chapter - 5 studies in the synthesis of...
TRANSCRIPT

179
CHAPTER - 5
STUDIES IN THE SYNTHESIS
OF
DILTIAZEM HYDROCHLORIDE

180
5.1 - INTRODUCTION
Diltiazem hydrochloride (Cardizem®) has been shown to produce
increases in exercise tolerance, probably due to its ability to reduce
myocardial oxygen demand. This is accomplished via reduction in heart
rate and systemic blood pressure at submaximal and maximal exercise
workloads.
In animal models, diltiazem interferes with the slow inward
(depolarizing) current in excitable tissue. It causes excitation-contraction
uncoupling in various myocardial tissues without changes in the
configuration of the action potential. Diltiazem produces relaxation of
coronary vascular smooth muscle and dilation of both large and small
coronary arteries at drug levels which cause little or no negative inotropic
effect. The resultant increases in coronary blood flow (epicardial and
subendocardial) occur in ischemic and nonischemic models and are
accompanied by dose-dependent decreases in systemic blood pressure
and decreases in peripheral resistance. 70

181
Table-5.1 - PRODUCT PROFILE OF DILTIAZEM HYDROCHLORIDE 69
Generic Name Diltiazem hydrochloride
Brand Name Cardizem®
Active Ingredient Diltiazem
Innovator Tanabe Seiyaku Co., Ltd., Osaka, Japan, a
corporation of Japan
Marketed by Biovail Labs international
Chemical Name (+)-cis-3(acetyloxy)-5-[2-(dimethylamino)ethyl]-2,3-
dihydro-2-(4-methoxyphenyl)-1,5-Benzothiazepin-
4(5H)-one, monohydrochloride
Chemical
Formula
C22H27ClN2O4S
Molecular Weight 450.98
Chemical
Structure
CAS Registry No 33286-22-5
Physical White to off-white crystalline powder

182
description
Solubility It is soluble in water, MeOH, and chloroform
Melting range 213 – 2140C
Approved
indication (US)
Cardizem® is indicated for the management of
chronic stable angina and angina caused by
coronary artery spasm.
Dosage strengths 120 mg, 180 mg, 200 mg & 240 mg capsules
5.2 – LITERATURE RIVEW
Many synthetic approaches are reported in literature for the
preparation of Diltiazem or its intermediates including asymmetric
synthetic approaches. Some of those reported synthetic approaches are
discussed here under as the starting point towards the objective of the
present work.
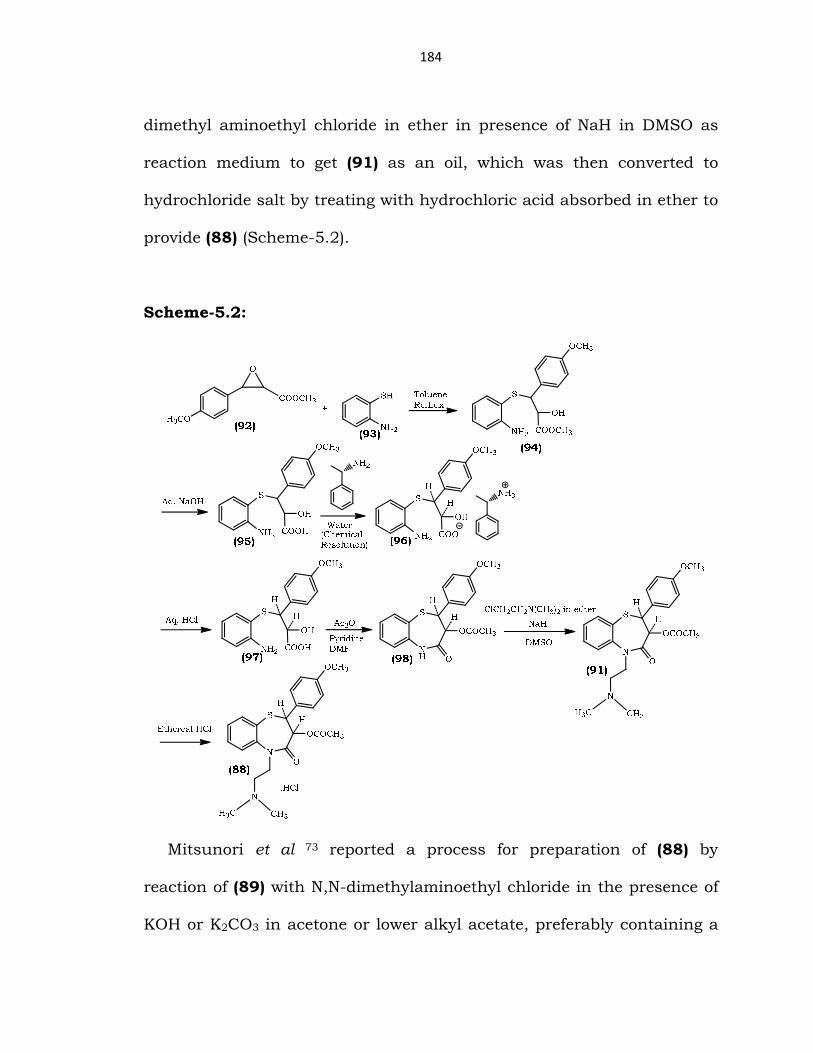
Hiroshi et al 71 reported a process for preparation of Diltiazem
hydrochloride (88) by reacting (+)-cis-3-hydroxy-2,3-dihydro2-(4-
methoxyphenyl)-1,5-benzothiazepin-4-(5H)-one (89) with N,N-
dimethylaminoethyl chloride, reaction of the resulting intermediate
(2S,3S)-5-(2-(dimethylamino)ethyl)-3-hydroxy-2-(4-methoxyphenyl)-2,3-
dihydrobenzo[b][1,4]thiazepin-4(5H)-one (90) with acetic anhydride
followed by conversion of the resulting Diltiazem (91) into hydrochloride
salt to afford (88) (Scheme-5.1).

