chapter-i - information and library network...
TRANSCRIPT

CHAPTER-I
1
CHAPTER-I
Introduction

CHAPTER-I
2
1.0. Introduction
eterocyclic chemistry persists an interesting psychology these
days. Commonly majority of the published work in organic
chemistry involves at least one heterocyclic ring. Moreover, in
providing us with a wealth of fascinating molecular arrays, over
half of which posses heterocyclic constitution, nature presents a most cogent
argument for developing an appreciation for this area. Heterocyclic chemistry is a vast
and expanding area of chemistry because of the obvious applications of compounds
derived from heterocyclic rings in pharmacy, medicine, agriculture and other fields.
The chemistry of heterocyclic compounds is as relevant as that of aliphatic or
aromatic compounds. Their study is of great interest both from the theoretical as well
as practical stand point. An important application of small molecule libraries is the
preparation of a directed or focused combinatorial library for assay against a specific
biological target. Heterocyclic compounds play a vital role in organic chemistry,
especially in the field of medicinal chemistry. More than half of the naturally
occurring compounds and a high proportion of drugs contain heterocycles.
Heterocyclic compounds may be classified in to aliphatic and aromatic heterocyclic
compounds. The aliphatic heterocycles are the cycles analogues of amines, ethers,
thioethers, amides etc. Heterocyclic compounds are organic compounds with
incorporation of nitrogen, sulfur, oxygen, or an atom of a related element into an
organic ring structure in place of a carbon atom gives rise to a heterocyclic
compound. A cyclic organic compound containing all carbon atoms in ring formation
is referred to as a carbolic compound. If at least one atom other than carbon forms a
part of the ring system then it is designed as a heterocyclic compound [1-4].
HHHH
CHAPTER-I
3
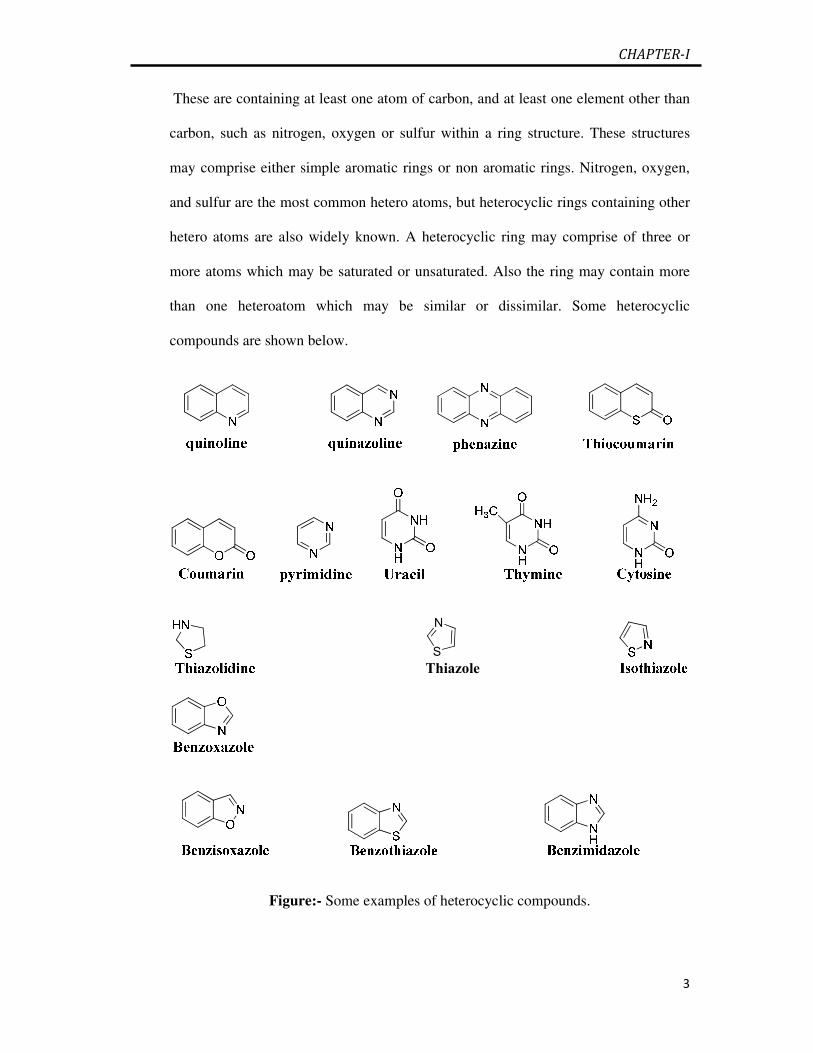
These are containing at least one atom of carbon, and at least one element other than
carbon, such as nitrogen, oxygen or sulfur within a ring structure. These structures
may comprise either simple aromatic rings or non aromatic rings. Nitrogen, oxygen,
and sulfur are the most common hetero atoms, but heterocyclic rings containing other
hetero atoms are also widely known. A heterocyclic ring may comprise of three or
more atoms which may be saturated or unsaturated. Also the ring may contain more
than one heteroatom which may be similar or dissimilar. Some heterocyclic
compounds are shown below.
S
N
Thiazole
Figure:- Some examples of heterocyclic compounds.

CHAPTER-I
4
1.1. Significance of heterocyclic compounds
There are a vast number of pharmacologically active heterocyclic compounds, many
of which are in regular clinical use. Some of these are natural products, for example
antibiotics such as penicillin and cephalosporin, alkaloids such as vinblastine,
ellipticine, morphine, and reserpine and cardiac glycosides such as those of digitals.
However, the large majority are synthetic heterocycles which found wide spread use,
for example as anticancer agents, analeptics, analgesis, hypnotics and vasopressin
modifiers and as pesticides, insecticides, weed killers and rodenticides. There are also
a large number of synthetic heterocyclic compounds with other important practical
applications, as dye stuffs, copolymers, solvents, photographic sensitizers and
developers, as antioxidants and vulcanization acceleration in the rubber industry, and
many are valuable intermediates in synthesis.
The successful application of heterocyclic compounds in these and many other ways
and their appeal as materials in applied chemistry and in more fundamental and
theoretical studies, stems form their very complexity; this ensures a virtually limitless
series of structurally novel compounds with a wide range of physical, chemical and
biological properties, spanning a broad spectrum of reactivity and stability. Another
consequence of their varied chemical reactivity, including the possible destruction of
the heterocyclic ring is their increasing use in the synthesis of specifically
functionalized non-heterocyclic structures.
More important is the recognition that, heterocycles can play a pivotal role not only as
goals in synthesis, but as mediators of synthetic transformations since the appearance
of the benchmark monograph by Meyers [5], which formally legitimized the use of
heterocycles as a viable and occasionally superior means of arriving at the

CHAPTER-I
5
functionalized materials. Just as the synthetic organic chemistry has blossomed since
that time, so has the use of heterocycles in synthesis.
The number of possible heterocyclic systems is almost limitless. An enormous
number of heterocyclic compounds are known and this number is increasing very
rapidly. The literature of the subject is correspondingly vast and of the three major
divisons of organic chemistry aliphatic, aromatic and heterocyclic, the last is much the
biggest. Over six million compounds are recorded in chemical abstracts and
approximately half of these are heterocyclic.
1.2. Introduction to pyrimidinones
One of the main objectives of organic and medicinal chemistry is the design, synthesis
and production of molecules having value as human therapeutic agents. During the
past decade, combinatorial chemistry has provided access to chemical libraries based
on privileged structures [6], with heterocyclic structures receiving special attention as
they belong to a class of compounds with proven utility in medicinal chemistry [7-
10]. Pyrimidine is the most important member of all the diazines as this ring system
occurs widely in living organisms. Purines, uric acid, alkoxan, barbutiric acid and a
mixture of anti-malarial and anti-bacterials also contain the pyrimidine ring. The
chemistry of pyrimidine has been widely studied [11, 12]. Pyrimidine was first
isolated by Gabriel and Colman in 1899. The derivatives of fused pyrimidinones have
been the focus of great interest over many years due to the fact that many compounds
containing a fused pyrimidinone ring play an important role in the biochemistry of
living cell [13-16]. Heterofused pyrimidines exhibit promising antiviral [17],
antibacterial [18], anti-AIDS [19], and antinociceptive [20] activities. The group of
pyrido [1,2-a] pyrimidin-4-ones is a well-known class of aza-bridgehead fused
heterocyclic compounds which have miscellaneous pharmaceutical applications [21].

