calculations of dipole moments, optical spectra, and...
TRANSCRIPT

J . Phys. Chem. 1988, 92, 2385-2390 2385
Conclusion In this study we have gained additional insight into the
mechanism of Hz production from water mediated by colloidal metal catalysts. The differing rates of decay of MV+ and of the conductiity signal observed in this investigation show that charging of the Ptc by electrons is followed by a slower uptake of protons and that the reducing species loading the Ptc above pH 7 cannot be assigned a simple formula like H-, although once equilibrium is established on the catalyst, the ratios of protons to electrons do not differ greatly from 0.5 predicted by this formula. Although both initial [MV'] and [Ptc] strongly affect the fractional decay of MV' , the concentration of MV2+ has little effect. An equi- librium with a Ptc surface saturated with adsorbed MV2+ is suggested. Slow changes in optical absorption and conductivity were found which can be attributed to equilibration of the Ptc with hydrogen (produced with G = 1.0). The ratio of protons to
electrons loading the Pt was found for this process to be similar to that in the case of reduction of the Ptc by MV+. The results described above therefore allow us to obtain a more complete understanding of the role played by colloidal metal catalysts in water reduction to hydrogen and have demonstrated the value of using the pulse radiolysis technique together with optical and conductivity detection methods in enabling us to probe such re- action systems.
Acknowledgment. The work described herein was supported by the Office of Basic Energy Sciences of the U S . Department of Energy as well as by the USA-Israel Binational Science Foundation and the Balfour Foundation.
Registry No. MV2+, 4685-14-7; MV*, 25239-55-8; H20, 7732-18-5;
propanol, 67-63-0; chloroplatinic acid, 16941-12-1; acetone, 67-64-1. Hz, 1333-74-0; Pt, 7440-06-4; 02, 7782-44-7; CH3C1, 74-87-3; 2-
Calculations of Dipole Moments, Optical Spectra, and Second-Order Hyperpolarizability Coefficients of Some Mono- and Disubstituted Stilbene Models for the Design of Nonlinear Optical Materials
Abraham Ulman Corporate Research Laboratories, Eastman Kodak Company, Rochester, New York 14650 (Received: July 1 , 1987; In Final Form: October 29, 1987)
The effects of substituents on the ground- and excited-state dipole moments, optical spectra, and second-order hyperpolarizability (@) in mono- and disubstituted stilbenes were studied. It was found that ground-state dipole moments as well as the charge on the 4'-position in 4-substituted stilbenes correlate with xu,. For optical spectra (Lx and oscillator strength), excited-state dipole moments, and fizz,, a correlation with both x u p and xu+/- was found.
Introduction The preparation and characterization of new materials for
nonlinear optics, with enhanced second- and third-order hyper- polarizabilities, and desired spectra properties are currently an area of intense interest.'s2 However, only with the pure material in hand can the molecular second-order hyperpolarizability coefficient (@) be determined. This EFISH (electric field induced second harmonic) experiment3 is also quite a complicated pro- cedure. Thus, a method is needed by which the organic chemistry could anticipate the properties of the suggested molecules prior to their synthesis. In this way, a better understanding of the relationships of structural properties to nonlinear optical properties can be obtained. Thus, synthesis of only the most promising molecules need be pursued.
Two molecular properties are important for nonlinear optics: (a) high hyperpolarizability density (@/V) (where Vis the volume per molecule) and (b) absorption spectra under 800 nm for electrooptic devices or under 400 nm for second-harmonic gen- eration (SHG) or parametric devices.
The requirements for good nonlinear optical materials will become clear only if the origin of nonlinear optical effects is well-understood. The macroscopic polarization [P( t ) ] can be comprised of the molecular nonlinear optical response, which is expressed as
P = N [ ( a ) E + (8) + ( y ) E E E + ...I (1)
where a is the linear polarizability, P and y are the second- and
(1) Li: D.; Marks, T. J.; Ratner, M. A. Chem. Phys. Lett. 1986,131,370. ( 2 ) D r k , C. W.; Twieg, R. J.; Wagniere, G . J. Am. Chem. Soc. 1986, 108,
(3) Levine, B. F.; Bethea, C . G . J . Chem. Phys. 1975, 63, 2666. 5381.
third-order nonlinear electronic susceptibilities, respectively, ( ) denotes an orientational average, and N is the concentration of chromophores. @ is a third-rank tensor (&, where ijk specify the Cartesian components). In molecules with strong intramo- lecular charge transfer (CT), the simplest approximation for describing fi is as a sum of two contributions4q5
(2) P = PADD + PCT where PADD can simply be due to the substituents inductive effects. In other words, PADD is the result of the asymmetry in the mystem due to the inductive effect of the substituent (I). On the other
I
hand, ficr is due to the resonance conjugation of the substituents, which may be expressed as the relative contributions of two va- lence-bond structures, i.e., one nonpolar ground state and one polar excited state (11). This resonance term, which is the dominant
D D+
I1
(4) Levine, B. F. Chem. Phys. Lett. 1976, 37, 516. (5) Oudar, J . L.; Chemla, D. S. J . Chem. Phys. 1977, 66, 2664
0022-3654/88/2092-2385$01.50/0 0 1988 American Chemical Society
2386
one, can be expressed, in a two-state model approximation, as a function of the difference between the ground- and excited-state dipole moments A W , , ~ by eq 35 and 4 where F(w) is a dispersion
The Journal of Physical Chemistry, Vol. 92, No. 8, 1988 Ulman
(3)
(4)
term: Wis the energy of the optical transition, and f is its oscillator strength. Thus, it is apparent that both f and Abgc are the elements which control (?, and when seeking new materials for NLO, one should look for ways to enhance these terms. The nature of the substituents, as well as the conjugation between them, is among the parameters which determine f as well as A P ~ , ~ .
