computational spectroscopy ii. ab initio methods chemistry 713 updated: january 29, 2008
Post on 21-Dec-2015
216 views
TRANSCRIPT

Computational SpectroscopyII. ab initio Methods
Chemistry 713
Updated: January 29, 2008

(b) Optimized Structures and Rotational Constants
Calculation of the ab initio energy at a single molecular geometry my involve significant computational effort.
To find a relative minimum, the geometry is changed along the line of steepest decent (fall line).
The program calculates the energies for slight changes in the geometry along each atomic coordinate to determine the fall line.
Second derivatives of the energy w.r.t. the coordinates are used to determine how far to go along the fall line. (Some methods allow analytic second derivatives: HF, B3LYP, MP2, CASSCF; for others the derivatives must be computed numerically).
Process is repeated until a minimum is found. (all 1st derivatives equal zeros and second derivatives are positive.)
Gaussian allows various convergence criteria to be set.

G03 example files Gaussian provides over 700 example input files and the corresponding
output files. Mac (UNIX)
Input files: /Applications/g03/tests/com/testxxx.com Output files: /Applications/g03/tests/rs6k/testxxx.log Test file index: /Applications/g03/tests/test.idx
Windows Input files: C:\G03W\tests\gif\testxxx.gif Output files: … missing Test file index: C:\G03W\tests\tests.idx
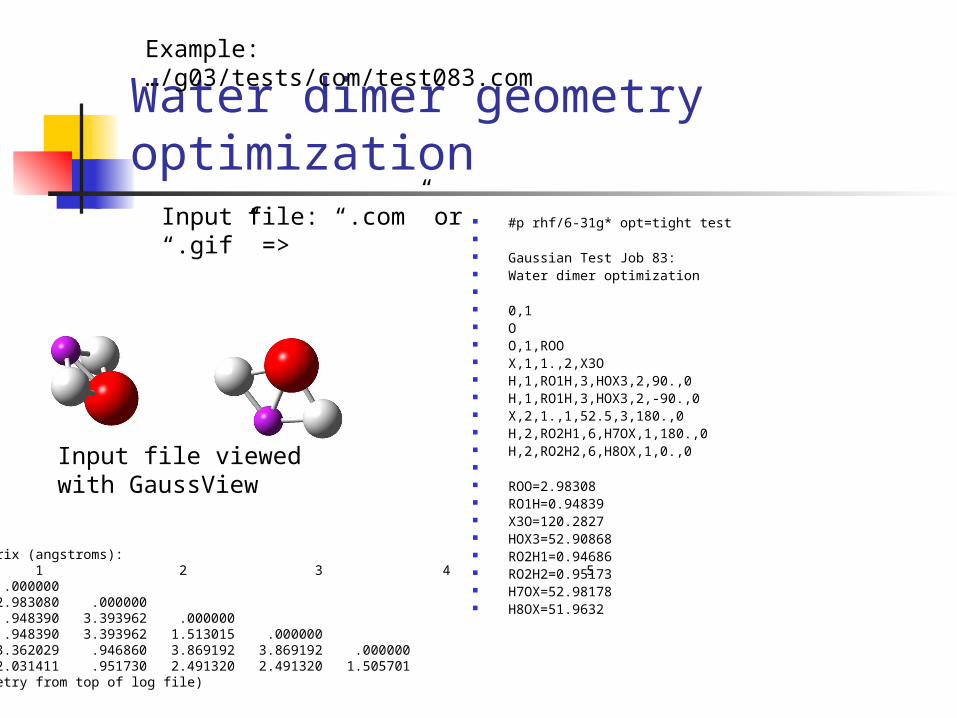
Water dimer geometry optimization #p rhf/6-31g* opt=tight test Gaussian Test Job 83: Water dimer optimization 0,1 O O,1,ROO X,1,1.,2,X3O H,1,RO1H,3,HOX3,2,90.,0 H,1,RO1H,3,HOX3,2,-90.,0 X,2,1.,1,52.5,3,180.,0 H,2,RO2H1,6,H7OX,1,180.,0 H,2,RO2H2,6,H8OX,1,0.,0 ROO=2.98308 RO1H=0.94839 X3O=120.2827 HOX3=52.90868 RO2H1=0.94686 RO2H2=0.95173 H7OX=52.98178 H8OX=51.9632
Example: …/g03/tests/com/test083.com
Input file: “.com” or “.gif” =>
Input file viewed with GaussView
Distance matrix (angstroms): 1 2 3 4 5 1 O .000000 2 O 2.983080 .000000 3 H .948390 3.393962 .000000 4 H .948390 3.393962 1.513015 .000000 5 H 3.362029 .946860 3.869192 3.869192 .000000 6 H 2.031411 .951730 2.491320 2.491320 1.505701(Initial geometry from top of log file)

