational product(s) or as a control.
Child in care: A child who has been placed under the control or protection of an agency, organization, institution or entity by the courts, the government or a government body, acting in accordance with powers conferred on them by law or regulation. The definition of a child in care can include a child cared for by foster parents or living in a care home or institution, provided that the arrangement falls within the definition above. The definition of a child in care does not include a child who is adopted or has an appointed LAR/s.
Eligible: Qualified for enrollment into the study based upon strict adherence to inclusion/exclusion criteria.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 29
Epoch: An epoch is a well defined part of a protocol that covers a set of consecutive timepoints. Generally, an epoch is self-contained and allows to perform a data analysis to address some of the trial objectives (e.g., primary, booster, yearly follow-ups,…).
eTrack: GSK’s tracking tool for clinical trials.
Evaluable: Meeting all eligibility criteria, complying with the procedures defined in the protocol, and, therefore, included in the according-to-protocol (ATP) analysis.
Medically Attended Adverse Event:
An event for which the child received medical attention defined as hospitalization, an emergency room visit or a visit to or from medical personnel (e.g., nurse practitioner or physician assistant or medical doctor) for any reason.
Protocol amendment:
The International Conference on Harmonisation (ICH) defines a protocol amendment as: ‘A written description of a change(s) to or formal clarification of a protocol.’ GSK Biologicals further details this to include a change to an approved protocol that affects the safety of subjects, scope of the investigation, study design, or scientific integrity of the study.
Protocol administrative change:
A protocol administrative change addresses changes to only logistical or administrative aspects of the study.
Note: Any change that falls under the definition of a protocol amendment (e.g. a change that affects the safety of subjects, scope of the investigation, study design, or scientific integrity of the study) MUST be prepared as an amendment to the protocol.
Randomization: Process of random attribution of treatment to children in order to reduce bias of selection.
SBIR: A central Internet randomization system.
Site Monitor: An individual assigned by the sponsor who is responsible for assuring proper conduct of clinical studies at one or more investigational sites.
Solicited adverse event:
Adverse events (AEs) to be recorded as endpoints in the clinical study. The presence/occurrence/intensity of these events is actively solicited from the child or an observer during a specified post-vaccination follow-up period.
Sub-cohort: A group of children for whom specific study procedures are used/performed relative to the other sub-cohorts.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a32919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 30
Subset: A group of children who have the same study procedures at a given timepoint, but the biological assays performed on that sample will be different, relative to the other subsets.
Treatment: Term used throughout the clinical study to denote a set of investigational product(s) or marketed product(s) or placebo intended to be administered to a child, identified by a unique number, according to the study randomization or treatment allocation.
Unsolicited adverse event:
Any adverse event (AE) reported in addition to those solicited during the clinical study. Also any ‘solicited’ symptom with onset outside the specified period of follow-up for solicited symptoms will be reported as an unsolicited adverse event.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 31
TRADEMARKS
The following trademarks are used in the present protocol.
Note: In the body of the protocol (including the synopsis), the names of the vaccines/products will be written without the superscript symbol ™ or ®.
Trademarks of the GlaxoSmithKline group of companies
Generic description
Priorix®* Measles, mumps, and rubella vaccine live
Havrix® Hepatitis A vaccine (HAV)
* Priorix is not licensed in the US.
Trademarks not owned by the GlaxoSmithKline group of companies
Generic description
M-M-RII (Merck & Co., Inc.) also calledM-M-R Vax Pro® (distributed by Sanofi Pasteur/Merck Sharp and Dohme [SPMSD] in certain countries)*
Measles, mumps and rubella virus vaccine live
Varivax® (Merck & Co., Inc.) Varicella virus vaccine live (VV)
Prevnar 13® (Pfizer Inc.) Pneumococcal 13-valent conjugate vaccine (diphtheria CRM197 protein) (PCV-13)
* The Merck & Co., Inc.’s combined measles-mumps-rubella virus vaccine live (lots 1 and 2) are referred to as Com_MMR (Com_MMR_L1 and Com_MMR_L2) for the comparator vaccine throughout the protocol.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 32
1. INTRODUCTION
1.1. Background
Please refer to the current Investigator Brochure for a review of the pre-clinical and clinical studies, and the potential risks and benefits of GSK's candidate combined measles, mumps and rubella vaccine (Inv_MMR).
Measles, mumps and rubella are common viral illnesses of childhood. These highly infectious diseases and their complications are responsible for considerable morbidity and mortality throughout the world [WHO, 2000; WHO, 2001; WHO, 2004]. Measles can cause pneumonia and encephalitis. Mumps is associated with complications including aseptic meningitis, deafness and orchitis. Rubella symptoms in children include fever, swollen lymph nodes and rash, while rubella infection in pregnant women can cause congenital rubella syndrome. When immunization against these diseases is routinely practiced, significant reduction in disease incidence and associated complications has been reported [MMWR, 1998]. The trivalent Inv_MMR vaccine is licensed outside the US under the trade name of Priorix in over 100 countries, including Canada, Australiaand all countries in the European Union.
1.2. Rationale for the study and study design
1.2.1. Rationale for the study
This study is intended to support licensure of GSK Biologicals' trivalent combined measles, mumps and rubella vaccine (referred to as Inv_MMR vaccine in this document)in the US by generating immunogenicity and safety data in children 12 to 15 months of age. The US Centers for Disease Control (CDC) Advisory Committee on Immunization Practices (ACIP) recommends routine two-dose vaccination with MMR vaccine at 12 to 15 months and at 4 to 6 years of age. However, two doses of MMR vaccine may be given at a minimum of a one month interval (i.e., minimum of 28 days) if the first dose is given on or after the first birthday [MMWR, 1998].
1.2.2. Rationale for the study design
This study is designed to establish the end of shelf-life potency of Inv_MMR vaccine. The Inv_MMR vaccine administered in this study will reflect a reasonable end of shelf-life potency for all antigens. In order to establish an end of shelf-life potency, the first dose of Inv_MMR vaccine will be given from one of two lots; one of a minimum potency, designated Inv_MMR_Min, and the other at a mid-range or medium potency designated Inv_MMR_Med. To maintain these Inv MMR lots at their specified potencies, these vaccine vials will remain frozen until reconstitution. Both of these Inv_MMR groups will then receive a second dose of the investigational vaccine which has a potency within the release potency range of the marketed vaccine. The primary endpoints of the study include: 1) evaluating the immunogenicity of a first dose of Inv_MMR_Min and/or the immunogenicity of a first dose of Inv_MMR_Med vaccine in contrast to a first dose of the US standard of care (MMR II vaccine referred to as Com_MMR within this document); and 2) demonstrating that the immune response for Inv_MMR_Min or
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 33
Inv_MMR_Med meet a pre-specified acceptability criteria. In order to obtain more representative data on the comparator, the Com_MMR vaccine used in this study will consist of two lots designated Com_MMR_L1 and Com_MMR_L2. Throughout the study Com_MMR_L1 and Com_MMR_L2 will be analyzed as pooled lots.
In the US, the primary dose of measles, mumps and rubella-containing vaccine is routinely co-administered with Varivax, Havrix and Prevnar 13 at 12 to 15 months of age. Hence, in the US the study will also evaluate the safety and immunogenicity of both Inv_MMR and Com_MMR vaccines when both are co-administered with these three vaccines. In other countries the study will evaluate safety and immunogenicity of Inv_MMR and Com_MMR vaccines when co-administered with Varivax and Havrix. Note: Given the heterogeneity in the use of pediatric pneumococcal vaccines globally, and administration of these vaccines generally begins in infancy, Prevnar 13 will be administered only to children enrolled in the US.
Six weeks after the first dose children randomized to receive either Inv_MMR_Min, or Inv_MMR_Med, will receive a second dose of the investigational vaccine, designated Inv_MMR_Release, which has a potency within the release range of the marketed vaccine. Children randomized to receive either Com_MMR_L1 or Com_MMR_L2 will receive a second dose of Com_MMR vaccine at the six week interval. Immune responses following a second dose of both Inv_MMR and Com_MMR will be evaluated in allchildren enrolled in the US. Thus, the study will provide data on the immunogenicity (in a sub-cohort of children) and safety of Inv_MMR when administered on a two-dose schedule to children in the second year of life.
2. OBJECTIVES
2.1. Primary objectives
To control the risk of erroneous conclusions, a hierarchical procedure will be used for the multiple study objectives with the possibility to conclude on objectives 6-10 associated to Inv_MMR_Med even though one or more of objectives 1-5 associated with Inv_MMR_Min are not met (see Section 10.3.1 for details).
Unless otherwise specified, all primary and secondary immunogenicity endpoints will be tested with an ELISA.
Minimum potency vaccine (Inv_MMR_Min):
1. To demonstrate non-inferiority of Inv_MMR_Min vaccine compared to pooled Com_MMR vaccine in terms of seroresponse rates (see Section 10.5 for definition) to measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% confidence interval (CI) on the group
difference (Inv_MMR_Min minus pooled Com_MMR) in seroresponse rate is ≥–5% for antibodies to measles, mumps and rubella viruses.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 34
2. To demonstrate non-inferiority of Inv_MMR_Min vaccine compared to pooled Com_MMR vaccine in terms of GMCs for antibodies to measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group ratio of GMCs
(Inv_MMR_Min over pooled Com_MMR) is ≥0.67 for antibodies to measles, mumps and rubella viruses.
3. To demonstrate an acceptable immune response of Inv_MMR_Min vaccine in terms of seroresponse rates for measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI for the seroresponse rate of
Inv_MMR_Min is ≥90% for antibodies to measles, mumps and rubella viruses.
4. To demonstrate non-inferiority of the Inv_MMR_Min vaccine compared to pooled Com_MMR vaccine in terms of seroresponse rates (see Section 10.5 for definition) for mumps virus (by Plaque Reduction Neutralization Test (PRNT)) at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group difference
(Inv_MMR_Min minus pooled Com_MMR) in seroresponse rate is ≥-10% for antibodies to mumps virus.
5. To demonstrate non-inferiority of the Inv_MMR_Min vaccine compared to pooled Com_MMR vaccine in terms of geometric mean titer (GMT) for antibodies to mumps virus (by PRNT) at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the GMT ratio (Inv_MMR_Min
over pooled Com_MMR) is ≥0.67 for antibodies to mumps virus.
Medium potency vaccine (Inv_MMR_Med) [To be statistically analyzed only if one or more of the objectives 1-5 related to the minimum potency vaccine are not met]:
6. To demonstrate non-inferiority of Inv_MMR_Med vaccine compared to pooled Com_MMR vaccine in terms of seroresponse rates to measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group difference
(Inv_MMR_Med minus pooled Com_MMR) in seroresponse rate is ≥–5% for antibodies to measles, mumps and rubella viruses.
7. To demonstrate non-inferiority of Inv_MMR_Med vaccine compared to pooled Com_MMR vaccine in terms of GMCs for antibodies to measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group ratio of GMCs
(Inv_MMR_Med over pooled Com_MMR) is ≥0.67 for antibodies to measles, mumps and rubella viruses.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 35
8. To demonstrate an acceptable immune response of Inv_MMR_Med vaccine in terms of seroresponse rates for measles, mumps and rubella viruses at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI for the seroresponse rate of
Inv_MMR_Med is ≥90% for antibodies to measles, mumps, and rubella viruses.
9. To demonstrate non-inferiority of the Inv_MMR_Med vaccine compared to pooled Com_MMR vaccine in terms of seroresponse rates for mumps virus (by PRNT) at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the group difference
(Inv_MMR_Med minus pooled Com_MMR) in seroresponse rate is ≥-10% for antibodies to mumps virus.
10. To demonstrate non-inferiority of the Inv_MMR_Med vaccine compared to pooled Com_MMR vaccine in terms of GMT for antibodies to mumps virus (by PRNT) at Day 42.
Criteria: The lower limit of the two-sided 97.5% CI on the GMT ratio (Inv_MMR_Med
over pooled Com_MMR) is ≥0.67 for antibodies to mumps virus.
Refer to Section 10.1 for the definition of the primary endpoint.
2.2. Secondary objectives
1. To assess the immunogenicity of Inv_MMR_Min followed by Inv_MMR_Releaseand pooled Com_MMR vaccine in terms of seroresponse rates and GMCs for antibodies to measles, mumps and rubella viruses at Day 84 (post Dose 2) (in a sub-cohort of children enrolled in the US).
2. To assess the immunogenicity of Inv_MMR_Med followed by Inv_MMR_Releaseand pooled Com_MMR vaccine in terms of seroresponse rate and GMCs for antibodies to measles, mumps and rubella viruses at Day 84 (post Dose 2) (in children enrolled in the US).
3. To assess the safety and reactogenicity of Inv_MMR_Min, Inv_MMR_Med, and Com_MMR when co-administered with Varivax, Havrix (to all children), and Prevnar 13 (only to children enrolled in the US).
Refer to Section 10.2 for the definition of the secondary endpoints.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 36
3. STUDY DESIGN OVERVIEW
Figure 1 Study design
Overall Randomization 2:1 (Inv_MMR:Com_MMR)
Group Randomization 2:2:1:1 (Inv_MMR_Min:Inv_MMR_Med:Com_MMR_L1:Com_MMR_L2)
Inv_MMR_Min 1500 children 12 - 15 months of age
Inv_MMR_Med 1500 children 12 - 15 months of age
Com_MMR_L1 750 children 12 - 15 months of age
Com_MMR_L2 750 children 12 - 15 months of age
Visit 1 (Day 0) Visit 2 (Day 42) Visit 3 (Day 84) Visit 4 (Day 222)
Blood sample
Vaccination 1:Inv_MMR_Min+
VV+HAV+(PCV-13*)or
Inv_MMR_Med+VV+HAV+(PCV-13*)
orCom_MMR_L1+
VV+HAV+(PCV-13*)or
Com_MMR_L2+VV+HAV+(PCV-13*)
Blood sample
Diary Card transcription
Vaccination 2:
Inv_MMR_Release
or
Com_MMR_L1
or
Com_MMR_L2
Blood sample**
Diary Card transcription
Safety Follow-up
Study conclusion
Primary Epoch
L= Lot; VV = Varicella Vaccine Varivax; HAV = Hepatitis A Vaccine Havrix; PCV-13 = Pneumococcal 13-Valent Conjugate Vaccine Prevnar 13* PCV-13 is only administered to children in the US** Blood sample collected at Day 84 is only for the children enrolled in the US (US subcohort)
Experimental design: Phase IIIA, observer-blind, randomized, controlled,multicenter, multi-country, end of shelf-life study with four parallel groups.
Duration of the study:
- Primary Epoch: The study period is approximately 7.5 months starting at Visit 1 (Day 0) and ending at Visit 4 (Day 222).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 37
Table 1 Study groups and epochs foreseen in the study
Study groups Number of children Age (Min/Max) Primary Epoch
Inv_MMR_Min 1500 12 – 15 months ●Inv_MMR_Med 1500 12 – 15 months ●Com_MMR_L1 750 12 – 15 months ●Com_MMR_L2 750 12 – 15 months ●
L = Lot
Control: active control Com_MMR_L1 and Com_MMR_L2 vaccines.
Co-administered vaccines: All children will receive Havrix and Varivax as study vaccines, concomitantly with MMR vaccine at 12 to 15 months of age. Prevnar 13 will only be administered to children recruited in the US. At the end of the study, GSK will provide a second dose of Havrix and/or varicella vaccine to selected non-US countries if local health departments do not routinely provide hepatitis A and varicella vaccination. The second dose of Havrix and varicella vaccine is not part of the study procedures. For information on administration of the 2nd dose of varicella vaccine in Malaysia, see Section 12.
Vaccination schedule: two-dose schedule
At Visit 1 children 12 to15 months of age will be administered a dose from either one of two Inv_MMR vaccine lots (Inv_MMR_Min or Inv_MMR_Med), or from one of two active control Com_MMR vaccine lots (Com_MMR_L1 and Com_MMR_L2). In addition, all children will be given two co-administered vaccines Varivax, and Havrix at Visit 1. All US children will also be given Prevnar 13.
Approximately 6 weeks later, at Visit 2, children in either the Inv_MMR_Min or Inv_MMR_Med groups will be administered a dose from a separate lot of the Inv_MMR at the release potency range(Inv_MMR_Release), for the second dose. Children who received Com_MMR_L1 or Com_MMR_L2 for the first dose will be administered a dose from one of two lots of Com_MMR vaccine for the second dose.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 38
Treatment groups:
Table 2 Treatment groups
Treatment nameVaccine / Product
name
Treatment groupsInv_MMR_Min
(active)Inv_MMR_Med
(active)Com_MMR_L1
(control)Com_MMR_L2 (control)
Inv_MMR_Min*Inv_MMR_Min xInv_MMR Diluent x
Inv_MMR_Med*Inv_MMR_Med xInv_MMR Diluent x
Inv_MMR_Release†
Inv_MMR_Release x xInv_MMR_ReleaseDiluent§
x x
Com_MMR_L1*†
Com_MMR_L1 xCom_MMR_L1
Diluent¥x
Com_MMR_L2*†
Com_MMR_L2 xCom_MMR_L2
Diluent¥x
Varivax*Varivax x x x xVarivax Diluent x x x x
Havrix* Havrix x x x xPrevnar 13*‡ Prevnar 13 x x x x
L= Lot* Administered during Visit 1 for Dose 1† Administered during Visit 2 for Dose 2§ The Inv_MMR_Release Diluent for Dose 2 is distinct from the Inv_MMR Diluent for Dose 1¥ The Com_MMR_L1 Diluent may be distinct from the Com_MMR_L2 Diluent‡ Prevnar 13 will only be given to children recruited in the US
Treatment allocation: Approximately 4500 healthy children 12 to 15 months of age will be randomized in a 2:2:1:1 ratio.
For the first dose, children will be given a dose from either one of two Inv_MMR vaccine lots designated Inv_MMR_Min or Inv_MMR_Med (1500 children each); or from one of two Com_MMR vaccine lots designated Com_MMR_L1 or Com_MMR_L2 (750 children each). For the second dose, both the Inv_MMR_Min and Inv_MMR_Med groups will be administered a separate lot of the investigational vaccine Inv_MMR_Release, which has a potency within the release potency range. Both the Com_MMR_L1 and Com_MMR_L2 groups will be administered one of two lots of Com_MMR vaccine for the second dose (refer to the Vaccination schedule section above). The treatment allocation at the investigator site will be performed using a central randomization system on the Internet (SBIR). Overall, the randomization is 2:2:1:1 (Inv_MMR_Min:Inv_MMR_Med:Com_MMR_L1:Com_MMR_L2).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 39
Blinding: observer-blind.
The study will be conducted in a double-blind fashion with regard to the two Inv_MMR vaccine lots (Inv_MMR_Min and Inv_MMR_Med) and in an observer-blind fashion for the lots of Inv_MMR vaccine versus the pooled Com_MMR vaccine lots.
Table 3 Blinding of study epochs
Study Epoch BlindingPrimary Epoch observer-blind
Visits: Children from each treatment group will participate in 4 study visits (Day 0, Day 42, Day 84 and Day 222).
Sampling:
Blood samples will be collected from each child at Day 0 and Day 42. In addition, a third blood sample will be collected from all US children at Day 84 (42 days post-dose 2).
Type of study: self-contained.
Data collection: Electronic Case Report Form (eCRF) encoded using remote data entry (RDE).
4. STUDY COHORT
4.1. Number of study children/centers
Target enrollment for the study will be 4500 children. The two combined Inv_MMRvaccine lot groups, Inv_MMR_Min and Inv_MMR_Med (N=1500 each) are twice the size of the two combined active control Com_MMR vaccine lot groups Com_MMR_L1 and Com_MMR_L2 (N=750 each).
Table 4 Sub-cohorts
Sub-cohort name Description Estimated number of childrenUS All children recruited in the US will have blood taken
and tested for antibodies to measles, mumps and rubella viruses at Day 84
1000 based on competitive enrollment
Non USAll children recruited outside the US
3500 based on competitive enrollment
Enrollment will be terminated when approximately 4500 children have been enrolled.
Refer to Section 10.3 for a detailed description of the criteria for the estimation of the sample size.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a33919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 40
4.2. Inclusion criteria for enrollment
All children must satisfy ALL the following criteria at study entry:
Male or female child between 12 and 15 months of age (e.g., from the 1 year birthday until the day before age 16 months) at the time of vaccination.
The investigator believes that the parent(s) or Legally Acceptable Representative(s) (LAR(s)) of the child, can, and will comply with the requirements of the protocol (e.g., completion of the diary cards, return for follow-up visits, respects intervals between visits).
Written informed consent obtained from the parent(s)/LAR(s) of the child.
Child is in stable health as determined by investigator’s clinical examination and assessment of child’s medical history.
For US children only:
Child that previously received a 3-dose series of Prevnar 13 with last dose at least 60 days prior to study entry.
4.3. Exclusion criteria for enrollment
The following criteria should be checked at the time of study entry. If ANYexclusion criterion applies, the child must not be included in the study:
Child in care (refer to the GLOSSARY OF TERMS for the definition of child in care).
Use of any investigational or non-registered product (drug or vaccine) other than the study vaccine(s) during the period starting 30 days before the day of study vaccination (i.e., 30 days prior to Day 0) or planned use during the entire study period.
Concurrently participating in another clinical study, in which the child has been or will be exposed to an investigational or a non-investigational product (pharmaceutical product or device).
Chronic administration (defined as 14 or more consecutive days) of immunosuppressants, or other immune-modifying drugs during the period starting 180 days prior to the first vaccine dose or any planned administration of immunosuppressive and immune-modifying drugs during the entire study.
For corticosteroids, this will mean prednisone ≥0.5 mg/kg/day or equivalent.
Inhaled and topical steroids are allowed.
Planned administration/ administration of a vaccine not foreseen by the study protocol during the period starting 30 days prior to study vaccination at Visit 1 and ending at Visit 2 (or ending at Visit 3 for the US sub-cohort). Please Note:
Inactivated influenza (Flu) vaccine and Haemophilus influenzae type b conjugate vaccine (Hib) vaccines may be given at any time, including the day of study vaccination (Flu and Hib vaccines must be administered at a different location than the study vaccine(s).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 41
Any age appropriate vaccine may be given starting at Visit 2 (or starting at Visit 3 for the US sub-cohort), and anytime thereafter.
Administration of immunoglobulins and/or any blood products during the period starting 180 days prior to study vaccination at Visit 1 or planned administration from the date of vaccination through the immunogenicity evaluation at Visit 2, or at Visit 3 for the US sub-cohort.
History of measles, mumps, rubella, varicella/zoster and/or hepatitis A diseases.
Known exposure to measles, mumps, rubella and/or varicella/zoster during the period starting 30 days prior to the first study vaccination.
Previous vaccination against measles, mumps, rubella, hepatitis A and/or varicella virus.
Any confirmed or suspected immunosuppressive or immunodeficient condition, based on medical history and physical examination (no laboratory testing required).
A family history of congenital or hereditary immunodeficiency.
History of allergic disease or reactions likely to be exacerbated by any component of the vaccines, including hypersensitivity to neomycin, latex or gelatin.
Blood dyscrasias, leukemia, lymphomas of any type, or other malignant neoplasms affecting the bone marrow or lymphatic systems.
Acute disease at the time of enrollment. (Acute disease is defined as the presence of a moderate or severe illness with or without fever). Fever is defined as temperature ≥38°C/100.4°F by any age appropriate route. All vaccines can be administered to persons with a minor illness such as diarrhea, mild upper respiratory infection without fever.
Active untreated tuberculosis based on medical history.
Any other condition which, in the opinion of the Investigator, prevents the child from participating in the study.
For US children only:
A child that previously received a fourth dose of any pneumococcal conjugate vaccine.
A list of criteria that may eliminate children from ATP analyses can be found in Sections 6.7.1, 6.8 and 10.4.
5. CONDUCT OF THE STUDY
5.1. Regulatory and ethical considerations, including the informed consent process
The study will be conducted in accordance with all applicable regulatory requirements.
The study will be conducted in accordance with the International Conference on Harmonization (ICH) Guideline for Good Clinical Practice (GCP), all applicable
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 42
participant privacy requirements and the guiding principles of the Declaration of Helsinki.
The study has been designed and will be conducted in accordance with the ICHHarmonized Tripartite Guideline for clinical investigation of medicinal products in the pediatric population (ICH E11) and all other applicable ethical guidelines.
GSK will obtain favorable opinion/approval to conduct the study from the appropriate regulatory agency, in accordance with applicable regulatory requirements, prior to a site initiating the study in that country.
Conduct of the study includes, but is not limited to, the following:
Institutional Review Board (IRB)/Independent Ethics Committee (IEC) review and favorable opinion/approval of study protocol and any subsequent amendments.
Child’s parent(s)/LAR(s) informed consent.
Investigator reporting requirements as stated in the protocol.
GSK will provide full details of the above procedures to the investigator, either verbally, in writing, or both.
Freely given and written informed consent must be obtained from each child’s parent(s)/LAR(s) as appropriate, prior to participation in the study.
GSK Biologicals will prepare a model Informed Consent Form (ICF) which will embody the ICH GCP and GSK Biologicals required elements. While it is strongly recommended that this model ICF is to be followed as closely as possible, the informed consent requirements given in this document are not intended to pre-empt any local regulations which require additional information to be disclosed for informed consent to be legally effective. Clinical judgment, local regulations and requirements should guide the final structure and content of the local version of the ICF.
The investigator has the final responsibility for the final presentation of the ICF, respecting the mandatory requirements of local regulations. The ICF generated by the investigator with the assistance of the sponsor’s representative must be acceptable to GSK Biologicals and be approved (along with the protocol, and any other necessary documentation) by the IRB/IEC.
5.2. Subject identification and randomization of treatment
The target will be to enroll 4500 children to be randomly assigned to four study groups (Inv_MMR_Min, Inv_MMR_Med, Com_MMR_L1 and Com_MMR_L2) in a 2:2:1:1 ratio. There will be approximately 1500 children in each of the Inv_MMR lot groups and approximately 750 children in each of the Com_MMR lot groups (see Figure 1).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 43
5.2.1. Subject identification
Subject numbers will be assigned sequentially to children participating in the study, according to the range of subject numbers allocated to each study center.
5.2.2. Randomization of treatment
5.2.2.1. Randomization of supplies
The randomization used to number the vaccines will be performed at GSK Biologicals, Rixensart, using MATEX, a program developed for use in SAS® (Cary, NC, USA) by GSK Biologicals.
A first list based on a randomization blocking scheme using a 2:2:1:1 randomization ratio will be used to number the MMR vaccines for Dose 1.
- Inv_MMR_Min
- Inv_MMR_Med
- Com_MMR_L1
- Com_MMR_L2
The vaccines from this list will be distributed to the study center while respecting the randomization block size.
A second list based on a randomization blocking scheme using a 4:1:1 randomizationratio will be used to number the MMR vaccines for Dose 2 (See Section 5.2.2.2. regarding treatment allocation to study children).
- Inv_MMR_Release
- Com_MMR_L1
- Com_MMR_L2
The vaccines from this list will be distributed to the study center while respecting the randomization block size.
To allow GSK Biologicals to take advantage of greater rates of recruitment than anticipated at individual centers in this multicenter study, and to thus reduce the overall study recruitment period, a surplus of supplies of at least 20% will be prepared.
5.2.2.2. Treatment allocation to the study children
The treatment allocation at the investigator site will be performed using SBIR. The treatment numbers will be allocated by dose. The randomization algorithm will be stratified by sub-cohort (US and Non US sub-cohorts) and within each sub-cohort, using a minimization procedure accounting for center.
When SBIR is not available, please refer to the SBIR user guide or the study procedures manual (SPM) for specific instructions.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 44
After having checked the eligibility of the child and obtaining the ICF, the site staff will access SBIR. Upon providing the child identification number and based on the sub-cohort, the randomization system will use the randomization algorithm to determine the group allocation and the treatment number(s) to be used for the child for MMR vaccination (Inv_MMR_Min, Inv_MMR_Med, Com_MMR_L1 or Com_MMR_L2). For the second MMR dose (Inv_MMR_Release, Com_MMR_L1 or Com_MMR_L2), the system will allocate a new treatment number according to the treatment received at Dose 1.
For allocation of co-administered vaccines (Havrix and Varivax to all children, and Prevnar 13 to the children in the US), the site staff will select a treatment number available for the vaccine dose to be used for the child. Please reference the SPM for further details.
All treatment numbers must be recorded in the eCRF on the ‘Vaccine Administration’screen.
5.3. Method of blinding
The study will be conducted in a double-blind fashion with regard to the two Inv_MMR vaccine lots (Inv_MMR_Min and Inv_MMR_Med) and in an observer-blind fashion for the lots of Inv_MMR vaccine versus the pooled Com_MMR vaccine lots.
