calculation of nmr chemical shifts - the third dimension of quantum chemistry
TRANSCRIPT

Calculation of NMR Chemical Shifts - The Third Dimensionof Quantum Chemistry
DIETER CREMER,* LARS OLSSON, FELIX REICHEL, AND ELFI KRAKA
Department of Theoretical Chemistry, University of Goteborg, Kemigarden 3, 5-41296 Goteborg, Sweden
(Received 15 February 1993)
Dedicated to Professor John A. Pople in recognition of his many excellent contributions to Quantum Chemistry.Both as a teacher and a scientific leader, he has strongly influenced our work over the years.
Abstract. NMR chemical shift calculations provide the basis for an intensivecollaboration between quantum chemists and experimentalists. Calculated shift datacan be used to describe the magnetic properties of a molecule, to identify unknowncompounds by comparison of experimental and theoretical shift values, to determineequilibrium geometries, to investigate conformational changes, to elucidate themechanism of molecular rearrangements, to determine solvent effects on NMR data,to identify complexation or coordination of soluted molecules by solvent molecules,to detect electronic structure changes caused by the medium, and to describe chemicalbonding. This is demonstrated by three examples, namely the determination of theequilibrium structure of the homotropylium cation, the description of BH
3NH3in
solution or condensed phases, and the investigation of stannyl cation complexes insolution. IGLO calculations of I3C, "B, 15N, and 119Sn chemical shifts with DZ+P orTZ+P basis sets lead to the following results: (I) The homotropylium cation possessesan equilibrium I ,7distance of2Athat is indicative of strong through-space interactionsand, as a consequence, homoaromatic character. (2) In solution, the charge transferfrom NH
3to BH3 is increased, which leads to a decrease of the BN bond length, an
increase of the dipole moment, and a shielding of both the B and the N nucleus. Theexperimental ~I1B)and l'eSN) values can be reproduced when the geometry effect andthe direct solvent effect are included in the shift calculations. (3) Stannyl cations formstrongly-bounded coordination complexes with solvent molecules (binding energy:~ 50 kcaVmol) that make the cation properties, in particular l'(1 19Sn) values, similar tothose of covalently-bounded stannyl compounds. An experimental detection ofstannyl cations in solution by NMR spectroscopy should only be possible by extensivesolvent variations.
Dieter Cremer is Professor of Theoretical Chemistry at theUniversity of Goteborg, Sweden. He is a native of Germany,where he received his Ph.D. at the University of Koln, From1972 to 1974 he was a postdoctoral research associarewith JohnPople at Carnegie-Mellon University, Pittsburgh, PA. Hisresearch interests are in the area of coupled cluster methods,perturbation theory, reaction mechanisms, calculation ofspectroscopic properties of molecules, and the theory of thechemical bond.Lars Olsson is presently carrying out his Ph.D. work intheoretical chemistry at the University of Goteborg,Felix Reichel received his Ph.D. under the supervision ofDieter Cremer at the University of Koln, and now works in theComputional Chemistry Department of Bayer Leverkusen,Germany.
INTRODUCTION
Quantum chemistry has always been judged on the basisof what solutions it can offer to the problems of generalchemistry. Therefore, it is not astonishing that the workof the pioneers in quantum chemistry between 1925 and1950, although well-appreciated arnong the experts, hadlittle or no impacton general chemistry. The beginningof
Elfi Kraka is Associate Professorat the University of'Goteborgwhere she works in quantum chemistry. She received her Ph.D.at the University of Koln and worked for two years at ArgonneNational Laboratory in Illinois. Her research interests are in thearea of ab initio theory and chemical reaction dynamics.
*Author to whom correspondence should be addressed.
Israel Journal of Chemistry Vol. 33 1993 pp. 369-385

370
modern quantum chemistry is generally dated from theearly fifties for three reasons: First, Boys introducedgaussian-type functions for electronic structurecalculations in 1950.1Second, in 1951 Roothaan and Hallindependently publishedtheir classic papers on theLCAOSCF approach.' And finally, one realized in these years,probably for the first time, the importance of electroniccomputers as valuable tools in quantum chemistry, whichwas emphasized at the first American conference ontheoretical chemistry at Shelter Island.'
