chapter 1 introduction -...
TRANSCRIPT

1
CHAPTER 1
INTRODUCTION
Catalysis is one of the most important and widely-spread concepts
of chemistry. Today, catalysis is the workhorse in the chemical industry with
approximately >90% of the products produced in catalytic processes. In
addition, life cannot exist without catalysis, as virtually all biochemical
processes that sustain life are reliant on enzymes, nature’s own catalysts. The
term catalysis was coined by Berzelius in 1836.
Catalysis is defined as the change in rate of a chemical reaction due
to the participation of a substance called catalyst, which is not consumed by
their action. However, the catalyst affects only the rate of the reaction; it
changes neither the thermodynamics of the reaction nor the equilibrium
composition. A catalyst can activate a definite molecule to lower its activation
energy of the corresponding transition state through the adsorption or the
coordination of the molecule on the catalyst (Chorkendorff and
Niemantsverdriet 2003).
More recently environmental concerns have driven the
development of specialized catalysts to prevent polluting substances from
being released into the environment. Since 1950s wide use of catalyst in the
petroleum and petrochemical industries was the most important factor
enabling man to tap into petroleum for energy as well as new raw materials
and new polymer materials resulting in a high level of living standard. After
late 1970 and through 1980, the technology of catalysis moved to better

2
understanding of the catalytic function and to improve the catalytic selectivity
in order to save the energy and chemicals for the better economy and for a
cleaner process to meet environmental regulation. In the last 10 years,
catalysts find greater importance in the development of fuel cell technology to
convert fossil fuels into hydrogen, in utilization of toxic gases in advanced
industry, and the gasification of solid wastes to restore natural environment.
1.1 PETROLEUM REFINING
A petroleum refinery is a manufacturing operation where crude
petroleum (the raw material) is converted into usable finished products. In
other words, it is the manufacturing phase of the oil industry. The function of
the refinery is to convert crude oil into the finished products required by the
market in the most efficient and hence most profitable manner. Crude oil is
complex mixtures containing many different hydrocarbon compounds that
vary in appearance and composition from one oil field to another. Crude oil
range in consistency from water to tar-like solids, and in color from clear to
black. An average crude oil contains about 84% carbon, 14% hydrogen, 1-3%
sulfur, and less than 1% each of nitrogen, oxygen, metals, and salts.
Crude oil is generally classified as paraffinic, naphthenic, or
aromatic, based on the predominant proportion of similar hydrocarbon
molecules. Mixed-base crudes have varying amounts of each type of
hydrocarbon. Refinery crude base stocks usually consist of mixtures of two or
more different crude oil. Crude oil that contains appreciable quantities of
hydrogen sulfide or other reactive sulfur compounds is called sour. Those
with less sulfur are called sweet.
1.2 HYDROCARBON COMPOUNDS IN CRUDE OIL
The paraffinic series of hydrocarbon compounds found in crude oil
have the general formula CnH2n+2 and can be either straight chains (normal) or

3
branched chains (isomers) of carbon atoms. The lighter, straight-chain
paraffin molecules are found in gases and paraffin waxes. Examples of
straight-chain molecules are methane, ethane, propane, and butane (gases
containing from one to four carbon atoms), and pentane and hexane (liquids
with five to six carbon atoms). The branched-chain (isomer) paraffins are
usually found in heavier fractions of crude oil and have higher octane
numbers than normal paraffins.
Aromatics are unsaturated ring-type (cyclic) compounds which
react readily because they have carbon atoms that are deficient in hydrogen.
All aromatics have at least one benzene ring as part of their molecular
structure. Naphthalenes are fused double-ring aromatic compounds. The most
complex aromatics, polynuclears (three or more fused aromatic rings) are
found in heavier fractions of crude oil.
Naphthenes are saturated hydrocarbon groupings with the general
formula CnH2n, arranged in the form of closed rings (cyclic) and found in all
fractions of crude oil except the very lightest fraction. Single-ring naphthenes
(monocycloparaffins) with five and six carbon atoms predominate, with two-
ring naphthenes (dicycloparaffins) found in the heavier ends of naphtha.
Alkenes are mono-olefins with the general formula CnH2n and
contain only one carbon-carbon double bond in the chain. The simplest alkene
is ethylene. Olefins are usually formed by thermal and catalytic cracking and
rarely occur naturally in unprocessed crude oil.
Dienes, also known as diolefins, have two carbon-carbon double
bonds. The alkynes, another class of unsaturated hydrocarbons, have a
carbon-carbon triple bond within the molecule. Both these series of
hydrocarbons have the general formula CnH2n-2. Diolefins such as
1,2-butadiene and 1,3-butadiene, and alkynes such as acetylene, occur in C5

4
and lighter fractions from cracking. These compounds are more reactive than
paraffins or naphthenes and readily combine with other elements such as
hydrogen, chlorine, and bromine.
1.3 NONHYDROCARBON COMPOUNDS IN CRUDE OIL
1.3.1 Sulfur Compounds
Sulfur may be present in crude oil as hydrogen sulfide (H2S), as
compounds (e.g. mercaptans, sulfides, disulfides, thiophenes, etc.), or as
elemental sulfur. Each crude oil has different amounts and types of sulfur
compounds, but as a rule the proportion, stability, and complexity of the
compounds are greater in heavier crude-oil fractions. Hydrogen sulfide is a
primary contributor to corrosion in refinery processing units. Other corrosive
substances are elemental sulfur and mercaptans. Moreover, the corrosive
sulfur compounds have an obnoxious odor.
Pyrophoric iron sulfide results from the corrosive action of sulfur
compounds on the iron and steel used in refinery process equipment, piping,
and tanks. The combustion of petroleum products containing sulfur
compounds produces undesirable products such as sulfuric acid and sulfur
dioxide. Catalytic hydrotreating processes such as hydrodesulfurization
remove sulfur compounds from refinery product streams. Sweetening
processes either remove the obnoxious sulfur compounds or convert them to
odorless disulfides, as in the case of mercaptans.
1.3.2 Nitrogen and Oxygen Compounds
Nitrogen is found in lighter fractions of crude oil as basic
compounds, and more often in heavier fractions of crude oil as nonbasic
compounds. The decomposition of nitrogen compounds in catalytic cracking
and hydrocracking processes forms ammonia and cyanides that can cause

5
corrosion. Oxygen compounds such as phenols, ketones, and carboxylic acids
occur in crude oil in varying amounts. The presence of such organic oxygen
containing molecule also affect the selectivity, stability and surface of the
sulfided hydrotreating catalysts (Furimsky 2000).
1.3.3 Trace Metals and Salts
Metals including nickel, iron, and vanadium are often found in
crude oil in small quantities and are removed during the refining process. The
burning of heavy fuel oil in refinery furnaces and boilers can leave deposits of
vanadium oxide and nickel oxide in furnace boxes, ducts, and tubes. It is also
desirable to remove trace amounts of arsenic, vanadium, and nickel prior to
processing as they can poison certain catalysts.
Crude oil often contains inorganic salts such as sodium chloride,
magnesium chloride, and calcium chloride in suspension or dissolved in
entrained water (brine). These salts must be removed or neutralized before
processing to prevent catalyst poisoning, equipment corrosion, and fouling.
Salt corrosion is caused by the hydrolysis of some metal chlorides to
hydrogen chloride (HCl) and the subsequent formation of hydrochloric acid
when crude is heated. Hydrogen chloride may also combine with ammonia to
form ammonium chloride (NH4Cl), which causes fouling and corrosion.
1.4 TYPES OF REACTION IN REFINERY
There are number of processes involved in petroleum refining as
shown in Figure 1.1. These include thermal, catalytic and hydroprocessing
upgrading processes. The hydroprocessing processes include three major
classes, namely, hydrotreating, hydrocracking, and hydrofinishing.

