chapter – 5 synthesis of new carbazole derivatives as...
TRANSCRIPT

168
CHAPTER – 5
Synthesis of new carbazole derivativesas -adrenoreceptor blockers

169
5.1 Introduction
β-Adrenergic receptors play a role in many diseases of the heart
and the brain. They have been shown to be altered in the thalamus of
patients suffering from Schizophrenia, Alzheimer’s disease, panic
disorder, aging, fear, anxiety, depression, and stress.1-7
The common structural features of -adrenoreceptor blockers
include either an arylethanolamine or an aryloxyisopropanolamine
moiety. The compounds differ in the nature of the aryl group, as well as
the group linked to the amine moiety. Carvedilol (-adrenoreceptor
blocker) contains an oxyisopropanolamine moiety with aromatic
substituents linked to both the oxy and amine ends of the molecule,
which provide its combined activity.8 It has been shown for a number of
carbazoles that contain phenoxypropanolamines that the β-blocking
activities of the two optical antipodes differ by a factor of at least 50.9
The (S)-enantiomer generally appears to be the most potent one.
Stereoselectivity of the carvedilol enantiomers has estabilished and found
that (S)-(-)enantiomer of carvedilol is effecting for -adrenoreceptor
blocker and vasodilation.10
The above studies prompted us to prepare compounds having (S)-
amino moiety in between the carbazole and methoxyphenoxyethylamine
that might exhibit higher activity.

170
NH
OHO
NH
O
H3CO
INSERTION OF AMINO ACID
NH
OOH
NH
R
O
HN O
H3CO
AMINO ACID
(S)
CARVEDILOL NEW CARBAZOLE ANALOGS
5.2. Literature Background
Berridge et al., reported11 the synthesis of carazolol (5) (Scheme
5.1), starting from the commercially available 4-hydroxy carbazole (1).
NH
O O
H2N
CH3
CH3
Dioxanereflux, N
H
OOH
NH
CH3
CH3
NH
OH
ClO
(1)(2)
(3)
(5)
KOH, Toluene
(4)
…..Scheme 5.1
Dubois et al., synthesized12 iodine containing analogue of carazolol
(CYBL8E) (11) (Scheme 5.2) by reaction of 4-hydroxy carbazole (1) with
the chiral auxiliary (6) results S-epoxide (7) and then treated with 1,1-
dimethylpropargylamine (8) followed by addition of tributyltinhydride in

171
the presence of azobisisobutyronitrile (AIBN) in toluene and halogenated
with iodine in chloroform.
NH
HO O2N
SO
OO O
K2CO3,Butanone
NH
OO
NH2H3CH3C CH
EthanolNH
OOH
NH
H3C CH3
CH nBu3SnH,AIBN, Toluene
NH
OOH
NH
H3CCH3
SnBu3 I2, CHCl3
NH
OOH
NH
H3CCH3
I
(1) (6)(7)
(9)
(10) (11)
(8)
…..Scheme 5.2
Zheng et al., reported13 the synthesis of fluorocarazolol (20)
(Scheme-5.3 & 5.4), starting from 4-hydroxy carbazole (1).

172
H3C
OOH H3C S
O
OCl
Pyridine,0-5°C
H3C SO
OO
CH3
O
18F-, Kryptofx 222,K2CO3
H3C
O18F
(12) (13) (14)
(15)
…..Scheme 5.3
NH
HOKOH,Toluene
NH
OKO
OTs
Acetonitrile
NH
OO
NH3,Methanol
NH
OOH
NH2
(1) (16)
(18) (19)
(15), NaCNBH3,Acetic acid
NH
OOH
NH CH3
18F
(20)
(17)
…..Scheme 5.4

173
Wiedemann et al., reported14 the synthesis of carvedilol (22)
(Scheme-5.5) by ring opening of 4-[(oxirane-2-yl)methoxy)-9H-carbazole
(3) with 2-(2-methoxyphenoxy)ethylamine (21) in ethyl acetate at reflux
temperature.
NH
O OEthy acetate, reflux
NH
O NH
(3)
(22)
H2NO
H3CO
OH
(21)
O
H3CO
…..Scheme 5.5
Crowell et al., reported15 the synthesis of (S)-4-[2-hydroxy-3-([4-(4-
carbamoylphenoxy)phenyl]-2-methylpropylamino)propoxy]carbazole (24)
(Scheme 5.6) by ring opening of chiral oxirane (18) with 4-(4-(2-amino-2-
methylpropyl)phenoxy)benzamide (23) in the presence of acetic acid in
methanol at 60°C.

174
NH
OO
(24)
(18)
H2N
O
NH2
O Acetic acid, water,methanol, 60°C
(23)
OHN
HO
O
NH2
O
(S)HN
…..Scheme 5.6
Thomas et al., described16 the synthesis of carvedilol (22) (Scheme
5.7), starting from 4-hydroxy carbazole (1) by the following reaction
sequence.
NH
O NO
O
H3CO
Ethanol, NaOH, reflux
NH
O NHOH
O
H3CO
(26) (22)
(1)
OCH3O
N O
O
Cl
(25)
K2CO3, KI,Sodium dithionite,Ethanol, reflux
O
NH
HO
…..Scheme 5.7
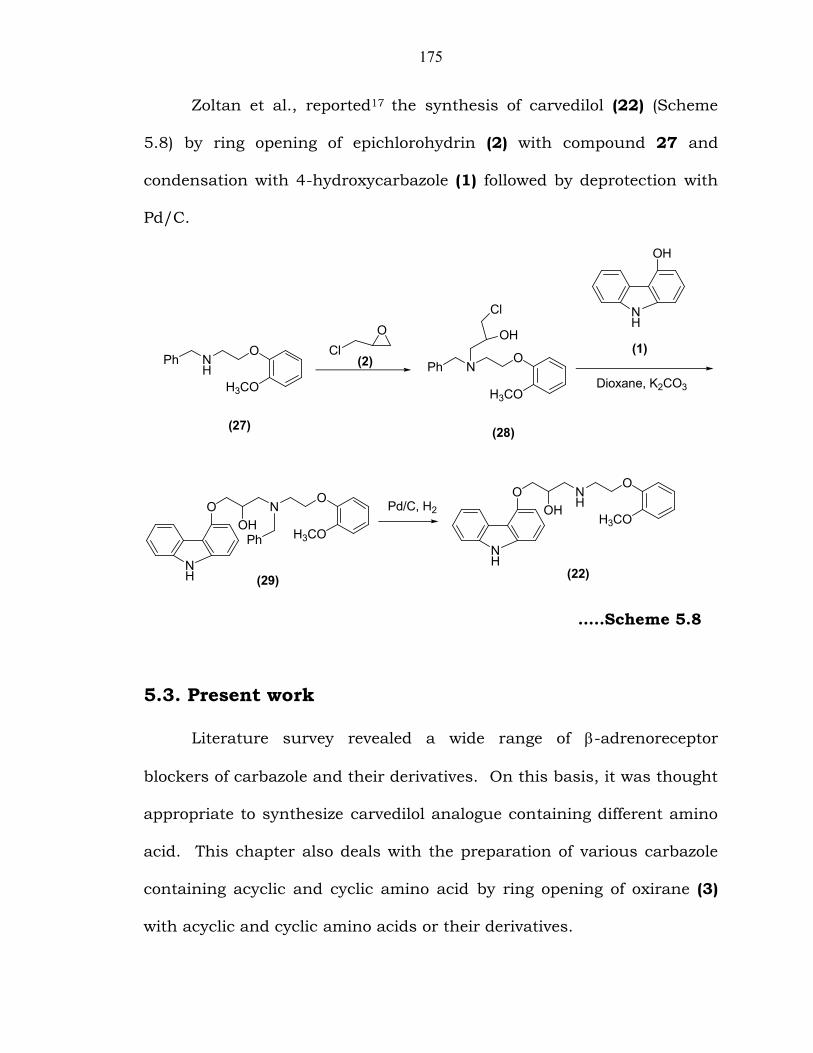
175
Zoltan et al., reported17 the synthesis of carvedilol (22) (Scheme
5.8) by ring opening of epichlorohydrin (2) with compound 27 and
condensation with 4-hydroxycarbazole (1) followed by deprotection with
Pd/C.
NH
OH
NH
O NHOH
O
H3CO
(29) (22)
(1)Ph N
HO
H3CO
(27)
ClO
Ph NO
H3CO
(28)
Cl
OH
Dioxane, K2CO3
Pd/C, H2
(2)
NH
O NOH
O
H3COPh
…..Scheme 5.8
5.3. Present work
Literature survey revealed a wide range of -adrenoreceptor
blockers of carbazole and their derivatives. On this basis, it was thought
appropriate to synthesize carvedilol analogue containing different amino
acid. This chapter also deals with the preparation of various carbazole
containing acyclic and cyclic amino acid by ring opening of oxirane (3)
with acyclic and cyclic amino acids or their derivatives.