One can look upon the past four decades of modemquantum chemistry in several ways; for example, on thebasisofhow its tools were developedor on the basisoftheimpact quantum chemistry had on general chemistry. Inthe first two decades quantum chemists essentially learnedhow to use computers and how to set up programs thatcould carry out Hartree-Fock (HF) and some simplecorrelation-corrected calculations. Programs such asPOLYATOM,4 IBMOL,s MOLE,6 and ALCHEMY,7characterized a development that culminated in thepublication of general purpose ab initio programs fornonspecialists. The best known among the latter is JohnPople's GAUSSIAN program package" that was startedin the late sixties and still continues with new updates andprogram improvements. In the last two decades ab initioprograms such as MOLECULEIMOLCAS,9 HONDO,lOGAMESS,II and CADPAC,12emerged. These, togetherwith the GAUSSIAN package, tremendously pushedforward the use of quantum chemical methods inchemistry.
It is fair to say that only after 1970 did quantumchemistry have a steadily increasing impacton chemistry.Three factors influenced this development: (a) a rapiddevelopmentofnew methods, algorithms, and techniquesfor electronic structure calculations; (b) the developmentof sophisticated ab initio programs that can be used forroutine as well as expert calculations of many molecularproperties; and (c) the availability of modernsupercomputers with steadily increasing computingpower.
Although any classification iscertainly very subjective,two majorphases in the development ofab initio methodsshould be stressed here. The seventies were the decade ofcorrelation-corrected methods that made it possible toaccurately calculate energy differences such as reactionenergies, activation energies, and ionization potentials.Many of the method developments of the seventies werealready started in the sixties or even in the fifties. Forexample, the Meller-Plesset (MP) perturbation methodsofPople and coworkers" were based on developments inmany-body perturbation theory that had already takenplace in theoretical physics in the fifties and wereintroduced into quantum chemistry by Kelly in 1963.14
Method development in the seventies focused on howto get better energies, while in the eighties the emphasiswas more on how to calculate molecular properties viaanalytical energy derivatives. Starting in the late seventiesanalytical energy gradients for the calculationofmoleculargeometriesbecameavailable for most ab initiomethods. ISThis development was already initiated in the late sixtieswhen Pulay" and Gerratt and Mills'? published theirlandmark papers on analytical gradient methods and thecoupled-perturbed HF approach.
Certainly, the calculation of molecular geometrieswas the second most important contribution quantumchemistry has given to chemistry. For small and mediumsized molecules, the ab inito calc:ulation of geometry andconformation is a reliable and economic alternative to Xray, electron diffraction, microwave, or other structuredeterminations. In the last decade, more structural datahave been determined by ab initio calculations than havebeen measured experimentally.Thecalculationofaccurategeometries and energies, in particular, for short-livedreaction intermediates, compounds not synthesized, ortransition states of reactions, had a large impact ongeneral chemistry. This continues and is obvious fromalmost any issue of a chemical journal.
Where analytical energy derivative methods wereimplemented into general purpose ab initio programs, thecalculation of molecular properties for both HF andcorrelation-correctedmethods becameroutine. Althoughthe computation of electrical moments, vibrationalfrequencies, infrared intensitie:s, zero-point energies,entropies, etc. clearly enhancedthe importanceofquantumchemistry, none of these possibilities turned out to be asimportant for general chemistry as the calculation ofmolecular energies and geometries. Actually, the next bigstep forward in this regard was only reachedwhen methodsfor the routine calculation of NMR parameters becameavailable.
CALCULATION OF NMR CHEMICAL SHIFTSIfa chemist synthesizes a new compound, one ofthe firstinvestigations to be carried out will be the measurementof the NMR spectrum. NMR spectral data provide in arelatively easy way useful information on the identity andstructure of the new compound. There exist in most casessufficient reference data that provide the basis for aqualitative structure determination or verification. Onlyafter these first NMR investigations have been done is itnormally decided whethera direct structuredeterminationwith other methods such as X-ray, electron diffraction, ormicrowave spectroscopy shouldbe performed or whetherother investigations for a spectroscopic, thermochemical,etc. characterization are desirable. However, all these
Israel Journal ofChemistry 33 1993

additional investigations require in most cases muchmore time and effort than the NMR measurements and,therefore, they are only done for interesting compounds.This is why for many compounds there is just an NMRspectrum available, which does not provide all theinformation and insights an experimentalist wants tohave on a new compound.
