center for drug evaluation and [email protected]. food and drug administration...
TRANSCRIPT

CENTER FOR DRUG EVALUATION AND RESEARCH
APPLICATION NUMBER:
211280Orig1s000
OTHER REVIEW(S)

1
****Pre-decisional Agency Information**** Memorandum Date: October 02, 2019 To: Nicholas Kozauer, M.D.
Division of Neurology Products (DNP) E. Andrew Papanastasiou, Regulatory Project Manager, DNP
Tracy Peters, Associate Director for Labeling, DNP
From: Dhara Shah, Regulatory Review Officer Office of Prescription Drug Promotion (OPDP) CC: Aline Moukhtara, Team Leader, OPDP Subject: OPDP Labeling Comments for REYVOW (lasmiditan) tablets, for oral use,
[controlled substance schedule pending] NDA: 211280
In response to the DNP consult request dated November 14, 2018, OPDP has reviewed the proposed product labeling (PI), Medication Guide, and carton and container labeling for the original NDA submissions for REYVOW (lasmiditan) tablets, for oral use, [controlled substance schedule pending]. PI and Medication Guide: OPDP’s comments on the proposed labeling are based on the draft PI received by electronic mail from DNP (E. Andrew Papanastasiou) on September 18, 2019, and are provided below. A combined OPDP and Division of Medical Policy Programs (DMPP) review was completed, and comments on the proposed Medication Guide was sent under separate cover on September 25, 2019. Carton and Container Labeling: OPDP has reviewed the attached proposed carton and container labeling submitted by the Sponsor to the electronic document room on September 17, 2019, and we do not have any comments. Thank you for your consult. If you have any questions, please contact Dhara Shah at (240) 402-2859 or [email protected].
FOOD AND DRUG ADMINISTRATION Center for Drug Evaluation and Research Office of Prescription Drug Promotion
Reference ID: 4500535
16 Pages of Draft Labeling have been Withheld in Full as B4(CCI/TS) Immediately Following this Page

--------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically. Following this are manifestations of any and allelectronic signatures for this electronic record.--------------------------------------------------------------------------------------------/s/------------------------------------------------------------
DHARA SHAH10/02/2019 01:17:07 PM
Signature Page 1 of 1
Reference ID: 4500535

Page 1 of 6
Department of Health and Human Services
Food and Drug Administration Center for Drug Evaluation and Research | Office of Surveillance and Epidemiology (OSE)
Epidemiology: ARIA Sufficiency Templates Version: 2018-01-24
Date: September 30, 2019
Reviewer: Catherine Callahan, PhD, MA Division of Epidemiology I
Team Leader: Kira Leishear, PhD, MS Division of Epidemiology I
Division Deputy Director: Sukhminder K. Sandhu, PhD, MS, MPH Division of Epidemiology I
Subject: ARIA Sufficiency Memo for Pregnancy Safety Concerns
Drug Name: REYVOW (Lasmiditan)
Application Type/Number: NDA 0211280
Applicant/sponsor: Eli Lilly and Company
OSE RCM #: 2019-1875
Reference ID: 4499125

Page 2 of 6
Expedited ARIA Sufficiency Template for Pregnancy Safety Concerns
1. BACKGROUND INFORMATION
1.1. Medical Product Lasmiditan is a new molecular entity that has no currently approved uses, it is a high-affinity, centrally penetrant, selective 5-hydroxytryptamine (serotonin) 1F (5-HT1F) receptor agonist with the proposed indication of treatment of acute migraine with or without aura. The proposed label’s warnings and precautions section has warnings for driving impairment;
dizziness; medication overuse headache; serotonin syndrome, . There are no Risk Evaluation and Mitigation Strategies
(REMS) planned for lasmiditan.
The proposed dosages of lasmiditan are 50 mg, 100 mg or 200 mg taken once daily, as needed, the maximum dose should not exceed 200 mg in 24 hours. 1.2. Describe the Safety Concern The Division of Neurology Products (DNP) requested that the Division of Epidemiology (DEPI) assess the sufficiency of ARIA for broad-based signal detection studies of lasmiditan during pregnancy. Safety during pregnancy due to drug exposure is a concern for women who are pregnant or of childbearing potential. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.1 In rat and rabbit studies, maternal exposure to lasmiditan was associated with embryofetal toxicity including decreased fetal weight and skeletal variations, which occurred at dosages causing maternal toxicity and were at 2 to 55-fold higher than the proposed human exposure. Women who were pregnant were excluded from lasmiditan clinical studies and birth control during participation was required for women of reproductive potential. Twenty-two women exposed to lasmiditan became pregnant during lasmiditan clinical studies. Nine of these 22 patients were last
Reference ID: 4499125
(b) (4)
(b) (4)
(b) (4) (b) (4)
(b) (4)
(b) (4)

Page 3 of 6
administered lasmiditan between 8 to 173 days prior to the last menstrual period (LMP) or estimated date of conception. Three of the nine pregnancies administered lasmiditan prior to pregnancy reported adverse pregnancy outcomes (two spontaneous abortion and one premature rupture of membranes). The subject with premature rupture of membranes underwent a caesarean section at 36 weeks and had a livebirth with no further complications. Lasmiditan has a mean half-life of 3.8 hours. Of the 13 pregnancies exposed to lasmiditan during the first trimester, there were five normal healthy infants (gestational ages between 36 and 38 weeks), three spontaneous abortions (gestational ages were 3.5, 6.5, and 7.5 weeks), one elective abortion (unknown reason), one preterm birth (gestational age of 31 weeks), and three are ongoing.
In the proposed labeling, as of September 30, 2019, lasmiditan will not be contraindicated in pregnant women and women of reproductive age will not be required to use contraception. Section 8.1 of the proposed labeling states: Risk Summary There are no adequate data on the developmental risk associated with the use of REYVOW in pregnant women.
In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations Disease-Associated Maternal and/or Embryo/Fetal Risk Published data have suggested that women with migraine may be at increased risk of preeclampsia and gestational hypertension during pregnancy.
Data
Reference ID: 4499125
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)


Page 5 of 6
☐ Exposures ☐ Outcomes ☐ Covariates ☒ Analytical Tools For any checked boxes above, please describe briefly:
Analytical Tools: ARIA analytic tools are not sufficient to assess the regulatory question of interest because data mining methods have not been tested for birth defects and other pregnancy outcomes. Because broad-based signal detection is not currently available, other parameters were not assessed.
2.5. Please include the proposed PMR language in the approval letter.
The Division of Neurology Products requests two PMRs related to pregnancy outcomes. As of September 6, 2019, the proposed PMR language, for these are:
Conduct prospective pregnancy exposure registry cohort analyses in the United States that compare the maternal, fetal, and infant outcomes of women with migraine exposed to Reyvow during pregnancy with two unexposed control populations: one consisting of women with migraine who have not been exposed to Reyvow before or during pregnancy and the other consisting of women without migraine. The registry will identify and record pregnancy complications, major and minor congenital malformations, spontaneous abortions, stillbirths, elective terminations, preterm births, small-for-gestational-age births, and any other adverse outcomes, including postnatal growth and development. Outcomes will be assessed throughout pregnancy. Infant outcomes, including effects on postnatal growth and development, will be assessed through at least the first year of life. Conduct a pregnancy outcomes study using a different study design than provided for in PMR XXXX-X (for example, a retrospective cohort study using claims or electronic medical record data or a case control study) to assess major congenital malformations, spontaneous abortions, stillbirths, preterm births, and small-for-gestational-age births in women exposed to Reyvow during pregnancy compared to an unexposed control population.
3. References
Reference ID: 4499125

Page 6 of 6
1. Dinatale M. Division of Pediatric and Maternal Health, FDA. The pregnancy and lactation labeling rule (PLLR). https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/PediatricAdvisoryCommittee/UCM520454.pdf. Accessed October 11, 2018.
Reference ID: 4499125

--------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically. Following this are manifestations of any and allelectronic signatures for this electronic record.--------------------------------------------------------------------------------------------/s/------------------------------------------------------------
CATHERINE L CALLAHAN09/30/2019 12:33:58 PM
KIRA N LEISHEAR09/30/2019 12:54:03 PM
SUKHMINDER K SANDHU09/30/2019 01:49:31 PM
JUDITH W ZANDER09/30/2019 03:15:50 PM
MICHAEL D NGUYEN09/30/2019 03:23:24 PM
ROBERT BALL09/30/2019 03:30:29 PM
Signature Page 1 of 1
Reference ID: 4499125

Department of Health and Human Services
Public Health Service
Food and Drug Administration
Center for Drug Evaluation and Research
Office of Medical Policy
PATIENT LABELING REVIEW
Date:
September 25, 2019
To:
William Dunn, MD
Director
Division of Neurology Products (DNP)
Through:
LaShawn Griffiths, MSHS-PH, BSN, RN
Associate Director for Patient Labeling
Division of Medical Policy Programs (DMPP)
From: Sharon W. Williams, MSN, BSN, RN
Senior Patient Labeling Reviewer
Division of Medical Policy Programs (DMPP)
Dhara Shah, PharmD, RAC
Regulatory Review Officer
Office of Prescription Drug Promotion (OPDP)
Subject:
Review of Patient Labeling: Medication Guide (MG)
Drug Name (established
name):
REYVOW (lasmiditan)
Dosage Form and Route: tablets, for oral use
Application
Type/Number:
Applicant:
NDA 211280
Eli Lilly and Company
Reference ID: 4497062

1 INTRODUCTION
On October 10, 2018, Eli Lilly and Company, Inc. submitted for the Agency’s
review an Orignal New Drug Application (NDA) for REYVOW (lasmiditan), tablets
for oral use. The purpose of the submission is to seek approval for marketing
REYVOW (lasmiditan), tablets for oral use for the acute treatment of migraine with
or without aura in adults.
This collaborative review is written by the Division of Medical Policy Programs
(DMPP) and the Office of Prescription Drug Promotion (OPDP) in response to a
request by the Division of Neurology Products (DNP) on October 25, 2018 for
DMPP and OPDP respectively to review the Applicant’s proposed MG for
REYVOW (lasmiditan) tablets, for oral use.
2 MATERIAL REVIEWED
• Draft REYVOW (lasmiditan) tablets, for oral use MG received on October 10,
2018, and received by DMPP and OPDP on September 18, 2019.
• Draft REYVOW (lasmiditan) tablets, for oral use Prescribing Information (PI)
received on October 10, 2018, revised by the Review Division throughout the
review cycle, and received by DMPP and OPDP on September 18, 2019.
3 REVIEW METHODS
To enhance patient comprehension, materials should be written at a 6th to 8th grade
reading level, and have a reading ease score of at least 60%. A reading ease score of
60% corresponds to an 8th grade reading level.
Additonally, in 2008, the American Society of Consultant Pharmacists Foundation
(ASCP) in collaboration with the American Foundation for the Blind (AFB)
published Guidelines for Prescription Labeling and Consumer Medication
Information for People with Vision Loss. The ASCP and AFB recommended using
fonts such as Verdana, Arial or APHont to make medical information more
accessible for patients with vision loss.
In our collaborative review of the MG we:
• simplified wording and clarified concepts where possible
• ensured that the MG is consistent with the Prescribing Information (PI)
• removed unnecessary or redundant information
• ensured that the MG is free of promotional language or suggested revisions to
ensure that it is free of promotional language
• ensured that the MG meets the Regulations as specified in 21 CFR 208.20
• ensured that the MG meets the criteria as specified in FDA’s Guidance for
Useful Written Consumer Medication Information (published July 2006)
Reference ID: 4497062

4 CONCLUSIONS
The MG is acceptable with our recommended changes.
5 RECOMMENDATIONS
• Please send these comments to the Applicant and copy DMPP and OPDP on the
correspondence.
• Our collaborative review of the MG is appended to this memorandum. Consult
DMPP and OPDP regarding any additional revisions made to the PI to determine
if corresponding revisions need to be made to the MG.
Please let us know if you have any questions.
Reference ID: 4497062
5 Pages of Draft Labeling have been Withheld in Full as B4(CCI/TS) Immediately Following this Page

--------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically. Following this are manifestations of any and allelectronic signatures for this electronic record.--------------------------------------------------------------------------------------------/s/------------------------------------------------------------
SHARON W WILLIAMS09/25/2019 12:36:21 PM
DHARA SHAH09/25/2019 12:48:49 PM
Signature Page 1 of 1
Reference ID: 4497062

Date: September 19, 2019
To: Billy Dunn, MD, Director
Division of Neurology Products
Through: Dominic Chiapperino, PhD, Director
Silvia Calderon, PhD, Senior Pharmacologist
Chad J Reissig, PhD, Supervisory Pharmacologist
Controlled Substance Staff
From: Shalini Bansil, MD, Medical Officer
Edward Hawkins, PhD, Pharmacologist
Controlled Substance Staff
Subject: Product name: Lasmiditan Trade Name: Reyvow
Dosages, formulations, routes: 50 mg and 100 mg, oral tablets with a
maximum dose of 200 mg in a 24 hour period
NDA number: 211280
IND Number: 103420
Indication(s): Acute treatment of migraine with or without aura in adults.
Sponsor: Eli Lilly and Company
PDUFA Goal Date: October 11, 2019
Materials Reviewed: All abuse-related data in Original NDA submission dated October 11,
2018, and subsequent amendments
Statistical review of human abuse potential study (Anna Sun PhD; Office of Biostatistics August 5, 2019)
Table of Contents
I. SUMMARY .........................................................................................................................................2
1. Background.......................................................................................................................................2
2. Conclusions.......................................................................................................................................3
M E M O R A N D U M
Department of Health and Human ServicesFood and Drug Administration
Center for Drug Evaluation and Research
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 2 of 40
3. Recommendations: ...........................................................................................................................4
II. DISCUSSION...................................................................................................................................5
1. Chemistry..........................................................................................................................................5
1.1 Substance Information.................................................................................................................5
1.2 In Vitro Manipulation and Extraction Studies ...........................................................................7
2. Nonclinical Pharmacology................................................................................................................7
2.1 Receptor Binding and Functional Assays .................................................................................8
2.2 Safety Pharmacology/Metabolites...........................................................................................10
2.3 Findings from Safety Pharmacology and Toxicology Studies ................................................14
2.4 Animal Behavioral Studies......................................................................................................16
2.5 Tolerance and Physical Dependence Studies in Animals .......................................................18
3. Clinical Pharmacology....................................................................................................................19
3. 1 Absorption, Distribution, Metabolism, Elimination (ADME) ................................................19
4. Clinical Studies ...............................................................................................................................22
4.1 Human Abuse Potential Studies ..............................................................................................22
4.2 Adverse Event Profile Through all Phases of Development ...................................................30
4.3 Safety Profile ...........................................................................................................................38
4.4 Evidence of Abuse, Misuse and Diversion in Clinical Trials .................................................38
4.5 Tolerance and Physical Dependence Studies in Humans........................................................39
5. Regulatory Issues and Assessment .................................................................................................39
III. References.....................................................................................................................................40
I. SUMMARY
1. BackgroundThis memorandum responds to a consult request by the Division of Neurology Products (DNP), dated
October 18, 2018, to evaluate abuse-related preclinical and clinical data submitted by Eli Lilly and
Company (Sponsor) under NDA 211280 and IND 103420 for Reyvow (lasmiditan hemisuccinate).
Lasmiditan is indicated for the acute treatment of migraine with or without aura in adults.
Lasmiditan is a new molecular entity that is not controlled and has no currently approved
therapeutic uses. Lasmiditan is a 5-hydroxytryptamine (5-HT, serotonin) 1F receptor agonist and
GABAA channel positive allosteric modulator that penetrates the central nervous system (CNS).
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 3 of 40
As a result of these mechanisms of action, lasmiditan was assessed for its abuse potential.
Lasmiditan is rapidly absorbed orally, has good bioavailability, and a half-life of approximately
5 hours in humans. Levels in the brain are nearly 2-fold higher than those in the plasma one hour
after oral administration in the rat, indicating the ability of the drug to cross the blood brain
barrier. Lasmiditan is metabolized into three major metabolites in humans, M7, M8, and M18,
and although M7 binds to GABAA channels, it does not appear to have relevant physiological
activity. Lasmiditan and its metabolites are excreted in the urine, bile, and feces.
A self-administration study conducted by the Sponsor, indicated that lasmiditan is weakly
reinforcing at the highest dose tested. In a drug discrimination study, lasmitidan did not produce
discriminative stimulus effects similar to the benzodiazepine lorazepam. The lack of
generalization to a benzodiapine cue, may be explained by the different mechanism of action of
lasmitidan when compared to that of a benzodiazepine.
In a human abuse potential (HAP) study conducted by the Sponsor, subjects responded to all doses of lasmiditan tested with significantly higher drug liking scores than for placebo, indicating that lasmiditan has abuse potential. In comparison to alprazolam (CIV), subjects reported significantly lower drug liking scores for lasmitidan than for alprazolam, indicating that lasmiditan has less abuse potential than alprazolam.
Phase 1 clinical studies indicate that more abuse-related adverse events (AEs) were reported by subjects receiving lasmitidan than in placebo group. Phase 2 and 3 studies indicate that, at therapeutic doses, lasmiditan displays abuse-related AEs (derealization, euphoric mood, hallucinations) to a greater extent than placebo. However, these AEs occur at a low frequency (about 1%) in lasmiditan treated patients versus placebo (0%)
The Sponsor proposes that lasmiditan be placed under Schedule V of the Controlled Substances Act (CSA) and we agree with that proposal based on our integrated review of all data provided in the NDA.
The following sections summarize the studies conducted by the Sponsor to characterize the
abuse potential of lasmiditan.
2. Conclusions
Lasmiditan is a new molecular entity and is not currently controlled in any schedule of the CSA
In vitro binding and functional studies indicate that lasmiditan is a 5-HT1F receptor agonist and
a GABAA channel positive allosteric modulator
Metabolism studies show that lasmiditan produces three major metabolites, M7, M8, and
M18
o In vitro binding studies indicate that M7 binds to the GABAA channel, however,
electrophysiological studies indicate no activity at the channel
A self-administration study in rats shows that lasmiditan is self-administered to a greater
degree than saline, but not to the same extent as heroin and may be similar to diazepam A drug discrimination study in rats shows that lasmiditan did not generalize to the lorazepam
discriminative cue, suggesting that animals did not recognize the effects of lasmiditan as a benzodiazepine-like drug
Reference ID: 4494251



Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 6 of 40
Table 1: General Chemical Properties of Lasmiditan hemisuccinate
NomenclatureInternational Non-proprietary Name
(INN)Lasmiditan hemisuccinate
Chemical Abstract Number (CAS) 439239-92-6
Chemical Name (IUPAC)
2,4,6-trifluoro-N-(6-(1-methylpiperidine-4-
carbonyl)pyridine-2-yl)benzamide
hemisuccinate
Substance codesCOL-144; LY683974; M026-A13 SUC; LSN683974; YKP
3089
StructureMolecular Formula C19H18F3N3O2∙0.5[C4H6O4]
Molecular mass 436.41 g mol-1
Structure
General PropertiesAppearance white powder
pKa 10.77 (acidic, amide) and 9.04 (basic)
Solubility (25°C)Soluble in water (23.6 mg/mL),
Freely soluble in methanol (61.9 mg/mL)
Melting point 198 °C
Chirality/Stereochemistry achiral
Isomerism No isomerism has been observed
Lasmitidan is synthesized
.
Drug Product Formulation
Reference ID: 4494251
(b) (4)
(b) (4)
(b) (4)

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 7 of 40
REYVOW is the Sponsor’s proposed tradename for the drug product that contains lasmitidan
hemisuccinate (lasmiditan) as the active pharmaceutical ingredient in tablets of 50 mg and 100 mg
quantities. The tablets are designed for oral consumption with a maximum dose of 200 mg in a 24 hour
period. The drug product will be marketed as oval 50 and 100 mg, debossed, film-coated,
immediate-release tablets. The 50 mg tablets will be light grey debossed with “4312” and “L-50” and
the 100 mg tablet will be light purple debossed with “4491” and “L-100.” The amount of lasmiditan in
the drug product and embossed on the tablets is the free base weight of the active ingredient (TABLE 2).
Excipients in the tablet
Lasmiditan tablets contain a series of excipients listed in Table 2 which shows their function in the
formulation. The excipients in lasmiditan do not have a known abuse liability.
Table 2: Composition of Excipients Used to Manufacture Lasmiditan
Component Function Quantity50 mg 100 mg
Lasmiditan hemisuccinate Active 57.82 115.65
Lasmiditan (free base) 50 100Pregelatinized Starch
Croscarmellose sodium
Microcrystalline cellulose
Sodium lauryl sulfate
Magnesium stearate
1.2 In Vitro Manipulation and Extraction Studies
The Sponsor did not conduct in vitro manipulation and extraction studies on the to-be-marketed
formulation. Dissolution and disintegration studies conducted by the Sponsor indicate that lasmitidan is
soluble to at least in water at pHs between 1 and 7.5. In conclusion, lasmitidan is partially
soluble in aqueous conditions and highly soluble in nonpolar solvents (i.e., methyl chloride). Extraction
of lasmitidan would likely be easy to perform.
Reference ID: 4494251
(b) (4)
(b) (4)
(b) (4)
(b) (4)

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 8 of 40
2. Nonclinical Pharmacology
Receptor binding and activity assays can give an indication as to whether or not a substance affects a
receptor pathway that is known to be associated with abuse potential. For active pharmaceutical
ingredients that are CNS active, Sponsors are required to determine if these substances and any major
metabolites, will bind to and have activity at these receptors. The Sponsor conducted binding and
activity studies on lasmitidan. The data, summarized below, indicate that lasmitidan is a 5-HT1F
receptor agonist.
2.1 Receptor Binding and Functional Assays
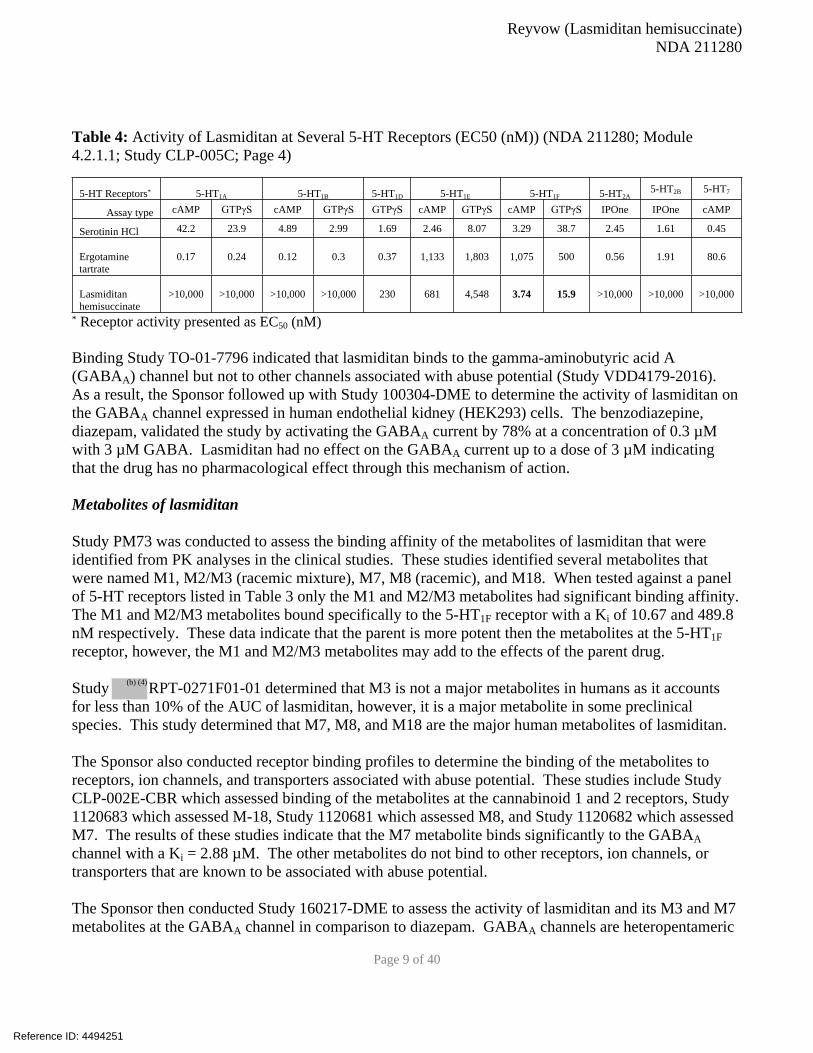
Study CLP-005C was conducted to determine the binding affinity and activity of lasmitidan to 5-HT
receptors in comparison to serotonin, ergotamine, and a panel of triptan analogs known to have activity
at these receptors. The data presented in Tables 3 and 4 indicate that lasmitidan has high affinity for the
5-HT1F receptor with a Ki1 of 1.85 nM. This is 10-fold higher then serotonin and 70-fold higher than
ergotamine. Several different studies were conducted to determine the activity of lasmitidan at various
human 5-HT receptors. Since, for the most part, serotonin receptors are part of the heteromeric G-
protein family of receptors, the study used the radiometric GTPγS and cAMP assays. To measure the
activity of lasmitidan at 5-HT receptors the Sponsor used a different second messanger analysis assay
called the IPOne assay. These studies indicate that lasmiditan is a highly specific agonist of the 5-HT1F
receptor with an EC502 between 3.74 nM (cAMP) and 15.9 nM (GTPγS) depending on the assay. These
data were further supported by in vitro Studies CNS558 and CNS571 which were also conducted on
human 5-HT receptors.
Table 3: Binding Affinity of Lasmiditan at Several 5-HT Receptors (Ki (nM)) (NDA 211280; Module
4.2.1.1; Study CLP-005C; page 3)
5-HT
Receptors* 5-HT1A 5-HT1B 5-HT1D 5-HT1E 5-HT1F 5-HT2A 5-HT2B 5-HT7
Serotonin
HCl0.076 0.53 0.3 6.64 11 87.5 13.7 0.45
Ergotamine
tartrate0.2 0.45 0.49 412 73.4 7.22 11.4 1.91
Lasmiditan
hemisuccinate228 1463 555 366 1.85 >30,000 5977 17,322
* Binding affinity expressed as Ki (nM)
1 Ki – The inhibitory constant is a measure of the binding affinity of a substance to its substrate or receptor
2 EC50 – The half maximal stimulatory concentration of a substance to produce a specific biological function
Reference ID: 4494251
Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 9 of 40
Table 4: Activity of Lasmiditan at Several 5-HT Receptors (EC50 (nM)) (NDA 211280; Module
4.2.1.1; Study CLP-005C; Page 4)
5-HT Receptors* 5-HT1A 5-HT1B 5-HT1D 5-HT1E 5-HT1F 5-HT2A5-HT2B 5-HT7
Assay type cAMP GTPγS cAMP GTPγS GTPγS cAMP GTPγS cAMP GTPγS IPOne IPOne cAMP
Serotinin HCl 42.2 23.9 4.89 2.99 1.69 2.46 8.07 3.29 38.7 2.45 1.61 0.45
Ergotamine
tartrate
0.17 0.24 0.12 0.3 0.37 1,133 1,803 1,075 500 0.56 1.91 80.6
Lasmiditan
hemisuccinate
>10,000 >10,000 >10,000 >10,000 230 681 4,548 3.74 15.9 >10,000 >10,000 >10,000
* Receptor activity presented as EC50 (nM)
Binding Study TO-01-7796 indicated that lasmiditan binds to the gamma-aminobutyric acid A
(GABAA) channel but not to other channels associated with abuse potential (Study VDD4179-2016).
As a result, the Sponsor followed up with Study 100304-DME to determine the activity of lasmiditan on
the GABAA channel expressed in human endothelial kidney (HEK293) cells. The benzodiazepine,
diazepam, validated the study by activating the GABAA current by 78% at a concentration of 0.3 µM
with 3 µM GABA. Lasmiditan had no effect on the GABAA current up to a dose of 3 µM indicating
that the drug has no pharmacological effect through this mechanism of action.
Metabolites of lasmiditan
Study PM73 was conducted to assess the binding affinity of the metabolites of lasmiditan that were
identified from PK analyses in the clinical studies. These studies identified several metabolites that
were named M1, M2/M3 (racemic mixture), M7, M8 (racemic), and M18. When tested against a panel
of 5-HT receptors listed in Table 3 only the M1 and M2/M3 metabolites had significant binding affinity.
The M1 and M2/M3 metabolites bound specifically to the 5-HT1F receptor with a Ki of 10.67 and 489.8
nM respectively. These data indicate that the parent is more potent then the metabolites at the 5-HT1F
receptor, however, the M1 and M2/M3 metabolites may add to the effects of the parent drug.
Study RPT-0271F01-01 determined that M3 is not a major metabolites in humans as it accounts
for less than 10% of the AUC of lasmiditan, however, it is a major metabolite in some preclinical
species. This study determined that M7, M8, and M18 are the major human metabolites of lasmiditan.
The Sponsor also conducted receptor binding profiles to determine the binding of the metabolites to
receptors, ion channels, and transporters associated with abuse potential. These studies include Study
CLP-002E-CBR which assessed binding of the metabolites at the cannabinoid 1 and 2 receptors, Study
1120683 which assessed M-18, Study 1120681 which assessed M8, and Study 1120682 which assessed
M7. The results of these studies indicate that the M7 metabolite binds significantly to the GABAA
channel with a Ki = 2.88 µM. The other metabolites do not bind to other receptors, ion channels, or
transporters that are known to be associated with abuse potential.
The Sponsor then conducted Study 160217-DME to assess the activity of lasmiditan and its M3 and M7
metabolites at the GABAA channel in comparison to diazepam. GABAA channels are heteropentameric
Reference ID: 4494251
(b) (4)

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 10 of 40
channels that are composed of alpha, beta, and gamma subunits. Currently, there are known to be 6
alpha subunits, 3 beta subunits, and 3 gamma subunits that are able to form multiple channels with
different expression patterns resulting in different behaviors. Those channels that express the alpha1
subunit are thought to be responsible for the addiction-related effects of GABAA receptor activating
drugs (i.e., benzodiazepines) (Tan et al., 2011). As a result, the Sponsor assessed the activity of
lasmiditan and the metabolites in multiple GABAA receptor isophorms. The EC50 for the GABAA
channels containing the following subunits α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2, and α5β3γ2 was
calculated to be greater than 100 µM. As a result, lasmiditan and the M3 and M7 metabolites are not
considered to have physiologically relevant GABAA agonist activity at these GABAA channels.
Conclusion
Lasmiditan is an agonist of the 5-HT1F receptor and does not bind to or activate other receptors, ion
channels, or transporters that are associated with abuse potential. Binding and activity studies indicate
that lasmiditan does not bind to or activate GABAA receptors at physiologically relevant doses. Studies
conducted on the major metabolites of lasmiditan indicate that the M7 metabolite binds to the GABAA
receptor, however, it has no appreciable activity at doses less than 100 µM. These data indicate that
lasmiditan and its major active metabolites do not appear to exert their effects through the GABAA
receptor.
2.2 Safety Pharmacology/Metabolites
Absorption
Initially, the absorption of lasmiditan was assessed in rats (Study 0226-2009) and dogs (Study # 0225-
2009). Rats were administered single doses of 6 mg/kg radio-labeled lasmitidan, equivalent to 60
µCi/kg, either orally or IV. Plasma samples were collected up to 48 hours after administration and
radioactivity levels were determined using liquid scintillation counting (LSC). The data presented in
Table 5 indicate no siginifcant sex differences in the PK of lasmiditan in rats when administered IV or
orally. Over 48 hours the animals had a mean oral exposure ranging from 9105 ng eq•h/g in males to
10280 ng eq•h/g in females. A half-life of approximately 31 hours was also calculated for rats when
both sexes were combined.
Table 5: PK of Radio-labeled Lasmiditan Administered Orally or IV in Rats (data expressed as mean
(±SD))
IV Oral
male female male female
Cmax (ng eq/g) 987 (62.99) 1142.8 (153.6) 1020 (77.44) 870 (321.54)
Half-life (h) 27.3 29.7 29.6 32.2
AUC 0-last (ng eq•h/g) 8764 10592 9105 10280
This study was followed by Study # B01-267 which was conducted to determine the PK of lasmiditan
HCl in beagle dogs in the fed state. Dogs were divided into two groups: Group 1 was fasted overnight
and Group 2 was fed 15 minutes before oral dosing of 2 mg/kg lasmiditan. Blood was collected at 5, 10,
15, 30, and 45 minutes, and 1, 1.5, 2, 4, 8, and 24 hours postdose and analyzed using LC/MS/MS. The
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 11 of 40
only parameter that was significantly different between the fed and the fasted state was the tmax.
Animals in the fasted state had a tmax of 1.25 hours and those in the fed state had a tmax of 3.5 hours.
These data indicate that food has a significant slowing effect on the absorption of lasmiditan when
consumed orally.
After assessing the PK parameters in the fed and fasted states the Sponsor determined the PK parameters
of lasmiditan and its major metabolites, M8, M7, (S,R)-M18, (S,S)-M18, and M3 in mice (Study #
RPT-0335R01-00), rats (Study # RPT-0334R01-01), and rabbits (Study # RPT-0333R01-01).
Since the animal abuse potential studies were conducted in rats the PK discussion will focus on the data
collected in rats. In this study, six male and female rats were dosed orally with 50 or 100 mg/kg
lasmiditan as a liquid formulation. Plasma samples were assessed using reversed-phase LC-MS/MS and
the PK parameters were calculated using a noncompartmental analysis. Unlike Study # 0226-2009
(above) the data presented in Table 6 indicate that lasmiditan, and its major active metabolites have
large sex differences in rats. The Cmax, and AUC of lasmiditan are twice as high in female rats
compared to their male counterparts, whereas the Tmax is two hours in males and one hour in females.
These data indicate that female rats appear to have a faster onset with a higher maximum plasma
concentration and exposure of lasmiditan compared to male rats.
Table 6: Rat PK Paramaters of Orally Administered Lasmiditan (NDA 211280, Module 4.2.2.2, Study #
RPT-0334R01-01, page 12)
Lasmiditan M8 M7 Total M18 M3
PK Parameter male female male female male female male female male female
Cmax (ng/mL) 1497 2963 61.4 98.1 459 715 316 838 794 866
Tmax, (hr) 2 1 2 4 2 1 8 4 1 1
AUClast (ng•hr/mL) 16090 29622 489 1481 5468 9024 4415 14927 7147 9189
Half-life (hr) 2.63 NC NC NC 2.51 NC 2.46 NC 2.18 NC
Data expressed as mean of 6 animals, NC – not calculated
The sex differences cannot be compared to the mouse and rabbit PK data because only male mice were
used in Study # RPT-0335R01-00 and only female rabbits were used in Study # RPT-
0333R01-01.
Study B01-144 was conducted to compare the plasma and brain PK parameters, as well as the
bioavailability of lasmiditan in male rats. Rats were administered a single dose of 1 mg/kg drug orally
or IV and blood or brain cortex was collected predosing or up to 24 hours postdose. The data presented
in Tables 7 and 8 indicate that lasmiditan crosses the blood brain barrier and collects in the brain
producing exposure levels 2.5 to 3-fold higher than those in plasma. The Tmax in both plasma and brain
was reached in 30 minutes, however, the Cmax was 2-fold higher in the brain with oral administration and
3-fold higher with IV administration compared to plasma levels. The oral bioavailability for oral
administration of the drug was calculated to be 63.3%. Overall, the data indicate that lasmiditan is
rapidly absorped, has good bioavailability, a relatively short half-life (2.5 hours), and rapidly
accumulates in the brain.
Reference ID: 4494251
(b) (4) (b) (4)
(b) (4)
(b) (4)
(b) (4) (b) (4)