176
5.4. Results and Discussion
5.4.1. Preparation of carvedilol derivatives containing (S)-amino acid
5.4.1.1. Preparation of compound 31a from compound 30a:
Boc-L-tert-leucine (30a) was prepared according to the method
described in the literature.15 Boc-L-tert-leucine (30a) was treated with 4-
nitrophenylchloro formate in the presence of 4-dimethylaminopyridine
(DMAP) at 0-5°C to gave mixed anhydride and treated with 2-(2-
methoxyphenoxy)ethylamine (21) at room temperature (Scheme 5.9) gave
tert-butyl-(S)-1-(2-(2-methoxyphenoxy)ethylcarbamoyl)-2,2-dimethyl-
propylcarbamate (31a).
NH
CH3H3C
O
OH
(30a)
O
OH3C
H3CH3C
H2NO
H3CO
( 21)NH
CH3H3C
O
HN
(31a)
O
OH3C
H3CH3C O
H3CO
4-Nitrophenylchloroformate,DMAP, ACN, 25-30°C
CH3 CH3
…..Scheme 5.9
The product 31a was characterized by its spectral data. Thus, its
IR (KBr) spectrum showed characteristic peaks at 3364 cm-1 (s, -NH str.),
and 1702 cm-1 (s, -C=O str.). Its 1H NMR (300 MHz, CDCl3) spectrum
showed signals at 0.97 (s, 9H, -C(CH3)3), 1.42 (s, 9H, -C(CH3)3), 3.59-
3.64 (m, 2H, -NHCH2-), 3.74 (s, 4H, -OCH3 & -NHCHCO-), 4.06-4.13 (m,
2H, -OCH2-), 5.28-5.31 (m, 1H, -CONHCH2-), 6.45-6.46 (m, 1H, -

177
CONHCH-), 6.89-6.99 (m, 4H, Ar-H) ppm. Its MS (ESI, m/z) showed the
[M + H]+ ion peak at 381 corresponding to a molecular mass of 380.
The above reaction has been found to be a general one and
extended to other amino acids (31b, 31c, 31d) whose structures were
assigned based on their spectral data (Table-5.1).
5.4.1.2. Preparation of compound 32a from compound 31a:
Deprotection of compound 31a in the presence of concentrated
hydrochloric acid in methylene chloride at room temperature (Scheme
5.10) gave corresponding (S)-N-(2-(2-methoxyphenoxy)ethyl)-2-amino-
3,3-dimethyl- butanamide (32a).
NH
CH3H3C
O
HN
(31a)
O
OH3C
H3CH3C
(32a)
Con. HCl,DCM, 25-30°C
O
H3CO
H2N
CH3H3C
O
HN
O
H3COCH3CH3
…..Scheme 5.10
The product 32a was characterized by its spectral data. Thus, Its
1H NMR (300 MHz, CDCl3) spectrum showed signals at 0.98 (s, 9H, -
C(CH3)3), 2.28 (br. 3H, -NH2 & -CONH), 3.07 (s, 1H, -CCHCO-), 3.62-3.69
(m, 2H, -NHCH2-), 3.88 (s, 3H, -OCH3), 4.03-4.13 (m, 2H, -OCH2-), 6.87-

178
6.99 (m, 4H, Ar-H) ppm. Its MS (ESI, m/z) showed the [M + H]+ ion peak
at 281 corresponding to a molecular mass of 280.
The above reaction was extended to other compounds (32b, 32c,
32d) whose structures were assigned by their spectral data (Table-5.2).
5.4.1.3. Preparation of compound 33a from compound 32a:
Compound 32a on condensation with 4-[(oxirane-2-yl)methoxy)-
9H-carbazole (3) in isopropyl alcohol at 75-80°C (Scheme 5.11) gave N-
(2-(2-methoxyphenoxy)ethyl)-1-(3-(9H-carbazol-4-yloxy)-2-hydroxypropyl)
-L-tert-leucine carboxamide (33a). The product 33a was characterized
by its spectral data. Thus, its IR (KBr) spectrum showed peak at 1654
cm-1 (s, -C=O str.). Its 1H NMR (300 MHz, CDCl3) spectrum showed
signals at 0.93 (s, 9H, -C(CH3)3), 2.62-2.73 (m, 4H, -NHCH2-), 3.49-
3.65 (m, 2H, -NHCHCO- & -CHOH-), 3.68-3.76 (s, 3H, -OCH3), 3.91-4.06
(m, 4H, -OCH2-), 6.76-8.27 (m, 12H, -CONH & Ar-H), 8.36 (s, 1H, -NH)
ppm. Its MS (ESI, m/z) showed the [M + H]+ ion peak at 520
corresponding to a molecular mass of 519.
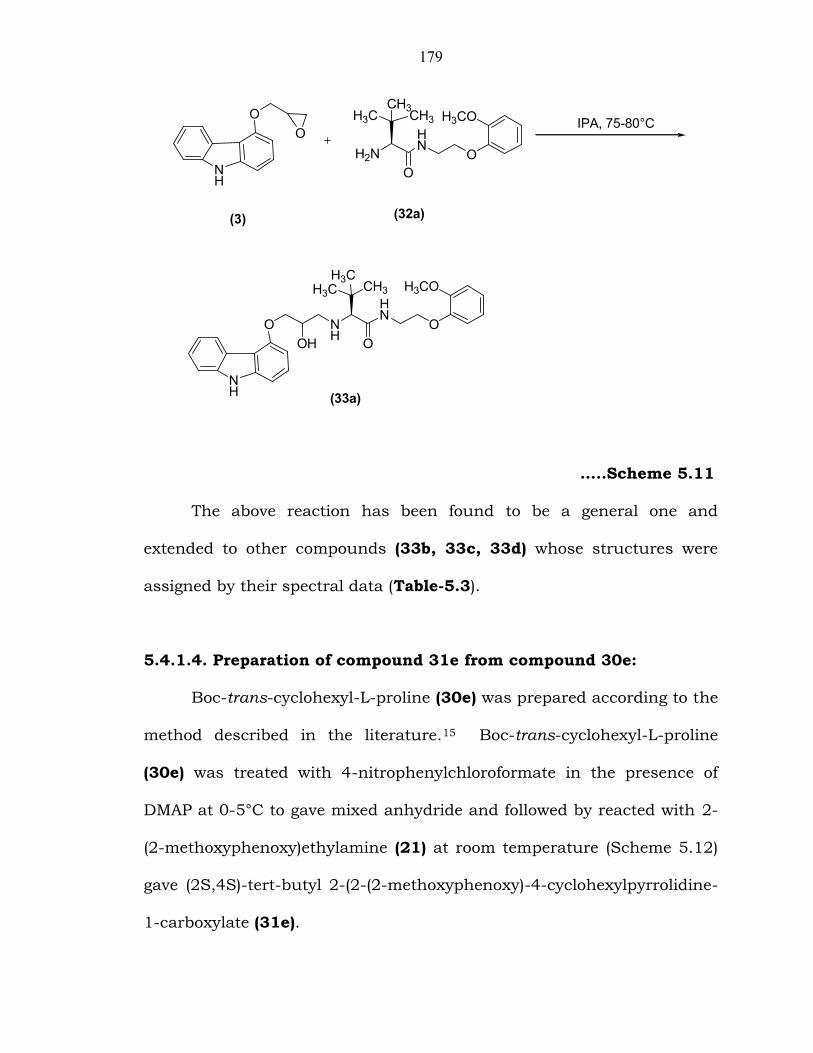
179
H2N
CH3H3C
O
HN
(32a)
NH
OO
NH
OOH
NH
CH3H3C
O
HN
O
H3CO
(33a)
IPA, 75-80°C
(3)
O
H3COCH3
H3C
…..Scheme 5.11
The above reaction has been found to be a general one and
extended to other compounds (33b, 33c, 33d) whose structures were
assigned by their spectral data (Table-5.3).
5.4.1.4. Preparation of compound 31e from compound 30e:
Boc-trans-cyclohexyl-L-proline (30e) was prepared according to the
method described in the literature.15 Boc-trans-cyclohexyl-L-proline
(30e) was treated with 4-nitrophenylchloroformate in the presence of
DMAP at 0-5°C to gave mixed anhydride and followed by reacted with 2-
(2-methoxyphenoxy)ethylamine (21) at room temperature (Scheme 5.12)
gave (2S,4S)-tert-butyl 2-(2-(2-methoxyphenoxy)-4-cyclohexylpyrrolidine-
1-carboxylate (31e).