183
Scheme-5.1:
In the above process, reaction in step-1 was carried out under critical
conditions using very strong bases, such as sodium hydride, metal
sodium or sodium amide in a solvent such as dimethylsulfoxide, dioxane,
toluene or xylene. Usage of sodium hydride as a base is in fact
dangerous, in view of its potential pyrophoric nature because of which
not preferable as it may create environment issues.
Susumu et al 72 reported a process for the preparation of (88) by
reaction of (92) with (93) in toluene at reflux temperature to get (94),
which up on hydrolysis with aqueous NaOH gave rise to an isolated
racemic intermediate (95). The said racemic acid intermediate was
subjected to chemical resolution using d-alpha-methyl benzyl amine by
separating diastereomeric salt (96) in water as solvent. The said
distereomeric salt was reacted with HCl in water as solvent by heating
whereby required (2S,3S) isomer (97) was isolated. The said acid was
reacted with Ac2O in presence of pyridine in DMF as reaction medium to
give rise to (98). The said intermediate was then reacted with N,N-
184
dimethyl aminoethyl chloride in ether in presence of NaH in DMSO as
reaction medium to get (91) as an oil, which was then converted to
hydrochloride salt by treating with hydrochloric acid absorbed in ether to
provide (88) (Scheme-5.2).
Scheme-5.2:
Mitsunori et al 73 reported a process for preparation of (88) by
reaction of (89) with N,N-dimethylaminoethyl chloride in the presence of
KOH or K2CO3 in acetone or lower alkyl acetate, preferably containing a

185
small amount of water. This method, even though overcoming some
drawbacks of the previous process, still it was not cost effective and eco-
friendly. In fact, the reaction takes place in heterogeneous system and
the recovery of solvent is very difficult, which is very important on
commercial scale to meet the cost. The product having the 3-hydroxy
group in free form or acetylated form was recovered and subsequently
transformed into the final hydrochloride salt. In a communication to EPO
(European patent office) during the prosecution of the patent application
Mitsunori et al, it was mentioned that N-alkylation of (89) does not take
place when the reaction solvent was toluene, though strong base such as
KOH and NaNH2 was used. In the same communication, it was further
stated that sodium carbonate in acetone also did not yield process
effective (Scheme-5.3).
Scheme-5.3:
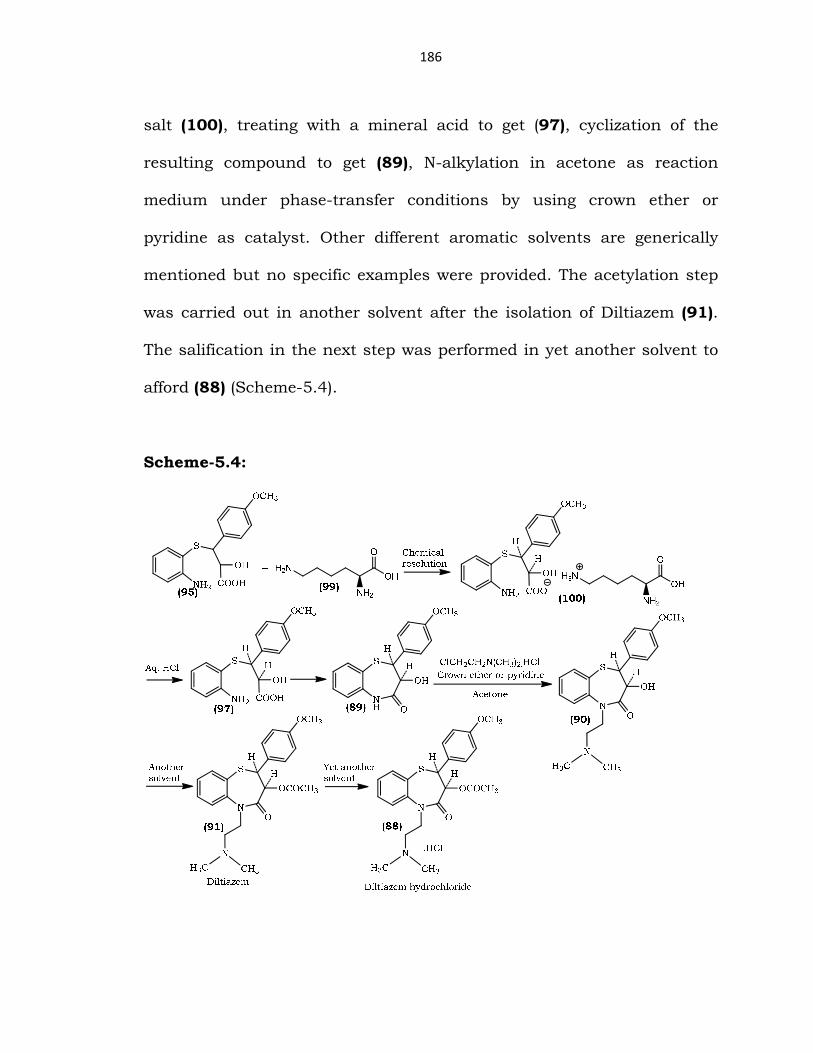
Nakamoto et al 74 reported the chemical resolution of racemic
propionic acid intermediate (95) with L-lysine (99) to get diastereomeric
186
salt (100), treating with a mineral acid to get (97), cyclization of the
resulting compound to get (89), N-alkylation in acetone as reaction
medium under phase-transfer conditions by using crown ether or
pyridine as catalyst. Other different aromatic solvents are generically
mentioned but no specific examples were provided. The acetylation step
was carried out in another solvent after the isolation of Diltiazem (91).
The salification in the next step was performed in yet another solvent to
afford (88) (Scheme-5.4).
Scheme-5.4:

187
Several other processes for preparation of intermediates of (88) were
reported in literature, which described synthesis of various achiral and
chiral intermediates and their conversion to (91) or its hydrochloride salt
(88).
5.2.1 - SUMMARY OF THE REPORTED SYNTHETIC APPROACHES
As discussed above and as per the synthetic processes described in
some more references75-91, although those reported synthetic procedures
seem to be practicable, they still need improvement in terms of cost and
commercial viability. For example, some of those procedures involve
costly and commercially less preferable reagents such as crown ethers,
pyridine, lipase (which requires a specialized technology to handle on
industrial scale), sodium hydride etc. Also some of those procedures
involve isolation of various intermediates that leads to lower yield
efficiency and lengthy time cycle for overall production on industrial
scale. Few more procedures involve synthesis of chiral intermediates
such as (2R,3S)-3-(4-methoxyphenyl)glycidic acid methyl ester for
example by enantioselective Mukaiyama aldol reaction involving the use
of very costly chiral reagents, which result in increase in cost of (88).