CHAPTER-I
6
This kind of structural pattern is present in the known psychotropic agents risperidone
and paliperidone [22, 23], the human leukocyte elastase inhibitor SSR69071 [24], the
antiallergic agent ramastine [25], and antioxidants [26]. Fused pyrimidines are
extensively used in neurology, particularly in the treatment of neurodegenerative
disorders such as Parkinson’s disease [27], antianxiety disorders [28] and depression
[29]. Fused pyrimidines are selective inhibitors for multidrug resistance (MDR) [30].
Folate metabolism has long been recognized as an attractive target for cancer
chemotherapy because of indispensable role of fused pyrimidines antifolates as
antitumor agents [31]. Risperidone a derivative of pyrido[1,2-a]pyrimidines [32, 33]
and derivatives showed antipsychotic activity [34] which were used as α-2 antagonists
[35-37]. They exhibits high affinity for α-2- adrenoceptor with high selectivity versus
the α- receptor and posses potent in vivo central activity [38, 39]. Many other
examples of pyrimidine based derivative have been investigated as potential antitumor
agents, including 2-phenyl amino derivatives [40-43], 4-phenyl amino derivatives[44,
45], 2.4-bis(phenylamino)derivatives [46], 4.6-bis(phenylamino)derivatives [47, 48],
and 4-aryl substituted derivatives [49]. All these observations point out that pyrimidin
ones occupies distant and remarkable place in medicinal chemistry. Keeping in view
of these above facts, it was thought of synthesizing some pyrimidinone derivatives to
explore their biological significance.
1.3. Biological significance of pyrimidinones
Signal transduction, angiogenesis and cancer activity
In 1996 Pfizer researchers reported the first selective Src-family inhibitors, named
PP1, (1) and PP2 (2) [50], endowed with good potency, but poor selectivity toward
the different members of SFKs. A very large amount of biological data is reported for
PP1 and PP2; they inhibited Lck, Fyn, c-Src and Hck activity kinase with IC50 values

CHAPTER-I
7
ranging from 4 to170 nM, while were essentially inactive against other non-Src
family TK, such as ZAP-70 or JAK 2. Some inhibition of EGFR was also observed
with IC50 values of 250 and 480 nM for PP1 and PP2, respectively. The compounds
appeared to be ATP competitive inhibitors. The library of compounds (3) and (4) 4-
amino-substtuted pyrazolo [3,4-d] pyrimidines bearing a 2-chloro-2-phenyl N1 side
chain showed a submicromolar to nanomolar activity toward isolated Src as well as
antiproliferative activity toward the epidermoid (A431) and breast cancer (BC-8701)
cell lines (that overexpress Src) blocking Src phosporylation and inducing apoptosis
with potency about two-fold higher than that of the reference compound 4-amino-5-
(4-chlorophenyl)-7-(tert-butyl)pyrazolo[3,4-d] pyrimidine (PP2) (2).
In 1997, Parke-Davis reported a family of pyrido[2,3-d]pyrimidines, belonging to the
important class of urea TK inhibitors [51], as potent, but non-selective inhibitors of
several kinases, acting with an ATP - competitive mechanism [52]. The most
interesting compounds are represented by the soluble 2-substituted aminopyrido
N
N
N
N
X
R
NHR1
SR2
4

CHAPTER-I
8
[2,3-d]pyrimidin-7-yl ureas, such as (5) that showed IC50 values of 18, 210 and 49
nM, toward Src, PDGFR, FGFR, respectively, in enzymatic assays. Compound (6)
(PD 166285) is a tyrosine kinase inhibitor with moderate selectivity for Src, PDGFR,
FGFR and EGFR, respectively [53, 54]. This derivative has been also reported to
inhibit tumor cell growth particularly of human leukemic cell lines and leukemic
blasts [55]. Compound (7) (PD 173955) has been found to be a more selective Src
inhibitor with IC50 values of 22, 1600 and 1600 nM, for Src, PDGFR, FGFR,
respectively. Treatment of MDA-MB-468 cells, a breast cancer line with this
compound causes a loss in Src activity, with a reduction in the tyrosine
phosphorylation of the enzymes; the cell growth of this tumor line resulted also
blocked, for the ability of (7) to cause mitotic arrest [56]. PD 173955 also inhibits
Bcr-Ab1 and c-Kit, with IC50 values of 2 and 50 nM, respectively [57].
Compound (8) (PD 166326) [58] is a potent dual inhibitor of Src and Abl with IC50
values of 6 and 8 nM, respectively moreover, it is a picomolar inhibitor of K562 cells
growth, showing an IC50 of 300 pM, and leading to apoptotic G1 arrest, PD 0166326
is also effective against imatinib-resistant Brc-Ab1 mutants and could represent a
prototype of a new generation of potential agents useful in the treatment of CMl [59].
Compound (9) (PD 180970) is one of the most recent member of this class, and shows
a similar biological behaviour to that of (8), inducing apoptosis in K562 cells and of
CD34-positive leukemic cells from patients with imatinib-resistant CMl [60, 61].

CHAPTER-I
9
Gangjee et al., [62] synthesized 2-amino-4-m-bromoanilino-6-arylmethyl-7H-pyrrolo
[2,3-d] pyrimidines (10). Embarked on the design of RTK inhibitors using the
pyrrolo[2,3-d] pyrimidine scaffold with an additional 2-NH2 moiety which is not
present in most other 6-6 or 6-5 ring system RTK inhibitors. The 2-NH2 group in our
compounds provides a third H-bonding moiety in the Hinge region of RTKs, and was
anticipated to increase binding and consequently potency against RTKs. This has
recently been shown to be true in most instances, but is dependent on the nature of the
scaffold and its substitutions [63].

CHAPTER-I
10
An initial series of 2-amino-4-(3-bromoanilino)-6-(arylmethyl)-pyrrolo[2,3-d]pyrimid
ine derivatives 10 (a-c) indicated that substitution on the 6-aryl moiety dictates both
the selectivity and potency against a variety of RTKs.
The pyrimidine moiety with some substitution shows promising antitumor activity as
there is large number of pyrimidine based antimetabolites. Early metabolite prepared
was 5-flurouracil (11) [64] a pyrimidine derivative followed by 5-thiouracil (12)
which also exhibits some useful antineoplastic activities [65].
Palwinder singh et al., [66] reported the synthesis of 5-substituted-1,3-dimethylpyri
midine-2,4,6-trione derivatives (13) reacted with amines provided enamines. The
investigation for anticancer activity of molecule with 59 human tumor cell lines was
done representing leukemia, melanoma and cancer of lung, colon, brain, ovary, breast
as well as kidney.

CHAPTER-I
11
Anti-inflammatory activity
Marylene Favre et al., [67] synthesized some substituted tetrahydrothieno[3, 2-
d]pyrimidin-4-one derivatives (14) and screened them for analgesic and anti-
inflammatory activity.
Cannito et al., [68] investigated analgesic and anti-inflammatory activity of some of
3,5,6-trisubstituted-2-thioxo-2,3-dihydrothieno[2,3-d]pyrimidine-4-one derivatives
(15). The i.p. administration of these products at a dose of 1000 mg/kg shows that
they are not toxic (one excepted). Some compounds showed analgesic and anti-
inflammatory activities equivalent to those of acetylsalicylic acid.
F. Russo et al., [69] described new thienopyrimido benzothiazole and thieno
pyrimidobenzo oxazoles (16) and screened them for analgesic and anti-inflammatory
activities.

CHAPTER-I
12
Nargund et al., [70] synthesised substituted 2-mercapto-3-(N-alkyl)pyrimido[5,4-C]
cinnolin-4-(3H)-ones (17) refluxing 4-aminocinnoline, 3-carboxylic acids, with aryl
isothiocyanates in anhydrous pyridine. These derivatives were evaluated for their
anti-microbial and anti-inflammatory activities. Some of the compounds possessed
potent anti-microbial activity.
Ishwaarsingh S. Rathod et al., [71] studied the analgesic activity of thieno[2,3-d]pyri
midine-4-(3H)-ones (18), 2-aryl amino-3-aryl-5-methyl-6-(substituted)thieno[2, 3- d]
pyrimidin-4(3H)-ones (19) by tail flick method in albino rats and by wriftling method
in albino mice.