It is important to emphasize that in a more complete pertur- bation description of the dipolar contribution to the hyperpolar- izability /3, contributions from all excited states can contribute to /3 and the charge-transfer resonance most heavily influencing @ could arise from states other than the first excited state. Thus, while the two-level model contributes to our understanding of P in a qualitative way, it is far from being adequate and the semiempirical SOS calculations as described in this work are a better approximation.
In our design of new molecules for nonlinear optics we adopt the following sequence of calculations:
1. Calculate structural parameters by using the AM1 semi- empirical method.’
2. Calculate the spectra and transition dipoles by using the INDO method proposed by Ridley and Zerner.* The configu- ration interaction (CI) calculations included 100 singly excited configurations built from the 10 highest occupied molecular or- bitals (HOMO) and the 10 lowest unoccupied molecular orbitals (LUMO). This amount of CI calculations is more than sufficient for our p ~ r p o s e . ~ ~ ~
3. Calculation of the molecular second-order hyperpolarizability coefficient (8) using a semiempirical SCF-CI basis set (described in step 2 ) and a second-order perturbation theory expression for /3 with a sum over all singly excited states method (SOS)1,2q10,21
W F(w) =
[ W - (2hw)2][W- (hw)2]
1 (glpile) (glP,le’) (e’lPklg) IBVk = 4hZS 5 (weg - 2W)(W,,$ - w )
where weg = we - wg, p i = wI - (glp,lg), p l is the dipole operator, and P is permute sets ((-2w,r)(w,s)(w,t)) (six terms). In this method @ADD as well as BcT is calculated, since all the valence electrons in the molecule are used. Thus, while in the EFISH experiment only P,, which is parallel to the molecular ground-state dipole moment, is determined,” the theoretical calculations provide more complete information on the various components of @ and their sources.
DANS (4-(dimethylamino)-4‘-nitrostilbene) has been widely investigated both for second- and third-order nonlinear effects.12 In fact, this molecule is a good example for other conjugated diaromatic systems that carry a strong electron-donor group on one side and a strong electron-acceptor group on the other. The
(6) Williams, D. J. Angew. Chem., Inr. Ed. Engl. 1984, 23, 690. (7) See, for example: Dewar, M. J. S.; Zoebjsch, E. G.; Healy, E. F.;
Steward, J. J. P. J . Am. Chem. Sac. 1985, 107, 3902 and references cited therein.
(8) Ridley, J. E.; Zerner, M. C. Theor. Chim. Acta 1973,32, 11 1. Theor. Chim. Acta 1976, 42, 223. Theor. Chim. Acra 1979, 53, 21, and references cited therein.
(9) We have used from 60 to 100 singly excited configurations in the INDO calculations and found that indeed, for all cases, the difference between the two extremes is negligible.
(10) Vincent, J.; Docherty, J.; Pugh, D.; Morley, J. 0. J. Chem. Sac., Faraday Trans. 2 1985, 81, 1179.
(1 1) In the EFISH experiment, the z component of the @,, is determined:
B z = 8 , z z + %(a, + @zxx + 2 L Z + 2bYY8,,,)
