cell, tissue, and gene therapies elizabeth read, md may 11, 2011
TRANSCRIPT

Cell, Tissue, andGene Therapies
Elizabeth Read, MDMay 11, 2011

Cell, Tissue & Gene TherapiesHeterogeneous group of (potential) products
Very few products on the market
Regulatory framework has evolved relatively recently (over past 20 years)
Special development considerations

Cell-based therapies originated with hematopoietic transplantation in 1970s
Bone marrow harvested, filtered, and transferred to blood bags in operating room
BM product carried directly to patient unit for infusion
Minimal donor & product testing, graft manipulation, quality systems
To date, FDA considers conventional autologous and allogeneic family-related BMT as “Practice of Medicine”

1980s – 2000s• Advances in science & technology spurred novel
approaches for development of cell-based therapies Hematopoietic transplants with “engineered” grafts starting
with bone marrow, peripheral blood, or cord blood sources Immunotherapies
T cells & subpopulations Dendritic cell tumor vaccines NK cells
Cellular gene therapies Cell therapies derived from bone marrow, other tissues, and
organs (e.g. mesenchymal stem cells, pancreatic islets)

During this period, clinical translation was facilitated by development of technologies for
collecting & handling cells in closed systems (often with single-use disposables)…
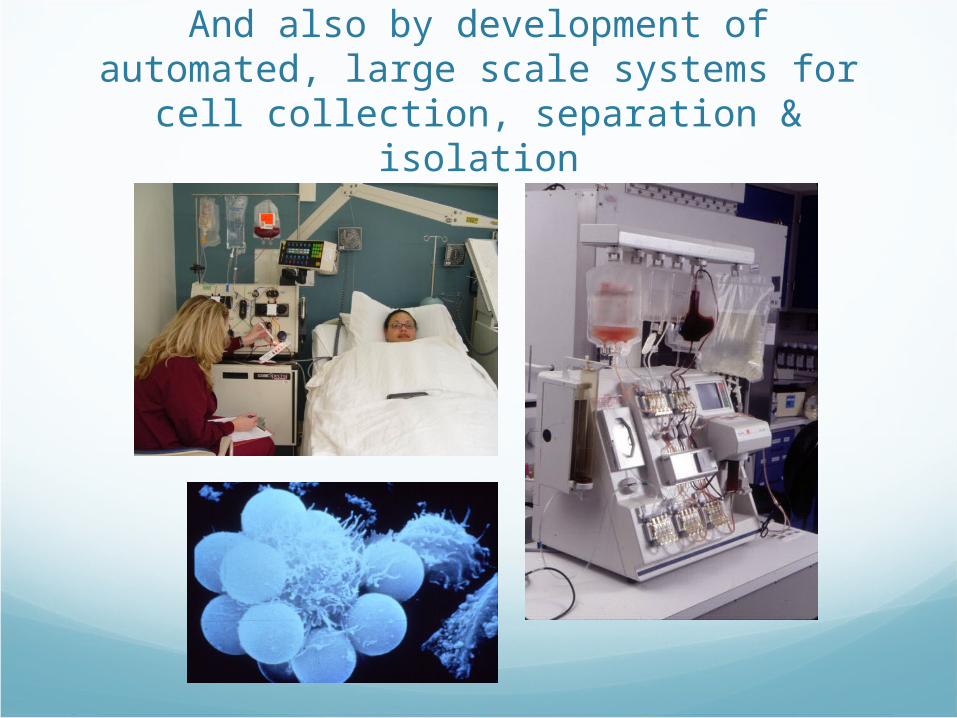
And also by development of automated, large scale systems for cell collection,
separation & isolation

2000s – PresentStem Cells & Regenerative Medicine
Explosion in stem cell science led to interest in use of stem cell-based therapies for many diseases and conditions, from cosmetic to life-threateningMultipotent
Adult stem cells from bone marrow, fat & other tissues/organs
Fetal stem cells & placental stem cells are usually considered “adult”
PluripotentEmbryonic stem (ES) cellsInduced pluripotent stem (iPS) cells