CHAPTER-I
13
A series of 3-substituted amino-2-methyl thieno[2,3-d]pyrimidin-4-(3H)-ones (20)
were synthesized and screened for acute (30mg/kg p.o) and chronic (10mg/kg p.o)
anti-inflammatory activity using carrageenan induced rat paw edema model and
carrageenan-induced granuloma air pouch model respectively. The tested compounds
have exhibited significant (P<0.05) anti-inflammatory activity in acute model while in
chronic model only compounds 20(a-g) have exhibited significant activity
(P < 0.05) [72].

CHAPTER-I
14
Proquazone (21) a non-steroidal anti-inflammatory agent (NSAID) which, unlike
most other NSAIDs, does not have a free acid group in its structure. It is advocated
for use in rheumatoid arthritis, ankylosing spondylitis, osteoarthritis, musculoskeletal
disorders, acute inflammatory conditions and acute pain states such as
dysmenorrhoea, post-operative pain and headache. Preliminary studies have
confirmed the efficacy of proquazone in acute inflammatory disorders, and shown that
it provides useful analgesic relief in acute pain states such as dysmenorrhoea,
Compound R
20(a) 4-methyl
20(b) 2-hydroxy
20(c) 4-hydroxy
20(d) 4-methoxy
20(e) 2-chloro
20(f) 4-chloro
20(g) N, N-dimethlamino
20(h) 2-nitro
20(i) 3-nitro
20(j) 3-methoxy,4-hydroxyl
20(k) 3,4-dimethoxy
20(l) 3,4,5-trimethoxy

CHAPTER-I
15
headache and after minor surgery produces a high incidence of gastrointestinal
symptoms such as diarrhoea [73].
Platelet aggregation inhibition activity
1,2,3,5-tetrahydroimidazo[l,2-a]thieno pyrimidin-2-one (22) derivatives have been
prepared and tested for platelet aggregation inhibitory activity in rats in vitro and ex
vivo. Few of the compounds are identified potent inhibitors of blood platelet
aggregation. Compounds 1,2,3,5,6,7,8,9-octahydro-[ l]benzothieno[2,3-d] imidazo[l,2
-a]pyrimidin-2-one exhibited the most favourable activity [74].
Antihypertensive activity
Mery Press et al., [75] synthesized of 3-substituted-thieno[2,3-d]pyrimidine-2,4-dione
(23) and for their antihypertensive activity.

CHAPTER-I
16
Russell et al., [76] evaluated the antihypertensive activity of thienopyrimidine
derivatives (24).
Atwal et al., [77] synthesized dihydropyrmidinone derivatives (25). Where as the
modifications of the substituent at N3 led to the development of orally long-lasting
antihypertensive activity. Furthermore, it has been established that the absolute
stereochemistry at the C4 carbon atom is very important. For example, the desired
antihypertensive effect was observed only with the (R)-enantiomer of the compound.

CHAPTER-I
17
Ketanserin (26) a serotonin receptor antagonist, reduces blood pressure appears as a
new and important alternative in the treatment of mild and moderate essential
hypertension [78].
CNS activity
Howard et al., [79] screened Risoperidone (27) for CNS activity, its structural hybrid
is of butyrophenone and can be used as anxiolytic, antidepresant and antiparkinsonian
drug.
Abott et al., [80] synthesized and checks the anesthetic activity of pyrimidine
analogue, thimylal (28).

CHAPTER-I
18
Sugiyama et al., [81] condensed thieno[3,2-d]-, [3,4-d], and [2,3-d]pyrimidin-5-one
derivatives (29) in which the oxygen atom of the oxazolidine moiety in 29 was
replaced by a sulfur atom or methylene groups, and evaluated for their gastric
antisecretory activity in pylorus-ligated rats.
Sedative/hypnotic/antiepileptic agents
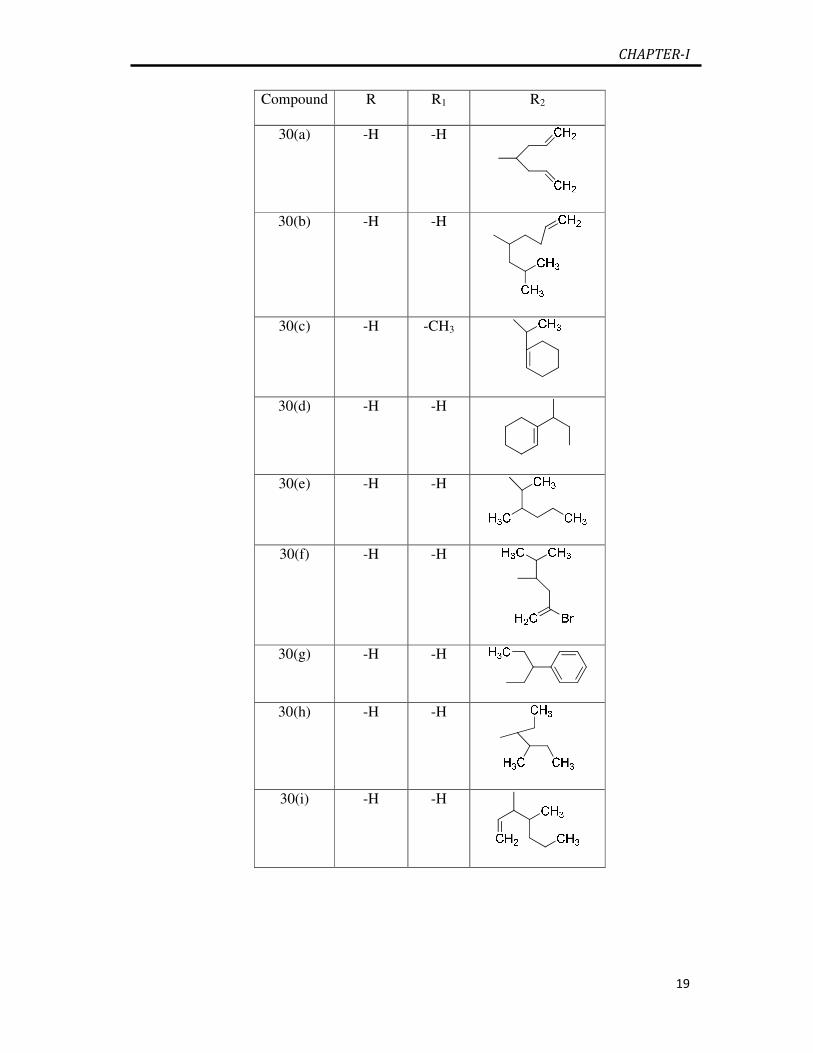
Agents of the anxiolytic, sedative and hypnotic group include a wide variety of
barbiturates 30(a-i) used as sedative and hypnotics and are classified as drugs having
short, intermediate and long duration of action [82]. Allobarbital 30(a), aprobarbital
30(b), pentobarbital 30(e), phenobarbital 30(g) and secobarbital 30(i) are frequently
used clinically as hypnotic barbiturates [83]. Hexobarbital 30(c), cyclobarbital 30(d)
and propallylonal 30(f) are some of the current drugs in the market used as sedative
hypnotics [84]. Barbiturates as sedative hypnotics hava a long and fascinating history.
Eli Lilly patented secbutabarbital 30(h) in 1932 [85] .
CHAPTER-I
19
Compound R R1 R2
30(a) -H -H
30(b) -H -H
30(c) -H -CH3
30(d) -H -H
30(e) -H -H
30(f) -H -H
30(g) -H -H
30(h) -H -H
30(i) -H -H

CHAPTER-I
20
Flucytosine (31) is a fluorinated pyrimidene used as nucleosidal anti-fungal agent for
the treatment of serious systemic infections caused by susceptible strains of candida
and Cryptococcus [86].
Non-peptide Luteinizing Hormone-Releasing Hormone (LHRH) Antagonists
Sasaki et al., [87] designed, synthesized, and evaluated a new class of non-peptide
luteinizing hormone-releasing hormone (LHRH) receptor antagonists, the 2-phenylimi
dazo[1,2-a]pyrimidin-5-ones, a heterocyclic 5-6 ring (32). This study resulted in the identi
fication of a new class of potent LHRH antagonists, represented by the methoxyurea
‘32 g’ possessing high binding affinity and potent in vitro antagonism, with IC50
values of 0.4 and 7 nM, respectively. These results conclude that the imidazopyrimidin
-5-one provide a new scaffold for small molecule LHRH antagonists devoid of a
thiophene ring. Taking this finding into consideration, it is suggested that the heterocycl
ic 5-6 ring system bearing a pendant phenyl group attached to the five-membered ring
is the key structural motif for a LHRH antagonist scaffold system (32) in which an
imidazole ring is embedded, as a novel scaffold for non-peptide LHRH antagonists.