188
5.3 - PRESENT WORK
In view of the draw backs of the reported synthetic procedures, the
present work has an objective to develop an improved process for the
preparation of (88) by modifying the conditions of the reported synthetic
procedures, replacing costly reagents and solvents with simple, cost
effective reagents and recoverable solvents, which are available
commercially and are inexpensive on commercial scale in industry to
produce low cost (88) when compared to reported synthetic procedures
discussed above.
As can be seen from Newport Premium database by Thomson Reuters,
the worldwide consumption of (88) by June 2011 is 408,931.9 kgs.
Therefore (88) is a tonnage molecule wherein the cost of the active
pharmaceutical ingredient is the key driver of the business for
formulators. Therefore, one need not look into a very novel and unique
synthetic pathway. The objective of the present work is achieved just by
tweaking the reported processes thereby giving cost effective and
commercially viable process for the manufacture of (88).
This chapter aimed at synthetic studies towards the efficient and
alternative synthesis of (88) to overcome the limitations of the reported

189
approaches. The detailed study aimed towards synthesis of (88) is as
follows.
5.3.1 - RETRO SYNTHETIC PATHWAY FOR DILTIAZEM
HYDROCHLORIDE:
Scheme-5.5:
Above scheme depicts different retro synthetic pathways possible for
(88).

190
5.4 - RESULTS AND DISCUSSION
After reviewing the various possible retro synthetic pathways given
above in scheme-5.5, normally it is believed that enantioselective process
leads to the cost-effective product, since we do not lose the 50% of the
other isomer as an unwanted product as in the case of chemical
resolution. However, based on the discussion of reported synthetic
schemes herein before, it is observed that asymmetric synthesis of
required isomer of (92) so as to afford (88) will be expensive as its
preparation involves expensive chiral auxiliaries, which ultimately leads
to an expensive (88), though there is no loss of 50% of the other isomer,
which defeats the objective of the present work. Therefore, chemical
resolution methodology is still believed to be inexpensive if we operate it
by carefully studying the reported procedures and by employing the
appropriate conditions to simplify and improve the process to reduce the
manufacturing cost of (88). Towards this objective, the present work
provides the following detailed study on the synthesis of (88).
Initially, (88) was prepared by (i) reacting (101) with methyl
chloroformate under Darzen’s reaction conditions using sodium
methoxide as the base in MeOH as the solvent to get (92), (ii) reaction of
the resulting glycidate with (93) by refluxing in toluene solvent to get
(94), (iii) hydrolysis of resulting propionate ester with NaOH in water as
solvent to get an isolated racemic intermediate (95), (iv) chemical
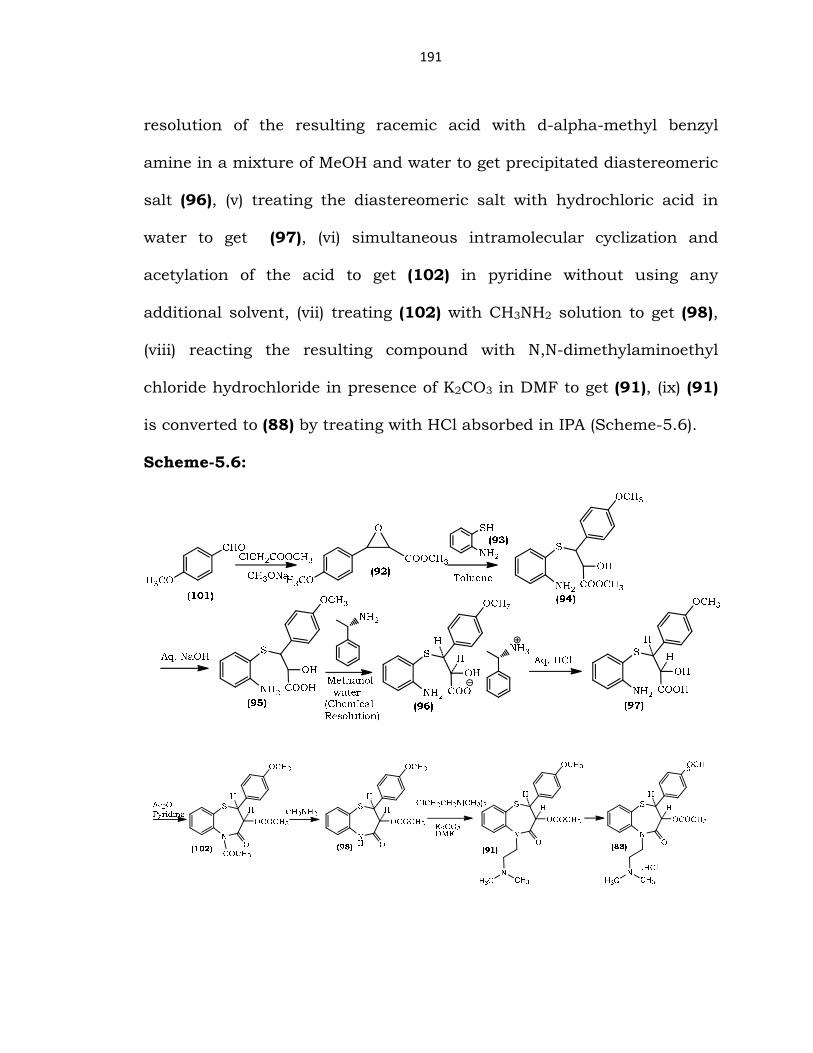
191
resolution of the resulting racemic acid with d-alpha-methyl benzyl
amine in a mixture of MeOH and water to get precipitated diastereomeric
salt (96), (v) treating the diastereomeric salt with hydrochloric acid in
water to get (97), (vi) simultaneous intramolecular cyclization and
acetylation of the acid to get (102) in pyridine without using any
additional solvent, (vii) treating (102) with CH3NH2 solution to get (98),
(viii) reacting the resulting compound with N,N-dimethylaminoethyl
chloride hydrochloride in presence of K2CO3 in DMF to get (91), (ix) (91)
is converted to (88) by treating with HCl absorbed in IPA (Scheme-5.6).
Scheme-5.6:

192
The draw backs in this process were the number stages, which results
in poor yield.
In order to provide an improved process, the synthetic scheme-5.7
was chosen for study, which was believed to be cost effective with
appropriate improvements by addressing the draw backs of the reported
procedures.
Scheme-5.7:
It was observed that the cost of the key intermediate cis-lactam (89)
looks to be the key cost driver in the synthetic scheme-5.7 selected for

193
study for the cost reduction of (88). Therefore, the objective of the study
was divided into two parts. First part dealt with cost reduction by
improvements in the synthesis of cis-lactam (89). The second part dealt
with cost reduction by improvements in the synthesis of (88) from (89).
5.4.1 - Studies in the synthesis of cis-lactam (89) - Results and
discussion:
In this study the starting point was the racemic (95), which was
prepared according to synthetic scheme-5.7 and subjected to chemical
resolution using d-alpha-methyl benzylamine to provide diastereomeric
salt (96). The resulting salt was directly converted to (89) without
isolating the intermediary D(+)-acid (97) by refluxing in toluene in
presence of slightly in excess of molar equivalents of conc. HCl and by
simultaneous azeotropic removal of water quantitatively.
Scheme-5.8:
In an effort towards further process improvements and cost reduction
of (89), the expensive d-alpha-methyl benzylamine was replaced by (99),
a very inexpensive reagent, for the chemical resolution of (95). (99), being
a naturally occurring product, is used in as food in poultry forms.

194
Therefore, it is a food product and is very safe and advantageous to use
in the process of the present work. Further, there is a huge cost
difference between d-alpha-phenylethylamine and L-lysine hydrochloride
(99). (99) as hydrochloride is inexpensive than its base. Therefore, (95)
was reacted with (99) in presence of NaOH in order to neutralize the
hydrochloride salt to free the amine group in (99) to participate in
resolution by salt formation with the acid group of (95). The resolution
step was carried out in a mixture of MeOH and water as solvent system.
The resulting (100) was directly converted to (89) without isolating the
intermediary (97) by refluxing in toluene in presence of slightly in excess
of molar equivalents of conc. HCl and by simultaneous quantitative
azeotropic removal of water (Scheme-5.9).
Scheme-5.9:

195
Though the process according to scheme-5.9 seems to be cost
efficient, still there is scope for improvement. In an endeavor to further
simplify the process, (94) was directly used in the step of resolution with
(99) without the need of isolating (97) and then subjecting it to chemical
resolution. (94) was first treated with molar equivalents of aqueous
NaOH. The product of the reaction would be sodium salt of acid and
MeOH. The reaction mixture containing these two products was directly
treated in-situ with (99) in a mixture of MeOH and water as solvent
system for resolution.
The advantage of this process modification was; (i) there was no need
of isolating (97), which results in saving of certain yield as there was no
isolation, and (ii) the sodium ions present in the resulting sodium salt of
acid within the reaction mixture after hydrolysis of (94) would be utilized
in neutralizing the hydrogen chloride associated with (99) so that there
was no need of using a base separately for neutralizing it.
Though this approach was just a combining of two stages by tweaking
the known processes, it has resulted in tremendous cost advantage and
made the process very simple and robust. This was a very novel
approach and not reported in literature before carrying out the present
work. Therefore, the starting point for this approach was (94) (Scheme-
5.10).

196
Scheme-5.10:
5.4.2 - Studies in the synthesis of Diltiazem hydrochloride from cis-
lactam (89) - Results and discussion:
(89) was N-alkylated with N,N-dimethylaminoethyl chloride in
presence of K2CO3 in acetone as solvent under reflux conditions to get
(89). The key observation in this reaction was that, the reaction did not
proceed at all in dry acetone, which was observed when laboratory grade
acetone was used. Whereas when commercial grade acetone was used,
the reaction was observed to some extent but still not to completion.
While investigating the reasons for this, the water content in both
laboratory and commercial grades of acetone were measured. Laboratory
grade acetone contained a water content of less than 0.1 percent weight
by volume whereas commercial grade acetone contained a water content
of about 0.5 percent weight by volume as measured by Karl Fischer
titration method. Then the quantity of water within the reaction medium
was optimized by deliberately adding varying quantities of water to the
system in the beginning of the reaction itself. As the quantity of water
was increased, the reaction was progressed towards completion. When

197
about 4% weight by volume of water was present in the quantity of
acetone used for the reaction, the reaction was proceeded to completion
comfortably. The said quantity of water in acetone came to around 3 to 4
molar equivalents with respect to the number of moles of (89) used.
Further improvement of the N-alkylation step lied in usage of toluene
instead of acetone. Surprisingly the reaction progressed comfortably even
in toluene as the reaction medium, which was contrary to the reported
procedure (Mitsunori et al). It was further advantageous that a solution of
(90) in toluene that was obtained after N-alkylation step could be directly
used in the next step of acetylation in situ without the isolation of N-
alkylated product. This would give a tremendous advantage for
simplification of the process on commercial scale.
The next step was the O-acetylation reaction to get (91). Ac2O was
used in many reported procedures. In this approach, first (91) was
obtained, which was often isolated as a solid by crystallization to avoid
excess Ac2O and the base viz. pyridine used for the O-acetylation
reaction. The isolated (91) needed to be converted to (88) in an additional
step. The first improvement of the present work involved the use of acetyl
chloride in presence of acetic acid for O-acetylation of (90) to directly get
(88) without the need of isolating the intermediary (91) base and its
further conversion to its hydrochloride salt thereby providing a simple
process without the need for isolation of (91). Though procedures were