Data will be collected in an observer-blind manner. By observer-blind, it is meant that during the course of the study, the vaccine recipient and those responsible for the evaluation of any study endpoint (e.g., safety, reactogenicity, and efficacy) will all be unaware of which vaccine was administered. To do so, vaccine preparation and administration will be done by authorized medical personnel who will not participate in any of the study clinical evaluation.
The laboratory in charge of the laboratory testing will be blinded to the treatment, and codes will be used to link the child and study (without any link to the treatment attributed to the child) to each sample.
5.4. General study aspects
Supplementary study conduct information, not mandated to be present in this protocol, is provided in the accompanying SPM. The SPM provides the investigator and the site personnel with administrative and detailed technical information that does not impact the safety of the study children.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34419-MAY-2015
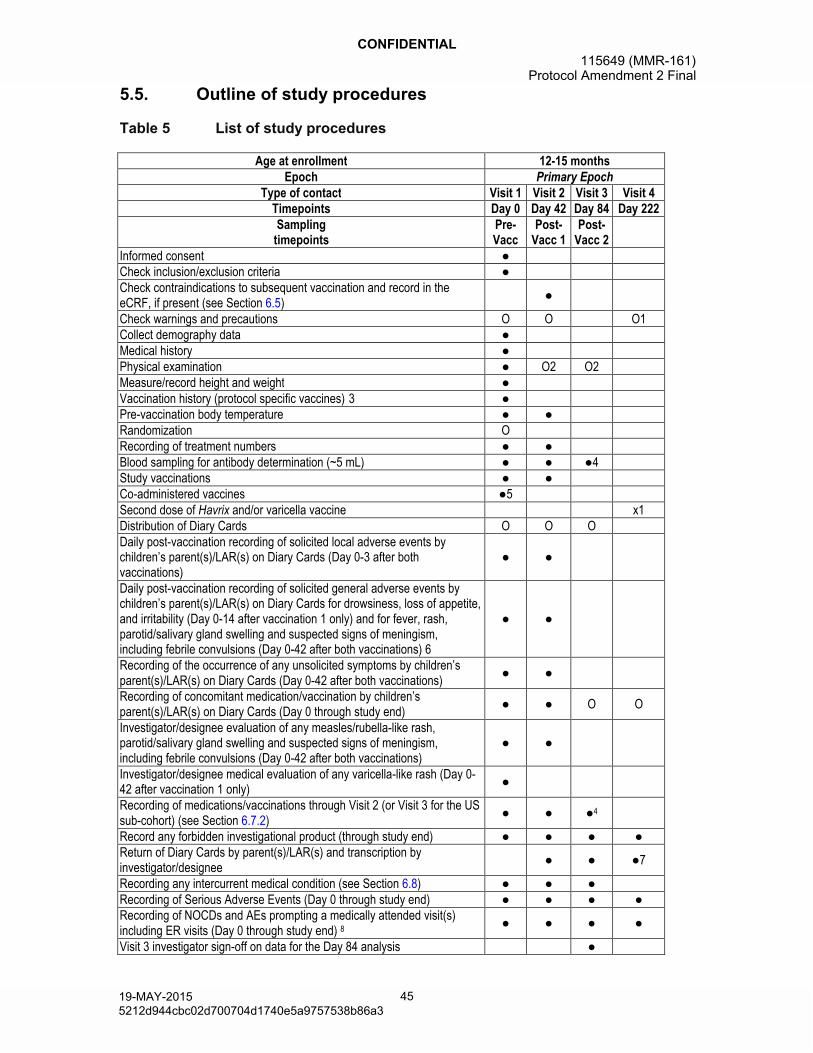
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 45
5.5. Outline of study procedures
Table 5 List of study procedures
Age at enrollment 12-15 monthsEpoch Primary Epoch
Type of contact Visit 1 Visit 2 Visit 3 Visit 4Timepoints Day 0 Day 42 Day 84 Day 222Sampling
timepointsPre-Vacc
Post-Vacc 1
Post-Vacc 2
Informed consent ●Check inclusion/exclusion criteria ●Check contraindications to subsequent vaccination and record in the eCRF, if present (see Section 6.5)
●
Check warnings and precautions O O O1Collect demography data ●Medical history ●Physical examination ● O2 O2Measure/record height and weight ●Vaccination history (protocol specific vaccines) 3 ●Pre-vaccination body temperature ● ●Randomization ORecording of treatment numbers ● ●Blood sampling for antibody determination (~5 mL) ● ● ●4Study vaccinations ● ●Co-administered vaccines ●5Second dose of Havrix and/or varicella vaccine x1Distribution of Diary Cards O O ODaily post-vaccination recording of solicited local adverse events by children’s parent(s)/LAR(s) on Diary Cards (Day 0-3 after both vaccinations)
● ●
Daily post-vaccination recording of solicited general adverse events by children’s parent(s)/LAR(s) on Diary Cards for drowsiness, loss of appetite, and irritability (Day 0-14 after vaccination 1 only) and for fever, rash, parotid/salivary gland swelling and suspected signs of meningism, including febrile convulsions (Day 0-42 after both vaccinations) 6
● ●
Recording of the occurrence of any unsolicited symptoms by children’s parent(s)/LAR(s) on Diary Cards (Day 0-42 after both vaccinations)
● ●
Recording of concomitant medication/vaccination by children’s parent(s)/LAR(s) on Diary Cards (Day 0 through study end)
● ● O O
Investigator/designee evaluation of any measles/rubella-like rash, parotid/salivary gland swelling and suspected signs of meningism, including febrile convulsions (Day 0-42 after both vaccinations)
● ●
Investigator/designee medical evaluation of any varicella-like rash (Day 0-42 after vaccination 1 only)
●
Recording of medications/vaccinations through Visit 2 (or Visit 3 for the US sub-cohort) (see Section 6.7.2)
● ● ●4
Record any forbidden investigational product (through study end) ● ● ● ●Return of Diary Cards by parent(s)/LAR(s) and transcription by investigator/designee
● ● ●7
Recording any intercurrent medical condition (see Section 6.8) ● ● ●Recording of Serious Adverse Events (Day 0 through study end) ● ● ● ●Recording of NOCDs and AEs prompting a medically attended visit(s)including ER visits (Day 0 through study end) 8 ● ● ● ●
Visit 3 investigator sign-off on data for the Day 84 analysis ●
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 46
Age at enrollment 12-15 monthsEpoch Primary Epoch
Type of contact Visit 1 Visit 2 Visit 3 Visit 4Timepoints Day 0 Day 42 Day 84 Day 222Sampling
timepointsPre-Vacc
Post-Vacc 1
Post-Vacc 2
Study Conclusion ●Final investigator signature ●
● is used to indicate a study procedure that requires documentation in the individual eCRF.x is used to indicate a procedure (that is not part of the study) that requires documentation in the individual eCRF○ is used to indicate a study procedure that does not require documentation in the individual eCRF.1 At the end of the study, GSK will provide a second dose of HAV and/or varicella vaccine in countries where they are not routinely provided. The second dose of HAV and varicella vaccine is not part of the study procedures. For information on administration of the 2nd dose of varicella vaccine in Malaysia, see Section 12.2 Physical exam at Visit 2 and 3 only required if deemed necessary based on child’s interim medical history. Findings are to be recorded only if symptom/abnormality is present.3 For US children only, child that previously received a 3-dose series of Prevnar 13 at least 60 days prior to study entry.4 Only the children enrolled in the US5 PCV-13 will only be administered to children enrolled in the US.6 Parent(s)/LAR(s) will contact the investigator immediately for any suspected varicella-like rash between Day 0 and Day 42 after vaccination 1 only; any measles/rubella-like rash, parotid/salivary gland swelling or suspected signs of meningism, including febrile convulsions occurring between Day 0 and Day 42 after each vaccination and will be asked to schedule a visit for evaluation by the investigator.7 Return of Diary Card for contraindicated medication / vaccination, any specific Adverse Events (AEs) for the 42 Days after each vaccination and medically attended events and AEs prompting Emergency Room (ER) visits until study end.8 Events to be reported will be new onset chronic disease(s) (NOCD(s)) (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies)and AEs prompting medically attended events including Emergency Room (ER) visits.
Table 6 Intervals between study visits
Visit Optimal Visit Day* Interval§ Allowed intervalVisit 1 Day 0Visit 2 Day 42 Visit 1 Visit 2 35 days – 77 days †
Visit 3 Day 84 Visit 2 Visit 3 35 days– 77 days †
Visit 4 Day 222 Visit 2 Visit 4 180 days – 210 days* Whenever possible the investigator should arrange study visits on this optimal visit day.§ Visit 1 serves as a reference date used to define the interval for Visit 2; Visit 2 serves as a reference date used to define the intervals for Visits 3 and 4.† Children will not be eligible for inclusion in the ATP immunogenicity analyses if study visits are outside the allowed interval.
5.6. Detailed description of study procedures
5.6.1. Procedures prior to study participation
5.6.1.1. Informed consent
Before performing any other study procedure, the signed informed consent of the child’sparent(s)/LAR(s) needs to be obtained.
Refer to Section 5.1 for the requirements on how to obtain informed consent.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 47
5.6.2. Procedures prior to the first vaccination
5.6.2.1. Check inclusion and exclusion criteria
Check all applicable inclusion and exclusion criteria as described in Sections 4.2 and 4.3, respectively, before enrollment.
5.6.2.2. Collect demographic data
Record demographic data such as date of birth, gender, ethnicity, geographic ancestry (race), in the child’s eCRF.
5.6.2.3. Medical history
Record any pre-existing conditions or signs and/or symptoms present in a child prior to the start of the study in the eCRF.
Prior vaccination history with respect to measles, mumps, rubella-virus containing vaccines should be obtained for all participants, as well as a history of pneumococcal conjugate containing vaccination (for children enrolled in the US only).
In addition, history of blood transfusions or human gamma globulin during the period starting 180 days prior to vaccination should also be obtained. Any chronicadministration (14 or more consecutive days) of immunosuppressants or other immune modifying drugs during the period starting 180 days prior to study entry must be recorded in the eCRF.
5.6.2.4. Physical examination
Perform a routine physical examination of the child, (as per local standard of care),including assessment of pre-vaccination body temperature, height and weight. Collected body system information, including any abnormal findings needs to be recorded in the eCRF.
Physical examination after the first visit will be performed only if the child’s parent(s)/LAR(s) indicate during questioning that there might be some underlying pathology(ies) or if deemed necessary by the Investigator or delegate. Treatment of any abnormality observed during this examination should be performed according to local medical practice outside this study or by referral to an appropriate health care provider.
5.6.3. Procedures during the study/primary epoch
Note that some of the procedures to be performed during the primary epoch (such as a physical exam as needed) are also performed prior to any subsequent vaccination(Section 5.6.2.).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 48
5.6.3.1. Check and record concomitant medication/vaccination and intercurrent medical conditions
Concomitant medication/vaccination must be recorded in the eCRF as described in Section 6.7. Refer also to Section 4.3 for details on the medication/vaccination forbidden and/or allowed during the study.
At Visit 2 for all subjects and also at Visit 3 for the US sub-cohort it must be verified if the child has experienced or is experiencing any intercurrent medical condition listed in Section 6.8. If it is the case, the condition(s) must be recorded in the eCRF. In addition, any concomitant medications/ vaccinations must be recorded in the eCRFthrough Visit 2 for the Non US sub-cohort and through Visit 3 for the US sub-cohort.Refer to Section 6.7.1.
At Visit 4 the use of any investigational or non-registered product (drug or vaccine) other than the study vaccine(s) must be recorded in the eCRF (see Section 6.7.1).
5.6.3.2. Check contraindications, warnings and precautions to vaccination
Contraindications, warnings and precautions to vaccination are to be checked at the beginning of each vaccination visit. Refer to Sections 6.5 and 6.6.
5.6.3.3. Assess pre-vaccination body temperature
The body temperature of all children needs to be measured prior to any study vaccine administration. If the child has fever on the day of vaccination (fever is defined as temperature 38°C/100.4°F by any age appropriate route [Marcy, 2004]), the vaccination visit will be rescheduled.
5.6.3.4. Randomization
At the first vaccination visit, randomization will occur as explained in Section 5.2.2.
5.6.3.5. Blood sampling for safety or immune response assessments
As specified in the List of Study Procedures in Section 5.5 (Table 5), blood samples are taken during certain study visits. Refer to the Module on Biospecimen Management in the SPM for general handling of blood samples.
A volume of approximately 5 mL of whole blood should be drawn from all childrenfor each analysis of humoral immune response at each pre-defined timepoint. After centrifugation, serum samples should be stored at or less than –20°C (–4°F).Alternatively storage at approximately –70°C to –80°C (–94°F to –112°F) until shipment is also acceptable.
5.6.3.6. Treatment number assignment
At the vaccination visit, the child will be assigned a treatment number defining the MMR treatment he/she will be receiving. The SBIR system will ensure, in a blinded manner, that the treatment assigned for Dose 2 matches the treatment assigned at Dose 1 (i.e., children who received Inv_MMR at Dose 1 will receive Inv_MMR at Dose 2 and children who received Com_MMR at Dose 1 will receive Com_MMR at Dose 2). Co-administered vaccine treatment will be assigned as described in Section
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 49
5.2.2.2. The treatment numbers must be recorded in the eCRF at each vaccination visit.
5.6.3.7. Vaccination
After completing the prerequisite procedures including blood sampling prior to MMR vaccination, one dose of study vaccine will be administered subcutaneously (SC)preferably in the triceps region of the left arm (refer to Section 6.3 for detailed description of the vaccine administration procedure) along with co-administered vaccines (at Visit 1 only). If the investigator or delegate determines that the child’s health on the day of vaccination temporarily precludes vaccination, the visit should be rescheduled.
The vaccinees will be observed closely for at least 30 minutes, with appropriate medical treatment readily available in case of anaphylaxis following the administration of vaccines.
5.6.3.8. Recording of AEs and SAEs
Refer to Section 8.2 for procedures for the investigator to record AEs and SAEs. Refer to Section 8.3 for guidelines on how to report AEs and SAEs to GSK Biologicals.
The child’s parent(s)/LAR(s) will be instructed to contact the investigator immediately should the child manifest any signs or symptoms they perceive as serious.
At Visits 1 and 2 diary cards will be provided to the child’s parent(s)/LAR(s) to record:
- Any solicited local AEs such as pain redness and swelling (from Day 0 – Day 3after each vaccination).
- Any solicited general AEs such as drowsiness, loss of appetite, and irritability (from Day 0 – Day 14 after first vaccination only).
- Any solicited general AEs such as fever, rash (measles/rubella-like rash and other rash), parotid/salivary gland swelling and suspected signs of meningism, including febrile convulsions (from Day 0 – Day 42 after each vaccination).
- Any occurrence of varicella-like rash will be reported (from Day 0 – Day 42 aftervaccination 1 only).
- Any unsolicited symptoms (from Day 0 – Day 42 after each vaccination).
- Any concomitant medications/vaccinations (from Day 0 – Day 42 after eachvaccination).
At Visit 3 diary cards will be provided to the child’s parent(s)/LAR(s) to record:
- Any medication / vaccination (from Day 84 to study end).
- Any AEs (from Day 84 to study end).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a34919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 50
The child’s parent(s)/LAR(s) will be instructed to return the completed diary card to the investigator at the next study visit.
Collection and verification of completed diary cards will occur during discussion with the child’s parent(s)/LAR(s) at post-vaccination visits. The investigator/ designee will transcribe the required collected information into the eCRF.
Collection and verification of completed diary cards will occur during discussion with the child’s parent(s)/LAR(s) at Visit 4. The investigator/ designee will transcribe the collected information on any NOCDs and AEs prompting a medically attended visit(s) including ER visits into the eCRF.
Any unreturned diary cards will be sought from the child’s parent(s)/LAR(s) through phone call(s) or any other convenient procedure.
5.6.3.9. Procedures during follow-up visits
Note that some of the procedures to be performed during the follow-up visits (such as blood sampling, recording of AEs) are also performed during the other visits of the primary epoch and are described in Section 5.6.3.1 up to Section 5.6.3.8.
5.6.3.10. Study conclusion
The investigator will review safety data collected to ensure accuracy and completeness and will complete the Study Conclusion screen in the eCRF.
A child who returns to the concluding Visit 4 (Day 222) will be considered to have completed the study.
5.7. Biological sample handling and analysis
Please refer to the SPM for details of biospecimen management (handling, storage and shipment).
Samples will not be labeled with information that directly identifies the child but will be coded with the identification number for the child (subject number).
Collected samples may be used in other assays, for test improvement or test development of analytical methods related to the study vaccine(s) and its constituents or the disease under study to allow to achieve a more reliable measurement of the vaccine response.Under these circumstances, additional testing on the samples may be performed by GSK Biologicals outside the scope of this protocol.
Information on further investigations and their rationale can be obtained from GSK Biologicals. Any sample testing will be done in line with the consent of the individual child’s parent(s)/LAR(s). Any human pharmacogenetic testing will require specific consent from the individual child’s parent(s)/LAR(s) and the ethics committee approval. Any human immunodeficiency virus (HIV) testing will also require specific consent and ethics committee approval.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 51
Refer also to the Investigator Agreement, where it is noted that the investigator cannot perform any other biological assays except those described in the protocol or its amendment(s).
If additional testing is performed, the marker priority ranking given in Section 5.7.4 may be changed.
Collected samples will be stored for up to 20 years (counting from when the last childperformed the last study visit), unless local rules, regulations or guidelines require different timeframes or different procedures, which will then be in line with the child’sconsent. These extra requirements need to be communicated formally to and discussed and agreed with GSK Biologicals.
5.7.1. Use of specified study materials
When materials are provided by GSK Biologicals, it is MANDATORY that all clinical samples (including serum samples) be collected and stored exclusively using those materials in the appropriate manner. The use of other materials could result in the exclusion of the child from the ATP analysis (See Section 10.4 for the definition of study cohorts to be evaluated). The investigator must ensure that his/her personnel and the laboratory(ies) under his/her supervision comply with this requirement. However, when GSK Biologicals does not provide material for collecting and storing clinical samples, appropriate materials from the investigator’s site must be used. Refer to the Module on Clinical Trial Supplies in the SPM.
5.7.2. Biological samples
Please refer to APPENDIX A and to Section 5.6.3.5 for a description of the assays performed in the study. Please refer to APPENDIX B for the address of the clinical laboratories used for sample analysis.
Biological samples will be collected as indicated in Table 7.
Table 7 Biological samples
Sample type Quantity Unit TimepointsBlood ~ 5 ml Visit 1Blood ~ 5 ml Visit 2Blood ~ 5 ml Visit 3*
ml = Milliliter* Only the US sub-cohort will have a blood sample taken at Visit 3 (Day 84)
5.7.3. Laboratory assays
Serological assays for the determination of antibodies against measles, mumps and rubella viruses will be performed by ELISA and/or PRNT at GSK Biologicals’ laboratory or in a validated laboratory designated by GSK Biologicals using standardized and validated procedures (refer to Table 8).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 52
Table 8 Humoral Immunity (Antibody determination) (Amended 19 May 2015)
System Component Method Kit / Manufacturer Unit Cut-off Laboratory*Serum Measles Virus Ab.IgG ELISA Dade Behring
EnzygnostmIU/mL 150 NÉOMED-
LABS Inc.Serum Rubella Virus Ab.IgG ELISA Dade Behring
EnzygnostIU/mL 4 NÉOMED-
LABS Inc.Serum Mumps Virus jeryl lynn
135 Ab.IgGELISA PPD EU/mL 5 PPD
Serum Mumps Virus Strain Mu90 Ab
PRNT In-house ED50 2.5 GSK Biologicals**
*Refer to APPENDIX B for the laboratory addresses.**GSK Biologicals laboratory refers to the Global Vaccines Clinical Laboratories (GVCL) in Rixensart, Belgium; Wavre, Belgium.Ab = Antibody; IgG = Immunoglobulin class GELISA = Enzyme-Linked Immunosorbent Assay; PRNT = Plaque Reduction Neutralization TestPPD = Pharmaceutical Product Development, Inc.IU = International Unit; mIU = Milli International Unit; EU = ELISA Unit; mL = Milliliter; ED50 = End-point Dilution 50%
Collected samples will be used for purposes related to the quality assurance of data generated within the scope of this protocol, such as for maintenance of assays described in this protocol and comparison between analytical methods and/or laboratories.
The addresses of the clinical laboratories for sample analysis in this study are:
GlaxoSmithKline BiologicalsClinical ImmunologyR & D Department/Building 7/44Rue de l'Institut, 89B-1330 Rixensart – Belgium
GSK Biologicals Global Vaccine Clinical Laboratory, Avenue Fleming, 20 –B-1300 Wavre - Belgium
NÉOMED-LABS Inc525 Cartier blvd West - Laval - Quebec - Canada - H7V 3S8(Amended 19 May 2015)
PPD, Inc.466 Devon Park DriveWayne, PA 19087-1816 – USA
The GSK Biologicals’ clinical laboratories have established a Quality System supported by procedures. The activities of GSK Biologicals’ clinical laboratories are audited regularly for quality assessment by an internal (sponsor-dependent) but laboratory-independent Quality Department.
The central laboratory responsible for sample handling/processing for sites in the United States (including Puerto Rico) is BARC USA Inc. 5, Delaware Drive, Lake Success, NY 11042-1114, U.S.A.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 53
Clinical laboratories contracted by GSK also conform to Good Laboratory Practice guidelines and operate in compliance with regulatory standards.
5.7.4. Biological samples evaluation
5.7.4.1. Immunological read-outs
Table 9 Immunological read-outs
Blood sampling timepoint
Sub-cohort NameNo. of
childrenComponent
Componentspriority rank
Type of contact and
timepoint
Sampling timepoint
Visit 1 (Day 0) Pre-VaccUS
Non US4500
Mumps Virus Strain Mu90 Ab(PRNT)*
1
Measles Virus Ab.IgG2
Rubella Virus Ab.IgGMumps Virus Jeryl Lynn
135 Ab.IgG3
Visit 2 (Day 42)
Post-Vacc 1US
Non US4500
Mumps Virus Strain Mu90 Ab (PRNT)*
1
Measles Virus Ab.IgG2
Rubella Virus Ab.IgGMumps Virus Jeryl Lynn
135 Ab.IgG3
Visit 3 (Day 84)
Post-Vacc 2 US 1000
Measles Virus Ab.IgG1
Rubella Virus Ab.IgGMumps Virus Jeryl Lynn
135 Ab.IgG2
Vacc = vaccination; Ab = Antibody; IgG = Immunoglobulin class G* The mumps Plaque Reduction Neutralization Test (PRNT)
In case of insufficient blood sample volume to perform assays for all antibodies, the samples will be analyzed according to priority ranking provided in Table 9.
All children will be tested for antibodies against measles, mumps and rubella virus by ELISA and mumps virus by PRNT with samples taken at Days 0 and 42. Only children in the US sub-cohort will be tested for antibodies against measles, mumps and rubella virus by ELISA at Day 84.
5.7.5. Immunological correlates of protection
The following antibody thresholds are accepted by the FDA as endpoints defining active immunization offering clinical benefit:
Measles ≥200 mIU/mL (ELISA)
Mumps ≥10 EU/mL (ELISA)
Rubella ≥10 IU/mL (ELISA).
For children enrolled in the US, the blood sample taken at Visit 3 will be the timepoint used to determine sub-optimal responders, i.e., an individual immunological assay result which is below the generally accepted immunological correlate of protection.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 54
A list of sub-optimal responders will be communicated to the investigators in a timely manner (no later than 12 months after the last blood sampling date of the study).
If immune response data (post dose 2 at Visit 3) indicate a suboptimal seroresponse to either the Inv_MMR or to Com_MMR vaccine, a rescue plan will be implemented and revaccination with M-M-R®II will be offered to children who fail to reach the seroresponse threshold for any MMR component.
For any child enrolled in the US who does not receive the indicated study vaccines at Visit 2, but for whom a Visit 2 blood sample is obtained; the sub-optimal responder listings will be based on the Visit 2 blood sample. If immune response data (post dose 1 for children who failed to get the second dose) indicates a suboptimal seroresponse to either the Inv_MMR or to Com_MMR vaccine, a rescue plan will be implemented and revaccination with M-M-R®II will be offered to children who fail to reach the seroresponse threshold for any MMR component.
For children enrolled outside the US, sub-optimal responder listings will not be provided based on Visit 2 laboratory data as all children should receive a second dose of study vaccine(s) at Visit 2. For any child that does not receive the indicated study vaccines at Visit 2, but for whom a Visit 2 blood sample is obtained, the sub-optimal responder listings will be based on the Visit 2 blood sample. If immune response data (post dose 1)for children who failed to get the second dose, indicates a suboptimal seroresponse to either the Inv_MMR or to Com_MMR vaccine, a rescue plan will be implemented and revaccination with Priorix will be offered to children who fail to reach the seroresponse threshold for any MMR.
6. STUDY VACCINES AND ADMINISTRATION
6.1. Description of study vaccines
The candidate live, attenuated Inv_MMR vaccine was developed and manufactured by GSK Biologicals. The Quality Control Standards and Requirements for the Inv_MMR vaccine are described in separate release protocols and the required approvals have been obtained.
The vaccines are labeled and packed according to applicable regulatory requirements.Commercial vaccines are assumed to comply with the specifications given in the manufacturer's Summary of Product Characteristics.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 55
Table 10 Study vaccines
Treatment nameVaccine/product
nameFormulation Presentation Volume
No. of doses
Inv_MMR_Min Inv_MMR_Min
Measles virus (Schwarz strain) 10 3.1 CCID50; Mumps virus (RIT4385 strain) 10 4.1
CCID50; Rubella virus (Wistar RA 27/3 strain) 10 2.9
CCID50; anhydrous lactose; sorbitol; mannitol; amino
acids; neomycin
Lyophilized pellet in a vial
for reconstitution with water for
injection
0.5 ml † 1*
Inv_MMR Diluent water for injection Vial 0.5 ml 1*
Inv_MMR_Med Inv_MMR_Med
Measles virus (Schwarz strain) 10 3.4 CCID50; Mumps virus (RIT4385 strain) 10 4.3
CCID50; Rubella virus (Wistar RA 27/3 strain) 10 3.2
CCID50; anhydrous lactose; sorbitol; mannitol; amino
acids; neomycin
Lyophilized pellet in a vial
for reconstitution with water for
injection
0.5 ml † 1*
Inv_MMR Diluent water for injection Vial 0.5 ml 1*
Inv_MMR_Release Inv_MMR_Release
Measles virus (Schwarz strain) ≥10 3.0 CCID50;
Mumps virus (RIT4385 strain) ≥10 4.3 CCID50; Rubella virus (Wistar RA 27/3 strain) ≥10 3.0
CCID50; anhydrous lactose; sorbitol; mannitol; amino
acids; neomycin
Lyophilized pellet in a vial
for reconstitution with water for
injection
0.5 ml † 1**
Inv_MMR_ReleaseDiluent***
water for injection Vial 0.5 ml 1**
Com_MMR_L1 Com_MMR_L1
Measles virus ≥1,000 TCID50; Mumps virus ≥12,500 TCID50; Rubella virus ≥1,000 TCID50;
sorbitol 14.5 mg; sodium phosphate, sucrose 1.9 mg; sodium chloride; hydrolyzed gelatin 14.5 mg; recombinant human albumin ≤0.3 mg; fetal bovine serum <1 ppm; other buffer and media ingredients;
neomycin approximately 25 mcg
Lyophilized pellet in a vial
for reconstitution with water for
injection
0.5 ml † 2
Com_MMR_L1 Diluent‡
water for injection
Vial or a Syringe
(depending on the country)
0.5 ml 2
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 56
Treatment nameVaccine/product
nameFormulation Presentation Volume
No. of doses
Com_MMR_L2 Com_MMR_L2
Measles virus ≥1,000 TCID50; Mumps virus ≥12,500 TCID50; Rubella virus ≥1,000 TCID50;
sorbitol 14.5 mg; sodium phosphate, sucrose 1.9 mg; sodium chloride; hydrolyzed gelatin 14.5 mg; recombinant human albumin ≤0.3 mg; fetal bovine serum <1 ppm; other buffer and media ingredients;
neomycin approximately 25 mcg
Lyophilized pellet in a vial
for reconstitution with water for
injection
0.5 ml † 2
Com_MMR_L2 Diluent‡
water for injection
Vial or a Syringe
(depending on the country)
0.5 ml 2
Varivax Varivax
A minimum of 1350 pfu of Oka/Merck varicella virus;
sucrose approximately 25 mg; hydrolyzed gelatin 12.5 mg,
sodium chloride 3.2 mg; monosodium L-glutamate 0.5
mg; sodium phosphate dibasic 0.45 mg; potassium phosphate monobasic 0.08 mg; potassium chloride 0.08 mg; residual components of MRC-5 cells including DNA
and protein; and trace quantities of sodium
phosphate monobasic; EDTA; neomycin; and fetal bovine
serum
Lyophilized pellet in a vial
for reconstitution with water for
injection
0.5 ml † 2§
Varivax Diluent water for injectionVial or Syringe (depending on
the country)0.5 ml 2§
Havrix Havrix
720 EU of hepatitis A virus antigen 0.25 mg aluminum (as
hydroxide) Amino acid supplement (0.3% w/v) in a phosphate-buffered saline
solution and polysorbate 20 (0.05 mg/mL)
Suspension in vial/ pre-filled
syringe0.5 ml 2§
Prevnar 13 Prevnar 13
2.2 mcg of the purified saccharides1, 3, 4, 5, 6A, 7F, 9V, 14, 18C, 19A, 19F, 23F
4.4 mcg of the purified saccharide 6B approximately 34 mcg CRM197 carrier protein
100 μg polysorbate 80, 295 μg succinate buffer and 125 μg aluminum as aluminum
phosphate adjuvant
Suspension in vial/ pre-filled
syringe0.5 ml 1¥
CCID = Cell Culture Infectious Dose; TCID = Tissue Culture Infectious Dose; EU = ELISA Unitppm = Parts Per Million; pfu = Plaque Forming Unit; mL = Milliliter; mg = Milligram; mcg = Microgram; g = Microgram
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 57
MRC = Medical Research Council; DNA = Deoxyribonucleic Acid; EDTA = Ethylenediamine-Tetraacetic AcidCRM197 = Cross-Reactive Material (mutant diphtheria toxin)† Volume after reconstitution* Administered during Visit 1 for Dose 1** Administered during Visit 2 for Dose 2 for the Inv_MMR_Min and Inv_MMR_Med groups ONLY*** The Inv_MMR_Release Diluent for Dose 2 is distinct from the Inv_MMR Diluent for Dose 1‡ The Com_MMR_L1 Diluent may be distinct from the Com_MMR_L2 Diluent§ At the end of the study, GSK will provide a second dose of HAV and/or varicella vaccine in countries where they are not routinely provided. Not applicable for subjects enrolled in US. For information on administration of the 2nd dose of varicella vaccine in Malaysia, see Section 12. The second dose of HAV and varicella vaccine is not part of the study procedures.¥ PCV-13 will only be administered to children in the US.