CHAPTER-I
21
Stadler et al., [88] described nitrofuryl-substituted DHPM derivative Nitracin (33) and
the compound displayed activity against viruses of the trachoma group and modest
antibacterial activity.
Calcium channel blocking activity
Rovnyak et al., [89] investigated 2-(methylthio)-4-(2-(methylthio)-3-nitrophenyl)-4,7-
dihydrofuro[3,4-d]pyrimidin-5(1H)-one (34), Alajarin et al., [90] investigated a
monocyclic dihydropyrimidine (35). Besides the monocyclic DHMP derivatives,
some fused analogs, which incorporate hetero- or carbocyclic rings attached to either
C2/N3 or the C5/C6 positions of dihydropyrimidine such as (34) and (35) possess
calcium channel blocking activity.

CHAPTER-I
22
1.4. Introduction to Thiazoles
Thiazoles (36) are structurally related to thiopene and pyridine, but in most of its
properties it resembles to the latter. Thiazole was first described by Hantzch and
Waber in 1889. The numbering in thiazole starts from the sulphur atom. Structure
(37) is benzothiazole. The basic structure of benzothiazole consists of benzene ring
fused with 4, 5 position of thiazole. The two rings together constitute the basic
nucleus 1,3-benzothiazole.
Thiazole is a five-membered ring containing one nitrogen and one sulfur.
Benzothiazole is a heterocyclic compound, weak base, having varied biological
activities and still of great scientific interest now a days. They are widely found in
bioorganic and medicinal chemistry with application in drug discovery. Benzothiazole
moieties are part of compounds showing numerous biological activities such as
antimicrobial [91-95], anticancer [96-98], anthelmentic [99], antidiabetic [100],
activities. They have also found application in industry as anti-oxidants, vulcanization
accelerators. Various benzothiazoles such as 2-arylbenzothiazole received much
attention due to unique structure and its uses as radioactive amyloid imaging agents
[101], and anticancer agents [102]. Benzothiazoles are bicyclic ring system with

CHAPTER-I
23
multiple applications. In the 1950’s, a number of 2-aminobenzothiazoles were
intensively studied, as the 2-aminobenzothiazole scaffold is one of privileged
structure in medicinal chemistry [101-103] and reported cytotoxic on cancer cells
[103]. It must be emphasized that combination of 2-aminobenzothiazoles with other
heterocyclic is a well known approach to design new drug like molecules, which
allows achieving new pharmacological profile, action, toxicity lowering. The 2-(4-
aminophenyl) benzothiazoles are novel class of potent and selective antitumor agents
and display characteristic profile of cytotoxic response across the cell lines. In
addition, benzothiazole ring is present in various marine or terrestrial natural
compounds, which have useful biological properties. Sulphur and nitrogen atoms
constitute the core structure of thiazole and many pharmacologically and biologically
active compounds.
1.5. Biological significance of Thiazoles
Antimicrobial activity
Yadav et al., [104] synthesized a series of fluoro, chloro 2-(α-substituted aryl amino
acetamido)benzothiazole (38) and screened for antibacterial activity against
B. subtilis, S. typhi, E. coli, and S. aureus bacterial strains. The fluoro, and chloro
substituents showed a significant antibacterial activity.
Basavaraja K. M. et al., [105] described 2-[1-aryl azo]methyleneimino-6-chloro
benzothiazole derivatives (39) and tested for antimicrobial activity against B. subtilis,

CHAPTER-I
24
S. typhi, E. coli, and S. aureus bacterial strains. The compounds showed a significant
antibacterial activity.
Murthy Y. et al., [106] evaluated some new 2-mercaptobenzothiazoles (40) and
correlated the effect on anti microbial potency by varying the substituents in benzene
part of the benzothiazole ring system. Anti-microbial screening was performed
against E. coli, S. aureus, C. albicans and antifungal activity against Aspergillus
flavus and Candida albicans at conc. 100 µg/mL using agar plate Kirby-Bauer disc
diffusion method.
Pandurangan A. et al., [107] reported N-2-benzothiazolylthiourea derivatives (41) as
good antimicrobial agents against Gram-positive and Gram-negative bacteria such as
S. aureus, P. aeruginosa, E. coli, and a yeast (C. albicans) and a mould (Microsporum
gypseum).

CHAPTER-I
25
Maharan M. et al., [108] discussed a series of benzothiazole-2-yl-dithiocarbamates
(42) along with copper complexes via reaction of suitable alkyl or hetroaryl halide
with sodium salt of benzothiazole-2-yl-dithiocarbamic acid followed by complexation
with copper sulphate and selected derivatives checked for their schistosomicidal
activity against Schistosoma mansoni.
Amir M. et al., [109] examined thiadiazole derivatives containing benzothiazole (43)
and screened for both antibacterial and antifungal activites using cup-plate agar
diffusion method. Anti-microbial screening was performed against E. coli, S. aureus,
C. albicans and antifungal activity against Aspergillus flavus and Candida albicans.
Soni B. et al., [110] reported newer Schiff bases of benzothiazole derivatives (44) and
they tested for antibacterial activity. The synthesized derivatives exhibited moderate
antibacterial activity against Gram-positive (S. aureus and S. pyrogenus),
Gram-negative bacteria (E. coli and P. aeruginasa) and Fungi (C. albicans, A. niger
and A. clavatus). The results demonstrated that compounds with a 4-hydroxy, 4-
dimethylamino and 3,4-dimethoxy group on the aromatic ring showed good
antibacterial activity.

CHAPTER-I
26
S
NNH
N N
N
SH
N
R
44
R= 4-OH, 2-Cl, 4-N(CH3)2 and 3, 4-OCH3
Alang et al., [111] studied seven new derivatives of 2-substituted benzothiazole (45)
and found them as good antibacterial agent against Gram-positive bacteria (S. aureus,
S. epidermis) and Gram-negative bacteria (P. aeruginasa and E. coli).
Malik et al.,[112] synthesized some new 2-amino substituted benzothiazoles (46) and
evaluated for in vitro antifungal activity against fungal strains such as C. albicans,
A. niger and A. flavus.
Barot H. K. et al., [113] evaluated nitrogen mustards of flurobenzothiazoles (47, 48)
and was determined for antibacterial and antifungal activites against S. aureus, B.
subtilis, C. tropicans, A. niger, and F. heterosporium. The synthesized compounds
showed an excellent inhibition at a conc. of 50µg/0.1mL.

CHAPTER-I
27
Anthelmentic activity
Sreenivasa M. et al., [114] synthesized fluorobenzothiazole comprising sulphanamide
pyrazole derivatives (49). The synthesized compounds were screened for anthelmentic
activity by using earthworms (Peritumaposthum). The compounds were evaluated by
time taken for complete paralysis and death of worms.
4-Fluoro-3-chloroaniline treated with potassium thiocyanate in presence of glacial
acetic acid and bromine was converted into 2-amino-6-fluoro-7-chlorobenzothiazole.
The synthesized compound in presence of p-dimethylaminobenzaldehyde refluxed in
ethanol to obtain a corresponding 6-fluoro benzothiazole Schiff’s base (50) the above
said compound was treated with ortho, meta and para nitroanilines, ortho, meta, para
chloroanilines, morpholino, piperazine, diphenylamine in the presence of DMF to
obtain different new moieties. Some new moieties showed promising anthelmintic
activity [115].