6
Figure 1.1 Stream of petroleum processes
1.4.1 Catalytic Reforming
Reforming is an important process used to convert straight-chain
alkane and alkenes into branched and aromatic hydrocarbons (low-octane into
high-octane) and catalysed by bifunctional platinum/Al2O3. Both the metal
and support are involved in the reaction, making the system bifunctional
catalyst. This process involves reaction such as, dehydrogenation,
aromatization, hydrogenation, isomerization and cracking. The first three
reactions are catalysed by the metal and last two reactions are catalysed by
acidic sites on the alumina support. Most of the reactions (except
isomerization and hydrogenation) are strongly endothermic, and a net

7
producer of hydrogen. Thermodynamics of this reaction therefore require high
temperature and low pressure. However, such conditions are favourable for
coke formation, which severely limits the life time of the catalysts. For this
reason, hydrogen is recycled at moderate pressures to limit the coke
formation. Hence, the actual reforming conditions (500 ºC, 5-20 bar, H2) are a
compromise between product quality and yield on one hand and reduce coke
formation on the other hand (Chorkendorff and Niemantsverdriet 2003).
Isomerisation is a reforming process in which the structure of the compound is
rearranged to give an isomer with desirable anti-knock properties.
Olefins obtained as by products during cracking can be
polymerized under acidic conditions using H2SO4 or H3PO4 to give high
molecular weight hydrocarbons which can be catalytically hydrogenated to
give branched alkanes.
1.4.2 Hydrocracking
Hydrocracking is a two-stage process combining catalytic cracking
and hydrogenation, wherein heavier feedstocks are cracked in the presence of
hydrogen to produce more desirable products. The process employs high
pressure, high temperature, a catalyst, and hydrogen.
Hydrocracking is used for feedstocks that are difficult to process by
either catalytic cracking or reforming, since these feedstocks are characterized
usually by a high polycyclic aromatic content and/or high concentration of the
two principal catalyst poisons, sulfur and nitrogen compounds. The
hydrocracking process largely depends on the nature of the feedstock and the
relative rates of the two competing reactions, hydrogenation and cracking.
Heavy aromatic feedstock is converted into lighter products very high
pressures (1,000-2,000 psi) and fairly high temperatures (750-1,500 °C), in
the presence of hydrogen and special catalysts. When the feedstock has a high
paraffinic content, the primary function of hydrogen is to prevent the formation of

8
polycyclic aromatic compounds. Another important role of hydrogen in the
hydrocracking process is to reduce tar formation and to prevent build-up of
coke on the catalyst. Hydrogenation also serves to convert sulfur and nitrogen
compounds present in the feedstock to hydrogen sulfide and ammonia.
Hydrocracking produces relatively large amounts of isobutane for
alkylation feedstock. Hydrocracking also performs isomerization for pour-
point control and smoke-point control, both of which are important in high-
quality jet fuel. Noble metals such as Pt, Pd, etc., supported on alumina or
zeolites are used as hydrocracking catalyst.
1.4.3 Hydrotreating
The crude oil contains S, N, O, and metals (Ni, V) and makes high
demands on the catalyst performance. The presence of nitrogen compounds
leads to poor color, smell and subsequently NOx causes pollution of the
atmosphere. N-compounds also act as poison for hydrocracking and reforming
catalysts in the later stages of oil refining. Furthermore, the intensified
protection of the environment has led to the sharpening of the norms for the S,
N and metal content of the petroleum products (Stinner et al 2001).
The basis for hydrotreating was laid in 1910 by Bergius who
renewed coal into gaseous and liquid fractions applying non-catalytic
hydrogenation and cracking at high temperatures and pressures. Although the
yield of Bergius' process (hydrogenation of coal) was high, the products were
not suitable because they mainly contained hydrocarbons with high molecular
weights and in addition contained large amounts of oxygen, nitrogen and
sulphur compounds. An improvement was seen when catalysts were used for
this process. Hydrotreating catalysts originated in the 1920s when German
researchers developed unsupported metal sulfide catalysts for liquefying coal.
However, it was not until 1970s that the structures of these catalysts and the
mechanisms of their catalytic action began to be understood. It was

9
established that under catalytic reaction conditions, most of the molybdenum
in industrial hydrotreating catalysts is present as small MoS2 particles in the
pores of -Al2O3. It was not until the 1980s that the location of the cobalt and
the nickel promoter ions in the hydrotreating catalysts was more or less
determined. The role of phosphate and fluorine additives is still under
investigation. Supports other than -Al2O3 like amorphous silica-alumina, are
also used in commercial units and their functions are topics of academic and
industrial research.
Hydrotreating is a generic name given to processes utilizing
hydrogen, which include Hydrodesulfurization (HDS), Hydrodenitrogenation
(HDN), Hydrodemetallization (HDM), hydrogenation and hydrofinishing.
Hydrotreating is the catalytic conversion of organic sulfur, nitrogen, oxygen,
and metal-containing molecules from crude oil at high hydrogen pressures
and includes the hydrogenation of unsaturated compounds and cracking of
petroleum feedstock to lower molecular hydrocarbons. As a consequence,
hydrotreating is the largest application of industrial catalyst on the basis of the
amount of material processed per year. Typically, hydrotreating is done prior
to processes such as catalytic reforming so that the catalyst is not
contaminated by untreated feedstock. Hydrotreating is also used prior to
catalytic cracking to reduce sulfur and improve product yields and to upgrade
middle-distillate petroleum fractions into finished kerosene, diesel fuel, and
heating fuel oil. In addition, hydrotreating converts olefins and aromatics to
saturated compounds.
HDS is a process employed to remove sulfur from petroleum and
other fossil fuel stocks through reaction with hydrogen in order to prevent
sulfur from poisoning the metal catalyst and also to remove the unpleasant
odour of lube oil.

10
HDN is a process in which organonitrogen compounds are removed
from hydrocarbon feed stocks to produce processible, stable and
environmentally acceptable liquid fuels and lube base stocks. HDN is an
important reaction in petroleum refining, because it removes nitrogen, which
causes pollution when fuels are burnt and also poisons the catalyst when
petroleum distillates are processed. The central reaction in HDN is the
breaking of the C-N bond of nitrogen containing hydrocarbons, in which the
nitrogen atom is removed as ammonia and thereby the hydrocarbons are free
from nitrogen. Hydrogenation followed by hydrogenolysis is found to be a
very effective and advantageous method for removing nitrogen and sulfur
from petroleum. Since the remaining fragments of the N- and S- containing
compounds are left as pure hydrocarbons in the products.
Oxygen containing compounds are acidic and their presence,
especially in commercial petroleum products, is unwelcome. Most of the
oxygen exists as hydroxyl groups and carbonyl groups. Water formed as a
result of hydrodeoxygenation (HDO) is known to act as a poison for catalysts.
These compounds interact with sulfur and nitrogen thereby reducing the rate
of HDN and HDS.
1.5 SULFUR CONTAINING COMPOUNDS
Organo sulfur compounds are usually present in almost all fractions
of crude oil distillation. Higher boiling fractions contain relatively more sulfur
compounds with high molecular weight. Therefore, a wide spectrum of sulfur-
containing compounds should be considered from the viewpoint of their
reactivity in the hydrotreating processes. The common types of sulfur
compounds in liquid fuels are mercaptans, sulfides, disulfides, thiophene and
benzothiophene.
Figure 1.2 presents a qualitative relationship between the type and
size of sulfur containing organic molecules in various distillate fuel fractions

11
and their relative reactivities (Song 2002). The reactivity ranking in
Figure 1.2 is based on well-known experimental observations and literature
information (Knudsen et al 1999, Whitehurst et al 1998 and Song and Ma
2003). HDS occurs directly through hydrogenolysis pathway for the sulfur
compounds without a conjugation structure between the lone pairs on S atom
and the -electrons on aromatic ring, disulfides, sulfides, thiols, and
tetrahydrothiophene.
Figure 1.2 Reactivity of organic sulfur compounds in
hydrodesulfurization (HDS)

