chapter 3: amino acids & polypeptidesduahn/teaching/neobiomaterials and... · define objectives...
TRANSCRIPT

Protein Purification

Protein Purification
•
In an organism, any one protein is present as a very small percentage of the total biomolecules.
•
In order to characterize a protein fully, it is necessary to purify it.
•
Many possible strategies to purify a protein.
•
In general, the more you know about the protein's properties, the better strategy you could design to purify it.
•
Purification strategy will be of several stages, each one taking advantage of different characteristics of the protein.

Protein Purification Principles•
Define objectives–
Purity, activity and quantity for the final product
•
Define properties of target protein and critical impurities–
to simplify technique selection and optimisation
•
Develop analytical assays–
for fast detection of protein activity/recovery and to work efficiently
•
Remove damaging contaminants early–
for example, proteases

Protein Purification Principles•
Use a different technique at each step–
to take advantage of sample characteristics which can be used for separation (size, charge, hydrophobicity, ligand specificity)
•
Minimize sample handling at every stage–
to avoid lengthy procedures which risk losing activity/reducing recovery
•
Minimize use of additives–
additives may need to be removed in an extra purification step or may interfere with activity assays
•
Minimize number of steps -
KEEP IT SIMPLE!–
extra steps reduce yield and increase time, combine steps logically

5
Number of Steps and the Yield at Each Step
Number of steps
Yield (%)
95% / step
90% / step
85% / step
80% / step75% / step
0
20
40
60
80
100
1 2 3 4 5 6 7 8
20% overallyield!

Protein Purification: Practical Aspects
•
You have to choose your source of material carefully–
unicellular organism
–
organ of a metazoan (or plant)? •
Where is the enzyme located?–
In cytosol?
–
Within membrane-bound organelle?–
Secreted?
•
You can use differential centrifugation to do a (crude) separation of various cellular compartments to get a starting point.
•
If the protein is in a specific organelle, density gradient centrifugation is usually used to purify the organelle first.

Protein Purification: Practical Aspects
•
All cells contain proteases –
enzymes that catalyze hydrolysis of peptide bonds
•
Upon breaking cells, these are released into the extract, where they can degrade the protein you want to purify
•
In order to inhibit proteolysis and denaturation, protein purification is usually carried out –
In the cold (on ice or in a cold room)
–
In the presence of protease inhibitors (small molecules that inhibit specific proteases)

Starting materials
•
Natural source or artificial expression system–
Bacteria, yeast, plants, transgenic animals
•
Abundance•
Lysis and clarification procedures–
Native or denaturing conditions
•
Selective precipitation–
Streptomycin Sulfate, polyethyleneimine (PEI) and
cetyltrimethylammonium bromide (CTAB) for RNA/DNA
–
Ammonium Sulfate for Proteins

9
Sample Preparation
General considerations:•
Select extraction procedure according to source and location of protein
•
Use gentle procedures to minimize acidification and release of proteolytic enzymes
•
Work quickly at sub-ambient temperatures•
Use buffer to maintain pH, ionic strength
Goal: To stabilize sample
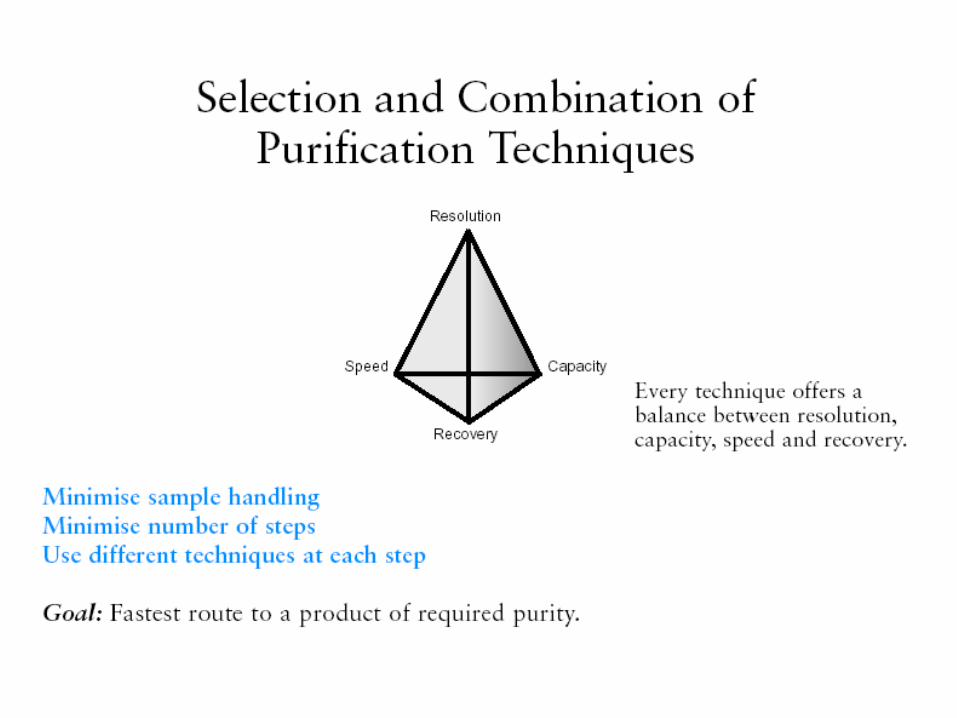

The basic techniques
•
Concentration (size)
–
precipitation–
ultrafiltration
–
dialysis–
centrifugation
•
Chromatography (size/charge/chemistry)
–
ion exchange–
size exclusion
–
affinity
•
Electrophoresis (size/charge)
–
"native"–
denaturing
–
isoelectric focusing–
2-dimensional
•
Immunological(size/charge/chemistry)
–
chromatography–
in situ imaging
–
immunoblotting
12
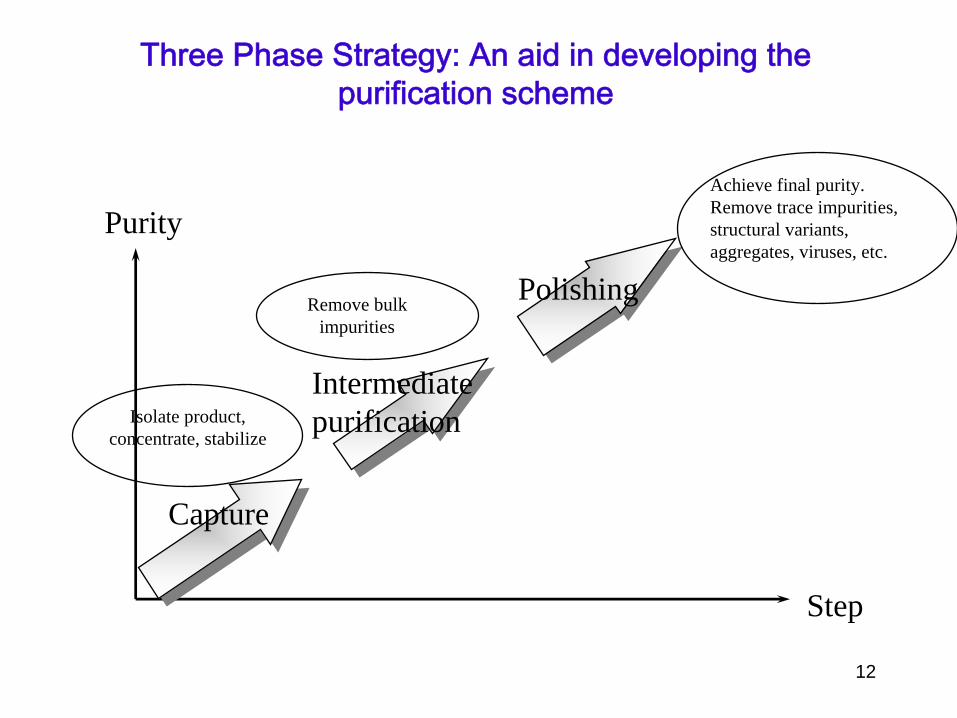
Purity
Step
Capture
Intermediatepurification
Polishing
Isolate product,concentrate, stabilize
Remove bulkimpurities
Achieve final purity.Remove trace impurities, structural variants,aggregates, viruses, etc.
Three Phase Strategy: An aid in developing the purification scheme