However, in a molecule with strong intramolecular charge transfer p, - p,,. (12) Levine, B. F.; Bethea, C. G. J . Chem. Phys. 1978,69, 6241. Koba-
yashi, Y.; Ohtani, H.; Kurokawa, K. Chem. Phys. Lett. 1985, 121, 356.
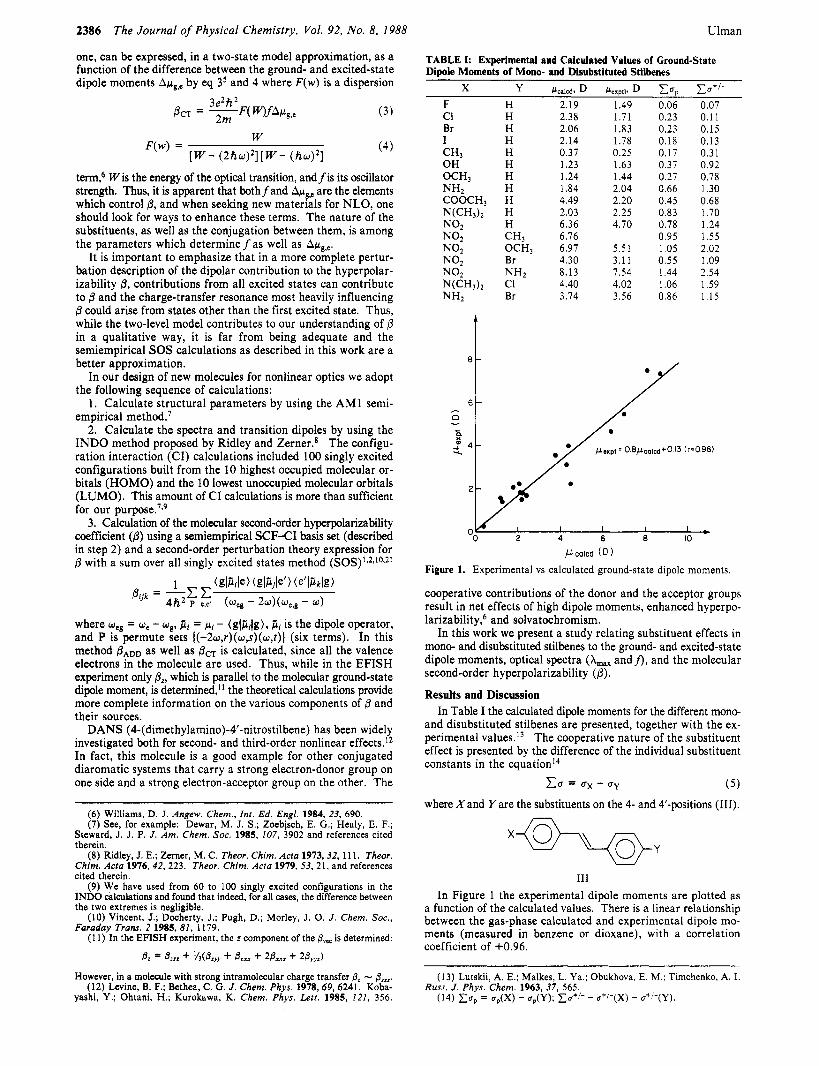
TABLE I: Experimental and Calculated Values of Ground-State Dipole Moments of Mono- and Disubstituted Stilbenes
X Y IL,.M, D I L ~ ~ I , D Can XU+/- H H H H H H H H H H H CH3 OCH, Br NH2 CI Br
2.19 1.49 0.06 0.07 2.38 1.71 0.23 0.11 2.06 1.83 0.23 0.15 2.14 1.78 0.18 0.13 0.37 0.25 0.17 0.31 1.23 1.63 0.37 0.92 1.24 1.44 0.27 0.78 1.84 2.04 0.66 1.30 4.49 2.20 0.45 0.68 2.03 2.25 0.83 1.70 6.36 4.70 0.78 1.24 6.76 0.95 1.55 6.97 5.51 1.05 2.02 4.30 3.11 0.55 1.09 8.13 7.54 1.44 2.54 4.40 4.02 1.06 1.59 3.74 3.56 0.86 1.15
OO s 2 4 6 8 IO
P calcd ( D )
Figure 1. Experimental vs calculated ground-state dipole moments.
cooperative contributions of the donor and the acceptor groups result in net effects of high dipole moments, enhanced hyperpo- larizability: and solvatochromism.
In this work we present a study relating substituent effects in mono- and disubstituted stilbenes to the ground- and excited-state dipole moments, optical spectra (A,,, a n d n , and the molecular second-order hyperpolarizability (P ) . Results and Discussion
In Table I the calculated dipole moments for the different mono- and disubstituted stilbenes are presented, together with the ex- perimental values.13 The cooperative nature of the substituent effect is presented by the difference of the individual substituent constants in the equation14
( 5 ) Cu = ux - CTy
where X and Yare the substituents on the 4- and 4’-positions (111). m
“Y I
111
In Figure 1 the experimental dipole moments are plotted as a function of the calculated values. There is a linear relationship between the gas-phase calculated and experimental dipole mo- ments (measured in benzene or dioxane), with a correlation coefficient of +0.96.
(13) Lutskii, A. E.; Malkes, L. Ya.; Obukhova, E. M.; Timchenko, A. I. Russ. J . Phys. Chem. 1963, 37, 565.
(14) x u p = u,(X) - u,(Y); XU*’- - u”-(X) - u’’-(Y).
Mono- and Disubstituted Stilbenes
t The Journal of Physical Chemistry, Vol. 92, No. 8. 1988 2387
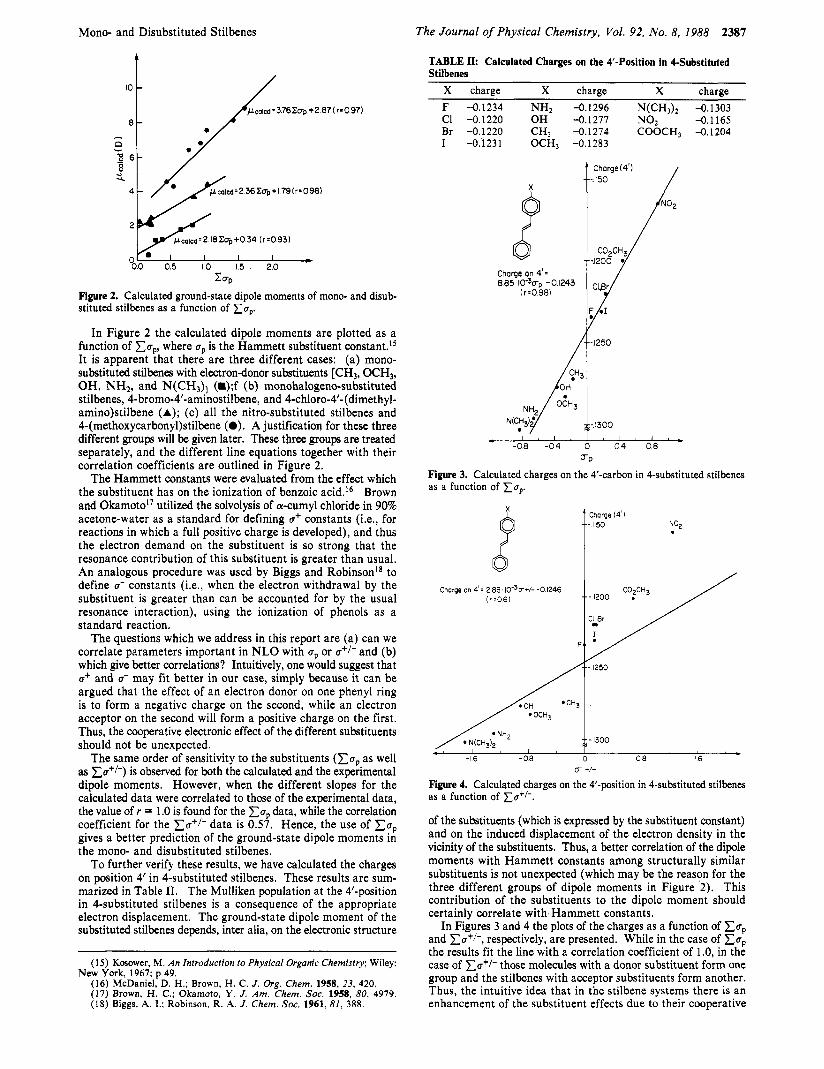
TABLE II: Calculated Charges on the 4'-Position in 4-Substituted Stilbenes
%? 015 I1O 115 2'0 * ZUP
Figure 2. Calculated ground-state dipole moments of mono- and disub- stituted stilbenes as a function of x u p
In Figure 2 the calculated dipole moments are plotted as a function of xu,, where up is the Hammett substituent con~tant . '~ It is apparent that there are three different cases: (a) mono- substituted stilbenes with electron-donor substituents [CH3, OCH3, OH, NH2, and N(CH,), (m);f (b) monohalogeno-substituted stilbenes, 4-bromo-4'-aminostilbene, and 4-chloro-4'-(dimethyl- amino)stilbene (A); (c) all the nitro-substituted stilbenes and 4-(methoxycarbony1)stilbene (0). A justification for these three different groups will be given later. These three groups are treated separately, and the different line equations together with their correlation coefficients are outlined in Figure 2.