12
These sulfur compounds exhibit higher HDS reactivity than that of
thiophene by an order of magnitude because they have high electron density
on the sulfur atom and weak C–S bond. The reactivities of the
1 to 3 ring sulfur compounds decrease in the order thiophenes >
benzothiophenes > dibenzothiophenes (Frye and Mosby 1967, Kilanowski et
al 1978, Houalla et al 1980, Girgis and Gates 1991, Vasudevan and Fierro,
1996). In naphtha, thiophene is much less reactive than thiols, sulfides, and
disulfides that the latter can be considered to be virtually infinitely reactive in
practical high-conversion processes (Gates and Topsoe 1997). Similarly, in
gas oil, the reactivities of (alkyl-substituted) 4-methyldibenzothiophene and
4,6-dimethyldibenzothiophene (4,6-DMDBT) are much lower than other
sulfur-containing compounds (Kabe et al 1992, Ma et al 1994 and 1995).
Consequently, in deep HDS, the conversion of these key substituted
dibenzothiophenes largely determines the required reaction conditions.
Gates and Topsoe (1997) pointed out that, the 4-methyldibenzothiophene and
4,6-DMDBT are the most appropriate compounds for investigations of
candidate catalysts and reaction mechanisms.
1.6 NITROGEN CONTAINING COMPOUNDS
Nitrogen-containing compounds in petroleum and coal-derived
liquids are normally divided into two groups: heterocyclic and non
heterocyclic. Noncyclic nitrogen compounds such as aliphatic amines and
nitriles are present in oil in small amounts and are easy to denitrogenate.
Among non-heterocyclic nitrogen compounds, aniline derivatives are the
most important ones in HDN because they are always formed in the HDN
network of heterocyclic nitrogen compounds and they are more difficult to
denitrogenate than aliphatic ones. Heterocyclic nitrogen containing
compounds are present in larger amount and are very difficult to remove.
They can be divided into basic and nonbasic compounds. Nonbasic
compounds consist of five-membered heterocycles such as pyrrole, indole,
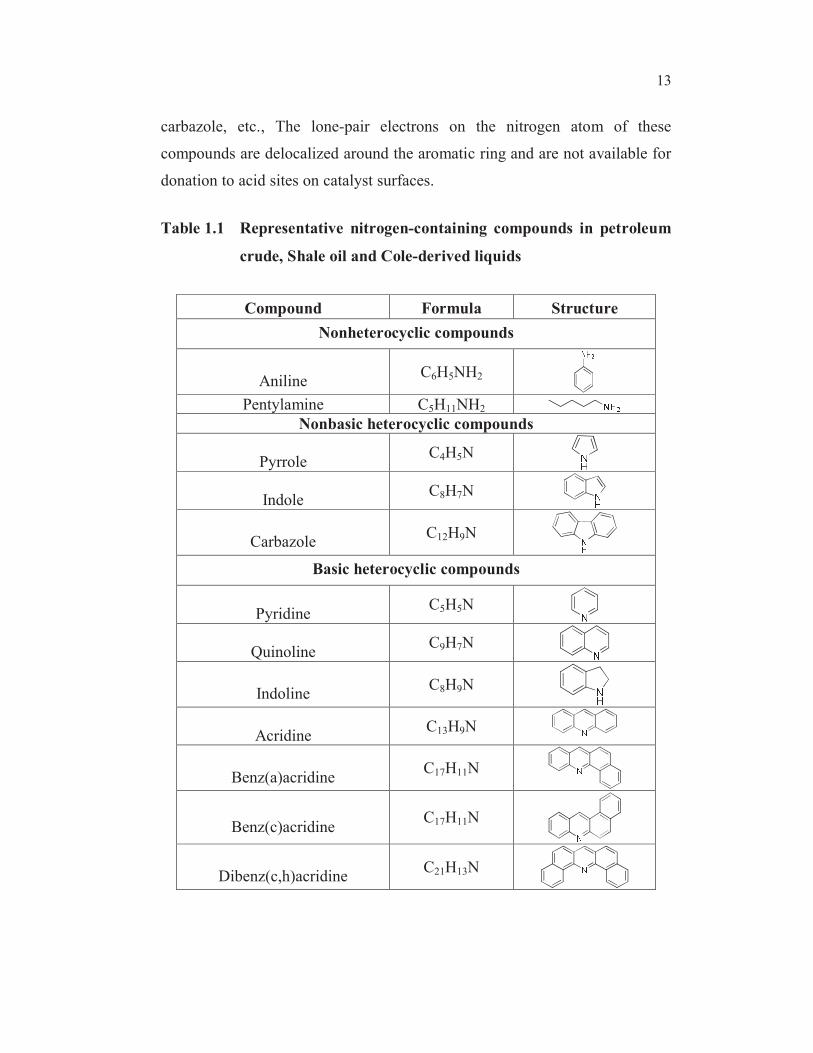
13
carbazole, etc., The lone-pair electrons on the nitrogen atom of these
compounds are delocalized around the aromatic ring and are not available for
donation to acid sites on catalyst surfaces.
Table 1.1 Representative nitrogen-containing compounds in petroleum
crude, Shale oil and Cole-derived liquids
Compound Formula Structure
Nonheterocyclic compounds
AnilineC6H5NH2
Pentylamine C5H11NH2
Nonbasic heterocyclic compounds
PyrroleC4H5N
IndoleC8H7N
CarbazoleC12H9N
Basic heterocyclic compounds
PyridineC5H5N
QuinolineC9H7N
IndolineC8H9N
AcridineC13H9N
Benz(a)acridineC17H11N
Benz(c)acridineC17H11N
Dibenz(c,h)acridineC21H13N

14
Basic compounds include six-membered heterocycles such as
pyridine, quinoline, acridine, etc. The lone-pair electrons on the nitrogen atom
of these compounds, on the contrary, are not tied up in the cloud of the
heterocyclic ring and are available for sharing with acid sites on catalyst
surfaces. The saturated five-membered heterocycles, like indoline are strongly
basic as aliphatic amines. Table 1.1 lists the various types of nitrogen-
containing compounds (without alkyl substitution).
1.7 HYDROTREATING CATALYSTS
Industrial hydrotreating catalyst contains molybdenum and cobalt
or nickel, supported on -Al2O3. MoO3/Al2O3 and Co/Ni/Al2O3 tested for the
removal of S, N and O atoms. It is found that MoO3/Al2O3 is highly active
than Co and Ni sulfide/Al2O3. Therefore, molybdenum sulfide is traditionally
considered to be the catalyst. As a consequence, cobalt and nickel are referred
to as the promoters of the Mo activity (Ho 1988 and Prins et al 1989). Cobalt
is used mainly as a promoter for sulfided Mo/Al2O3 in HDS, and nickel in
HDN. In addition to molybdenum and cobalt or nickel, hydrotreating catalysts
often contain additives such as phosphorus, boron, fluorine or chlorine, which
may influence the catalytic as well as the mechanical properties of the catalyst
(Lewandowski and Sarbak 2000, Jian et al 1995, Sun et al 2003 and Gioia and
Murena 1998). Typical hydrotreating catalysts need to be sulfided to activate
the reacting sites. Sulfiding is traditionally done during the start-up phase by
exposing the catalyst to the sulfur-containing liquid feed or to the gaseous
mixture of H2S and hydrogen at high temperature. The sulfiding procedure
has significant influence on the catalytic activity and stability.