•
Ammonium sulfate precipitation
•
Sedimentation (rare)
•
Detergent extraction
•
Heat treatment (especially for recombinant thermophile proteins expressed in E. coli)
Non-chromatographic
protein purification techniques
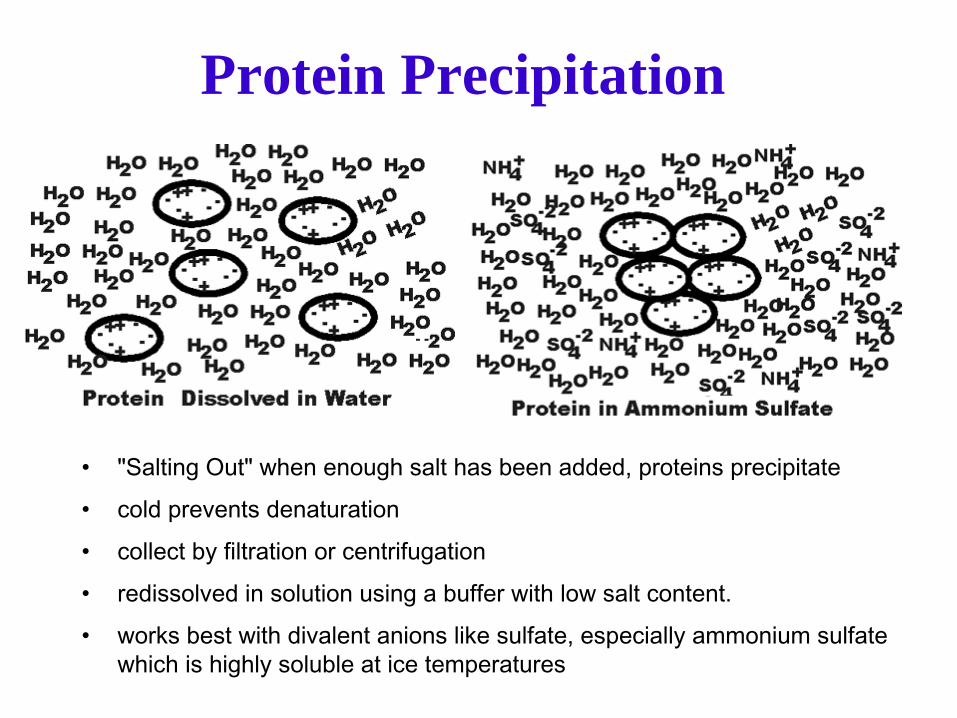
Protein Precipitation
•
"Salting Out" when enough salt has been added, proteins precipitate •
cold prevents denaturation
•
collect by filtration or centrifugation•
redissolved in solution using a buffer with low salt content.
•
works best with divalent anions like sulfate, especially ammonium sulfate which is highly soluble at ice temperatures

Salting out methods–
Solubility is sensitive to ionic strength.
–
Initial “salting in”
and then “salting out”–
At high salt, solvent tied up with interacting with salts so that it is insufficient to solubilize proteins.
–
1st go to maximal salt that the target protein is soluble, centrifuge and discard pellet.
–
Now add just enough salt to bring down the target protein
–
Collect pellet–
Redissolve the pellet in low-salt buffer

Ammonium sulfate precipitationExample:•
Your protein will remain soluble at 30% (w/v) ammonium sulfate, but precipitates at 40% ammonium sulfate.
•
Slowly add the salt to your protein extract until it reaches a concentration of 30%, allow precipitation to occur, and then centrifuge the solution to remove the precipitate.
•
Take the supernatant, and add ammonium sulfate until it reaches a concentration of 40%
•
Allow precipitation to occur, and then centrifuge the solution to remove the precipitate.
•
Dissolve the precipitate in a buffer and dialyze it to remove the salt.

Solubilities of several proteins in ammonium sulfate solutions

Other solubility techniques•
Organic solvents–
Same principle as salting out; taking advantage of different solubilities. Avoid totally denaturing proteins
•
pH–
Proteins have many ionizable groups with range of pKs.
–
When net charge of protein is zero, this is the isoelectric point or pI.
–
Proteins are typically least soluble at their pI due to minimizing charge-charge interactions
•
Crystallization–
The solubility methods where proteins are ppt can be used to grow crystals of proteins. This is only done when the protein is relatively pure.

Solubility of lactoglobin as a function of pH at several NaCl concentrations

Solubility of hemoglobin at its pI as a function of ionic strength and ion type

Buffer Exchanges
•
Almost all purification steps will use a buffer with specific pH and/or ionic strength
•
The buffer used impacts the protein's biophysical characteristics
•
Why exchange?–
If you have just precipitated a protein with ammonium sulfate, you now have that protein in a high salt environment.
How can you remove salt?

Dialysis
•
The protein is put in a bag of cellulose membranes having small pores of controlled size.
•
Proteins bigger than the pores are retained, while smaller molecules may diffuse out.
•
As the volume of the buffer surrounding the bag is many times (100-1000x) the volume within the bag, the smaller molecules can be effectively removed after several changes of the outer buffer.