6.2. Storage and handling of study vaccines
All study vaccines to be administered to the children must be stored in a safe and locked place with no access by unauthorized personnel.
The study vaccines will be stored at the defined temperature ranges:
Inv_MMR_Min and Inv_MMR_Med lots will be stored from (-15°C to -25°C (5F to -13°F)).
Diluents for Inv_MMR_Min and Inv_MMR_Med will be stored at 2°C to 8°C (36Fto 46F).
Inv_MMR_Release, Com_MMR_L1 and Com_MMR_L2 lots and all the respective diluents will be stored at 2°C to 8°C (36F to 46F).
Outside the US Havrix, Varivax and Varivax diluent will be stored at 2°C to 8°C(36F to 46F).
In the US Varivax will be stored at -15°C to -50°C (5F to -58°F). The Havrix, Prevnar 13 and Varivax diluent will be stored at 2°C to 8°C (36F to 46F).
(Note: Refrigerated Varivax vaccine will be used at non-US sites, while Frozen Varivaxvaccine will be used at US sites).
Please refer to the Module on Clinical Trial Supplies in the SPM for more details on storage of the study vaccines. The storage temperature of the vaccines will be monitored daily with validated temperature monitoring device(s) and will be recorded as specified in the SPM.
The storage conditions will be assessed during pre-study activities under the responsibility of the sponsor study contact.
Any temperature excursion, outside the range of 0°C to +8°C (32F to 46F) for refrigerated vaccines, outside the range of -15°C to -25C (5°F to -13F) for Inv_MMR_Min and Inv_MMR_Med vaccines, or -15°C to -50°C (5F to -58F) for the Varivax vaccine (in the US), must be reported to the sponsor as soon as detected. Following an exposure to a temperature excursion, vaccines will not be used until written approval has been given by the Sponsor.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 58
Outside the US, adequate actions must be taken for refrigerated vaccines in case of temperature excursion between 0°C and +2°C (32°F and 36°F) which go back to the defined range +2°C to +8°C (36°C to 46°F). The impacted study vaccine(s)/product(s) can still be administered, but the site should avoid re-occurrence of temperature excursion.
Please refer to the Module on Clinical Trial Supplies in the SPM for details and instructions on the Temperature excursion process as well as packaging and accountability of the study vaccines.
6.3. Dosage and administration of study vaccines
Table 11 Dosage and administration
Type of contact and timepoint
Dose Treatment Group Vaccine/Product Route 1 Site Side 2
Visit 1 (Day 0) 1 Inv_MMR_MinInv_MMR_Min
SC Tricep region LeftInv_MMR_Diluent
Visit 1 (Day 0) 1 Inv_MMR_MedInv_MMR_Med
SC Tricep region LeftInv_MMR_Diluent
Visit 1 (Day 0) 1 Com_MMR_L1Com_MMR_L1
SC Tricep region LeftCom_MMR_L1-Diluent
Visit 1 (Day 0) 1 Com_MMR_L2Com_MMR_L2
SC Tricep region LeftCom_MMR_L2-Diluent
Visit 1 (Day 0) 1
Inv_MMR_MinInv_MMR_MedCom_MMR_L1Com_MMR_L2
VarivaxSC Tricep region Right
Varivax-Diluent
Visit 1 (Day 0) 1
Inv_MMR_MinInv_MMR_MedCom_MMR_L1Com_MMR_L2
Havrix IMAnterolateral
thighRight
Visit 1 (Day 0) 1
Inv_MMR_MinInv_MMR_MedCom_MMR_L1Com_MMR_L2
Prevnar 13* IMAnterolateral
thighLeft
Visit 2 (Day 42) 1Inv_MMR_MinInv_MMR_Med
Inv_MMR_ReleaseSC Tricep Region LeftInv_MMR_Release
-Diluent
Visit 2 (Day 42) 1 Com_MMR_L1Com_MMR_L1
SC Tricep Region LeftCom_MMR_L1-Diluent
Visit 2 (Day 42) 1 Com_MMR_L2Com_MMR_L2
SC Tricep Region LeftCom_MMR_L2-Diluent
1 Intramuscular (IM)/ Subcutaneous (SC)2 Left (L)/ Right (R)* PCV-13 will only be administered to children in the US.
Inv_MMR, Com_MMR and Varivax should be given subcutaneously in the upper arm (“Triceps” region) by inserting the needle in a pinched-up fold of skin and subcutaneous tissue (to prevent injection into muscle). The Inv_MMR and Com_MMR vaccines will be
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 59
injected in the left arm and the Varivax will be injected in the right arm. A 25-gauge needle, 5/8 inch (1.6 cm) long, is recommended. The needle should be changed between drawing the vaccine into the syringe and injecting it into the child.
The dose of Inv_MMR, Com_MMR for any age is 0.5 mL administered subcutaneously. These vaccines should be reconstituted by first withdrawing the entire volume of diluent to be used for reconstitution into the syringe. Note that some diluents may already be provided in pre-filled syringes. The Com_MMR_L1 diluent may be distinct from the Com_MMR_L2 diluent and for countries outside the US, Com_MMR diluents will be pre-packaged in a syringe. Inject all of the diluent in the syringe into the vial of lyophilized vaccine and gently agitate to mix thoroughly. The vial must be shaken well until the pellet is completely dissolved. If the lyophilized vaccine cannot be dissolved, discard. Withdraw the entire contents of the reconstituted vaccine into the syringe and change the needle. Using a new needle, inject the total volume of reconstituted vaccine subcutaneously.
After reconstitution, the MMR vaccines should be injected promptly (within 30 minutes).
Due to potential color differences between the reconstituted Inv_MMR and Com_MMR, dedicated unblinded staff in each investigational site will be accountable for reconstitution and administration to each child.
Varivax should be reconstituted by first withdrawing 0.7 mL of diluent into a syringe. Inject all the diluent in the syringe into the vial of lyophilized vaccine and gently agitate to mix thoroughly. Withdraw the entire contents into a syringe and inject the total volume (about 0.5 mL) of reconstituted vaccine subcutaneously.
Havrix and Prevnar 13 will be administered by intramuscular injection in the right and left thigh, respectively. The dosage for these co-administered vaccines is 0.5 mL.
Attenuated viruses in these vaccines are rapidly inactivated by alcohol and detergents, and care should be taken to avoid contact between the reconstituted vaccine and these substances. Ensure the disinfected area of skin and/or vaccine vial(s) are completely dry prior to administering any vaccines.
All vaccine recipients will be observed closely for at least 30 minutes following the administration of vaccines with appropriate medical treatment readily available in case of a rare anaphylactic reaction.
6.4. Replacement of unusable vaccine doses
Additional vaccine doses will be provided to replace those that are unusable (see the Module on Clinical Trial Supplies in the SPM for details).
In addition to the vaccine doses provided for the planned number of children (including at least 20% over-supply when applicable), additional doses will be supplied to replace those that are unusable.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a35919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 60
The investigator or designee will use SBIR to obtain a new treatment number (for Com_MMR and Inv_MMR only). The new treatment numbers will be allocated by doseto replace an unusable dose. The system will ensure, in a blinded manner, that the new vial matches the formulation the child was assigned to by randomization.
6.5. Contraindications to subsequent vaccination
The following events constitute absolute contraindications to further administration of the study vaccine (Inv_MMR and Com_MMR). If any of these events occur during the study, the child must not receive additional doses of vaccine but may continue other study procedures at the discretion of the investigator (see Section 8.4).
Anaphylaxis following the administration of vaccines.
Any confirmed or suspected immunosuppressive or immunodeficient condition, based on medical history and physical examination (no laboratory testing required).
Discovery of any health condition which, in the investigator’s opinion, places the child at increased risk from receipt of further study vaccine(s); or discovery of a change in the child’s status which renders him/her unable to comply with protocol-mandated safety follow-up.
The following events constitute contraindications to administration of study vaccine (Inv_MMR and Com_MMR) at that point in time; if any of these events occur at the time scheduled for vaccination, the child may be vaccinated at a later date, within the time window specified in the protocol.
Acute disease at the time of vaccination (defined as the presence of a moderate or severe illness with or without fever. All vaccines can be administered to persons with a minor illness such as diarrhea, mild upper respiratory infection without fever).
Fever i.e., temperature ≥38°C/100.4°F by any age appropriate route at the time of vaccination.
Children should not receive a second dose of study vaccine if any of the following events occur:
Receipt of a second dose of MMR prior to the second scheduled study dose (i.e., a third dose of MMR should not be given).
Receipt of immunoglobulin at any time during the study, as this may impact the immune response to study vaccines. In this case, the second dose should be deferred until at least three months after administration of immunoglobulin, outside the context of the current protocol.
Individuals who experience thrombocytopenia with the first dose of MMR vaccines may develop thrombocytopenia with repeat doses. For children who experience thrombocytopenia following the first dose of MMR vaccine, the risk-benefit of vaccination with a second dose of MMR vaccine should be considered.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 61
6.6. Warnings and precautions
Refer to the approved product label/package insert for licensed vaccine(s).
It is recommended that members of the investigational team be immune to measles, mumps, rubella and varicella viruses. The measles and mumps components of investigational and commercial vaccines are produced in chick embryo cell culture and may therefore contain traces of egg protein. Persons with a history of anaphylactic, anaphylactoid, or other immediate reactions (e.g., generalized urticaria, swelling of the mouth and throat, difficulty breathing, hypotension, or shock) subsequent to egg ingestion may be at an enhanced risk of immediate-type hypersensitivity reactions after vaccination, although these types of reactions have been shown to be very rare. Individuals who have experienced anaphylaxis after egg ingestion should be vaccinated with extreme caution, with adequate treatment for anaphylaxis on hand should such a reaction occur.
If tuberculin testing has to be done it should be carried out before or simultaneously with vaccination since it has been reported that combined measles, mumps and rubella vaccines may cause a temporary depression of tuberculin skin sensitivity. As this anergy may last up to a maximum of six weeks, tuberculin testing should not be performed within that period after vaccination to avoid false negative results.
Individuals with current thrombocytopenia may develop more severe thrombocytopenia following vaccination with MMR vaccines. In addition, individuals who experienced thrombocytopenia with the first dose MMR may develop thrombocytopenia with repeat doses. The potential risk to benefit ratio should be carefully evaluated before considering vaccination in such cases.
Post-marketing experience with Varivax suggests that transmission of vaccine virus may occur rarely between healthy vaccinees that develop a varicella-like rash and healthy susceptible contacts. Transmission of vaccine virus from a mother who did not develop a varicella-like rash to her newborn infant has also been reported. Information will be given to the parent(s)/LAR(s) in the event that during the study period a household member becomes a susceptible high-risk individual. Susceptible high-risk individuals should avoid close association with vaccine recipients, and especially those vaccine recipients who develop a vaccine-associated rash, for up to six weeks. In circumstances where contact with high-risk individuals is unavoidable, the potential risk of transmission of vaccine virus should be weighed against the risk of acquiring and transmitting natural varicella virus. Susceptible high-risk individuals include:
Residence in the same household as the following:
- Newborn infants (0-4 weeks of age).
- Pregnant women with a negative history of chickenpox disease and without recorded vaccination against chickenpox.
- Pregnant women at or beyond 28 weeks gestation regardless of varicella vaccination status or varicella disease history.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 62
- Persons with known immunodeficiency.
The use of salicylates by vaccine recipients should be avoided for six weeks after vaccination with Varivax as Reye’s Syndrome has been reported following the use of salicylates during natural varicella infection.
6.7. Concomitant medication/vaccination
At each study visit, the investigator should question the child’s parent(s)/LAR(s) about any medication taken and vaccination received by the child.
All concomitant medication/vaccination, with the exception of vitamins and/or dietary supplements, are to be recorded in the eCRF (in the window defined by the protocol (see Section 6.7.2)). This also applies to concomitant medication administered prophylactically in anticipation of reaction to the vaccination and any medication intended to treat an AE.
A prophylactic medication is a medication administered in the absence of ANY symptom and in anticipation of a reaction to the vaccination (e.g., an anti-pyretic is considered to be prophylactic when it is given in the absence of fever and any other symptom, to prevent fever from occurring [fever is defined as temperature 38.0°C/100.4°F]).
Similarly, concomitant medication administered for the treatment of a SAE, at any time, must be recorded on the SAE Report / SAE screens in the eCRF, as applicable. Refer to Section 8.1.2 for the definition of a SAE.
6.7.1. Medications/products that may lead to the elimination of a childfrom ATP analyses
The following criteria should be checked at each visit. If any become applicable during the study, it will not require withdrawal of the child from the study but may determine a child’s evaluability in the according-to-protocol (ATP) analysis. See Section 10.4 for definition of study cohorts to be evaluated.
Use of any investigational or non-registered product (drug or vaccine) other than the study vaccine(s) during the entire study period through study end.
Chronic administration (defined as 14 or more consecutive days) of immunosuppressants or other immune-modifying drugs during the period starting at vaccination and ending at the immunogenicity evaluation at Visit 2 (or at Visit 3 for the US sub-cohort).
- For corticosteroids, this will mean prednisone 0.5 mg/kg/day, or equivalent.
- Inhaled and topical steroids are allowed.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 63
Planned administration/ administration of a vaccine not foreseen by the study protocol during the period starting at vaccination and ending at Visit 2 (or ending at Visit 3 for the US sub-cohort). Please Note:
- Inactivated Flu and Hib vaccines may be given at any time , including the day of study vaccination (Flu and Hib vaccines must be administered at a different location than the study vaccine/s).
- Any age appropriate vaccine may be given starting at Visit 2, (or starting at Visit 3 for the US sub-cohort) and any time thereafter.
Administration of immunoglobulins and/or any blood products during the period starting at vaccination and ending at the last blood draw at Visit 2 (or at Visit 3 for the US sub-cohort).
6.7.2. Time window for recording concomitant medication/vaccinationin the eCRF
At each study visit, the investigator should question the child’s parent(s)/LAR(s) about any medication taken and vaccination(s) received by the child.
All concomitant medications, with the exception of vitamins and/or dietary supplements, administered at ANY time during the period starting 30 days preceding the administration of the first dose of study vaccine (i.e., 30 days prior to Day 0) and ending at Visit 2 (Non US sub-cohort) or Visit 3 (US sub-cohort) must be recorded in the eCRF. Concomitant medication/vaccination should be recorded by the child’s parent(s)/LAR(s) in the subject diary between Day 0 and Day 42 after each vaccination. The investigator will transcribe this information into the eCRF as indicated below.
Any medications specifically contraindicated, e.g., any immune-globulins, other blood products and any immune modifying drugs administered during the period starting 180 days prior to the vaccination to Visit 2 (Non US sub-cohort) or Visit 3 (US sub-cohort) are to be recorded in the eCRF with the generic name of the medication (trade names are allowed for combination drugs only), medical indication, total daily dose, route of administration, start and end dates of treatment.
Any vaccine administered in the period starting 30 days before study vaccination through Visit 2 (or Visit 3 for the US sub-cohort), must be recorded in the eCRF. The second dose of HAV and/or varicella vaccine administered at Visit 4 must be recorded in the eCRF (see Section 5.5). In addition, any previous administration of measles, mumps, and rubella virus containing vaccines, regardless of when they were given, must be recorded in the eCRF.
Any medication administered for the treatment of an AE should be recorded in the child’s eCRF.
Any investigational medication or vaccine administered throughout the entire study must be recorded in the eCRF.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 64
6.8. Intercurrent medical conditions that may lead to eliminationfrom an ATP cohort
Children may be eliminated from the ATP cohort if they are confirmed to have an immunodeficiency condition, or if they develop measles, mumps, or rubella, in the interval between vaccination and the collection of the blood specimen for immunogenicity at the last blood draw at Visit 2 (or at Visit 3 for the US sub-cohort).
7. HEALTH ECONOMICS
Not applicable
8. ADVERSE EVENTS AND SERIOUS ADVERSE EVENTS
The investigator or site staff is responsible during the study for the detection and documentation of events meeting the criteria and definition of an adverse event (AE) or serious adverse event (SAE) as provided in this protocol.
Each child’s parent(s)/LAR(s) will be instructed to contact the investigator immediately should their child manifest any signs or symptoms they perceive as serious.
8.1. Safety definitions
8.1.1. Definition of an adverse event
An AE is any untoward medical occurrence in a clinical investigation subject, temporally associated with the use of a medicinal product, whether or not considered related to the medicinal product.
An AE can therefore be any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease (new or exacerbated) temporally associated with the use of a medicinal product. For marketed medicinal products, this also includes failure to produce expected benefits (i.e., lack of efficacy), abuse or misuse.
Examples of an AE include:
Significant or unexpected worsening or exacerbation of the condition/indication under study.
Exacerbation of a chronic or intermittent pre-existing condition including either anincrease in frequency and/or intensity of the condition.
New conditions detected or diagnosed after investigational product administration even though they may have been present prior to the start of the study.
Signs, symptoms, or the clinical sequelae of a suspected interaction.
Signs, symptoms, or the clinical sequelae of a suspected overdose of either investigational product or a concurrent medication (overdose per se should not be reported as an AE/SAE).
Signs, symptoms temporally associated with vaccine administration.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 65
Significant failure of expected pharmacological or biological action.
Examples of an AE DO NOT include:
Medical or surgical procedures (e.g., endoscopy, appendectomy); the condition that leads to the procedure is an AE.
Situations where an untoward medical occurrence did not occur (e.g., convenience admission to a hospital, admission for routine examination).
Anticipated day-to-day fluctuations of pre-existing disease(s) or condition(s) present or detected at the start of the study that do not worsen.
AEs may include pre- or post-treatment events that occur as a result of protocol-mandated procedures (i.e., invasive procedures, modification of child’s previous therapeutic regimen).
NB: AEs to be recorded as endpoints (solicited AEs) are described in Section 8.1.3. All other AEs will be recorded as UNSOLICITED AEs.
Example of events to be recorded in the medical history section of the eCRF:
Pre-existing conditions or signs and/or symptoms present in a child prior to the start of the study (i.e., prior to the first study vaccination).
8.1.2. Definition of a serious adverse event
A serious adverse event is any untoward medical occurrence that:
a. Results in death,
b. Is life-threatening,
NB: The term ‘life-threatening’ in the definition of ‘serious’ refers to an event in which the child was at risk of death at the time of the event. It does not refer to an event, which hypothetically might have caused death, had it been more severe.
c. Requires hospitalization or prolongation of existing hospitalization,
NB: In general, hospitalization signifies that the child has been admitted at the hospital or emergency ward for observation and/or treatment that would not have been appropriate in the physician’s office or in an out-patient setting. Complications that occur during hospitalization are also considered AEs. If a complication prolongs hospitalization or fulfils any other serious criteria, the event will also be considered serious. When in doubt as to whether ‘hospitalization’ occurred or was necessary, the AE should be considered serious.
Hospitalization for elective treatment of a pre-existing condition (known or diagnosed prior to informed consent signature) that did not worsen from baseline is NOT considered an AE.
d. Results in disability/incapacity.
NB: The term disability means a substantial disruption of a person’s ability to conduct normal life functions. This definition is not intended to include experiences
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 66
of relatively minor medical significance such as uncomplicated headache, nausea, vomiting, diarrhea, influenza like illness, and accidental trauma (e.g., sprained ankle) which may interfere or prevent everyday life functions but do not constitute a substantial disruption.
Medical or scientific judgment should be exercised in deciding whether reporting is appropriate in other situations, such as important medical events that may not be immediately life-threatening or result in death or hospitalization but may jeopardize the child or may require medical or surgical intervention to prevent one of the other outcomes listed in the above definition. These should also be considered serious. Examples of such events are invasive or malignant cancers, intensive treatment in an emergency room or at home for allergic bronchospasm, blood dyscrasias or convulsions that do not result in hospitalization.
8.1.3. Solicited adverse events
The following local (injection-site) adverse events will be solicited at the Inv_MMR and Com_MMR injection sites only (Table 12):
Table 12 Solicited local adverse events at Inv_MMR and Com_MMR injection sites
Adverse event Solicited follow-up periodPain at injection site from Day 0 to Day 3 after each vaccinationRedness at injection site from Day 0 to Day 3 after each vaccinationSwelling at injection site from Day 0 to Day 3 after each vaccination
These local injection site reactions will be recorded from Day 0 to Day 3 on the Diary Cards following each vaccination in both groups. Redness and swelling will be measured in millimeters and the greatest surface diameter will be recorded. Pain at injection site will be assessed.
The following solicited general adverse events are shown in Table 13:
Table 13 Solicited general adverse events
Adverse Event Solicited follow-up periodDrowsiness
from Day 0 to Day 14 after dose 1Loss of appetiteIrritabilityVaricella-like rash* from Day 0 to Day 42 after dose 1
Fever (defined as temperature ≥38C/100.4F)**
from Day 0 to Day 42 after each vaccinationMeasles/rubella-like rash*Other Rash (not measles/rubella-like nor varicella-like)Parotid gland / Salivary gland swelling*Meningism including febrile convulsions*
* To be confirmed by a physician.** Fever is defined as temperature ≥38C/100.4F [Marcy, 2004]. The preferred route for recording temperature in this study will be axillary. Temperature will be recorded daily from Day 0 to Day 42 after each vaccination. The child’s parent(s)/LAR(s) are requested to record the child’s body temperature each evening at bedtime. Should additional temperature measurements be performed at other times of day, the highest temperature will be recorded.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 67
NB: Temperature will be recorded in the evening. Should additional temperature measurements be performed at other times of day, the highest temperature will be recorded in the eCRF.
8.1.4. Clinical laboratory parameters and other abnormal assessments qualifying as adverse events or serious adverse events
Abnormal laboratory findings (e.g., clinical chemistry, hematology, urinalysis) or other abnormal assessments that are judged by the investigator to be clinically significant will be recorded as AEs or SAEs if they meet the definition of an AE, as defined in Section 8.1.1 or of a SAE, as defined in Section 8.1.2. Clinically significant abnormal laboratory findings or other abnormal assessments that are detected during the study or are present at baseline and significantly worsen following the start of the study will be reported as AEs or SAEs.
The investigator will exercise his or her medical and scientific judgment in deciding whether an abnormal laboratory finding or other abnormal assessment is clinically significant.
8.1.5. Adverse events of specific interest
AEs of specific interest for safety monitoring include NOCDs (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies) and AEs prompting ER visits. Occurrences of AEs of specific interest, including NOCDs and AEs prompting ER visits,will be reported throughout the study, through a minimum of six months after the last dose of MMR vaccine, whether or not they are considered to be possibly related to the treatment administration. Medical documentation of the events will be reported in appropriate targeted follow-up forms included in the eCRF. These events also have to be reported as AE or SAE as appropriate in the eCRF.
8.2. Detecting and recording adverse events and serious adverse events
8.2.1. Time period for detecting and recording adverse events andserious adverse events
All AEs starting from Day 0 through 42 days after the administration of each dose of study vaccine must be recorded on the Adverse Event screen in the child’s eCRF, irrespective of intensity or whether or not they are considered vaccination-related. The standard time period for collecting and recording SAEs will begin at the first receipt of study vaccines and will be collected throughout the entire study. See Section 8.3 for instructions on reporting of SAEs.
SAEs that are related to the study vaccine(s) will be collected and recorded from the time of the first study vaccination until the child is discharged.
In addition to the above-mentioned reporting requirements and in order to fulfillinternational reporting obligations, SAEs that are related to study participation (i.e.,
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36719-MAY-2015
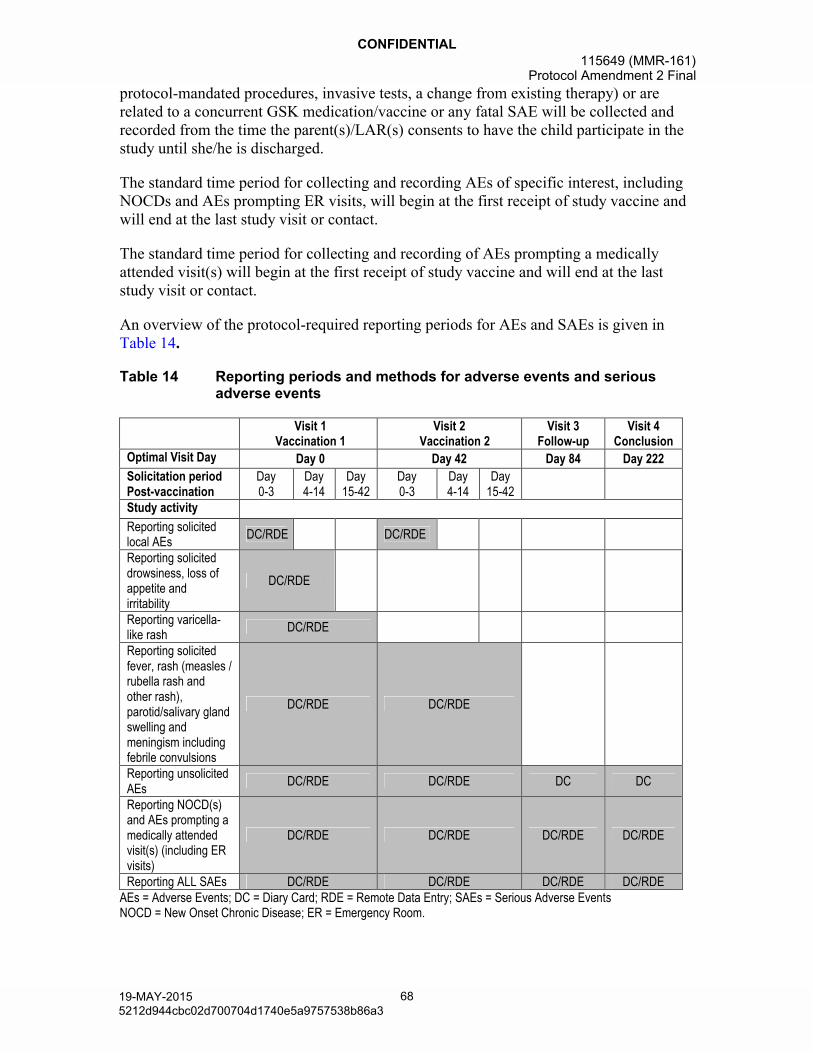
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 68
protocol-mandated procedures, invasive tests, a change from existing therapy) or are related to a concurrent GSK medication/vaccine or any fatal SAE will be collected and recorded from the time the parent(s)/LAR(s) consents to have the child participate in the study until she/he is discharged.
The standard time period for collecting and recording AEs of specific interest, including NOCDs and AEs prompting ER visits, will begin at the first receipt of study vaccine and will end at the last study visit or contact.
The standard time period for collecting and recording of AEs prompting a medically attended visit(s) will begin at the first receipt of study vaccine and will end at the last study visit or contact.
An overview of the protocol-required reporting periods for AEs and SAEs is given in Table 14.