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 12 of 40
Table 7: Rat Plasma PK Parameters for Lasmiditan, Oral or IV (1 mg/kg) (NDA 211280, Module
4.2.2.3, Study B01-144, page 6)
Plasma PK ParameterIV
(1 mg/kg)
Oral
(1mg/kg)
Cmax (ng/mL) 109 59.7
Tmax, (hr) -- 0.5
AUClast (ng•hr/mL) 250 158
Half-life (hr) 1.95 2.65
Clearance (mL/min/kg) 66.8 --
Vol. Dist (L/kg) 11.3 --
Bioavailability (%) -- 63.3
Table 8: Rat Brain PK Parameters for Lasmiditan, Oral or IV (1 mg/kg) (NDA 211280, Module 4.2.2.3,
Study B01-144, page 6)
Brain PK ParameterIV (1
mg/kg)
Oral
(1mg/kg)
Cmax (ng/mL) 307 107
Tmax, (hr) 0.25 0.5
AUClast (ng•hr/mL) 731 409
Half-life (hr) 2.57 2.56
Ratio AUC
(brain/plasma)2.93 2.59
Distribution
Study 0227-2009 was conducted to determine the distribution of radio-labeled lasmiditan in the body
after oral administration. Lasmiditan (6 mg/kg or 60 µCi/kg) was given to male and female rats that
were sacrified 0.5, 2, 6, 24, or 48 hours after dosing. The distribution of radioactivity was determined
using Quantitative Whole Body Autoradioluminography (QWBA). This study determined that there
were no sex differences in the distribution of the drug in the rat. In the CNS, detectable levels of
radioactivity were measured up to 24 hours in the brain (Cmax ≤ 1.05 µg eq/g) and up to 6 hours in the
spinal cord (Cmax ≤ 0.99 µg eq/g). The highest levels of radioactivity were measured in the urinary
bladder (Cmax ≤ 532.86 µg eq/g) indicating that lasmiditan or its metabolites are most likely excreted
renally. The CNS, liver, lung, and kidney all showed higher levels of radioactivity than blood, however
low levels of radioactivity were measured in the testis, epididymis, ovaries, and uterus.
In order to confirm and further eludcidate the distribution of lasmiditan in the CNS the Sponsor
conducted Study 0172-2010. In this study, drug (6 mg/kg or 846 µCi/kg) was orally administered to
male albino rats who were sacrificed one hour after dosing. Distribution in the brain was measured
using QWBA using both sagittal and coronal sections. After one hour, the whole brain concentration of
lasmiditan was 1.1 µg eq/g with concentrations in the midbrain of 0.8 µg eq/g and the cortex of 1.2 µg
eq/g. Generally, these are the areas of the brain associated with addiction and these data indicate that
lasmiditan is able to penetrate into these brain regions in significant concentrations.
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 13 of 40
Metabolism
The metabolism of lasmiditan was determined using an in vitro assay and in animal studies in rats and
dogs through oral and IV routes of administration.
Study RPT-0104-2 was conducted to determine if rats produced the same major metabolites as
those detected in humans. Rat liver was ground and fractionated to concentrate the metabolic enzymes.
Lasmiditan (5 µM) was incubated with the rat liver S9 fraction (3.58 mg/mL protein) for 0, 30, and 60
minutes. Samples were analyzed using LC-MS/MS to detect the metabolism of lasmiditan and the
appearance of any metabolites. Each of the major metabolites was identified and shown to be present in
the incubates with maximum concentrations of 1.3 ng/mL for M18, 7.1 ng/mL for M8, and 25.2 ng/mL
for M7. The minor metabolite, M3 (81.8 ng/mL) was detected at the 30 minute timepoint but not at the
60 minute timepoint suggesting that it undergoes further metabolism.
After assessing the metabolism of lasmiditan HCl in an in vitro study the Sponsor conducted two in vivo
studies to detect the metabolites in the blood (Study # 0222-2009) and in the feces and urine (Study #
0221-2009) of rats. In both of the studies male and female rats were given oral or IV doses of radio-
lableled lasmiditan (6 mg/kg) and blood samples were collected at 1, 4, and 12 hours, and feces and
urine were collected over 24 hours. The samples were analyzed using liquid chromatography with on-
line UV, radioactivity and mass spectrometry detection (LC-UV-RAD-MS). After IV administration the
parent drug accounted for 80%, 50%, and 30% of the circulating radioactivity after 1, 4, and 12 hours
respectively in the plasma. The numbers were similar for oral administration in which the parent drug
was 60%, 40%, and 35% after 1, 4, and 12 hours respectively in the plasma. After 24 hours 60% of the
urine sample was composed of the parent compound and the M1 metabolite. The M8 and M18
metabolites were detected at greater than 5% of the total urinary radioactivity and all other metabolites
were below this amount. In the fecal samples, 50% of the radioactivity in the sample was composed of
the parent compound and the M1 metabolite, all other metabolites were below 5%. The main metabolic
pathways for generation of the metabolites were determined to be N-desmethylation (M1, M6, M10), N-
oxidation (M2, M3, and M16), aliphatic oxidation on the piperidine moiety (M6, M7, M18) and ketone
reduction (M8, M10, M16, M18) (the major human metabolites are in bold).
Since the data in the previous studies was collected on the HCl salt, the Agency required that the
metabolite studies be redone on the hemisuccinate salt that is the API in the final formulation. As a
result the Sponsor conducted Study 8377-180-MET to analyze the biliary, fecal, and urinary excretion of
radiolabeled lasmiditan in male rats following oral administration. The rats were given [14C]lasmiditan
(6 mg/kg) with a collection interval spanning 0 – 72 hours. Samples were analyzed using LC/MS/MS
and metabolite identification was confirmed using retention times of available reference standards. In
total, 14% of the excreted radioactivity was parent drug and 29 metabolites were detected in the samples
that were tested. The major human metabolites detected in rats were M7 (0.1%), M8 (3.1%), and M18
(1.4%) with the percentages equating to the ratio of that metabolite detected compared over all
metabolites. In the rat, the major metabolites detected were M1 (21.3%), M2 (9.1%), and M3 (6.5%).
These data are similar to that seen with the lasmiditan hydrochloride salt measured in Study # 0222-
2009. From these studies it is concluded that rats do not metabolise lasmiditan in a similar fashion as
humans and therefore the metabolites will not be properly evaluated in animal studies in which rats are
administered lasmiditan. However, binding and activity studies indicate that the major human
Reference ID: 4494251
(b) (4)

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 14 of 40
metabolites, M7, M8, and M18 do not have activity at receptors or ion channels associated with abuse
potential.
The rat study was followed by a similar study (Study 011D03-MET) in which female beagle dogs were
administered [14C]lasmiditan hemisuccinate IV or oral at 6 mg/kg (11µCi/kg). Plasma, urine, and feces
were collected over a time course of 24 hours and analyzed using HPLC and the metabolites were
identified using LC/MS/MS. The majority of the radioactivity was excreted in the urine with 52.1%
measured after the IV dose and 47.9% following the oral dose. Similar to the rat data, the most
abundant circulating metabolites (plasma) were M1, M2, and M3 accounting for 7%, 7%, and 7% of the
total radioactivity IV and 15%, 10%, and 12% orally. The major metabolites detected in the dog were
M1, M5, M8, and M10 representing 12% of the dose following IV and about 16% of the dose following
oral administration.
Excretion
Similar to the metabolism studies, the Sponsor first performed their excretion studies using lasmiditan
HCl (Study # 0224-2009 (discussed above)) and Study 11D03-EX (discussed above), however, since the
hemisuccinate salt is the version that is used in the to-be-marketed formulation, the Agency asked the
Sponsor to conduct their excretion studies using lasmiditan hemisuccinate. As a result, the Sponsor
conducted Study # 8377-180 which is described above in the metabolism section. This study analyzed
the biliary, fecal, and urinary excretion of radiolabeled lasmiditan hemisuccinate (6 mg/kg) in male rats
following oral administration. In this study, greater than 96% of the radioactivity was collected with
mean values of 48%, 28%, and 14% recovered in the urine, bile, and feces respectively. These data
indicate that the majority of lasmiditan and its metabolites are excreted renally, however, a substantial
amount is still excreted in the feces.
Conclusion
The animal pharmacology of lasmiditan hemisuccinate indicate that the drug is readily abosorbed orally
although food does slow the Tmax from 1.25 hrs in the starved state to 3.5 hrs in the fed state in rats.
Female rats have a faster onset with a higher Cmax and significantly greater exposure than their male
counterparts, however, the half-life calculation was combined to yield a T1/2 of 2.63 h. The differences
in PK parameters were also seen in the major metabolites M3, M7, M8, and M18 (Table 6). The major
human metabolites are M7, M8, and M18 and dos not include M3 seen in rats. Furthermore, in rats,
CNS exposure of lasmiditan is approximately 3-fold higher in the brain compared to plasma exposure
whether the drug is given orally or IV (Tables 7 and 8). The drug distributes mainly to the CNS, liver,
lungs, and kidneys where it is metabolized and excreted renally (48%), in the bile (28%), or in the feces
(14%).
2.3 Findings from Safety Pharmacology and Toxicology Studies
Safety Studies
The Sponsor conducted three in vitro studies and two animal studies to assess the cardiovascular and
respiratory safety pharmacology of lasmiditan and its metabolites (CNS safety assessments are described
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 15 of 40
in the next section). Study LLY01_21 assessed the ability of lasmiditan HCl to block the heart’s
repolarizing current (IKr) which is mediated by the human Ether-a-go-go-Related-Gene (hERG)
potassium channel. The hERG channel mediates repolarization of the cardiac muscle which helps to
coordinate the heart’s beating, blocking this channel leads to long QT syndrome or tosades do pointes
which can be fatal. In this study, human embryonic kidney (HEK) cells were stably transfected with the
hERG channel and the patch clamp technique was used to measure the K+ current across the membrane
in the presence of increasing drug concentrations. Lasmiditan HCl was found to dose dependently block
the hERG current with an IC50 value of 3.1 µM. This study was followed by Studies 090731-DME and
100127-DME which determined the effects of the M7, M8, and the M18 metabolites on the hERG
current. The metabolites have little effect at these ion channels with IC50s of 129 µM, 40 µM, and 30-
100 µM respectively. As a result, lasmiditan hemisuccinate was administered to beagle dogs to
determine the cardiovascular effects in animals. Doses of 0.6, 2, and 6 mg/kg IV were infused over 20
minutes and the results indicated that there were no observable effects on left ventricular ionotropic
state, systemic arterial pressure, heart rate, and electrocardiograms.
Study MN103025 was then conducted to assess the respiratory effects of lasmiditan hemisuccinate in
male rats. A single dose of drug at 1, 4, or 12 mg/kg was orally administered and the rats were
monitored for respiratory rate, tidal volume, and minute volume for 24-hours post dose. The rats did not
show any significant changes in any of these respiratory parameters over the time measured.
Toxicity Studies
The Sponsor also conducted a series of single and repeat dose toxicological studies using both the
hydrochloride and the hemisuccinate salts of lasmiditan. Table 9 presents an overview of the
toxicological studies conducted using lasmiditan hemisuccinate because that is the API used in the to-
be-marketed formulation. In single dose studies the Sponsor determined that a lethal dose in rats
equated to 100 mg/kg (killed two of six animals) and there was also a 20% reduction in body weight in
rats given 30 mg/kg. In beagle dogs, animals given single doses of 90 or 120 mg/kg vomited and
developed ataxia, tremors, changes in fecal consistency (diarrhea), and hypoactivity. These adverse
events were also seen in rats and dogs in the repeat dose studies that lasted 28 days, 13 weeks, 26 weeks,
or 39 weeks. In the repeat dose studies a single dose of 450 mg/kg in rats was established as an acute
lethal dosing killing all of the rats. Orally administered repeat doses of 100 and 200 mg/kg produced the
same adverse events as those recorded above in the single dose studies. Other renal and liver toxicity
issues were noted but are not relevant to abuse potential. Doses below 100 mg/kg orally were deemed to
produce no serious adverse events or behaviors. All of these adverse events were temporary and
diminished upon cessation of the administration of lasmiditan.
Table 9: Overview of Toxicological Studies in Animals using Lasmiditan Hemisuccinate
Study Single/Repeat Dose (mg/kg) Species (N) Salt Adverse Events
R036025 Single10, 30, 100
(IV)
Fischer 344
Rat (6)hemisuccinate
2 deaths were noted at the 100 mg/kg dose;
20% reduction in body weight in female
rats given 30 mg/kg
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 16 of 40
7874-101 Single10, 30, 60,
90, 120 (oral)
Beagle dogs
(4)hemisuccinate
2 animals developed convulsion at 120
mg/kg dose; 60 mg/kg each animal
vomited, and ataxia and tremors were
noted in one animal; 90 and 120 mg/kg
vomiting, tremors, changes in fecal
consistency, and hypoactivity
8302173Repeat
(28 days)
100, 200,
250
(oral)
RasH2 mice
(10-36)hemisuccinate
450 mg/kg was established as acute lethal
dose; At all doses hypoactivity, ataxia, low
carriage, and diarhea were noted
7874-126Repeat
(13 week)
30, 100, 200
(oral)
ICR mice
(10)hemisuccinate
there were no significant test article related
effects in this study
7874-116Repeat
(13 week)
50, 100, 200
(oral)rats (10) hemisuccinate
100 and 200 mg/kg decreased body weight
by 29%
8202968Repeat
(26 week)
10, 30, 50,
100, 200
(oral)
Sprague-
Dawley
rats (9)
hemisuccinate100 and 200 mg/kg developed convulsions
and myoclonic jerking, decreases in body
weight
7874-125Repeat
(13 week)
5, 10, 20, 50
(oral)
Beagle dogs
(4)hemisuccinate
20 and 50 mg/kg doses resulted in transient
decreases in body weight, tremors,
twitching, and hypoactivity
8204496Repeat
(39 week)
5, 10, 20, 40,
50 (oral)
Beagle dogs
(4-6)hemisuccinate
tremors, hypoactivity, dehydration,
salivation, vomitus, reduced body weight,
no or liquid feces, and squinting
Conclusion
Lasmiditan appears to have little effect on cardiovascular or respiratory function when tested up to 6
mg/kg IV in rats. Table 9 demonstrates that there were severe toxic effects of high doses of lasmiditan,
however, they appear to be drug related adverse events and do not indicate clear signs of abuse potential.
2.4 Animal Behavioral Studies
Several types of in vivo behavioral studies are used to ascertain the reinforcing effects as well as the
pharmacodynamic effects of a drug. Taken together these studies help to determine whether or not a
substance has abuse potential and to what pharmacological class of drugs the substance is most similar.
General CNS effects
The general CNS effects of a drug substance are typically measured in an Irwin screen. In this type of
study, increasing doses of a drug are administered to a group of animals which are then observed and
evaluated for CNS-mediated behaviors such as locomotion, body temperature, and convulsions. The
Sponsor conducted two studies (Study PN0216 and Study PN0217) to evaluate the behavioral and CNS
mediated properties elicited by doses of 1, 4, and 12 mg/kg IV lasmiditan in CD-1 mice. The 12 mg/kg
IV dose of lasmiditan produced a large number of behavioral effects that were not reported at the 1 and
4 mg/kg dose. The high dose (12 mg/kg) produced analgesia in the acetic acid writing test, significantly
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 17 of 40
decreased amubulatory and nonambulatory locomotion at 15 and 30 minutes, produced a significant
decrease in body temperature with a mean difference of -1.6°C, an increase in auditory sensitivity, and
an increase in the convulsive threshold as determined by electroshock and pentylenetetrazole
administration. The 1 and 4 mg/kg doses did not produce any significant effects in any of the measured
parameters resulting in a no-observed-effect level (NOEL) of 4 mg/kg in mice. In conclusion, this study
determined that lasmiditan produces analgesia, sedation, hypothermia, and increases convulsive
thresholds.
Self-administration
A self-administration assay is an experimental paradigm in which animals identify if a substance has
positive reinforcing effects. Positive reinforcement occurs when the presentation of a desired stimulus
results in an increase in behavior that is associated with the administration of the desired stimulus
(Gauvin et al., 2017). For example, for abuse assessment purposes, animals are first trained to press a
lever (behavior) resulting in the administration (typically IV) of a training drug (desired stimulus)
known to be a drug of abuse (e.g. cocaine). Once properly trained, the animals undergo an extinction
test to confirm that the training drug is the stimulus responsible for the reinforcing effects and not some
other cue in the assay. Animals then receive test drug, and rates of lever pressing and rates of injections
are measured. If the rates of administered drug are significantly different from placebo and the animals
are not motor impaired by the drug, as measured by rates of lever pressing, the drug is said to be self-
administered (Gauvin et al., 2017).
Study RS1732 was a self-administration study conducted to assess the reinforcing potential of
lasmiditan hemisuccinate compared to midazolam and diazepam in heroin maintained rats. In this study,
Sprague-Dawley rats were initially trained to lever-press for food reward to a fixed ratio (FR) three
schedule of reinforcement. Rats were then surgically implanted with an indwelling jugular catheter and
trained to self-administer a low dose of heroin (0.015 mg/kg/injection IV) to an FR3 schedule of
reinforcement. Animals then underwent extinction in which saline was readministered to confirm that
the lever pressing was a drug response. In the test phase, saline (0.5 mL/kg/injection IV) was used as
the negative control, lasmiditan was tested at doses of 0.05, 0.2, and 0.8 mg/kg/injection IV, and the
positive controls were midazolam (0.001 and 0.0015 mg/kg/injection) and diazepam (0.001 and 0.0015
mg/kg/injection). Heroin produced significantly greater responding than the saline controls across
experiments: heroin 19.1 ± 0.5 versus 5.0 ± 0.3 injections/session, as did diazepam with 6.4 ± 1.5,
however, midazolam treated animals did not maintain statistically significant positive reinforcement
with 6.3 ± 1.6 injections/session. The study is validated by the use of the heroin and diazepam positive
controls. The response rates for lasmiditan were dose-dependent with the highest dose producing a
statistically significant reinforcing effect and the two lower doses did not when compared to saline. The
response rates were 9.5 ± 2.1 injections/session for the highest dose of 0.8 mg/kg/injection IV, and 4.5 ±
1.3 and 6.5 ± 2.4 injections/session for the two lowest doses of 0.05 and 0.2 mg/kg/injection IV of
lasmiditan.
The PK was also tested across a 40-fold range of doses in order to confirm that an appropriate dose
range of 2- to 3-fold the therapeutic Cmax was tested in this study. Lasmiditan at doses of 0.1 to 4 mg/kg
IV produced Cmax values in rats that ranged from 0.08 – 3.4 times an estimated clinical Cmax of ~ 300
ng/mL after a 200 mg oral dose of drug in humans. The estimated cumulative dose received by the rats
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 18 of 40
in the self-administration study produced a Cmax range of 160 – 2420 ng/mL which spans the clinical
therapeutic range to 8-fold the highest therapeutic Cmax.
Drug Discrimination
Drug discrimination is an experimental method in which animals identify whether a test drug produces
physical or behavioral effects (an interoceptive response) similar to those produced by another drug with
known pharmacological properties. If the known drug is one with abuse potential, drug discrimination
can be used to predict if a test drug will have abuse potential in humans (Balster and Bigelow, 2003).
For abuse assessment purposes, an animal is first trained to press one bar when it receives a known drug
of abuse (the training drug) and another bar when it receives placebo. A challenge session with the test
drug determines which of the two bars the animal presses more often, as an indicator of whether the test
drug is more like the known drug of abuse or more like placebo. A test drug is said to have "full
generalization" to the training drug when the test drug produces bar pressing >80% on the bar associated
with the training drug (Sannerud and Ator, 1995; Doat et al., 2003). Thus, a test drug that generalizes to
a known drug of abuse will likely be abused by humans (Balster and Bigelow, 2003).
Study VPT4730 was conducted to compare the discriminative stimulus effects of lorazepam to
lasmiditan. In this study, male Sprague-Dawley rats were trained to discriminate lorazepam 1 mg/kg IP
from saline in a two-lever food-reinforced task under an FR10 schedule of reinforcement. Animals were
placed in the operant chambers 1 hour after drug dosing at the Tmax of lasmiditan and near the Tmax of
the M7 metabolite (ranged from 1 - 1.5 hours). Before the discrimination phase, the rats demonstrated
discrimination of the training drug by responding with ≥ 80% on the drug appropriate lever over three
consecutive trials. In the training phase, the animals produced ≥ 98.62% responding on the lorazepam
appropriate lever. Once training was demonstrated, the discrimination phase commenced in which
multiple doses of the test drug were tested (10, 30, and 100 mg/kg, N = 10). Treatment with lasmiditan
at 10, 30, and 100 mg/kg PO engendered ≤ 0.3% on the drug appropriate lever, indicating that the
interoceptive cue produced by lasmiditan does not generalize to lorazepam at the doses tested. The rates
of responding at the 10 and 30 mg/kg dose (115.81 and 108.52 respectively) increased slightly, however
they decreased slightly at the 100 mg/kg dose (90.75).
The mean Cmax at the 10, 30, and 100 mg/kg PO doses were 565, 998, 2530 ng/mL when measured at
the end of the study. These data indicate that Cmax levels that are 2 to 3 fold higher than the desired
therapeutic levels (TABLE 10) were tested in this study.
Conclusion
The animal abuse studies indicate that lasmiditan is reinforcing at the highest dose tested of 0.8
mg/kg/injection producing 9.5 ± 2.1 injections/session. It was significantly lower than the reinforcing
effect produced by heroin (CII) and similar to that produced by diazepam (CIV). However, the drug
discrimination data indicate that lasmiditan does not generalize to the lorazepam discrimantive stimulus
cue at any of the doses tested.
2.5 Tolerance and Physical Dependence Studies in Animals
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 19 of 40
Study VPT5005 was an animal study that was conducted to assess withdrawal in Sprague-Dawley rats
administered lasmiditan at 10, 25, and 75, mg/kg/day orally for 21-days followed by a 14-day treatment
free period. The vehicle for lasmiditan was used as a negative control and chlordiazepoxide (CDP) at
doses of 20 to 200 mg/kg BID orally was used as a positive control. physiological parameters were used
to assess dependence and withdrawal such as body weight, food consumption, and body temperature, as
well as locomotor activity and other behavioral observations. Discontinuation of CDP produced an
increase in body temperature, a decrease in body weight and food consumption, and an increase in
locomotor activity, all of which are signs indicative of physical withdrawal. On the other hand,
lasmiditan produced vehicle like increases in body weight, no change in body temperature, and slight
but significant increases in locomotor activity (total distance traveled and vertical activity) at all doses
(10, 25, and 75 mg/kg). At the time of discontinuation, the rats had a mean Cmax of lasmiditan of 2070
ng/mL and an AUC0-24 of 30500 ng•hr/mL at the 75 mg/kg dose. These plasma values far exceed those
of the expected clinical therapeutic values seen in section 3. In conclusion, this animal study indicates
that lasmiditan did not produce signs consistent with physical dependence.
3. Clinical Pharmacology
Determining the clinical pharmacology of a drug is an important aspect in understanding the mechanism
of action of a drug of abuse. Understanding of the PK parameters can give an indication as to how a
drug will be abused and therefore, how it should be tested in a human abuse potential study.
3. 1 Absorption, Distribution, Metabolism, Elimination (ADME)
Distribution
The distribution of a drug is heavily affected by the extent to which the drug binds to plasma proteins.
As a result, the Sponsor conducted Study 7874-123 to determine the extent of plasma binding of [14C]-
lasmiditan at concentrations of 15, 75, 150, 250, and 500 ng/mL in mouse, rat, dog, monkey, and human
plasma. In humans the mean binding ranged from 55% to 60% at all of the doses tested indicating that
there is no dose effect on the amount of drug bound. The study indicates that lasmiditan is not highly
plasma protein bound indicating that it may have a high distribution throughout the body.
A similar plasma binding study (Study # RPT-0077.2) was conducted on the lasmiditan
metabolites M7, M8, and M18. In this study the concentrations for each metabolite were 1200, 400, and
15 ng/mL in pooled human plasma samples. The M7 metabolite was found to be approximately 85%
protein bound, the M8 metabolite was approximately 55% protein bound, and the M18 metabolite
ranged from 42 – 58% protein bound depending on dose.
Metabolism
Study 7874-117 was conducted to analyze the metabolism of lasmiditan in isolated hepatocytes from rat,
dog, monkey, and humans. The hepatocytes, at a concentration of either 1 or 5 µM were incubated at 37
°C for up to 120 minutes and the samples were analyzed by HPLC. The results determined that 13
possible metabolites are formed with one major metabolite being found in the human samples. This was
followed up with Studies 6180-505 and 7874-118 which determined that lasmiditan does not
significantly inhibit the P450s isozymes that are responsible for the majority of drug metabolism. As a
Reference ID: 4494251
(b) (4)