Scope of cell & tissue therapies Bone marrow and other hematopoietic stem cell transplantation
Cellular immunotherapies (dendritic cell vaccines, NK cells, T cells, etc)
Cell therapies derived from stem cells
Adult (including fetal) stem cells
Induced pluripotent stem cells
Embryonic stem cells
Cellular gene therapies
Conventional organ transplantation (e.g., kidney, heart, liver)
Conventional tissue transplantation (e.g., tendons, bone)
Reproductive tissue (sperm, oocytes, embryos)
Tissue engineering (autologous, allogeneic) – may include synthetic or natural biomaterials, or decellularized tissues
Xenotransplantation

How does FDA regulate these products?

Development pathway for cell & tissue therapies is similar to drugs & conventional
biologics
But with many important exceptions

Exception #1Transplantation of vascularized whole organs is
regulated by HRSA, not FDA

Exception #2Xenotransplantation is regulated by its own
separate set of FDA regulations

Exception #3Bone marrow transplantation using autologous
or family-related allogeneic donors is not regulated at all (practice of medicine)

Exception #4Bone marrow transplantation from unrelated
donors is regulated by HRSA, not FDA

Exception #5Some tissue products have been regulated by
CDRH as devices, with less stringent requirements and minimal involvement of CBER
This is historical – CBER will be involved going forward

What’s left in scope?

What’s left falls into FDA definition ofHCT/Ps
• Human cells, tissues, and cellular and tissue-based products (HCT/Ps) are articles containing human cells or tissues that are intended for implantation, transplantation, infusion, or transfer into a human recipient

FDA’s Risk-Based Approach forHCT/Ps
Lower risk “361”Autologous or family related donors and minimally
manipulated and homologous useRegulated under section 361 of Public Health
Service Act
Higher risk “351”Allogeneic unrelated donors and/or more than
minimally manipulated and/or non-homologous use
Regulated under section 351 of Public Health Service Act, and subject to same rules as drugs & other biologics for IND and premarket approval

FDA regulations for HCT/Ps361
HCT/Ps
351
HCT/Ps
(Tissue) Establishment registration √ √
(Tissue) Donor eligibility √ √
(Tissue) CGTP manufacturing √ √
cGMP regulations √
IND / IDE regulations √
Premarket approval (BLA or PMA) √

What about stem cells?
Cellular products derived from multipotent or pluripotent stem cells are
regulated as HCT/Ps


HCT/Ps derived from pluripotent stem cells: FDA concerns
CMC Donor source Consistency of differentiation & expansion process Detection of residual pluripotent stem cells Genetic and epigenetic stability
Preclinical studies Case-by-case approach “hybrid” efficacy/safety studies – much attention to
modeling ROA and biodistribution Tumorigenicity

HCT/Ps derived from pluripotent stem cells: FDA concerns
Clinical Protocol: for novel stem cell products, the risk : benefit assessment is difficult; therefore:Rationale for clinical trial must be justified by
especially strong proof of conceptGreater emphasis placed on product
characterization and preclinical testing

Gene Therapies

Gene therapy approaches IN VIVO: Vector administered directly to patient, and
transfers genetic information to patient cells in vivo Intravenously administered vector delivers gene for
factor IX to patient with hemophilia B
EX VIVO: Vector used to transfer genetic information to cells ex vivo, then cells are administered to patient Vector that delivers gene for enzyme adenosine
deaminase is incubated ex vivo with autologous lymphocytes of patient with ADA-deficient form of SCID (severe combined immunodeficiency), and genetically modified cells are infused to patient