Table 14 Reporting periods and methods for adverse events and serious adverse events
Visit 1Vaccination 1
Visit 2Vaccination 2
Visit 3Follow-up
Visit 4Conclusion
Optimal Visit Day Day 0 Day 42 Day 84 Day 222
Solicitation periodPost-vaccination
Day0-3
Day 4-14
Day15-42
Day0-3
Day 4-14
Day15-42
Study activity
Reporting solicited local AEs
DC/RDE DC/RDE
Reporting solicited drowsiness, loss of appetite andirritability
DC/RDE
Reporting varicella-like rash
DC/RDE
Reporting solicited fever, rash (measles /rubella rash and other rash), parotid/salivary gland swelling and meningism including febrile convulsions
DC/RDE DC/RDE
Reporting unsolicited AEs
DC/RDE DC/RDE DC DC
Reporting NOCD(s) and AEs prompting a medically attended visit(s) (including ER visits)
DC/RDE DC/RDE DC/RDE DC/RDE
Reporting ALL SAEs DC/RDE DC/RDE DC/RDE DC/RDEAEs = Adverse Events; DC = Diary Card; RDE = Remote Data Entry; SAEs = Serious Adverse EventsNOCD = New Onset Chronic Disease; ER = Emergency Room.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 69
A post-study AE/SAE is defined as any event that occurs outside of the AE/SAE reporting period defined in .
Table 14. Investigators are not obligated to actively seek AEs or SAEs in former study participants. However, if the investigator learns of any SAE at any time after a child has been discharged from the study, and he/she considers the event reasonably related to the investigational product, the investigator will promptly notify the Study Contact for Reporting SAEs.
8.2.2. Evaluation of adverse events and serious adverse events
8.2.2.1. Active questioning to detect adverse events and serious adverse events
As a consistent method of soliciting AEs, the child’s parent(s)/LAR(s) should be asked a non-leading question such as:
‘Has your child acted differently or felt different in any way since receiving the vaccine or since the last visit?’
When an AE/SAE occurs, it is the responsibility of the investigator to review all documentation (e.g., hospital progress notes, laboratory, and diagnostics reports) relative to the event. The investigator will then record all relevant information regarding an AE/SAE on the eCRF or SAE Report screens as applicable. It is not acceptable for the investigator to send photocopies of the child’s medical records to GSK Biologicals instead of the appropriate completed AE/SAE screens in the eCRF. However, there may be instances when copies of medical records for certain cases are requested by GSK Biologicals. In this instance, all participant identifiers will be blinded on the copies of the medical records prior to submission to GSK Biologicals.
The investigator will attempt to establish a diagnosis pertaining to the event based on signs, symptoms, and/or other clinical information. In such cases, the diagnosis should be documented as the AE/SAE and not the individual signs/symptoms.
8.2.2.2. Assessment of adverse events
8.2.2.2.1. Assessment of intensity
Intensity of the following AEs will be assessed as described in Table 15.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a36919-MAY-2015
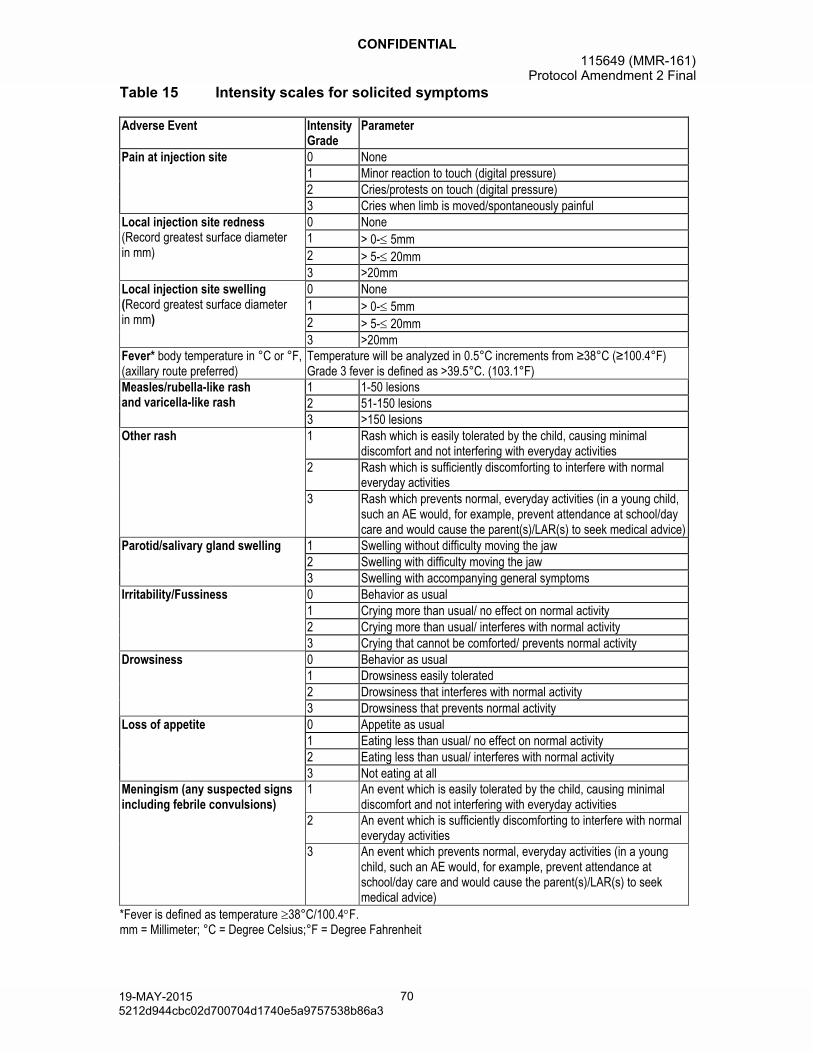
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 70
Table 15 Intensity scales for solicited symptoms
Adverse Event Intensity Grade
Parameter
Pain at injection site 0 None1 Minor reaction to touch (digital pressure)2 Cries/protests on touch (digital pressure)3 Cries when limb is moved/spontaneously painful
Local injection site redness(Record greatest surface diameterin mm)
0 None1 > 0- 5mm2 > 5- 20mm3 >20mm
Local injection site swelling(Record greatest surface diameterin mm)
0 None1 > 0- 5mm2 > 5- 20mm3 >20mm
Fever* body temperature in °C or °F, (axillary route preferred)
Temperature will be analyzed in 0.5°C increments from ≥38°C (≥100.4°F)Grade 3 fever is defined as >39.5°C. (103.1°F)
Measles/rubella-like rashand varicella-like rash
1 1-50 lesions2 51-150 lesions3 >150 lesions
Other rash 1 Rash which is easily tolerated by the child, causing minimal discomfort and not interfering with everyday activities
2 Rash which is sufficiently discomforting to interfere with normal everyday activities
3 Rash which prevents normal, everyday activities (in a young child, such an AE would, for example, prevent attendance at school/day care and would cause the parent(s)/LAR(s) to seek medical advice)
Parotid/salivary gland swelling 1 Swelling without difficulty moving the jaw2 Swelling with difficulty moving the jaw3 Swelling with accompanying general symptoms
Irritability/Fussiness 0 Behavior as usual1 Crying more than usual/ no effect on normal activity2 Crying more than usual/ interferes with normal activity3 Crying that cannot be comforted/ prevents normal activity
Drowsiness 0 Behavior as usual1 Drowsiness easily tolerated2 Drowsiness that interferes with normal activity3 Drowsiness that prevents normal activity
Loss of appetite 0 Appetite as usual1 Eating less than usual/ no effect on normal activity2 Eating less than usual/ interferes with normal activity3 Not eating at all
Meningism (any suspected signs including febrile convulsions)
1 An event which is easily tolerated by the child, causing minimaldiscomfort and not interfering with everyday activities
2 An event which is sufficiently discomforting to interfere with normal everyday activities
3 An event which prevents normal, everyday activities (in a young child, such an AE would, for example, prevent attendance at school/day care and would cause the parent(s)/LAR(s) to seek medical advice)
*Fever is defined as temperature 38°C/100.4F.mm = Millimeter; °C = Degree Celsius;°F = Degree Fahrenheit
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 71
The maximum intensity of local injection site redness/swelling will be graded based on child’s parent(s)/LAR(s) measurements recorded on the Diary Card.
Temperature/ fever: Temperature should be recorded daily, from Day 0 through Day 42after each vaccination. Parent(s)/LAR(s) are requested to record on the diary card the child’s body temperature measured each evening at bedtime (while at home ONLY one temperature route [axillary preferred] should be used consistently for all measurements in a given child). Should additional temperature measurements be performed at other times of day, the highest temperature will be recorded.
Rash and parotid/salivary gland swelling and signs of meningism including febrile convulsions:
IMPORTANT: If any of the following symptoms below are suspected during the follow-up period after each vaccination (Day 0-42), the parent(s)/LAR(s) must perform daily post-vaccination recording on the Diary Card and contact the investigator’s center.
Rash/ exanthem:
Parent(s)/LAR(s) will be provided with a description of rashes. Parent(s)/LAR(s) will be asked to contact the study site immediately if their child develops a rash that could be a measles/rubella-like rash (Day 0-42 after each vaccination, as applicable) or a varicella-like rash (Day 0-42 after Vaccination 1 only). If the investigator/designee also suspects that the rash could possibly be a measles/rubella-like rash or a varicella-like rash, a visit should be arranged as soon as possible; ideally within 48 hours after rash onset, or as soon as possible thereafter.
The investigator will classify cases of the rash as follows:
(1) Measles/ rubella rashes (macular or maculo-papular rashes): presence of macules,discolored small patches or spots of the skin, neither elevated nor depressed below the skin's surface and/or papules, raised bumps on the skin usually <1 cm in diameter.
(2) Varicella-like rash (papulo-vesicular): simultaneous presence of papules and vesicles raised above the skin's surface.
(3) Or other types of rash (heat rash, etc…).
Duration of rash episode will be indicated, as well as the locality of the rash:
Measles/rubella-like or varicella-like rash:
rash onset, i.e., date when the first lesion appears
rash ended, i.e., date of the first day when no new lesion appears.
Other Rash:
start date, i.e., date when first symptom appears
end date, i.e., date when the rash disappears/resolves.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 72
Intensity of measles/rubella-like, or varicella-like rash will be assessed according to the maximum number of overall lesions as described in Table 15.
The outcome of each rash will be assessed as: recovered/resolved, recovering/resolving, not recovered/not resolved, recovered with sequelae/resolved with sequelae.
The investigator will assess the causality of rash episode.
Parotid/salivary gland swelling:
If parotitis is clinically suspected (swelling / tenderness in the mandibular / submandibular region) during Day 0 to Day 42 after each vaccination, the child will be seen by the investigator as soon as possible for an additional visit.
Only those cases diagnosed by the investigator/designee as parotitis are to be recorded as a solicited symptom in the eCRF. Other cases of swelling/tenderness in the mandibular/submandibular region that do not meet the criteria for parotitis clinically are to be reported as an unsolicited AE in the eCRF. Intensity will be recorded as described in Table 15. The outcome of each case will be assessed as described in 8.2.2.2.
Any suspected signs of meningism including febrile convulsions/seizures:
If the child experiences febrile convulsions or presents any other neurological signs or symptoms indicative of meningism (i.e., neck stiffness with or without light intolerance (photophobia) and headache; or convulsion/seizure) during Day 0 to Day 42 follow-up after each vaccination, the parent(s)/LAR(s) should contact the investigator immediately for follow-up. The investigator is requested to write a descriptive assessment of the case and evaluate the relationship to the study vaccination.
The investigator will be requested to document full case details and to provide a diagnosis. In the case of all seizures, including febrile seizures, the investigator should classify the level of diagnostic certainty regarding the seizure as guided by The BrightonCollaboration Seizure Working Group’s case definitions of generalized convulsive seizure as an adverse event following immunization, as described in Table 16[Bonhoeffer, 2004].
Table 16 Brighton Collaboration levels of diagnostic certainty
Level 1 of diagnostic certaintywitnessed sudden loss of consciousness ANDgeneralized, tonic, clonic, tonic-clonic, or atonic motor manifestationsLevel 2 of diagnostic certaintyhistory of unconsciousness ANDgeneralized, tonic, clonic, tonic-clonic, or atonic motor manifestationsLevel 3 of diagnostic certaintyhistory of unconsciousness ANDother generalized motor manifestationsLevel 4 of diagnostic certaintyreported generalized convulsive seizure with insufficient evidence to meet the case definitions for Level 1, 2 or 3 of diagnostic certainty aboveLevel 5 of diagnostic certaintyNot a case of generalized convulsive seizure
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 73
For any case of febrile convulsion, the investigator is also asked to classify the eventbased on the definitions below [American Academy of Pediatrics, 2008]:
Simple Febrile Convulsion/Seizure: defined as a brief (<15 minutes in duration) generalized tonic-clonic seizure that occurs once during a 24 hour period in a febrile child who does not have intracranial infection, metabolic disturbance or history of afebrile seizures. The neurologic examination in the post-ictal period should be no different from baseline, i.e., there should be no newly acquired neurologic deficits.
Complex Febrile Convulsion/Seizure: defined as a generalized tonic-clonic seizure that is not a simple febrile seizure. For instance, any seizure that has at least one of the following characteristics should be defined as a complex seizure:
lasts 15 minutes or longer, or
has a focal/localized component, or
recurs within 24 hours, or
is associated with an abnormal post-ictal neurologic examination.
The investigator will assess the maximum intensity that occurred over the duration of the event for all other AEs, i.e., unsolicited symptoms, including SAEs reported during the study. The assessment will be based on the investigator’s clinical judgment. The intensity of each AE and SAE recorded in the eCRF or SAE Report screens, as applicable, should be assigned to one of the following categories:
1 (mild) = An AE which is easily tolerated by the child, causing minimal discomfort and not interfering with everyday activities.
2 (moderate) = An AE which is sufficiently discomforting to interfere with normal everyday activities.
3 (severe) = An AE which prevents normal, everyday activities (in a young child, such an AE would, for example, prevent attendance at school/day-care and would cause the parent(s)/LAR(s) to seek medical advice.)
An AE that is assessed as Grade 3 (severe) should not be confused with a SAE. Grade 3 is a category utilized for rating the intensity of an event; and both AEs and SAEs can be assessed as Grade 3. An event is defined as ‘serious’ when it meets one of the pre-defined outcomes as described in Section 8.1.2.
AEs of specific interest:
AEs of specific interest for safety monitoring include NOCDs (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies) and AEs prompting ER visits.
Occurrences of AEs of specific interest, including NOCDs and AEs prompting ER visits, will be reported throughout the study, up to six months after the dose of MMR vaccine,
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 74
whether or not they are considered to be possibly related to the treatment administration. Medical documentation of the events will be reported in appropriate targeted follow-up forms included in the eCRF. These events also have to be reported as AE or SAE as appropriate in the eCRF.
8.2.2.2.2. Assessment of causality
The investigator is obligated to assess the relationship between study vaccine(s) and the occurrence of each AE/SAE. The investigator will use clinical judgment to determine the relationship. Alternative plausible causes, based on natural history of the underlying diseases, concomitant therapy, other risk factors and the temporal relationship of theevent to the investigational product will be considered and investigated. The investigator will also consult the Investigator Brochure and/or Product Information for marketed products, in the determination of his/her assessment.
There may be situations when a SAE has occurred and the investigator has minimal information to include in the initial report to GSK Biologicals. However, it is very important that the investigator always makes an assessment of causality for every event prior to submission of the SAE to GSK Biologicals. The investigator may change his/heropinion of causality in light of follow-up information, amending the SAE information accordingly. The causality assessment is one of the criteria used when determining regulatory reporting requirements.
In case of concomitant administration of multiple vaccines, it may not be possible to determine the causal relationship of general AEs to the individual vaccines administered. The investigator should, therefore, assess whether the AE could be causally related to vaccination rather than to the individual vaccines/products.
All solicited local (injection site) reactions will be considered causally related to vaccination. Causality of all other AEs should be assessed by the investigator using the following question:
Is there a reasonable possibility that the AE may have been caused by the investigational product?
NO : The AE is not causally related to administration of the study vaccine(s). There are other, more likely causes and administration of the study vaccine(s) is not suspected to have contributed to the AE.
YES : There is a reasonable possibility that the vaccine(s) contributed to the AE.
Non-serious and serious AEs will be evaluated as two distinct events. If an event meets criteria to be determined ‘serious’ (see Section 8.1.2 for definition of serious adverse event), additional examinations/tests will be performed by the investigator in order to determine ALL possible contributing factors applicable to each SAE.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 75
Possible contributing factors include:
Medical history.
Other medication.
Protocol required procedure.
Other procedure not required by the protocol.
Lack of efficacy of the vaccine(s), if applicable.
Erroneous administration.
Other cause (specify).
The investigator should assess the causality of each SAE. The investigator will use clinical judgment to determine the relationship of SAEs to study procedures or to GSK concomitant medication. Alternative causes, such as natural history of the underlying diseases, concomitant therapy and other risk factors will be considered and investigated.
There may be situations when a SAE has occurred and the investigator has minimal information to include in the initial report to GSK Biologicals. However it is very important that the investigator always makes an assessment of causality for every event prior to submission of the SAE to GSK Biologicals. The investigator may change his/her opinion of causality in light of follow-up information, amending the SAE information accordingly.
If an event meets the criteria to be determined ‘serious’ (refer to Section 8.1.2), it will be examined by the investigator to the extent to enable determination of all contributing factors applicable to each SAE.
Possible contributing factors include:
Medical history.
Concomitant medication.
Protocol required procedure.
Other procedure not required by the protocol.
Other cause (specify).
8.2.2.3. Assessment of outcomes
Outcome of any non-serious AE occurring within 42 days post each vaccination (i.e.,unsolicited AE) or any SAE reported during the entire study will be assessed as:
Recovered/resolved.
Recovering/resolving.
Not recovered/not resolved.
Recovered with sequelae/resolved with sequelae.
Fatal (SAEs only).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 76
8.2.2.4. Medically attended visits
For each solicited and unsolicited symptom the child experiences, the child’s parent(s)/LAR(s) will be asked if the child received medical attention defined as hospitalization, an emergency room visit or a visit to or from medical personnel (e.g., medical doctor) for any reason and this information will be recorded in the eCRF.Routine “well child” visit information will not be recorded in the eCRF.
8.3. Reporting of serious adverse events
8.3.1. Prompt reporting of serious adverse events to GSK Biologicals
SAEs that occur in the time period defined in Section 8.2 will be reported promptly to GSK within the timeframes described in Table 17, once the investigator determines that the event meets the protocol definition of a SAE.
Table 17 Timeframes for submitting serious adverse event reports to GSK Biologicals
Type of EventInitial Reports
Follow-up of Relevant Information on a Previous Report
Timeframe Documents Timeframe DocumentsAll SAEs 24 hours* SAE report/SAE screen 24 hours* SAE report/SAE screen
* Timeframe allowed after receipt or awareness of the information.
8.3.2. Contact information for reporting serious adverse events to GSK Biologicals
Please see the Sponsor Information Sheet for contact details.
Study Contact for Reporting SAEs
Refer to the local study contact information document.
Back-up Study Contact for Reporting SAEs
24/24 hour and 7/7 day availability:GSK Biologicals Clinical Safety & PharmacovigilanceFax: or
8.3.3. Completion and transmission of SAE reports to GSK Biologicals
Once an investigator becomes aware that a SAE has occurred in a study child, the investigator (or designate) must complete the information in the SAE screens of the eCRF WITHIN 24 HOURS. The SAE screens will always be completed as thoroughly as possible with all available details of the event. Even if the investigator does not have all information regarding a SAE, the SAE screens should still be completed within 24 hours. Once additional relevant information is received, the SAE screens in the eCRF should be updated WITHIN 24 HOURS.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 77
The investigator will always provide an assessment of causality at the time of the initial report.
8.3.3.1. Back-up system in case the electronic SAE reporting system does not work
If the electronic SAE reporting system does not work, the investigator (or designate) mustcomplete, then date and sign a SAE Report Form and fax it to the GSK Biologicals Clinical Safety and Pharmacovigilance department within 24 hours.
This back-up system should only be used if the electronic SAE reporting system is not working and NOT if the system is slow. As soon as the electronic SAE reporting system is working again, the investigator (or designate) must complete the SAE screens in the eCRF within 24 hours. The final valid information for regulatory reporting will be the information reported through the electronic SAE reporting system.
8.3.3.2. Updating of SAE information after freezing of the child’s eCRF
When additional information is received on a SAE after freezing of the child’s eCRF, new or updated information should be recorded on a SAE Report Form, with all changes signed and dated by the investigator. The updated form should be faxed to the GSK Biologicals Clinical Safety and Pharmacovigilance department or to the Study Contact for Reporting SAEs (refer to the Sponsor Information Sheet) WITHIN 24 HOURS of receipt of the follow-up information.
8.3.4. Regulatory reporting requirements for serious adverse events
The investigator will promptly report all SAEs to GSK in accordance with the procedures detailed in Section 8.3.1. GSK Biologicals has a legal responsibility to promptly notify, as appropriate, both the local regulatory authority and other regulatory agencies about the safety of a product under clinical investigation. Prompt notification of SAEs by the investigator to the Study Contact for Reporting SAEs is essential so that legal obligations and ethical responsibilities towards the safety of other children are met.
Investigator safety reports are prepared according to the current GSK policy and are forwarded to investigators as necessary. An investigator safety report is prepared for a SAE(s) that is both attributable to the investigational product and unexpected. The purpose of the report is to fulfill specific regulatory and Good Clinical Practice (GCP) requirements, regarding the product under investigation.
8.4. Follow-up of adverse events and serious adverse events
8.4.1. Follow-up of adverse events and serious adverse events
After the initial AE/SAE report, the investigator is required to proactively follow each child within the study and provide further information on the child’s condition to GSK Biologicals.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 78
All SAEs documented at a previous visit and designated as not recovered/not resolved or recovering/resolving will be reviewed at subsequent visits/contacts until the end of the study.
All AEs documented at a previous visit and designated as not recovered/not resolved or recovering/resolving will be reviewed at subsequent visits/contacts until study end.
AEs of specific interest (including NOCDs such as autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies; and AEs prompting ER visits) documented at a previous visit and designated as not recovered/not resolved or recovering/resolving will be reviewed at subsequent visits until study end.
Investigators will follow-up children:
With SAEs or children withdrawn from the study as a result of an AE, until the event has resolved, subsided, stabilized, disappeared, or until the event is otherwise explained, or the child is lost to follow-up.
Or, in the case of other non-serious AEs, until resolution or study end, or they are lost to follow-up.
Clinically significant laboratory abnormalities will be followed up until they have returned to normal, or a satisfactory explanation has been provided. Additional information (including but not limited to laboratory results) relative to the subsequent course of such abnormalities noted for any child must be made available to the Site Monitor.
GSK Biologicals may request that the investigator performs or arranges for the conduct of additional clinical tests or evaluations to elucidate as fully as possible the nature and/or causality of the AE or SAE. The investigator is obliged to assist. If a child dies during participation in the study or during a recognized follow-up period, GSK Biologicals will be provided with a copy of any available post-mortem findings, including histopathology.
8.5. Treatment of adverse events
Treatment of any AE is at the sole discretion of the investigator and according to current good medical practice. Any medication administered for the treatment of an AE should be recorded in the child’s eCRF. Refer to Section 6.7.
8.6. Unblinding
GSK Biologicals’ policy (which incorporates ICH E2A guidance, EU Clinical Trial Directive and US Federal Regulations) is to unblind the report of any SAE which is unexpected and attributable or suspected to be attributable to the investigational product, prior to regulatory reporting. The GSK Biologicals’ Central Safety Physician is responsible for unblinding the treatment assignment in accordance with the specified timeframes for expedited reporting of SAEs (refer to Section 8.3.1).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 79
8.7. Emergency unblinding
Unblinding of a subject’s individual treatment code should occur only in the case of a medical emergency, or in the event of a serious medical condition, when knowledge of the study treatment is essential for the clinical management or welfare of the subject, as judged by the investigator.
The emergency unblinding process consists of the automated Internet-based system SBIRthat allows the investigator to have unrestricted, immediate and direct access to the subject’s individual study treatment.
The investigator has the option of contacting a GSK Biologicals’ On-call Central Safety Physician (or Backup) if he/she needs medical advice or needs the support of GSK to perform the unblinding (i.e., he/she cannot access the automated Internet-based system).
Any emergency unblinding must be fully documented by using the Emergency Unblinding Documentation Form, which must be appropriately completed by the investigator and sent within 24 hours to GSK Biologicals.
GSK Biologicals’ Contact information for Emergency Unblinding
24/24 hour and 7/7 day availability
GSK Biologicals’ Central Safety Physician:
Outside US/Canada: (GSK Biologicals Central Safety Physician on-call)
US/Canada only: (GSK Biologicals Central Safety Physician on-call)
GSK Biologicals’ Central Safety Physician Back-up:
Outside US/Canada
US/Canada only:
Emergency Unblinding Documentation Form transmission:
Outside US & Canada:Fax: or
US/Canada only:Fax:
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a37919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 80
8.8. Subject card
Study child’s parent(s)/LAR(s) must be provided with the address and telephone number of the main contact for information about the clinical study.
The investigator (or designate) must therefore provide a “subject card” to each child’s parent(s)/LAR(s). In an emergency situation this card serves to inform the responsible attending physician that the child is in a clinical study and that relevant information may be obtained by contacting the investigator.
The child’s parent(s)/LAR(s) must be instructed to keep subject cards in their possession at all times.
9. SUBJECT COMPLETION AND WITHDRAWAL
9.1. Subject completion
A child who returns for the concluding Visit 4 (Day 222) will be considered to have completed the study.
9.2. Child withdrawal
Children who are withdrawn because of SAEs/AEs must be clearly distinguished from children who are withdrawn for other reasons. Investigators will follow children who are withdrawn as result of a SAE/AE until resolution of the event (see Section 8.3).
Withdrawals will not be replaced.
9.2.1. Child withdrawal from the study
From an analysis perspective, a ‘withdrawal’ from the study refers to any child who did not come back for the concluding visit or was not available for the concluding contact foreseen in the protocol.
All data collected until the date of withdrawal or last contact of the child will be used for the analysis.
A child is considered a ‘withdrawal’ from the study when no study procedure has occurred, no follow-up has been performed and no further information has been collected for this child from the date of withdrawal or last contact.
Investigators will make an attempt to contact the parent(s)/LAR(s) of those children who do not return for scheduled visits or follow-up.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 81
Information relative to the withdrawal will be documented in the eCRF. The investigator will document whether the decision to withdraw a child from the study was made by the child’s parent(s) or LAR(s), or by the investigator, as well as which of the following possible reasons was responsible for withdrawal:
Serious adverse event.
Non-serious adverse event.
Protocol violation (specify).
Consent withdrawal, not due to an adverse event.
Moved from the study area.
Lost to follow-up.
Other (specify).
9.2.2. Child withdrawal from investigational vaccine
A ‘withdrawal’ from the investigational vaccine refers to any child who does not receive the complete treatment, i.e., when no further planned dose is administered from the date of withdrawal. A child withdrawn from the investigational vaccine may not necessarily be withdrawn from the study as further study procedures or follow-up may be performed (safety or immunogenicity) if planned in the study protocol.
Information relative to premature discontinuation of the investigational vaccine will be documented on the Vaccine Administration screen of the eCRF. The investigator will document whether the decision to discontinue further vaccination/treatment was made bythe child’s parent(s) or LAR(s), or by the investigator, as well as which of the following possible reasons was responsible for withdrawal:
Serious adverse event.
Non-serious adverse event.
Other (specify).
10. DATA EVALUATION: CRITERIA FOR EVALUATION OF OBJECTIVES
Throughout this section, endpoints for the two lots of comparator Com_MMR vaccine (Com_MMR_L1 and Com_MMR_L2) will be analyzed as pooled lots.
10.1. Primary endpoint
Immunogenicity of the MMR vaccines at Day 42.
- Seroresponse to measles, mumps and rubella viruses (by ELISA) and to mumps virus (by PRNT) (see Section 10.5 for definition).
- Measles, mumps and rubella virus antibody concentrations (by ELISA) and mumps virus antibody titers (by PRNT).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 82
10.2. Secondary endpoints
Immunogenicity of the MMR vaccines at Day 84 post-dose 2 (US sub-cohort).
Seroresponse to measles, mumps and rubella viruses (by ELISA).
Measles, mumps and rubella virus antibody concentrations (by ELISA).
Solicited local and general symptoms.
Occurrence of solicited local symptoms in terms of injection site redness, pain and swelling from Day 0 to Day 3 after each vaccination.
Occurrence of solicited general symptoms in terms of drowsiness, loss of appetite and irritability from Day 0 to Day 14 after first vaccination.