Dialysis

Capture
•
Quickly remove most damaging contaminants•
Concentrate, adsorption methods–
Ion Exchange most general
–
Affinity chromatography can combine capture, intermediate and polishing steps
–
This step should remove most unwanted contaminants

Intermediate purification
•
Use a different technique•
Affinity chromatography, Hydrophobic interaction chromatography
•
Starting conditions are specific for each technique–
Buffer must be compatible with adsorption
–
Can change buffer by dialysis or desalting by GFC•
Adsorption techniques result in small volume concentrated sample

Polishing
•
Final removal of trace contaminants•
Often size exclusion chromatography–
Buffer exchange is a part of the process
–
Sample volume always increases need to start with a concentrated sample
•
Sample can be concentrated by–
Precipitation (selective or nonselective)
–
Ultrafiltration (dialysis under pressure)

Assays, Quantitation and Documentation
•
Assay enzyme activity at every step–
Contaminants at early stages can mask or inhibit activity
–
Inactivation can occur at high temperatures, because of proteolysis, oxidation, aggregation, etc.
•
Assay total protein •
Run an SDS gel to visualize specific contaminants
•
Specific activity is defined as units of enzymatic activity per unit of total protein -
•
Yield can be defined in terms of total protein mass, and total enzyme units
•
Goal is a high yield and high specific activity.

Detection
•
Spectroscopy–
A280 e 1%
280
= 14.5 g-1Lcm-1
•
Protein Assay–
Bradford (coomassie)
–
Biuret (copper)–
Lowry (modified biuret -
phosphomolybdotungstate mixed
acid reduced by Cu2+ and F,Y,W to form heteropolymolybdenum blue A575
•
Enzyme Assay
A550

Absorption spectra of Trp & Tyr
Beer’s law: A = εcl. Used to estimate protein concentration

Biuret Reaction
•
Primarily measure peptide bonds•
Cupric acid react with the peptide groups of proteins and form a cupric ion complex
•
Generate purplish-violet color with Amax
at 540 nm
•
Lowry method•
Low sensitivity

Ninhydrin Reaction•
The most widely used reagents for amino acids, peptides, and proteins
•
Reacts with amino groups•
The major step is oxidative deamination of an amino acid to CO2, NH3, and an aldehyde containing one less carbon atom than the original amino acid
•
Ninhydrin is simultaneously reduced to ninhydrindantin•
Ninhydrindantin reacts with NH3 and produce purple color (Amax
570 nm)
•
Destroy amino acid or peptide and the material detected cannot be used for further characterization

Assays
•
Enzymatic assays–
PNPP is hydrolyzed to PNP and Pi
–
p-Nitrophenyl Phosphate (PNPP) is a non-proteinaceous, non-specific substrate used to assay protein, alkaline and acid phosphatases
–
Fixed time assay•
Mix enzyme and substrate, react for a fixed time, s
•
top the reaction with a strong base, •
read the concentration of PNP at pH>10
–
Continuous assay•
Monitor PNP production directly in the spec at ph 8
•
SDS page for the distribution of proteins by size.

Purification tableStep Protein
(mg)Activity
(U)SpecificActivity(U/mg)
Yield Purificationfactor
1 1200 800 0.67 – –2 600 600 1 75% 1.53 200 400 2 67% 24 30 300 10 75% 5
Activity: definition of 1 unit (U) will vary, depending upon enzymeSpecific activity: measure of enzyme’s purity = activity/proteinYield
of a step = Units retained after that step/Units input
(Measure of how well enzyme activity is preserved by the step.)Purification factor
= specific activity after the step/before
(Measure of how effective the step is.)

Purification table
•
Yield and purification factor can be expressed –
for the individual step or
–
as cumulative yield or purification factor, taking into account all steps up to to that point
•
You need to be careful to distinguish between them.
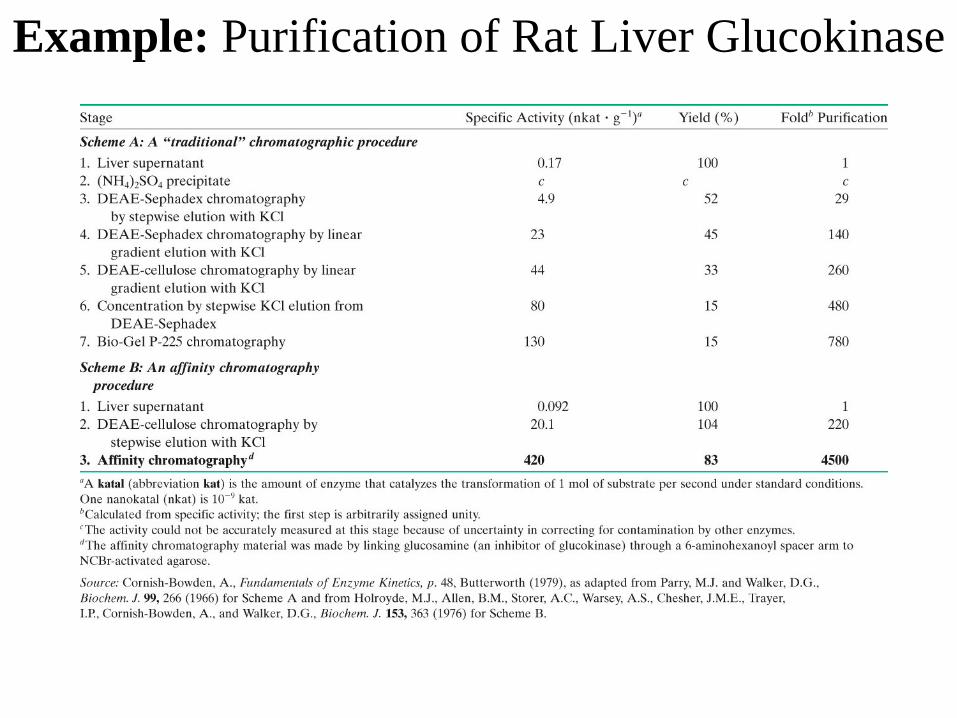
Example: Purification of Rat Liver Glucokinase

Criteria for purity
When is protein pure or pure enough?•
Homogeneity–
protein complexes?
•
Constant specific activity •
Practical: further attempts at purification are futile since the only material left in the fraction is the material that actually is responsible for the activity being assayed.

Characterization of Purified Proteins
-
SDS PAGE (both reducing and non-reducing)-
Bioassay (if you have one)
-
Total protein determination-
UV spectrophotometry
-
CD spectrometry-
Amino acid analysis
-
N-terminal (& C-terminal?) sequencing- HPLC?-
Metal analysis
-
Mass spectrometry-
NMR spectrometry & X-ray crystallography*

38
A. Differential Precipitation
•
Precipitation:
the process of formation of a solid that was previously held in solution. The solid is separated from the solution.
•
When NH4 SO4 or polyethylene glycol are added to a protein solution
•
A precipitate forms and it can be separated from the solution after centrifugation.
•
If the concentrations of NH4 SO4 or polyethylene glycol are increased, more precipitate forms.