Gene therapy: history1974: NIH established Recombinant DNA Advisory
Committee (RAC) NIH Guidelines on recombinant DNA research
1980s: New subcommittee of RAC to oversee clinical gene therapy Appendix M to NIH Guidelines – covered design of
preclinical & clinical research, consent issues, AE reporting
PUBLIC review of gene transfer protocols
1989: First clinical gene transfer study (gene marking) using retroviral vector
1990: First clinical gene transfer study (therapeutic intent) using retroviral vector

Gene therapy: history1995: No real clinical efficacy demonstrated, and
NIH report concluded that enthusiasm had outstripped knowledge Back to the bench for research on improved gene
delivery methods (e.g., higher titer vectors, use of stromal feeder layer or fibronectin for HSC transductions)
By 1995, NIH RAC Had approved 149 GT clinical protocols No dire consequences Policy change: public review & approval only for GT
protocols that presented novel or unresolved issues
1997: Role of NIH RAC modified – still required public review, but not “approval” of novel GT protocols

Jessie Gelsinger (1999) 18 y.o. with clinically mild form of
ornithine transcarbamlase defiency
Volunteered for clinical trial of gene therapy at U of Pennsylvania
Adenoviral vector caused massive immune response, muti-organ failure, and death within 4 days
All gene therapy trials placed on hold
Multiple ethical issues raised Adverse events in primate studies Adverse events in 2 previous human
subjects Informed consent Principal investigator conflict of interest

Insertional Oncogenesis2000-2007: X-linked SCID trials, using gamma
retroviral vectors to deliver the corrective gene (IL2RG) to autologous hematopoietic progenitor cells5 of 20 pts developed T cell leukemia-like
proliferative disorder, caused by INSERTIONAL ONCOGENESISRetroviral vector integrated adjacent to one or more
cellular proto-oncogenes (LMO-2 in 4 of the cases), which increased their expression, leading to malignant transformation and outgrowth of clonal population of T cells

Gene delivery methodsVector = an agent used to introduce genetic
material into cells
Vectors can beViralNon-viral
Plasmid DNALiposomes or other agents that facilitate entry into
cell

Viral vectorsRetrovirus and lentivirus (developed to
overcome inability of γ-retroviral vectors to infect non-dividing cells)
Adenovirus
Parvovirus (adeno-associated virus or AAV)
Herpes simplex virus
Poxvirus
Togavirus

Vector selection depends on… Disease state
Route of administration
Size of payload genetic sequences, regulatory elements
Cell cycling Lentivirus, adenovirus, AAV do not require cycling cells
Intended duration of expression Retrovirus and lentivirus give stable integration Plasmid used for transient expression
Target cells Poor expression of adenoviral CAR receptor on hematopoietic
cells

More advanced vector design features Conditional replication-competence
Control of gene expression Tissue-specific promoters Drug-responsive promoters
To reduce risk of insertional oncogenesis ofγ-retroviral and lentiviral vectors Self-inactivating (SIN design) Insulators
Suicide genes Ganciclovir administered to patient will kill cells with
thymidine kinase gene

Safety issuesObserved to date
Insertional mutagenesis/oncogenesis Immunogenicity
VectorTransgeneFBS (bovine protein used to manufacture vector)
Potential Inadvertent transmission & expression in non-
target cells (including germline, transplacental)

FDA regulations & guidance for gene therapies
Overall similar to biotechnology products ICH guidances
Gene therapy CMC guidance 2008Vector description, map, sequence analysisCell banks, viral banks, cell lines (packaging,
producer, feeder) Vector production/purificationDocumentation of RAC reviewFor ex vivo gene therapy, cell requirements same
as HCT/Ps (i.e. CMC guidance, tissue rules)

FDA guidance on GT delayed AEs Recommends preclinical study
designs to assess clinical risk Requires long term clinical follow up,
based on preclinical studies, for In vivo gene therapy with
persistence of vector sequences, when sequences are integrated
Ex vivo gene therapy with sequences integrated, or not integrated but have potential for latency & reactivation
Specific follow up observations yearly for at least 10 years, and reporting to FDA
Informed consent for long term follow up, and for use of retroviral vectors