180
N OH
OOH3C
H3CH3C O
H2NO
H3CO
(21)
(30e)
NHN
O
OH3C
CH3H3C O
O
H3CO
(31e)
4-Nitrophenylchloroformate,DMAP, ACN, 25-30°C
…..Scheme 5.12
The product 31e was characterized by its spectral data. Thus, its
IR (KBr) spectrum showed characteristic peaks at 3325 (m, -NH str.), and
1682 (s, -C=O str.). Its 1H NMR (300 MHz, CDCl3) spectrum showed
signals at 0.88-0.92 (m, 2H, -CH2), 1.09-1.16 (m, 4H, -CH2CH2-), 1.35
(s, 9H, -C(CH3)3), 1.59-1.76 (m, 8H, -3 x CH2 & -2 x CH), 2.23-2.35 (m,
1H, -NCH2), 2.95-3.02 (m, 1H, -NCH2), 3.65-3.68 (m, 3H, -NHCH2 & -
CHCO), 3.85 (s, 3H, -OCH3), 4.05-4.08 (m, 2H, -OCH2), 4.24-4.28 (m,
1H, - CONH), 6.90-6.98 (m, 4H, Ar-H) ppm. Its MS (ESI, m/z) showed
the [M + H]+ ion peak at 446 corresponding to a molecular mass of 445.
5.4.1.5. Preparation of compound 32e from compound 31e:
Deprotection of compound 31e in the presence of acid at room
temperature (Scheme 5.13) gave corresponding amino (2S,4S)-N-(2-(2-
methoxyphenoxy)ethyl)-4-cyclohexylpyrrolidine-2-carboxamide (32e).

181
NHN
O
OH3C
CH3H3C O
O
H3CO
(31e)
HNHN
OO
H3CO
(32e)
Con. HCl,DCM, 25-30°C
…..Scheme 5.13
The product 32e was characterized by its spectral data. Thus, Its
1H NMR (300 MHz, CDCl3) spectrum showed signals at 0.82-0.86 (m,
2H, -CH2), 1.07-1.09 (m, 4H, -CH2CH2-), 1.52-1.65 (m, 7H, 3 x CH2 & -
CH), 2.01-2.06 (m, 1H, -CH), 2.52-2.58 (m, 1H, -NHCH2), 3.04-3.07 (m,
1H, -NHCH2), 3.56-3.69 (m, 3H, -NCH2 & -COCH), 3.80 (s, 3H, -OCH3),
4.01-4.05 (m, 2H, -OCH2), 6.83-6.89 (m, 4H, Ar-H) 8.06 (br, 1H, -
CONHCH2) ppm. Its MS (ESI, m/z) showed the [M + H]+ ion peak at 347
corresponding to a molecular mass of 346.
5.4.1.6. Preparation of compound 33e from compound 32e:
Compound 32e on condensed with oxirane (3) in isopropyl alcohol
at 75-80°C (Scheme 5.14) gave (2S,4S)-N-(2-(2-methoxyphenoxy)ethyl)-1-
(3-(9H-carbazol-4-yloxy)-2-hydroxypropyl)-4-cyclohexylpyrrolidine-2-
carboxamide (33e). The product 33e was characterized by its spectral
data.

182
NNH
OO
H3CO
(33e)
HNHN
OO
H3CO
(32e)
NH
OO
(3)
IPA, 75-80°C
OOH
NH
…..Scheme 5.14
Thus, it’s IR (KBr) spectrum showed typical peak at 1654 cm-1 (s, -
C=O str.). Its 1H NMR (300 MHz, CDCl3) spectrum showed signals at
0.9 (s, 2H, -CH2), 1.15-1.98 (m, 4H, -CH2), 1.57-1.65 (m, 5H, -CH2 & -
CH), 1.99-2.25 (m, 3H, -CH & -CH2), 3.05-3.10 (m, 1H, -CHCO), 3.46-
3.61 (m, 4H, N(CH2)2), 3.71 (s, 3H, -OCH3), 3.88-4.04 (m, 3H, -NHCH2 &
-OCH), 4.14-4.54 (m, 4H, -OCH2), 6.67-8.26 (m, 11H, Ar-H), 11.37 (s,
1H, -NH) ppm. Its MS (ESI, m/z) showed the [M + H]+ ion peak at 586
corresponding to a molecular mass of 585.

183
5.4.2. Preparation of carbazole derivative containing acyclic (S)-
amino acid
5.4.2.1. Preparation of compound 35a from compound 34a:
Oxirane (3) was prepared according to the method described in the
literature.14 Oxirane (3) on condensation with L-valine (34a) in the
presence of potassium carbonate in water at 75-80°C (Scheme 5.15) gave
(S)-2-(3-(9H-carbazol-4-yloxy)-2-hydroxypropylamino)-3-methylbutanoic
acid (35a).
The product 35a was characterized by its spectral data. Thus, its
IR (KBr) spectrum showed characteristic peaks at 3406 cm-1 (s, -NH str.),
and 1606 cm-1 (s, -C=O str.). Its 1H NMR (300 MHz, DMSO-d6) spectrum
showed signals at 0.91-0.96 (m, 6H, -CH(CH3)2), 1.98-2.02 (m, 1H, -
CH(CH3)2), 2.93-3.17 (m, 3H, -NHCHCO- & -CHCH2NH-), 4.15-4.22 (m,
3H, -OCH2CHCH2-), 6.66-8.21 (m, 7H, Ar-H) 11.28 (s, 1H, NH) ppm.
Its 13C NMR (75 MHz, DMSO-d6) spectrum showed signals at
18.6, 18.7, 18.8, 29.7, 50.9, 51.2, 66.9, 67.4, 67.6, 69.9, 70.1, 100.4,
104.0, 110.4, 111.5, 118.6, 121.7, 122.6, 124.5, 126.4, 138.9, 141.1,
154.7, 172.0, 172.3. Its MS (ESI, m/z) showed the [M + H]+ ion peak at
357 corresponding to a molecular mass of 356.