CHAPTER-I
28
Anti-inflammatory activity
Shashank et. al., [116] described synthesis of 2-substituted benzothiazole derivatives
(51) and evaluated for anti-inflammatory activity. The maximum activity may be due
to presence of -F and -OCH3 groups. The same series of compounds also showed good
anticancer activity.
A series of 2-(benzo[d]thiazol-2-ylthio)-N-(2-oxoindolin-3-ylidene)acetohydrazide
52(a-g), 2(benzo[d]thiazol-2-ylthio)-N-(2,4-dioxospiro[indolin-3,2-thiazolidin]-3’-yl)
acetamide 53(a-g) and 2'-((benzo[d]thiazol-2-ylthio)methyl)spiro[indoline-3,5'-thiazolo
[4,3-b][1,3,4]oxadiazol]-2-ones 54(a-g) have been synthesized and screened for their
anti-inflammatory, analgesic and anti-bacterial activities. The most potent anti-inflamm

CHAPTER-I
29
atory and anti-bacterial compound of this series was compound 54d and most potent
analgesic compound was compound 54e [117].
Oketani et al., [118] studied the in vitro pharmacological profiles of 6-hydroxy- 5, 7-
dimethyl-2-(methylamino)-4-(3-pyridylmethyl)benzothiazole (55) against the 5 lipooxy
genase activity of rat basophilic leukemia cells. This result indicated that the compound
effectively inhibited 5-lipoxygenase and thromboxane B2 production in rat peritoneal
and human blood vessels.
Cyclooxygenase inhibitor activity
Gurupadaya B. et al., [119] synthesized azatidin-2-ones and thiazoline-4-ones (56)
encompassing benzothiazole derivatives and evaluated for anti-inflammatory activity
using carrageenan induced rat hind paw edema method.

CHAPTER-I
30
Anti-cancer activity
The 2-arylsubstituted benzothiazole derivatives (57) were synthesized by refluxing o-
aminothiophenol with substituted benzoic acids in the presence of polyphosphoric
acid at 220 0C 2-Mercaptothiazole was used along with thionyl chloride to get the
carbothioates. The screening for antitumour activity was done as per the National
Cancer Institute drug screening strategy 3 compounds were found to be significantly
cytotoxic as compared to [2-(3-bromo-4-aminophenyl)benzothiazole] against the
human cervical cancer cell lines [120].
Fluorinated 2-arylbenzothiazoles (58) are new potential antitumor drugs, which show
potent and selective inhibitory activity against breast, lung, and colon cancer cell
lines. Carbon-11 labeled fluorinated 2-arylbenzothiazoles may serve as novel probes
for positron emission tomography (PET) to image tyrosine kinase in cancers [121].
Gupta S. et al., [122] synthesized benzothiazole derivatives (59) and screened for in
vitro cytotoxic activity against HL-60 and U-937 cell lines. In silico pharmacokinetic
study revealed that benzothiazole dimere were free from teratoginicity, irritation and
sensitivity properties than monomers. The QSAR study showed that increase in

CHAPTER-I
31
hydrogen donor count is conductive for cytotoxic activity of benzothiazole derivatives
against HL-60 cell lines.
MTP inhibition activity
A series of triamide derivatives bearing a benzothiazole (60) core is shown to be
potent microsomal triglyceride transfer protein (MTP) inhibitors. In order to minimize
liver toxicity, these compounds have been optimized to have activity only in the
enterocytes and have limited systemic bioavailability. Upon oral administration,
selected analogs within this series have been further demonstrated to reduce food
intake along with body weight and thereby improve glucose homeostasis and insulin
sensitivity in a 28 day mice induced obesity (Dio) model [123].
Amyloid imaging agent in Alzheimer’s disease
Serdons K. et al., [124] examined F-labelled 2-(4-fluorophenyl)benzo[d]thiazoles (61)
evaluated it as amyloid imaging agent in Alzheimer’s disease and they showed good
affinity for amyloid plaques present in human Alzheimer’s disease.

CHAPTER-I
32
Antimalarial activity
Hout S. et al., [125] synthesized 2-substituted-6-nitro (62) and 6-aminobenzothiazoles
(63) and their anthranilic acids were carried out on W2 and 3D7 strains of
P. falciparum. The results revealed the potency of compounds 62 and 63 as the
antimalarial agents of clinical and biological research.
PPAR δ activity
To find novel PPAR δ-selective agonists, Fujida et al., [126] designed and
synthesized phenylpropanoic acid derivatives bearing 6-substituted benzothiazole
derivatives. Optimization of this series led to the identification of a potent and
selective PPAR δ-agonist. Among the synthesized compounds, compound (65) has
shown PPAR δ activity and selectivity. The introduction of Cl group at the C-6
position of the benzothiazole ring and methyl group at the ortho position of phenyl
propanoic acid further improved PPAR δ transcriptional activity. Compound (65) was
found to be the most potent and selective PPAR δ agonist.

CHAPTER-I
33
Anti convulsive activity
Several benzothiazoles containing sulphanamide derivatives (66) [127], benzothiazolyl
guanidones (67) [128], benzothiazolamines (68) [129], were synthesized and evaluated
for their activity against electro shock and phenyl tetrazolone induced seizures. The review
of these literatures revealed the benzothiazole moiety as a dynamic agent against convulsive
seizures.
Siddiqui et al., [130] reported N-(6-substituted-1, 3-benzothiazole-2-yl)-4-sulphanami
des (69) and screened for anticonvulsive activity. The tested compound showed an
anticonvulsive effect in MES and PTZ induced seizures.
Amnerkar N. et al., [131] investigated prop-2-eneamido and 1-acetyl-pyrazolin
derivative of aminobenzothiazole (70) and the compound was evaluated for anticonvu
lsive activity. The screened compound showed anticonvulsive activity in MES and
PTZ induced seizures.

CHAPTER-I
34
Anti-diabetic activity
Pattan S. R. et al., [132] described a series of 2-amino[5’(4-sulphonylbenzylidene)-2,
4-thiazolidinone]-6-flurobenzothiazoles (71) and examined for anti-diabetic activity.
All the compounds of this series showed promising anti-diabetic activity.
Paoli and co-workers [133] prepared a small library of 2-arylsulphonyl aminobenzothi
azoles (72, 73) and screened them for protein tyrosine phosphatase 1B inhibition. The
most active compounds (72) and (73) were observed as rapid reversible inhibitors of
PTP-1B and significantly lowered plasma glucose concentration.

CHAPTER-I
35
1.6. Scope of the present work
Heterocyclic chemistry is a vast and expanding area of chemistry, because of the
obvious applications of compounds derived from heterocyclic rings in pharmacy,
medicine, agriculture and other fields. This study is of great interest both from
theoretical as well as practical standpoint. In five-membered and six-membered ring
systems, the presence of nitrogen, oxygen, and sulphur heteroatom defines an
interesting class of compounds. An important application of small molecule libraries
is the preparation of a directed or focused combinatorial library for assay against a spe
cific biological target. Pyrimidinone and thiazole is among the most important heteroc
yclic compounds, which exhibit their therapeutic activity is due to their
conformationally flexible nature. The accentuated interest in the pyrimidinone and
thiazoles class of opiate analgesics continues to be expressed in the pharmaceutical
community and biological properties of these agents have been the subject of ongoing
investigations. In this connection the present investigation is undertaken and is
divided in to three chapters. Chapter-I gives the introduction to pyrimidinone and
benzothiazole derivatives. Pyrimidinones and benzothiazoles were proven to be
biologically very potent and selective. A wide spectrum of pharmacological activities
has been reported for these compounds. These include anti-cancer, anti-inflammatory,
platelet aggregation inhibition, C.N.S activity, antimicrobial, anthelmentic, anti-
inflammatory, cyclooxygenase inhibitor activity, anti-diabetic, antimalarial, MTP
inhibition activity, PPAR-δ activity. Such varieties of interesting biological activity
for this class of compounds prompted us to synthesize pyrimidinone and thiazole
derivatives and investigate their biological importance. It is expected that these would
result in highly potent and selective pharmaceutical agents.