RCR/RCL testing(FDA 2006 supplemental guidance)

Case Study
Cellular gene therapy for sickle cell disease
PI - Donald Kohn MD (UCLA)
Funded by CIRM

Sickle Cell Disease (SCD)Autosomal recessive disorder
Approx 8% of African Americans have mutation
Approx 1 in 500 African Americans is homozygous and has SCD
Clinical coursehemolytic anemiavaso-occlusive episodes (pain), strokes, acute
chest syndrome, progressive organ dysfunction

Molecular basis of SCD
Substitution of T for A in 6th codon of human β-globin gene
Results in non-polar valine instead of polar glutamic acid on the surface of HbS tetramer (α2βS2)

Molecular basis of SCD During partial deoxygenation,
valine creates hydrophobic pocket that fits into natural hydrophilic pocket on HbS tetramers, leading to HbS polymerization
This causes red blood cells to become rigid and poorly deformable, leading to hemolysis and impaired blood flow through microcirculation

Treatment of SCD Supportive for vaso-occlusive crisis
Pain medication, hydration, oxygen
Blood transfusions For some acute complications Prophylaxis for stroke and other complications Complications: iron overload, alloimmunization
Hydroxyurea Key mechanism: raises Hb F, which has anti-sickling effect Complications: pancytopenia
Allogenic bone marrow transplantation Potential for cure, but only 14% have HLA-matched sibling
donor

Potential Gene Therapy Strategies for SCD
Correct HbS mutation
But sickle β-globin acts in a dominant manner, and you would
need very high levels of expression to achieve a state similar
to sickle trait
Insert genes for normal HbF γ-globin into HSCs, in order to increase expression of HbF (α2γ2), to inhibit Hb S polymerization
and sickling
But fetal γ-globin gene is poorly expressed in adult RBCs, due
to absence of fetal-specific positive regulatory factors in
adult cells
Modify HbS β-globin gene to have anti-sickling properties
of γ-globin while retaining the adult HSC expression pattern
inherent in the β-globin gene

Townes βAS3 vectorSelf-inactivating (SIN) lentiviral vector
Carries and expressesβAS3, a β-globin gene with 3 amino acid substitutionsExpression product has biophysical anti-sickling
properties equivalent to fetal γ-globin AND advantage over βS–globin for dimerization with α-globin
Incorporates β-globin transcriptional regulatory elements

Preclinical Proof of Concept(Levasseur 2003)
In murine model of SCD, transduction of HSC with the lenti/βAS3 vectorExpression: 2-3 gm Hb/dl/vector copy Correction of hematological and clinical
manifestations of SCD

IND development for SCD gene therapy

Clinical protocol considerations Phase 1 trial
Risks: known and unknown Benefits: unlikely in first trial
SCD patient population Adults (ethical considerations for children) Should not be candidates for allo BMT (i.e., matched sibling
donor available) Severity of disease may impact
feasibility of cell collection endpoint assessment
Myeloablation with busulfan to create “space” in marrow

Product considerations Vector: based on Townes SIN lentiviral vector
Additional engineering underway to further reduce risk of insertional oncogenesisTAT independent backbone, insulators, etc.
Cell source Autologous Ideally want most primitive hematopoietic stem cells (HSCs)
that will differentiate into erthyroid cells HSCs vs iPS cells
iPS cells not quite ready for prime timeHSCs have track record, CD34+ selection isolates stem &
progenitor cells

Product considerationsHSC options
Placental/umbilical cord bloodmost proliferative source, but not useful for
autologous protocol in adultsG-CSF mobilized peripheral blood HSCs
SCD patients have had serious adverse events, including death, associated with G-CSF
Bone marrowWill require general anesthesiaAvailable cell dose will be an issue