Density Gradients
•
An equilibrium sedimentation experiment can be set up with linear gradients of sucrose or glycerol.
•
The protein is loaded on top, and centrifugation is started.
•
The advantage of this technique is that the gradients tend to be rather stable and allow one to remove the protein of interest from them.
•
The disadvantage is that they are not so accurate for determination of S.

Zonal ultracentrifugation

Zonal
Centrifugation•
Zonal centrifugation: Mixture to be separated is layered on top of a gradient (e.g. sucrose or ficoll) increasing concentration down the tube -
can be continuous or discontinuous (layers)
-
provides gravitational stability as different species move down
tube at different rates forming separate bands. –
Species are separated by differences in SEDIMENTATION COEFFICIENT
(S) = Rate
of movement down tube/Centrifugal
force–
S is increased for particle of LARGER MASS
(because sedimenting force a M(1-vr)–
S is also increased for MORE COMPACT STRUCTURES of equal particle mass (frictional coefficient is less)

Zonal ultracentrifugation

Isopycnic
Centrifugation•
Isopycnic (equal density) centrifugation: Molecules separated on
EQUILIBRIUM POSITION, NOT by RATES of sedimentation.
•
Each molecule floats or sinks to position where density equals density of solution (e.g. CsCl gradient for nucleic acid separation).

45
Gel Filtration Chromatography
•
Gel Permeation Chromatography.•
Separates protein molecules according to their molecular size.
•
The solution is inserted to the top of a specialized column.•
This column consists of specialized porous beads.
•
Small molecules of protein enter the beads while large molecules can’t
and stay in the space between the beads.
•
Therefore, large molecules flow more rapidly through the column and emerge first from the bottom of the column.
•
Advantage: larger quantities of proteins can be separated.•
Disadvantage: Lower resolution.

Sephadex
In Sepadex, the dextran chains are covalently linked to a highly cross-linked agarose matrix. The figure shows a
schematic of a section through a Sepadex particle
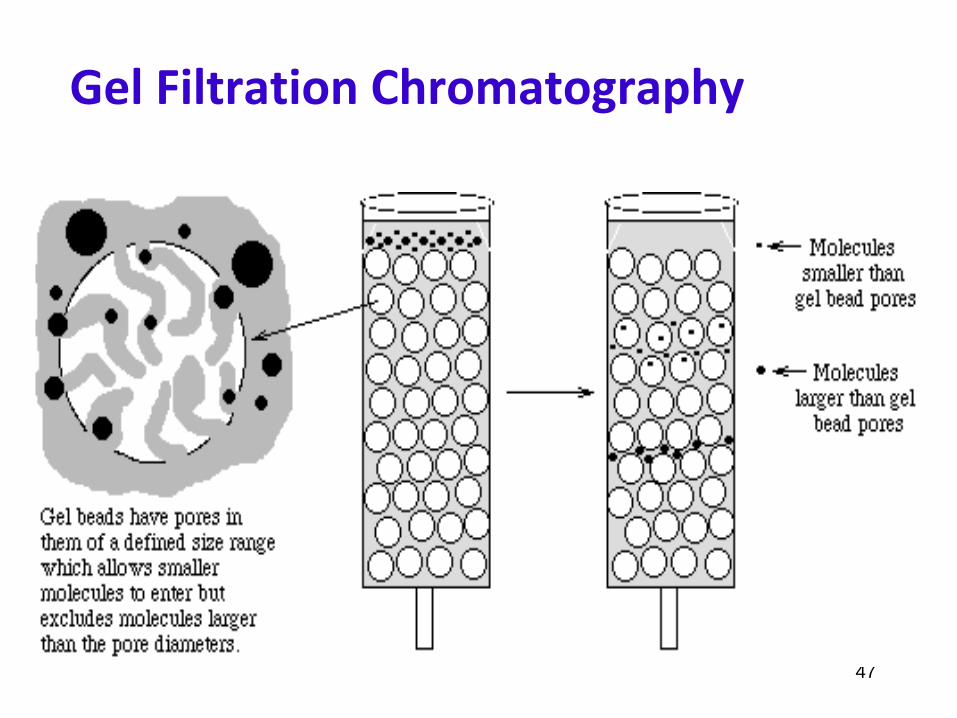
47
Gel Filtration Chromatography
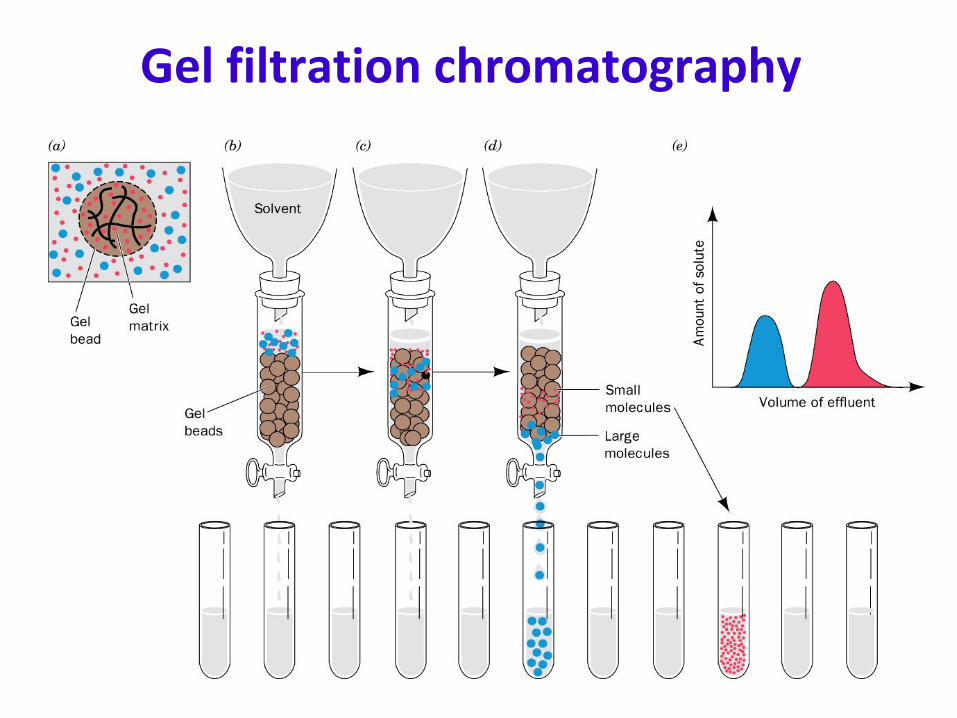
Gel filtration chromatography

Gel filtration chromatography•
Vbed
= Vbeads
+ Vvoid
–
The void volume
is the volume surrounding the beads.
–
The bed volume
is the total volume of the column.–
Vvoid
is typically about Vbed
/3•
A protein can be characterized by its elution volume (Velution
), which is the volume of solvent required to elute it from the column. The relative elution volume (= Velution
/ Vvoid
) is a quantity independent of the particular column used.
•
By standardizing the column with proteins of known size, one can use this technique to estimate molecular weight (assuming that the shape is close to that of the standards –
more or less spherical).