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 20 of 40
result, lasmiditan is not expected to have a significant effect on the metabolism of other drugs if they are
co-administered.
Study 7874-119 was an in vitro study that was conducted to identify the human cytochrome P450
isozymes responsible for the in vitro metabolism of lasmiditan. In this study, human hepatic
microsomes were incubated with 1, 5, or 10 µM lasmiditan for up to 60 minutes. Interestingly,
incubation in the microsomes did not produce any significant metabolites as they did in the studies listed
in the previous paragraph. As a result, the Sponsor incubated 1 µM lasmiditan in liver cytosol and was
able to measure the M8 metabolite using HPLC. The Sponsor concluded that this metabolite is formed
by a family of enzymes named the aldo keto reductases, which are found in the cytosol and not in liver
microsomes. To determine how the M7 and M18 human metabolites are formed the Sponsor conducted
Study RPT-0104-1 which was designed similarly to the above study. However, in this study, the
Sponsor used inbitors to specific P450 enzymes to determine which are responsible for producing which
metabolite. This type of reaction phenotyping of the human cytochrome P450 component of the
metabolism of COL-144 using these selective inhibitors indicated some possible involvement of
CYP1A2 in the production of metabolites M3, M7, M8, and M18, CYP2D6 and CYP2C9 in the
production of M7 and M18, as well as CYP2C19 and CYP3A4 in the production of M3, M7, and M18.
Elimination
The Sponsor conducted a phase 1 study to investigate the absorption, metabolism, and excretion of
[14C]-lasmiditan following single oral administration in healthy male and female subjects (Study #
110/LAHH). Eight subjects (5 males and 3 females) were fasted before receiving an oral dose of 200
mg lasmiditan (the proposed maximum daily dose). They were confined for up to 9 days during which
plasma, urine, and fecal samples were collected. This study determined that lasmiditan is rapidly
absorped and eliminated. In this study, the M7, M8, and (S,R)-M18 metabolites were identified as the
major circulating metabolites in humans. A majority of the drug/metabolites were excreted in the urine
(86.8%) after 312 hours, with the parent accounting for 2.91% and the M8 metabolites accounting for
66.1% of the total radioactivity. In terms of PK, the drug produced a median Tmax of 2.02 hours, a
mean half-life of 4.12 hours, and an AUC0-last of 2100 h*ng/mL. The PK parameters of lasmiditan and
its major metabolites are presented in Table 10.
The Sponsor then conducted a second phase 1 study to assess the safety, tolerability, and potential
withdrawal symptoms of multiple once daily dosing of 200 mg and 400 mg lasmiditan orally in healthy
subjects. The subjects received drug for seven days after which they were assessed using the
Benzodiazepine Withdrawal Symptom Questionnaire from day 2 to day 14 post dosing. The PK
parameters presented in Table 11 indicated that there is no significant difference in peak drug
concentration, exposure, half-life, or clearance when comparing single or repeated daily administration
of lasmiditan at 200 mg.
According to PK study report PK 02 the PK of lasmiditan was not appreciably affected by age, body
weight, sex, race, ethnicity, or population. Thus there is no reason differences in these factors need to be
considered for abuse of lasmiditan.
Reference ID: 4494251
(b) (4)

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 21 of 40
Table 10: Human PK Parameters for Lasmiditan and its Metabolites After Single Oral Administration
of 200 mg (NDA 211280; Module 5.3.3.1; Study 110/LAHH - Study report body, Pg 33)
Table 11: Human PK Parameters of Lasmiditan Following Daily Dosing of 200 or 400 mg for Seven
Days (NDA 211280; Module 5.3.3.1; Study H8H-MC-LAHE; Study Report Body, pg 31)
Lasmiditan
PK Parameter 200 mg 400 mg
Cmax (ng/mL) 353 808
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 22 of 40
Tmax (hr) 2 2
half-life (hr) 4.34 4.05
CL/F (L/h) 92.8 85.4
Vz/F (L) 581 498
In human abuse potential study # H8H-MC-LAHB subjects were given single oral doses of 100, 200, or
400 mg lasmiditan. Table 12 indicates that these doses produced steadily increasing Cmax values from
therapeutic to supra therapeutic plasma levels. The Tmax, AUC, half-life, and clearance values are
consistent with those calculated from previous studies and indicate that this study was conducted at
therapeutic to supratherapeutic doses.
Table 12: Pharmacokinetic Parameters of Lasmiditan Following a Single Oral Dose of 100, 200, or 400
mg (NDA 211280; Module 5.3.5.4; Study H8H-MC-LAHB; Study report, pg 27)
Lasmiditan
PK Paramter 100 mg 200 mg 400 mg
Cmax (ng/mL) 132 (37%) 299 (35%) 689 (34%)
Tmax (hr) 1.42 1.42 1.42
AUC0-last (ng•hr/mL) 831 (32%) 1760 (35%) 3830 (28%)
half-life (hr) 4.61 4.4 4.28
CL/F (L/h) 117 111 102
Vz/F (L) 777 704 629
4. Clinical Studies
4.1 Human Abuse Potential StudiesA Randomized, Subject- and Investigator-Blind, Placebo- and Active-Controlled Study to Assess
the Abuse Potential of Lasmiditan H8H-MC-LAHB
This was a Phase 1, randomized, double-blind, placebo- and active-controlled,
crossover clinical trial in adult subjects who were recreational poly-drug users to assess the abuse
potential of lasmiditan compared to the positive control alprazolam and placebo.
The primary objective was to assess the abuse potential of lasmiditan compared to alprazolam and
placebo using the maximal effect score (Emax) of the at-the-moment 100-mm bipolar Drug Liking
VAS.
The secondary objectives of the study were:
Further characterize the abuse potential of lasmiditan with additional Drug Effects and
Drug Similarity Visual Analog Scale (VAS) measures
Safety evaluations
Qualification Phase: Subjects were randomized to a test dose of 1 mg alprazolam and placebo in
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 23 of 40
a crossover manner with a washout period of at least 72 hours between each dose. “Drug Liking” was
assessed before and after alprazolam and placebo administration using a 100-mm bipolar Drug Liking
VAS. To qualify for the Treatment Period of the study, subjects must have met the following criteria:
Acceptable placebo response ranging from 40 to 60 (inclusive) on the 100-mm bipolar
VAS for Drug Liking “at this moment” ≥15-mm increase in Drug Liking “at this moment” alprazolam more than placebo
Treatment Phase: This phase had a subject- and investigator-blind, placebo- and active-controlled, 5-
period crossover design. Subjects were randomized to 1 of 10 dosing sequences. Each dosing sequence
consisted of 5 dosing periods that evaluated the abuse liability of 1 of the 5 study treatments: placebo, 2
mg alprazolam, 100 mg lasmiditan, 200 mg lasmiditan, and 400 mg lasmiditan. The washout period
between each dose was at least 72 hours.
Safety was assessed by recording AEs, clinical laboratory tests, physical examinations, vital
signs, electrocardiograms (ECGs), and Columbia Suicide Severity Rating Scale and Self-Harm
Form.
Inclusion Criteria: Subjects were recreational drug users, defined as follows:
≥10 lifetime non-therapeutic experiences (i.e., for psychoactive effects) with CNS depressants
(e.g., benzodiazepines, barbiturates, zolpidem, eszopiclone, propofol/fospropofol, gamma
hydroxybutyrate); and
≥1 non-therapeutic use of a CNS depressant/sedative drug within the 12 weeks prior to
screening; and
≥1 lifetime non-therapeutic use of another drug class of abuse (e.g., opioids, stimulants,
dissociatives, or hallucinogens)
A randomized, double-blind, placebo- and active-controlled, crossover clinical trial in adult subjects
who were recreational poly-drug users with experience with sedative drugs, is an appropriate study
design to assess the abuse potential of lasmiditan. This type of study evaluates the responses of
individuals experienced with the psychoactive effects of drugs. The cross-over design is suitable for
assessing the effects of the test drug, positive control, and placebo in the same subject.
Alprazolam was an appropriate positive control for this study due to similarities in the AE profiles of
alprazolam and lasmiditan (somnolence, sedation). The wash out period was appropriate (72 hours) as it
was about 5 half-lives of the drug with the longest half-life (alprazolam half -life 15 hours) . The doses
studied for lasmiditan represent therapeutic (100 mg, 200 mg) and supratherapeutic (400 mg) doses,
typical for a HAP study.
A total of 58 subjects, 48 male and 10 female, between the ages of 19 and 50 years qualified for and
participated in the Treatment Phase. Of these, 53 subjects completed all 5 periods of the Treatment
Phase, and 5 subjects withdrew their consent before completing the study
Results:
Reference ID: 4494251
Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 24 of 40
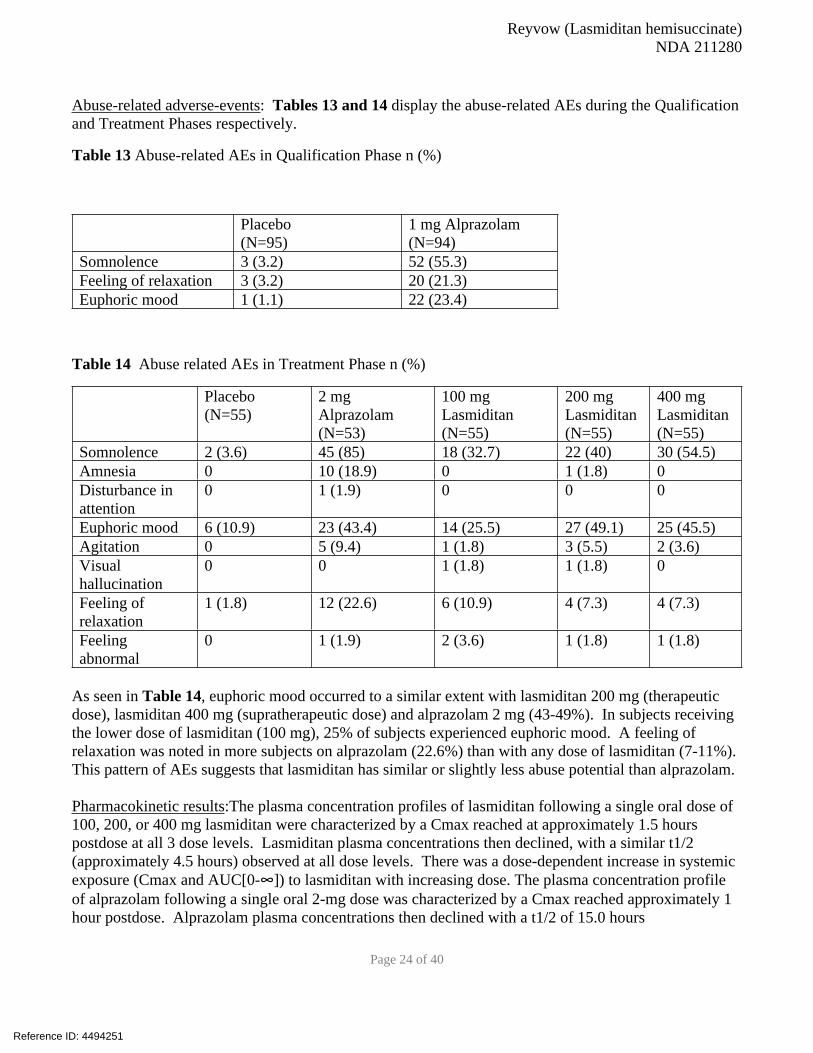
Abuse-related adverse-events: Tables 13 and 14 display the abuse-related AEs during the Qualification
and Treatment Phases respectively.
Table 13 Abuse-related AEs in Qualification Phase n (%)
Placebo
(N=95)
1 mg Alprazolam
(N=94)
Somnolence 3 (3.2) 52 (55.3)
Feeling of relaxation 3 (3.2) 20 (21.3)
Euphoric mood 1 (1.1) 22 (23.4)
Table 14 Abuse related AEs in Treatment Phase n (%)
Placebo
(N=55)
2 mg
Alprazolam
(N=53)
100 mg
Lasmiditan
(N=55)
200 mg
Lasmiditan
(N=55)
400 mg
Lasmiditan
(N=55)
Somnolence 2 (3.6) 45 (85) 18 (32.7) 22 (40) 30 (54.5)
Amnesia 0 10 (18.9) 0 1 (1.8) 0
Disturbance in
attention
0 1 (1.9) 0 0 0
Euphoric mood 6 (10.9) 23 (43.4) 14 (25.5) 27 (49.1) 25 (45.5)
Agitation 0 5 (9.4) 1 (1.8) 3 (5.5) 2 (3.6)
Visual
hallucination
0 0 1 (1.8) 1 (1.8) 0
Feeling of
relaxation
1 (1.8) 12 (22.6) 6 (10.9) 4 (7.3) 4 (7.3)
Feeling
abnormal
0 1 (1.9) 2 (3.6) 1 (1.8) 1 (1.8)
As seen in Table 14, euphoric mood occurred to a similar extent with lasmiditan 200 mg (therapeutic
dose), lasmiditan 400 mg (supratherapeutic dose) and alprazolam 2 mg (43-49%). In subjects receiving
the lower dose of lasmiditan (100 mg), 25% of subjects experienced euphoric mood. A feeling of
relaxation was noted in more subjects on alprazolam (22.6%) than with any dose of lasmiditan (7-11%).
This pattern of AEs suggests that lasmiditan has similar or slightly less abuse potential than alprazolam.
Pharmacokinetic results:The plasma concentration profiles of lasmiditan following a single oral dose of
100, 200, or 400 mg lasmiditan were characterized by a Cmax reached at approximately 1.5 hours
postdose at all 3 dose levels. Lasmiditan plasma concentrations then declined, with a similar t1/2
(approximately 4.5 hours) observed at all dose levels. There was a dose-dependent increase in systemic
exposure (Cmax and AUC[0-∞]) to lasmiditan with increasing dose. The plasma concentration profile
of alprazolam following a single oral 2-mg dose was characterized by a Cmax reached approximately 1
hour postdose. Alprazolam plasma concentrations then declined with a t1/2 of 15.0 hours
Reference ID: 4494251
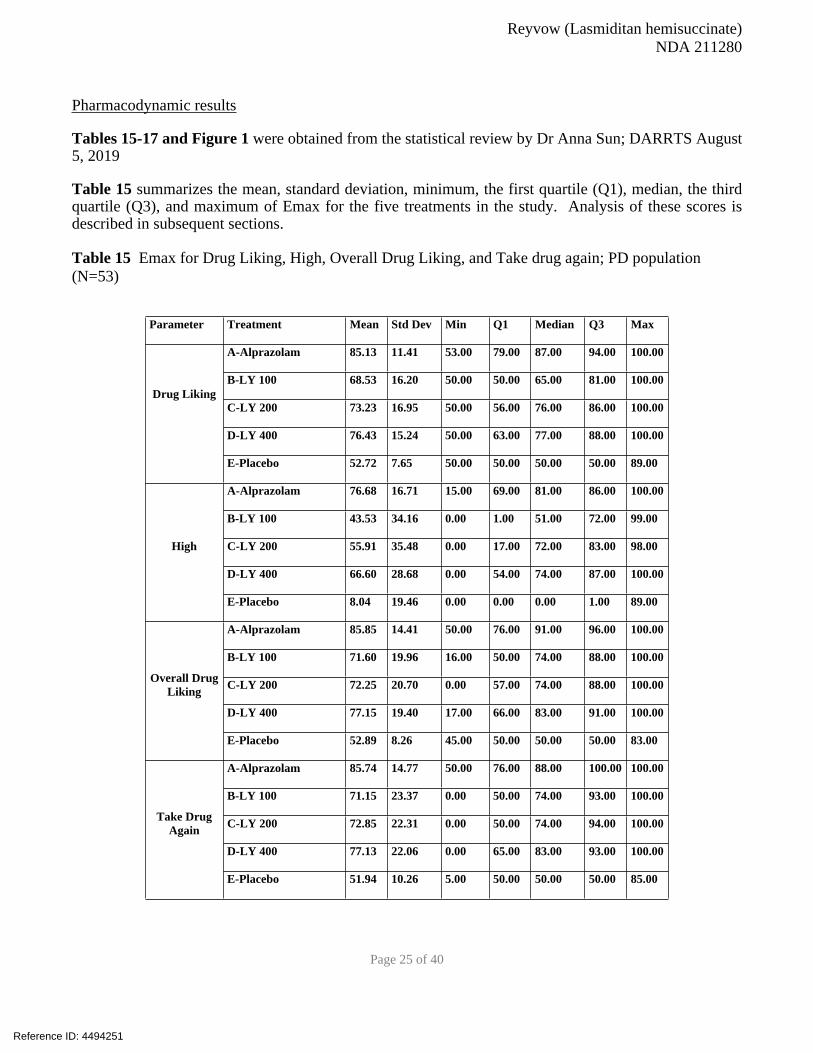
Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 25 of 40
Pharmacodynamic results
Tables 15-17 and Figure 1 were obtained from the statistical review by Dr Anna Sun; DARRTS August 5, 2019
Table 15 summarizes the mean, standard deviation, minimum, the first quartile (Q1), median, the third quartile (Q3), and maximum of Emax for the five treatments in the study. Analysis of these scores is described in subsequent sections.
Table 15 Emax for Drug Liking, High, Overall Drug Liking, and Take drug again; PD population
(N=53)
Parameter Treatment Mean Std Dev Min Q1 Median Q3 Max
A-Alprazolam 85.13 11.41 53.00 79.00 87.00 94.00 100.00
B-LY 100 68.53 16.20 50.00 50.00 65.00 81.00 100.00
C-LY 200 73.23 16.95 50.00 56.00 76.00 86.00 100.00
D-LY 400 76.43 15.24 50.00 63.00 77.00 88.00 100.00
Drug Liking
E-Placebo 52.72 7.65 50.00 50.00 50.00 50.00 89.00
A-Alprazolam 76.68 16.71 15.00 69.00 81.00 86.00 100.00
B-LY 100 43.53 34.16 0.00 1.00 51.00 72.00 99.00
C-LY 200 55.91 35.48 0.00 17.00 72.00 83.00 98.00
D-LY 400 66.60 28.68 0.00 54.00 74.00 87.00 100.00
High
E-Placebo 8.04 19.46 0.00 0.00 0.00 1.00 89.00
A-Alprazolam 85.85 14.41 50.00 76.00 91.00 96.00 100.00
B-LY 100 71.60 19.96 16.00 50.00 74.00 88.00 100.00
C-LY 200 72.25 20.70 0.00 57.00 74.00 88.00 100.00
D-LY 400 77.15 19.40 17.00 66.00 83.00 91.00 100.00
Overall Drug Liking
E-Placebo 52.89 8.26 45.00 50.00 50.00 50.00 83.00
A-Alprazolam 85.74 14.77 50.00 76.00 88.00 100.00 100.00
B-LY 100 71.15 23.37 0.00 50.00 74.00 93.00 100.00
C-LY 200 72.85 22.31 0.00 50.00 74.00 94.00 100.00
D-LY 400 77.13 22.06 0.00 65.00 83.00 93.00 100.00
Take Drug Again
E-Placebo 51.94 10.26 5.00 50.00 50.00 50.00 85.00
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 26 of 40
Figure 1 shows that following oral administration of a single dose of lasmiditan, there was a dose-
dependent increase in Drug Liking score during the first 2 hours post-dose, which gradually returned to
pre-dose levels by approximately 8 hours post-dose. For placebo, the mean Drug Liking score remained
at approximately 50 (neither like nor dislike) at all time points. Following the positive control, 2 mg
alprazolam, Drug Liking score increased at a similar rate to that following lasmiditan dosing but reached
a greater score than any dose of lasmiditan (with Emax reached at approximately 2 hours post-dose) and
the score remained elevated for longer before returning to 50’s by 24 hours postdose.
Figure 1. Mean Drug Liking VAS Scores over time (PD Population, N=53)
Reference ID: 4494251
APPEARS THIS WAY ON ORIGINAL