198
reported for the O-acetylation using acetyl chloride, those procedures
involve the use of acetic acid or Ac2O as the solvent, which is not
preferable on commercial scale. The present work dealt this problem by
using stoichiometric quantity of acetyl chloride and acetic acid in toluene
as solvent for the reaction. (88) was directly obtained comfortably in
quantitative yield with good quality. The said process is schematically
represented.
Scheme-5.11:
ClCH2CH2N(CH3)2.HCl
K2CO3, TolueneWater, 700C N
S
O
OCH3
H
OH
H
NH3C CH3
N
S
O
OCH3
H
OCOCH3
H
NH3C CH3
.HCl
CH3COCl,Acetic acid,Toluene,450C
NH
S
O
OCH3
H
OH
H
(89) (90) (88)
5.4.3 – Results and discussion on related impurities of Diltiazem
hydrochloride (88):
The following compounds were identified as impurities in (88)
obtained in the present work.

199
Out of these impurities; (89), (94) & (90) are intermediates of (88) and
their characterization data is in agreement with the data give for those
compounds herein above. (98) was prepared as per the procedure given
in experimental section.
5.5 - EXPERIMENTAL SECTION
Preparation of 3-(4-Methoxyphenyl)-2,3-epoxymethylpropionate (92):
A stirred solution of 39.7 grams of (0.735 mol) of sodium methoxide
powder in 1000 ml of MeOH was cooled to -10 to -50C. 100 grams (0.735
mol) of (101) was added slowly over a period of 60 minutes. The mixture
was aged at the same temperature for 25 minutes. Then 87.1 grams
(0.803 mol) of methyl chloro acetate was added drop wise slowly in about
2 hours. After reaction completion (monitored by TLC; mobile phase -
ethyl acetate : hexanes – 1:5), pH of the reaction mixture was adjusted to
7 with very dilute hydrochloric acid very slowly at -10 to -50C. The
separated solid was filtered, washed with chilled water and dried under

200
reduced pressure at 250C for 30 minutes to afford 132 grams (86.2%) of
(92).
Preparation of methyl 3-((2-aminophenyl)thio)-2-hydroxy-3-(4-
methoxyphenyl)propanoate (94):
To a stirred solution of 100 grams (0.48 mol) of (92) in 1000 ml of
toluene was added 72.0 grams (0.576 mol) of (93) and heated reflux.
Reaction mixture was aged at refluxing for 5 hours. The reaction mixture
was slowly cooled to room temperature and aged for 3 hours. Separated
solid was filtered and dried at 600C to afford 134 grams (84%) of (94).
Characterization of (94):
IR spectrum of (94):
(cm-1) 1680 (C=O str, amide); 3369 (OH str); 3189 (N-H str).
Mass spectrum of (94):
m/z 334 (M++1).
1H-NMR spectrum of (94):
(δ ppm) 2.6 (s, 1H, OH), 3.7 (m, 3H, CH3 of ester), 3.9 (m, 3H, CH3 of
p-OMe Ph), 4.4 & 4.8 (m, 2H, CH), 6.2 (s, 2H, NH2), 6.4-7.3 (m, 8H, Ar-
H).
Preparation of L-Lysine salt of (2S,3S)-3-((2-aminophenyl)thio)-2-
hydroxy-3-(4-methoxyphenyl) propanoic acid (100):
To a stirred solution of 100 grams (0.299 mol) of (94) in 500 ml of
MeOH was added 11.9 grams (0.299 mol) of NaOH and stirred for 30

201
minutes. 65.3 grams (0.359 mol) of L-Lysine hydrochloride (99) dissolved
in 100 ml of water was added in about 30 minutes. Stirring was
continued at room temperature for about one hour. Separated solid was
filtered and washed with a 5:1 mixture of MeOH and water. The resulting
wet compound was dried at a temperature of 500C to yield 56 grams of
(100).
Preparation of cis-lactam (89):
Dean-Stark apparatus was arranged to a round bottom flask
containing 1000 ml of toluene. 100 grams (0.215 mol) of (100) was
charged and stirring was started. 50 ml of concentrated HCl was added
slowly. The mixture was heated to reflux. The reaction mixture was aged
under reflux condition by simultaneously collecting water azeotropically
in Dean-Stark apparatus. Reflux was continued until water collection
ceased. The reaction mixture was slowly cooled to room temperature. The
separated solid was filtered and washed with toluene. The resulting wet
compound was taken into 1000 ml of water and stirred at room
temperature for about 30 minutes. The solid was filtered and dried at
700C to yield 62 grams (Yield: 95%) of (89) (Purity by HPLC: 98.6 %).
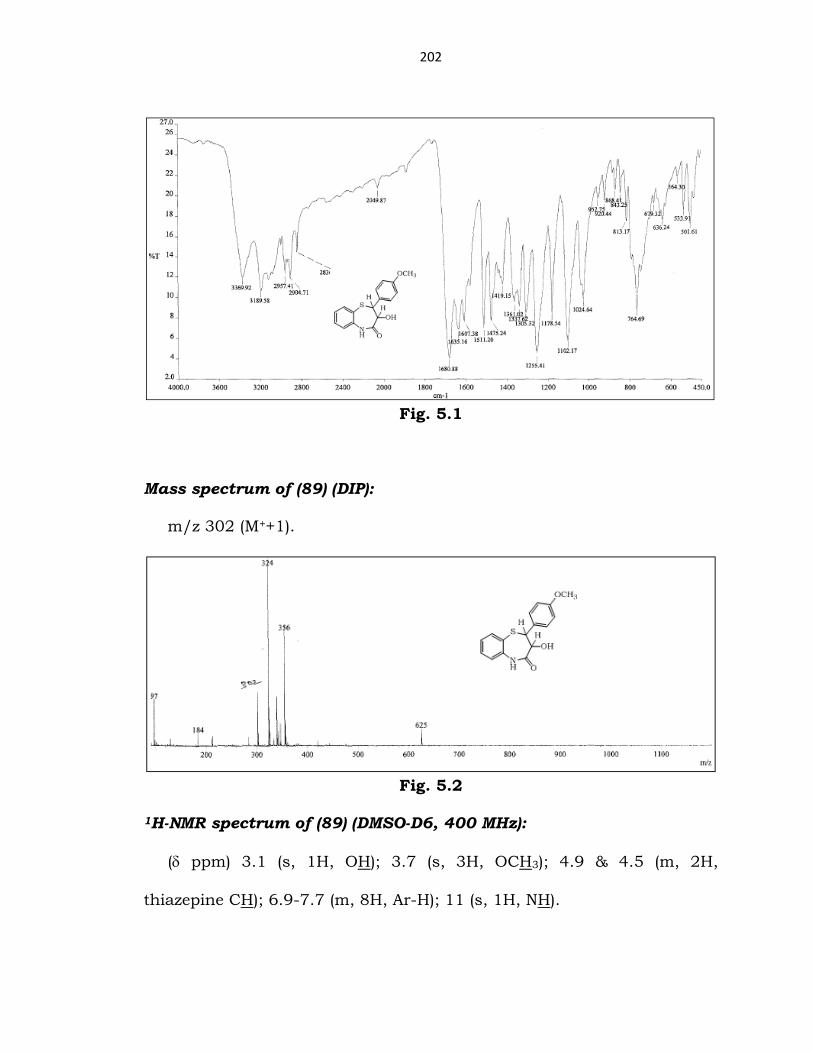
Characterization of cis-lactam (89):
IR spectrum of (89):
(cm-1) 3189 (N-H str); 3369 (OH str); 1680 (C=O str).
202
Fig. 5.1
Mass spectrum of (89) (DIP):
m/z 302 (M++1).
Fig. 5.2
1H-NMR spectrum of (89) (DMSO-D6, 400 MHz):
(δ ppm) 3.1 (s, 1H, OH); 3.7 (s, 3H, OCH3); 4.9 & 4.5 (m, 2H,
thiazepine CH); 6.9-7.7 (m, 8H, Ar-H); 11 (s, 1H, NH).