Principles of gel chromatography (con’d)

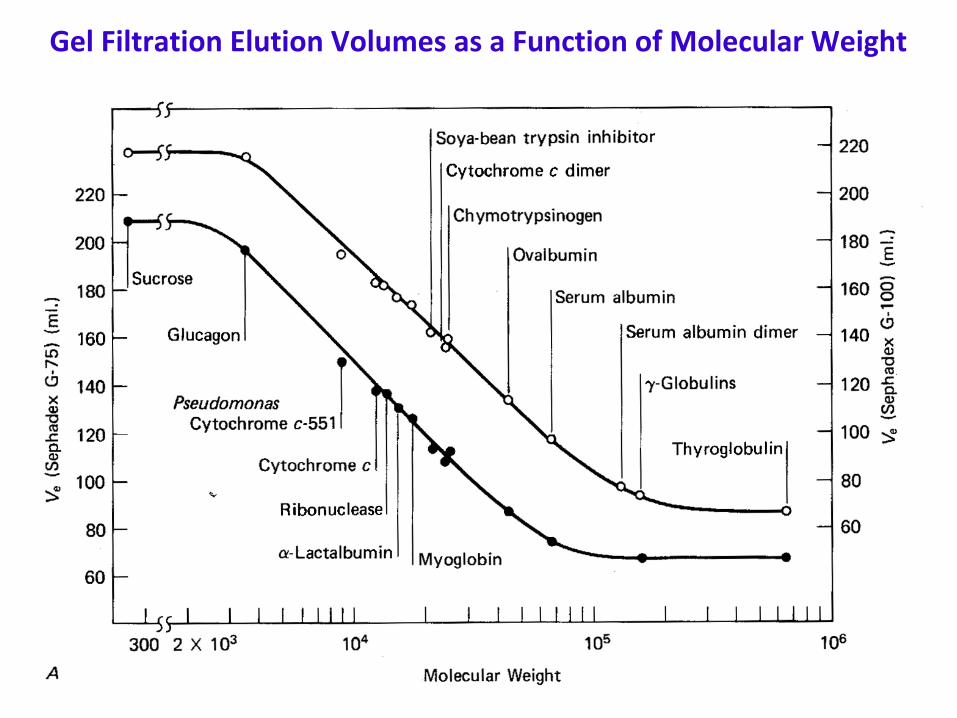
Gel Filtration Elution Volumes as a Function of Molecular Weight

Commonly Used Gel Filtration Materials

Ion exchange chromatography•
This technique uses the charge on a protein to separate it from other proteins.
•
In the process of ion exchange, ions in solution replace ions that are electrostatically bound to an inert support carrying groups with the opposite charge.
•
Polyelectrolytes, such as proteins, can bind to either cation or anion exchangers, depending on their net charge (i.e. depending on the pH).
•
The protein can be eluted from the column either by changing the pH or by increasing the salt concentration, which shields the charges and thus decreases their attraction.
•
Elution is most often carried out by applying a salt gradient to the column.

Ion –
Exchange Chromatography•
A cation or an anion is attached to the resin beads.
•
Depending upon the electrical properties of the proteins, they may attach to the column. –
positively charged proteins will stick to a negatively charged column.
•
These proteins can then be removed by washing the column with either a strong salt solution or changing the pH of the wash buffer.
•
Anion exchangers such as DEAE ( Diethyl aminoethyl) are used.•
Attraction of proteins at a pH above the isolectric point of the
protein.•
Cation exchangers such as CM ( Carboxy methyl) are used.Attraction of protein at a pH below the isoelectric point of the
protein.
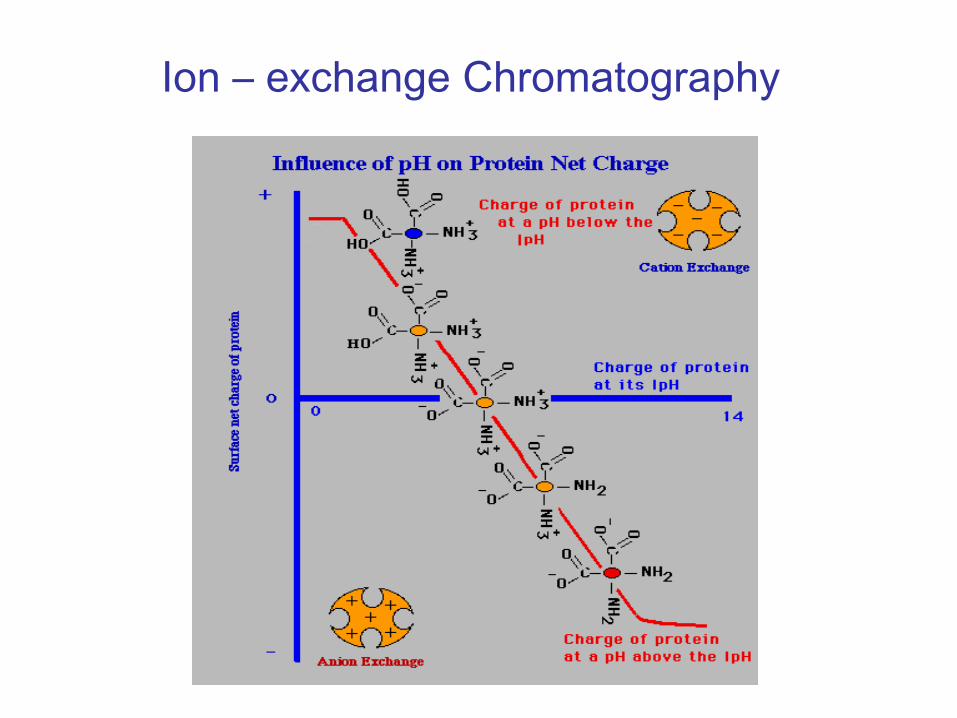
Ion –
exchange Chromatography