The Hammett constants were evaluated from the effect which the substituent has on the ionization of benzoic acid.16 Brown and Okamotol' utilized the solvolysis of a-cumyl chloride in 90% acetone-water as a standard for defining u+ constants (Le., for reactions in which a full positive charge is developed), and thus the electron demand on the substituent is so strong that the resonance contribution of this substituent is greater than usual. An analogous procedure was used by Biggs and Robinson18 to define u- constants (Le., when the electron withdrawal by the substituent is greater than can be accounted for by the usual resonance interaction), using the ionization of phenols as a standard reaction.
The questions which we address in this report are (a) can we correlate parameters important in NLO with up or a+/- and (b) which give better correlations? Intuitively, one would suggest that u+ and u- may fit better in our case, simply because it can be argued that the effect of an electron donor on one phenyl ring is to form a negative charge on the second, while an electron acceptor on the second will form a positive charge on the first. Thus, the cooperative electronic effect of the different substituents should not be unexpected.
The same order of sensitivity to the substituents ( x u p as well as xu+/-) is observed for both the calculated and the experimental dipole moments. However, when the different slopes for the calculated data were correlated to those of the experimental data, the value of r = 1 .O is found for the x u p data, while the correlation coefficient for the xu+/- data is 0.57. Hence, the use of x u p gives a better prediction of the ground-state dipole moments in the mono- and disubstituted stilbenes.
To further verify these results, we have calculated the charges on position 4' in 4-substituted stilbenes. These results are sum- marized in Table 11. The Mulliken population at the 4'-position in 4-substituted stilbenes is a consequence of the appropriate electron displacement. The ground-state dipole moment of the substituted stilbenes depends, inter alia, on the electronic structure
(15) Kosower, M. An Introduction to Physical Organic Chemistry: Wiley:
(16) McDaniel, D. H.; Brown, H. C. J . Org. Chem. 1958, 23, 420. (17) Brown, H. C.; Okamoto, Y . J. Am. Chem. SOC. 1958, 80, 4979. (18) Biggs, A. I.: Robinson, R. A. J . Chem. SOC. 1961, 81, 388.
New York, 1967; p 49.
X charge X charge X charge F -0.1234 NH2 -0.1296 N(CH,)2 -0.1303 CI -0.1220 OH -0.1277 NO2 -0.1165
I -0.'1231 OCH, -0.1283 Br -0.1220 CH, -0.1274 COOCH, -0.1204
x
0 Charge on 4'. 885 I 0 d r p -01243
(r=0981
-4200
i1 -4250
* ' I ' I ' -08 -04 0 0 4 0 8
UP Figure 3. Calculated charges on the 4'-carbon in 4-substituted stilbenes as a function of x u P .
Charge 14'1 - 1150 NO2 f
Charge on 4'; 2 88 10-3P+/- -0 1246 (rz06)
1 . -16 -08 0 0 8 16
u +/-
Figure 4. Calculated charges on the 4'-position in 4-substituted stilbenes as a function of xu+/-.
of the substituents (which is expressed by the substituent constant) and on the induced displacement of the electron density in the vicinity of the substituents. Thus, a better correlation of the dipole moments with Hammett constants among structurally similar substituents is not unexpected (which may be the reason for the three different groups of dipole moments in Figure 2 ) . This contribution of the substituents to the dipole moment should certainly correlate with Hammett constants.