203
Fig. 5.3
13C-NMR spectrum of (89) (DMSO-D6, 400 MHz):
(δ ppm) 55 (S-C); 57 (OCH3); 69 (C-OH); 113-134 (10 remaining
carbons of phenyl rings); 141 (Ph-C-N of benzothiazepine); 159 (Ph-C-
OMe); 172 (C=O thiazepinone).
NH
S
O
OCH3
H
OHH
Fig. 5.4

204
DEPT spectrum of (89) (DMSO-D6 200 MHz):
Methyl and methyne groups as positive peaks and methylene groups
as negative peaks.
Fig. 5.5
Preparation of (2S,3S)-5-(2-(dimethylamino)ethyl)-3-hydroxy-2-(4-
methoxyphenyl)-2,3-dihydrobenzo[b][1,4]thiazepin-4(5H)-one (90):
To a stirred mixture of 50 grams (0.166 mol) of cis-lactam (89) in 500
ml of toluene was added 22 grams (0.166 mol) of K2CO3. 12 ml of water
was added and the reaction mixture was heated to 700C. The reaction
mixture was aged at that temperature for 6 hours. Reaction completion
was monitored by TLC (Mobile phase: Chloroform : MeOH – 4:1). The
reaction mixture was cooled to room temperature and filtered. Filtrate
was washed with water. The resulting solution of (90) in toluene was

205
heated to reflux and water was removed by azeotropic distillation.
Solution was cooled to room temperature.
Characterization of (90):
IR spectrum of (90):
(cm-1) 1660 (C=O str); 3469 (OH str); 3189 (N-H str) of (2) is absent.
Fig. 5.6
Mass spectrum of (90) (DIP):
m/z 373 (M++1).
Fig. 5.7
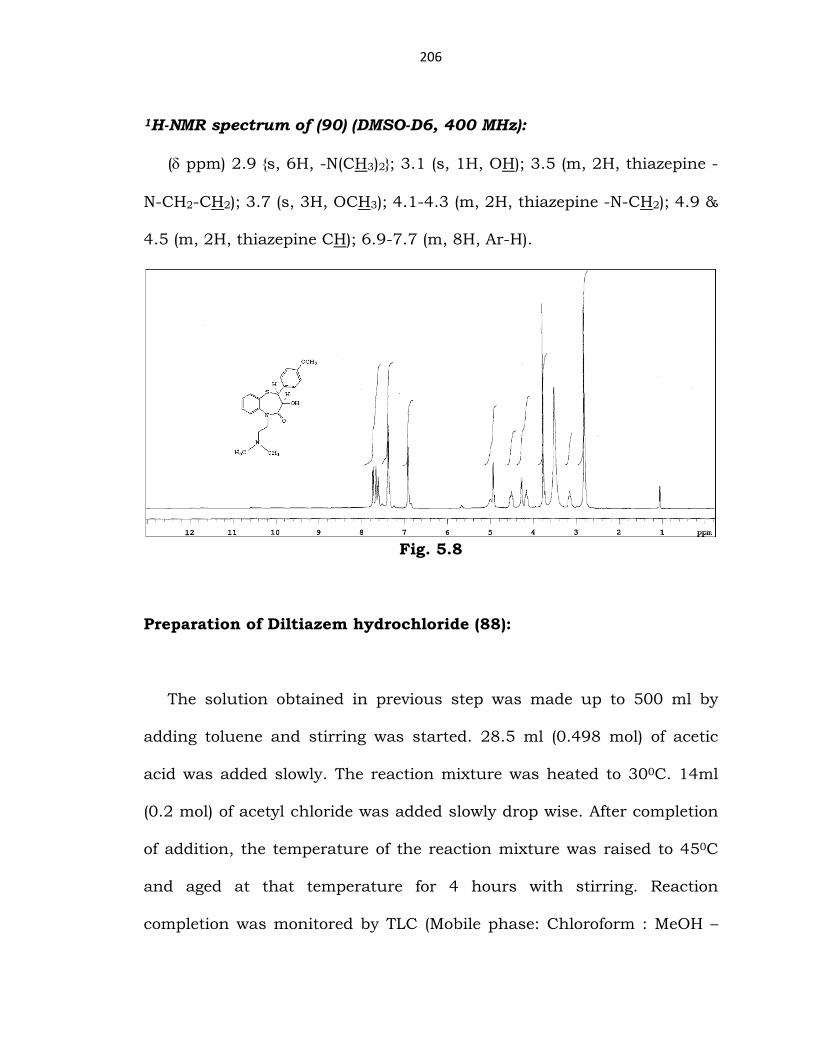
206
1H-NMR spectrum of (90) (DMSO-D6, 400 MHz):
(δ ppm) 2.9 {s, 6H, -N(CH3)2}; 3.1 (s, 1H, OH); 3.5 (m, 2H, thiazepine -
N-CH2-CH2); 3.7 (s, 3H, OCH3); 4.1-4.3 (m, 2H, thiazepine -N-CH2); 4.9 &
4.5 (m, 2H, thiazepine CH); 6.9-7.7 (m, 8H, Ar-H).
Fig. 5.8
Preparation of Diltiazem hydrochloride (88):
The solution obtained in previous step was made up to 500 ml by
adding toluene and stirring was started. 28.5 ml (0.498 mol) of acetic
acid was added slowly. The reaction mixture was heated to 300C. 14ml
(0.2 mol) of acetyl chloride was added slowly drop wise. After completion
of addition, the temperature of the reaction mixture was raised to 450C
and aged at that temperature for 4 hours with stirring. Reaction
completion was monitored by TLC (Mobile phase: Chloroform : MeOH –