Water dimer geometry optimization #p rhf/6-31g* opt=tight test … Gaussian Test Job 83: Water dimer optimization … Berny optimization. Internal Forces: Max .000871642 RMS .000359230 Search for a local minimum. Step number 1 out of a maximum of 23 … Item Value Threshold Converged? Maximum Force .000851 .000015 NO RMS Force .000275 .000010 NO Maximum Displacement .013238 .000060 NO RMS Displacement .005575 .000040 NO Predicted change in Energy=-5.784113D-06 … GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradG Berny optimization. Internal Forces: Max .000000441 RMS .000000186 Search for a local minimum. Step number 18 out of a maximum of 23 …. Item Value Threshold Converged? Maximum Force .000000 .000015 YES RMS Force .000000 .000010 YES Maximum Displacement .000008 .000060 YES RMS Displacement 000004 .000040 YES Predicted change in Energy=-2.034343D-12 Optimization completed. -- Stationary point found
Example: …/g03/tests/rs6k/test083.log
Output file: “.log” or “.out” =>
Distance matrix (angstroms): 1 2 3 4 5 1 O .000000 2 O 2.971263 .000000 3 H .948352 3.360777 .000000 4 H .948352 3.360777 1.512866 .000000 5 H 3.422316 .946706 3.905657 3.905657 .000000 6 H 2.025317 .951801 2.481287 2.481287 1.510657(from bottom of log file)

THE BEST WAY TO CONVINCE A FOOL THAT HE IS WRONG IS TO LET HIM HAVE HIS OWN WAY. JOSH BILLINGS
Hints: How to cope with Gaussian output “.log” or “.out” are plain text files. may be over 100 pages long! If your input file was “MyMolecule1.com” (or “… .gif”), then
the ouput file is named “MyMolecule1.log” (or “… .out”). ALWAYS us a different file name for every job, so that you
don’t over-write any of your old outputs. The next job might be “MyMolecule2.com”, etc.
NEVER delete any of your input or output files! Use a spreadsheet (or lab notebook) to keep a log of your
Gaussian (& Spartan) jobs. One line of a spreadsheet can summarize the essential info:
Date, File name, Molecule, Purpose, Command lne (e.g., #p rhf/6-31g* opt=tight test), Any problem encountered.
If you don’t keep meticulous records of your calculations, you will risk drowning in your data!

Hints: How to read Gaussian output Open your “.log” or “.out” files
with a simple text editor such as NotePad (Windows) or TextEdit (Mac). Microsoft Word is NOT
recommended because, for this purpose, it is big, clumsy, and slow.
It you find a cryptic bit of philosophy at the bottom of the file, the Gaussian thinks it completed the job successfully.
You will also find the CPU time and a densely packed summary of the results.
… from file test083,log
1\1\GINC-JERRY\FOpt\RHF\6-31G(d)\H4O2\FRISCH\07-Apr-2004\0\\#P RHF/6-3 1G* OPT=TIGHT TEST\\Gaussian Test Job 83: Water dimer optimization\\0, 1\O,-0.0029670399,0.,-1.4285923793\O,-0.0012160547,0.,1.5426696978\H,0 .504461364,0.7564331492,-1.6925928276\H,0.504461364,-0.7564331492,-1.6 925928276\H,-0.8866727134,0.,1.8776611243\H,-0.0887852583,0.,0.5949059 822\\Version=IBM64-G03RevC.02\State=1-A'\HF=-152.0304565\RMSD=7.932e-0 9\RMSF=1.778e-07\Dipole=0.0596938,0.,-1.0577951\PG=CS [SG(H2O2),X(H2)] \\@
THERE'S SMALL CHOICE IN A BOWL OF ROTTEN APPLES. SHAKESPEARE Job cpu time: 0 days 0 hours 0 minutes 36.7 seconds. File lengths (MBytes): RWF= 18 Int= 0 D2E= 0 Chk= 10 Scr= 1 Normal termination of Gaussian 03 at Wed Apr 7 15:20:15 2004.