Occurrence of solicited general symptoms in terms of fever (temperature ≥38.0°C / 100.4°F), rash, parotid/salivary gland swelling, any sign of meningism (including febrile convulsions) from Day 0 to Day 42 after each vaccination.
Unsolicited adverse events.
Occurrence of unsolicited symptoms according to the Medical Dictionary for Regulatory Activities (MedDRA) classification from Day 0 to Day 42 after each vaccination.
Adverse events of specific interest.
Occurrence of new onset chronic disease (NOCD) (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies) and AEs prompting emergency room (ER) visits from Day 0 through the end of study.
Serious adverse events.
Occurrence of serious adverse events from Day 0 through the end of study.
10.3. Estimated sample size
The target will be to enroll 4500 children to be randomly assigned to four study groups (Inv_MMR_Min, Inv_MMR_Med, Com_MMR_L1 and Com_MMR_L2) in a 2:2:1:1 ratio. There will be approximately 1500 children in each of the Inv_MMR lot groups and approximately 750 children in each of the Com_MMR lot groups. Assuming approximately 20% non-evaluable children, there will be 3600 evaluable children (1200 in each Inv_MMR lot group and 600 in each Com_MMR lot group).
10.3.1. Control on type I error
To control the global type I error of this study below 2.5% a hierarchical procedure with adjustment of the nominal type I error used at the level of a study objective is required (see Figure 2). More specifically:
(1) To be able to conclude on objectives 6-10 associated to Inv_MMR_Med if one or more of objectives 1-5 associated to Inv_MMR_Min are not met, a Bonferroni adjustment will be used for the set of objectives 1-5 and for the set of objectives 6-10.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 83
(2) To control the type I error within each set of objectives, 1-5 and 6-10 respectively, a hierarchical procedure will be used. Namely the primary objective 5 can only be reached if all the associated criteria are met and the previous primary objectives 1, 2, 3 and 4 have been reached. Likewise, the primary objective 10 can only be reached if all the associated criteria are met and the previous primary objectives 6, 7, 8 and 9have been reached.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 84
Figure 2 Sequence for evaluating the study objectives in order to control the overall type I error below 2.5%
An objective will be reached if its associated criterion is met and the previous objectives were reached. Objectives for Inv_MMR_Med will only be tested if one or more of the Inv_MMR_Min objectives are not met.(Unless otherwise specified, all primary and secondary immunogenicity endpoints will be tested with an ELISA.)
Acceptable seroresponse rates of Inv_MMR_Min 90% for anti-
measles, anti-mumps and anti-rubella virus antibodies
Non-inferiority of Inv_MMR_Min vs. pooled Com_MMR lots in terms of seroresponse rates for anti-measles, anti-mumps and anti-rubella virus
antibodies
Non-inferiority of Inv_MMR_Min vs. pooled Com_MMR lots in terms of GMCs of anti-measles, anti-mumps
and anti-rubella virus antibodies
Nominal = 1.25%
Nominal = 1.25%
Nominal = 1.25%
Yes
Yes
Acceptable seroresponse rates of Inv_MMR_Med 90% for anti-
measles, anti-mumps and anti-rubella virus antibodies
Non-inferiority of Inv_MMR_Med vs. pooled Com_MMR lots in terms of seroresponse rates for anti-measles, anti-mumps and anti-rubella virus
antibodies
Non-inferiority of Inv_MMR_Med vs. pooled Com_MMR lots in terms of GMCs of anti-measles, anti-mumps
and anti-rubella virus antibodies
Nominal = 1.25%
Nominal = 1.25%
Nominal = 1.25%
Yes
Yes
No
No
NoYes
Non-inferiority of Inv_MMR_Min vs. pooled Com_MMR lots in terms of seroresponse rates for anti-mumps
virus antibodies (by PRNT)
Nominal = 1.25%
NoYes
Non-inferiority of Inv_MMR_Min vs. pooled Com_MMR lots in terms of
GMT of anti-mumps virus antibodies (by PRNT)
Nominal = 1.25%
No
Non-inferiority of Inv_MMR_Med vs. pooled Com_MMR lots in terms of
GMT of anti-mumps virus antibodies (by PRNT)
Non-inferiority of Inv_MMR_Med vs. pooled Com_MMR lots in terms of seroresponse rates for anti-mumps
virus antibodies (by PRNT)
Nominal = 1.25%
Nominal = 1.25%
Yes
Yes
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38419-MAY-2015
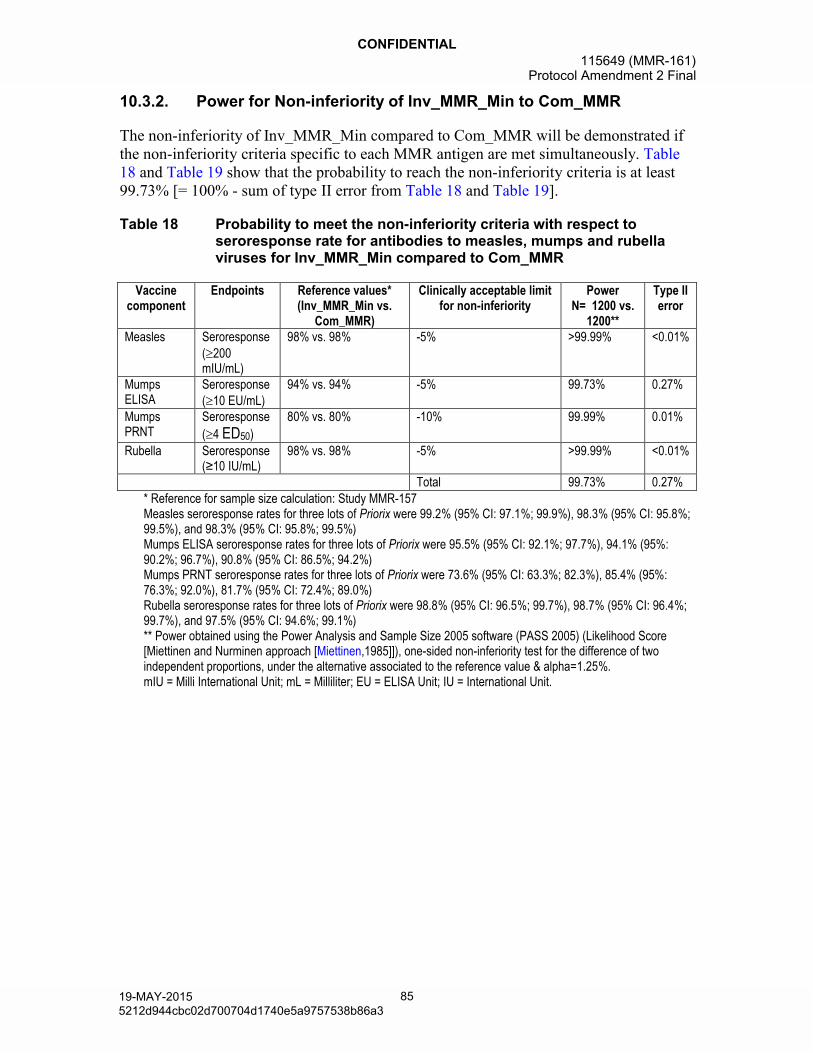
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 85
10.3.2. Power for Non-inferiority of Inv_MMR_Min to Com_MMR
The non-inferiority of Inv_MMR_Min compared to Com_MMR will be demonstrated if the non-inferiority criteria specific to each MMR antigen are met simultaneously. Table 18 and Table 19 show that the probability to reach the non-inferiority criteria is at least 99.73% [= 100% - sum of type II error from Table 18 and Table 19].
Table 18 Probability to meet the non-inferiority criteria with respect to seroresponse rate for antibodies to measles, mumps and rubella viruses for Inv_MMR_Min compared to Com_MMR
Vaccine component
Endpoints Reference values*(Inv_MMR_Min vs.
Com_MMR)
Clinically acceptable limit for non-inferiority
PowerN= 1200 vs.
1200**
Type II error
Measles Seroresponse(200 mIU/mL)
98% vs. 98% -5% >99.99% <0.01%
MumpsELISA
Seroresponse(10 EU/mL)
94% vs. 94% -5% 99.73% 0.27%
Mumps PRNT
Seroresponse
(4 ED50)
80% vs. 80% -10% 99.99% 0.01%
Rubella Seroresponse(≥10 IU/mL)
98% vs. 98% -5% >99.99% <0.01%
Total 99.73% 0.27%* Reference for sample size calculation: Study MMR-157Measles seroresponse rates for three lots of Priorix were 99.2% (95% CI: 97.1%; 99.9%), 98.3% (95% CI: 95.8%; 99.5%), and 98.3% (95% CI: 95.8%; 99.5%)Mumps ELISA seroresponse rates for three lots of Priorix were 95.5% (95% CI: 92.1%; 97.7%), 94.1% (95%: 90.2%; 96.7%), 90.8% (95% CI: 86.5%; 94.2%)Mumps PRNT seroresponse rates for three lots of Priorix were 73.6% (95% CI: 63.3%; 82.3%), 85.4% (95%: 76.3%; 92.0%), 81.7% (95% CI: 72.4%; 89.0%)Rubella seroresponse rates for three lots of Priorix were 98.8% (95% CI: 96.5%; 99.7%), 98.7% (95% CI: 96.4%; 99.7%), and 97.5% (95% CI: 94.6%; 99.1%)** Power obtained using the Power Analysis and Sample Size 2005 software (PASS 2005) (Likelihood Score [Miettinen and Nurminen approach [Miettinen,1985]]), one-sided non-inferiority test for the difference of two independent proportions, under the alternative associated to the reference value & alpha=1.25%.mIU = Milli International Unit; mL = Milliliter; EU = ELISA Unit; IU = International Unit.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 86
Table 19 Probability to meet the non-inferiority criteria with respect to GMCs of antibodies to measles, mumps and rubella viruses for Inv_MMR_Min compared to Com_MMR
Vaccine component
Endpoints Reference ratio
(Com_MMR over
Inv_MMR_Min)
Reference SD*
Clinically acceptable
limit for non-
inferiority
PowerN= 1200 vs.
1200
Type II error**
Measles GMC 1 0.50 0.67 >99.99% <0.01%Mumps ELISA GMC 1 0.50 0.67 >99.99% <0.01%Mumps PRNT GMT 1 0.60 0.67 >99.99% <0.01%Rubella GMC 1 0.44 0.67 >99.99% <0.01%
Total >99.99% <0.01%* Reference for sample size calculation: Study MMR-157SD = Standard Deviation (of the log10 transformed titer); GMC = Geometric Mean ConcentrationObserved SDs for measles for three lots of Priorix were 0.33, 0.34, and 0.34Observed SDs for mumps (PPD ELISA) for three lots of Priorix were 0.41, 0.40, and 0.44Observed SDs for mumps (PRNT) for three lots of Priorix were 0.63, 0.61, and 0.58Observed SDs for rubella for three lots of Priorix were 0.33, 0.34, and 0.34** Type II error is obtained using PASS 2005, one-sided non-inferiority test for 2 means, under the alternative of equal means & alpha=1.25%.
10.3.3. Power for a minimal seroresponse rate of Inv_MMR_Min lots
With an estimated sample size of 1200 evaluable children, Table 20 shows the probability to reach the immunogenicity criterion that the MMR seroresponse rate is at least 90% for measles, mumps, and rubella.
Considering this is conditional to previous primary objectives the power to conclude on minimal MMR seroresponse rate is at least 99.59% [= 100% - sum of type II error from Table 18, Table 19 and Table 20].
Table 20 Probability to meet the minimum level of seroresponse rate for antibodies to measles, mumps and rubella for Inv_MMR_Min
Vaccine component
Endpoints Reference values*
Acceptablelower limit
PowerN= 1200**
Type II error
Measles Seroresponse(200 mIU/mL)
98% 90% >99.99% <0.01%
Mumps Seroresponse(10 EU/mL)
94% 90% 99.87% 0.13%
Rubella Seroresponse(≥10 IU/mL)
98% 90% >99.99% <0.01%
Total 99.87% 0.13%* Reference for sample size calculation: Study MMR-157Measles seroresponse rates for three lots of Priorix were 99.2% (95% CI: 97.1%; 99.9%), 98.3% (95% CI: 95.8%; 99.5%), and 98.3% (95% CI: 95.8%; 99.5%)Mumps ELISA seroresponse rate for three lots of Priorix were 95.5% (95% CI: 92.1%; 97.7%), 94.1% (95%: 90.2%; 96.7%), 90.8% (95% CI: 86.5%; 94.2%)Rubella seroresponse rates for three lots of Priorix were 98.8% (95% CI: 96.5%; 99.7%), 98.7% (95% CI: 96.4%; 99.7%), and 97.5% (95% CI: 94.6%; 99.1%)** PASS 2005, 1-sided non-inferiority test on one proportion alpha = 1.25%, Ho: p ≤ margin, Ha: p = reference value.mIU = Milli International Unit; mL = Milliliter; EU = ELISA Unit; IU = International Unit.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38619-MAY-2015
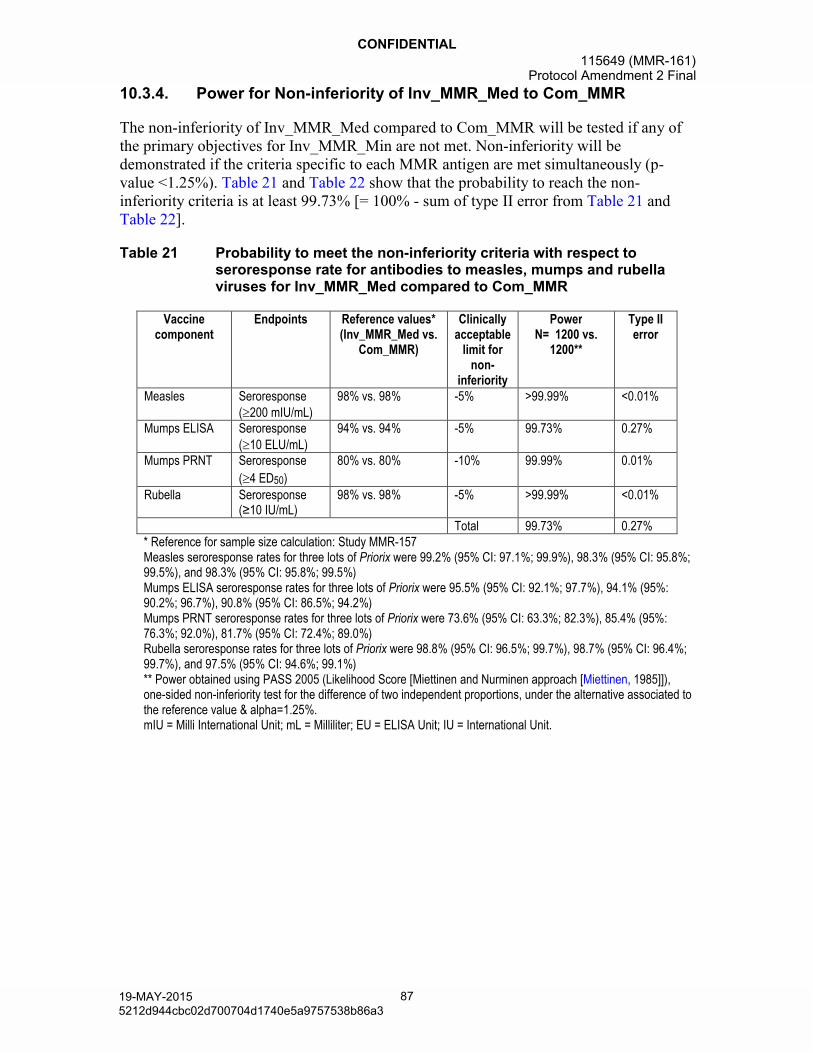
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 87
10.3.4. Power for Non-inferiority of Inv_MMR_Med to Com_MMR
The non-inferiority of Inv_MMR_Med compared to Com_MMR will be tested if any of the primary objectives for Inv_MMR_Min are not met. Non-inferiority will be demonstrated if the criteria specific to each MMR antigen are met simultaneously (p-value <1.25%). Table 21 and Table 22 show that the probability to reach the non-inferiority criteria is at least 99.73% [= 100% - sum of type II error from Table 21 and Table 22].
Table 21 Probability to meet the non-inferiority criteria with respect to seroresponse rate for antibodies to measles, mumps and rubella viruses for Inv_MMR_Med compared to Com_MMR
Vaccine component
Endpoints Reference values*(Inv_MMR_Med vs.
Com_MMR)
Clinically acceptable
limit for non-
inferiority
PowerN= 1200 vs.
1200**
Type II error
Measles Seroresponse(200 mIU/mL)
98% vs. 98% -5% >99.99% <0.01%
Mumps ELISA Seroresponse(10 ELU/mL)
94% vs. 94% -5% 99.73% 0.27%
Mumps PRNT Seroresponse
(4 ED50)
80% vs. 80% -10% 99.99% 0.01%
Rubella Seroresponse(≥10 IU/mL)
98% vs. 98% -5% >99.99% <0.01%
Total 99.73% 0.27%* Reference for sample size calculation: Study MMR-157Measles seroresponse rates for three lots of Priorix were 99.2% (95% CI: 97.1%; 99.9%), 98.3% (95% CI: 95.8%; 99.5%), and 98.3% (95% CI: 95.8%; 99.5%)Mumps ELISA seroresponse rates for three lots of Priorix were 95.5% (95% CI: 92.1%; 97.7%), 94.1% (95%: 90.2%; 96.7%), 90.8% (95% CI: 86.5%; 94.2%)Mumps PRNT seroresponse rates for three lots of Priorix were 73.6% (95% CI: 63.3%; 82.3%), 85.4% (95%: 76.3%; 92.0%), 81.7% (95% CI: 72.4%; 89.0%)Rubella seroresponse rates for three lots of Priorix were 98.8% (95% CI: 96.5%; 99.7%), 98.7% (95% CI: 96.4%; 99.7%), and 97.5% (95% CI: 94.6%; 99.1%)** Power obtained using PASS 2005 (Likelihood Score [Miettinen and Nurminen approach [Miettinen, 1985]]), one-sided non-inferiority test for the difference of two independent proportions, under the alternative associated to the reference value & alpha=1.25%.mIU = Milli International Unit; mL = Milliliter; EU = ELISA Unit; IU = International Unit.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 88
Table 22 Probability to meet the non-inferiority criteria with respect to GMCs of antibodies to measles, mumps and rubella viruses for Inv_MMR_Med compared to Com_MMR
Vaccine component
Endpoints Reference ratio (Com_MMR
over Inv_MMR_Med)
Reference SD*
Clinically acceptable
limit for non-
inferiority
PowerN= 1200 vs.
1200
Type II error**
Measles GMC 1 0.50 0.67 >99.99% <0.01%Mumps ELISA GMC 1 0.50 0.67 >99.99% <0.01%Mumps PRNT GMT 1 0.60 0.67 >99.99% <0.01%Rubella GMC 1 0.44 0.67 >99.99% <0.01%
Total >99.99% <0.01%* Reference for sample size calculation: Study MMR-157SD = Standard Deviation (of the log10 transformed titer); GMC = Geometric Mean ConcentrationObserved SDs for measles for three lots of Priorix were 0.33, 0.34, and 0.34Observed SDs for mumps (PPD ELISA) for the three lots of Priorix were 0.41, 0.40, and 0.44Observed SDs for mumps (PRNT) for three lots of Priorix were 0.63, 0.61, and 0.58Observed SDs for rubella for three lots of Priorix were 0.33, 0.34, and 0.34** Type II error is obtained using PASS 2005, one-sided non-inferiority test for 2 means, under the alternative of equal means & alpha=1.25%.
10.3.5. Power for a minimal seroresponse rate of Inv_MMR_Med lots
With an estimated sample size of 1200 evaluable children, Table 23 shows the probability to reach the immunogenicity criterion that the MMR seroresponse rate is at least 90% for measles, mumps, and rubella.
Considering this is conditional to previous primary objectives the power to conclude on minimal MMR-seroresponse rate is at least 99.59% [= 100% - sum of type II error from Table 21, Table 22, and Table 23].
Table 23 Probability to meet the minimum level of seroresponse rate for antibodies to measles, mumps and rubella for Inv_MMR
Vaccine component
Endpoints Reference values*
Acceptablelower limit
PowerN= 1200**
Type II error
Measles Seroresponse(200 mIU/mL)
98% 90% >99.99% <0.01%
Mumps Seroresponse(10 ELU/mL)
94% 90% 99.87% 0.13%
Rubella Seroresponse(≥10 IU/mL)
98% 90% >99.99% <0.01%
Total 99.87% 0.13%* Reference for sample size calculation: Study MMR-157Measles seroresponse rates for three lots of Priorix were 99.2% (95% CI: 97.1%; 99.9%), 98.3% (95% CI: 95.8%; 99.5%), and 98.3% (95% CI: 95.8%; 99.5%)Mumps ELISA seroresponse rates for three lots of Priorix were 95.5% (95% CI: 92.1%; 97.7%), 94.1% (95%: 90.2%; 96.7%), 90.8% (95% CI: 86.5%; 94.2%)Rubella seroresponse rates for three lots of Priorix were 98.8% (95% CI: 96.5%; 99.7%), 98.7% (95% CI: 96.4%; 99.7%), and 97.5% (95% CI: 94.6%; 99.1%)** PASS 2005, 1-sided non-inferiority test on one proportion alpha = 1.25%, Ho: p ≤ margin, Ha: p = reference value.mIU = Milli International Unit; mL = Milliliter; EU = ELISA Unit; IU = International Unit.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 89
10.4. Study cohorts to be evaluated
10.4.1. Total vaccinated cohort
The Total Vaccinated cohort will include all vaccinated children.
An immunogenicity analysis based on the Total Vaccinated cohort will include all vaccinated children for whom immunogenicity data are available.
A safety analysis based on the Total Vaccinated cohort will include all vaccinated children with at least one vaccine administration of either Inv_MMR lots or one of the Com_MMR lots documented.
10.4.2. According-to-protocol cohort for analysis of safety
The ATP cohort for analysis of safety will include eligible children:
who have received at least one MMR study vaccine/comparator as per protocol
who have not received a vaccine leading to exclusion from the ATP cohort in the protocol as described in Section 6.7.1. up to Visit 2 for the non US sub-cohort and up to Visit 3 for the US sub-cohort.
for whom the randomization code has not been broken
for whom the administration route of study vaccine(s) is known/correct.
10.4.3. According-to-protocol cohort for analysis of immunogenicity
The ATP cohort for analysis of immunogenicity post-dose 1 will include all eligible children in the ATP cohort for safety:
with pre-vaccination and post-dose 1 serology results available for at least one antigen of measles, mumps, or rubella
who are below the assay cut-off for at least one vaccine antigen for MMR at pre-vaccination
who do not meet any elimination criteria up to the Visit 2 blood sample (see Sections 6.7.1 and 6.8)
who complied with the post-dose 1 blood sample schedule, i.e., 35-77 days between vaccination 1 at Visit 1 and the blood draw at Visit 2.
Likewise, the ATP cohort for analysis of immunogenicity post-dose 2 will include all eligible children in the ATP cohort for safety:
who are in the US sub-cohort
who have received two doses of MMR study vaccine/comparator as per protocol
with pre-vaccination and post-dose 2 serology results available for at least one antigen of measles, mumps, or rubella
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a38919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 90
who do not meet any elimination criteria up to the Visit 3 blood sample (see Sections 6.7.1 and 6.8)
who complied with the post-dose 2 blood sample schedule, i.e., 35-77 days between Dose 2 at Visit 2 and the blood draw at Visit 3.
10.5. Derived and transformed data
Immunogenicity
- Seroresponses for MMR are defined as follows:
o For measles virus, a seroresponse is defined as post-vaccination anti-measles virus antibody concentration 200 mIU/mL (ELISA, Enzygnost) among children who were seronegative (antibody concentration <150 mIU/mL) before Dose 1.
o For mumps virus, a seroresponse is defined as post-vaccination anti-mumps virus antibody concentration 10 EU/mL (ELISA, PPD) among children who were seronegative (antibody concentration <5 EU/mL) before Dose 1.
o For mumps virus as measured by PRNT, a seroresponse is defined as post-vaccination anti-mumps virus antibody concentration 4 ED50 (PRNT) among children who were seronegative (antibody concentration <2.5 ED50) beforeDose 1.
o For rubella virus, a seroresponse is defined as post-vaccination anti-rubella virus antibody concentration 10 IU/mL (ELISA, Enzygnost) among children who were seronegative (antibody concentration <4 IU/mL) before Dose 1.
GMC/T calculation:
The GMC/T calculation for antibodies to measles virus, mumps virus and to rubella virus, using the respective ELISA assays, will be performed by taking the anti-log of the mean of the log concentration transformations. Antibody concentrations below the cut-off of the assay will be given an arbitrary value of half the cut-off for the purpose of GMC/Tcalculation.
Handling of missing immune results
For a given child and a given immunogenicity measurement, missing or non-evaluable measurements will not be replaced. Therefore, an analysis will exclude children with missing or non-evaluable measurements.
Handling of missing recording for reactogenicity and safety
For the analysis of solicited symptoms, only children with a completed solicited AE section of the eCRF will be considered.
Missing or non-evaluable measurements will not be replaced. Therefore the analysis of the solicited symptoms based on the Total vaccinated cohort will include only children/doses with documented safety data (i.e., symptom screen/sheet completed).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 91
In the primary analysis of solicited symptoms, missing daily recordings will be replaced by the maximum value recorded for the child. For children reporting a symptom without a single daily recording (i.e., reporting fever as present in the absence of temperature measurement), missing daily recordings will be replaced by grade 1. A sensitivity analysis of the impact of missing data on the endpoints will be conducted if the percentage of children reporting a symptom without a single daily recording is above 1%.
For the analysis of unsolicited adverse events/serious adverse event/concomitant medication, all vaccinated children will be considered and children who did not report an event will be considered as children without an event.
10.6. Conduct of analyses
Any deviation(s) or change(s) from the original statistical plan outlined in this protocol will be described and justified in the final study report.
10.6.1. Sequence of analyses (Amended 19 May 2015)
The analysis will be performed in two steps:
A summary of post dose 1 (Day 42) and post dose 2 (Day 84) GMCs and seroresponse rates for measles, mumps and rubella (by ELISA) will be generated by independent statisticians once all CRF study data are available and cleaned and measles, mumps, and rubella ELISA testing has been fully completed.
A final analysis, including full immunogenicity analysis for post dose 1 (Day 42) and post dose 2 (Day 84) including mumps PRNT results post dose 1 and all safety data (including all immunogenicity and safety analyses), will be performed once the final serology data are available.
These two analyses will be combined in the final clinical report.
For the first analysis step, access to group attribution and individual laboratory results will be limited to statisticians at an independent data analysis center. Members of the GSK study team will only be able to view summary tables (GMCs and seroresponse rates per group, and GMC ratios and differences in seroresponse rates between groups), thereby ensuring that they remain blinded to the treatment group attribution until all data are available and the final analysis is carried out.
A separate charter will be written to provide further details of the scope of the first analysis step, and to clarify the unblinding procedure and attribution of elimination codes by the independent statisticians.
10.6.2. Statistical considerations for interim analyses
No interim analyses are planned.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 92
10.7. Statistical methods
10.7.1. Methodology for computing CI
The exact CIs for a proportion within a group will be calculated using SAS. SAS will also be used to derive the standardized asymptotic 95% CI for the group difference in proportions (method 6 in [Newcombe, 1998]).
The CI for GMCs will be obtained within each group separately. The 95% CI for the mean of log-transformed titer/concentration will be first obtained assuming that log-transformed values were normally distributed with unknown variance. The 95% CI for the GMCs will then be obtained by exponential-transformation of the 95% CI for the mean of log-transformed titer/concentration.
The CI for GMC ratio will be obtained using an ANOVA model on the logarithm-transformed titers. The ANOVA model will include the vaccine group as the fixed effect (3 groups) and the country effect. The GMC ratio and their 95% CI will be derived by exponential-transformation of the corresponding group contrast in the model.
10.7.2. Analysis of demographics/baseline characteristics
Demographic characteristics (age at Visit 1 in months, geographic ancestry (race), ethnicity and gender) will be tabulated as a whole for the Inv_MMR_Min and Inv_MMR_Med groups and for the pooled Com_MMR groups.
The distribution of children enrolled among the study sites/countries will be tabulated as a whole and per group (Inv_MMR_Min, Inv_MMR_Med, and for the pooled Com_MMR groups).
10.7.3. Analysis of immunogenicity
The analysis of immunogenicity will be based on the ATP cohort for immunogenicity. If, for any vaccine group, the percentage of enrolled children with serological results excluded from this ATP cohort is higher than 5%, a second analysis based on the Total vaccinated cohort will be performed to complement the ATP analysis.
Post vaccination 1 and 2 analyses will be based on children who were seronegative for that assay prior to first vaccination.