207
4:1). Reaction mixture was cooled to 200C and stirred at that
temperature for one hour. Separated solid was filtered and dried at 600C
to yield 59 grams (Yield: 80%) of Diltiazem hydrochloride (1) {Purity by
HPLC: 99 %; SR: + 1140 (c=10mg/ml water)}.
Characterization of Diltiazem hydrochloride (88):
IR spectrum of (88):
(cm-1) 3056 (Ar C-H str); 2966 (aliphatic C-H); 2389 (+N-H str); 1743
& 1680 (C=O str); 1475 (C-N str); 1255 & 1026 {C-O-C (aryl alkyl ether)}.
Fig. 5.9
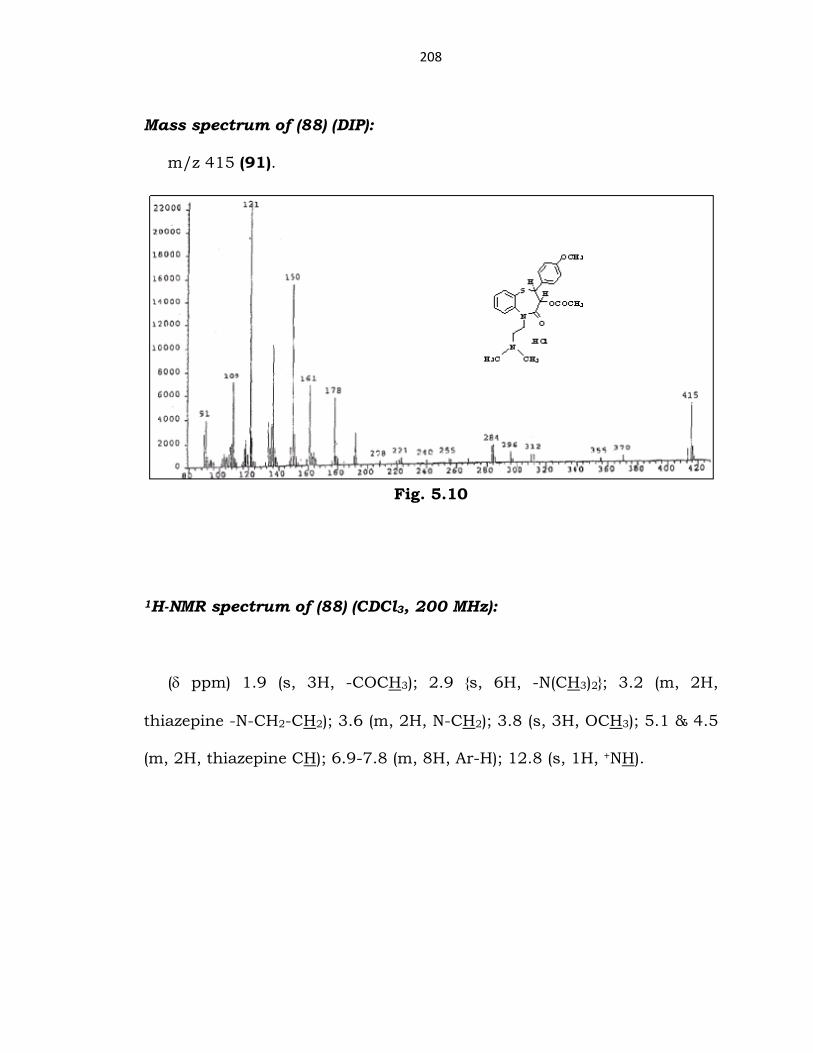
208
Mass spectrum of (88) (DIP):
m/z 415 (91).
Fig. 5.10
1H-NMR spectrum of (88) (CDCl3, 200 MHz):
(δ ppm) 1.9 (s, 3H, -COCH3); 2.9 {s, 6H, -N(CH3)2}; 3.2 (m, 2H,
thiazepine -N-CH2-CH2); 3.6 (m, 2H, N-CH2); 3.8 (s, 3H, OCH3); 5.1 & 4.5
(m, 2H, thiazepine CH); 6.9-7.8 (m, 8H, Ar-H); 12.8 (s, 1H, +NH).