Initial definition of product candidateThe investigational product is autologous human
CD34+ hematopoietic stem cells (HSC) from the bone marrow of patients with sickle cell disease (SCD) modified by ex vivo transduction using the βAS3 lentiviral vector

Quantitative targetsInitial quantitative targets for product
CD34+ : minimum 2 x 106/kgBack up BM MNCs: 5 x 107/kgVector in cells: 1-3 copies/cell
Based on estimates of Hb produced per VCN, and data showing benefit from Hb F of 10-20%

CMC: Beginning with the end in mind

CMC Development:Basic Manufacturing Process

CMC Development:Basic Manufacturing Process

Bone Marrow SourceAutologous - SCD
Is cell content (MNC, CD34) of bone marrow of SCD patients comparable to normal BM? PILOT STUDIES SAY YES
How much marrow to harvest? ENOUGH TO YIELD AT LEAST 1-2
x 106 CD34/kg IN FINAL PRODUCT, PLUS BACK UP OF 5 x 107 MNCs/kg

CMC Development:Basic Manufacturing Process
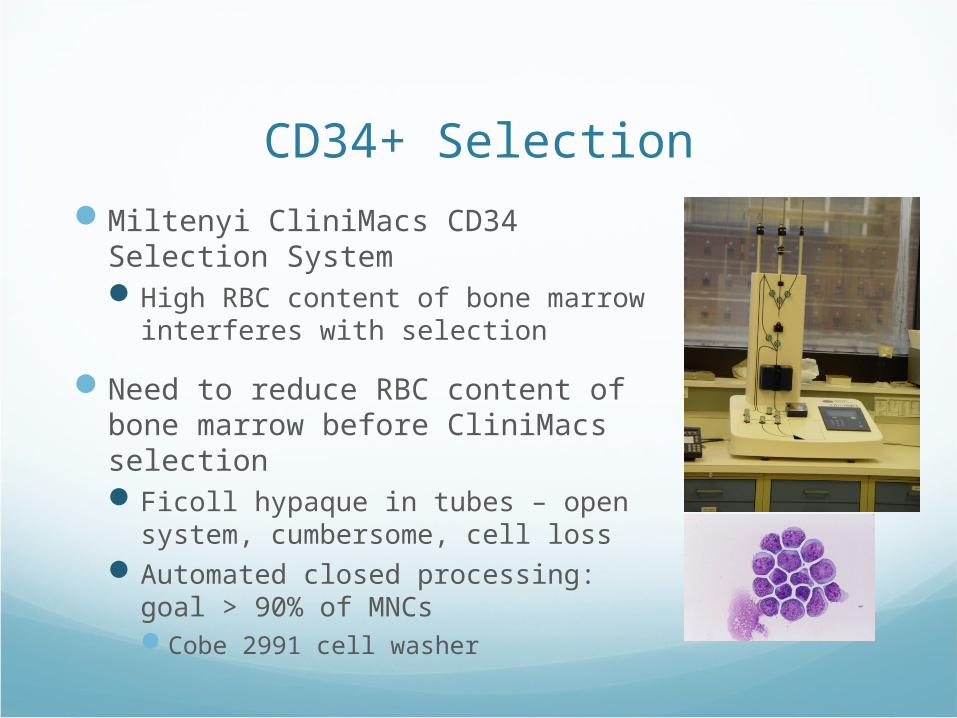
CD34+ Selection
Miltenyi CliniMacs CD34 Selection SystemHigh RBC content of bone marrow
interferes with selection
Need to reduce RBC content of bone marrow before CliniMacs selectionFicoll hypaque in tubes – open
system, cumbersome, cell lossAutomated closed processing: goal >
90% of MNCsCobe 2991 cell washer