184
NH
OO
(3)
H2N
CH3H3C
O
OH
K2CO3,H2O,75-80°C
NH
O
(35a)
OH
NH
H3C CH3
O
OH
(34a)
…..Scheme 5.15
The above reaction has been found to be a general one and
extended to other compounds (35b, 35c, 35d, 35e, 35f, 35g, 35h, 35i,
35j, 35k) whose structures were assigned based on their spectral data
(Table-5.4).
5.4.3. Preparation of carbazole derivatives containing cyclic (S)-
amino acid
5.4.3.1. Preparation of compound 37a from compound 36a:
Ring opening of oxirane (3) with 4-hydroxy-L-proline (36a) in the
presence of potassium carbonate in water at 75-80°C (Scheme 5.16) gave
(2S,4R)-1-(3-(9H-carbazol-4-yloxy)-2-hydroxypropyl)-4-hydroxy-
pyrrolidine-2-carboxylic acid (37a). The product 37a was characterized
by its spectral data. Thus, its IR (KBr) spectrum showed a characteristic
peak at 1625 cm-1 (s, -C=O str.). Its 1H NMR (300 MHz, DMSO-d6)
spectrum showed signals at 2.04-2.09 (m, 2H, -CHCH2CH-), 2.74-2.81
(m, 1H, -CHCH2N-), 3.04-3.15 (m, 2H, -NCH2CH-), 3.58-3.59 (m, 1H, -
CHCH2N-), 3.80-3.84 (m, 1H, -CH2CHCO -), 4.16-4.30 (m, 4H, -
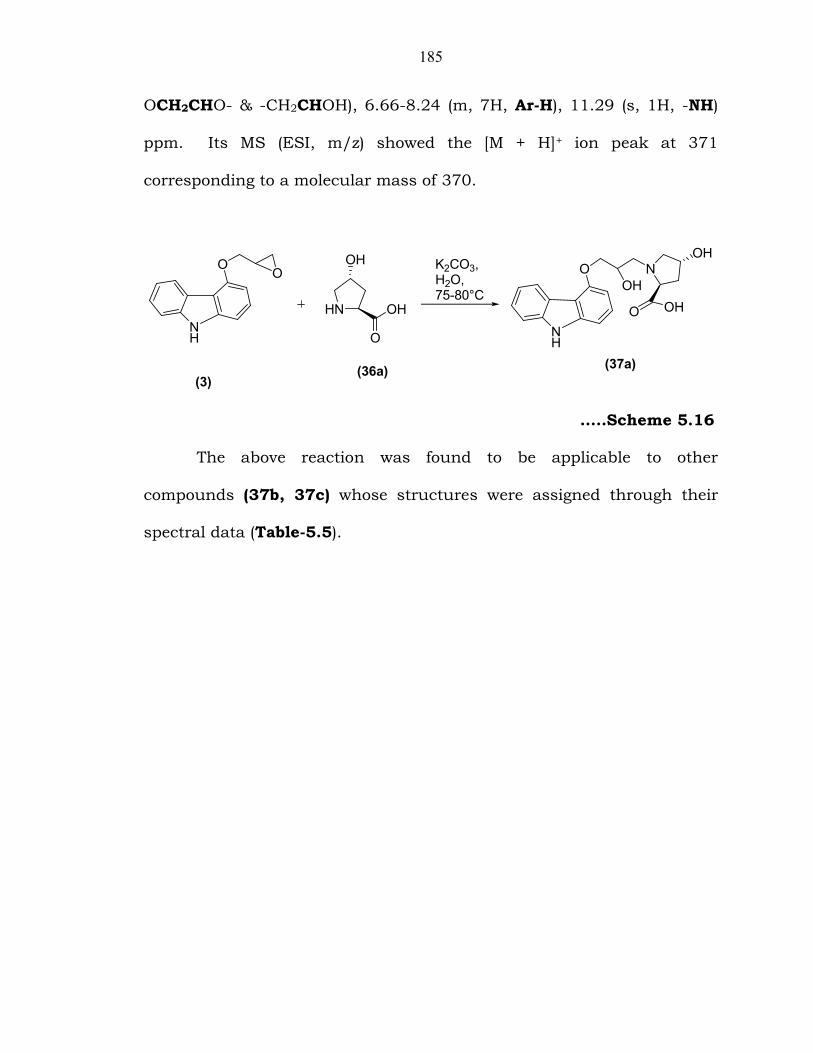
185
OCH2CHO- & -CH2CHOH), 6.66-8.24 (m, 7H, Ar-H), 11.29 (s, 1H, -NH)
ppm. Its MS (ESI, m/z) showed the [M + H]+ ion peak at 371
corresponding to a molecular mass of 370.
NH
OO
(3)
K2CO3,H2O,75-80°C
NH
O
(37a)
OHN
(36a)
HN
O
OH
OH
OHO
OH
…..Scheme 5.16
The above reaction was found to be applicable to other
compounds (37b, 37c) whose structures were assigned through their
spectral data (Table-5.5).
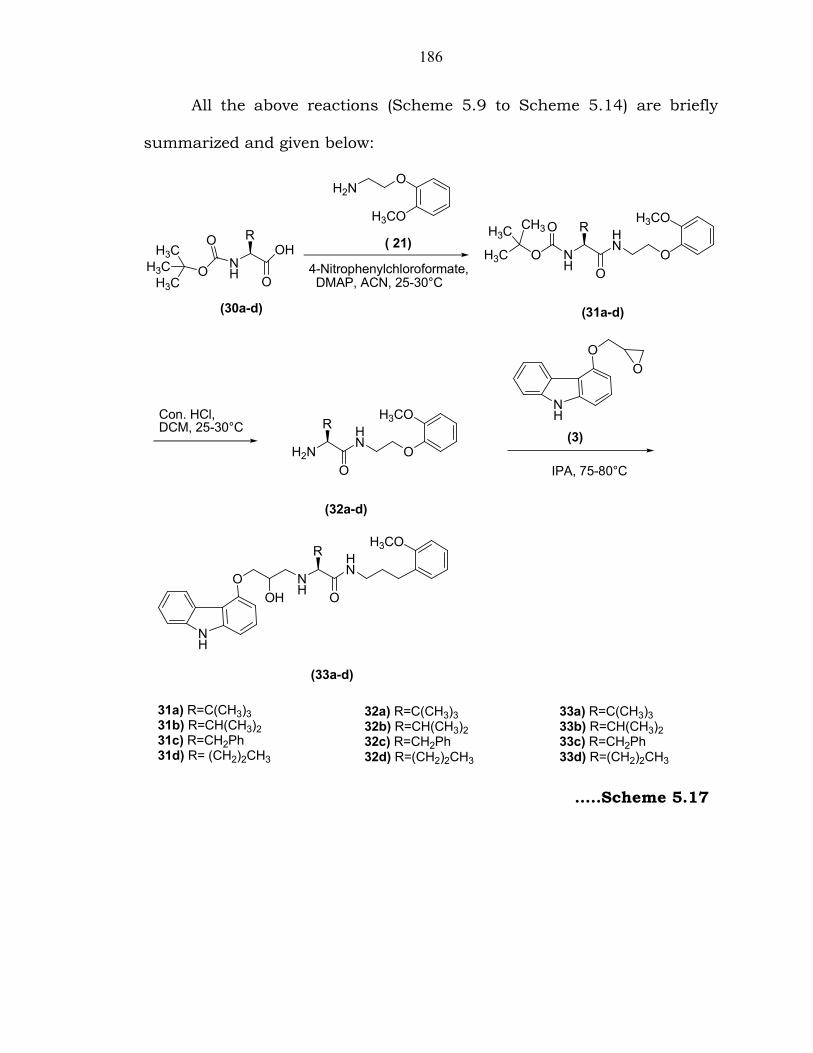
186
All the above reactions (Scheme 5.9 to Scheme 5.14) are briefly
summarized and given below:
NH
R
O
OH
(30a-d)
O
OH3C
H3CH3C
H2NO
H3CO
( 21)
4-Nitrophenylchloroformate,DMAP, ACN, 25-30°C
NH
R
O
HN
(31a-d)
O
O
CH3
H3C
H3C
O
H3CO
H2N
R
O
HN
(32a-d)
O
H3COCon. HCl,DCM, 25-30°C
NH
OO
NH
OOH
NH
R
O
HN
H3CO
(33a-d)
IPA, 75-80°C
(3)
31a) R=C(CH3)331b) R=CH(CH3)231c) R=CH2Ph31d) R= (CH2)2CH3
32a) R=C(CH3)332b) R=CH(CH3)232c) R=CH2Ph32d) R=(CH2)2CH3
33a) R=C(CH3)333b) R=CH(CH3)233c) R=CH2Ph33d) R=(CH2)2CH3
…..Scheme 5.17

187
The above reaction in scheme 5.15 is briefly summarized and
depicted below:
NH
OO
(3)
H2N
R1
O
OR2
K2CO3,H2O,75-80°C
HN
O
(35a-k)
OHNH
R1
O
OR2
(34a-k)
35a) R1=CH(CH3)2, R2=H,35b) R1=H, R2=H,35c) R1=CH3, R2=H,35d) R1=C(CH3)3, R2=H,35e) R1=CH2Ph, R2=H,35f) R1=(CH2)3CH3, R2=H,
35g) R1=CH(CH3)CH2CH3, R2=H,35h) R1=CH2CH(CH3)2, R2=H,35i) R1=(CH2)2CH3, R2=H,35j) R1=CH2CH(CH3)2, R2=CH3,35k) R1=CH(CH3)2, R2=CH3,
…..Scheme 5.18
The above reaction in scheme 5.16 is briefly summarized and
depicted below:
NH
O O
(3)
K2CO3,H2O, 75-80°C
NH
O
(37a-c)
OHN
(36a-c)
HN
O
OH
R
37a) R=OH,37b) R=C6H5,37c) R=C6H11
OHO
R
…..Scheme 5.19

188
5.5. Experimental Section
All the chemicals and reagents used were of LR grade. 1H NMR
and 13C NMR spectra were recorded on a Bruker 300 spectrometer at
300 MHz and 75 MHz, respectively and the chemical shifts were reported
as δ values in parts per million relative to TMS as an internal standard.
Infrared spectra were recorded in the solid state as KBr dispersion using
a Perkin-Elmer spectrophotometer. Mass spectra were recorded on API
2000 Perkin Elmer PE-SCIEX mass spectrometer.
1) General procedure for preparation of compound 31a from
compound 30a: Compound 30a (3.0 g, 0.01299 mol) was dissolved in
ACN (30 mL) at room temperature and cooled to 5-10°C. DMAP (1.46 g,
0.01364 mol) was added at 5-10°C and stirred for 10 min. Solution of 4-
nitrophenychloroformate, prepared by dissolving 4-
nitrophenychloroformate (2.75 g, 0.01364 mol) in ACN (20 mL) was
added at 5-10°C and stirred for 30 min. Compound 21 (2.28 g, 0.01364
mol) was added and then the reaction mixture temperature was slowly
raised to 20-25°C and stirred at the same temperature till completion of
reaction. Thereafter, the reaction mixture was filtered to remove
insoluble. The clear filtrate was concentrated at 40°C under reduced
pressure. The resulting oily mass was dissolved in methylene chloride
(50 mL) and washed with 10% w/w aqueous sodium carbonate solution
(3 x 20 mL). The organic layer was separated and washed with 2.5%
w/w aqueous ammonia (5 x 30 mL) followed by DM water (30 mL).