Ion –
exchange Chromatography
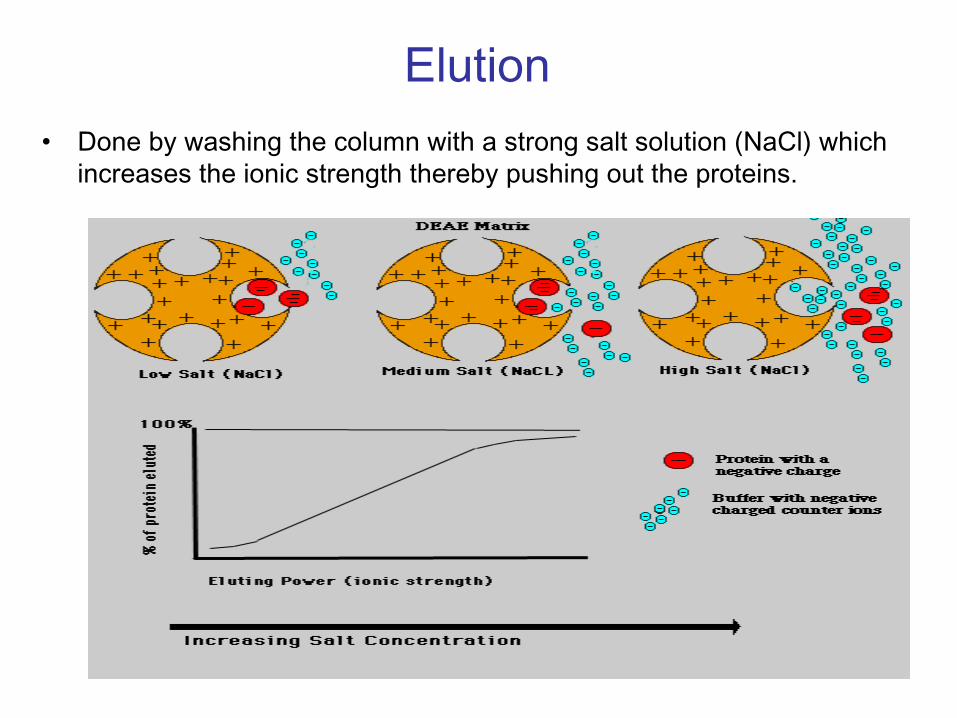
Elution•
Done by washing the column with a strong salt solution (NaCl) which increases the ionic strength thereby pushing out the proteins.


Typical Gradient Elution

Device for generating a linear
concentration gradient.

Ion exchange chromatography using stepwise elution
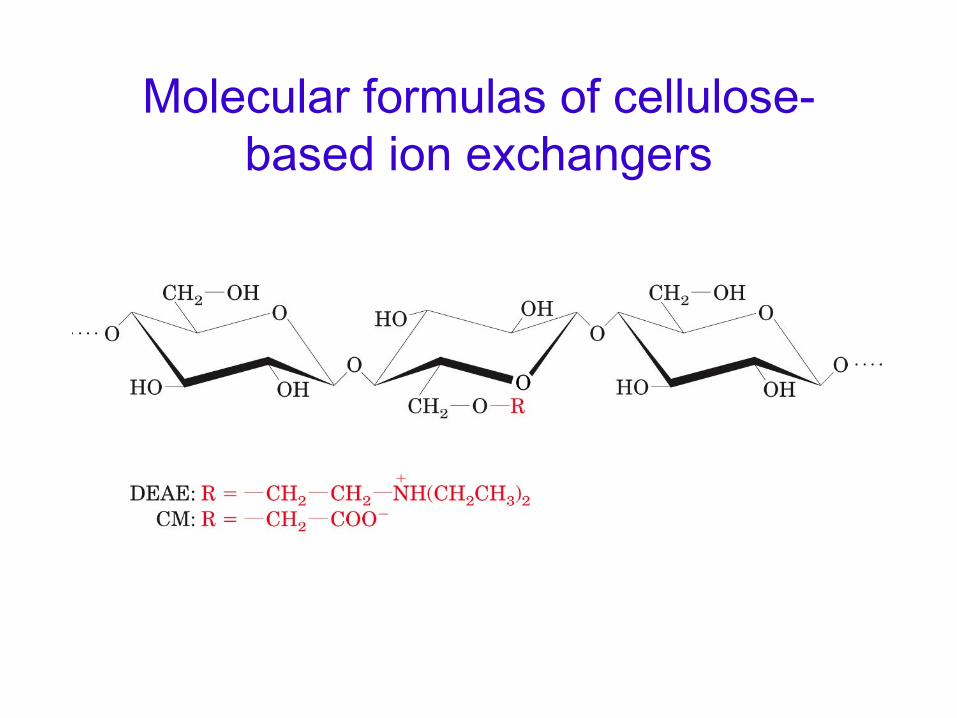
Molecular formulas of cellulose- based ion exchangers

Some common ion exchangers.

Ion Exchange Chromatography (con’d)

Hydrophobic interaction chromatography
•
Principle–
Proteins are separated by hydrophobic interaction on columns with hydrophobic groups attached (e.g. phenyl-, octyl groups)
•
Surface hydrophobicity–
Hydrophobicity of amino acid sidechains
–
Tryptofan > Isoleucine, Phenylalanine > Tyrosine > Leucine > Valine > Methionine
–
Most hydrophobic sidechains are buried in interior of protein, but some (clusters of) hydrophobic groups occur at surface of protein.
–
Surface hydrophobic sidechains can interact with hydrophobic groups of a column.

Hydrophobic interaction chromatography
•
TemperatureIncreasing temperature --> stronger hydrophobic interactions
•
Sample (application)Column having high concentration of a salt promotes binding (for example ammonium sulfate just below the concentration that starts to precipitate protein).
•
Elution of bound proteinsNegative gradient of salting-out ions (from high to low concentration).

Hydrophobic interaction chromatography
•
Hydrophobic group bound to solid phase•
Binding –
high salt (increases water surface tension, decreases available water molecules, increases hydrophobic interactions)
•
Elution–
decrease salt
–
add detergent–
decrease polarity
of mobile phase

Hydrophobic Interaction Separation (HIC)

70
•
Purifies proteins according to their hydrophobicity•
The stationary phase is hydrophobic and the mobile phase is more
hydrophilic than the stationary phase -
“Reversed" from normal phase chromatography
•
Silica beads with attached hydrocarbon chain groups.•
The hydrocarbon groups may be octacecyl, butyl, propyl, phenyldimethyl
•
The hydrophobic groups of the protein will bind to the beads. •
The hydrophilic proteins will emerge from the column first.
•
Many different sizes of columns with varying widthsPreperative (2.5-5cm) Analytical (0.4-1cm) Microbore (0.1-0.2cm)
•
The solvents used:–
Aqueous Phase: Water + 0.1% Triflouroacetic acid–
Organic Phase: Acetonitrile
Reversed Phase HPLC

“Reversed Phase”
Chromatography (RPC)(elution with organic solvents)