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 27 of 40
Comparison of the Drug Liking VAS- Emax for lasmiditan versus alprazolam and placebo are displayed in Table 16.
Table 16. Comparison of Drug Liking VAS-Emax (Primary end point) PD population
Treatments LS Mean StdE Lower UpperA-Alprazolam 85.38 1.61 82.15 88.61
B-LY 100 68.59 2.22 64.14 73.04
C-LY 200 73.33 2.30 68.71 77.95
D-LY 400 76.60 2.05 72.50 80.70
E-Placebo 53.00 1.13 50.70 55.30
Contrasts LS Mean StdE P-value Lower Upper
Positive Controls vs. Placebo (Trial Validity, H0: µC - µP≤ 15)
A-Alprazolam vs. E-Placebo32.38 1.79 <.0001 29.39 Infty
Positive Controls vs. Lasmiditan (Relative Abuse Potential, H0: µC - µT≤ 0)
A-Alprazolam vs B-LY 100 16.79 2.62 <.0001 12.43 Infty
Reference ID: 4494251
APPEARS THIS WAY ON ORIGINAL

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 28 of 40
A-Alprazolam vs C-LY 200 12.05 2.69 <.0001 7.57 Infty
A-Alprazolam vs D-LY 400 8.78 2.47 0.0003 4.67 Infty
Lasmiditan vs. Placebo (Absolute Abuse Potential, H0: µT - µP ≥ 11)
B-LY 100 vs. E-Placebo 15.59 2.35 0.9722 -Infty 19.52
C-LY 200 vs. E-Placebo 20.33 2.44 0.9999 -Infty 24.39
D-LY 400 vs. E-Placebo 23.60 2.19 1 -Infty 27.25
The validity of the study was determined from the comparison of Drug Liking Emax between positive
control and placebo. Emax of alprazolam was significantly higher than placebo (exceeding the margin of
15 with p value < 0.0001, thereby confirming study validity.
Lasmiditan was evaluated by the comparison of Drug Liking Emax scores of positive control versus
each dose of lasmiditan. All doses of lasmiditan were statistically significantly lower than alprazolam on
mean Emax. It should be noted that the Sponsor analyzed these results using a prespecified margin of 5;
by this analysis there was no significant difference between alprazolam and lasmiditan 400 mg (P
value=0.065). Typically, a margin of 0 is used for this comparison and reanalysis of this data (Dr
Anna Sun) using a margin of 0, showed that Mean Drug Liking scores were significantly greater for
alprazolam than for all doses of lasmiditan.
For the absolute abuse potential test, lasmiditan was evaluated by the comparison of Drug Liking Emax between each dose of lasmiditan and placebo. The null hypothesis was defined as a mean difference in Drug Liking Emax of ≥ 11 points. If the null hypothesis was not rejected then the results support that the test drug was not similar to placebo. All doses of lasmiditan (100 mg, 200 mg and 400 mg) were significantly higher than placebo (P value close to 1) indicating that lasmiditan has abuse potential.
Table 17. Comparison of High VAS Emax PD population
Treatments LS Mean StdE Lower UpperA-Alprazolam 77.15 2.31 72.50 81.81
B-LY 100 43.65 4.71 34.21 53.08
C-LY 200 56.10 4.66 46.75 65.44
D-LY 400 66.90 3.64 59.61 74.20
E-Placebo 8.56 3.04 2.41 14.72
Contrasts LS Mean StdE P-value Lower Upper
Positive Controls vs. Placebo (Trial Validity, H0: µC - µP≤ 15)
A-Alprazolam vs. E-Placebo68.59 3.24 <.0001 63.20 Infty
Positive Controls vs. Lasmiditan (Relative Abuse Potential, H0: µC - µT≤ 0)
A-Alprazolam vs B-LY 100 33.51 4.84 <.0001 25.42 Infty
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 29 of 40
A-Alprazolam vs C-LY 200 21.06 4.80 <.0001 13.04 Infty
A-Alprazolam vs D-LY 400 10.25 3.81 0.0046 3.89 Infty
Lasmiditan vs. Placebo (Absolute Abuse Potential, H0: µT - µP ≥ 11)
B-LY 100 vs. E-Placebo 35.08 5.22 1 -Infty 43.78
C-LY 200 vs. E-Placebo 47.53 5.18 1 -Infty 56.16
D-LY 400 vs. E-Placebo 58.34 4.29 1 -Infty 65.48
Table 17 shows that for High VAS:
• For the validation test: Emax of Alprazolam was significantly higher than placebo (p value< 0.01), thereby confirming study validity.
• For the relative abuse potential test: All lasmiditan doses (100 mg, 200 mg, and 400 mg) were significantly lower than Alprazolam on mean Emax (P value <0.01).
• For the absolute abuse potential: Lasmiditan was evaluated by the comparison of High Emax between lasmiditan and placebo. All doses of lasmiditan (100 mg, 200 mg, and 400 mg) were significantly higher than placebo (P values are all equal 1), indicating that lasmiditan has abuse potential.
Overall Drug Liking VAS:
• For the validation test: Emax of alprazolam was significantly higher than placebo (p value< 0.01), thereby confirming study validity.
• For the relative abuse potential test: All lasmiditan doses (100 mg, 200 mg, and 400 mg) were significantly lower than alprazolam on mean Emax (all P values <0.01).
• For the absolute abuse potential: Lasmiditan was evaluated by the comparison of Overall Drug Liking Emax between lasmiditan and placebo. All doses of Lasmiditan (100 mg, 200 mg, and 400 mg) were significantly higher than placebo (P values are close to 1), indicating that lasmiditan has abuse potential.
Take Drug Again:
• For the validation test: Emax of alprazolam was significantly higher than placebo (p value< 0.01), thereby confirming study validity.
• For the relative abuse potential test: All lasmiditan doses (100 mg, 200 mg, and 400 mg) were significantly lower than alprazolam on mean Emax (all P values <0.01).
• For the absolute abuse potential: Lasmiditan was evaluated by the comparison of Take Drug Again Emax between lasmiditan and placebo. All doses of lasmiditan (100 mg, 200 mg, and 400 mg) were significantly higher than placebo (P values are close to 1), indicating that lasmiditan has abuse potential.
Conclusions:
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 30 of 40
• The HAP study was appropriately designed, with appropriate controls, wash out periods, and doses studied
• The validity of the study was shown by the positive control, alprazolam, showing statistically significantly higher Emax scores than placebo for primary and all secondary end points by a margin of 15
• All doses of lasmiditan showed statistically significantly higher Emax scores than placebo by a margin of 11 for the primary and all secondary end points. This indicates that lasmiditan has abuse potential
• In comparison to alprazolam, lasmiditan had statistically significantly lower Emax scores on all
primary and secondary end points. However, euphoric mood occurred to a similar extent with
lasmiditan 200 mg (therapeutic dose), lasmiditan 400 mg (supratherapeutic dose) and alprazolam
2 mg (43-49%). A feeling of relaxation was noted in more subjects on alprazolam (22.6%) than
with any dose of lasmiditan (7-11%).
• Overall, the HAP data indicate that lasmiditan has less abuse potential than alprazolam (a
Schedule IV drug)
4.2 Adverse Event Profile Through all Phases of Development Phase 1 Studies: The Sponsor conducted 18 Phase 1 studies in which AEs, including abuse-related AEs
were evaluated.
Table 18 displays the abuse-related AEs in Phase 1, single dose studies with healthy subjects. As
shown in Table 18 somnolence occurred in 0-63% lasmiditan subjects and greater than the control
groups (0-5.6%), feeling drunk 0-12.5% lasmiditan, none in control group; Euphoric mood 0-12.5%
lasmiditan, 0-10% in controls. Euphoric mood occurred in 5/12 studies in lasmiditan and 1/7 studies
which had a control group
Table 18. Abuse-related AEs Lasmiditan (L) single dose, Phase 1 studies n (%) healthy subjects
Study/dose Somnolence Feeling
drunk
Euphoric
mood
Feeling
abnormal
Hypersomnia Hallucination Control group
COL MIG-
104 L200mg
N=30
19 (63.3) 0 0 0 0 0 No control
group
COL MIG-
103 L50-
400mg N=57
5 (9) 0 0 0 0 0 No control
group
COL MIG-
110 L200
N=8
0 1(12.5) 1(12.5) 0 0 0 No control
group
COL MIG-
113 L200
N=16
(normal and
5 (31.2) 0 0 1 (6.3) 0 0 No control
group
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 31 of 40
impaired
renalfunction
COL MIG-
114 L200mg
N=24 normal
and impaired
hepatic
function
7 (29.2) 0 0 0 0 0 No control
group
H8H-MC-
LAHA
L200mg
N=35
4 (11.4) 0 1 (2.9) 0 0 0 Placebo:no
abuse-related
AEs
H8H-MC-
LAIG L100-
200 mg
N=36
0 0 0 0 0 0 Placebo:no
abuse-related
AEs
COL-MIG-
118 L200
N=41
4 (9.8) 0 2 (4.9) 0 9 (22) 0 Sumatriptan:
10%
hypersomnia
2.5%
somnolence
H8H-MC
LAHT L200
N=20
2 (10) 0 2 (10) 0 0 0 Placebo:10%
euphoric
mood;
topiramate:
3.3% euphoric
mood
H8H-MC
LAHU L200
N=39
0 0 0 0 0 0 Placebo: no
abuse-related
AEs
COL-MIG
105
L100:N=52
L400: N=55
L100 13
(25)
L400 22
(40)
L100
1(1.9)
L400 1
(1.8)
L100 0
L400
1(1.8)
0 0 L100 1(1.9)
L400 2 (3.6)
Placebo: 5.6%
somnolence
H8H-MC
LAHD L200
N=44
1(2.3) 0 0 0 0 0 Propranolol:
no abuse
related AEs
The Sponsor conducted two Phase1 single dose driving performance studies in healthy subjects in which
they compared AEs occurring with lasmiditan to positive controls and placebo (Tables 19 and 20)
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 32 of 40
As shown in Table 19, the number of subjects reporting an AE of feeling abnormal was similar between
lasmiditan and alprazolam (2-3%), and both drugs were higher than placebo (1.2%). Alprazolam had a
higher incidence of the AE of feeling drunk and euphoric mood than lasmiditan (3.5% vs 1%; 3.5% vs
0% respectively). Overall at therapeutic doses lasmiditan is associated with a lower incidence of abuse-
related AEsthan alprazolam in this study
Table 19: Abuse related AEs, single dose, COL MIG-106, Phase 1, healthy subjects, driving study n
(%)
Placebo N=85 Lasmiditan 50
mg N=87
Lasmiditan
100 mg N=86
Lasmiditan
200mg N=89
Alprazolam
1mg N=85
Feeling
abnormal
1(1.2) 2 (2.3) 2 (2.3) 3 (3.4) 3 (3.5)
Feeling drunk 0 0 0 1 (1.1) 3 (3.5)
Feeling of
relaxation
0 0 0 1 (1.1) 0
Somnolence 2 (2.4) 10 (11.5) 23 (26.7) 38 (42.7) 45 (53)
Euphoric
mood
0 0 0 0 3 (3.5)
Table 20 displays the abuse related AEs in the second driving study compared to diphenhydramine ( a
non-controlled drug). Somnolence was increased in lasmiditan treated subjects, disturbance in attention,
feeling abnormal was slightly increased with lasmiditan. This study showed possible increased abuse
potential of lasmiditan compared to placebo and diphenhydramine
Table 20: Abuse related AEs, single dose, H8H MC LAIF, Phase 1, healthy subjects, driving study n
(%)
Placebo N=67 Lasmiditan 100
mg N=68
Lasmiditan 200
mg N=68
Diphenhydramine
50 mg N=68
Somnolence 0 5 (7.4) 7 (10.3) 4 (5.9)
Euphoric mood 1 (1.5) 1(1.5) 1(1.5) 1(1.5)
Disturbance in
attention
0 2 ( 2.9) 1 (1.5) 0
Feeling abnormal 0 1(1.5) 2 (2.9) 0
Feeling of
relaxation
0 1(1.5) 0 0
H8H MC LAHE: This was a Phase 1, multiple ascending dose study study in healthy subjects. In
Cohort 1 subjects received 200 mg of lasmiditan or placebo daily for 7 days; Cohort 2 subjects received
400mg/day lasmiditan or placebo for 7 days. Based on the benzodiazepine withdrawal symptom
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 33 of 40
questionnaire and Penn Physician withdrawal check list, no withdrawal symptoms occurred upon abrupt
withdrawal of lasmiditan after 7 days of administration.
The abuse-related AEs in this study are displayed in Tables 21 and 22
Table 21. Abuse-related AEs Study H8H MC LAHE Cohort 1 n (%)
Lasmiditan 200mg N=28 Placebo N=12
Feeling abnormal 1 (3.6) 0
Euphoric mood 1 (3.6) 0
Hyperhidrosis 1 (3.6) 0
Table 22. Abuse-related AEs Study H8H MC LAHE Cohort 2 n (%)
Lasmiditan 400mg N=15 Placebo N=15
Euphoric mood 2 (13.3) 0
H8H MC LAIE: This was a Phase 1 study in healthy subjects who received single doses of lasmiditan
(50-400mg) or placebo on multiple days. The abuse-related AEs are displayed in Table 23.
Table 17 Abuse-related AEs Study H8H MC LAIE n (%)
Placebo N=20 Lasmiditan N=27
Somnolence 1 (5) 9 (33.3)
Euphoric mood 0 2 (7.4) 200 mg and 400mg
group
Amnesia 0 1 (3.7)
COL MIG-102: This was a Phase1 single ascending dose study in healthy subjects in which somnolence
(16-50%) was the only abuse-related AE.
H8H MC LAHC: This was a Phase 1 single dose (200mg) study in migraine patients. Somnolence
occurred in 6.3% of patients.
In summary the Phase 1 studies indicate that a higher number of abuse-related adverse events were
reported when subjects received lasmitidan than when receiving placebo. In one study comparing
lasmiditan to alprazolam, the latter showed a greater incidence of abuse -related adverse events than
lasmitidan.
Phase 2 and 3 studies
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 34 of 40
A Placebo-Controlled, Group Sequential, Adaptive Treatment Assignment Study of Intravenous COL-
144 in the Acute Treatment of Migraine: COL MIG 201 Phase 2.
The primary study objective was to evaluate the efficacy of a range of intravenous doses of COL-144 in
order to select a dose range for further evaluation in the acute treatment of moderate or severe migraine.
This was a prospective, randomized, double-blind, placebo-controlled study. Patients were allocated to a