CHAPTER-I
36
Chapter-II deals with the synthesis of 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-
4H-pyrido[1,2-a]pyrimidin-4-one derivatives 6(a-s) and investigate for antimicrobial
activity. From the results obtained compounds 6c and 6i showed inhibitory activity
against tested bacterial strains. Synthesis of 2-methyl-3-(2-(piperazin-1-yl)ethyl)-6,7,8
,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one derivatives 8(a-g) and 9(a-f) and inve
stigate for antimicrobial activity. From the results obtained compounds 8a and 9d
showed inhibitory activity against tested bacterial strains. Synthesis of 2-methyl-3-(2-
(piperazin-1-yl)ethyl)-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one derivatives
8(a-d) and 9(a-e) and screened for anti-angiogenesis by EAT cells in vivo. From the
results obtained compounds 8d and 9a showed potent activity against EAT cells in
vivo.
Chapter III contains the evalution of 1-(substituted phenyl)-3-(6-ethoxybenzo[d]thiaz
ol-2-yl)thiourea derivatives 5(a-i) and evaluated for anti-angiogenesis by EAT cells in
vivo. From the results obtained compounds 5e and 5g can be considered as anti-angi
ogenic compounds against EAT cells in vivo.
Synthesis of N-(benzyl)-6-ethoxybenzo[d]thiazol-2-amine derivatives 7(a-i) and scree
ned for anti-angiogenesis by EAT cells in vivo. From the results obtained compounds
7b and 7g have shown good anti-angiogenic effects against mouse Ehrlich Ascites
Tumour. Synthesis of N-(benzylidene)-6-ethoxybenzo[d]thiazol-2-amine derivatives
9(a-i) and tested for anti-diabetic agents. From the results obtained compounds 9a and
9d can be considered as anti-diabetic agents against sucrase, maltase, glucosidase, and
amylase of in vitro activity.

CHAPTER-I
37
References
[1]. R. Katritzky, J. A. Boultan (Eds), Academic press, New York, Advances in
Heterocyclic Chemistry., 1963, 1.
[2]. Weissberger, Wiley Interscience, New York, The Chemistry of Heterocyclic
Compounds., 1950, 1.
[3]. R. Katritzky, Academic press, New York, Physical methods in Heterocyclic
Chemistry., 1963, 1.
[4]. G. R. Newkome, W. W, Pandler, Contemporary Heterocyclic Chemistry., John
Wiley, New York, 1982.
[5]. G. Bertram. Katzung, Basic and Clinical Pharmacology., 6th
edition, 1995, 671.
[6]. D. A. Horton, G. T. Bourne, M. L. Smyth, Chem. Rev., 2003, 103, 893.
[7]. L. A. Thompson, J. A. Ellman, Chem. Rev., 1996, 96, 555.
[8]. J. S. Fruchtel, G. Jung Angew. Chem. Int. Ed. Engl., 1996, 35, 17.
[9]. A. Nefzi, J. M. Ostresh, R. A. Houghten, Chem. Rev., 1997, 97, 449.
[10]. R. G. Frazen, J. Comb. Chem., 2000, 2, 195.
[11]. D. J. Brown, Revs. Pure Appl. Chem., 1953, 3, 115
[12]. D. J. Brown, “The Pyridines” (Ed.), A. Weissberger, Interscience, NewYork
1962, pp. 183.
[13]. K. Haraguchi, Y. Kubota, H. Tanaka, J. Org. Chem., 2004, 69, 1831.
[14]. M. Cushman, T. Sambaiah, G. Jin, B. Illarinov, M. Bacher, A. J. Org. Chem.,
2004, 69, 601.
[15]. G. Depecker, N. Patino, C. D. Giorgio, R. Terreux, D. Carbol- Bass, C. Bailly,
A. M. Aubertin, R. Condom, Org. Biomol. Chem., 2004, 2, 74.
[16]. S. F. Wnuk, E. Lewandowska, D. R. Garcia, P. I. Jr, J. A. Secrist, III Org.
Biomol. Chem., 2004, 2, 120.

CHAPTER-I
38
[17]. N. Hossain, J. Rozenski, E. De Clercq, P. Herdewijn, J. Org.
Chemistry.1997, 62, 8, 2442.
[18]. R. W. Sabnis, D. W. Rangnekar, Indian Journal of Technology., 1990, 28, 2, 54.
[19]. S. Joseph, J. M. Burke, The Journal of Biological Chemistry., 1993, 268, 33,
24515.
[20]. B. C. Bookser, B. G. Ugarkar, M. C. Matelich, Journal of Medicinal Chemistry.,
2005, 48, 24, 7808.
[21]. A. R. Katritzky, J. W Rogers, R. M Witek, S. K Nair, Arkivoc., 2004, 52.
[22]. B. U. Khan, J. Autism. Dev. Disord., 1997, 27, 479.
[23]. D. V. Jeste, A. Okamoto, J. Napolitano, J. M. Kane, R. A. Martinez, Am. J.
Psychiatry., 2000, 157, 1150.
[24]. Z. Kapui, M. Varga, K. Urban-Szabo, E. Mikus, T. Szabo, J. Szeredi, S. Batori,
O. Finance, P. Armani, J. Pharmacol. Exper. Therap., 2003, 305, 451.
[25]. F. Awouters, J. Vermeire, F. Smeyers, P. Vermote, R. Van Beek, C. J. E.
Niemegeers, Drug. Dev. Res., 1986, 8, 95.
[26]. C. L. Motta, S. Sartini, L. Mugnaini, F. Simorini, S. Taliani, S. Salerno, A. M.
Marini, F. D. Settimo, A. Lavecchia, E. Novellino, M. Cantore, P. Failli, M.
Ciuffi, J. Med. Chem., 2007, 50, 4917.
[27]. P. G. Baraldi, B. Cacciari, R. Romagnoli, Journal of Medicinal Chemistry.,
2002, 45, 1, 115.
[28]. S. C. Goodacre, L. J. Street, D. J. Hallett, J. Med Chem., 2006, 49, 1, 35.
[29]. C. Chen, K. M. Wilcoxen, C. Q. Huang, J. Med Chem., 2004, 47, 19, 4787.
[30]. S. Wang, A. Folkes, I. Chuckowree, J. Med Chem., 2004, 47, 6, 1329.
[31]. A. Gangjee, H. D. Jain, J. Phan, J. Med Chem., 2006, 49, 3, 1055.
[32]. O. Bruno, C. Brullo, S. Schenone, Bioorg. Med. Chem., 2006, 14, 1, 121.
[33]. G. La Vielle, T. Dubuffet, O. Muller, M. Millan, A. Dekeyne, M. Brocco,
(Les Laboratories Servier, France) 2002, FR 2823752 Al20021025.

CHAPTER-I
39
[34]. R. T. Venkatasubramanian, D. G. Sathe, D. Govind, C. V. Suryavanshi, (RRG
Life Science s Limited, India) PCT International Application., 2001,
WO 2001085731 A20011115.
[35]. K. L. E. Josephine, P. S. M. Aloysius, B. F. Paul, (Janssen Pharmaceutica N.V.
Belg.) PCT International Application., 2000, WO 2000020422 Al 20000413,
Chemical Abstracts, 132, 26, 5187c.
[36]. K. L. E. Josehine, V. D. Keybus, F. M. Alfons, M. J. Carolus, PCT Internation
Application 2000, WO 2000020421 A2 200002413, Chemical Abstracts., 132,
265187c.
[37]. L. E. J. Kennis, F. P. Bischoff, C. J. Mertens, Bioorg. Med. Chem. Lett., 2000,
10, 1, 71.
[38]. K. L. E. Josephine, C. J. Love, PCT InternationalApplication WO/1998/045297
Al 199810 15, Chemical Abstracts., 1998, 129, 302654c.
[39]. C. J. Coulson, R. E. Ford, E. Hunt, Eur. J. Med. Chem., 1974, 9, 13.
[40]. P. J. Manley, A. E. Balitza, M. T. Biolodeau, K. E. Coll, G. D. Hartman, R. C.
Mc Fall, K.W. Ricket, L. D. Rodman, K. A.Thomas, Bioorg. Med. Chem .lett.,
2003, 13, 1673.
[41]. M. Anderson, J. F. Beattie, G. A. Breault, J. Breed, K. F. Byth, J. D. Culshaw,
R. P. A. Ellston, S. Green, C. A. Minshull, R. A. Norman, R. A. Pauptit, J.
Stanway, A. P. Thomas, P. J. Jewsbury, Bioorg. Med. Chem. lett ., 2003,
13, 3021.
[42]. K. F. Byth, J. D. Culshaw, S. Green, S. E. Oakes, A. P. Thomas, Bioorg. Med.
Chem. lett ., 2004, 14, 2245.
[43]. S. Emanuel, R. H. Gruninger, A. Fuentes-Pesquera, P. J. Connolly, J. A.
Seamon, S. Hazel, R. Tominovich, B. Hollister, C. Napier, M. R. D. Andrea,
M. Reuman, G. Bignan, S. A. Middleton, Molecular pharmacology., 2004, 66,
635.