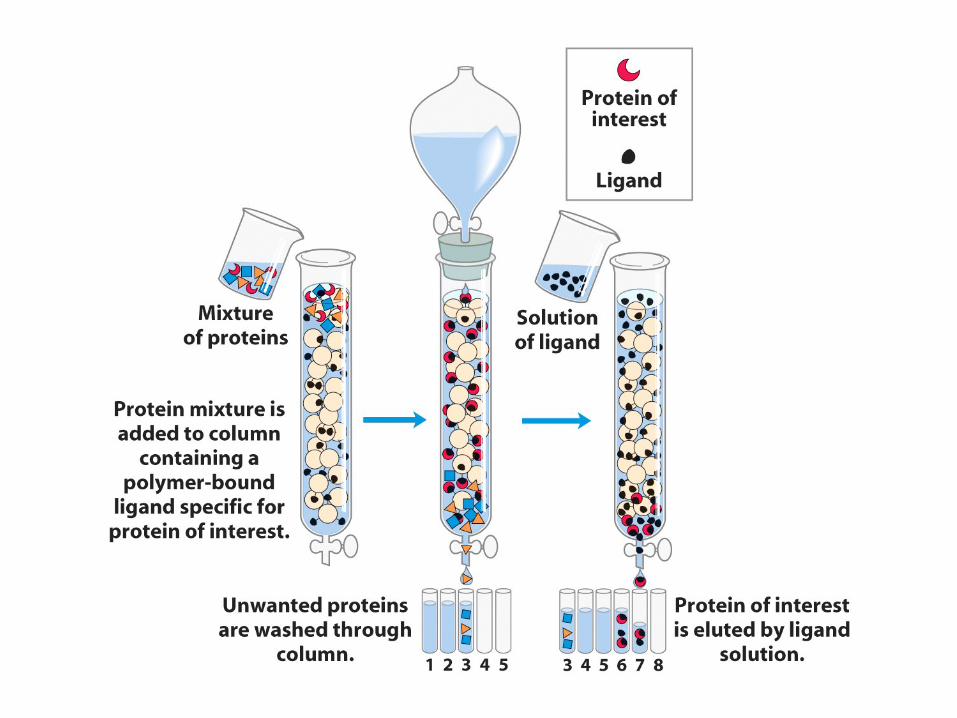
Affinity chromatography•
Takes advantage of the fact that many proteins specifically bind other molecules as part of their function.
•
Construct a column containing the ligand covalently attached to a matrix.
•
Upon passing the protein solution through such a column, only the proteins that can bind the ligand will be retained on the column.
•
Then the conditions can be adjusted to effect release from the ligand.
•
Often this can be done simply by eluting with the soluble version of the ligand.

Covalent linking of ligand to agarose

Derivatization of epoxy-activated agarose

•
Other proteins can be separated by this method based on their affinity for specific groups or compounds.
•
Examples:
Antigen Antibody
Antibody Antigen
Substrate Enzyme
Concavalin A Glycoprotein
Hormone Binding Protein/Receptor
Affinity Chromatography

Example: purification of staphylococcal nuclease by affinity chromatography on
bisphosphothymidine-linked agarose

Immuno-Affinity Chromatography
•
Antibody fixed to matrix
•
Protein binds to antibody
•
Wash unbound and loosely bound proteins off column
•
Elute protein with change in salt/pH

Typical Affinity Separation

Purification schemes

Gel electrophoresis•
General technique to analyze mixtures of proteins, and for limited purification of proteins.
•
The protein is driven through a viscous solvent by an applied electric field (E), due to the charge of the protein (z):
v = Ez/f (f = the protein's frictional coefficient)
•
A protein’s electrophoretic mobility (µ) is defined: µ
= v/E
•
Typically this is performed in the presence of a gel support, such as polyacrylamide–
prevents convection currents
–
enhances separation by serving as a molecular sieve

Native gel electrophoresis
•
The native protein migrates through the gel according to the charge of the protein at
the pH of the buffer system.
•
Useful for getting relatively pure protein
•
Not used very often.

SDS‐polyacrylamide gel electrophoresis (SDS‐PAGE)
•
Most common form of PAGE, but not useful for purification of proteins in their native conformation.
•
Proteins are solubilized with the detergent SDS (sodium dodecyl sulfate):–
binds polypeptide at a ratio of ~1 SDS per 2 amino acid residues
–
denatures protein → converts to roughly rod-like shape –
protein has a negative charge roughly proportional to its mass
–
The additional negative charge is much greater than the protein's intrinsic charge, which can usually be ignored.

SDS‐polyacrylamide gel electrophoresis (SDS‐PAGE)
•
As charge/mass ratio is almost constant and the molecular shapes are all similar, separation is on the basis of size.
•
Smaller polypeptides migrate faster and larger ones migrate slower, due to the gel filtration effect.
•
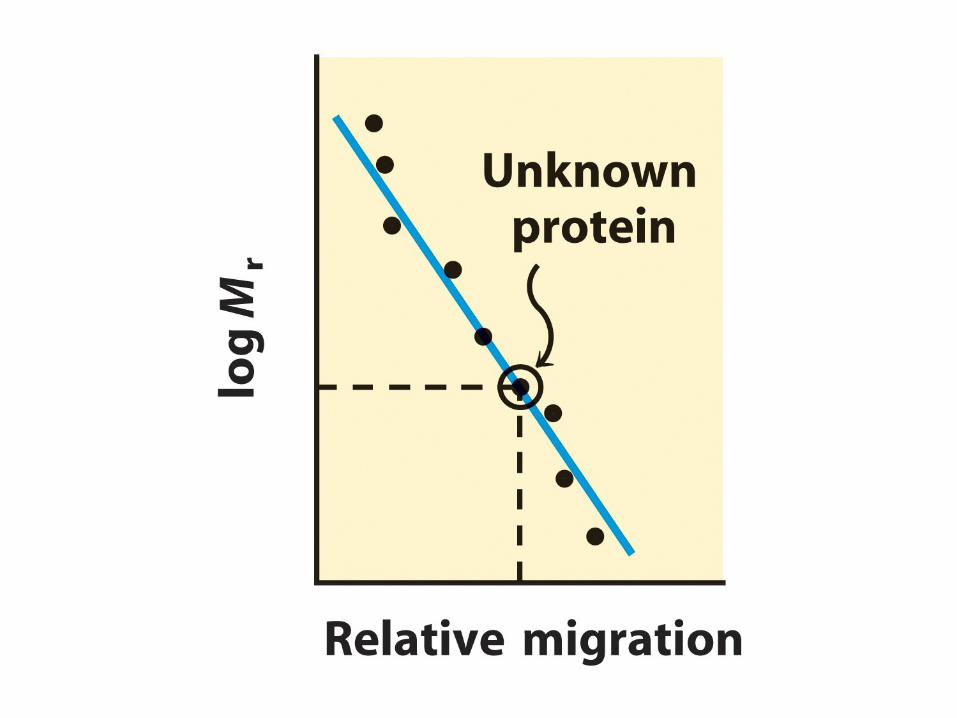
There is an empirical relationship between mobility and molecular weight:
µ
∝
1/log Mr
•
Average pore size of the gel can be controlled by varying the concentration of acrylamide before initiating polymerization.
•
Higher percentage polyacrylamide gels (smaller pore sizes) will result in better resolution of smaller polypeptides.