189
Finally, the clear solution was washed with 3% w/w aqueous
hydrochloric acid solution (30 mL) followed by DM water (30 mL). The
organic layer was separated and concentrated completely at 40°C under
reduced pressure to yield 31a [4.04 g, 82%].
2) General procedure for preparation of compound 32a from
compound 31a: Compound 31a (4.0 g, 0.0105 mol) was dissolved in
methylene chloride (10 mL) at room temperature. Con. HCl (3.0 g,
0.02625 mol) was added and stirred at 25-30°C until completion of
reaction. After completion of reaction, DM water (20 mL) was added and
stirred for 10 min. The aqueous layer was separated and washed with
methylene chloride (10 mL). The aqueous layer pH was adjusted to 8.5-
9.0 with 10% w/w aqueous sodium carbonate and concentrated
completely at 60°C under reduced pressure. The resulting solid was
dissolved in methylene chloride (15 mL) and filtered insoluble. The clear
filtrate was concentrated completely at 35°C under reduced pressure to
afford 32a [2.26 g,.77%].
3) General procedure for preparation of compound 33a from
compound 32a: Compound 32a (1.52 g, 0.005434 mol) was dissolved in
isopropyl alcohol (10 mL) at 25-30°C and heated to 75°C. Compound 3
(1.0 g, 0.00418 mol) was added in one lot at 75°C and stirred at the same
temperature till completion of reaction. Thereafter, the reaction mass
was concentrated completely and cooled to room temperature. The
resulting oily mass was dissolved in methylene chloride (20 mL) and

190
washed with DM water (2 x 30 mL). The organic layer was separated and
concentrated completely at 35°C under reduced pressure to afford 33a
[1.6 g,.74%]
4) Preparation of compound 31e from compound 30e: Compound 30e
(3.0 g, .0101 mol) was dissolved in ACN (30 mL) at room temperature and
cooled to 5-10°C. DMAP (1.13 g, 0.010605 mol) was added at 5-10°C and
stirred for 10 min. Solution of 4-nitrophenychloroformate, prepared by
dissolving 4-nitrophenychloroformate (2.14 g, 0.010605 mol) in ACN (20
mL) was added at 5-10°C and stirred for 30 min. Compound 21 (1.77 g,
0.010605 mol) was added and then the reaction mixture temperature
was slowly raised to 20-25°C and stirred at the same temperature till
completion of reaction. Thereafter, the reaction mixture was filtered to
remove insoluble. The clear filtrate was concentrated at 40°C under
reduced pressure. The resulting oily mass was dissolved in methylene
chloride (50 mL) and washed with 10% w/w aqueous sodium carbonate
solution (3 x 20 mL). The organic layer was separated and washed
successively with 2.5% w/w aqueous ammonia (5 x 30 mL) followed by
DM water (30 mL). Finally, the clear solution was washed with 3% w/w
aqueous hydrochloric acid solution (30 mL) followed by DM water (30
mL). The organic layer was separated and concentrated completely at
40°C under reduced pressure to yield 31e [3.82 g,.85%].
5) Preparation of compound 32e from compound 31e: Compound 31e
(3.5 g, 0.00785 mol) was dissolved in methylene chloride (10 mL) at room

191
temperature. Con. HCl (2.23 g, 0.019625 mol) was added and stirred at
25-30°C until completion of reaction. After completion of reaction, DM
water (20 mL) was added and stirred for 10 min. The aqueous layer was
separated and washed with methylene chloride (10 mL). The aqueous
layer pH was adjusted to 8.5-9.0 with 10% w/w aqueous sodium
carbonate and concentrated completely at 40°C under reduced pressure.
The resulting solid was dissolved in methylene chloride (15 mL) and
filtered insoluble. The clear filtrate was concentrated completely at 35°C
under reduced pressure to afford 32e [2.12 g,.78%].
6) Preparation of compound 33e from compound 32e: Compound 32e
(1.88 g, 0.005434 mol) was dissolved in isopropyl alcohol (10 mL) at 25-
30°C and heated to 75°C. Compound 3 (1.0 g, 0.00418 mol) was added
in one lot at 75°C and stirred at the same temperature till completion of
reaction. Thereafter, the reaction mass was concentrated completely and
cooled to room temperature. The resulting oily mass was dissolved in
methylene chloride (20 mL) and washed with DM water (2 x 30 mL). The
organic layer was separated and concentrated completely at 35°C under
reduced pressure to afford 33e [1.83 g,.75%].
7) General procedure for preparation of compound 35a from
compound 34a: Compound 34a (7.31 g, 0.0624 mol), was dissolved in
11% w/w aq. potassium carbonate (60 mL) at room temperature and
heated to 75°C. Compound 3 (3.0 g, 0.0125 mol) was added in one lot at
75-80°C and stirred at same temperature until completion of reaction.

192
Thereafter, the reaction mixture was cooled to RT and filtered through
hyflo bed. The clear filtrate pH was adjusted to 6.2-6.5 with 10% w/w
hydrochloric acid (25 mL). The separated solid was filtered, washed with
water and dried under reduced pressure to yield 35a [3.7 g,.84%].
8) General procedure for preparation of compound 37a from
compound 36a: Compound 36a (8.2 g, 0.0627 mol), was dissolved in
11% w/w aq. potassium carbonate (60 mL) at room temperature and
heated to 80°C. Compound 3 (3.0 g, 0.0125 mol) was added in one lot at
80-85°C and stirred at same temperature until completion of reaction.
Thereafter, the reaction mixture was cooled to RT and filtered through
hyflo bed. The clear filtrate pH was adjusted to 6.2-6.5 with 10% w/w
hydrochloric acid (27 mL). The separated solid was filtered, washed with
water and dried under reduced pressure to yield 37a [3.84 g,.83%].

193
Table-5.1: Reaction condition and yield of compounds 31b-d.
S. No. Startingmaterial Reagent /solvent Reaction
conditionsProductobtained Yield (%)
1 30b 4-Nitrophenychloro formate/DMAP/ACN
20-25°C 31b(R=CH(CH3)2,)
80
2 30c 4-Nitrophenychloro formate/DMAP/ ACN
20-25°C 31c(R=CH2Ph)
86
3 30d 4-Nitrophenychloro formate/DMAP/ ACN
20-25°C 31d(R=(CH2)2CH3)
84
Table-5.2: Reaction condition and yield of compounds 32b-d.
S. No. Startingmaterial Reagent / solvent Reaction
conditionsProductobtained Yield (%)
1 31b Con.HCl / DCM 20-25°C 32b(R=CH(CH3)2,)
80
2 31c Con.HCl / DCM 20-25°C 32c(R=CH2Ph) 77
3 31d Con.HCl / DCM 20-25°C32d
(R=(CH2)2CH3) 79

194
Table-5.3: Reaction condition and yield of compounds 33b-d.
S. No. Startingmaterial Solvent Reaction
conditions Product obtained Yield (%)
1 32b IPA 75-80°C 33b(R=CH(CH3)2,)
74
2 32c IPA 75-80°C 33c(R=CH2Ph)
76
3 32d IPA 75-80°C 33d(R=(CH2)2CH3)
74
Table-5.4: Reaction condition and yield of compounds 35b-k.
S. No. Startingmaterial Reagent used Reaction
conditions Product obtained Yield (%)
1 34b K2CO3/ H2O 75-80°C 35b(R1= H, R2=H)
85
2 34c K2CO3/ H2O 75-80°C 35c(R1= CH3, R2=H)
86
3 34d K2CO3/ H2O 75-80°C 35d(R1=C(CH3)3, R2=H)
86

195
4 34e K2CO3/ H2O 75-80°C 35e(R1=CH2Ph, R2=H)
85
5 34f K2CO3/ H2O 75-80°C 35f(R1=(CH2)3CH3, R2=H)
84
6 34g K2CO3/ H2O 75-80°C 35g(R1=CH(CH3)CH2CH3,
R2=H)
85
7 34h K2CO3/ H2O 75-80°C 35h(R1=CH2CH(CH3)2,
R2=H)
82
8 34i K2CO3/ H2O 75-80°C 35i(R1=(CH2)2CH3, R2=H)
88
9 34j K2CO3/ H2O 75-80°C 35j(R1=CH2CH(CH3)2,
R2=CH3)
85
10 34k K2CO3/ H2O 75-80°C 35k(R1=CH(CH3)2,
R2=CH3)
83
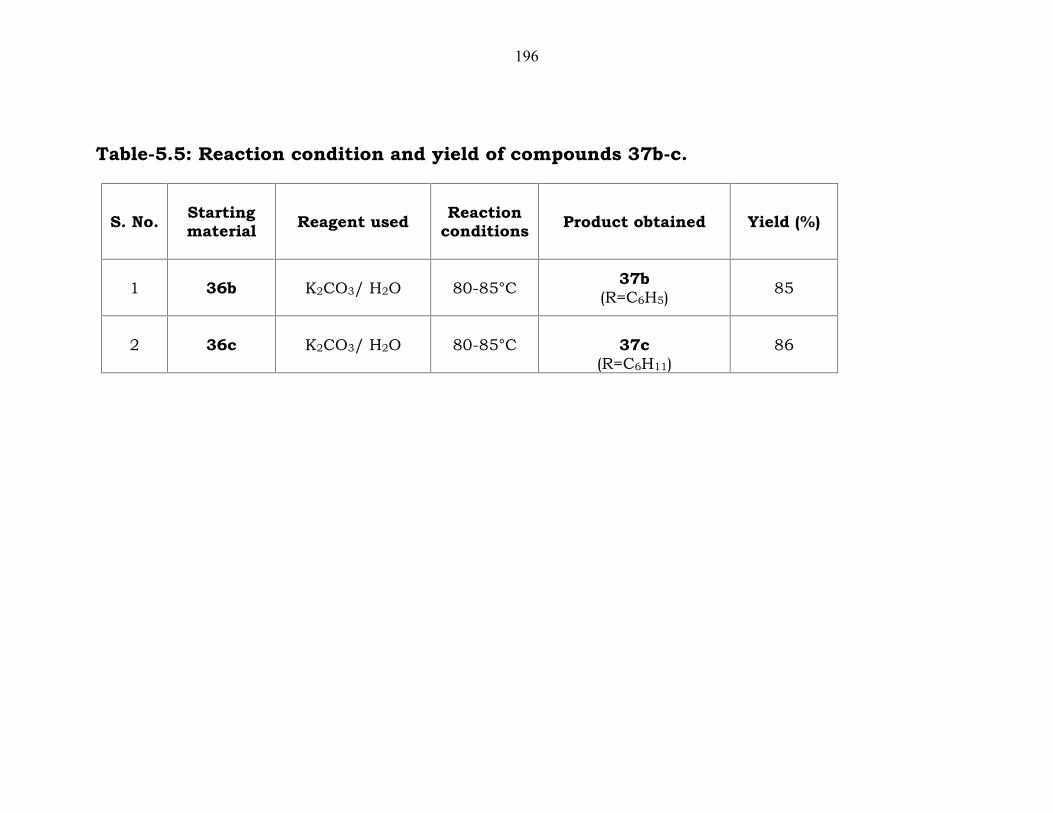
196
Table-5.5: Reaction condition and yield of compounds 37b-c.
S. No. Startingmaterial Reagent used Reaction
conditions Product obtained Yield (%)
1 36b K2CO3/ H2O 80-85°C 37b(R=C6H5)
85
2 36c K2CO3/ H2O 80-85°C 37c(R=C6H11)
86