Therefore, an experimentalist expects additionalinformation from quantum chemical calculations that usethe existing NMR data in some way. This is best done iffor some appropriate trial structure NMR chemical shiftsare calculated and compared with the existing NMR data.If the two sets of NMR data agree, the compound inquestion must possess the molecular structure used in thecalculation and, therefore, is clearly identified. All themolecular properties that come out ofthe calculation canbe assigned to the new compound and, accordingly, theexperimentalist gets a detailed description of thiscompound. If the quantum chemical description of themolecule can be obtained in a faster and more economicalway than additional experimental investigations, an idealbasis for the collaboration between experimentalists andquantum chemists will exist.
For a long time, this information could not be providedby quantum chemists since routine calculations ofNMRparameters with sufficient accuracy were not possible.There were several approaches to calculating NMRchemical shift data, of which the best known is probablythe method based on gauge independent atomic orbitals(GlAOS),18 originally suggested by London." GlAOswere used at the semiempiricallevel by Pople'" and laterat the ab initio level by Ditchfield" and other authors.22However, the computer-time-consuming integralevaluation over GIAOs prevented applications to largermolecules. Computer timings only improved when Pulayand coworkers" improved the GIAO method byimplementingmodem techniques for integraland integralderivative evaluation. However, this development tookplace years after the routine calculationofNMRchemicalshifts at the ab initio level had been mastered by Kutzelniggand Schindler." These authors solved the gauge probleminherent in all calculations of magnetic properties withthehelp oflocalizedMOs rather than GIAOs. Accordingly,they coined their method individual gauge for localizedorbitals (IGLO).24,2s
The workby Kutzelnigg and Schindler triggeredfurtherdevelopments in the field of NMR chemical shiftcalculations. Beside the IGLO program, several othercomputer programs are presently available for routinecalculations of magnetic properties ofmolecules: (1) TheLORG (localized orbital/local origin) method by Hansenand Bouman;" (2) GlAO-HF in the version ofPulay andcoworkers;" (3) GIAO-MBPT2 by Gauss to calculate
371
correlation-corrected NMR chemical shifts with secondorder many-body perturbation theory:" (4) MC-IGLO byKutzelnigg et al. for problems that require an MCSCFwave functionf" In addition, other methods to obtaincorrelation-corrected NMR chemical shift values havebeen described in the literature."
The use of ab initio methods for the calculation ofNMR chemical shifts was pushed forward by Schleyerand his coworkers who, either in collaboration with theKutzelnigg group or other groups, published more than40 papers in the last five years on the investigation ofNMR chemical shifts. The success of this researchtremendously increasedthe acceptanceofab initio resultsin general, and ab initio NMR results in particular, amongexperimental chemists.
Calculations with IGLO, LORG, or GIAO have led toa wealth of NMR chemical shift data and to a newdimension in the cooperation between quantum chemistsand experimentalists, as is amply documented in theliterature.P" Besides energies and geometries, quantumchemists can nowadays offer experimentalists detailedNMR chemical shift data which provide a direct linkbetween theory andexperiment so that calculated energiesand geometries become more meaningful for theexperimentalist. NMR chemical shift calculations havebeen used not only to describe the magnetic properties ofmolecules, but also to identify unknown compounds bycomparison ofexperimental and theoretical shift values,to determine equilibrium geometries, to investigateconformational changes, to elucidate the mechanism ofmolecular rearrangements, to determine solvent effectson NMR data, to identify complexation or coordinationof soluted molecules by solvent molecules, to detectelectronic structure changes caused by the medium, andto describe chemical bonding - to mention just some ofthe many possibilities that have opened to quantumchemists.2S-32.
In the following, three examples will be discussed thatdemonstrate the usefulness and applicability of NMRchemical shift calculations. These examples deal with (a)the determination of the equilibrium structure of thehomotropylium cation, (b) the description ofBH3NH3insolution, and (c) the identification and investigation oforganotin cations in solution.