Hints: How to read Gaussian output Use your text editor to search for certain words or phrases that will
take you directly to the part of the output that you need. E.g., “#p”, “Job CPU time”, “Optimization completed” Some phases like “Converged?” occur after each step of the optimization. With 100 pages of output, this is MUCH faster than scrolling through the
file.
You can also open any Gaussian input or output or . chk file with Gaussview, but less information is accessible that way.
Item Value Threshold Converged? Maximum Force .000000 .000015 YES RMS Force .000000 .000010 YES Maximum Displacement .000008 .000060 YES RMS Displacement .000004 .000040 YES Predicted change in Energy=-2.034343D-12 Optimization completed. -- Stationary point found. ----------------------------

Rotational constants The rotational Hamiltonian is
with A = 1/2Ia, etc., where Ia, Ib, and Ic are the principal monents of inertia. (Beware of units!)
where i labels the atoms in the molecule.
Gaussian calculates the rotational constants, A, B, and C: Standard orientation: ---------------------------------------------------------------------------- Center Atomic Atomic Coordinates (Angstroms) Number Number Type X Y Z ---------------------------------------------------------------------------- 1 8 0 .003321 -1.427751 .000000 2 8 0 .003321 1.555329 .000000 3 1 0 -.490596 -1.716172 .756507 4 1 0 -.490596 -1.716172 -.756507 5 1 0 .915824 1.808076 .000000 6 1 0 .012237 .603641 .000000 ----------------------------------------------------------------------------- Rotational constants (GHZ): 215.8670694 6.1508112 6.1482646
€
H = AJa2 + BJb
2 + CJa2
€
Ia = mi ri2 − zi
2( )
i
∑
A B C
R.N. Zare, Angular Momentum, pp 253-258.

(c) Infrared and Raman Spectra
Vibrations of Diatomic Molecules Vibration in Polyatomic Molecules: Normal
Modes Infrared Spectra Raman Spectra “Frequency” Calculations in Gaussian

Vibrations of diatomic molecules
Chemical bonds will break at large interatomic distances. (red curve)
A harmonic potential approximates the true potential near the equilibrium geometry. (orange)
The real potential is ANHARMONIC.€
V = 12 k(x − xe )2

The Harmonic Oscillator Harmonic oscillator wavefunctions are shown
in red, n = 0, 1,2, …. Allowed optical (infrared) transitions are
n = ±1. (thick arrows) Real molecules are anharmonic, therefore
overtone transitions (n = ±2, 3, …) are also allowed, but are 1 or 2 orders of magnitude weaker for each higher overtone. (thinner arrows)
At room temperature, most of the molecules are in n = 0, so the n = 1 0 “fundamental” transition dominates the observed IR spectrum.
Note that anharmonicity shifts the observed IR fundamental transition to a frequency lower than the harmonic frequency by about 1 - 5%.

Vibrations in polyatomic molecules A nonlinear molecule with N atoms has 3N-6 vibrations:
3N degrees of freedom minus 3 translations and 3 rotations
Each vibration is assumed to be a harmonic oscillator and treated as an independent “normal” mode.
For each mode, all atoms move in hase with the same frequency.
The commonly used functional group vibrations (e.g., C-F stretch) are only rough descriptions.
The n = 1 0 “fundamental” transitions (one for each normal mode) dominate the IR spectrum.
Overtone and combination bands are often observed as small peaks in the experimental spectrum, but are NOT included in the simulated spectra produced by Spartan and Gaussian.
As with diatomics, anharmocity shifts the obesrved IR fundamentals are shift to lower frequency.Movies created by GaussView,
captured by Snapz Pro X, andedited by QuickTime.
Similar movies can be obtained from Spartan 04 or 06 without additional software.
Methyl fluoride

Calculation of molecular vibrations In the harmonic approximation, the vibrational
frequencies depend on: all of the second derivatives of the potential, including the
diagonal and mixed second derivatives The molecular geometry, including all bond lengths,
angles, and dihedral angles The atmoic masses
The calculation is only valid at a stationary point A place where all of the first derivatives are zero Local minima, maxima, saddlepoints MUST optimize the structure first, and then run the
frequency calculation at ezacly the same level of theory and same basis set.