10.7.3.1. Within groups assessment
The following descriptive analyses will be performed for each treatment group(Inv_MMR_Min, Inv_MMR_Med, and the pooled Com_MMR groups) at each blood sampling timepoint for which a serological result is available:
Percentage of children above each specific cut-off and their exact 95% CIs will be tabulated. The specific cut-offs will be 150 and 200 mIU/mL for measles, 5 and 10 EU/mL for mumps by PPD ELISA, 2.5 and 4 ED50 for mumps by PRNT and 4 and 10 IU/mL for rubella, respectively.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 93
Antibody concentrations also will be summarized by GMCs with their 95% CIs after vaccination.
Antibody concentration distributions will be displayed as reverse cumulative curves for each antigen and each treatment group after vaccination.
A descriptive summary by sub-cohort will also be provided.
10.7.3.2. Between group assessment post-dose1
The asymptotic standardized 97.5% CIs for the difference in seroresponse rate and percentage of children with titer/concentration above each specific cut-off [Inv_MMR_Min vaccine minus pooled Com_MMR vaccine and Inv_MMR_Med vaccine minus pooled Com_MMR vaccine] will be computed.
The 97.5% CI for the group GMC ratio [Inv_MMR_Min vaccine over pooled Com_MMR vaccine and Inv_MMR_Med vaccine over pooled Com_MMR vaccine] will be computed using an ANOVA model on the logarithm-transformed concentrations. The ANOVA model will include the vaccine group and the country as fixed effects.
10.7.3.3. Interpretation of analyses
With respect to primary objectives involving pre-specified criteria, the interpretation must be done according to a hierarchical procedure described in Section 10.3.1. For instance, the primary objective 3 can only be reached if all the associated criteria are met and the previous primary objectives have been reached.
Except for analyses addressing criteria specified in the objectives, comparative analyses will be exploratory with the aim to characterize the difference between groups in immunogenicity. These exploratory analyses should be interpreted with caution since there is no adjustment for multiplicity of endpoints.
10.7.4. Analysis of safety
The analysis of safety will be based on the Total Vaccinated cohort. If more than 5% of the vaccinated children are excluded from the ATP cohort for the analysis of safety, a second analysis based on the ATP cohort will be performed to complement the Total Vaccinated cohort analysis.
For each group (Inv_MMR_Min, Inv_MMR_Med, and pooled Com_MMR lots), the following results will be tabulated:
the number and percentage of children (with exact 95% CIs) with at least one local, general, and any adverse event (solicited and/ or unsolicited) reported from Day 0 -Day 42 after each vaccination;
the number and percentage of children (with exact 95% CIs) reporting each local solicited symptom from Day 0 - Day 3 after each vaccination for the following categories: any grade, grade 2 or 3, grade 3, and resulting in a medically attendedvisit;
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 94
the number and percentage of children (with exact 95% CIs) reporting general symptom (drowsiness, irritability and loss of appetite) from Day 0 - Day 14 after first vaccination for the following categories: any grade, grade 2 or 3, grade 3, and resulting in a medically-attended visit;
the number and percentage of children reporting fever by half degree (°C) cumulative increments from Day 0 - Day 14, from Day 0 - Day 42 and from Day 5 -Day 12 after each vaccination. Similar tabulations will be performed for any fever with a causal relationship to vaccination and for any fever resulting in a medically attended visit . In addition, the prevalence of any and grade 3 fever will be presented graphically over time after vaccination(s); (Amended 19 May 2015)
the number and percentage of children (with exact 95% CIs) reporting measles/rubella-like rash (Days 0-42, according to number of lesions), varicella-like rash (Days 0-42 post vaccination 1 only, according to number of lesions), any rash (Days 0-42), parotitis/salivary gland swelling (Days 0-42, according to intensity) and any sign of meningism (convulsions/seizures) (Days 0-42, according to intensity after each vaccination. Similar tabulations will be performed for general solicited adverse events with a causal relationship to vaccination and for solicited AEs resulting in a medically-attended visit;
the number and percentage of children (with exact 95% CIs) for whom unsolicited adverse events are reported from Days 0-42. The verbatim reports of unsolicited symptoms will be reviewed by a physician and the signs and symptoms will be coded according to MedDRA, the Medical Dictionary for Regulatory Activities. Every verbatim term will be matched with the appropriate Preferred Term. Similar tabulations will be done for grade 3 unsolicited adverse events, for unsolicited adverse events related to vaccination and for unsolicited adverse events resulting in a medically attended visit;
the number and percentage of children who received at least one concomitant medication from Day 0 - Day 14 and from Day 0 - Day 42 after each vaccination. Additionally, the number and percentage of children receiving antipyretic drugs will also be calculated by group from Day 0 - Day 14 and from Day 0 - Day 42 after each vaccination.
Discontinuations due to AEs will be described in detail.
The percentages of children who experienced at least one serious adverse event, or who reported a NOCD (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies), or an AE leading to an ER visit, or an AE leading to a medically attended visit from Day 0 to study end, will be calculated.
Descriptive listing of SAEs, any related SAEs to study participation, study withdrawals and deaths post-vaccination(s) for the entire study.
A descriptive summary by sub-cohort will also be provided.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 95
11. ADMINISTRATIVE MATTERS
To comply with ICH GCP administrative obligations relating to data collection, monitoring, archiving data, audits, confidentiality and publications must be fulfilled.
11.1. Case Report Form/Remote Data Entry instructions
The investigator will keep a paper copy of each diary card and any data query forms of the final version of the data generated at the investigational site.
A validated computer application, will be used as the method for data collection.
In all cases, the study child’s initials will not be collected nor transmitted to GSK. Study participant data necessary for analysis and reporting will be entered/transmitted into a validated database or data system. Clinical data management will be performed in accordance with applicable GSK standards and data cleaning procedures.
While completed eCRFs are reviewed by a Site Monitor at the study site, omissions or inconsistencies detected by subsequent eCRF review may necessitate clarification or correction of omissions or inconsistencies with documentation and approval by the investigator or appropriately qualified designee. In all cases, the investigator remains accountable for the study data.
eCRFs and diary cards, will be supplied by GSK for recording all data. Any questions or comments related to the eCRF should be directed to the assigned Site Monitor.
The investigator will be provided with a CD-ROM of the final version of the data generated at the investigational site once the database is archived and the study report is complete and approved by all parties.
11.2. Monitoring by GSK Biologicals
Monitoring visits by a GSK Site Monitor are for the purpose of confirming that GSK Biologicals’ sponsored studies are being conducted in accordance with the ethical principles that have their origins in the Declaration of Helsinki and that are consistent with Good Clinical practice (GCP) and the applicable regulatory requirement(s) (verifying continuing compliance with the protocol, amendment(s), reviewing the investigational product accountability records, verifying that the site staff and facilities continue to be adequate to conduct the study).
The investigator must ensure provision of reasonable time, space and qualified personnel for monitoring visits.
Direct access to all study-site related and source data is mandatory for the purpose of monitoring review. The monitor will perform an eCRF review and a Source Document Verification (SDV). By SDV we understand verifying eCRF entries by comparing them with the source data that will be made available by the investigator for this purpose.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 96
The Source Documentation Agreement Form describes the source data for the different data in the eCRF. This document should be completed and signed by the site monitor and investigator and should be filed in the monitor’s and investigator’s study file. Any data item for which the eCRF will serve as the source must be identified, agreed and documented in the source documentation agreement form.
For the eCRF, the monitor will mark completed and approved screens at each visit.
In accordance with applicable regulations, GCP, and GSK procedures, GSK monitors will contact the site prior to the start of the study to review with the site staff the protocol, study requirements, and their responsibilities to satisfy regulatory, ethical, and GSK requirements. When reviewing data collection procedures, the discussion will also include identification, agreement and documentation of data items for which the eCRFentries will serve as the source document.
GSK will monitor the study to verify that, amongst others, the:
Data are authentic, accurate, and complete.
Safety and rights of the study children are being protected.
Study is conducted in accordance with the currently approved protocol and any amendments, any other study agreements, GCP and all applicable regulatory requirements.
The investigator and the head of the medical institution (where applicable) agrees to allow the monitor direct access to all relevant documents.
Upon completion or premature discontinuation of the study, the monitor will conduct site closure activities with the investigator or site staff, as appropriate, in accordance with applicable regulations, GCP, and GSK procedures.
11.3. Archiving of data at study sites
Following closure of the study, the investigator must maintain all site study records in a safe and secure location. The records must be maintained to allow easy and timely retrieval, when needed (e.g., audit or inspection), and, whenever feasible, to allow any subsequent review of data in conjunction with assessment of the facility, supporting systems, and staff. Where permitted by applicable laws/regulations or institutional policy, some or all of these records can be maintained in a validated format other than hard copy (e.g., microfiche, scanned, electronic for studies with an eCRF); however, caution needs to be exercised before such action is taken. The investigator must assure that all reproductions are legible and are a true and accurate copy of the original and meet accessibility and retrieval standards, including re-generating a hard copy, if required. Furthermore, the investigator must ensure there is an acceptable back-up of these reproductions and that an acceptable quality control process exists for making these reproductions.
GSK will inform the investigator/institution of the time period for retaining these records to comply with all applicable regulatory requirements. However, the
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 97
investigator/institution should seek the written approval of the sponsor before proceeding with the disposal of these records. The minimum retention time will meet the strictest standard applicable to that site for the study, as dictated by ICH GCP, any institutional requirements or applicable laws or regulations, or GSK standards/procedures; otherwise, the minimum retention period will default to 20 years.
The investigator/institution must notify GSK of any changes in the archival arrangements, including, but not limited to, the following: archival at an off-site facility, transfer of ownership of the records in the event the investigator leaves the site.
11.4. Audits
To ensure compliance with GCP and all applicable regulatory requirements, GSK may conduct a quality assurance audit. Regulatory agencies may also conduct a regulatory inspection of this study. Such audits/inspections can occur at any time during or after completion of the study. If an audit or inspection occurs, the investigator and institution agree to allow the auditor/inspector direct access to all relevant documents and to allocate his/her time and the time of his/her staff to the auditor/inspector to discuss findings and any relevant issues.
11.5. Posting of information on Clinicaltrials.gov
Study information from this protocol will be posted on clinicaltrials.gov before enrollment of children begins.
11.6. Ownership, confidentiality and publication
11.6.1. Ownership
All information provided by GSK and all data and information generated by the site as part of the study (other than a study child’s medical records) are the sole property of GSK.
All rights, title, and interests in any inventions, know-how or other intellectual or industrial property rights which are conceived or reduced to practice by site staff during the course of or as a result of the study are the sole property of GSK, and are hereby assigned to GSK.
If a written contract for the conduct of the study which includes ownership provisions inconsistent with this statement is executed between GSK and the study site, that contract’s ownership provisions shall apply rather than this statement.
11.6.2. Confidentiality
Documented evidence that a potential investigator is aware and agrees to the confidential nature of the information related to the study must be obtained by means of a confidentiality agreement.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 98
All information provided by GSK and all data and information generated by the site as part of the study (other than a child’s medical records) will be kept confidential by the investigator and other site staff. This information and data will not be used by the investigator or other site personnel for any purpose other than conducting the study. These restrictions do not apply to: (i) information which becomes publicly available through no fault of the investigator or site staff; (ii) information which it is necessary to disclose in confidence to an IEC or IRB solely for the evaluation of the study; (iii) information which it is necessary to disclose in order to provide appropriate medical care to a study child; or (iv) study results which may be published as described in the next paragraph. If a written contract for the conduct of the study which includes confidentiality provisions inconsistent with this statement is executed, that contract’s confidentiality provisions shall apply rather than this statement.
11.6.3. Publication
For multicenter studies, the first publication or disclosure of study results shall be a complete, joint multicenter publication or disclosure coordinated by GSK. Thereafter, any secondary publications will reference the original publication(s).
Prior to submitting for publication, presentation, use for instructional purposes, or otherwise disclosing the study results generated by the site (collectively, a ‘Publication’), the investigator shall provide GSK with a copy of the proposed Publication and allow GSK a period to review the proposed Publication (at least twenty-one working days, or at least fifteen working days for abstracts/posters/presentations). Proposed Publications shall not include either GSK confidential information other than the study results or personal data on any child, such as name or initials.
At GSK’s request, the submission or other disclosure of a proposed Publication will be delayed a sufficient time to allow GSK to seek patent or similar protection of any inventions, know-how or other intellectual or industrial property rights disclosed in the proposed Publication.
If a written contract for the conduct of the study, which includes publication provisions inconsistent with this statement is executed, that contract’s publication provisions shall apply rather than this statement.
11.6.4. Provision of study results to investigators, posting to the clinical trials registers and publication
Where required by applicable regulatory requirements, an investigator signatory will be identified for the approval of the clinical study report. The investigator will be provided reasonable access to statistical tables, figures, and relevant reports and will have the opportunity to review the complete study results at a GSK site or other mutually-agreeable location.
GSK will also provide the investigator with the full summary of the study results. The investigator is encouraged to share the summary results with the study children’s parent(s)/LAR(s), as appropriate.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 99
The results summary will be posted to the GSK Clinical Study Register no later than 12 months after the last subject’s last visit (LSLV) or sooner if required by legal agreement, local law or regulation. In addition, a manuscript will be submitted to a peer-reviewed journal for publication within 18 months of LSLV. When manuscript publication in a peer-reviewed journal is not feasible, further study information will be posted to the GSK Clinical Study Register to supplement the results summary.
A manuscript will be progressed for publication in the scientific literature if the results provide important scientific or medical knowledge.
12. COUNTRY SPECIFIC REQUIREMENTS
The use of varicella vaccines as a 2-dose vaccine is now under regulatory review in Malaysia. If varicella vaccines are not yet approved for a 2 dose indication at the time the second dose of varicella-containing vaccine is to be administered, the second dose of varicella-containing vaccine will be administered as part of the study procedures. If regulatory approval is granted, the 2nd dose of varicella-containing vaccine will still be administered but it will not be a part of the study procedures.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a39919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 100
13. REFERENCES
American Academy of Pediatrics AAP. Steering Committee of Quality Improvement and Management, Subcommittee on Febrile Seizures. Febrile seizures: Clinical practice guideline for the long-term management of the child with simple febrile seizures. Pediatrics.2008; 121(6):1281-1286.
Bonhoeffer J, Menkes J, Gold MS, et al. The Brighton Collaboration Seizure Working Group - Generalized convulsive seizure as an adverse event following immunization: case definition and guidelines for data collection, analysis, and presentation. Vaccine. 2004; 22:557-562.
Marcy SM, Kohl KS, Dagan R, et al. The Brighton Collaboration Fever Working Group -Fever as an adverse event following immunization: case definition and guidelines of data collection, analysis, and presentation. Vaccine. 2004; 22:551-556.
Miettinen O, Nurminen M, Comparative analysis of two rates. Stat Med. 1985; 4:213-226.
Morbidity and Mortality Weekly Report (MMWR). MMR Vaccine Use and strategies for elimination of measles, rubella, and congenital rubella syndrome and control of mumps. Recommendations of the ACIP, May 22, 1998.
Newcombe RG. Interval estimation for the difference between independent proportions: comparison of eleven methods. Statist. Med. 1998;17:873-890.
World Health Organization (WHO). (2000) WHO Position Paper on Rubella Vaccines. Weekly Epidemiological Record 75:161-172.
World Health Organization (WHO) (2001) WHO Position Paper on Mumps Vaccines. Weekly Epidemiological Record 76:345-356.
World Health Organization (WHO). (2004) WHO Position Paper on Measles Vaccines. Weekly Epidemiological Record 79:129-144.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 101
APPENDIX A LABORATORY ASSAYS (Amended 19 May 2015)
Instructions for collection and handling of blood/serum samples:
For all children, approximately 5 mL of whole blood will be collected at the appropriate visit using serum separator tubes. After blood centrifugation and serum separation, samples will be stored at or less than -20°C/ -4°F (alternatively at approximately -70°C/-80°C is also acceptable) until sent to the sponsor. Serological assays will be performed at GlaxoSmithKline Biologicals central laboratory or PPD, a contract research organization, or any GSK validated lab using standardized, validated procedures with adequate controls. The samples will be tested in a blinded fashion for the presence of antibodies.
Anti-measles ELISA: Anti-measles Ab concentrations will be measured using a commercial ELISA kit (Enzygnost/Dade Behring), which is based on a single point quantification Ab method. The assay technical cut-off is 150 milli-international units (mIU)/mL. The assay will be performed on human serum at GSK Biologicals’ laboratory or another laboratory designated by GSK Biologicals
Anti-rubella ELISA: Anti-VZV Ab concentrations will be measured using a commercial ELISA kit (Enzygnost/Dade Behring), which is based on a single point quantification Ab method. The assay technical cut-off is 4 milli-international units (mIU)/mL. The assay will be performed on human serum at GSK Biologicals’ laboratory or another laboratory designated by GSK Biologicals
Anti-mumps ELISA: Anti-mumps wild type IgG Ab concentrations are measured by using the PPD ELISA assay. Solid phase of the assay (96 wells microplate) consist of wells coated with Mumps Jeryl Lynn® 135 antigen (used in the M-M-R® II and ProQuad™ vaccines and is considered by CBER to be a WT-like strain). Single dilutions of serum sample are tested. The assay technical cut-off is 5 antibody units (AbU)/mL.
Mumps antibody concentration will be determined at PPD.
The GSK Mumps PRNT is an in vitro functional assay based on the indirect quantification of neutralized virus (MU90/Lo1 mumps virus). Serial dilutions of sera are mixed with challenge virus and incubated with cultured Vero cells that are sensitive to the virus. Anti-Mumps antibodies present in the human sera bind on the surface of the virus and block viral attachment to receptors of susceptible cells. The infectivity of the challenge virus is reduced as measured by the reduction of a viral cytopathic effect (CPE) in Vero cells. This decrease is measured by detecting the reduction of viral plaques in the assay. Plaques are counted (visually or automatically) after immunostaining of the infected cells. The neutralization titer corresponds to the reciprocal serum dilution that reduces the number of plaques by 50% when compared to plaques induced by the challenge virus in the absence of serum. The assay technical cut-off is 2.5 End-point Dilution 50% (ED50). The GSK Mumps PRNT is performed in 96 well plates and is derived from the National Institute for Biological Standards and Control (NIBSC) mumps PRNT procedure.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 102
If additional testing is performed, the marker priority ranking may be changed.
Other laboratory assays may be performed to supplement determination of responses to the components of the vaccines administered in this study.
APPENDIX B CLINICAL LABORATORIES
Table 24 GSK Biologicals’ laboratories (Amended 19 May 2015)
Laboratory AddressGSK Biologicals Global Vaccine Clinical Laboratory, Rixensart
Biospecimen Reception - B7/44Rue de l'Institut, 89 - B-1330 Rixensart - Belgium
GSK Biologicals Global Vaccine Clinical Laboratory, Wavre-Nord Noir Epine
Avenue Fleming, 20 - B-1300 Wavre - Belgium
Table 25 Outsourced laboratories (Amended 19 May 2015)
Laboratory AddressBARC USA Inc. 5, Delaware Drive
Lake SuccessNY 11042-1114USA
NÉOMED-LABS Inc. 525, Cartier Ouest Laval, Quebec Canada H7V 3S8
PPD, Inc. 466 Devon Park DriveWayne, PA 19087-1816 – USA
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 103
APPENDIX C AMENDMENTS AND ADMINISTRATIVE CHANGES TO THE PROTOCOL
GlaxoSmithKline Biologicals
Clinical Research & Development
Protocol Amendment 1
eTrack study number and Abbreviated Title(s)
115649 (MMR-161)
Amendment number: Amendment 1
Amendment date: 17 February 2014Co-ordinating author: , Scientific Writer, XPE Pharma for GSK
Biologicals
Rationale/background for changes: At CBER’s request, the US non post dose 2 subcohort was eliminated. This
subcohort was previously defined as any child enrolled in the US after the target enrollment of 1000 US children was met. These children would not have been required to have a blood sample taken post dose 2 as post dose 2 immunogenicity testing was only required in 1000 children. At CBER’s request, the serologic response after each dose of both Merck’s MMR-II and GSK’s investigational vaccine will be evaluated in all US children. In the event that the pre-specified criteria for seroresponse are not met, U.S. children will be offered vaccination with MMR-II to assure protection from measles, mumps and rubella diseases.
Vaccination against influenza and haemophilus influenza type B can be given at any time before, during, or after the study, including the day of study vaccination. This is to correct a prior exclusion criterion which stated that it could be given at any time during the study. The intent has always been to allow these vaccinations at any time regardless of whether or not it was before, during or after the study period.
For other MMR US Phase III studies, CBER has requested that all conditions leading to non-routine medically attended visits be collected for the entire study in the eCRF. It has been clarified throughout the protocol that From Visit 1 (Day 0) to study end (Day 222), all medically attended events will be recorded in the eCRF. Subjects’ routine ‘well child’ doctor visits will not be recorded in the eCRF.
Other minor corrections.
Amended text has been included in bold italics and deleted text in strikethrough in the following sections:
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 104
Cover page
Co-ordinating author , Scientific Writer, XPE Pharma for GSK Biologicals
Contributing authors , Director, Clinical Development Manager
(SynteractHCR for GSK Biologicals), Study Delivery Lead
, Study Delivery Lead
, Global Study Manager
(Keyrus Biopharma for GSK Biologicals), Study Data Manager
, , Clinical Safety and Pharmacovigilance
, Global Regulatory Affairs
, Director, Clinical Research and Development Leader, Vaccines for MMR and Varicella
, Vice President, Global Vaccine Development, GSK Biologicals
Protocol Amendment 1 Sponsor Signatory Approval page
Sponsor signatory , M.D.Director, Global Vaccine Development, MMR/VVaccines, GSK Biologicals
Vice President, Global Vaccine Development, GSK Biologicals
SynopsisSecondary objectives
1. To assess the immunogenicity of Inv_MMR_Min followed by Inv_MMR_Releaseand pooled Com_MMR vaccine in terms of seroresponse rates and GMCs for antibodies to measles, mumps and rubella viruses at Day 84 (post Dose 2) (in a sub-cohort of children enrolled in the US).
2. To assess the immunogenicity of Inv_MMR_Med followed by Inv_MMR_Releaseand pooled Com_MMR vaccine in terms of seroresponse rate and GMCs for antibodies to measles, mumps and rubella viruses at Day 84 (post Dose 2) (in a sub-cohort of children enrolled in the US).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310419-MAY-2015
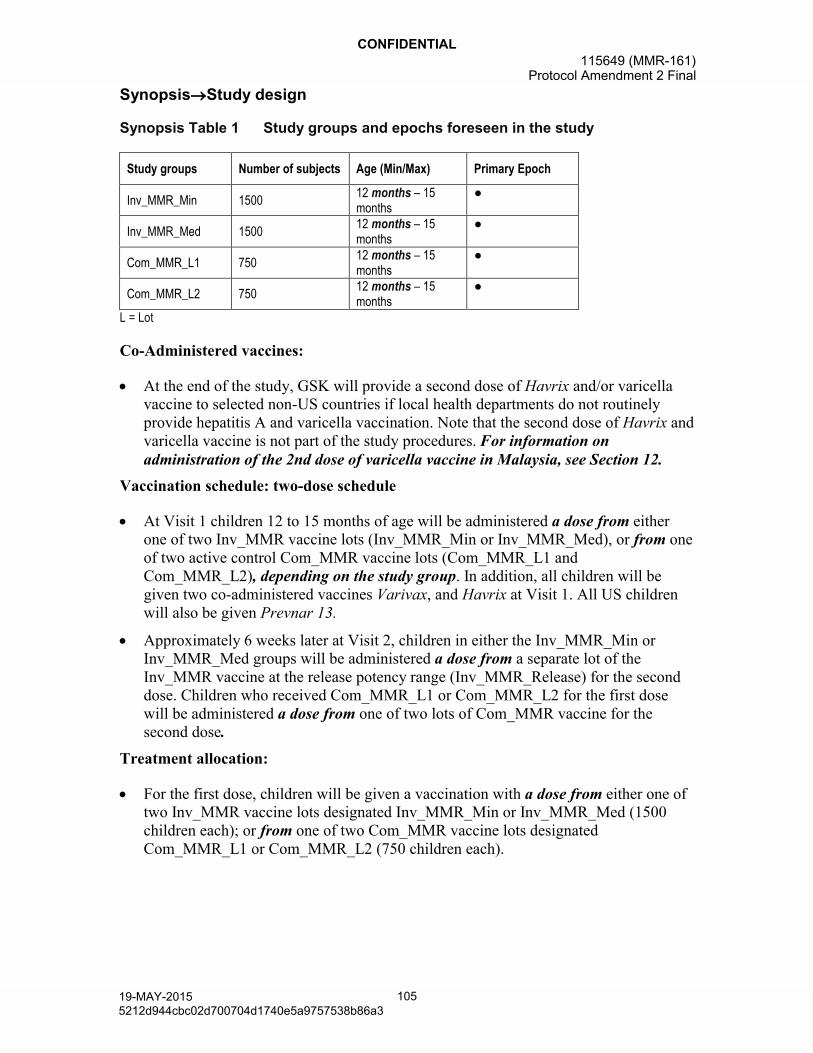
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 105
SynopsisStudy design
Synopsis Table 1 Study groups and epochs foreseen in the study
Study groups Number of subjects Age (Min/Max) Primary Epoch
Inv_MMR_Min 150012 months – 15months
●
Inv_MMR_Med 150012 months – 15months
●
Com_MMR_L1 75012 months – 15months
●
Com_MMR_L2 75012 months – 15months
●
L = Lot
Co-Administered vaccines:
At the end of the study, GSK will provide a second dose of Havrix and/or varicella vaccine to selected non-US countries if local health departments do not routinely provide hepatitis A and varicella vaccination. Note that the second dose of Havrix and varicella vaccine is not part of the study procedures. For information on administration of the 2nd dose of varicella vaccine in Malaysia, see Section 12.
Vaccination schedule: two-dose schedule
At Visit 1 children 12 to 15 months of age will be administered a dose from either one of two Inv_MMR vaccine lots (Inv_MMR_Min or Inv_MMR_Med), or from one of two active control Com_MMR vaccine lots (Com_MMR_L1 and Com_MMR_L2), depending on the study group. In addition, all children will be given two co-administered vaccines Varivax, and Havrix at Visit 1. All US children will also be given Prevnar 13.
Approximately 6 weeks later at Visit 2, children in either the Inv_MMR_Min or Inv_MMR_Med groups will be administered a dose from a separate lot of the Inv_MMR vaccine at the release potency range (Inv_MMR_Release) for the second dose. Children who received Com_MMR_L1 or Com_MMR_L2 for the first dose will be administered a dose from one of two lots of Com_MMR vaccine for the second dose.
Treatment allocation:
For the first dose, children will be given a vaccination with a dose from either one of two Inv_MMR vaccine lots designated Inv_MMR_Min or Inv_MMR_Med (1500 children each); or from one of two Com_MMR vaccine lots designated Com_MMR_L1 or Com_MMR_L2 (750 children each).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 106
Sampling:
- Blood samples will be collected from each child at Day 0 and Day 42. In addition, a third blood sample will be collected from all US children at Day 84 (42 days post-dose 2(Day 84) in a group of US children, referred to as the US post-dose 2 sub-cohort.)
Secondary endpoints:
Immunogenicity of the MMR vaccines at Day 84 post-dose 2 (US post-dose 2 sub-cohort).
Section 1.2.2.Rationale for the study design
This study is designed to establish the end of shelf-life potency of Inv_MMR vaccine. The Inv_MMR vaccine administered in this study will reflect a reasonable end of shelf-life potency for all antigens. In order to establish an end of shelf-life potency, the first dose of Inv_MMR vaccine will be given from one of two lots; one of a minimum potency, designated Inv_MMR_Min, and the other at a mid-range or medium potency designated Inv_MMR_Med. To maintain these Inv MMR lots at their specified potencies, these vaccine vials will remain frozen until reconstitution. Both of these Inv_MMR groups will then receive a second dose of the investigational vaccine which has a potency within the release potency range of the marketed vaccine. The primary endpoints of the study include: 1) evaluating the immunogenicity of a first dose of Inv_MMR_Min and/or the immunogenicity of a first dose of Inv_MMR_Med vaccine in contrast to a first dose of the US standard of care (MMR II vaccine referred to as Com_MMR within this document); and 2) demonstrating that the immune response for Inv_MMR_Min or Inv_MMR_Med meet a pre-specified acceptability criteria. In order to obtain more representative data on the comparator, the Com_MMR vaccine used in this study will consist of two lots designated Com_MMR_L1 and Com_MMR_L2. Throughout the study Com_MMR_L1 and Com_MMR_L2 will be analyzed as pooled lots.