209
Fig. 5.11
13C-NMR spectrum of (88) (CDCl3, 50 MHz):
(δ ppm) 19.8 (COCH3); 42.5 (-N(CH3)2); 44.3 (thiazepine-N-CH2-CH2);
53.6 (thiazepine-N-CH2-CH2); 53.3 (thiazepine-S-C); 54.7 (OCH3); 70.5
(benzothiazepine-C-COCH3); 113-135 (10 remaining carbons of phenyl
rings); 143 (phenylic-C-N of benzothiazepine ring); 159 (Ph-C-OMe); 167
(OCOCH3); 169 (C=O thiazepinone).
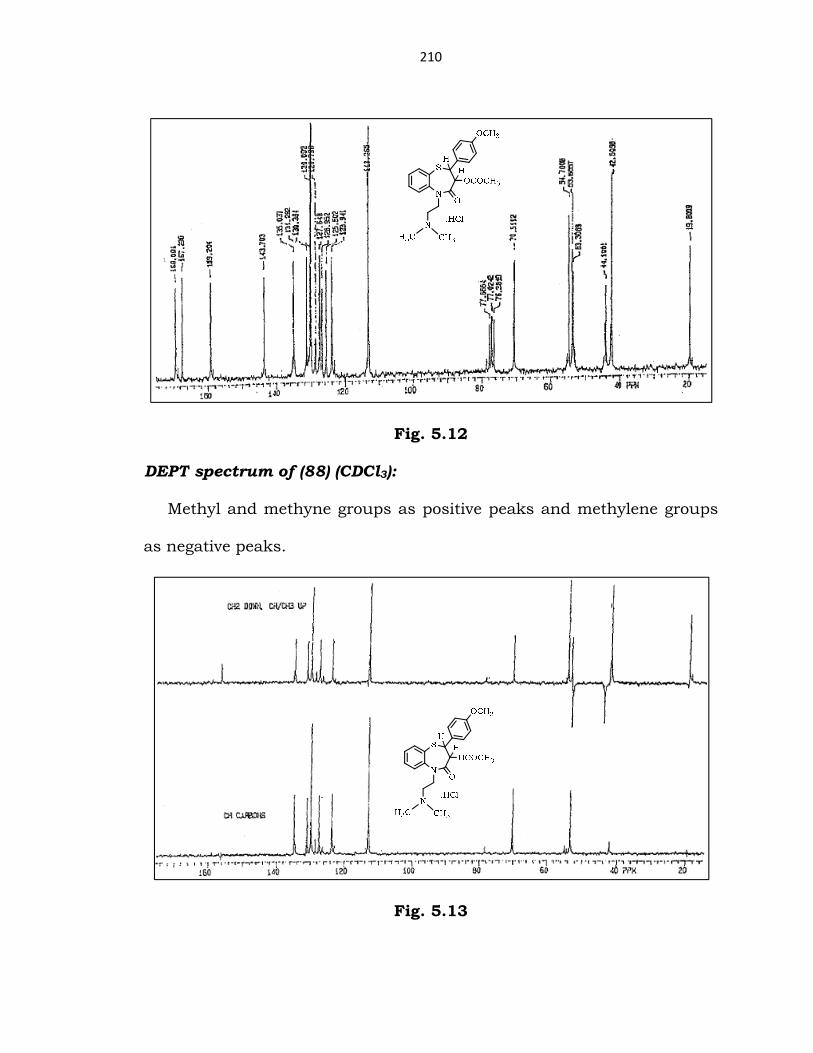
210
Fig. 5.12
DEPT spectrum of (88) (CDCl3):
Methyl and methyne groups as positive peaks and methylene groups
as negative peaks.
Fig. 5.13

211
Preparation of (2S,3S)-2-(4-methoxyphenyl)-4-oxo-2,3,4,5-
tetrahydrobenzo[b][1,4]thiazepin-3-yl acetate (98): (98) is prepared according to the procedure reported in Susumu et al.
Step-1:
To a stirred mixture of 5 g (0.01 mol) of (100) in 50 ml water was
added dil. HCl {prepared by mixing 3 ml (0.02 mol) of conc. HCl in 10 ml
of water} slowly. The mixture was stirred for 1 hour at room temperature.
Filtered and solid was washed with water to afford 4.3 g of (97).
Step-2:
To a stirred mixture of 4.0 g (0.0125 mol) of (97) in 100 ml of toluene
was added 6.4 g (0.0625 mol) of acetic anhydride and heated to reflux.
The mixture was aged at the same temperature for 3 hours by azetropic
removal of acetic acid along with toluene. After reaction completion, the
reaction mixture was cooled to room temperature and the separated solid
was filtered, washed with toluene and dried at 700C to afford 4.5 g of
(102). Melting point: 156-1580C.
Step-3:
To a stirred solution of 4.0 g (0.0103 mol) of (102) in 50 ml of
DCM was added (0.015 mol) of diethylamine and aged at room
temperature for 2 hours. After reaction completion, the solvent was
distilled off under reduced pressure at 450C. The resulting compound

212
was triturated with 10 ml of IPA to afford 2.0 g of (98). Melting point:-
196-1980C.
Characterization of (98):
IR spectrum of (98):
(cm-1) 1680 (C=O str, amide); 3369 (OH str); 3189 (N-H str).
Mass spectrum of (98) (ESI):
m/z 345 (M+2).
1H-NMR spectrum of (98):
(δ ppm) 2.1 (s, 3H, CH3); 3.7 (s, 3H, OCH3); 4.8 & 4.4 (m, 2H,
thiazepine ring hydrogens); 6.8-7.6 (m, 8H, Ar-H); 11 (s, 1H, NH).
5.6 - CONCLUSION
The objective of the present work is achieved by providing cost
effective, eco-friendly process, which is well suited for commercial scale
up. The Diltiazem hydrochloride (88) obtained in the present novel
process has >99.0% purity as determined by HPLC (as required by ICH
lmits) and resulted in a crystalline form as characterized by X-ray powder
diffraction. The Diltiazem hydrochloride obtained in the present process
is free flowing and non-solvated solid; hence it is well suited for
pharmaceutical applications. The process of the present work is cost
effective, eco-friendly and amenable for scale up.
The resultant improved easily scaleable and cost effective process for
preparation of Diltiazem hydrochloride (88) of the present work is under
evaluation for patent filing at Dr.Reddy’s Laboratories Ltd, Hyderabad.