15
1.8 DIFFICULTIES OF HDN
HDN is typically more difficult than HDS. This can be partially
explained by the relative bond strength as shown in Table 1.2. The C N bond
energy (615 kJ/mol) is higher than the C N bond (308 kJ/mol), and the C N
bond is also stronger than the C S bond (259 kJ/mol). That is why the
aromatic nitrogen containing compounds, especially the aromatic nitrogen-
containing heterocycles must be saturated before they can be further
denitrogenated, unless some materials with metallic properties such as metal
carbides and nitrides are used as HDN catalysts. Heavier feedstocks such as
VGO, residue, light cycle oil from VGO and residue, and coal-derived liquids
usually contain much higher concentration of nitrogen containing compounds
than straight-run streams. They are more complex in structure and difficult to
denitrogenate. Nitrogen containing compounds often have a strong adsorption
capacity on the catalyst surface. Self-Inhibition by the high concentration of
these compounds as well as the secondary compounds leads to a much lower
HDN rate. More severe catalyst deactivation is caused by the high molecular
weight-aromatic compounds in the feedstock. During HDN, coke is also
formed, mainly by condensation of the basic nitrogen-containing compounds
on Lewis acid sites of the catalyst.
Table 1.2 Bond Energies between various atoms
BondEnergy,
kJ/molBond
Energy,
kJ/mol
436 391
413 308
348 615
614 891
839 259
358 577
799 347

16
1.9 STRUCTURE OF HYDROTREATING CATALYSTS
1.9.1 Structure of the Oxidic Catalyst
Hydrotreating catalysts are usually prepared by a sequential pore
volume impregnation procedure or by co-impregnation (Prins et al 1989 and
Chianelli et al 1994). In the former method, -Al2O3 support is first
impregnated with an aqueous solution of ammonium heptamolybdate
(NH4)6Mo7O24, and then with an aqueous solution of nickel nitrate Ni(NO3)2
or cobalt nitrate Co(NO3)2. In the latter method, both the Mo and Ni
precursors are impregnated simultaneously. Massoth (1979) explained the
formation of monolayer structure of MoO3 over Al2O3 surface. The results
concluded that there is a strong interaction between molybdenum with the
hydroxyl groups on the Al2O3 surface has been assumed to result in a MoO3
monolayer structure. A similar conclusion was drawn in a combined 1H-NMR
and low temperature chemisorption study (Kraus and Prins 1996) and in
several EXAFS studies (Chiu et al 1984, Parham and Merrill 1984 and
Clausen et al 1986). Infrared emission spectroscopy could not detect bands
due to a MoO3 phase even at high loadings (15 wt.%) of molybdenum on
Mo/Al2O3 catalysts, indicating that the molybdenum is present in a highly
dispersed phase (Li et al 1991). Investigations on Ni and Co promoted
catalysts confirmed an interaction between molybdenum and nickel or cobalt
in the catalyst in the oxidic state. The infrared absorption bands of NO
adsorbed on CoMo/Al2O3 are shifted from those of NO on Co/Al2O3 (Topsoe
and Topsoe 1982), and Raman bands due to polymeric molybdenum oxide
species decrease in intensity with increasing cobalt loading in an oxidic
CoMo/Al2O3 catalyst (Gao and Xin 1993). The results suggested that nickel
or cobalt cations interact especially with the most highly polymerized
molybdenum oxide species. In this way the promoter cations stay at the

17
surface and close to the molybdenum cations and are well positioned to form
the active Ni-Mo-S structure during sulfidation.
1.9.2 Structure of the Sulfidic Catalyst
The oxidic catalyst precursors are transformed into the actual
hydrotreating catalyst by sulfidation in a mixture of H2 and one or more
compounds containing sulfur. H2S, thiophene, CS2, dimethyl disulfide or the
oil fraction to be hydrotreated can be used for the sulfidation. The properties
of the sulfided catalyst depend to a great extent on the calcination and
sulfidation conditions. Calcination at high temperature induces a strong
interaction between molybdenum and cobalt or nickel cations and the Al2O3
support. Consequently, it is difficult to transform completely the oxidic
species into sulfides. Mossbauer spectroscopy of CoMo/Al2O3 catalysts
showed that, at increasingly high calcination temperatures, more Co2+ ions are
incorporated into the bulk of the alumina (Wivel et al 1984). The higher the
calcination temperature, the higher the sulfidation temperature needed to
bring these cations back to the surface to provide a high catalytic activity for
hydrotreating. At temperatures that are too high, however, the metal sulfide
particles sinter or do not form the catalytically active Co-Mo-S structure.
Optimum calcination and sulfidation temperatures are in the range of
673-773 K for Al2O3-supported catalysts (Prada Silvy et al 1989a). MoS2 has
a layer lattice, and the sulfur-sulfur interaction between successive MoS2
layers is weak (Van der Waals force). Crystals grow as platelets with
relatively large dimensions parallel to the basal sulfur planes and small
dimensions perpendicular to the basal planes. Investigations of model
catalysts consisting of MoS2 grown on -Al2O3 films on the surfaces of
MgAl2O4 supports have shown that MoS2 grows with its basal plane parallel

18
to the (111) surface of -Al2O3 and perpendicular to the (100) -Al2O3 surface
(Sakashita and Yoneda 1999). This observation also suggested that the edges
of the MoS2 platelets are bonded to the (100) surface of -Al2O3 by Mo-O-Al
bonds (Figure 1.3).
(a) (111) - Al2O3 (b) (100) - Al2O3
Figure 1.3 Orientation of small MoS2 particles on (a) (111) and
(b) (100) -Al2O3 surface
Nickel may be present in three forms after sulfidation: as Ni3S2
crystallites on the support, as nickel atoms adsorbed on the edges of the MoS2
crystallites (the so-called Ni-Mo-S phase) and as nickel cations at octahedral
or tetrahedral sites in the -Al2O3 lattice (Figure 1.4). Analogously, cobalt can
be present as segregated Co9S8, as Co-Mo-S and as cobalt cations on the
support. Depending on the relative concentrations of nickel (or cobalt) and
molybdenum and on the pre-treatment conditions, a sulfided catalyst may
contain a relatively large amount of either Ni3S2 (or Co9S8) or the
Ni-Mo-S (or Co-Mo-S) phase.

19
Figure 1.4 Three forms of nickel present in a sulfided NiMo/Al2O3
catalyst: as active sites on the MoS2 edges (the so-called
Ni-Mo-S phase), as segregated Ni3S2 and as Ni2+
ions in the
support lattice
Several in-situ characterization techniques such as Mossbauer,
infrared, and EXAFS confirmed the Ni-Mo-S (or Co-Mo-S) edge decoration
model. The infrared spectra of NO molecules adsorbed on sulfided
CoMo/Al2O3 catalysts indicated that as the cobalt content increased at fixed
molybdenum content, the number of NO molecules adsorbed on cobalt sites
increased and the number of NO molecules adsorbed on molybdenum sites
decreased (Topsoe and Topsoe 1983). Cobalt atoms at edge-decoration sites
cover molybdenum atoms and block adsorption of NO on these molybdenum
atoms. The observed behavior is therefore in accordance with the edge-
decoration location. EXAFS studies showed that a nickel atom in a sulfided
NiMo catalyst supported on -Al2O3 or on carbon is surrounded by four or
five sulfur atoms at a distance of 2.2 Å, by one or two molybdenum atoms at a
distance of 2.8 Å and by one nickel atom at a distance of 3.2 Å (Louwers and
Prins 1992). These data are consistent with a model in which the nickel atoms
are located at the MoS2 edges in the molybdenum plane in a square pyramidal
coordination. The nickel atoms are connected to the MoS2 by four sulfur
atoms and depending on the H2S partial pressure, a fifth sulfur atom may be