Six weeks after the first dose children randomized to receive either Inv_MMR_Min, or Inv_MMR_Med, will receive a second dose of the investigational vaccine, designated Inv_MMR_Release, which has a potency within the release range of the marketed vaccine. Children randomized to receive either Com_MMR_L1 or Com_MMR_L2 will receive a second dose of Com_MMR vaccine at the six week interval. Immune responses following a second dose of both Inv_MMR and Com_MMR will be evaluated in a sub-cohort ofall children enrolled in the US. Thus, the study will provide data on the immunogenicity (in a sub-cohort of children) and safety of Inv_MMR when administeredon a two-dose schedule to children in the second year of life.
Section 2.2.Secondary objectives
1. To assess the immunogenicity of Inv_MMR_Min followed by Inv_MMR_Release and pooled Com_MMR vaccine in terms of seroresponse rates and GMCs for antibodies to measles, mumps and rubella viruses at Day 84 (post Dose 2) (in a sub-cohort of children enrolled in the US).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 107
2. To assess the immunogenicity of Inv_MMR_Med followed by Inv_MMR_Releaseand pooled Com_MMR vaccine in terms of seroresponse rate and GMCs for antibodies to measles, mumps and rubella viruses at Day 84 (post Dose 2) (in a sub-cohort of children enrolled in the US).
Section 3.Study design overview
Figure 1 Study design
Overall Randomization 2:1 (Inv_MMR:Com_MMR)
Group Randomization 2:2:1:1 (Inv_MMR_Min:Inv_MMR_Med:Com_MMR_L1:Com_MMR_L2)
Inv_MMR_Min 1500 children 12 - 15 months of age
Inv_MMR_Med 1500 children 12 - 15 months of age
Com_MMR_L1 750 children 12 - 15 months of age
Com_MMR_L2 750 children 12 - 15 months of age
Visit 1 (Day 0) Visit 2 (Day 42) Visit 3 (Day 84) Visit 4 (Day 222)
Blood sample
Vaccination 1:Inv_MMR_Min+
VV+HAV+(PCV-13*)or
Inv_MMR_Med+VV+HAV+(PCV-13*)
orCom_MMR_L1+
VV+HAV+(PCV-13*)or
Com_MMR_L2+VV+HAV+(PCV-13*)
Blood sample
Diary Card transcription
Vaccination 2:
Inv_MMR_Release
or
Com_MMR_L1
or
Com_MMR_L2
Blood sample**
Diary Card transcription
Safety Follow-up
Study conclusion
Primary Epoch
L= Lot; VV = Varicella Vaccine Varivax; HAV = Hepatitis A Vaccine Havrix; PCV-13 = Pneumococcal 13-Valent Conjugate Vaccine Prevnar 13* PCV-13 is only administered to children in the US** Blood sample collected at Day 84 is only for the first 1000 children enrolled in the US (the US post dose 2 sub-cohort subcohort)
Control: active control Com_MMR_L1 and Com_MMR_L2 vaccines.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 108
Co-administered vaccines: All children will receive Havrix and Varivax as study vaccines, concomitantly with MMR vaccine at 12 to 15 months of age. Prevnar 13 will only be administered to children recruited in the US. At the end of the study, GSK will provide a second dose of Havrix and/or varicella vaccine to selected non-US countries if local health departments do not routinely provide hepatitis A and varicella vaccination. The second dose of Havrix and varicella vaccine is not part of the study procedures. For information on administration of the 2nd dose of varicella vaccine in Malaysia, see Section 12.
Vaccination schedule: two-dose schedule
At Visit 1 children 12 to15 months of age will be administered a dose from either one of two Inv_MMR vaccine lots (Inv_MMR_Min or Inv_MMR_Med), or from one of two active control Com_MMR vaccine lots (Com_MMR_L1 and Com_MMR_L2). In addition, all children will be given two co-administered vaccines Varivax, and Havrix at Visit 1. All US children will also be given Prevnar 13.
Approximately 6 weeks later, at Visit 2, children in either the Inv_MMR_Min or Inv_MMR_Med groups will be administered investigational vaccinea dose from a separate lot designated of the Inv_MMR_Release, which has a potency within at the release potency range of the marketed vaccine. Children in either the Com_MMR_L1 or Com_MMR_L2 groups will be administered one of two lots of Com_MMR vaccine (Inv_MMR_Release), for the second dose. Children who received Com_MMR_L1 or Com_MMR_L2 for the first dose will be administered a dose from one of two lots of Com_MMR vaccine for the second dose.
Treatment allocation:
For the first dose, children will be given a dose from either one of two Inv_MMR vaccine lots designated Inv_MMR_Min or Inv_MMR_Med (1500 children each); or from one of two Com_MMR vaccine lots designated Com_MMR_L1 or Com_MMR_L2 (750 children each). For the second dose, both the Inv_MMR_Min and Inv_MMR_Med groups will be administered a separate lot of the investigational vaccine Inv_MMR_Release, which has a potency within the release potency range. Both the Com_MMR_L1 and Com_MMR_L2 groups will be administered one of two lots of Com_MMR vaccine for the second dose (refer to the Vaccination schedule section above). The treatment allocation at the investigator site will be performed using a central randomization system on the Internet (SBIR). Overall, the randomization is 2:2:1:1 (Inv_MMR_Min:Inv_MMR_Med:Com_MMR_L1:Com_MMR_L2).
Sampling:
Blood samples will be collected from each child at Day 0 and Day 42. In addition, a third blood sample will be collected from all US children at Day 84 (42 days post-dose 2 (Day 84) in a group of US children, referred to as the US post-dose 2 sub-cohort.).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 109
Section 4.1.Number of study children/centers
Table 4 Sub-cohorts
Sub-cohort name Description Estimated number of childrenUS post dose 2 The first 1000All children enrolledrecruited in the US
will have blood taken and tested for antibodies to measles, mumps and rubella viruses at Day 84
1000 based on competitive enrollment
US non post dose 2
All children recruited in the US (excluding those in the US post dose 2 sub cohort)
Based on competitive enrollment
Non US All children recruited outside the US 3500 based on competitive enrollment
Enrollment will be terminated when approximately 4500 children have been enrolled. For US centers, once the enrollment into the US post-dose 2 sub-cohort is fulfilled, enrollment into the US non post-dose 2 sub-cohort will be initiated.
Section 4.2.Inclusion criteria for enrollment
For US children only:
Child that previously received a 3-dose series of Prevnar 13 with last dose at least 60 days prior to study entry.
Section 4.3.Exclusion criteria for enrollment
Planned administration/ administration of a vaccine not foreseen by the study protocol during the period starting 30 days prior to study vaccination at Visit 1 and ending at Visit 2 (or ending at Visit 3 for the US post-dose 2 sub-cohort). Please Note:
- Inactivated influenza (Flu) vaccine and Haemophilus influenzae type b conjugate vaccine (Hib) vaccines may be given at any time during the study, including the day of study vaccination (Flu and Hib vaccines must be administered at a different location than the study vaccine/s).
- Any age appropriate vaccine may be given starting at Visit 2 (or starting at Visit 3 for the US post-dose 2 sub-cohort), and anytime thereafter.
Administration of immunoglobulins and/or any blood products during the period starting 180 days prior to study vaccination at Visit 1 or planned administration from the date of vaccination through the immunogenicity evaluation at Visit 2, or at Visit 3 for the US post-dose 2 sub-cohort.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a310919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 110
Section 5.2.2.2.Treatment allocation to the study children
The treatment allocation at the investigator site will be performed using SBIR. The treatment numbers will be allocated by dose. The randomization algorithm will be stratified by sub-cohort (US post-dose 2, US non post-dose 2 and Non US sub-cohorts) and within each sub-cohort, using a minimization procedure accounting for center.
After having checked the eligibility of the child and obtaining the ICF, the site staff in charge of the vaccination will access SBIR. Upon providing the child identification number and based on the sub-cohort, the randomization system will use the randomization algorithm to determine the group allocation and the treatment number(s) to be used for the child for MMR vaccination (Inv_MMR_Min, Inv_MMR_Med, Com_MMR_L1 or Com_MMR_L2). For the second MMR dose (Inv_MMR_Release, Com_MMR_L1 or Com_MMR_L2), the system will allocate a new treatment number according to the treatment received at Dose 1.
For allocation of co-administered vaccines (Havrix and Varivax to all children, and Prevnar 13 to the children in the US), the site staff in charge of the vaccination will select a treatment number available for the vaccine dose to be used for the child. Please reference the SPM for further details.
Section 5.5.Outline of study procedures
Table 5 List of study procedures
Age at enrollment 12-15 monthsEpoch Primary Epoch
Type of contact Visit 1 Visit 2 Visit 3 Visit 4Timepoints Day 0 Day 42 Day 84 Day 222Sampling
timepointsPre-Vacc
Post-Vacc 1
Post-Vacc 2
Informed consent ●Check inclusion/exclusion criteria ●Check contraindications to subsequent vaccination and record in the eCRF, if present (see Section 6.5)
●
Check warnings and precautions O O O1Collect demography data ●Medical history ●Physical examination ● O2 O2Measure/record height and weight ●Vaccination history (protocol specific vaccines) 3 ●Pre-vaccination body temperature ● ●Randomization ORecording of treatment numbers ● ●Blood sampling for antibody determination (~5 mL) ● ● ●4Study vaccinations ● ●Co-administered vaccines ●5Second dose of Havrix and/or varicella vaccine O x1
Distribution of Diary Cards O O ODaily post-vaccination recording of solicited local adverse events by children’s parent(s)/LAR(s) on Diary Cards (Day 0-3 after both vaccinations)
● ●
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311019-MAY-2015
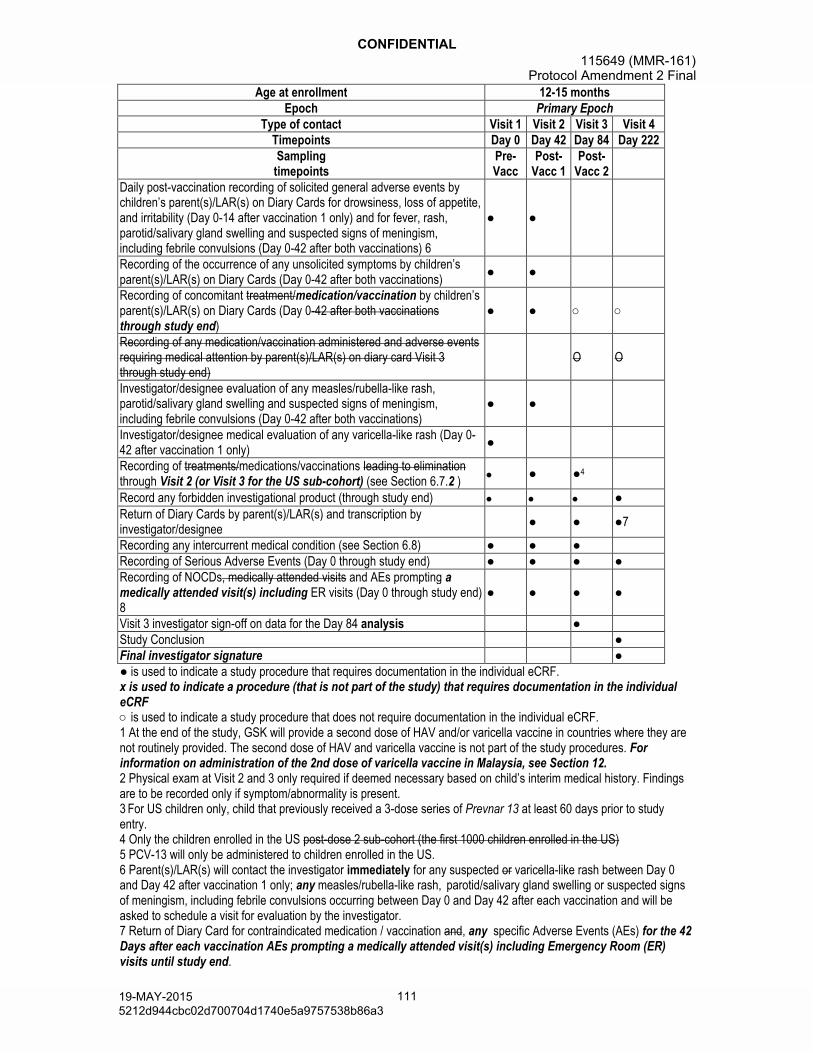
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 111
Age at enrollment 12-15 monthsEpoch Primary Epoch
Type of contact Visit 1 Visit 2 Visit 3 Visit 4Timepoints Day 0 Day 42 Day 84 Day 222Sampling
timepointsPre-Vacc
Post-Vacc 1
Post-Vacc 2
Daily post-vaccination recording of solicited general adverse events by children’s parent(s)/LAR(s) on Diary Cards for drowsiness, loss of appetite, and irritability (Day 0-14 after vaccination 1 only) and for fever, rash, parotid/salivary gland swelling and suspected signs of meningism, including febrile convulsions (Day 0-42 after both vaccinations) 6
● ●
Recording of the occurrence of any unsolicited symptoms by children’s parent(s)/LAR(s) on Diary Cards (Day 0-42 after both vaccinations)
● ●
Recording of concomitant treatment/medication/vaccination by children’s parent(s)/LAR(s) on Diary Cards (Day 0-42 after both vaccinationsthrough study end)
● ● ○ ○
Recording of any medication/vaccination administered and adverse events requiring medical attention by parent(s)/LAR(s) on diary card Visit 3 through study end)
O O
Investigator/designee evaluation of any measles/rubella-like rash, parotid/salivary gland swelling and suspected signs of meningism, including febrile convulsions (Day 0-42 after both vaccinations)
● ●
Investigator/designee medical evaluation of any varicella-like rash (Day 0-42 after vaccination 1 only)
●
Recording of treatments/medications/vaccinations leading to eliminationthrough Visit 2 (or Visit 3 for the US sub-cohort) (see Section 6.7.2 )
● ●4
Record any forbidden investigational product (through study end) ●Return of Diary Cards by parent(s)/LAR(s) and transcription by investigator/designee
● ● ●7
Recording any intercurrent medical condition (see Section 6.8) ● ● ●Recording of Serious Adverse Events (Day 0 through study end) ● ● ● ●Recording of NOCDs, medically attended visits and AEs prompting a medically attended visit(s) including ER visits (Day 0 through study end) 8
● ● ● ●
Visit 3 investigator sign-off on data for the Day 84 analysis ●Study Conclusion ●Final investigator signature ●● is used to indicate a study procedure that requires documentation in the individual eCRF.x is used to indicate a procedure (that is not part of the study) that requires documentation in the individual eCRF ○ is used to indicate a study procedure that does not require documentation in the individual eCRF.1 At the end of the study, GSK will provide a second dose of HAV and/or varicella vaccine in countries where they are not routinely provided. The second dose of HAV and varicella vaccine is not part of the study procedures. For information on administration of the 2nd dose of varicella vaccine in Malaysia, see Section 12.2 Physical exam at Visit 2 and 3 only required if deemed necessary based on child’s interim medical history. Findings are to be recorded only if symptom/abnormality is present.3 For US children only, child that previously received a 3-dose series of Prevnar 13 at least 60 days prior to study entry. 4 Only the children enrolled in the US post-dose 2 sub-cohort (the first 1000 children enrolled in the US)5 PCV-13 will only be administered to children enrolled in the US.6 Parent(s)/LAR(s) will contact the investigator immediately for any suspected or varicella-like rash between Day 0 and Day 42 after vaccination 1 only; any measles/rubella-like rash, parotid/salivary gland swelling or suspected signs of meningism, including febrile convulsions occurring between Day 0 and Day 42 after each vaccination and will be asked to schedule a visit for evaluation by the investigator. 7 Return of Diary Card for contraindicated medication / vaccination and, any specific Adverse Events (AEs) for the 42 Days after each vaccination AEs prompting a medically attended visit(s) including Emergency Room (ER) visits until study end.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 112
8 Events to be reported will be new onset chronic disease(s) (NOCD(s)) (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies)and AEs prompting medically attended visit s including Emergency Room (ER) visits.
Table 6 Intervals between study visits
Visit Optimal Visit Day* Interval§ Allowed intervalVisit 1 Day 0Visit 2 Day 42 Visit 1 Visit 2 35 days – 77 days †
Visit 3 Day 84 Visit 2 Visit 3 35 days– 77 days †
Visit 4 Day 222 Visit 2 Visit 4 180 days – 210 days* Whenever possible the investigator should arrange study visits on this optimal visit day.§ Visit 1 serves as a reference date used to define the interval for Visit 2; Visit 2 serves as a reference date used to define the intervals for Visits 3 and 4.† Children will not be eligible for inclusion in the ATP immunogenicity analyses if study visits are outside the allowed interval.
Section 5.6.3.1.Check and record concomitant medication/vaccination and intercurrent medical conditions
At Visits 2 for all subjects and also at Visit 3 for the US sub-cohort it must be verified if the child has experienced or is experiencing any intercurrent medical condition listed in Section 6.8. If it is the case, the condition(s) must be recorded in the eCRF. In addition, any concomitant treatment/medications/vaccinations medications/products that may lead to the elimination of a child from ATP analysesmust be recorded in the eCRF through Visit 2 for the Non US sub-cohort and through Visit 3 for the US sub-cohort. Refer to Section 6.7.1.
Section 5.6.3.5.Blood sampling for safety or immune response assessments
As specified in the List of Study Procedures in Section 5.5 (Table 5), blood samples are taken during certain study visits. Blood samples should be obtained prior to vaccination during visits and at a different anatomical location from the sites of vaccination. Refer to the Module on Biospecimen Management in the SPM for general handling of blood samples.
Section 5.6.3.8.Recording of AEs and SAEs
At Visit 3 diary cards will be provided to the child’s parent(s)/LAR(s) to record:
- Any medication / vaccination (from Day 84 to study end).
- Any AEs prompting a medically attended visit(s) (from Day 84 to study end).
Collection and verification of completed diary cards will occur during discussion with the child’s parent(s)/LAR(s) at post-vaccination visits. The investigator/ designee will transcribe the required collected information into the eCRF.
Collection and verification of completed diary cards will occur during discussion with the child’s parent(s)/LAR(s) at Visit 4. The investigator/ designee will transcribe the collected information on any NOCDs, investigational products and AEs prompting a medically attended visit(s) including ER visits into the eCRF.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 113
Section 5.7.2.Biological samples
Table 7 Biological samples
Sample type Quantity Unit TimepointsBlood ~ 5 ml Visit 1Blood ~ 5 ml Visit 2Blood ~ 5 ml Visit 3*
ml = Milliliter* Only the US post-dose 2 sub-cohort will have a blood sample taken at Visit 3 (Day 84)
Section 5.7.3.Laboratory assays
The central laboratory responsible for sample handling/processing for sites in the United States (including Puerto Rico)is BARC USA Inc. 5, Delaware Drive, Lake Success, NY 11042-1114, U.S.A.
Section 5.7.4.1.Immunological read-outs
Table 9 Immunological read-outs
Blood sampling timepointSub-cohort Name
No. of children
ComponentComponentspriority rank
Type of contact and timepoint
Sampling timepoint
Visit 1 (Day 0) Pre-VaccUS post-dose 2
US Non post dose 2 Non US
4500
Mumps Virus Strain Mu90 Ab(PRNT)*
1
Measles Virus Ab.IgG2
Rubella Virus Ab.IgGMumps Virus Jeryl Lynn
135 Ab.IgG3
Visit 2 (Day 42) Post-Vacc 1US post-dose 2 +
US Non post dose 2 +Non US
4500
Mumps Virus Strain Mu90 Ab (PRNT)*
1
Measles Virus Ab.IgG2
Rubella Virus Ab.IgGMumps Virus Jeryl Lynn
135 Ab.IgG3
Visit 3 (Day 84) Post-Vacc 2 US post dose 2 1000
Measles Virus Ab.IgG1
Rubella Virus Ab.IgGMumps Virus Jeryl Lynn
135 Ab.IgG2
Vacc = vaccination; Ab = Antibody; IgG = Immunoglobulin class G* The mumps Plaque Reduction Neutralization Test (PRNT)
All children will be tested for antibodies against measles, mumps and rubella virus by ELISA and mumps virus by PRNT with samples taken at Days 0 and 42. Only the first 1000 children in the US (US post-dose 2 sub-cohort)sub-cohort will be tested for antibodies against measles, mumps and rubella virus by ELISA at Day 84.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 114
Section 5.7.5.Immunological correlates of protection
The blood sample taken at Visit 3 (for only those children in the US post-dose 2 immunogenicity sub-cohort) will be the timepoint used to determine sub-optimal responders, i.e., an individual immunological assay result which is below the generally accepted immunological correlate of protection. A list of sub-optimal responders will be communicated to the investigators in a timely manner (no later than 12 months after the last child of the US post-dose 2 sub-cohort is sampled at Visit 3). Sub-optimal responder listings will not be provided based on Visit 2 laboratory data as all children should receive a second dose of study vaccine(s) at Visit 2. The only exception to this will be any child who does not receive the indicated study vaccines at Visit 2, but for whom a Visit 2 blood sample is obtained. For these children, sub-optimal responder listings will be based on the Visit 2 blood sample. If immune response data (post dose 2 at Day 84, or for children who failed to get the second dose, then the post dose 1 sample at visit 2) indicate a suboptimal seroresponse to either the Inv_MMR or to Com_MMR vaccine a rescue plan will be implemented and revaccination with M-M-R®II (or Priorix in ex-US settings where the vaccine is licensed) will be offered to children who fail to reach the seroresponse threshold for any MMR component.
For children enrolled in the US, the blood sample taken at Visit 3 will be the timepoint used to determine sub-optimal responders, i.e., an individual immunological assay result which is below the generally accepted immunological correlate of protection. A list of sub-optimal responders will be communicated to the investigators in a timely manner (no later than 12 months after the last blood sampling date of the study).
If immune response data (post dose 2 at Visit 3) indicate a suboptimal seroresponse to either the Inv_MMR or to Com_MMR vaccine, a rescue plan will be implemented and revaccination with M-M-R®II will be offered to children who fail to reach the seroresponse threshold for any MMR component.
For any child enrolled in the US who does not receive the indicated study vaccines at Visit 2, but for whom a Visit 2 blood sample is obtained; the sub-optimal responder listings will be based on the Visit 2 blood sample. If immune response data (post dose 1 for children who failed to get the second dose) indicates a suboptimal seroresponse to either the Inv_MMR or to Com_MMR vaccine, a rescue plan will be implemented and revaccination with M-M-R®II will be offered to children who fail to reach the seroresponse threshold for any MMR component.
For children enrolled outside the US, sub-optimal responder listings will not be provided based on Visit 2 laboratory data as all children should receive a second dose of study vaccine(s) at Visit 2. For any child that does not receive the indicated study vaccines at Visit 2, but for whom a Visit 2 blood sample is obtained, the sub-optimal responder listings will be based on the Visit 2 blood sample. If immune response data (post dose 1) for children who failed to get the second dose, indicates a suboptimal seroresponse to either the Inv_MMR or to Com_MMR vaccine, a rescue plan will be implemented and revaccination with Priorix will be offered to children who fail to reach the seroresponse threshold for any MMR component.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 115
Section 6.1.Description of study vaccines
Table 10 Study vaccines
Treatment nameVaccine/product
nameFormulation Presentation Volume
No. of doses
Inv_MMR_Min Inv_MMR_Min
Measles virus (Schwarz strain) 10 3.1 CCID50; Mumps virus (RIT4385 strain) 10 4.1
CCID50; Rubella virus (Wistar RA 27/3 strain) 10 2.9 CCID50; anhydrous lactose; sorbitol; mannitol; amino acids; neomycin
Lyophilized pellet in a vial for reconstitution with water for injection
0.5 ml † 1*
Inv_MMR Diluent water for injection Vial 0.5 ml 1*
Inv_MMR_MedInv_MMR_Med
Measles virus (Schwarz strain) 10 3.4 CCID50; Mumps virus (RIT4385 strain) 10 4.3
CCID50; Rubella virus (Wistar RA 27/3 strain) 10 3.2 CCID50; anhydrous lactose; sorbitol; mannitol; amino acids; neomycin
Lyophilized pellet in a vial for reconstitution with water for injection
0.5 ml † 1*
Inv_MMR Diluent water for injection Vial 0.5 ml 1*
Inv_MMR_Release Inv_MMR_Release
Measles virus (Schwarz strain) ≥10 3.0 CCID50; Mumps virus (RIT4385 strain) ≥10 4.3 CCID50; Rubella virus (Wistar RA 27/3 strain) ≥10 3.0 CCID50; anhydrous lactose; sorbitol; mannitol; amino acids; neomycin
Lyophilized pellet in a vial for reconstitution with water for injection
0.5 ml † 1**
Inv_MMR_Release Diluent***
water for injection Vial 0.5 ml 1**
Com_MMR_L1Com_MMR_L1
Measles virus ≥1,000 TCID50; Mumps virus ≥12,500 TCID50; Rubella virus ≥1,000 TCID50; sorbitol 14.5 mg; sodium phosphate, sucrose 1.9 mg; sodium chloride; hydrolyzed gelatin 14.5 mg; recombinant human albumin ≤0.3 mg; fetal bovine serum <1 ppm; other buffer and media ingredients; neomycin approximately 25 mcg
Lyophilized pellet in a vial for reconstitution with water for injection
0.5 ml † 2
Com_MMR_L1 Diluent‡
water for injection
Vial or a Syringe (depending on the country)
0.5 ml 2
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 116
Treatment nameVaccine/product
nameFormulation Presentation Volume
No. of doses
Com_MMR_L2Com_MMR_L2
Measles virus ≥1,000 TCID50; Mumps virus ≥12,500 TCID50; Rubella virus ≥1,000 TCID50; sorbitol 14.5 mg; sodium phosphate, sucrose 1.9 mg; sodium chloride; hydrolyzed gelatin 14.5 mg; recombinant human albumin ≤0.3 mg; fetal bovine serum <1 ppm; other buffer and media ingredients; neomycin approximately 25 mcg
Lyophilized pellet in a vial for reconstitution with water for injection
0.5 ml † 2
Com_MMR_L2 Diluent‡
water for injection
Vial or a Syringe (depending on the country)
0.5 ml 2
Varivax Varivax
A minimum of 1350 pfu of Oka/Merck varicella virus; sucrose approximately 25 mg; hydrolyzed gelatin 12.5 mg, sodium chloride 3.2 mg; monosodium L-glutamate 0.5 mg; sodium phosphate dibasic 0.45 mg; potassium phosphate monobasic 0.08 mg; potassium chloride 0.08 mg; residual components of MRC-5 cells including DNA and protein; and trace quantities of sodium phosphate monobasic; EDTA; neomycin; and fetal bovine serum
Lyophilized pellet in a vial for reconstitution with water for injection
0.5 ml † 2§
Varivax Diluent water for injection
Vial or Syringe (depending on the country)
0.5 ml 2§
Havrix Havrix
720 EU of hepatitis A virus antigen 0.25 mg aluminum (as hydroxide) Amino acid supplement (0.3% w/v) in a phosphate-buffered saline solution and polysorbate 20 (0.05 mg/mL)
Suspension in vial/ pre-filled syringe
0.5 ml 2§
Prevnar 13 Prevnar 13
2.2 mcg of the purified saccharides1, 3, 4, 5, 6A, 7F, 9V, 14, 18C, 19A, 19F, 23F 4.4 mcg of the purified saccharide 6B approximately 34 mcg CRM197 carrier protein 100 μg polysorbate 80, 295 μg succinate buffer and 125 μg aluminum as aluminum phosphate adjuvant
Suspension in vial/ pre-filled syringe
0.5 ml 1¥
CCID = Cell Culture Infectious Dose; TCID = Tissue Culture Infectious Dose; EU = ELISA Unit
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 117
ppm = Parts Per Million; pfu = Plaque Forming Unit; mL = Milliliter; mg = Milligram; mcg = Microgram; g = MicrogramMRC = Medical Research Council; DNA = Deoxyribonucleic Acid; EDTA = Ethylenediamine-Tetraacetic AcidCRM197 = Cross-Reactive Material (mutant diphtheria toxin)† Volume after reconstitution* Administered during Visit 1 for Dose 1** Administered during Visit 2 for Dose 2 for the Inv_MMR_Min and Inv_MMR_Med groups ONLY*** The Inv_MMR_Release Diluent for Dose 2 is distinct from the Inv_MMR Diluent for Dose 1‡ The Com_MMR_L1 Diluent may be distinct from the Com_MMR_L2 Diluent§ At the end of the study, GSK will provide a second dose of HAV and/or varicella vaccine in countries where they are not routinely provided. Not applicable for subjects enrolled in US. For information on administration of the 2nd dose of varicella vaccine in Malaysia, see Section 12. The second dose of HAV and varicella vaccine is not part of the study procedures.¥ PCV-13 will only be administered to children in the US.
Section 6.2.Storage and handling of study vaccines
Any temperature excursion, outside the range of 0°C to +8°C (32F to 46F) for refrigerated vaccines, above outside the range of -15°C to -25C (5°F to -13F) for Inv_MMR_Min and Inv_MMR_Med vaccines, or -15°C to -50°C (5F to -58F) for the Varivax vaccine (in the US), must be reported to the sponsor as soon as detected. Following an exposure to a temperature excursion, vaccines will not be used until written approval has been given by the Sponsor.