Polymerization of acrylamide and N,N¢‐methylenebisacrylamide

Electrophoresis
•
Tris‐glycine buffer
•
10% SDS



Western blotting
•
Separate proteins by electrophoresis
•
Transfer to membrane (e.g. nitrocellulose)
•
Bind primary antibody
•
Bind secondary antibody
•
Detection

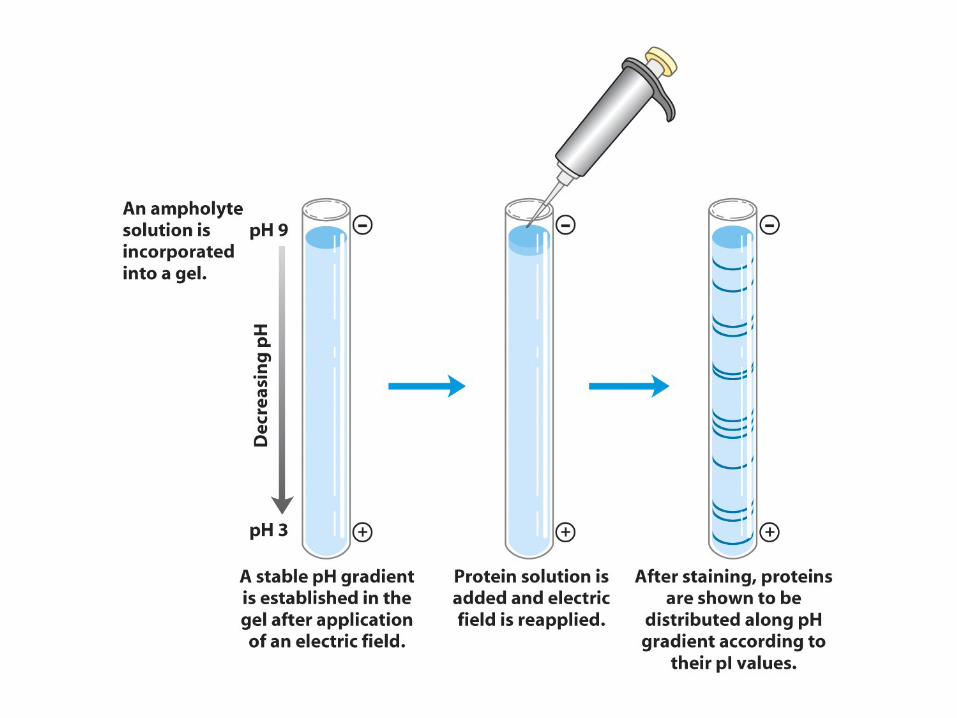
Isoelectric focusing• A pH gradient is set up by electrophoresing
polyampholytes (300-600 Da oligomers bearing amino and carboxylate groups in varying rations) in a gel tube.
• The more basic ones (cationic) will accumulate near the cathode and the more acidic (anionic) will accumulate near the anode, thereby establishing a continuous pH gradient.
• When a protein is applied to the gel, it will migrate toward the anode if it is negatively charged or toward the cathode if positively charged, until it reaches the region corresponding to its pI, where it will stop.
• Proteins are often denatured in 6M urea, which does not change the protein's charge (unlike SDS).

General formula of the ampholytes used in isoelectric focusing.

Example
•
A protein with a pI of 5.2 is introduced near the cathode end, which has pH of 9.5 (in this gel).
•
It is then negatively charged and will migrate toward the anode.
•
As it migrates in this direction, the pH will steadily decrease, and the amount of negative charge carried by the protein will
also decrease, as more and more ionizable groups become protonated.
•
Thus it will slow down, until it hits the region where pH = 5.2, where it will experience no further force.
•
If it diffuses in either direction, it will pick up charge and the electric field will force it back.
•
This results in the protein being concentrated (focused) in a very narrow region of the gel.


2‐Dimensional Gels
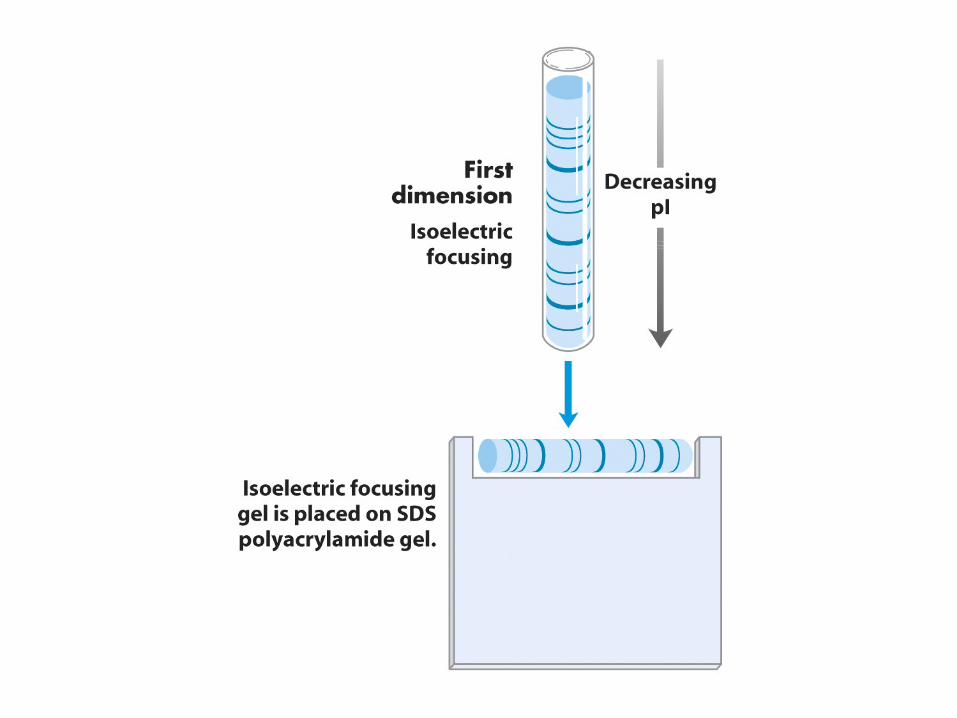
•
This is a powerful technique that combines isoelectric focusing with SDS‐PAGE.
•
Proteins are separated in the first dimension by isoelectric focusing (IEF).
•
Then this tube is attached to the side of an SDS‐ polyacrylamide gel. SDS‐PAGE provides the second
dimension.
•
Each protein migrates to a semi‐unique spot according to its pI and molecular weight (MW).


Two‐dimensional (2D) gel electrophoresis

Using antibodies
Antibodies (immunoglobulins) bind specific antigens/epitopesmonoclonal - all bind same epitopepolyclonal - mixture that binds several epitopes
Secondary antibodies - anit-immunoglobulins (antibodies to antibodies)

Using antibodies