dose level of COL-144 in small cohorts. The first cohort was allocated to the 2.5 mg dose level. The
dose allocation for subsequent cohorts depended on the response of the previous cohort. If two or less of
the active-treated patients in that cohort responded, then the next cohort received the next higher dose. If
3 or more active treated patients responded then the next cohort received the next lower dose. Female
and male patients aged 18 to 65 years, with a diagnosis of migraine were included. Doses of 1, 2.5, 5,
10, 20, 30, 45, 60 mg, and placebo were administered intravenously. The starting dose for the first
cohort was 2.5 mg.
Duration of treatment: One single dose for a single migraine attack.
A total of 130 patients (22 cohorts) were randomized to either COL-144 or placebo (COL-144: N=88,
placebo: N=42). Table 23 displays the abuse-related AEs in this study. As shown in the table, a feeling
of relaxation occurred in more subjects (7-19%) with higher IV doses of lasmiditan compared with
placebo (0%)
Table 18. Percentage of subjects reporting abuse related AEs COL MIG-201(I.V. lasmitidan)
Placebo
N=42
2.5 mg
COL-
144
N=4
5 mg
COL-
144
N=12
10 mg
COL-
144
N=24
20 mg
COL-
144
N=28
30 mg
COL-
144
N=16
45 mg
COL-
144
N=4
COL-
144
(total)
N=88
Feeling
abnormal
2.4
(n=1)
0 0 8.3
(n=2)
0 6.3
(n=1)
0 3.4
(n=3)
Feeling of
relaxation
0 0 0 0 7.1
(n=2)
18.8
(n=3)
0 5.7
(n=5)
Formication 0 0 0 0 0 18.8
(n=3)
0 3.4
(n=3)
Somnolence 2.4
(n=1)
0 0 4.2
(n=1)
3.6
(n=1)
0 0 2.3
(n=2)
A double-blind randomized placebo-controlled parallel group dose-ranging study
of oral COL-144 in the acute treatment of migraine COL MIG-202 Phase:2
The objective of this study was to evaluate the efficacy (headache response at 2 hours) of a range of oral
doses of COL-144 in order to select a dose or doses for further evaluation. This was a prospective,
randomized, double-blind, placebo-controlled, dose-ranging study in patients with migraine. Patients were
asked to treat a single migraine attack with study medication at home.
Five hundred and twelve (512) patients were randomized to receive either placebo or one of the four COL-
144 doses. A total of 121 patients did not use study medication and were therefore not included in any
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 35 of 40
analyses. Table 24 displays the abuse-related AEs in this study. Somnolence was the primary AE noted to a
greater extent in the lasmiditan -treated groups vs placebo.
Table 19. Abuse-related AEs COL MIG-202 n (%)
Placebo N=86 COL-144 (50
mg)
(N= 82)
COL-144 (100
mg)
(N= 82)
COL-144 (200
mg)
(N= 71)
COL-144 (400
mg)
(N= 70)
Disturbance in
attention
0 1 (1.2) 3 (3.7) 2 (2.8) 2 (2.9)
Somnolence 2 (2.3) 8 (9.8) 10 (12.2) 8 (11.3) 8 (11.4)
Agitation 0 0 0 1 (1.4) 2 (2.9)
Derealisation 0 0 1 (1.2) 0 1 (1.4)
Euphoric
mood
0 1( 1.2) 0 0 0
Visual
hallucination
0 0 0 1 (1.4) 0
Illusion 0 0 0 0 1 (1.4)
Restlessness 0 1 (1.2) 0 1 (1.4) 2 (2.9)
A Study of Two Doses of Lasmiditan (100 mg and 200 mg) Compared to Placebo in the AcuteTreatment of
Migraine: A randomized, double-blind, placebo-controlled parallel group study (SAMURAI) COL MIG-301
Phase: 3
The primary objective of this study was to evaluate the efficacy at 2 hours of lasmiditan 100 mg and 200 mg
compared to placebo on migraine headache pain. This was a prospective randomized, double-blind, placebo-
controlled study in subjects with disabling migraine. Subjects were asked to treat a migraine attack with
study drug on an outpatient basis. Subjects were provided with a dosing card containing a dose for initial
treatment and a second dose to be used for rescue or recurrence of migraine.
Duration of Treatment: Up to 2 doses to treat a single migraine during a period of 8 weeks. A total of 2231
subjects were randomized; 1856 (83.2%) subjects used at least 1 dose of study drug (safety population).
Table 25 displays the abuse-related AEs in this study. Somnolence was noted to a greater extent in drug vs
placebo. Table 26 shows that a low number of patients who took a second dose had abuse-related AEs.
Table 20. Abuse-related AEs COL MIG-301 n (%)
L100 mg
N=630
L200 mg
N=609
Placebo
N=617
Feeling abnormal 6 ( 1) 4 (0.7) 0
Feeling jittery 3 (0.5) 3 (0.5) 1 (0.2)
Feeling drunk 1 (0.2) 1 (0.2) 0
Feeling of relaxation 1 (0.2) 1 (0.2) 0
Somnolence 36 ( 5.7) 33 (5.4) 14 (2.3)
Sedation 2 (0.3) 7 (1.1) 1 (0.2)
Disturbance in
attention
2 (0.3) 2 (0.3) 0
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 36 of 40
Hypersomnia 1 (0.2) 3 (0.5) 0
Confusional state 2 (0.3) 2 (0.3) 0
Euphoric mood 2 (0.3) 2 (0.3) 0
Depersonalisation 1(0.2) 1(0.2) 0
Table 21. Abuse-related AEs COL MIG-301after 2nd dose n (%)
L100 mg/
L100 mg
N=203
L100 mg/
Placebo
N=86
L200 mg/
L200 mg
N=159
L200 mg/
Placebo
N=79
Placebo/
Placebo
N=401
Feeling jittery 1 (0.5) 1 (1.2) 1 (0.6) 1 (1.3) 0
Feeling
abnormal
2 (1) 0 1 (0.6) 0 0
Feeling of
relaxation
1 (0.5) 0 0 0 0
Somnolence 7 (3.4) 2 (2.3) 1 (0.6) 1 (1.3) 3 (0.7)
Sedation 0 0 1 (0.6) 0 1 (0.2)
A Study of Three Doses of Lasmiditan (50 mg, 100 mg, and 200 mg) Compared to Placebo in the Acute
Treatment of Migraine: A randomized, double-blind, placebo-controlled parallel group study (SPARTAN)
COL MIG-302 Phase: 3
The primary objective of this study was to evaluate the efficacy at 2 hours of lasmiditan 50 mg, 100 mg, and
200 mg compared to placebo on migraine headache pain. This was a prospective randomized, double-blind,
placebo-controlled study in subjects with disabling migraine. Subjects were asked to treat a single migraine
attack with study drug on an outpatient basis. Subjects were provided with a dosing card containing a dose
for initial treatment and a second dose to be used for rescue or recurrence of the migraine.
Duration of Treatment: Up to 2 doses, as needed, for the treatment of a single migraine attack. A total of
2583 subjects took at least 1 dose and were included in the safety population. A total of 1141 subjects took a
second dose and were included in the safety-2nd dose population. Table 27 displays the abuse-related AEs in
this study. Somnolence was present in more subjects on lasmiditan than placebo. Table 28 shows that a
low number of patients who took a second dose had abuse-related AEs.
Table 22. Abuse-related AEs COL-MIG-302 n (%)
L50 mg
N=654
L100 mg
N=635
L200 mg
N=649
Placebo
N=645
Feeling abnormal 2 (0.3) 3 (0.5) 5 (0.8) 1 (0.2)
Feeling jittery 0 0 4 (0.6) 0
Somnolence 35 (5.4) 29 ( 4.6) 42 (6.5) 13 (2.0)
Sedation 1 (0.2) 4 (0.6) 4 (0.6) 0
Formication 0 3 (0.5) 1 (0.2) 0
Euphoric mood 2 (0.3) 4 (0.6) 3 (0.5) 0
Restlessness 2 (0.3) 4 (0.6) 3 (0.5) 0
Hallucination 0 1 (0.2) 1 (0.2) 0
Depersonalisation 0 1 (0.2) 0 0
Reference ID: 4494251
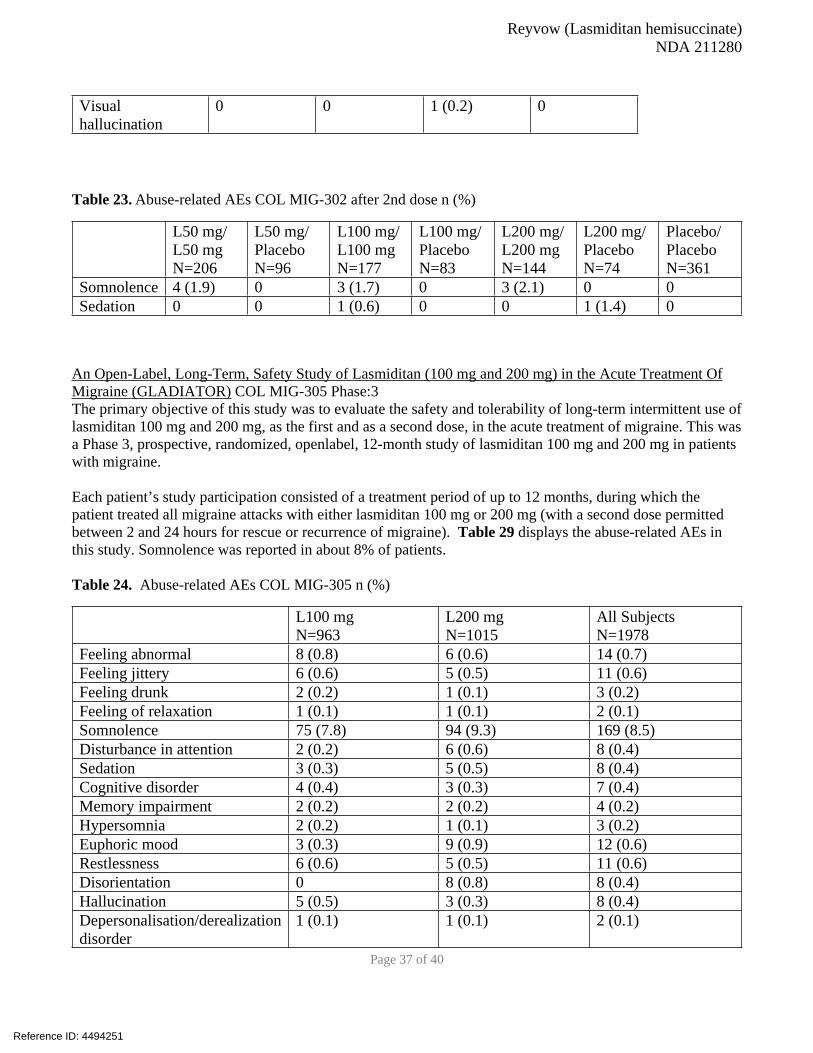
Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 37 of 40
Visual
hallucination
0 0 1 (0.2) 0
Table 23. Abuse-related AEs COL MIG-302 after 2nd dose n (%)
L50 mg/
L50 mg
N=206
L50 mg/
Placebo
N=96
L100 mg/
L100 mg
N=177
L100 mg/
Placebo
N=83
L200 mg/
L200 mg
N=144
L200 mg/
Placebo
N=74
Placebo/
Placebo
N=361
Somnolence 4 (1.9) 0 3 (1.7) 0 3 (2.1) 0 0
Sedation 0 0 1 (0.6) 0 0 1 (1.4) 0
An Open-Label, Long-Term, Safety Study of Lasmiditan (100 mg and 200 mg) in the Acute Treatment Of
Migraine (GLADIATOR) COL MIG-305 Phase:3
The primary objective of this study was to evaluate the safety and tolerability of long-term intermittent use of
lasmiditan 100 mg and 200 mg, as the first and as a second dose, in the acute treatment of migraine. This was
a Phase 3, prospective, randomized, openlabel, 12-month study of lasmiditan 100 mg and 200 mg in patients
with migraine.
Each patient’s study participation consisted of a treatment period of up to 12 months, during which the
patient treated all migraine attacks with either lasmiditan 100 mg or 200 mg (with a second dose permitted
between 2 and 24 hours for rescue or recurrence of migraine). Table 29 displays the abuse-related AEs in
this study. Somnolence was reported in about 8% of patients.
Table 24. Abuse-related AEs COL MIG-305 n (%)
L100 mg
N=963
L200 mg
N=1015
All Subjects
N=1978
Feeling abnormal 8 (0.8) 6 (0.6) 14 (0.7)
Feeling jittery 6 (0.6) 5 (0.5) 11 (0.6)
Feeling drunk 2 (0.2) 1 (0.1) 3 (0.2)
Feeling of relaxation 1 (0.1) 1 (0.1) 2 (0.1)
Somnolence 75 (7.8) 94 (9.3) 169 (8.5)
Disturbance in attention 2 (0.2) 6 (0.6) 8 (0.4)
Sedation 3 (0.3) 5 (0.5) 8 (0.4)
Cognitive disorder 4 (0.4) 3 (0.3) 7 (0.4)
Memory impairment 2 (0.2) 2 (0.2) 4 (0.2)
Hypersomnia 2 (0.2) 1 (0.1) 3 (0.2)
Euphoric mood 3 (0.3) 9 (0.9) 12 (0.6)
Restlessness 6 (0.6) 5 (0.5) 11 (0.6)
Disorientation 0 8 (0.8) 8 (0.4)
Hallucination 5 (0.5) 3 (0.3) 8 (0.4)
Depersonalisation/derealization
disorder
1 (0.1) 1 (0.1) 2 (0.1)
Reference ID: 4494251
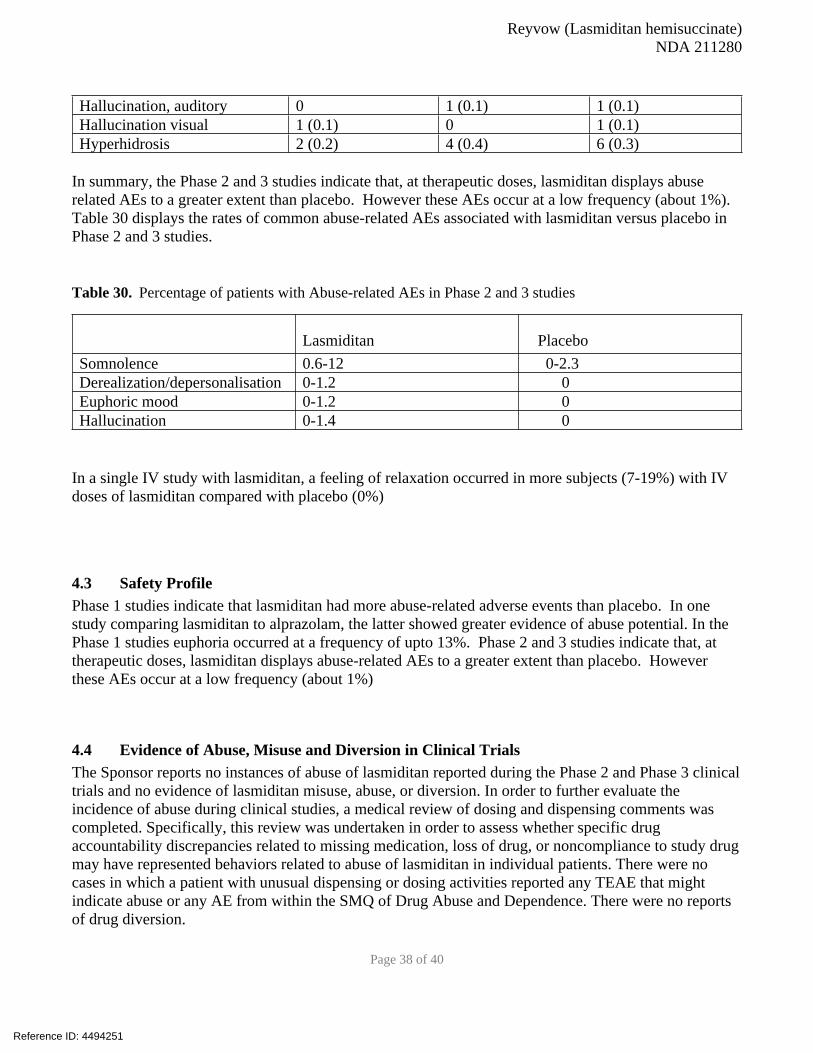
Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 38 of 40
Hallucination, auditory 0 1 (0.1) 1 (0.1)
Hallucination visual 1 (0.1) 0 1 (0.1)
Hyperhidrosis 2 (0.2) 4 (0.4) 6 (0.3)
In summary, the Phase 2 and 3 studies indicate that, at therapeutic doses, lasmiditan displays abuse
related AEs to a greater extent than placebo. However these AEs occur at a low frequency (about 1%).
Table 30 displays the rates of common abuse-related AEs associated with lasmiditan versus placebo in
Phase 2 and 3 studies.
Table 30. Percentage of patients with Abuse-related AEs in Phase 2 and 3 studies
Lasmiditan Placebo
Somnolence 0.6-12 0-2.3
Derealization/depersonalisation 0-1.2 0
Euphoric mood 0-1.2 0
Hallucination 0-1.4 0
In a single IV study with lasmiditan, a feeling of relaxation occurred in more subjects (7-19%) with IV
doses of lasmiditan compared with placebo (0%)
4.3 Safety Profile Phase 1 studies indicate that lasmiditan had more abuse-related adverse events than placebo. In one
study comparing lasmiditan to alprazolam, the latter showed greater evidence of abuse potential. In the
Phase 1 studies euphoria occurred at a frequency of upto 13%. Phase 2 and 3 studies indicate that, at
therapeutic doses, lasmiditan displays abuse-related AEs to a greater extent than placebo. However
these AEs occur at a low frequency (about 1%)
4.4 Evidence of Abuse, Misuse and Diversion in Clinical Trials The Sponsor reports no instances of abuse of lasmiditan reported during the Phase 2 and Phase 3 clinical
trials and no evidence of lasmiditan misuse, abuse, or diversion. In order to further evaluate the
incidence of abuse during clinical studies, a medical review of dosing and dispensing comments was
completed. Specifically, this review was undertaken in order to assess whether specific drug
accountability discrepancies related to missing medication, loss of drug, or noncompliance to study drug
may have represented behaviors related to abuse of lasmiditan in individual patients. There were no
cases in which a patient with unusual dispensing or dosing activities reported any TEAE that might
indicate abuse or any AE from within the SMQ of Drug Abuse and Dependence. There were no reports
of drug diversion.
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 39 of 40
4.5 Tolerance and Physical Dependence Studies in Humans Assessment of withdrawal symptoms indicative of physical dependence was not possible in the
placebo-controlled Phase 2 or Phase 3 studies, as these were single migraine attack studies where
patients administered study drug during a single migraine attack. One long-term open-label Phase 3
study allowed for multiple attacks to be treated over the course of 12 months, however, study drug was
administered intermittently to treat migraine attacks. This intermittent use did not allow for a specific
evaluation of withdrawal symptoms indicative of physical dependence.
A multiple-ascending dose study (Study LAHE) where healthy subjects took a daily dose of lasmiditan
for 7 consecutive days allowed for evaluation of the potential for withdrawal symptoms indicative of
physical dependence. In Study LAHE, the assessment of abrupt withdrawal was evaluated following 7
days of once-daily dosing with lasmiditan 200 mg or 400 mg. No evidence of withdrawal symptoms was
identified based upon review of Benzodiazepine Withdrawal Symptom Questionnaire and Penn
Physician Withdrawal Checklist scores.
5. Regulatory Issues and Assessment
Based on the preclinical data, the HAP study, the abuse-related AE profile in clinical studies, and the
physical dependence studies, we agree with the Sponsor, that lasmiditan should be placed under
ScheduleV of the CSA.
CSS recommendations regarding the label are addressed in the Recommendations section.
Reference ID: 4494251