20
present in the apical position in front of the nickel atom (Figure 1.5). Density
functional theory (DFT) calculations suggested a different edge-decoration
model (Byskov et al 1999). Instead of substituting molybdenum atoms at the
molybdenum edge, cobalt atoms were claimed to prefer to substitute
molybdenum atoms at the sulfur edge. Other DFT calculations
indicated that these particular edge positions are an artifact of the too small
MoS2 clusters used to model MoS2 in the calculations (Byskov et al 1999).
DFT calculations with larger MoS2 clusters showed that the most favorable
location of the promoter atoms is the substitutional position at the
molybdenum edge. The nickel and cobalt atoms extend, as it were, the MoS2
lattice by taking up molybdenum positions (Raybaud et al 2000). This
conclusion is in good agreement with the EXAFS results (Figure 1.5)
(Louwers and Prins 1992).
Figure 1.5 Structure of Ni-Mo-S phase as determined by EXAFS studies
The structure of the active phase has been a matter of great debate.
Voorhoeve and Stuive (1971) proposed the intercalation of co-promoter atoms
between alternating MoS2 layers (intercalation model), while Farragher and
Cossee (1973) suggested that the promoter ions are located in alternate layers
at the edges (pseudo–intercalation or decoration model). The remote control

21
or contact synergy model of Delmon (1990), the physical contact between
separate Co9S8 and MoS2 crystallites offers an explanation for the
promotional effect, the first one causing spill–over hydrogen that enhancing
the activity of the MoS2 phase. Ratnasamy and Sivasanker (1980) suggested
the promoter ions to be located at the edges of the MoS2 layers. Topsoe and
Topsoe (1983) produced convincing experimental evidence for this on the
basis of IR studies of NO molecules adsorbed on these catalysts. Using
Mössbauer Emission Spectroscopy (MES), Topsoe et al (1981) and Wivel et
al (1984) assigned a specific Co signal to a so–called ‘Co–Mo–S’ interaction
phase different from Co9S8. Wivel et al (1981) observed a linear correlation
between the amount of co-promoter ions present in this ‘Co–Mo–S’ phase and
thiophene HDS activity. Later, the promoter atoms were found present at the
edges of MoS2 crystals was obtained by Analytical Electron Microscopy
(Sorensen et al 1985). The term ‘Co–Mo–S’ structures has been used
exclusively to describe the local structures at the edges of MoS2 slabs
involving Co, Mo, and S atoms. It was commonly proposed that the
catalytically active sites in hydrotreating catalyst are the molybdenum atoms
at the surfaces of the MoS2 crystallites, with at least one sulfur vacancy at a
site to allow the reacting molecule to bond chemically to the molybdenum
atom.
1.10 EFFECT OF MoO3 LOADING ON HYDROTREATING
PROCESS
It has been understood from the literature that an optimum level of
MoO3 loading is required for a particular support. The catalytic activity has
been found to increase as the MoO3 loading increased. Many reports are
available in this context. Muralidhar et al (1984 and 2000) studied the effect
of Mo loading on metal oxides supported hydrotreating catalyst by varying
the percentage of MoO3 from 2 to 16 wt.%. It is found that no XRD pattern

22
resembling that of MoO3 up to 14 wt. % and 8 wt.% of MoO3 on ZrO2-TiO2,
and MoS2/TiO2–Al2O3 (1:1) respectively, indicating that MoO3 is well
dispersed. In another study by Landau et al (2001), on AlMCM-41 supported
CoO-MoO3 only at 45 wt. % formation of MoO3 was observed when the
catalysts was synthesised by ultrasonic. However, on H-Al-MCM-41, up to
24 wt.% of Mo loading no crystallites due to MoO3 was observed
(Sardhar Basha et al 2006). Over SBA-15 and Al-SBA-15 supported NiMo
catalysts 8 wt.% MoO3 has been reported to be the optimum for better
dispersion. (Muthu Kumaran et al 2007). However, in another study it is
reported that until 17 wt.% of molybdenum, there is complete dispersion of
MoO3 without any crystalline species on SBA-15 support (Sundaramurthy et
al 2008b).
1.11 ROLE OF ADDITIVES IN HYDROTREATING
To increase activity and stability of the hydrotreating catalysts,
elements such as fluorine, phosphorous and boron are used as additives to the
support. Fluorinated hydroprocessing catalysts are used in refineries for
processing lubricant oil (Stanulonis et al 1982). With increasing demand for
hydrotreating, fluorine is widely studied as an additive in hydrotreating and
hydrocracking catalysts containing Mo and W supported on alumina, silica–
alumina, and zeolites. The enhancement of the activity by fluorine addition
has been attributed to high acidity, good dispersion, and high chemisorption
capacity for hydrogen. In most cases, fluorination of the catalyst is done by
impregnating the support with a fluoride salt, like NH4F (Ramirez et al 1990
and Lewandowski et al 1997). When fluorine is incorporated in CoMo-Al2O3
an increase in the rate of cyclohexane hydrogenation and decrease in the rate
of cyclohexene isomerization was reported (Boorman et al 1987).
Jiratova and Kraus (1986) reported that the addition of 3 wt.% F to
a raw CoMo-Al2O3 catalyst increased substantially both hydrogenation and

23
isomerization of cyclohexene. Ramirez et al (1990) showed that fluorine
incorporation enhanced appreciably and moderately the hydrogenation
activity of Mo and CoMo catalysts, respectively; this increase being only
roughly related to Mo dispersion in both types of catalysts.
The changes induced by fluorination on the surface distribution of
the oxidic tungsten species led to improvements in the sulfidation, significant
changes in the morphology of the WS2 structures, and increased activity for
both gas oil HDS and pyridine HDN. Moreover, it was found that the
promotion induced by fluoride was relatively higher for HDN than for HDS
(Benitez et al 1995).
The order of fluorine addition changes the dispersion of the nickel
and the tungsten species, incorporation of nickel with the tungsten edge sites,
and consequently the HDS activity of the catalysts. The order of fluorine
addition does not affect the catalyst surface area; neither does it affect the
amount of fluorine retained in the catalyst after calcination. The dispersion of
the metal species and incorporation of nickel with the tungsten edge sites also
increase in the order of W-Ni-F> W-F-Ni> F-W-Ni. Fluorine addition
enhances the thiophene HDS activity in the order W-Ni-F> W-F-Ni> F-W-Ni.
The HDS activity of NiW/A12O3 is enhanced more by the repartition of the
metal species than by partial solubilization of alumina in the fluorine-addition
step (Kwak and Moon 1999).
Guemez et al (1995) studied the effect of fluorine addition to
molybdena-alumina catalyst doubly promoted by Zn and Co for
hydrodenitrogenation of quinoline and found that HDN activity increased by
the addition of fluorine and attained maximum in the region 1.0-1.3 wt.% F.
The increased activity has been attributed to the increase in the number of
catalytically active metal sulfide sites. Marques et al (2011) studied the effect
of addition of Fluorine on acidity of alumina supported Ni-Mo catalyst.

24
Phosphorous, as phosphate, mainly alters the acid–base properties
of the support, which is directly correlated to the active phase dispersion
(Lewis et al 1992). Therefore phosphorus is frequently used to modify the
activity of hydrotreating catalyst. (Eijsbouts et al 1991, Lewis et al 1992). The
addition of phosphorus has been found to increase the formation of
polymolybdate species which are more active for hydrotreating reaction. It is
also observed that reducibility of these species is increased with phosphorous
loading. However, at high phosphorous loading the formation of crystalline
MoO3 species is reported (Maity et al 2008).
Addition of boron and phosphorous caused the formation of
extremely strong acid sites on the catalyst. Though the addition of boron and
phosphorous to NiMo/Al2O3 catalyst did not show any significant effect on
sulfur conversion, the HDN and HDS activities of the catalyst containing
1.7 wt.% boron and the one containing 2.7 wt.% phosphorous are comparable
to those of a commercial hydrotreating catalyst (Ferdous et al 2004).
It is reported in the literature that the addition of phosphorous to
NiMo/ -Al2O3-carbide catalyst has enhanced the dispersion of
Ni-Mo carbide phase, changed the nature of surface active sites and increased
HDN activity through accelerated C–N bond breaking, compared to the
NiMo/ -Al2O3 catalysts. HDN activity of phosphorous modified NiMo nitride
catalyst was enhanced by addition of phosphorous, with maximum activity at
1.6 wt.% of phosphorous loading. Also the high activity of phosphorous
modified catalyst has been attributed due to increased acidity of the catalyst
(Sundaramurthy et al 2007, 2008a).