197
Table-5.1: Spectral data for compounds 31b-d.
S. No. Productobtained
IR (KBr)cm-1
1H-NMRδ, ppm
Massm/z
(M+1)
1 31b(R=CH(CH3)2,)
3302 (s, -NHstr.), 1664 (s, -C=O str.)
DMSO-d6; 0.79-0.84 (m, 6H, -CH(CH3)2), 1.37 (s,9H, -C(CH3)3), 1.87-1.94 (m, 1H, -CH(CH3)2),3.35-3.50 (m, 2H, -NHCH2-), 3.74 (s, 4H, -OCH3& -NHCHCO-), 3.90-3.97 (m, 2H, -OCH2-), 6.60-6.63 (m, 1H, -CONHCH-), 6.83-6.97 (m, 4H, Ar-H), 8.03-8.05 (m, 1H, -CONHCH2-).
367
2 31c(R=CH2Ph)
3360 (s, -NHstr.), 1665 (s, -C=O str.)
CDCl3; 1.35 (s, 9H, -C(CH3)3), 2.96-2.98 (m, 2H,-CH2Ph), 3.51-3.54 (m, 2H, -NHCH2-), 3.75 (s,3H, -OCH3), 3.82-3.84 (m, 1H, -NHCHCO-), 3.89-3.94 (m, 1H, -OCH2), 4.27-4.29 (m, 1H, -OCH2),5.00 (br, 1H, -CONHCH), 6.36 (br, 1H, -CONHCH2), 6.75-6.90 (m, 4H, Ar-H), 7.04-7.19(m, 5H, Ar-H).
415
3 31d(R=(CH2)2CH3)
3314 (s, -NHstr.), 1659 (s, -C=O str.)
CDCl3; 0.87-0.92 (t, 3H, -CH3), 1.31-1.36 (m, 2H,-CH2CH3), 1.38 (s, 9H, -C(CH3)3), 1.52-1.79 (m,2H, -CHCH2), 3.63-3.69 (m, 2H, -NHCH2), 3.87(s, 3H, -OCH3), 4.06-4.09 (m, 2H, -OCH2), 5.05(br, 1H, -CONHCH-), 6.75 (br, 1H, -CONHCH2),6.89-7.27 (m, 4H, Ar-H).
367

198
Table-5.2: Spectral data for compounds 32b-d.
S. No. Productobtained
IR (KBr)cm-1
1H-NMRδ, ppm
Massm/z
(M+1)
1 32b(R=CH(CH3)2,)
3348 (s, -NHstr.), 1618 (s, -C=O str.)
CDCl3; 0.83-0.99 (m, 6H, -CH(CH3)2), 2.26-2.28(m, 1H, -CH(CH3)2), 3.36-3.38 (m, 1H, NH2CHCO-),3.65-3.70 (m, 2H, -NHCH2-), 3.85 (s, 3H, -OCH3),4.04-4.12 (m, 2H, -OCH2-), 6.86-6.96 (m, 4H, Ar-H), 7.81-7.83 (m, 1H, -CONHCH2-).
267
232c
(R=CH2Ph)3486 (s, -NHstr.), 1667 (s, -C=O str.)
DMSO-d6; 3.04-3.06 (m, 2H, -CH2Ph), 3.37-3.53(m, 2H, -NHCH2-), 3.74 (s, 3H, -OCH3), 3.85-3.92(m, 1H, - OCH2), 3.99-4.03 (m, 1H, - NHCHCO-),6.88-6.99 (m, 4H, Ar-H), 7.20-7.23 (m, 5H, Ar-H),8.77 (m, 1H, -CONHCH2).
315
3 32d(R=(CH2)2CH3)
3395 (s, -NHstr.), 1666 (s, -C=O str.)
CDCl3; 0.82-0.87 (t, 3H, -CH3), 1.29-1.42 & 1.71-1.76 (m, 2H, -CH2CH2CH3), 3.29-3.33 (m, 1H, -NH2CH-), 3.56-3.61 (m, 2H, -NHCH2), 3.79 (s, 3H,-OCH3), 4.01-4.04 (m, 2H, -OCH2), 6.89-7.27 (m,4H, Ar-H), 7.69 (br, 1H, -CONHCH2).
267
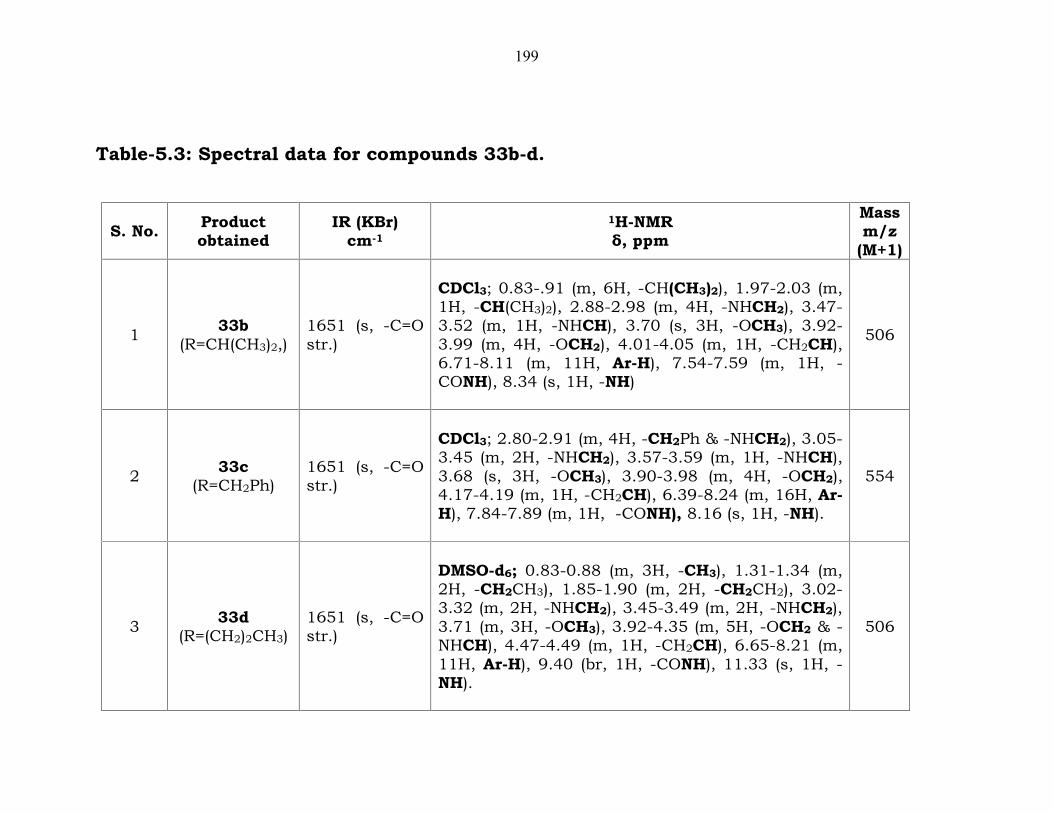
199
Table-5.3: Spectral data for compounds 33b-d.
S. No. Productobtained
IR (KBr)cm-1
1H-NMRδ, ppm
Massm/z
(M+1)
1 33b(R=CH(CH3)2,)
1651 (s, -C=Ostr.)
CDCl3; 0.83-.91 (m, 6H, -CH(CH3)2), 1.97-2.03 (m,1H, -CH(CH3)2), 2.88-2.98 (m, 4H, -NHCH2), 3.47-3.52 (m, 1H, -NHCH), 3.70 (s, 3H, -OCH3), 3.92-3.99 (m, 4H, -OCH2), 4.01-4.05 (m, 1H, -CH2CH),6.71-8.11 (m, 11H, Ar-H), 7.54-7.59 (m, 1H, -CONH), 8.34 (s, 1H, -NH)
506
2 33c(R=CH2Ph)
1651 (s, -C=Ostr.)
CDCl3; 2.80-2.91 (m, 4H, -CH2Ph & -NHCH2), 3.05-3.45 (m, 2H, -NHCH2), 3.57-3.59 (m, 1H, -NHCH),3.68 (s, 3H, -OCH3), 3.90-3.98 (m, 4H, -OCH2),4.17-4.19 (m, 1H, -CH2CH), 6.39-8.24 (m, 16H, Ar-H), 7.84-7.89 (m, 1H, -CONH), 8.16 (s, 1H, -NH).
554
3 33d(R=(CH2)2CH3)
1651 (s, -C=Ostr.)
DMSO-d6; 0.83-0.88 (m, 3H, -CH3), 1.31-1.34 (m,2H, -CH2CH3), 1.85-1.90 (m, 2H, -CH2CH2), 3.02-3.32 (m, 2H, -NHCH2), 3.45-3.49 (m, 2H, -NHCH2),3.71 (m, 3H, -OCH3), 3.92-4.35 (m, 5H, -OCH2 & -NHCH), 4.47-4.49 (m, 1H, -CH2CH), 6.65-8.21 (m,11H, Ar-H), 9.40 (br, 1H, -CONH), 11.33 (s, 1H, -NH).
506