In Figures 3 and 4 the plots of the charges as a function of Cup and ZCT+/-, respectively, are presented. While in the case of Cup the results fit the line with a correlation coefficient of 1.0, in the case of xu+/- those molecules with a donor substituent form one group and the stilbenes with acceptor substituents form another. Thus, the intuitive idea that in the stilbene systems there is an enhancement of the substituent effects due to their cooperative
2388 The Journal of Physical Chemistry, Vol. 92, No. 8 , 1988
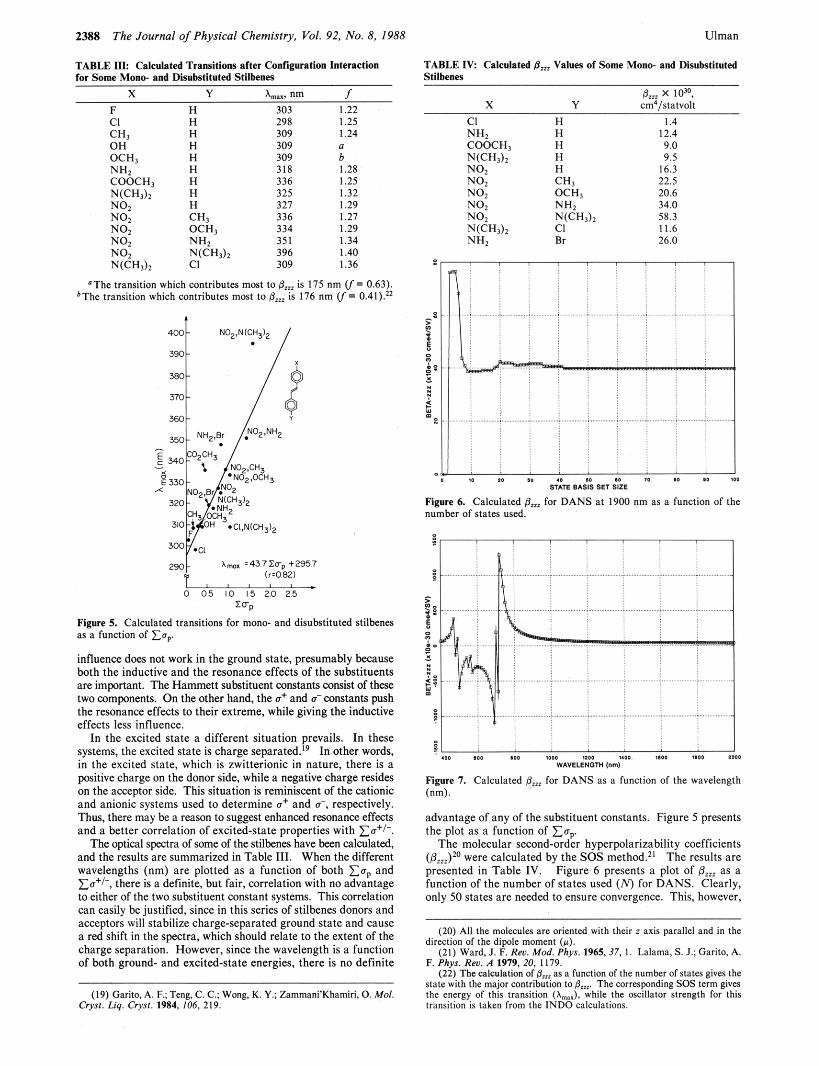
TABLE III: Calculated Transitions after Configuration Interaction for Some Mono- and Disubstituted Stilbenes
Ulman
TABLE IV: Calculated 8,, Values of Some Mono- and Disubstihlted Stilbenes
8, x lo=, X Y cm'/statvolt
CI H I .4 NH2 H 12.4 COOCH, H 9.0
NO, H 16.3
NO2 OCH, 20.6 NO2 NH2 34.0
N(CHJ2 c1 11.6 NH, Br 26.0
N ( C H 3 ) 2 H 9.5
NO, CHI 22.5
NO, N(CHd2 58.3
X Y Amax. nm f F H 303 1.22 CI H 298 1.25 ~~
H H H H H H H CH,
309 309 309 318 336 325 327 336
I .24
b 1.28 1.25 1.32 1.29 1.27
a
NO; OCH, 334 1.29 NO. NH. 351 1.34
'The transition which contributes most to &, is 175 nm (f= 0.63). bThe transition which contributes most to 8, is 176 nm (f= 0.41)."
I
390
380
370
Figure 6. Calculated 8, for DANS at 1900 nm as a function of thc number of states used.
0 05 10 15 2 0 2 5
= U P
Figure 5. Calculated transitions for mono- and disubstituted stilbenes as a function of Eav
influence does not work in the ground state, presumably because both the inductive and the resonance effects of the substituents are important. The Hanunett substituent wnstants wnsist of these two wmponents. On the other hand, the a+ and d wnstants push the resonance effects to their extreme, while giving the inductive effects less influence.
In the excited state a different situation prevails. In these systems, the excited state is charge separated.19 In other words, in the excited state, which is zwitterionic in nature, there is a positive charge on the donor side, while a negative charge resides on the acceptor side. This situation is reminiscent of the cationic and anionic systems used to determine a+ and 6, respectively. Thus, there may be a reason to suggest enhanced m n a n c e effects and a better correlation of excited-state properties with xo+l-.
The optical spectra of some of the stilbenes have been calculated, and the results are summarized in Table 111. When the different wavelengths (nm) are plotted as a function of both x u D and ~ c + ~ - , there is a definite, hut fair, correlation with no advantage to either of the two substituent constant systems. This correlation can easily be justified, since in this series of stilbenes donors and acceptors will stabilize charge-separated ground state and cause a red shift in the spectra, which should relate to the extent of the charge separation. However, since the wavelength is a function of both ground- and excited-state energies, there is no definite
(19) Garita,A. F.:Teng. C.C.; W0ng.K. Y.: Zammani'Khamiri.0. Mol. Cryst. Lzq. Cryst. 1984. 106, 219.
Figure 7. Calculated 8,, for DANS as a function of the wavelength (nm).
advantage of any of the substituent constants. Figure 5 presents the plot as a function of Zap.