For refrigerated vaccinesOutside the US, adequate actions must be taken for refrigerated vaccines in case of temperature excursion between 0°C and +2°C (32°F and 36°F) towhich go back to the defined range +2°C to +8°C (36°C to 46°F). The impacted study vaccine(s)/product(s) can still be administered, but the site should avoid re-occurrence of temperature excursion.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311719-MAY-2015
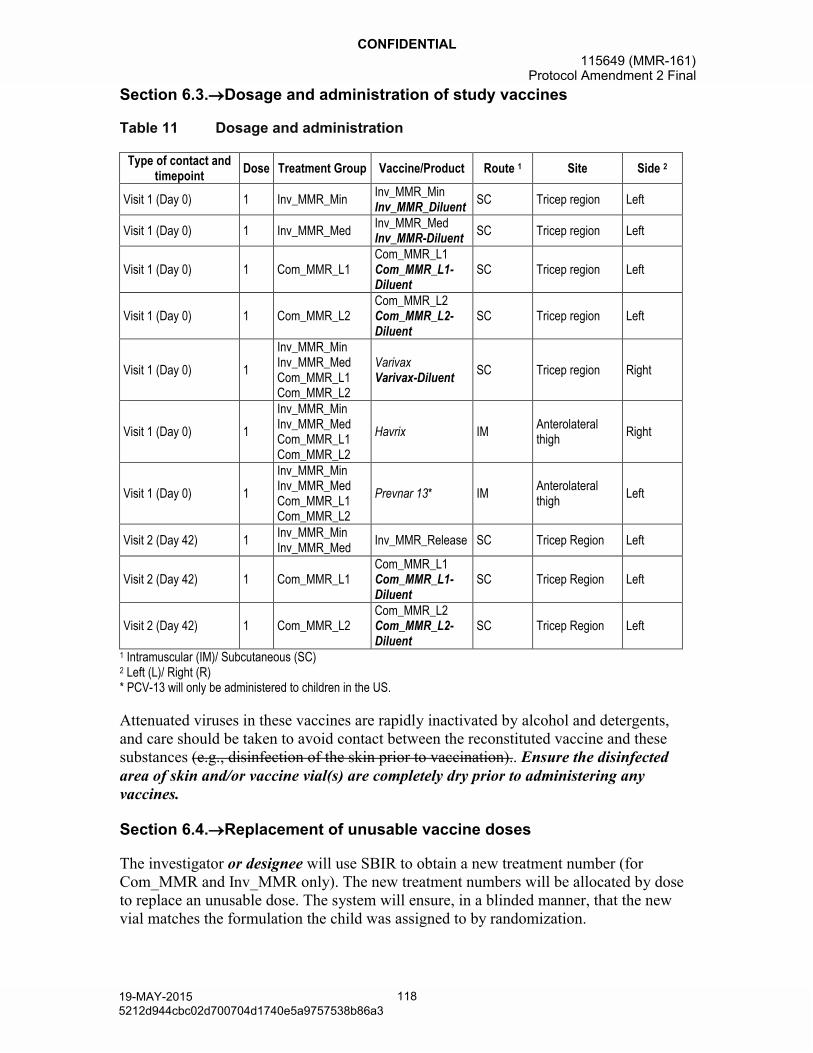
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 118
Section 6.3.Dosage and administration of study vaccines
Table 11 Dosage and administration
Type of contact and timepoint
Dose Treatment Group Vaccine/Product Route 1 Site Side 2
Visit 1 (Day 0) 1 Inv_MMR_MinInv_MMR_MinInv_MMR_Diluent
SC Tricep region Left
Visit 1 (Day 0) 1 Inv_MMR_MedInv_MMR_MedInv_MMR-Diluent
SC Tricep region Left
Visit 1 (Day 0) 1 Com_MMR_L1Com_MMR_L1Com_MMR_L1-Diluent
SC Tricep region Left
Visit 1 (Day 0) 1 Com_MMR_L2Com_MMR_L2Com_MMR_L2-Diluent
SC Tricep region Left
Visit 1 (Day 0) 1
Inv_MMR_Min Inv_MMR_Med Com_MMR_L1 Com_MMR_L2
VarivaxVarivax-Diluent
SC Tricep region Right
Visit 1 (Day 0) 1
Inv_MMR_Min Inv_MMR_Med Com_MMR_L1 Com_MMR_L2
Havrix IMAnterolateral thigh
Right
Visit 1 (Day 0) 1
Inv_MMR_Min Inv_MMR_Med Com_MMR_L1 Com_MMR_L2
Prevnar 13* IMAnterolateral thigh
Left
Visit 2 (Day 42) 1Inv_MMR_Min Inv_MMR_Med
Inv_MMR_Release SC Tricep Region Left
Visit 2 (Day 42) 1 Com_MMR_L1Com_MMR_L1Com_MMR_L1-Diluent
SC Tricep Region Left
Visit 2 (Day 42) 1 Com_MMR_L2Com_MMR_L2Com_MMR_L2-Diluent
SC Tricep Region Left
1 Intramuscular (IM)/ Subcutaneous (SC)2 Left (L)/ Right (R)* PCV-13 will only be administered to children in the US.
Attenuated viruses in these vaccines are rapidly inactivated by alcohol and detergents, and care should be taken to avoid contact between the reconstituted vaccine and these substances (e.g., disinfection of the skin prior to vaccination).. Ensure the disinfected area of skin and/or vaccine vial(s) are completely dry prior to administering any vaccines.
Section 6.4.Replacement of unusable vaccine doses
The investigator or designee will use SBIR to obtain a new treatment number (for Com_MMR and Inv_MMR only). The new treatment numbers will be allocated by dose to replace an unusable dose. The system will ensure, in a blinded manner, that the new vial matches the formulation the child was assigned to by randomization.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 119
Section 6.7.1.Medications/products that may lead to the elimination of a child from ATP analyses
Chronic administration (defined as 14 or more consecutive days) of immunosuppressants or other immune-modifying drugs during the period starting at vaccination and ending at the immunogenicity evaluation at Visit 2 (or at Visit 3 for the US post-dose 2 sub-cohort).
- For corticosteroids, this will mean prednisone 0.5 mg/kg/day, or equivalent.
- Inhaled and topical steroids are allowed.
Planned administration/ administration of a vaccine not foreseen by the study protocol during the period starting at vaccination and ending at Visit 2 (or ending at Visit 3 for the US post-dose 2 sub-cohort). Please Note:
- Inactivated Flu and Hib vaccines may be given at any time during the study, including the day of study vaccination (Flu and Hib vaccines must be administered at a different location than the study vaccine/s).
- Any age appropriate vaccine may be given starting at Visit 2, (or starting at Visit 3 for the US post-dose 2 sub-cohort) and any time thereafter.
Administration of immunoglobulins and/or any blood products during the period starting at vaccination and ending at the last blood draw at Visit 2 (or at Visit 3 for the US post-dose 2 sub-cohort).
Section 6.7.2.Time window for recording concomitant medication/vaccination in the eCRF
All concomitant medications, with the exception of vitamins and/or dietary supplements, administered at ANY time during the period starting 30 days preceding the administration of the first dose of study vaccine (i.e., 30 days prior to Day 0) and ending at Day 42 after the second dose of study vaccine (Visit 3Visit 2 (Non US sub-cohort) or Visit 3 (US sub-cohort) must be recorded in the eCRF. Concomitant medication/vaccination should be recorded by the child’s parent(s)/LAR(s) in the subject diary between Day 0 and Day 42 after each vaccination. The investigator will transcribe this information into the eCRF as indicated below.
Any treatments and/or medications specifically contraindicated, e.g., any immune-globulins, other blood products and any immune modifying drugs administered during the period starting 180 days prior to the vaccination throughto Visit 2 (Non US sub-cohort) or Visit 3 (US sub-cohort) are to be recorded in the eCRF with the generic name of the medication (trade names are allowed for combination drugs only), medical indication, total daily dose, route of administration, start and end dates of treatment.
Any vaccine not foreseen in the study protocol administered in the period starting 30 days before the first dose of study vaccine and ending at vaccination through Visit 2 (or at Visit 3 for the US post-dose 2 sub-cohort), must be recorded in the eCRF. This includes any vaccines permitted to be given at or after Visit 2.The second dose of HAV and/or varicella vaccine administered at Visit 4 must be recorded in the eCRF (see Section
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a311919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 120
5.5). In addition, any previous administration of measles, mumps, and rubella virus containing vaccines, regardless of when they were given, must be recorded in the eCRF
Any medication administered for the treatment of an AE should be recorded in the child’s eCRF.
Section 6.8.ntercurrent medical conditions that may lead to elimination from an ATP cohort
Children may be eliminated from the ATP cohort if they are confirmed to have an immunodeficiency condition, or if they develop measles, mumps, or rubella, in the interval between vaccination and the collection of the blood specimen for immunogenicity at the last blood draw at Visit 2 (or at Visit 3 for the US post-dose 2 sub-cohort).
Section 8.1.3.Solicited adverse events
Table 13 Solicited general adverse events
Adverse Event Solicited follow-up periodDrowsiness
from Day 0 to Day 14 after vaccination dose 1Loss of appetiteIrritabilityVaricella-like rash* from Day 0 to Day 42 after vaccination dose 1
Fever (defined as temperature ≥38C/100.4F)**
from Day 0 to Day 42 after each vaccinationMeasles/rubella-like rash**Other rash (not measles/rubella-like nor varicella-like)Parotid gland / Salivary gland swelling**Meningism including febrile convulsions**
* To be confirmed by a physician.** Fever is defined as temperature ≥38C/100.4F. The preferred route for recording temperature in this study will be axillary. Temperature will be recorded daily from Day 0 to Day 42 after each vaccination. The child’s parent(s)/LAR(s) are requested to record the child’s body temperature each evening at bedtime. Should additional temperature measurements be performed at other times of day, the highest temperature will be recorded.
Section 8.1.5.Adverse events of specific interest
AEs of specific interest for safety monitoring include NOCDs (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies) and AEs prompting ER visits. Occurrences of AEs of specific interest, including NOCDs and AEs prompting ER visits, will be reported throughout the study, through a minimum of six months post after the last dose 2of MMR vaccine, whether or not they are considered to be possibly related to the treatment administration. Medical documentation of the events will be reported in appropriate targeted follow-up forms included in the eCRF. These events also have to be reported as AE or SAE as appropriate in the eCRF.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312019-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 121
Section 8.2.1.Time period for detecting and recording adverse events and serious adverse events
All AEs starting from Day 0 through 42 days after the administration of each dose of study vaccine (at Day 42 or 84) must be recorded on the Adverse Event screen in the child’s eCRF, irrespective of intensity or whether or not they are considered vaccination-related. The standard time period for collecting and recording SAEs will begin at the first receipt of study vaccines and will be collected throughout the entire study. See Section 8.3 for instructions on reporting of SAEs.
The standard time period for collecting and recording of AEs of specific interest, including NOCDs and AEs prompting ER visits, will begin at the first receipt of study vaccine and will end at the last study visit or contact following administration of the last dose of study vaccine. An overview of the protocol-required reporting periods for AEs, and SAEs is given in Table 14.
The standard time period for collecting and recording of AEs prompting a medically attended visit(s) will begin at the first receipt of study vaccine and will end at the last study visit or contact.
Table 14 Reporting periods and methods for adverse events and serious adverse events
Visit 1Vaccination 1
Visit 2Vaccination 2
Visit 3Follow-up
Visit 4Conclusion
Optimal Visit Day Day 0 Day 42 Day 84 Day 222
Solicitation period Post-vaccination
Day0-3
Day 4-14
Day15-42
Day0-3
Day 4-14
Day15-42
Study activity
Reporting solicited local AEs
DC/RDE
DC/RDE
Reporting solicited drowsiness, loss of appetite and irritability
DC/RDE
Reporting varicella-like rash
DC/RDE
Reporting solicited fever, rash (measles / rubella rash and other rash), parotid/salivary gland swelling and meningism including febrile convulsions
DC/RDE DC/RDE
Reporting unsolicited AEs
DC/RDE DC/RDE DC DC
Reporting NOCD(s)* and AEs prompting a medically attended visit(s) (including ER visits)
DC/RDE DC/RDE DC/RDE DC/RDE
Reporting ALL SAEs DC/RDE DC/RDE DC/RDE DC/RDE
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312119-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 122
AEs = Adverse Events; DC = Diary Card; RDE = Remote Data Entry; SAEs = Serious Adverse EventsNOCD = New Onset Chronic Disease; ER = Emergency Room.* Occurrence of NOCD(s) (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies) up to 6 months after vaccination.
Section 8.2.2.2.1.Assessment of intensity
Temperature/ fever: Temperature should be recorded daily, from Day 0 through Day 42 after each vaccination. Parent(s)/LAR(s) are requested to record on the diary card the child’s body temperature measured each evening at bedtime (while at home ONLY one temperature route [axillary preferred] should be used consistently for all measurements in a given child). Should additional temperature measurements be performed at other times of day, the highest temperature will be recorded.
Rash/ exanthem:
Parent(s)/LAR(s) will be provided with a description of rashes. Parent(s)/LAR(s) will be asked to contact the study site immediately if their child develops a rash that could be a measles/rubella-like rash (Day 0-42 after both each vaccinations, as applicable) or a varicella-like rash (Day 0-42 after Vaccination 1 only). If the investigator/designee also suspects that the rash could possibly be a measles/rubella-like rash or a varicella-like rash, a visit should be arranged as soon as possible; ideally within 48 hours after rash onset, or as soon as possible thereafter.
AEs of specific interest:
Occurrences of AEs of specific interest, including NOCDs and AEs prompting ER visits, will be reported throughout the study, up to six months post-after the dose 2of MMR vaccine, whether or not they are considered to be possibly related to the treatment administration. Medical documentation of the events will be reported in appropriate targeted follow-up forms included in the eCRF. These events also have to be reported as AE or SAE as appropriate in the eCRF.
Section 8.2.2.4.Medically attended visits
For each solicited and unsolicited symptom the child experiences, the child’s parent(s)/LAR(s) will be asked if the child received medical attention defined as hospitalization, an emergency room visit or a visit to or from medical personnel (e.g., medical doctor) for any reason and this information will be recorded in the eCRF. Routine “well child” visit information will not be recorded in the eCRF.
Section 8.3.2.Contact information for reporting serious adverse events to GSK Biologicals
See Sponsor Information Sheet for contact details Refer to the local study contact information document.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312219-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 123
Section 8.4.1.Follow-up of adverse events and serious adverse events
Investigators will follow-up children:
With SAEs or children withdrawn from the study as a result of an AE, until the event has resolved, subsided, stabilized, disappeared, or until the event is otherwise explained, or the child is lost to follow-up.
Or, in the case of other non-serious AEs, until resolution or study end,or they are lost to follow-up.
Section 10.2.Secondary endpoints
Immunogenicity of the MMR vaccines at Day 84 post-dose 2 (US post-dose 2 sub-cohort).
Section 10.4.2.According-to-protocol cohort for analysis of safety
who have not received a vaccine leading to exclusion from the ATP cohort in the protocol as described in Section 6.7.1. up to Visit 2 for all children (except for the non US post-dose 2 sub-cohort whereand up to Visit 3 is usedfor the US sub-cohort.
Section 10. 4.3.According-to-protocol cohort for analysis of immunogenicity
who are in the US post-dose 2 sub-cohort
Section 10.7.1.Methodology for computing CI
The exact CIs for a proportion within a group will be calculated from Proc StatXact 8.1 [Clopper, 1934]. Proc StatXact 8.1 using SAS. SAS will also be used to derive the standardized asymptotic 95% CI for the group difference in proportions.
Section 10.7.4.Analysis of safety
The percentages of children who experienced at least one serious adverse event from Day 0 - study end, or who reported a NOCD (e.g., autoimmune disorders, asthma, type I diabetes, vasculitis, celiac disease, conditions associated with sub-acute or chronic thrombocytopenia and allergies), or an AE leading to an ER visit, or an AE leading to a medically attended visit from Day 0 to study end, will be calculated.
Section 12.Country specific requirements
Not applicable.
The use of varicella vaccines as a 2-dose vaccine is now under regulatory review in Malaysia. If varicella vaccines are not yet approved for a 2 dose indication at the time the second dose of varicella-containing vaccine is to be administered, the second dose of varicella-containing vaccine will be administered as part of the study procedures. If regulatory approval is granted, the 2nd dose of varicella-containing vaccine will still be administered but it will not be a part of the study procedures.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312319-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 124
Section 13.REFERENCES
Clopper CJ, Pearson, E. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 1934;26:404-413.
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312419-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 125
GlaxoSmithKline Biologicals
Clinical Research & Development
Protocol Administrative Change 1
eTrack study number and Abbreviated Title(s)
115649 (MMR-161)
Administrative changenumber:
Administrative change 1
Administrative changedate:
28 May 2014
Co-ordinating author: , Scientific Writer, XPE Pharma for GSK Biologicals
Rationale/background for changes: This Protocol Administrative Change corrects a few typing errors in Protocol Amendment1:
error in serological cut-off
a few minor errors in Appendix C describing the rationale and changes made in Amendment 1.
Amended text has been included in bold italics and deleted text in strikethrough in the following sections:
LIST OF ABBREVIATIONS
Protocol administrative change:
A protocol administrative change addresses changes to only logistical or administrative aspects of the study.
Note: Any change that falls under the definition of a protocol amendment (e.g. a change that affects the safety of subjects, scope of the investigation, study design, or scientific integrity of the study) MUST be prepared as an amendment to the protocol.
Section 4.3. Exclusion criteria for enrollment
Inactivated influenza (Flu) vaccine and Haemophilus influenzae type b conjugate vaccine (Hib) vaccines may be given at any time, including the day of study vaccination (Flu and Hib vaccines must be administered at a different location than the study vaccine/(s).
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312519-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 126
Section 5.7.3. Laboratory assays
Table 8 Humoral Immunity (Antibody determination)
System Component Method Kit / Manufacturer Unit Cut-off Laboratory*Serum Measles Virus Ab.IgG ELISA Dade Behring
EnzygnostmIU/mL 150 GSK
Biologicals**Serum Rubella Virus Ab.IgG ELISA Dade Behring
EnzygnostIU/mL 4 GSK
Biologicals**Serum Mumps Virus jeryl lynn
135 Ab.IgGELISA PPD EU/mL 105 PPD
Serum Mumps Virus Strain Mu90 Ab
PRNT In-house ED50 42.5 GSK Biologicals**
*Refer to APPENDIX B for the laboratory addresses.**GSK Biologicals laboratory refers to the Global Vaccines Clinical Laboratories (GVCL) in Rixensart, Belgium; Wavre, Belgium; Laval, Canada.Ab = Antibody; IgG = Immunoglobulin class GELISA = Enzyme-Linked Immunosorbent Assay; PRNT = Plaque Reduction Neutralization TestPPD = Pharmaceutical Product Development, Inc.IU = International Unit; mIU = Milli International Unit; EU = ELISA Unit; mL = Milliliter; ED50 = End-point Dilution 50%
Section APPENDIX C
Amendment date: 1713 February 2014Rationale/background for changes: For other MMR US Phase III studies, CBER has requested that all conditions
leading to non-routine medically attended visits be collected for the entire study in the eCRF. It has been clarified throughout the protocol that From Visit 1 (Day 0) to study end (Day 180222), all medically attended events will be recorded in the eCRF. Subjects’ routine ‘well child’ doctor visits will not be recorded in the eCRF.
Contributing authors , Director, Clinical Research and Development Leader, Vaccines for MMR and Varicella
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312619-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 127
GlaxoSmithKline Biologicals
Clinical Research & Development
Protocol Amendment 2
eTrack study number and Abbreviated Title(s)
115649 (MMR-161)
Amendment number: Amendment 2
Amendment date: 19 May 2015
Co-ordinating author: , Scientific Writer, XPE Pharma for GSK Biologicals
Rationale/background for changes:
Serological assays for the determination of antibodies against measles, rubella and varicella viruses will now be performed by a new 3rd party Contract Research Organization (CRO) named NEOMED-LABS Inc. Initially the testing was planned to be performed by GSK Biologicals’ laboratory in Rixensart. The assays have been transferred to GSK Biologicals’ laboratory in Laval. As of April 1st 2015, the GSK Biologicals’ laboratory in Laval became part of Neomed. The only change between GSK Biologicals’ laboratory in Laval and NEOMED-LABS Inc. is the name of the laboratory: assays and facilities remain the same.
Due to a delay in the availability of serologic data for the mumps Plaque Reduction Neutralization Test (PRNT) data analysis for this study will be conducted as follows: Part 1 will include a summary of measles, mumps and rubella Enzyme-Linked Immunosorbent Assay (ELISA) results post dose 1 (Day 42) and post dose 2 (Day 84). Part 2 will include a full immunogenicity analysis for post dose 1 (Day 42) and post dose 2 (Day 84) including mumps PRNT results post dose 1, and all safety data.
ELISA details were added in Appendix A.
Amended text has been included in bold italics and deleted text in strikethrough in the following sections:
Cover page
Contributing authors , Study Manager Study Data Manager , Clinical Safety and
Pharmacovigilance , GVCL Clinical Readout Lead , GVCL Study Manager
Abbreviations (added)
CRO Contract Research Organization
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312719-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 128
Section 5.7.3 Laboratory Assays
Table 8. Humoral Immunity (Antibody determination)
System Component Method Kit / Manufacturer Unit Cut-off Laboratory*
Serum Measles Virus Ab.IgG ELISA Dade Behring Enzygnost
mIU/mL 150 GSK Biologicals**NÉOMED-LABS Inc.
Serum Rubella Virus Ab.IgG ELISA Dade Behring Enzygnost
IU/mL 4 GSK Biologicals**NÉOMED-LABS Inc.
Serum Mumps Virus jeryl lynn 135 Ab.IgG
ELISA PPD EU/mL 5 PPD
Serum Mumps Virus Strain Mu90 Ab
PRNT In-house ED50 2.5 GSK Biologicals**
*Refer to APPENDIX B for the laboratory addresses.**GSK Biologicals laboratory refers to the Global Vaccines Clinical Laboratories (GVCL) in Rixensart, Belgium; Wavre,
Belgium; Laval, Canada.Ab = Antibody; IgG = Immunoglobulin class GELISA = Enzyme-Linked Immunosorbent Assay; PRNT = Plaque Reduction Neutralization TestPPD = Pharmaceutical Product Development, Inc.IU = International Unit; mIU = Milli International Unit; EU = ELISA Unit; mL = Milliliter; ED50 = End-point Dilution 50%
GSK Biologicals Global Vaccine Clinical Laboratory, Avenue Fleming, 20 –B-1300 Wavre - Belgium
NÉOMED-LABS Inc.525 Cartier blvd West - Laval - Quebec - Canada - H7V 3S8
Section 10.6.1 Sequence of analyses
The analysis will be performed in two steps:
A summary of post dose 1 (Day 42) and post dose 2 (Day 84) GMCs and seroresponse rates for measles, mumps and rubella (by ELISA) will be generated by independent statisticians once all CRF study data are available and cleaned and measles, mumps, and rubella ELISA testing has been fully completed.
A final analysis, including full immunogenicity analysis for post dose 1 (Day 42) and post dose 2 (Day 84) including mumps PRNT results post dose 1 and all safety data (including all immunogenicity and safety analyses), will be performed once the final serology data are available.
A final analysis of immunogenicity data and solicited symptoms up to Day 84 will be performed as soon as all immunogenicity data and reactogenicity data (i.e. solicited
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312819-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 129
symptoms) up to Visit 2 are available and cleaned. No clinical report will be written at this time.
A final analysis of unsolicited AEs from Day 0 to Day 42 following each vaccination, and of SAEs and specific AEs covering the period from Day 0 to study end (including the 6-months safety follow-up) will be performed at the end of the study.
These two analyses will be combined in the final clinical report.
For the first analysis step, access to group attribution and individual laboratory results will be limited to statisticians at an independent data analysis center. Members of the GSK study team will only be able to view summary tables (GMCs and seroresponse rates per group, and GMC ratios and differences in seroresponse rates between groups), thereby ensuring that they remain blinded to the treatment group attribution until all data are available and the final analysis is carried out.
A separate charter will be written to provide further details of the scope of the first analysis step, and to clarify the unblinding procedure and attribution of elimination codes by the independent statisticians.
Following unblinding for the analysis up to Day 84, accessibility to group attribution will be limited to the statisticians until all study procedures pertaining to the active phase and the 6-months safety follow-up are completed for all children.
Section 10.7.4. Analysis of safety
the number and percentage of children reporting fever by half degree (°C) cumulative increments from Day 0 - Day 14, from Day 0 - Day 42 and from Day 5 - Day 12 after each vaccination. Similar tabulations will be performed for any fever with a causal relationship to vaccination and for any fever resulting in a medically attended visit . In addition, the prevalence of any and grade 3 fever will be presented graphically over time after vaccination(s). The distribution of maximum temperature recorded over 43 days after vaccination(s) will be displayed as reverse cumulative curves for each Inv_MMR vaccine lot and pooled Com_MMR lots;
APPENDIX A LABORATORY ASSAYS
Measles virus and rubella virus antibody concentrations will be determined by commercial immunoassay kits according to GSK standard operating procedures (SOPs).Anti-measles ELISA: Anti-measles Ab concentrations will be measured using a commercial ELISA kit (Enzygnost/Dade Behring), which is based on a single point quantification Ab method. The assay technical cut-off is 150 milli-international units (mIU)/mL. The assay will be performed on human serum at GSK Biologicals’ laboratory or another laboratory designated by GSK Biologicals
Anti-rubella ELISA: Anti-VZV Ab concentrations will be measured using a commercial ELISA kit (Enzygnost/Dade Behring), which is based on a single point quantification Ab method. The assay technical cut-off is 4 milli-international units (mIU)/mL. The assay will be performed on human serum at GSK Biologicals’ laboratory or another laboratory designated by GSK Biologicals
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a312919-MAY-2015

CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
19-MAY-2015 130
Anti-mumps ELISA: Anti-mumps wild type IgG Ab concentrations are measured by using the PPD ELISA assay. Solid phase of the assay (96 wells microplate) consist of wells coated with Mumps Jeryl Lynn® 135 antigen (used in the M-M-R® II and ProQuad™ vaccines and is considered by CBER to be a WT-like strain). Single dilutions of serum sample are tested. The assay technical cut-off is 5 5 antibody units (AbU)/mL.
Mumps antibody concentration will be determined at PPD using a validated ELISA, which measures reactivity to the Jeryl Lynn strain of the mumps virus.
The GSK Mumps PRNT is an in vitro functional assay based on the indirect quantification of neutralized virus (MU90/Lo1 mumps virus). Serial dilutions of sera are mixed with challenge virus and incubated with cultured Vero cells that are sensitive to the virus. Anti-Mumps antibodies present in the human sera bind on the surface of the virus and block viral attachment to receptors of susceptible cells. The infectivity of the challenge virus is reduced as measured by the reduction of a viral cytopathic effect (CPE) in Vero cells. This decrease is measured by detecting the reduction of viral plaques in the assay. Plaques are counted (visually or automatically) after immunostaining of the infected cells. The neutralization titer corresponds to the reciprocal serum dilution that reduces the number of plaques by 50% when compared to plaques induced by the challenge virus in the absence of serum. The assay technical cut-off is 2.5 End-point Dilution 50% (ED50). The GSK Mumps PRNT is performed in 96 well plates and is derived from the National Institute for Biological Standards and Control (NIBSC) mumps PRNT procedure.
APPENDIX B CLINICAL LABORATORIES
Table 24 GSK Biologicals’ laboratories
Laboratory AddressGSK Biologicals Global Vaccine Clinical Laboratory, Rixensart
Biospecimen Reception - B7/44Rue de l'Institut, 89 - B-1330 Rixensart - Belgium
GSK Biologicals Global Vaccine Clinical Laboratory, North America-Laval
Biospecimen Reception - Clinical Serology525, Cartier Ouest Laval, Quebec Canada H7V 3S8
GSK Biologicals Global Vaccine Clinical Laboratory, Wavre-Nord Noir Epine
Avenue Fleming, 20 - B-1300 Wavre - Belgium
Table 25 Outsourced laboratories
Laboratory AddressBARC USA Inc. 5, Delaware Drive
Lake SuccessNY 11042-1114USA
NÉOMED-LABS Inc. 525, Cartier Ouest Laval, Quebec Canada H7V 3S8
PPD, Inc. 466 Devon Park DriveWayne, PA 19087-1816 – USA
CONFIDENTIAL115649 (MMR-161)
Protocol Amendment 2 Final
5212d944cbc02d700704d1740e5a9757538b86a313019-MAY-2015