200
Table-5.4: Spectral data for compounds 35b-k.
S. No. Product obtained IR (KBr)cm-1
1H-NMR (DMSO-d6)δ, ppm
Massm/z
(M+1)
1 35b(R1= H, R2=H)
1603(s, -C=Ostr.)
2.90-3.25 (m, 4H, -NHCH2CO & -NHCH2CH), 4.13-4.15 (m, 3H, -OCH2CHOH), 6.65-8.25 (m, 7H, Ar-H), 11.30 (s, 1H, -NH).
315
2 35c(R1= CH3, R2=H)
1605(s, -C=Ostr.)
1.18-1.28 (m, 3H, -CHCH3), 2.85-3.12 (m, 3H, -NHCH2- & -NHCHCO), 4.09-4.15 (m, 3H, -OCH2CHOH), 6.64-8.23 (m, 7H, Ar-H), 11.37 (s,1H, -NH).
329
3 35d(R1=C(CH3)3, R2=H)
1606(s, -C=Ostr.)
0.93-0.97 (m, 9H, -C(CH3)3), 2.50-2.81 (m, 2H, -NHCH2), 2.91-2.94 (m,1H, -NHCHCO), 4.12-4.23(m, 3H, -OCH2CHOH), 6.65-8.21 (m, 7H, Ar-H),11.29 (s, 1H, -NH).
371
4 35e(R1=CH2Ph, R2=H)
1606(s, -C=Ostr.)
2.89-3.13 (m, 3H, -NHCH2- & -NHCHCO), 3.56-3.60 (m, 2H, -CH2Ph), 4.4.04-4.16 (m, 3H, -OCH2CHOH), 6.69-8.23 (m, 12H, Ar-H), 11.28 (s,1H, -NH).
405
535f
(R1=(CH2)3CH3,R2=H)
1604(s, -C=Ostr.)
0.81-0.85 (m, 3H, -CH2CH3), 1.22-1.35 (m, 4H, -CH2CH2CH3), 1.65-1.69 (m, 2H, -CHCH2CH2), 2.94-3.22 (m, 3H, -NHCH2- & -NHCHCO), 4.16-4.27 (m,3H, -OCH2CHOH), 6.65-8.26 (m, 7H, Ar-H), 11.36(s, 1H, -NH).
371
635g
(R1=CH(CH3)CH2CH3, R2=H)
1606(s, -C=Ostr.)
0.82-0.92 (m, 6H, -CH2CH3 &, -CHCH3), 1.23-1.28(m, 1H, -CH2CH3), 1.53-1.57 (, 1H, -CH2CH3), 1.73-1.76 (m, 1H, -CHCH3), 2.87-3.18 (m, 3H, -NHCH2-& -NHCHCO), 4.16-4.22 (m, 3H, -OCH2CHOH),6.65-8.21 (m, 7H, Ar-H), 11.30 (s, 1H, -NH).
371

201
735h
(R1=CH2CH(CH3)2,R2=H)
1607 (s, -C=O str.)
0.83-0.90 (m, 6H, -CH(CH3)2), 1.47-1.65 (m, 3H, -CHCH2 & -CH(CH3)2), 2.67-3.15 (m, 3H, -NHCH2- &-NHCHCO), 4.07-4.23 (m, 3H, -OCH2CHOH), 6.65-8.22 (m, 7H, Ar-H), 11.29 (s, 1H, -NH).
371
835i
(R1=(CH2)2CH3,R2=H)
1607 (s, -C=O str.)
0.82-0.88 (m, 3H, -CH2CH3), 1.39-1.44 (m. 2H, -CH2CH3), 1.67-1.69 (m, 2H, -CHCH2CH2), 2.99-3.28 (m, 3H, -NHCH2- & -NHCHCO), 4.17-4.31 (m,3H, -OCH2CHOH), 6.65-8.26 (m, 7H, Ar-H), 11.39(s, 1H, -NH).
357
935j
(R1=CH2CH(CH3)2,R2=CH3)
1738 (s, -C=O str.)
0.91-0.93 (m, 6H, -CH(CH3)2), 1.66-1.79 (m, 3H, -CHCH2 & -CH(CH3)2), 3.21-3.24 (m, 1H, -NHCH2-),3.41-3.44 (m, 1H, -NHCH2-), 3.77 (s, 3H, -OCH3),4.14-4.27(m, 3H, -NHCHCO & -OCH2), 4.43-4.45(m, 1H, -CHOH), 6.68-8.22 (m, 7H, Ar-H), 11.34 (s,1H, -NH).
385
1035k
(R1=CH(CH3)2,R2=CH3)
1728 (s, -C=O str.)
CDCl3; 0.97-1.02 (m, 6H, -CH(CH3)2), 2.04-2.09 (m,1H, -CH(CH3)2), 2.91-3.19 (m, 3H, -NHCH2- & -NHCHCO), 3.73 (s, 3H, -OCH3), 4.22-4.31 (m, 3H, -OCH2CHOH), 6.65-8.26 (m, 7H, Ar-H), 8.23 (s, 1H,-NH).
371

202
Table-5.5: Spectral data for compounds 37b-c.
S. No. Productobtained
IR (KBr)cm-1
1H-NMR (DMSO-d6)δ, ppm
Massm/z
(M+1)
1 37b(R=C6H5)
1620 (s, -C=Ostr.)
2.13-2.34 (m, 2H, -CHCH2), 2.60-2.63 (m, 1H, -CHCH2), 2.95-3.02 (m, 2H, -NCH2), 3.35-3.39 (m,2H, -NCH2), 3.58-3.63 (m, 1H, -NCHCO), 4.06-4.17(m, 3H, -OCH2CHOH), 6.66-8.21 (m, 12H, Ar-H),11.29 (s, 1H, -NH).
431
2 37c(R=C6H11)
1584 (s, -C=Ostr.)
0.84-0.99 (m, 6H, -(CH2)3), 1.47-1.63 (m, 6H, -(CH(CH2)2 & -CHCH2), 1.96-2.05 (m, 2H, -(CH(CH2)2 & -CHCH2), 2.05-2.08 (m, 1H, -NCH2CHC), 2.65-2.69 (m, 1H, -NCH2CHC), 2.83-2.95 (m, 2H, NCH2CHOH), 3.06-308 (m, 1H, -NCHCO), 3.95-4.11 (m, 3H, -OCH2CHOH), 6.62-8.20 (m, 7H, Ar-H), 11.33 (s, 1H, -NH).
437

203
5.6. Spectral data
IR spectrum of compound 31a:
1H NMR spectrum (300MHz, CDCl3) of compound 31a:
NH
CH3H3C
O
HN
(31a)
O
OH3C
H3CH3C O
H3COCH3
NH
CH3H3C
O
HN
(31a)
O
OH3C
H3CH3C O
H3COCH3

204
Mass spectrum of compound 31a:
1H NMR spectrum (300MHz, CDCl3) of compound 32a:
NH
CH3H3C
O
HN
(31a)
O
OH3C
H3CH3C O
H3COCH3
(32a)
H2N
CH3H3C
O
HN
O
H3COCH3

205
Mass spectrum of compound 32a:
IR spectrum of compound 33a:
(32a)
H2N
CH3H3C
O
HN
O
H3COCH3
NH
OOH
NH
CH3H3C
O
HN
O
H3CO
(33a)
H3C

206
1H NMR spectrum (300MHz, CDCl3) of compound 33a:
Mass spectrum of compound 33a:
NH
OOH
NH
CH3H3C
O
HN
O
H3CO
(33a)
H3C
NH
OOH
NH
CH3H3C
O
HN
O
H3CO
(33a)
H3C