The molecular second-order hyperpolarizability coefficients (&sJ20 were calculated by the SOS method.z' The results are presented in Table IV. Figure 6 presents a plot of paZz as a function of the number of states used (N) for DANS. Clearly, only 50 states are needed to ensure convergence. This, however,
(20) All the molecules are oriented with their z axis parallel and in the direction of the dipole moment (w).
(21) Ward. J. F. Reo. Mod. Phys. 1965. 37, 1. Lalama, S. J.:Garito. A. F. Phys. Reo. A 1979. 20, 1179.
(22) The calculation of O,, as a function of the number of states gives the state with the major contribution to &,. The corresponding SOS term gives the energy of this transition (Am,"), while the oscillator strcnath for this transition is taken from the INDO calculations.
Mono- and Disubstituted Stilbenes
(r = 095) ( r = O 8 4 ) /!
-.-.-ALL
I' OCL
C \ap 05 1 0 1 5 2 0
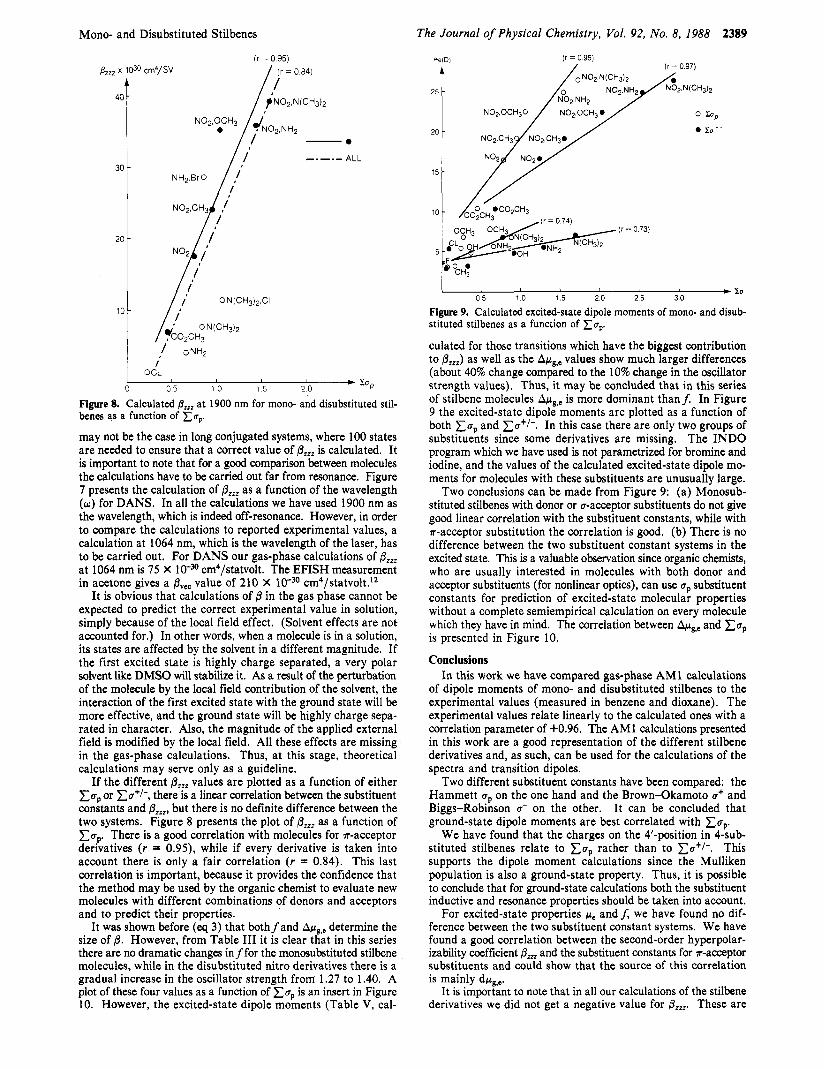
Figure 8. Calculated @,,, at 1900 nm for mono- and disubstituted stil- benes as a function of x u p .
may not be the case in long conjugated systems, where 100 states are needed to ensure that a correct value of p,,, is calculated. It is important to note that for a good comparison between molecules the calculations have to be carried out far from resonance. Figure 7 presents the calculation of p,,, as a function of the wavelength (w) for DANS. In all the calculations we have used 1900 nm as the wavelength, which is indeed off-resonance. However, in order to compare the calculations to reported experimental values, a calculation at 1064 nm, which is the wavelength of the laser, has to be carried out. For DANS our gas-phase calculations of @,, at 1064 nm is 75 X cm4/statvolt. The EFISH measurement in acetone gives a p,,, value of 210 x
It is obvious that calculations of p in the gas phase cannot be expected to predict the correct experimental value in solution, simply because of the local field effect. (Solvent effects are not accounted for.) In other words, when a molecule is in a solution, its states are affected by the solvent in a different magnitude. If the first excited state is highly charge separated, a very polar solvent like DMSO will stabilize it. As a result of the perturbation of the molecule by the local field contribution of the solvent, the interaction of the first excited state with the ground state will be more effective, and the ground state will be highly charge sepa- rated in character. Also, the magnitude of the applied external field is modified by the local field. All these effects are missing in the gas-phase calculations. Thus, a t this stage, theoretical calculations may serve only as a guideline.
If the different p,,, values are plotted as a function of either x u p or xu+/-, there is a linear correlation between the substituent constants and pZzB,,,, but there is no definite difference between the two systems. Figure 8 presents the plot of p,,, as a function of Cup. There is a good correlation with molecules for a-acceptor derivatives ( r = 0.95), while if every derivative is taken into account there is only a fair correlation ( r = 0.84). This last correlation is important, because it provides the confidence that the method may be used by the organic chemist to evaluate new molecules with different combinations of donors and acceptors and to predict their properties.