25
1.12 SUPPORT EFFECT IN HYDROTREATING
Alumina has been widely used to as a support for industrial
hydrotreating catalyst. Alumina supported CoMo and NiMo hydrotreating
catalysts have been investigated extensively and the literature is exhaustive.
There has been considerable interest to study the use of other supports such as
silica, carbon, mixed metal oxides, zeolites and mesoporous materials. These
supports have been used either alone or mixed with alumina. The effect of
these materials as support for Ni-Mo and Co-Mo is discussed with reference
to hydrotreating process. Thus, it is appropriate to discuss in the following
section, the influence of supports on the surface area, dispersion of active
phases, the interaction of the active component with the support, and the
degree of sulfidation as these factors significantly affect the HDN activity and
selectivity.
1.12.1 Carbon
Carbon has been considered as a good support with regard to the
interaction of metals with support for hydrotreating catalyst. (Topsoe et al
1986, Vissers et al 1987, Boorman and Chong 1992). Carbon supported
Ni-Mo catalyst is less likely to be poisoned by basic nitrogen-containing
compounds during HDS (Hillerova et al 1991) and more resistant to coke
formation (Boorman and Chong 1992). The high activity of carbon-supported
tungsten oxide catalyst is attributed to the weak interaction of the active
metals with the support (Topsoe et al 1986) and complete sulfidation of
tungsten oxide (Louwers and Prins 1993).
Recently, mesoporous carbon substituted hydrotreating catalysts are
used as support for Ni-Mo catalyst. This catalyst has been reported to be
better than the commercial alumina supported catalysts. Hussain and Ihm

26
(2007) studied the mesoporous carbon supported Mo catalyst as a function of
molybdenum loading with respective pore structure of the carbon material and
found that CMK-3 supported Mo catalysts are superior to CMK-1 and Al2O3
supported catalysts due to its larger pore size and higher acidic functional
groups.
To improve the low density and low mechanical strength of
activated carbon, it was combined with alumina support. Liu et al (2011)
studied the alumina activated carbon (AAC) composite as the support for
NiMo HDS catalyst and found that AAC supported catalyst is better than
NiMo supported individually on Al2O3 and activated carbon in terms of
activity and stability . The outstanding activity of NiMo/AAC is attributed to
the mesoporous structure of the AAC support and good dispersion of Ni and
Mo species on the AAC.
1.12.2 Oxides as Support
Silica was often used as support because of its inert character and
its weak interaction with the sulfided phase which permits a better
understanding of the sulfided Mo phase on the support surface. NiMo/SiO2
and NiW/SiO2 catalysts were prepared by impregnation of the support with
dimethylformamide solution of molybdophosphoric and tungstophosphoric
acids and found to be more active for the HDN of a model mixture containing
pyridine (2000 ppm N) and thiophene (6000 ppm S) in cyclohexane than the
corresponding NiMo/Al2O3 and NiW/Al2O3 catalysts (Rives et al 2001). This
has been attributed to the weak interaction of the heteropoly acids with SiO2
than with Al2O3. This has also favored the formation of the more active type
II structure upon sulfidation of the former. However, there are also reports
stating that the activity is low when silica alone is used as support than when
SiO2 is used in combination with Al2O3 for hydrotreating (Massoth et al

27
1984). The increased activity in mixed oxide catalyst is due to the formation
of uniform monolayer with fine dispersion of active metal.
Oxides like titania and zirconia have been studied as supports for
hydrotreating catalyst. The high activity of titania supported catalyst is due to
the uniform distribution of hydroxyl groups on the surface which can provide
a homogenous surface for the adsorption of molybdate anions with the
tetrahedrally coordinated Ti4+. On the other hand, on Al2O3, A13+ ions are
octahedrally and tetrahedrally coordinated and hydroxyl groups are ordered in
parallel rows (Knozinger and Ratnasamy 1978). Such preferential
arrangement in rows requires an arrangement of the molybdate anions during
the impregnation process, which leads to molybdenum trioxide even at sub
monolayer loadings. These observations are in agreement with XPS data
recorded on the oxidic precursors by Nag (1987) and Caceres et al (1990).
The reducibility of molybdate species depends on the support. Molybdenum
on Mo/TiO2 is more easily reduced than on Mo/Al2O3; this result was
confirmed using high pressure and high temperature ESR technique. Kohno et
al (1986)
ZrO2 has been attracting attention of researchers as catalyst as and
as support due to its high thermal stability, extreme hardness, and high
specific mass of zirconia. It has both acidic and basic properties. Nag (1987)
reported that Mo oxidic species on ZrO2 are homogenously distributed and
chemically coordinated better than on Al2O3. According to Zaki et al (1986),
there cannot be the formation of monolayer of Mo on the ZrO2 and MoO3 can
only form aggregates. Large differences in CO hydrogenation, observed by
Mauchausse et al (1998) for Mo/ZrO2 and Mo/CeO2, compared to Mo/Al2O3
The variations in the density of the active sites depending on residual
Mo-O-Al bonds is responsible for the hydrogenation properties. This
property has been exploited for the hydrogenation of pyridine and

28
hydrodenitrogenation of piperidine (Portefaix et al 1989) on transition metals
sulfide hydrotreating catalyst. The high activity of Mo/ZrO2 has been
attributed to the increase in the number of active sites Mauge et al (1991).
However, pure titania and zirconia supports generally have low surface area
and porosity; their commercial applications are limited due to lack of thermal
stability and mechanical properties. With an aim to overcome these
disadvantages mixed oxides of these materials with -Al2O3 have been used as
supports to take advantage of favorable characteristics of both the systems.
The Hydrocracking catalyst is bifunctional, having both hydro-
dehydrogenation and acidic function. The hydro-dehydrogenation component
is usually a sulfide phase, while the acidic component is generally a zeolite or
an amorphous silica alumina (ASA). To obtain a good selectivity towards gas
oil and kerosene, the hydrocracking catalyst should be moderate and
significantly lower than that of zeolites. Nevertheless, too weak acidic
properties lead to a catalyst of low activity. Consequently, balanced acidity is
crucial to obtain high conversion and appropriate selectivity. The use of
oxides presenting moderate acidic properties as ASA is preferred to maximize
middle distillate products.
The influence of chelating agents on the hydrodesulfurization
(HDS) activity of -Al2O3 and SiO2-Al2O3 supported CoMo catalysts was
studied by Al-Dalama and Stanislaus (2006) and found that the addition of a
complexing agent (EDTA or nitriloacetic acid (NTA) to the impregnation
solution resulted in a remarkable improvement in the HDS activity of both
-Al2O3 and SiO2-Al2O3 supported CoMo catalyst.The HDS reaction of
dibenzothiophene was studied over Alumina-Silica-Alumina (ASA) supported
CoMo hydrotreating catalyst (Hensen et al 2007) and pointed out that the
surface acidity play a prime role. The high activity of ASA supported catalyst