Reyvow (Lasmiditan hemisuccinate)
NDA 211280
Page 40 of 40
III. References
Balster RL and Bigelow GE (2003) Guidelines and methodological reviews concerning drug abuse
liability assessment. Drug and alcohol dependence 70:S13-40.
Doat MM, Rabin RA and Winter JC (2003) Characterization of the discriminative stimulus properties of
centrally administered (-)-DOM and LSD. Pharmacol Biochem Behav 74:713-721.
Gauvin DV, Zimmermann ZJ and Baird TJ (2017) Method of data interpretation for the determination of
abuse liability in rodent self-administration studies under the FDA guidance document. Journal of pharmacological and toxicological methods 86:44-59.
Sannerud CA and Ator NA (1995) Drug discrimination analysis of midazolam under a three-lever
procedure: I. Dose-dependent differences in generalization and antagonism. J Pharmacol Exp Ther 272:100-111.
Tan KR, Rudolph U and Luscher C (2011) Hooked on benzodiazepines: GABAA receptor subtypes and
addiction. Trends in neurosciences 34:188-197.
Reference ID: 4494251

--------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically. Following this are manifestations of any and allelectronic signatures for this electronic record.--------------------------------------------------------------------------------------------/s/------------------------------------------------------------
SHALINI M BANSIL09/19/2019 12:56:14 PM
EDWARD G HAWKINS09/19/2019 12:58:15 PM
CHAD REISSIG09/20/2019 11:38:29 AM
SILVIA N CALDERON09/20/2019 11:42:50 AM
DOMINIC CHIAPPERINO09/20/2019 12:37:03 PM
Signature Page 1 of 1
Reference ID: 4494251


Page 2 Clinical Inspection Summary NDA #211280 Lasmiditan Protocol 301/LAHJ (COL-MG-301, SAMURAI)
Title: “A study of two doses of lasmiditan (50 mg and 100 mg) compared to placebo in the acute treatment of migraine: a randomized, double-blind, placebo-controlled parallel group study”
Subjects: 2231 randomized
Sites: 98 Sites in the U.S.
Study Initiation and Completion Dates: 4/27/2015 – 8/12/2016
Database Lock: 8/21/2016 This was a randomized, double-blind, placebo-controlled study in subjects with migraine. Included were male or female subjects > 18 years of age, diagnosis of migraine with or without aura, history of disabling migraine for at least one year, history of 3 to 8 migraine attacks per month, Migraine Disability Assessment (MIDAS) score > 11, and migraine onset before 50 years of age. Concomitant medications to reduce the frequency of migraine episodes were allowed during the study if doses were stable for the three months prior to screening. The study consisted of a screening visit, an 8-week double-blind treatment period, and an end-of-study visit within 7 days of treating a single migraine attack. At the screening visit, subjects were randomized and provided study drug but instructed not to treat a migraine attack until their eligibility had been confirmed by telephone. Subjects were randomized to one of 5 treatment sequences (subjects were stratified for use of concomitant migraine medications):
1. First dose: Lasmiditan 100 mg; second dose (if needed): lasmiditan 100 mg 2. First dose: Lasmiditan 100 mg; second dose (if needed): placebo 3. First dose: Lasmiditan 200 mg; second dose (if needed): lasmiditan 200 mg 4. First dose: Lasmiditan 200 mg; second dose (if needed): placebo 5. First dose: Placebo; second dose (if needed): placebo
Subjects were instructed to treat their next migraine attack within 4 hours of onset provided that the headache severity was at least moderate and not improving. If the migraine did not “respond” (become pain free) at 2 hours, a second dose of study drug could be taken up to 24 hours after the first dose as long as no other rescue medication had been taken. If the migraine responded within 2 hours but recurred after 2 hours, a second dose of study drug could be taken up to 24 hours after the first dose. Subjects recorded their response to the first and second dose over the next 48 hours using an electronic diary (e-Diary). Subjects were to contact the clinic to schedule the end-of-study visit within 7 days after one migraine attack had been treated, or after 8 weeks if no migraine attacks had occurred. The primary efficacy endpoint was the proportion of subjects who were headache pain-free at 2 hours post-first dose, comparing lasmiditan 200 mg vs. placebo. The key secondary efficacy endpoint was the proportion of subjects who were most bothersome symptom (MBS)-free at 2 hours post-first dose, comparing lasmiditan 200 mg vs. placebo.
Reference ID: 4475507

Page 3 Clinical Inspection Summary NDA #211280 Lasmiditan Protocol 302/LAHK (COL-MG-302, SPARTAN)
Title: “A study of three doses of lasmiditan (50 mg, 100 mg, and 200 mg) compared to placebo in the acute treatment of migraine: a randomized, double-blind, placebo-controlled parallel group study”
Subjects: 3005 randomized
Sites: 97 sites in the United States, 16 sites in Germany, and 12 sites in the United Kingdom
Study Initiation and Completion Dates: 5/19/2016 – 6/29/2017
Database Lock: 7/21/2017 The study design was similar to 301/LAHJ except that subjects were randomized to one of 7 treatment sequences:
1. First dose: Lasmiditan 50 mg; second dose (if needed): lasmiditan 50 mg 2. First dose: Lasmiditan 50 mg; second dose (if needed): placebo 3. First dose: Lasmiditan 100 mg; second dose (if needed): lasmiditan 100 mg 4. First dose: Lasmiditan 100 mg; second dose (if needed): placebo 5. First dose: Lasmiditan 200 mg; second dose (if needed): lasmiditan 200 mg 6. First dose: Lasmiditan 200 mg; second dose (if needed): placebo 7. First dose: Placebo; second dose (if needed): placebo
Rationale for Site Selection The clinical sites were chosen primarily based on numbers of enrolled subjects, site efficacy, and prior inspectional history.
III. RESULTS
For these protocols, subjects were screened, randomized, and provided investigational product to take home on the same day. Labs were drawn at this screening visit, but results were not available at the time these subjects were randomized. Subjects were informed that they were not to take investigational product until they received a telephone call from the site confirming their eligibility (after screening labs were reviewed). Therefore, subjects could be randomized but determined not to be eligible for the study based on results of screening labs. In the clinical investigator inspection summaries below, information is provided for number of subjects who were screened, randomized, confirmed to be eligible, and completed the studies.
1. Mira Baron, M.D. 2277 Palm Beach Lakes Blvd. West Palm Beach, FL 33409 At this site for Protocol 302/LAHK (Site #314), 111 subjects were screened, 81 were randomized, 73 were confirmed to be eligible and continued in the study, and 57 subjects
Reference ID: 4475507



Page 6 Clinical Inspection Summary NDA #211280 Lasmiditan reported to the IRB and are included in sponsor line listings. Of note, Subjec ended up being a screen failure due to meeting exclusion criterion 17 (use of >3 doses/month of either opiates or barbiturates).
Reviewer comment: Under-reporting of adverse events was noted at this site for two of 50 (4%) subjects enrolled and randomized to lasmiditan. It is recommended that the review division include these additional events when evaluating the safety profile of lasmiditan.
3. William Kirby, M.D. 832 Princeton Avenue SW Birmingham, AL 35211 At this site for Protocol 301/LAHJ (Site #131), 60 subjects were screened, 49 subjects were randomized, 47 were confirmed to be eligible and continued in the study, and 44 subjects completed the study. Three subjects discontinued the study due to noncompliance (n = 2) and loss to follow-up (n = 1). Five of the 44 subjects completing the study did not have a headache of sufficient severity during the study and so did not take the study drug. Signed informed consent forms, dated prior to participation in the study, were present for 58 of 60 subjects who were screened (see below). An audit of the study records of all 48 subjects enrolled was conducted. Records reviewed included, but were not limited to, source documents, case report forms (paper), monitoring documents, IRB/sponsor communications, financial disclosure, test article accountability, inclusion/exclusion criteria, adverse event reports, laboratory results, concomitant medications, protocol deviations, and the primary efficacy endpoint data (headache). For this clinical site, e-Diary data was available in a computer database and included audit trails. The FDA field investigator was provided access to the database to verify headache data against sponsor line listings. Data reviewed included date and time of dosing of study drug and 2-hour post dose symptoms (headache severity, disability, most bothersome symptom). A review of e-Diary data for all 39 subjects who took study drug was performed. There were no discrepancies identified. There was no evidence of under-reporting of adverse events at this site. One SAE was reported by the site, exacerbation of asthma, occurring in Subject who was randomized to lasmiditan. A narrative for this subject is included in the NDA submission. One inspectional observation was discussed with the clinical investigator. Specifically, a review of ICFs for the 60 subjects who were screened noted that two ICFs could not be located (Subjects ). The clinical investigator stated that the process for packing of study records for storage was not followed and that these ICFs were missing and could not be located. Subjects were contacted but were unable to provide a copy of the ICF they had signed. Other corroborating evidence was available at the site indicating that these subjects had been consented, including monitoring visit reports stating that ICFs had been reviewed for these subjects.
Reference ID: 4475507
(b) (6)
(b) (6)
(b) (6)

Page 7 Clinical Inspection Summary NDA #211280 Lasmiditan
4. David Larsen, M.D. 4001 S 700 E Suite 105 Salt Lake City, UT 84107 At this site for Protocol 302/LAHK (Site #395), 93 subjects were screened and randomized, 91 were confirmed to be eligible and continued in the study, and 83 subjects completed the study. Five of the 83 subjects who completed the study did not have a headache of sufficient severity during the study and did not take study drug. Eight subjects discontinued the study due to loss to follow-up (n = 5), withdrawal by subject (n = 2) and investigator request (n = 1). Signed informed consent forms (ICFs), dated prior to participation in the study, were present for all subjects who were screened. An audit of the study records of all 48 subjects enrolled was conducted. Records reviewed included, but were not limited to, source documents, case report forms (paper), monitoring documents, IRB/sponsor communications, financial disclosure, test article accountability, inclusion/exclusion criteria, adverse event reports, laboratory results, concomitant medications, protocol deviations, and the primary efficacy endpoint data (headache). For this clinical site, e-Diary data was available on a password protected CD. E-Diary data for all enrolled subjects was verified against sponsor line listings. There were no discrepancies identified. There was no evidence of under-reporting of adverse events.
{See appended electronic signature page} Cara Alfaro, Pharm.D. Clinical Analyst Good Clinical Practice Assessment Branch Division of Clinical Compliance Evaluation Office of Scientific Investigations
CONCURRENCE:
{See appended electronic signature page}
Phillip Kronstein, M.D.
Team Leader Good Clinical Practice Assessment Branch Division of Clinical Compliance Evaluation Office of Scientific Investigations
CONCURRENCE:
Reference ID: 4475507

Page 8 Clinical Inspection Summary NDA #211280 Lasmiditan
{See appended electronic signature page}
Kassa Ayalew, M.D., M.P.H Branch Chief Good Clinical Practice Assessment Branch Division of Clinical Compliance Evaluation Office of Scientific Investigations
cc: Central Document Room/NDA #211280 DNP/Division Director/Billy Dunn DNP/Medical Team Leader/Heather Fitter DNP/Medical Officer/Viveca Livezey DNP/Project Manager/Emilios (Andrew) Papanastasiou OSI/Office Director/David Burrow OSI/DCCE/ Division Director/Ni Khin OSI/DCCE/GCPAB/Branch Chief/Kassa Ayalew OSI/DCCE/GCPAB/Team Leader/Phillip Kronstein OSI/DCCE/GCPAB/Reviewer/Cara Alfaro OSI/ GCPAB Program Analyst/Yolanda Patague
Reference ID: 4475507

--------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically. Following this are manifestations of any and allelectronic signatures for this electronic record.--------------------------------------------------------------------------------------------/s/------------------------------------------------------------
CARA L ALFARO08/09/2019 05:31:53 PM
PHILLIP D KRONSTEIN08/09/2019 07:30:33 PM
KASSA AYALEW08/13/2019 09:39:18 AM
Signature Page 1 of 1
Reference ID: 4475507