CHAPTER-I
40
[44]. J. T. Sisko, T. J. Tucker, M. T. Bilodeau, C. A. Buser, P. A. Ciecko, K. E.Coll,
C. Fernandes, J. B. Gibbs, T. J. Koester, N. Kohl, J. J. Lynch, X. Mao, D.
McLoughin, C. M. Miller-Stein, L. D. Rodman, K. W. Rickert, L. Seep-
Lorenzino, J. M. Shipman, K. A. Thomas, B. K. Wong, G. D. Hartman, Bioorg.
Med. Chem. Lett., 2004, 16, 1146.
[45]. A. G. Waterson, K. L. Stevens, M. J. Reno, Y. M. Zhang, E. E. Bros, F. Bouvi
A. Rast agar, D. E. Uehling, S. H. Dickerson, B. Reep, O. B. Mc Donald, E.
Wood, D. W. Rusnak K. J. Alligood, S. K. Rudolph, Bioorg. Med. Chem. Let.,
2006, 16, 2419.
[46]. G. A. Breault, R. P. A. Ellston, S. Green, S. R. James, P. J. Jewsbury,
C. J. Midgley, R. A. Pauptit, C. A. Minshull, J. A. Tucker, Pease. Bioorg. Med.
Chem. Lett., 2003, 13, 2961.
[47]. J. F. Beattie, G. A. Breault, R .P. A. Ellston, S. Green, P. J. Jewsbury, C. J. Mid
gley, R. T. Naven, C. A. Minshull, R. A. Pauptit, J. A.Tucker, J. E. Pease,
Bioorg. Med. Chem.Lett ., 2003, 13, 2955.
[48]. Q. Zhang, Y. Liu, F. Gao, Q. Ding, C. Cho, W. Hur, Y. Jin, T. C. Uno, A. P.
Joazeiro, N. Gray, Journal of the American Chemical Society., 2006, 128, 2182.
[49]. V. J. Cee, B. K. Albrecht, S. Geuns-Meyer, P. Hughes, S. Bellon, J. Bready,
S. Caenepeel, S. C. Chaffee, A. Coxon, M. Emery, J. Fretland, P. Gallant, Y.
Gu, B. L. Hodous, D. Hoffman, R. E. Johnson, R. Kendall, J. L. Kim,
A. M. Long, D. Mcgowan, M. Morrison, P. R. Oliveri, V. F. Patel, A.
Polverino, D. Powers, P. Rose, L. Wang, H. Zhao, J. Med. Chem. 2007, 50,
627.
[50]. J. H. Hanke J. P. Gardner, R. L. Dow, J. Biol. Chem., 1996, 271, 695.
[51]. J. Dumas, R. A. Smith, T. B. Lowinger, Curr.Opin. Drug. Discov. Dev., 2004,
7, 600.
[52]. C. J. C. Connolly, J. M. Hamby, M. C. Schroeder, Bioorg. Med. Chem. Lett.,
1997, 7, 2415.
[53]. S. R. Klutchko, J. M. Hamby, D. H. Boschelli, J. Med. Chem., 1998, 41, 3276.

CHAPTER-I
41
[54]. R. L. Panek, G. H. Lu, S. R. Klutchko J. Pharmacol. Exp.Ther., 1997, 283,
1433.
[55]. V. Roginskaya, S. Zuo, E. Caudell, G. Nambudiri, A. J. Kraker, S. J. Corey,
Leukemia., 1999, 13, 855.
[56]. M. M. Moasser, M. Srethapakdi, K. S. Sachar, A. J Kraker, N. Rosen, Cancer
Res., 1999, 59, 6145.
[57]. D. Wisniewski, C. I. Lambek, C. Liu, Cancer Res., 2002, 62, 4244.
[58]. C. J. Dimitroff, W. Klohs, A. Sharma, Invest. New Drugs., 1999, 17, 121.
[59]. D. R. Huron, M. E. Gorre, A. J. Kraker, Clin Cancer Res., 2003, 9, 1267.
[60]. A. J. Kraker, B. J. Hartl, A. Amar, Biochem. Pharmacol., 2000, 60, 885.
[61]. P. La Rosée, A. S. Corbin, E. P. Stoffregen, M. W. Deininger, B. J. Druker,
Cancer Res., 2002, 62, 7149.
[62]. A. Gangjee, J. Yang, M. Ihnat, S. Kamat, Bioorg. Med. Chem., 2003, 11, 5155.
[63]. A. Gangjee, O. Namjoshi, J. Yu, M. A. Ihnat, J. E. Thorpe, L. A. Warnke,
Bioorg.Med. Chem., 2008, 16, 5514.
[64]. P. Callery, P. Gannett, Cancer and Cancer Chemotherapy, In Foye’s Principles
of Medicinal Chemistry (eds Williams DA and Lemke TL), Lippincott
Williams and Wilkins, Philadelphia, 2002, 934.
[65]. O. N. Al Safarjalani, X. J. Zhou, R. H. Ras, J. Shi, R. F. Schinazi, F. N. Naguib,
M. H. ElKouni, Cancer Chemotherapy Pharmacology., 2005, 55, 541.
[66]. P. Singh, J. Kaur, K. Paul, Indian. J. Chem., 2008, 47B, 291.
[67]. Marylenefavre, Cuong Luu Duu, Francios Huguet, Chantal Gaultier, J. Med.
Chem., 1998, 23, 453.
[68]. A. Cannito, M. Perrissin, C. Luu-Duc, F. Huguet, C. Gaultier, G. Narcisse, Eur.
J. Med. Chem., 1990, 25, 635.
[69]. F. Russo, G. Romeo, A. Caruso, V. Cutuli, D. Amore, N. A. Santagati, Eur. J.
Med. Chem., 1999, 29, 569.
[70]. L.V.G. Nargund, V. V. Badiger, S. M. Yarnal, J. Pharm. Sci., 1992, 81, 365.

CHAPTER-I
42
[71]. S. Ishwaarsingh Rathod, S. Ajay Pillai, S. Vikas Shirsath, Indian J. Heterocyclic
Chem., 2000, 10, 93.
[72]. Srinath Rangappa, Venkataranganna marikunte, Saravanan Janardhanan, Int. J.
Pharmacy and pharmaceutical science research., 2011, 1(2), 35.
[73]. S. P. Clissold, R. Beresford, Drugs., 1984, 33, 478.
[74]. Fumiyoshi Ishikawa, Akira Kosasayama, Hitoshi Yamaguchi, Yoshifumi
Watanabe, Juji Saegusa, Seiichi Shibamura, Kyoko Sakuma, Shinichiro
Ashida, Yasushi Abiko, J. Med. Chem., 1981, 24, 376.
[75]. B. Mery Press, J. James Mc Nally, A. Joan Keisher, J. Steve Offord, B.
Laurence Katz, Robert Falotico, J. Alfonso Tobaia, Eur. J. Med. Chem., 1989,
24, 627.
[76]. K. Ronald Russell, B. Jeffery Press, A. Richard Rampulla, J. James Mcnally,
Robert Falotico, A. Joan Keiser, A. David Bright, Alfonso Tobia, J. Med.
Chem., 1988, 31, 1786.
[77]. K. S. Atwal, B. N. Swanson, S. E. Unger, D. M. Floyd, S. Moreland, A.
Hedberg, B. C. O’Reilly, J. Med. Chem., 1991, 34, 806.
[78]. J. Mileij, J. Lemui, E. Bernardiner, Acta Cardiol., 1986, 41(6), 427.
[79]. H. R. Howard, T. F. Seeger, Annu, Rep. Med. Chem., 1993, 28, 39.
[80]. Abott, US Patent., 1934, 2153729.
[81]. M. Sugiyama, T. Sakamoto, K. Tabata, H. Fuumi, Chem. Pharm. Bull., 1989,
37(10), 2717.
[82]. Bristol-Myer, US Patent., 1978, 4, 12274.
[83]. B. Gauthier, Ann. Pharm. Fr., 1963, 21, 655.
[84]. Takeda, US Patent., 1962, 3016380.
[85]. J. Tani, J. Med. Chem., 1979, 22, 95.
[86]. A. Polak, H. J. Schole, Chemotherapy., 1975, 21, 113.
[87]. Satoshi Sasaki, Toshihiro Imaeda, Yoji Hayase, Yoshiaki Shimizu, Shizuo
Kasai, Nobuo Cho, Masataka Harada, Nobuhiro Suzuki, Shuichi Furuya,
Masahiko Fujino, Bioorg. Med. Chem. Lett., 2002, 12, 2073.