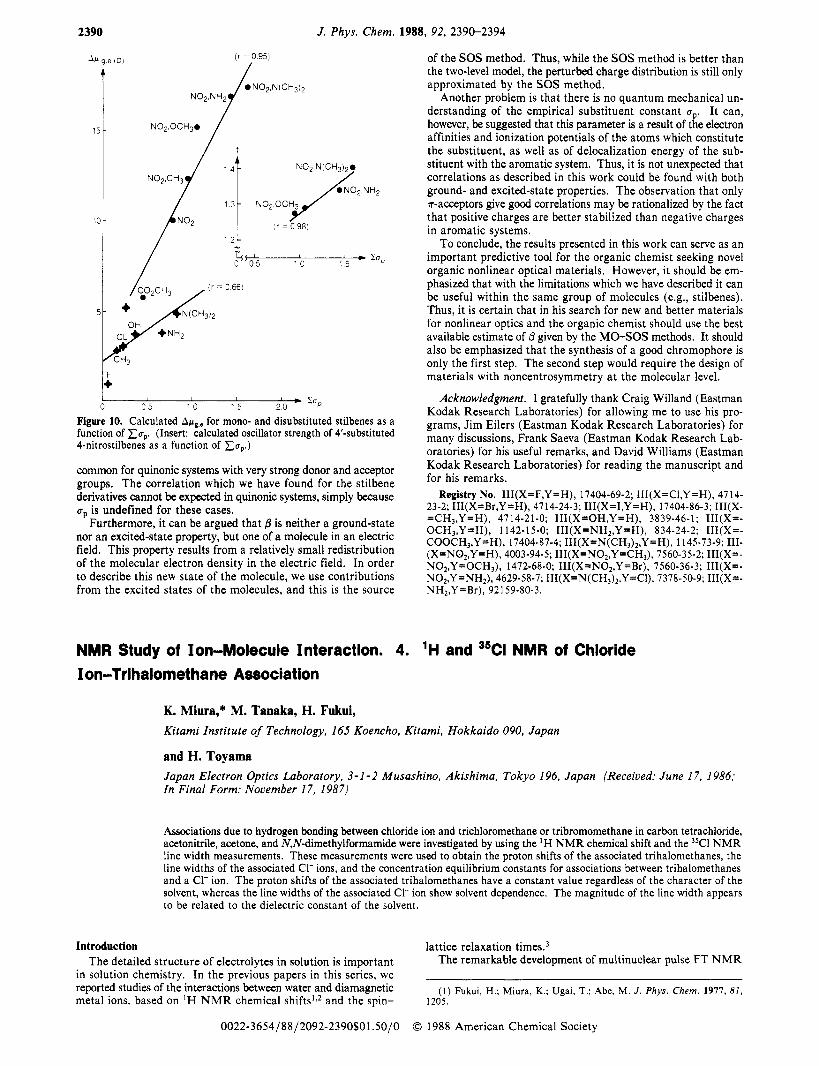
It was shown before (eq 3) that bothfand Apg,e determine the size of p. However, from Table I11 it is clear that in this series there are no dramatic changes in f for the monosubstituted stilbene molecules, while in the disubstituted nitro derivatives there is a gradual increase in the oscillator strength from 1.27 to 1.40. A plot of these four values as a function of Cap is an insert in Figure 10. However, the excited-state dipole moments (Table V, cal-
~ m ~ / s t a t v o ~ t . ' ~
The Journal of Physical Chemistry, Vol. 92, No. 8, 1988 2389
25:
(r = 0.74) ( r = 0 73)
0 Zo'
I - To 0 5 1 0 1 5 2 0 2 5 30
Figure 9. Calculated excited-state dipole moments of mono- and disub- stituted stilbenes as a function of x u p
culated for those transitions which have the biggest contribution to p,,,) as well as the Apg,e values show much larger differences (about 40% change compared to the 10% change in the oscillator strength values). Thus, it may be concluded that in this series of stilbene molecules Apg,e is more dominant than$ In Figure 9 the excited-state dipole moments are plotted as a function of both x u p and xu+/-. In this case there are only two groups of substituents since some derivatives are missing. The INDO program which we have used is not parametrized for bromine and iodine, and the values of the calculated excited-state dipole mo- ments for molecules with these substituents are unusually large.
Two conclusions can be made from Figure 9: (a) Monosub- stituted stilbenes with donor or u-acceptor substituents do not give good linear correlation with the substituent constants, while with ?r-acceptor substitution the correlation is good. (b) There is no difference between the two substituent constant systems in the excited state. This is a valuable observation since organic chemists, who are usually interested in molecules with both donor and acceptor substituents (for nonlinear optics), can use up substituent constants for prediction of excited-state molecular properties without a complete semiempirical calculation on every molecule which they have in mind. The correlation between Apg,e and x u p is presented in Figure 10.
Conclusions In this work we have compared gas-phase AM1 calculations
of dipole moments of mono- and disubstituted stilbenes to the experimental values (measured in benzene and dioxane). The experimental values relate linearly to the calculated ones with a correlation parameter of +0.96. The AM 1 calculations presented in this work are a good representation of the different stilbene derivatives and, as such, can be used for the calculations of the spectra and transition dipoles.
Two different substituent constants have been compared: the Hammett up on the one hand and the Brown-Okamoto u+ and Biggs-Robinson u- on the other. It can be concluded that ground-state dipole moments are best correlated with xup .