1
MEMORANDUM REVIEW OF REVISED LABEL AND LABELING
Division of Medication Error Prevention and Analysis (DMEPA) Office of Medication Error Prevention and Risk Management (OMEPRM)
Office of Surveillance and Epidemiology (OSE)Center for Drug Evaluation and Research (CDER)
Date of This Memorandum: July 22, 2019
Requesting Office or Division: Division of Neurology Products (DNP)
Application Type and Number: NDA 211280
Product Name and Strength: Reyvow (lasmiditan) tablet, 50 mg, 100 mg
Applicant/Sponsor Name: Eli Lilly and Company (Eli Lilly)
FDA Received Date: July 16, 2019
OSE RCM #: 2018-2213-2
DMEPA Safety Evaluator: Chad Morris, PharmD, MPH
DMEPA Team Leader (Acting): Briana Rider, PharmD
1 PURPOSE OF MEMORANDUMThe Applicant submitted revised container labels and carton labeling received on July 16, 2019 for Reyvow. The Division of Neurology Products (DNP) requested that we review the revised container labels and carton labeling for Reyvow (Appendix A) to determine if they are acceptable from a medication error perspective. The revisions are in response to recommendations that we made during a previous label and labeling review.a
2 CONCLUSIONThe Applicant implemented all of our recommendations, and we have no additional recommendations at this time.
a Morris, C. Label and Labeling Review for Reyvow (lasmiditan) NDA 211280. Silver Spring (MD): FDA, CDER, OSE, DMEPA (US); 2019 JUN 26. RCM No.: 2018-2213-1.
Reference ID: 4465858
4 Pages of Draft Labeling have been Withheld in Full as B4(CCI/TS) Immediately Following this Page

--------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically. Following this are manifestations of any and allelectronic signatures for this electronic record.--------------------------------------------------------------------------------------------/s/------------------------------------------------------------
JOHN C MORRIS07/22/2019 02:33:03 PM
BRIANA B RIDER07/22/2019 05:55:07 PM
Signature Page 1 of 1
Reference ID: 4465858

1
MEMORANDUM REVIEW OF REVISED LABEL AND LABELING
Division of Medication Error Prevention and Analysis (DMEPA) Office of Medication Error Prevention and Risk Management (OMEPRM)
Office of Surveillance and Epidemiology (OSE)Center for Drug Evaluation and Research (CDER)
Date of This Memorandum: June 26, 2019
Requesting Office or Division: Division of Neurology Products (DNP)
Application Type and Number: NDA 211280
Product Name and Strength: Reyvow (lasmiditan) tablet, 50 mg, 100 mg
Applicant/Sponsor Name: Eli Lilly and Company (Eli Lilly)
FDA Received Date: June 17, 2019
OSE RCM #: 2018-2213-1
DMEPA Safety Evaluator: Chad Morris, PharmD, MPH
DMEPA Team Leader (Acting): Briana Rider, PharmD
1 PURPOSE OF MEMORANDUMThe Applicant submitted revised container labels and carton labeling received on June 17, 2019 for Reyvow. The Division of Neurology Products (DNP) requested that we review the revised container labels and carton labeling for Reyvow (Appendix A) to determine if they are acceptable from a medication error perspective. The revisions are in response to recommendations that we made during a previous label and labeling review.a
2 CONCLUSIONThe revised container labels and carton labeling are unacceptable from a medication error perspective for the following reasons:
1. The format for the expiration date should be further clarified. 2. The prominence of the established name should be further revised.3. A placeholder for the machine-readable 2D data matrix barcode is not present on the
carton labeling. 4. The presentation of the human-readable product identifiers should be improved to
ensure readability.
a Morris, C. Label and Labeling Review for Reyvow (lasmiditan) NDA 211280. Silver Spring (MD): FDA, CDER, OSE, DMEPA (US); 2019 MAY 14. RCM No.: 2018-2213.
Reference ID: 4454767

2
3 RECOMMENDATIONS FOR ELI LILLY AND COMPANYWe recommend the following be implemented prior to approval of this NDA:
1. The format for the expiration date should be further clarified. As currently presented, the format for the expiration date on the on the container labels and carton labeling, is “MM YYYY”. However, it is unclear whether the month (i.e., MM) will be displayed using numerical or alphabetical characters. Please clarify whether you propose to use only numerical characters for the expiration date, or whether you proposed to use alphabetical characters for the month.We reiterate: FDA recommends that the human-readable expiration date on the drug package label include a year, month, and non-zero day. FDA recommends that the expiration date appear in YYYY-MM-DD format if only numerical characters are used or in YYYY-MMM-DD if alphabetical characters are used to represent the month. If there are space limitations on the drug package, the human-readable text may include only a year and month, to be expressed as YYYY-MM if only numerical characters are used or YYYY-MMM if alphabetical characters are used to represent the month. FDA recommends that a hyphen or a space be used to separate the portions of the expiration date.
2. The established name should be further revised. The established name for drug products should include the finished dosage form. The prominence of the dosage form ‘tablets’ as part of the established name, is not commensurate with the proprietary name on the principal display panel. Increase the prominence of the established name, (lasmiditan) tablets, taking into account all pertinent factors, including typography, layout, contrast, and other printing features in accordance with 21 CFR 201.10(g)(2).
3. As currently presented on the carton labeling, a placeholder for the machine-readable 2D data matrix barcode is not present. We understand you will add the exact 2D matrix barcode “live”. However, without identifying the exact placement on the carton labeling we cannot assess the product identifier from a medication safety perspective. We recommend you identify the location of the 2D data matrix barcode on the carton labeling with a placeholder
4. The exp/lot numbers and SN are displayed on a single line separated by the “/” symbol. The presentation of the human-readable product identifier should be improved to ensure readability. FDA’s draft guidance on product identifiers required under the Drug Supply Chain Security Actb recommends the following format for the human-readable product identifier:
NDC: [insert product’s NDC]SERIAL: [insert product’s serial number]LOT: [insert product’s lot number]EXP: [insert product’s expiration date]
b The draft guidance is available from: https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm621044.pdf
Reference ID: 4454767
3 Pages of Draft Labeling have been Withheld in Full as B4(CCI/TS) Immediately Following this Page

--------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically. Following this are manifestations of any and allelectronic signatures for this electronic record.--------------------------------------------------------------------------------------------/s/------------------------------------------------------------
JOHN C MORRIS06/26/2019 05:34:04 PM
BRIANA B RIDER06/26/2019 07:56:32 PM
Signature Page 1 of 1
Reference ID: 4454767

1
LABEL AND LABELING REVIEWDivision of Medication Error Prevention and Analysis (DMEPA)
Office of Medication Error Prevention and Risk Management (OMEPRM)Office of Surveillance and Epidemiology (OSE)
Center for Drug Evaluation and Research (CDER)
*** This document contains proprietary information that cannot be released to the public***
Date of This Review: May 14, 2019
Requesting Office or Division: Division of Neurology Products (DNP)
Application Type and Number: NDA 211280
Product Name and Strength: Reyvow (lasmiditan) tablet, 50 mg, 100 mg
Product Type: Single Ingredient Product
Rx or OTC: Prescription (Rx)
Applicant/Sponsor Name: Eli Lilly and Company (Eli Lilly)
FDA Received Date: October 11, 2018 and April 15, 2019
OSE RCM #: 2018-2213
DMEPA Safety Evaluator: Chad Morris, PharmD, MPH
DMEPA Team Leader (Acting): Briana Rider, PharmD
Reference ID: 4433095

2
1 REASON FOR REVIEWAs part of the approval process for Reyvow (lasmiditan) tablet, the Division of Neurology Products (DNP) requested that we review the proposed Reyvow prescribing information (PI), trade and sample carton labeling and container labels, and medication guide (MG) for areas of vulnerability that may lead to medication errors.
2 MATERIALS REVIEWED
Table 1. Materials Considered for this Label and Labeling ReviewMaterial Reviewed Appendix Section
(for Methods and Results)
Product Information/Prescribing Information A
Previous DMEPA Reviews B (N/A)
ISMP Newsletters C (N/A)
FDA Adverse Event Reporting System (FAERS)* D (N/A)
Other E (N/A)
Labels and Labeling F
N/A=not applicable for this review*We do not typically search FAERS for our label and labeling reviews unless we are aware of medication errors through our routine postmarket safety surveillance
3 FINDINGS AND RECOMMENDATIONS
Tables 2 and 3 below include the identified medication error issues with the submitted prescribing information (PI), trade and sample carton labeling and container labels, our rationale for concern, and the proposed recommendation to minimize the risk for medication error.
Table 2. Identified Issues and Recommendations for Division of Neurology Products (DNP)
IDENTIFIED ISSUE RATIONALE FOR CONCERN RECOMMENDATION
General Recommendations for the Prescribing Information
1. As presented in Section 2 Dosage and Administration and throughout the PI, the driving impairment statement lacks clarity.
This statement may be misinterpreted to mean
Throughout the PI, consider removing the terms “between dosing” and “each dose” from the driving impairment statements. Consider revising the statements to read, “patients should not take REYVOW unless they can wait after taking REYVOW to drive or operate machinery.” and “Advise patients not to drive or operate machinery until after taking REYVOW…”
Reference ID: 4433095
(b) (4)
(b) (4)
(b) (4)
(b) (4)

3
Table 2. Identified Issues and Recommendations for Division of Neurology Products (DNP)
IDENTIFIED ISSUE RATIONALE FOR CONCERN RECOMMENDATION
Full Prescribing Information – Section 16 How Supplied/Storage and Handling
1. The temperature statement does not contain the temperature scale designation (i.e., “C” or “F”) after each numeric value.
Lack of clarity. Revise the statement to read “Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].”
Table 3. Identified Issues and Recommendations for Eli Lilly and Company (entire table to be conveyed to Applicant)
IDENTIFIED ISSUE RATIONALE FOR CONCERN RECOMMENDATION
Container Labels and Carton Labeling
1. As currently presented, the established name is not enclosed in parentheses.
This layout is not consistent with the presentation of the proprietary name, established name, dosage form, and strength for drug products.
The established name and proprietary name should be displayed in a manner consistent with FDA regulations, taking into account all pertinent factors including typography, layout, contrast, and other printing features (see 21 CFR 201.10(g)).
Revise the display of the established name as follows:
(lasmiditan) tablets
2. The format for expiration date is not defined.
We are unable to assess the expiration date format from a medication safety perspective.
Identify the expiration date format you intend to use and ensure the expiration date is clearly differentiated from the lot number. FDA recommends that the human-readable expiration date on the drug package label include a year, month, and non-zero day. FDA recommends that the expiration date appear in YYYY-MM-DD format if only numerical characters are used or in YYYY-MMM-DD if alphabetical characters are used to represent the month. If there are space limitations on the drug package, the human-readable text may
Reference ID: 4433095
4
Table 3. Identified Issues and Recommendations for Eli Lilly and Company (entire table to be conveyed to Applicant)
IDENTIFIED ISSUE RATIONALE FOR CONCERN RECOMMENDATIONinclude only a year and month, to be expressed as YYYY-MM if only numerical characters are used or YYYY-MMM if alphabetical characters are used to represent the month. FDA recommends that a hyphen or a space be used to separate the portions of the expiration date.
3. The proprietary and established names should be the most prominent information on the principle display panel (PDP).
Lack of adequate prominence of the proprietary and established names may increase the risk for selection errors.
Revise the PDP to ensure the proprietary name, established name, and product strength are the most prominent information presented on the PDP, taking into account all pertinent factors, including typography, layout, contrast, and other printing features in accordance with 21 CFR 201.10(g)(2).
4. The established name lacks prominence commensurate with the proprietary name on the principle display panel.
The prominence of the established name is not in accordance with 21 CFR 201.10(g)(2).
Increase the prominence of the established name taking into account all pertinent factors, including typography, layout, contrast, and other printing features in accordance with 21 CFR 201.10(g)(2).
Container Labels
1. The “Rx only” statement is not present on the container labels.
The “Rx” only statement is required on the drug label by Section 503(b)(4)(A) of the Federal Food, Drug, and Cosmetic Act.
Include the “Rx only” statement on the container label and ensure the “Rx only” statement appears less prominent than other important information (e.g., proprietary name, established name, strength).
Carton Labeling
1. The product strength is not expressed in milligram per single unit.
The carton labeling does not immediately make it clear that the designated strength is per one unit (one tablet), which could lead to dosing errors.
Revise the strength statement to make it clear that the designated strength is per unit (i.e., 50 mg per tablet and 100 mg per tablet) so there is no confusion as to
Reference ID: 4433095

5
Table 3. Identified Issues and Recommendations for Eli Lilly and Company (entire table to be conveyed to Applicant)
IDENTIFIED ISSUE RATIONALE FOR CONCERN RECOMMENDATIONhow much product is contained in a single unit as compared to the total contents of the entire blister card.
2. As currently presented, the graphic design is the most prominent information on the principal display panel (PDP).
The graphic design competes in prominence with other important information, distracts the reader from important information, and contributes to visual clutter.
Revise the PDP to ensure the graphic design does not compete in prominence with the proprietary name, established name, and product strength.
3. The “Rx only” statement is more prominent than other important information on the principle display panel (PDP).
The “Rx only”” statement is more prominent than the established name, which may increase the risk for selection errors.
Ensure the proprietary name, established name, and product strength are the most prominent information presented on the PDP, taking into account all pertinent factors, including typography, layout, contrast, and other printing features in accordance with 21 CFR 201.10(g)(2).
4. A machine-readable 2D data matrix barcode is not present.
The Drug Supply Chain Security Act (DSCSA) requires certain prescription drugs to have a human-readable and machine-readable (2D data matrix barcode) product identifier on the smallest saleable unit (usually the carton) for tracking and tracing purposes.
In September 2018, FDA released draft guidance on product identifiers required under the Drug Supply Chain Security Act.
The Act requires manufacturers and repackagers, respectively, to affix or imprint a product identifier to each package and homogenous case of a product intended to be introduced in a transaction in(to) commerce beginning November 27, 2017, and November 27, 2018, respectively.
We recommend that you review the draft guidance to determine if the product identifier requirements apply to your product’s labeling.
Reference ID: 4433095

6
Table 3. Identified Issues and Recommendations for Eli Lilly and Company (entire table to be conveyed to Applicant)
IDENTIFIED ISSUE RATIONALE FOR CONCERN RECOMMENDATION
The draft guidance is available from: https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm621044.pdf
5. The “usual dosage” statement is missing from the proposed carton labeling.
The “usual dosage” statement is required per 21 CFR 201.55.
Revised the statement
to read “Usual dosage: see prescribing information”, or a similar statement, in accordance with 21 CFR 201.55.
6. The NDC number and net quantity statement are located in close proximity to the product strength on the principal display panel (PDP).
We are concerned the presence of multiple numbers located in close proximity to one another on the PDP may contribute to medication errors. Additionally, when displayed on the PDP, the NDC number is customarily presented at the top of the PDP, above the proprietary name and the net quantity statement is customarily located at the bottom of the PDP.
Consider relocating the NDC number and net quantity statement away from the product strength on the PDP.
7. The current temperature statement does not contain the temperature scale designation (i.e., “C” or “F”) after each numeric value.
We are concerned the temperature statement could be misinterpreted and should therefore be revised for clarity.
Revise the temperature statement to include the temperature scale (i.e., “C” or “F”) after each numeric value.
8. The side panel is visually cluttered.
It is difficult to readily locate and understand critical safety information (e.g., equivalency statement, storage statement, warnings) presented on the side panel.
Reformat the information located on the side panel to ensure critical safety information can be readily located and understood, taking into account the font size and style, color contrast, and other design elements.
Reference ID: 4433095
(b) (4)
(b) (4)
(b) (4)

7
4 CONCLUSION
Our evaluation of the proposed Reyvow prescribing information (PI), trade and sample carton labeling and container labels, and medication guide (MG) identified areas of vulnerability that may lead to medication errors. Above, we have provided recommendations in Table 2 for the Division and Table 3 for the Applicant. We ask that the Division convey Table 3 in its entirety to Eli Lilly and Company so that recommendations are implemented prior to approval of this NDA.
Reference ID: 4433095
8
APPENDICES: METHODS & RESULTS FOR EACH MATERIAL REVIEWED APPENDIX A. PRODUCT INFORMATION/PRESCRIBING INFORMATION
Table 4 presents relevant product information for Reyvow that Eli Lilly and Company submitted on October 11, 2018 and April 15, 2019.
Table 4. Relevant Product Information for Reyvow
Initial Approval Date N/A
Active Ingredient Lasmiditan
Indication Acute treatment of migraine with or without aura in adults
Route of Administration Oral
Dosage Form Tablet
Strength 50 mg, 100 mg
Dose and Frequency 50 mg, 100 mg, or 200 mg, the maximum dose should not exceed 200 mg in 24 hours. If the migraine has resolved after taking 50 mg or 100 mg and then returns, a second dose may be administered.
How Supplied 50 mg:Carton
100 mg:Carton
Storage Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Container Closure card
Reference ID: 4433095
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)

9
APPENDIX F. LABELS AND LABELING F.1 List of Labels and Labeling Reviewed
Using the principles of human factors and Failure Mode and Effects Analysis,a along with postmarket medication error data, we reviewed the following Reyvow labels and labeling submitted by Eli Lilly and Company.
Container label(s) received on October 11, 2018 Carton labeling received on October 11, 2018 Professional Sample Blister cards received on October 11, 2018 Professional Sample Carton Labeling received on October 11, 2018 Medication Guide (Image not shown) received on October 11, 2018 Prescribing Information (Image not shown) received on April 15, 2019
a Institute for Healthcare Improvement (IHI). Failure Modes and Effects Analysis. Boston. IHI:2004.
Reference ID: 4433095
3 Pages of Draft Labeling have been Withheld in Full as B4(CCI/TS) Immediately Following this Page

--------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically. Following this are manifestations of any and allelectronic signatures for this electronic record.--------------------------------------------------------------------------------------------/s/------------------------------------------------------------
JOHN C MORRIS05/14/2019 11:40:22 AM
BRIANA B RIDER05/14/2019 03:02:09 PM
Signature Page 1 of 1
Reference ID: 4433095