CHAPTER-I
43
[88]. A. Stadler, C. O. Kappe, J. Comb. Chem., 2001, 3, 624.
[89]. G. C. Rovnyak, S. D. Kimball, B. Beyer, G. Cucinotta, J. D. DiMarco, J.
Gougoutas, A. Hedberg, M. Malley, J. P. McCarthy, R. A. Zhang, S. Moreland,
J. Med.Chem., 1995, 38, 119.
[90]. R. Alajarin, J. J. Vaquero, J. Alvarez-Builla, M. Faude Casa-Juana, C. Sunkel, J.
G. Priego, P. Gomez-Sal, R. Torres, Bioorg. Med. Chem., 1994, 2, 323.
[91]. S. Gupta, N. Ajmera, N. Gautam, R. Sharma, D. Gauatam, Ind. J. Chem., 2009,
48B, 853.
[92]. R. M. Kumbhare, V. N. Ingle, Ind. J. Chem., 2009, 48B, 996.
[93]. Y. Murthi, D. Pathak, J. Pharm. Res., 2008, 7(3), 153.
[94]. B. Rajeeva, N. Srinivasulu, S. Shantakumar, E-J. Chem., 2009, 6(3), 775.
[95]. M. Maharan, S. William, F. Ramzy, A. Sembel, Molecules., 2007, 12, 622.
[96]. S. Kini, S. Swain, A. Gandhi, Ind. J. Pharm Sci., 2007, 46.
[97]. H. L. K. Stanton, R. Gambari, H. C. Chung, C.O.T. Johny, C. Filly, S. C.
Albert, Bioorg. Med. Chem., 2008, 16, 3626.
[98]. M. Wang, M. Gao, B. Mock, K. Miller, G. Sledge, G. Hutchins, Q. Zheng,
Bioorg. Med. Chem., 2006, 14, 8599.
[99]. M. Sreenivasa, E. Jaychand, B. Shivakumar, K. Jayrajkumar, J. Vijaykumar,
Arch. Pharm Sci. and Res., 2009, 1(2), 150.
[100]. S. Pattan, C. Suresh, V. Pujar, V. Reddy, V. Rasal, B. Koti, Ind. J. Chem.,
2005, 44B, 2404.
[101]. P. Reddy, Y. Lin, H. Chang, Arcivoc., 2007, xvi, 113.
[102]. Y. Heo, Y. Song, B. Kim, J. Heo, Tetrahedron Letters., 2006, 47, 3091.
[103]. F. Piscitelli, C. Ballatore, A. Smith, Bioorg. Med. Chem. Lett., 2010, 20,
644.
[104]. A. Yadav, P. Sharma, V. Ranjeeta, S. Sunder, U. Nagaich, The Pharma
Research., 2009, 1, 182.

CHAPTER-I
44
[105]. K. M. Basavaraja, B. Somashekhar, B. Shivakumar, Int. J. of PharmTech Res.,
2010, 2(2), 1139.
[106]. Y. Murthi, D. Pathak, J. Pharm. Res., 2008, 7(3), 153.
[107]. A. Pandurangan, A. Sharma, N. Sharma, P. K. Sharma, S. Visht, Der pharma
Chemica., 2010, 2(3), 316.
[108]. M. Maharan, S. William, F. Ramzy, A. Sembel, Molecules., 2007, 12, 622.
[109]. M. Amir, A. Kumar, I. Ali, S. Khan, Ind. J. Chem., 2009, 48B, 1288.
[110]. B. Soni, M. Ranawat, R. Sharma, A. Bhandari, S. Sharma, Eur. J. Chem., 2010,
45, 2938.
[111]. G. Alang, R. Kaur, G. Kaur, A. Singh, P. Singla, Acta. Pharmaceutica.
Sciencia., 2010, 52, 213.
[112]. J. Malik, F. V. Manvi, B. K. Nanjwade, S. Singh, Drug Invention Today., 2009,
1(1), 32.
[113]. H. K. Barot, G. Mallika, B. B. Sutariya, J. Shukla, L.V.G. Nargund, Research
Journal of Pharmaceutical, Biological and Chemical Sciences., 2010, 1(1),
124.
[114]. M. Sreenivasa, E Jaychand, B. Shivakumar, K. Jayrajkumar, J. Vijaykumar,
Arch Pharm Sci and Res., 2009, 1(2), 150.
[115]. B. S. Sathe, E. Jayachandran, V. A. Jagtap, G. M. Sreenivasa, Research Journal
of Pharmceutical, Biological and Chemical Sciences., 2011, 2(1), 510.
[116]. D. Shashank, T. Vishawanth, A. Prasha Md, V. Balasubramaniam, A.Nagendra,
P.Perumal, R. Suthakaran, International Journal of ChemTech Research.,
2009, 1(4), 1224.
[117]. H. Kaur, S. Kumar, I. Singh, K. K. Saxena, A. Kumar, Digest Journal of
Nanomaterials and Biostructures., 2010, 5(1), 67.
[118]. K. Oketan, N. Nagakura, K. Harada, T. Inour, EUR. J. Pharmacol., 2001, 422,
209.
[119].B. Gurupadaya, M. Gopal, B. Padmashali, Y. Manohara, Indian. J. Pharm. Sci.,
2008, 70(5), 572.

CHAPTER-I
45
[120]. S. Kini, S. Swain, A. Gandhi, Ind. J. Pharm. Sci., 2007, 46.
[121]. M. Wang, M. Gao, B. Mock, K. Miller, G. Sledge, G. Hutchins, Q. Zheng,
Bioorg. Med. Chem., 2006, (14), 8599.
[122]. S. Gupta, N. Moorthi, U. Sanyal, Ind. J. Pharmacy Pharma Sci., 2010, 2(3), 57.
[123]. B. Chi, C. Jill, P. David, J. Song, W. Choy, P. Lambert, D. Gagne, Bioorg.
Med. Chem. Lett. 2009, 19, 1416.
[124]. K. Serdons, T. Verduyckt, D. Vanderghinste, J. Cleynhens, P. Borghgraef,
Vermaelen, Bioorg. Med. Chem. Lett., 2009, (17), 602.
[125]. S. Hout, N. Azas, A. Darque, M. Robin, C. Giorgio, M. Gasquet, J. Galy,
P. David Parasitology., 2004, 129, 525.
[126]. H. Fujeda, S. Usui, T. Suzuki, H. Nakaqawa, M. Oqura, M. Makishima, N.
Miyata, Bioorg. Med. Chem. Lett., 2007, 1, 17(15), 4351.
[127]. N. Siddiqui, M. Alam, Ind. J. Het. Chem., 2004, 13, 361.
[128]. N. Siddiqui, S. N. Pandeya, A. P. Sen, G. S. Singh, Pharmak. Eftiki., 1992, 4,
121.
[129]. P. Jimonet, A. Francois, M. Barreau, J. C. Blanchard, A. Boirean, Ind. J.
Med. Chem., 1991, 42, 2828.
[130]. N. Siddiqui, S. N. Pandeya, S. Khan, J. Stables, A. Rana, M. Alam, M. Arshad,
M. Bhat, Bioorg. Med. Chem. Lett., 2007, 17, 255.
[131]. N. Amnerkar, K. P. Bhusari, Eur. J. Med. Chem., 2010, 45, 149.
[132]. S. R. Pattan, V. Suresh Ch, D. Pujar, V. V. K. Reddy, V. P. Rasal, B. C. Koti,
Indian Journal of Chemistry., 2005, 44B, 2404.
[133]. G. N. Vazquez, P. Paoli, I. L. Rivera, R.V Molina, J. Franco, R. O. Andrade,
S. Estrada- Soto, G. Camici, D. Coutino, I. Ortiz, K. Mayorga, H. Diaz,
Bioorg. Med. Chem., 2009, 17, 3332.