29
is due to preferential adsorption of the dibenzothiophene molecule on the
active Co-Mo-S sites.
The effect of preparation methods of supports on hydroprocessing
of Maya heavy crude was studied over Al2O3-TiO2 binary oxide. The catalyst
supported on binary oxide was found to exhibit high activity and stability with
time-on-stream (Maity et al 2006).
Srinivas et al (1998) and Muralidhar et al (2003) have studied the
activity of MoO3 and WO3 catalysts supported on Al2O3-TiO2 oxide, pure
TiO2 and pure Al2O3 for HDS of thiophene. The catalyst supported on
Al2O3-TiO2 (1:1) showed 5 times higher activity than Al2O3-supported
catalysts and 2 times higher than TiO2 supported catalysts. From the low-
temperature oxygen chemisorption and temperature-programmed reduction
results, it was found that the presence of large number of easily reducible
MoO3 species on binary oxide compared with Al2O3-supported catalyst.
Pophal et al (1997) also found that alumina-titania supported catalysts were
highly efficient for hydrodesulfurization of 4,6-dimethyldibenzothiophene.
Maity et al (2003a and 2003b) have observed that catalyst supported on
Al2O3-TiO2 showed high activity compared with catalysts supported on Al2O3
and Al2O3-SiO2
The beneficial effect of Al2O3, zeolite mixed-Al2O3 and Al2O3-
TiO2, ZrO2-Al2O3 was discussed for the deep hydrodesulfurization of
4, 6-dimethyl dibenzothiophene (Bej et al 2004) and reported that removal of
sulfur atom was easier on mixed oxide supported catalyst than the others.
Titania-alumina of various compositions (atomic ratios: 1/9, 1/1
and 9/1) were prepared from the sulfates by co precipitation with aqueous
ammonia or urea. A maximum acidity was measured for TiO2-Al2O3 (1/9)
prepared with ammonia while no acid sites were found for TiO2-Al2O3 (1/9)

30
prepared with urea. Basic property appeared only for TiO2-Al2O3 (1/1)
prepared with ammonia when it was exposed to water vapor. The maximum
activities were observed on TiO2-Al2O3 (1/9 and 1/1) prepared with ammonia
for the isomerization of 1-butene and on TiO2-Al2O3 (1/9) prepared with
ammonia for the dehydration of 2-butanol, respectively (Rodenas et al 1981).
Zhang et al (2010) prepared Al2O3–ZrO2 composite supported
NiMo catalysts with various ZrO2 content and tested for hydrotreating. From
the results it is found that the composite supports prepared by the chemical
precipitation method existed as amorphous phase in the samples with
insufficient amount of ZrO2, and further incorporation of ZrO2 into supports
provided a better dispersion of NiMo species and easy reduction. The Lewis
acid sites of catalysts increased significantly by the introduction of ZrO2 into
alumina. The results showed that the ZrO2–Al2O3 supported NiMo catalysts
with suitable ZrO2 content exhibited much higher catalytic activity than Al2O3
supported catalyst.
1.12.3 Mesoporous MCM-41 as Support
Zeolites and related molecular sieves are largely being used for
various catalytic applications because of their unique structural and textural
properties. However, the performance of the zeolitic systems is limited by
diffusional limitations of bigger molecules for catalytic conversions within
the zeolitic channels. Hydrodenitrogenation being slower than HDS, may not
be affected by diffusional limitations, in which case variations in pore size
within a reasonable range may not affect the catalytic activity. However, with
high molecular weight nitrogen containing compounds, diffusional limitations
become promising with decreasing pore size (Katzer and Sivasubramanian
1979).

31
The major breakthrough came when Mobil reported the successful
synthesis of mesoporous M41S materials in 1992 (Beck et al 1992, Kresge
et al 1992). Since then, hexagonal mesoporous silica [HMS] (Tanev and
Pinnavaia 1995), mesoporous aluminophosphate (Zhao et al 1997), different
types of Mesoporous metallophosphates (Jones et al 2000) and other
mesoporous molecular sieves such as FSM-16 (Inagaki et al 1993), KIT
(Ryoo et al 1996) and so forth, were also synthesized successively. Among
the other members of the M41S family, the synthesis and catalytic
applications of MCM-41 type mesoporous materials have been investigated
extensively. This material exhibits hexagonal array of unidimensional
mesopores, the diameter of which can be tuned in the range between 20 and
100 Å with a narrow pore size distribution, through the proper choice of
surfactants as the template, auxiliary chemicals, and reaction conditions. The
unique properties of MCM-41 are exploited for the transformation of bulky
molecules. The large pore channels may reduce pore diffusion limitation and
allow the effective use of the active sites on the surface of the pore wall by the
reactants (Ying et al 1999).
After the discovery of MCM-41, this material has been used as a
carrier for mild hydrocracking (MHC). The MHC performance of a
NiMo/MCM-41 catalyst was compared with that of amorphous silica-alumina
and ultrastable low unit cell size Y zeolites (USY) having same Ni and Mo
content. From the point of the acidity of the carrier, USY zeolite showed the
highest amount of Bronsted acidity, most of the sites having medium-strong
acid strength. The acidity of MCM-41 was similar to that of amorphous silica-
alumina, both in number and acid strength distribution. Moreover, most of the
Bronsted acid sites in mesoporous MCM-41 and silica-alumina carriers are of
weak-medium strength, as it is required for producing diesel in MHC
operation. However, it should be taken into account that the acid
characteristics of the supports are modified when supporting the metals, and

32
in this particular case, the Mo strongly interacts with the acid sites, making
the strongest acid sites disappear from MCM-41 and amorphous silica-
alumina samples (Corma et al 1995). In case of MHC, selectivity to middle
distillates was equally important as the total conversion. The product
distribution showed that the NiMo/MCM-41 catalyst could produce the
lowest amount of gases and consequently the highest amount of gasoline.
Since, the acidities are very similar on the two catalysts; the differences in
selectivities were related to the regularity, size, and dimensionality of the
pores present in MCM-41(Corma et al 1996).
Further SiMCM-41 has been exchanged with proton and used as a
support for Ni-Mo catalyst and tested for HDS of dibenzothiophene (DBT).
Proton exchanged SiMCM-41 performs better than untreated
SiMCM-41.Most of the sodium cations contained in MCM-41 were removed
by proton exchange. This proton exchange has little effect on the structural
and acidic properties of SiMCM-41. It seems that using HNO3 for proton
exchange clears the pore channels of MCM-41 during the ion exchange and
thus the reconstruction of the mesostructure has contributed only to the
increase of the crystallinity (Li et al 2002). Wang et al (2001) reported that
the SiMCM-41 supported Co-Mo or Ni-W sulfides are more active for the
HDS of DBT, than conventional -Al2O3 supported catalyst. This is attributed
to the high surface area and mild acidity of support which lead to high
dispersion of the metal species on the surface. The HDS of vacuum gas oil
was studied over MCM-41 supported NiMo catalysts as a function of shape of
mesoporous material (tubular shaped MCM-41, non-tubular shaped MCM-41)
and compared with alumina and silica supported NiMo catalysts. The results
clearly showed that tubular shaped MCM-41 supported catalyst is better than
the others (Ling et al 2009).