€
V (q1,q2,...) ≈ Veq +∂V
∂qi eq
qi − qieq( )i=1
3N−6
∑ + 12
∂ 2V
∂qi∂q j eq
qi − qieq( ) q j − q jeq( )i, j=1
3N−6
∑ + anharmonic terms
Expand the potential in a multidimensional Taylor series”:
Calculation of molecular vibrations:More on the harmonic approximation
The energy of the molecule at it equilibrium geometry.
The the 1st derivatives (forces) are all zero at the equilibrium geometry.
These 2nd derivatives (force constants) are used to determine the normal mode frequencies in the harmonic approximation.
The higher derivatives are neglected in most calculations causing most calculated frequencies to be too high.

Porcedure for calculating vibrational frequencies
In Gaussian, optimized the molecular geometry first (OPT) Then run a frequency calculation (FREQ) at the same level of theory. You can run both at the same time, but it is not recommended until
you have had some experience. Analytical second derivatives are available for the following methods:
HF, B3LYP, MP2, CASSCF 2nd derivatives are calculated numericaly for other methods, and are
MUCH more time consuming.

Porcedure for calculating vibrational frequencies
In Gaussview: Build the molecule. From the Edit menu, click “Symmetrize” Job type is “OPT” ,
then “FREQ”. From the Results menu, select
“Vibrations …”.

Vibrations in Gaussian output
Check that the Point Group is what you expect.
The information for each vibrational mode is given.
Gaussian assumes that each atom is the most abundant isotope of each element, but other istoppes can be set with the ReadIsotopes option to te FREQ command.
Thermochemical data, e.g., U, CV, S are included. The default temperature is 298 K, but this can be changed
# opt freq hf/3-21g geom=connectivity. . . Full point group D2H NOp 8. . . Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering activities (A**4/AMU), depolarization ratios for plane and unpolarized incident light, reduced masses (AMU), force constants (mDyne/A), and normal coordinates: 1 2 3 B2U B3U B2G Frequencies -- 941.6104 1114.4412 1156.0372 Red. masses -- 1.0439 1.1607 1.5215 Frc consts -- .5453 .8494 1.1980 IR Inten -- 1.3937 133.9954 .0000 Raman Activ -- .0000 .0000 4.9745 Depolar (P) -- .3940 .0000 .7500 Depolar (U) -- .5653 .0000 .8571 Atom AN X Y Z X Y Z X Y Z 1 6 .00 -.04 .00 .08 .00 .00 .15 .00 .00 2 6 .00 -.04 .00 .08 .00 .00 -.15 .00 .00 3 1 .00 .24 -.44 -.50 .00 .00 -.49 .00 .00 4 1 .00 .24 .44 -.50 .00 .00 -.49 .00 .00 5 1 .00 .24 -.44 -.50 .00 .00 .49 .00 .00 6 1 .00 .24 .44 -.50 .00 .00 .49 .00 .00 4 5 6 AU B3G AG Frequencies -- 1164.9569 1386.3705 1521.8389 Red. masses -- 1.0078 1.5294 1.2818 Frc consts -- .8059 1.7319 1.7491 IR Inten -- .0000 .0000 .0000 Raman Activ -- .0000 .9300 45.6702 Depolar (P) -- .0000 .7500 .4294 Depolar (U) -- .0000 .8571 .6008 Atom AN X Y Z X Y Z X Y Z 1 6 .00 .00 .00 .00 .15 .00 .00 .00 .11 2 6 .00 .00 .00 .00 -.15 .00 .00 .00 -.11 3 1 .50 .00 .00 .00 -.15 .47 .00 -.19 .46 4 1 -.50 .00 .00 .00 -.15 -.47 .00 .19 .46 5 1 .50 .00 .00 .00 .15 -.47 .00 .19 -.46 6 1 -.50 .00 .00 .00 .15 .47 .00 -.19 -.46. . . E (Thermal) CV S KCal/Mol Cal/Mol-Kelvin Cal/Mol-Kelvin Total 36.426 7.387 52.029 Electronic .000 .000 .000 Translational .889 2.981 35.927 Rotational .889 2.981 15.788 Vibrational 34.649 1.425 .313. . .