CMC Development:Basic Manufacturing Process

Culture & Gene TransductionSmall scale experiments
Minimize differentiation of HSCsCytokines (SCF, Flt-3L, IL-3, Tpo)Overall culture duration
Optimize transduction efficiencyTiming: pre-stimulation in culture improves
transductionHow many hits?Recombinant human fibronectin fragment
Preserve vectorVector titers are not highQuantity will be limited

Culture & Gene TransductionAssays
Vector sequence in HSCsqPCR (vector copy number per cell)
Gene-modified HSCs are capable of erythroid differentiationIn vitro erythroid differentiation modelFold expansion & flow phenotype
RBC progeny have appropriate functionRheology and morphology
Gene-modified HSCs still contain stem cellsNOD/SCID/γc(null), primary/secondary transplants

CMC Development:Basic Manufacturing Process

A hitch: Timing of product manufacturing vs clinical protocol• Bone marrow harvest to obtain HSCs must
occur BEFORE busulfan starts
Busulfan schedule = 4 days + 2 days washout
Final gene-modified CD34 cell product cannot be given until after busulfan washout
Extended culture of cells is likely to result in differentiation of HSCs
THEREFORE WILL NEED TO CRYOPRESERVE EITHER INTERMEDIATE PRODUCT (CD34+ CELLS) OR FINAL GENE-MODIFIED PRODUCT

CMC Development:Manufacturing Process – Option A

CMC Development:Manufacturing Process – Option B

Cryopreservation & Thaw
Evaluate effects of cryopreservation & thawCD34+ cells vs final gene-modified CD34+
cellsOptimal cryomediumControlled rate device vs Mr. FrostyReadouts
Recovery of viable cellsIn vitro clonigenic assaysVector in cells and expression of gene product

Assay developmentAssay For preclinical studies For product in
clinical TrialCell counts, flow phenotype (CD34), viability
Research lab methods Clinical lab methods
Gene/vector in cells qPCR for vector copy number Same
Gene expression product (Hb AS3)
Isoelectric focusing (IEF): Hb AS3 migrates with Hb A, not with Hb S
Need another assay – patients transfused (Hb A)
Characterization & function of transduced cells (in vitro)
Erythroid differentiation culture• Currently being optimized• Assess expansion, differentiation (flow phenotype), transduction• Generate enucleated RBCs to evaluate rheology
No
Function of transduced cells (in vivo)
SCID-repopulating cells by LDA in NOD/SCID/γc(null) mouse model
• Clinical endpoints• Rheology of RBCs generated in vivo
Safety/Toxicity Assess risk of insertional mutagenesis and clonal imbalance• in vitro “clonal dominance” assay• in vivo mouse transplants
Micro cultures, endotoxin(RCL not needed if cells in culture < 4 days)

Project statusClinical protocol in draft
CMC development in progress
Preclinical studies in progress
Pre-IND meeting this summer

FDA CMC guidances

Cell & tissue therapiesapproved by FDA to date
Product Company Description; indication Year approved
FDA Center-Mechanism
Carticel Genzyme Autologous cultured chondrocytes; repair of traumatic knee injury
2000 CBER(BLA)
Provenge Dendreon Autologous dendritic cell tumor vaccine; prostate cancer
2010 CBER(BLA)
TransCyte Advanced Biohealing
Human fibroblast-derived temporary skin substitute; severe burns (now off market)
1997 CDRH(PMA)
Apligraf Organogenesis
Human keratinocytes + human fibroblasts in bovine collagen matrix; venous stasis leg ulcers, diabetic foot ulcers)
1998 CDRH(PMA)
Dermagraft
Advanced Biohealing
Human fibroblasts + extracellular matrix + bioabsorbable scaffold; diabetic foot ulcers
2001 CDRH(PMA)
Orcel Forticell Keratinocytes + dermal fibroblasts + bovine collagen; epidermolysis bullosa, burns
1998 CDRH(HDE)
Epicel Genzyme Autologous keratinocytes grown w/ murine fibroblasts; deep dermal or full thickness burns
2007 CDRH(HDE)