33
The activity of AlMCM-41 supported Co-Mo catalyst was studied
for deep HDS of light cycle oil (LCO). The activity of this catalyst has been
reported to be much higher than commercial -Al2O3. (Kostova et al 2002).
AlMCM-41 supported CoO-MoO3 catalyst at normal metal loading level
(3 wt.% CoO-12 wt.% MoO3) showed less activity in converting DBT than
-Al2O3 supported catalyst. However, at high metal loading it was
substantially more active than -Al2O3 supported catalyst in DBT conversion
(Song and Madhusudan Reddy 1999). It is clearly evident from the results
MCM-41 can be a more suitable support for hydrotreating if proper amount of
CoO and MoO3 are loaded.
The effect of lanthanum addition to the mesoporous
Al-MCM-41 supported catalyst was studied by Song et al (2006), the results
showed higher HDS conversion in the lanthanum modified catalyst than
unmodified catalyst. The increased activity is due to the presence of more
acidic sites created by the addition of lanthanum.
The HDN activity of NiO-MoO3/H-AlMCM-41 catalysts as a
function of effect of molybdenum loading was reported by Sardhar Basha
et al (2006). The results showed that the impregnation of MoO3 up to
24 wt.% over H-AlMCMA-41 resulted in fine dispersion and high activity
towards HDN. It is also found that the protonation of the support remarkably
affected the activity of the catalysts. The same authors (2009) studied the
effect of order of impregnation of active and promoter metals over high
surface area Al-MCM-41 support and concluded that the catalyst prepared by
reverse order impregnation showed higher catalytic activity than prepared by
the other methods of impregnation. The effect of addition of TiO2 into
MoP/MCM-41 catalyst was studied towards the HDN of quinoline and
decahydroquinoline and found that the catalyst containing 5 wt.% of TiO2
showed maximum hydrodenitrogenation activity (Duan et al 2010).

34
1.12.4 Mesoporous SBA-15 as Support
Though MCM-41 type material has high surface area, because of its
small wall thickness the hydrothermal stability is much less. Due to this
reason, there is serious limitation to its practical application (Kooyman et al
2003). The discovery of SBA-15 has opened new possibilities for the
preparation of efficient catalysts for hydrotreating. SBA-15 presents several
advantages such as high surface area (800 m2g-1) and a hexagonal
arrangement of mesopores with sizes from 4 to 30 nm. Pore wall thicknesses
of around 3–6 nm in SBA-15 is responsible for high thermal and
hydrothermal stability (Zhao et al 1998a and 1998b). In recent years different
research groups have employed SBA-15 as a promising support for
hydrotreating catalysts. Vradman et al (2003) reported high activities for HDS
and hydrogenation using Ni-W-S/SBA-15 as catalyst compare to commercial
-Al2O3 supported catalyst.
Ni and Fe supported on SBA-15 catalyst showed high
hydrocracking activity because of the good hydrogenation properties of nickel
Byambajav and Ohtsuka (2003). AlSBA-16 has been reported to be an
excellent support for NiMo catalysts in the conversion of the refractory sulfur
compound (Klimova et al 2004). It is inferred from the detailed study of
method of incorporation of aluminium in to SBA-16 that, the post-synthetic
alumination of SBA-16 by reaction with AlCl3 or Al (i-PrO)3 has number of
advantages than with sodium aluminate or SBA-16 synthesized by direct
method. It is concluded that grafting with AlCl3 or Al(i-PrO)3 improves the
activity of SBA-15; this can be attributed to good dispersion of Ni and Mo
active phases and to the bifunctional character of these catalyst namely, to the
participation of both types of sites, coordinatively unsaturated sites of NiMoS
active phase and Bronsted acids sites of the support. Carlos Amezcua et al
(2005) studied the addition of TiO2 to NiMo supported SBA-16 catalyst by

35
different methods (post-synthetic, chemical grafting and incipient wetness
impregnation) and found that, the best dispersion of TiO2 in the SBA-16 pore
channels was obtained by grafting methods.
The activity of SBA-15 supported Mo, CoMo and NiMo for
thiophene hydrodesulfurization and cyclohexene hydrogenation reaction was
compared with that of alumina Murali Dhar et al (2005). Further (Shelu et al
2008) the effect of addition of ZrO2 on SBA-15 was studied for hydrotreating.
A significant increase in the activity was reported up to 25 wt.%. A
comparative study indicated that ZrO2-SBA-15 and TiO2-SBA-15 supported
CoMo are better than pure SBA-15 supported catalyst for
hydrodesulfurization.
A systematic study of effect of variation of molybdenum and
promoter content and catalytic functionalities over Al-SBA-15 support was
made and reported that molybdenum is dispersed well up to 8 wt.% and
maximum activity at 8 wt.% (Muthu Kumaran et al 2007). The effect of
MoO3 loading and the addition of boron and aluminium over SBA-15 and its
hydrotreating activity towards the gas oil were studied by Sundaramurthy et al
(2008b). 17 wt.% of molybdenum loaded catalyst was found to have fine
dispersion and also the incorporation of Boron and Aluminium increases the
activity. The activity of all the catalyst found to be higher than that of
commercial Al2O3 supported catalysts.
A detailed systematic study was made on Mo, CoMo and NiMo
supported on Al-SBA-15 with various Si/Al ratios for HDS (Klimova et al
2008). The activity of Mo and NiMo/SBA-15 catalysts found to increase with
Al incorporation into the support.
The effect of Si/Al ratio and activity of Al-SBA-15 supported
hydrotreating catalysts was studied by Muthu Kumaran et al (2006) and found

36
that the molybdenum dispersion and anion vacancies and catalytic activities
are significantly influenced by the aluminium content. The result concluded
that the anion vacancy on molybdenum sulfide phase as well as its dispersion
decreases with increasing Si/Al ratio.
There are numerous studies reported the use of various supports
with and without modifications for hydrotreating catalyst. SBA-15 support is
found to be a suitable support towards hydrotreating process with a main
focus on the hydrodesulfurization activity of the model compound at high
pressure condition. However, studies on hydrodenitrogenation using SBA-15
supported NiMo catalyst is very much limited and not explored fully. Hence,
the studies on NiMo supported on Si-SBA-15 and modified SBA-15 is
focused in depth with relevance to hydrodenitrogenation of some nitrogen
containing compounds as model compounds.
1.13 SCOPE OF THE PRESENT STUDY
The work described in this thesis is principally aimed at studying
SBA-15 supported NiO-MoO3 catalysts with respect to their physical and
chemical properties and their activity towards HDN of methylcyclohexylamine
(MCHA). The following are the main objectives:
Synthesis of mesoporous Si-SBA-15 and Al-SBA-15 with
varying Si/Al ratio values (Si/Al = 10, 20, 30 and 40).
Synthesis of mesoporous NiO-MoO3 catalysts supported on
Al-SBA-15 by wet impregnation of catalytic components such
as NiO and MoO3.
Synthesis of series of catalysts with various weight % of
MoO3 (8, 12, 18 and 24 wt.%) at constant weight % of
NiO (3 wt.%) on Al-SBA-15(10) support.

37
Synthesis of catalysts by changing the order of
impregnation of the active components NiO and MoO3
(normal, reverse and co-impregnation).
Synthesis of series of NiO-MoO3 catalysts supported on
Al-SBA-15 with varying Si/Al ratio values (Si/Al = 10, 20, 30
and 40).
Synthesis of AlMCM-41 and - Al2O3 supports.
Synthesis of mesoporous AlMCM-41 and -Al2O3 supported
NiO-MoO3 by wet impregnation of catalytic components such
as NiO (3 wt.%) and MoO3 catalysts (12 wt.%).
Synthesis of fluorine and phosphorus modified SBA-15
supports.
Synthesis of mesoporous P-SBA-15 and F-SBA-15 supported
NiO-MoO3 by wet impregnation of catalytic components such
as NiO (3 wt.%) and MoO3 (12 wt.%) catalysts.
Characterization of the synthesized NiO-MoO3/SBA-15
catalysts using XRD, Nitrogen adsorption-desorption
measurements, DRS, TGA, ICP-OES, 27Al MAS NMR,31P MAS NMR, SEM, TEM, FT-IR, TPD, TPR and RAMAN
techniques.
Evaluation of effect of sulfiding agents on the
hydrodenitrogenation of methylcyclohexylamine.
Evaluation of the catalytic activity of all the catalysts for
hydrodenitrogenation of methylcyclohexylamine.
Correlation of catalytic activity with their physico-chemical
properties.