We have found that the charges on the 4'-position in 4-sub- stituted stilbenes relate to x u p rather than to xu+/-. This supports the dipole moment calculations since the Mulliken population is also a ground-state property. Thus, it is possible to conclude that for ground-state calculations both the substituent inductive and resonance properties should be taken into account.
For excited-state properties pe andf , we have found no dif- ference between the two substituent constant systems. We have found a good correlation between the second-order hyperpolar- izability coefficient @,,, and the substituent constants for a-acceptor substituents and could show that the source of this correlation is mainly dpg,e.
It is important to note that in all our calculations of the stilbene derivatives we did not get a negative value for p,,,. These are
J. Phys. Chem. 1988, 92, 2390-2394 2390
AIJ 9 e ID1 (r = 0 951
N02.0CH30 /
II
05 1 0 1 5 2'0 LOP
Figure 10. Calculated AwglC for mono- and disubstituted stilbenes as a function of x u p . (Insert: calculated oscillator strength of 4'-substituted 4-nitrostilbenes as a function of x u p )
common for quinonic systems with very strong donor and acceptor groups. The correlation which we have found for the stilbene derivatives cannot be expected in quinonic systems, simply because up is undefined for these cases.
Furthermore, it can be argued that j3 is neither a ground-state nor an excited-state property, but one of a molecule in an electric field. This property results from a relatively small redistribution of the molecular electron density in the electric field. In order to describe this new state of the molecule, we use contributions from the excited states of the molecules, and this is the source
NMR Study of Ion-Molecule Interaction. 4. Ion-Trlhalomethane Association
of the SOS method. Thus, while the SOS method is better than the two-level model, the perturbed charge distribution is still only approximated by the SOS method.
Another problem is that there is no quantum mechanical un- derstanding of the empirical substituent constant up. It can, however, be suggested that this parameter is a result of the electron affinities and ionization potentials of the atoms which constitute the substituent, as well as of delocalization energy of the sub- stituent with the aromatic system. Thus, it is not unexpected that correlations as described in this work could be found with both ground- and excited-state properties. The observation that only s-acceptors give good correlations may be rationalized by the fact that positive charges are better stabilized than negative charges in aromatic systems.
To conclude, the results presented in this work can serve as an important predictive tool for the organic chemist seeking novel organic nonlinear optical materials. However, it should be em- phasized that with the limitations which we have described it can be useful within the same group of molecules (e.g., stilbenes). Thus, it is certain that in his search for new and better materials for nonlinear optics and the organic chemist should use the best available estimate of /3 given by the MO-SOS methods. It should also be emphasized that the synthesis of a good chromophore is only the first step. The second step would require the design of materials with noncentrosymmetry at the molecular level.
Acknowledgment. I gratefully thank Craig Willand (Eastman Kodak Research Laboratories) for allowing me to use his pro- grams, Jim Eilers (Eastman Kodak Research Laboratories) for many discussions, Frank Saeva (Eastman Kodak Research Lab- oratories) for his useful remarks, and David Williams (Eastman Kodak Research Laboratories) for reading the manuscript and for his remarks.
Registry No. III(X=F,Y=H), 17404-69-2; III(X=Cl,Y=H), 4714- 23-2; III(X=Br,Y=H), 4714-24-3; III(X=I,Y=H), 17404-86-3; III(X- =CH,,Y=H), 4714-21-0; III(X=OH,Y=H), 3839-46-1; III(X=- OCH,,Y=H), 1142-15-0; III(X=NH,,Y=H), 834-24-2; III(X=- COOCH,,Y=H), 17404-87-4; III(X=N(CH,)z,Y=H), 1145-73-9; III- (X=NQ,,Y=H), 4003-94-5; III(X=NOz,Y=CHJ, 7560-35-2; III(X=-
NOZ,Y=NHJ, 4629-58-7; III(X=N(CH3)Z,Y=CI), 7378-50-9; III(X=- NO,,Y=OCH,), 1472-68-0; III(X=N02,Y=Br), 7560-36-3; III(X=-
NH2,Y=Br), 92159-80-3.
'H and 35CI NMR of Chloride
K. Miura,* M. Tanaka, H. Fukui, Kitami Institute of Technology, I65 Koencho, Kitami, Hokkaido 090, Japan
and H. Toyama Japan Electron Optics Laboratory, 3- 1-2 Musashino, Akishima, Tokyo I96, Japan (Received: June 17, 1986; In Final Form: November 17, 1987)
Associations due to hydrogen bonding between chloride ion and trichloromethane or tribromomethane in carbon tetrachloride, acetonitrile, acetone, and N,N-dimethylformamide were investigated by using the 'H NMR chemical shift and the 35Cl NMR line width measurements. These measurements were used t9 obtain the proton shifts of the associated trihalomethanes, the line widths of the associated CI- ions, and the concentration equilibrium constants for associations between trihalomethanes and a CI- ion. The proton shifts of the associated trihalomethanes have a constant value regardless of the character of the solvent, whereas the line widths of the associated C1- ion show solvent dependence. The magnitude of the line width appears to be related to the dielectric constant of the solvent.
Introduction lattice relaxation timesS3 The detailed structure of electrolytes in solution is important
in solution chemistry. In the previous papers in this series, we
metal ions, based on 'H N M R chemical shifts's2 and the spin-
The remarkable development of multinuclear pulse FT N M R
reported studies of the interactions between water and diamagnetic (1) Fukui, H.; Miura, K.; Ugai, T.; Abe, M. J . Phys. Chem. 1977, 81, 1205.
0022-3654/88/2092-2390$01.50/0 0 1988 American Chemical Society