Infrared Intensities Infrared intensities are proportional to
The transition moment vectorsare represented graphically by GaussView (amber).
The infrared fundamental bands are allowed or forbidden according to the irreducible representation (e.g., B2u) in the character table for the point group.
€
Iξ =∂μ
∂ξ
2
ψ upper ξ ψ lower
2, ξ = x,y,z
€
I = Ix,Iy,Iz( )QuickTime™ and a
decompressorare needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
1: 941.6 cm-1 B2u
Peter Bernath, Spectra of Atoms and Molecules, Oxford University Press, New York, 1995, p 381.
Infraredallowedfundamentals
Y
Z
X

Scaling calculated infrared spectra Calculated harmonic frequencies may be higher or lower than the true harmonic
frequencies, according to the level of theory and basis set. The true harmonic frequencies are higher than the experimental frequencies because of
the neglect of anharmonicity. To compare with experiment, the calculated frequencies are SCALED empirically
(fudged) according to the level of theory and basis set. It is advisable to check the literature on what scaling people have done for similar
systems.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
Foresman and Frisch, p 64.

Conventions of vibrational spectroscopy By convention, the highest frequency mode of the highest symmetry (most
symmetric irreducible representation) is 1, the next highest of that symmetry is 2 etc., through all th emodes of that symmetry.
Then the modes of the next greatest symmetry get the next available mode numbers from highest frequenct to lowest, until all modes have a number.
Gaussian does NOT follow this convention, but the literature does, and you should.
The z-axis is always the highest symmetry axis.
Lesssymmetry
Conventionally numbered
Ethylene modes
Ag 1 3329.6 cm-1
2 1841.8 cm-1
3 1521.8 cm-1
B2g 4 1156.0 cm-1
B3g 5 3373.1 cm-1
6 1386.3 cm-1
Au 7 1164.9 cm-1
B1u 8 3307.8 cm-1
9 1604.0 cm-1
B2u 10 3404.5 cm-1
11 941.6 cm-1
B2u 12 1114.4 cm-1
The frequencies are from our calculation above.

Raman Spectroscopy
Figures from Peter Bernath, Spectra of Atoms and Molecules, Oxford University Press, New
York, 1995, p 284, 285.
Most routine Raman spectra are Q-branches in the Stokes vibrational Raman band.
The scattered light is shifted to lower frequencies. The dfference between the scattered and incident
frequencies corresponds to a vibrational excitation of the molecules
Many nonlinear Raman rechniques are also available.

Selection rules for Raman spectroscopy Raman spectra derive their intensity from the dependence of the molecular polarizability
on the various vibrational coordinates. The polarizability at the equilibrium geometry gives rise to Rayleigh scattering (light scattered
at the same frequency as the incident light), which is MUCH more intense that the Raman scattered light.
The polarizability components multiply bilinear combinations of the the coordinates, which are listed in point group character tables.
For molecules with a center of symmetry, vibrational bands that are allowed in Raman are forbidden in the infrared, and vice versa.
Raman allowed
Infrared allowed
QuickTime™ and a decompressor
are needed to see this picture.
Ag: Raman allowed;
Infrared forbidden

Calculation of Raman spectra Calculated in Gaussian by default with FREQ
command. (No Raman intensities in Spartan). Raman spectra can be polarized or depolarized, or both
according to whether the scattered light has the same or different polarization as the incident laser beam.
A useful aid in assignments The same scalling as for IR spectra may be applied.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.

Advantages and disadvantages of Raman spectroscopy*
Advantages Complements IR spectroscopy: Provides access to vibrations that are weak or forbidden in IR.
Systematic studies of molecular vibrations always involve both IR and Raman spectra. The effective resolution of routine spectra is higher.
Gas phase rotational structure: The Q-branch (J=0) is strong and produces as a sharp, narrow peak for each vibration, whereas in the infrared, the P, Q, and R branches (J=-1,0,+1) all have substantial intensity resulting in broad bands that may overlap the bands of other vibrations. O and S branches (J=-2, +2) are allowed in Raman, but are weak and very speard out, and so usually disappear into the baseline.
Condensed phase: Rotations become librations, but the qualitative effect is the same. Disadvantages
The Raman effect is weak, so the signal levels are low. It is much harder to observe overtone and combination bands. Raman spectra are less routinely available because a Raman spectrometer requires a laser light
source and so is more expensive. (For both IR and Raman, current spectrometers are usually Fourier transform spectrometers.)
* Common Old-fashioned Ordinary Raman Spectroscopy (COORS). Other specialized forms include Coherent Antistokes Raman Spectroscopy (CARS), Coherent Stokes Raman Spectroscopy (CSRS), Box-CARS, and Resonance Raman Spectroscopy.