207
1H NMR spectrum (300MHz, DMSO-d6) of compound 35a:
13C NMR spectrum (75MHz, DMSO-d6) of compound 35a:
NH
O
(35a)
OH
NH
H3C CH3
O
OH
NH
O
(35a)
OH
NH
H3C CH3
O
OH
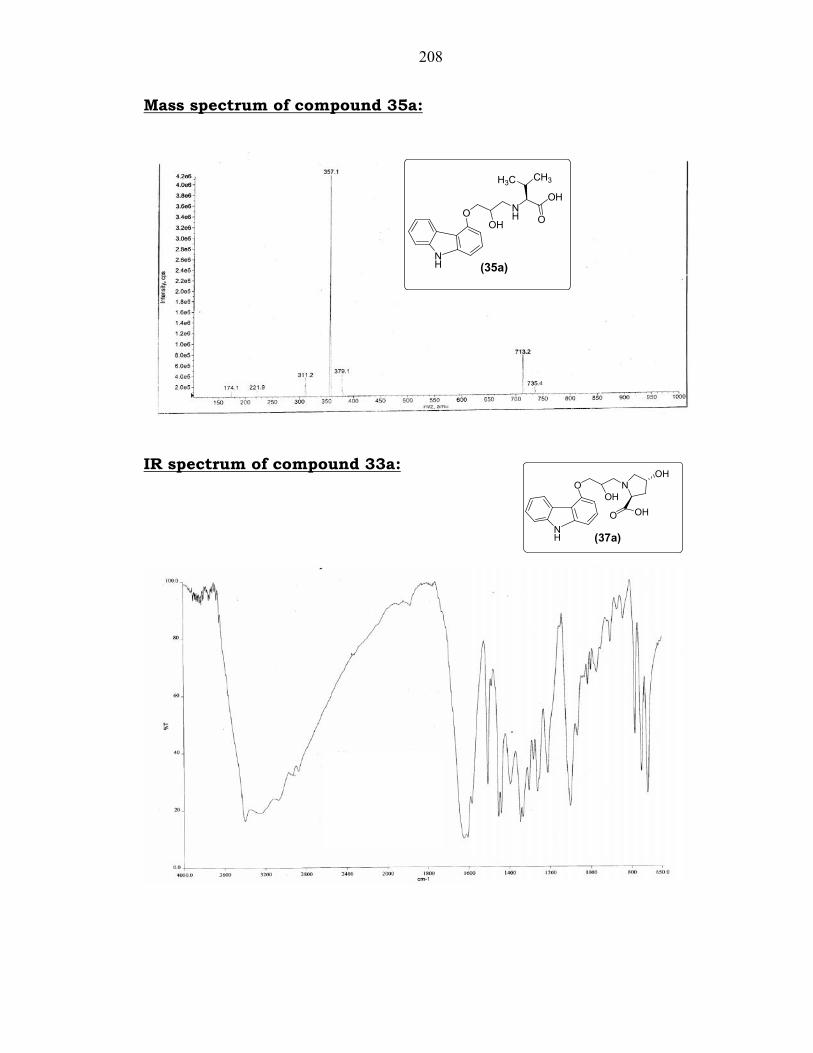
208
Mass spectrum of compound 35a:
IR spectrum of compound 33a:
NH
O
(35a)
OH
NH
H3C CH3
O
OH
NH
O
(37a)
OHN
OHO
OH
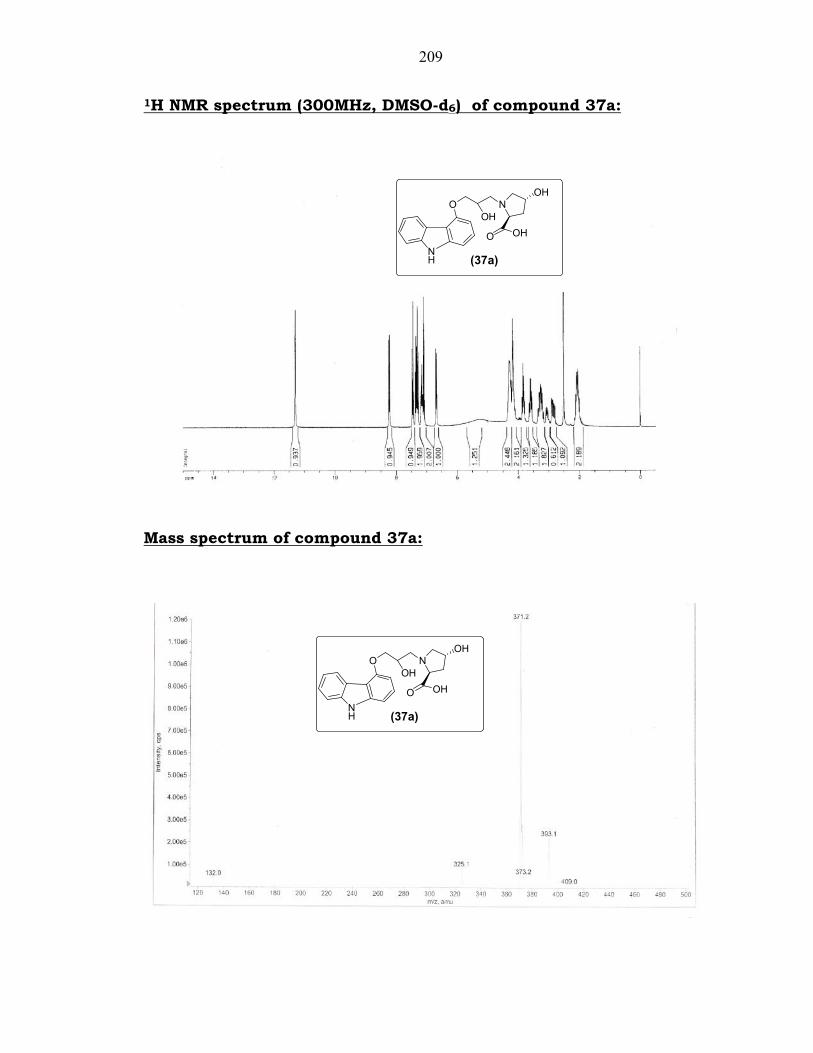
209
1H NMR spectrum (300MHz, DMSO-d6) of compound 37a:
Mass spectrum of compound 37a:
NH
O
(37a)
OHN
OHO
OH
NH
O
(37a)
OHN
OHO
OH

210
5.7. References
1. Joyce, J. N., Lexow, N., Kim, S. J., Artymyshyn, R., Senzon, S.,
Lawrence, D., Cassanova, M. F., Kleinman, J. R., Bird, E. D.,
Winokur, A. Synapse, 10, 1992, 228.
2. Kalaria, R. N., Andorn, A. C., Tabaton, M., Whitehouse, P. J.,
Karik, S. I., Unnerstall, J. R. J. Neurochem. 53, 1989, 1772.
3. Lemmer, B., Langer, L., Ohm, T., Bohl, J. Naunyn-Schmiedeberg’s
Arch. Pharmacal. 347, 1993, 214.
4. Corwin, J., Peselow, E., Feenan, K., Rotrosen, J., Fieve, R. Biol.
Psychiatry, 27, 1990, 813.
5. Cole, B. J., Koob, G. F. J. Pharmacal. Exp. Ther. 247, 1988, 902.
6. Laverdure, B., Boulenger, J. P. Encephale, 17, 1991, 481.
7. Berridge, C. W., Dunn, A. J. J. Neurosci. 9, 1989, 3513.
8. Otto-Erich, B. Pharm. Rev. 43, 1988, 204.
9. Lefkowitz, R. J., Caron, M. G., Stiles, G. L. New Eng. J. Med.
307, 1984, 1570.
10. Morris, T. H., Kanumann, A. J. Naunyn-Schmiedebergs Arch.
Pharmacol. 327, 1984, 176.
11. Berridge, M. S., Cassidy, E. H., Terris, A. H., Vesselle, J. M. Nucl.
Med. Biol. 19, 1992, 563.
12. Dubois, E. A., Van den Bos, J. C., Doornbos, T., Van Doremalen,
P. A. P. M., Somsen, G. A., Vekemans, J. A. J. M., Janssen, A. G.

211
M., Batink, H. D., Boer, G. J., Pfaffendorf, M., Royen, E. A. V.,
Van Zwieten, P. A. J. Med. Chem. 39, 1996, 3256.
13. Zheng, L., Berridge, M. S., Ernsberger, P. J. Med. Chem. 37,
1994, 3219.
14. Wiedemann, F., Kampe, W., Thiel, M., Spaner, G., Roesch, E.,
Dietmann, K. US 4503067, 1985.
15. Crowell, T. A., Evrard, D. A., Jones, C. D., Muehj, B. S., Rito, C.
J., Shuker, A. J., Thorpe, A. J., Thrasher, K. J. US 6686372,
2004.
16. Thomas, P., Erik, F., Peter, T. S. EP 1282601, 2004.
17. Zoltan, R., Jozsef, B., Gyula, S., Tamas, G., Donath, G. V.,
Norbert, N., Kalman, N., Judit, C., Tibor, S., Laszlo, B., Imre, D.,
Zoltan, G., Kotay, P. N., Peter, S. EP 0918055, 2003.