THE USE OF NMR CHEMICAL SHIFTS FORSTRUCTURE DETERMINATION - THE
HOMOTROPYLIUM CATIONThe determination of NMR chemical shifts either byIGLO, LORG, or GlAO turns out to be very sensitivewith regard to the geometry used.30-34 Experimentalgeometries are not that useful in this connection since
Cremer et al. / NMR Chemical Shift Calculations

372
Ia Ib Ie
The normal way of solving such a problem is to applycorrelation-corrected methods for an accurate geometrydetermination. For example, MP2,13 which coversimportant pair correlation effects, can give a betteraccountofcritical geometry parameters than HF or semiempirical
very often they are not accurate enough, representdifferentgeometries (r, r., r, r, r., r, etc.), or suffer fromz S 0 a g v
intermolecular interactions in condensedphases. Ab initiogeometries provide a consistent description ofmoleculesthat does not suffer from the ambiguities of experimentalgeometries. Many calculations have shown that reasonableNMR chemical shifts are obtained if the geometry of themolecule in question has been optimized at a correlationcorrected level oftheory such as second-order MP (MP2)using DZ, DZ+P, or better basis sets. Since the calculatedNMR chemical shifts clearly depend on the geometry, anagreement between experimental and theoretical shiftsnot only provides a clear identification, but also a geometrydetermination of the molecule in question. On the otherhand, if theoretical and experimental shifts differconsiderably, other possible geometries or structureshave to be tested.
Schleyer was the first who fully realized the sensitivityof calculated NMR chemical shifts with regard tomolecular geometry, and he used this for ab initiolIGLOINMR-based structural determinations in many casesincluding carbocations, boron, and organolithiumcompounds.v-" A recent assessment of this approachsuggests that "structural assignments based on the abinitiolIGLOINMR method are quickly approaching aconfidence level that rivals modem-day X-ray diffractiondeterminations of molecular structures.?" To illustratethis method we describe in the following our investigationof the equilibrium structure of the homotropyliumcation (I).
HF/STO-3G, as well as semiempirical calculations,predict for I the bicyclic structure la while HF/6-31 G*calculations suggest the open structure Ie. 33.34ExperimentalNMR chemical shift data, on the other hand, are in linewith a nonclassical (homoaromatic) structure Ib with adelocalized 61tsystem.35 X-ray structure determination ofsubstituted homotropylium cations makes the situationeven more confusing.v" For example, 2-hydroxy-1 wasfound to possess a relatively short 1,7 distance, in linewith the bicyclic structure Ia, 36 while l-ethoxy-I has a 1,7distance of 2.4 A., in line with the open structure Ic."
& C1
-100+...........,..~.............,..~..,.............,.~·...,............,...........,...~........................1,441,54 1.64 1,74 1,841.942.042.142,242,342.442,54
-80
-80
100
80
E SOQ.
~
() 40
'iiiuMc::; 20
Q:)( 0CllMc::;
-20II
<:]
-40
r(1,7) Distance [AJ
methods. However, MP2 is known to overestimate theimportanceofbiradical states and structures with elongatedbond Iengths.P-" Because of this, the MP2 geometry of amolecule with nonclassical structure should be betterconfirmed at higher levels of theory. Third-order MP(MP3)13 is not sufficient in this regard since it onlypartially corrects the exaggeration of pair correlationeffects without introducing new correlation correctionsand, therefore, leads to results that are normally shifted inthe direction ofthe corresponding HF data. Both MP2 andMP3 results have to be tested by fourth-order MP (MP4)calculations,13 which introduce correlation effects resultingfrom single (S), triple (T), and quadruple (Q) excitations.But with MP4, there is the danger that, for example, Teffects are overestimated since 'ITcoupling as well as ST,DT, and QT couplings do not come in before fifth-orderMP (MP5).13.40 MPn geometries oscillate very oftenbetween MPI (= HF) and MP2 values, and in criticalcases it is difficult to predict at what level oscillations aredampened out. 39.40
Of course, one can avoid problems with MPngeometries by using coupled-cluster (CC) methods."They include infinite-order effects and, therefore, results
Fig. 1. Difference (6) between experimental and IGLO/63IG(d)//HF/6-3IG(d) I3C NMR chemical shifts of thehomotropylium cationasafunctionofthe 1,7distance. Thezeroline (6 = 0) indicates that theoretical and experimental shiftvaluesare identica1.
d
Israel Journal ofChemistry 33 1993

373
b7,0
6,5
6,0
5,5
0 MP2I6·31G(d)5,0 0 MP3/6·31G(d)
• MP4(SOQ}/6-31 G(d}4,5
"0~ 4.0
Oiu
3,5~
a:- 3,0
::;:W 2,5<l
2,0
1,5
1,0
0,5
0,0
1,6 1,7 1,8 1,9 2,0 2,1 2,2 2,3 2,4
r(1,7) Distance [AI
1
I'>E(HF/6-31G(d»
Mean DevIation
of 13C Shifts
o
5
10
15
·5+---........,~""'"T"~'"T'"""~r-......,..~.,...,.....~,....-r-e-r ~...,.,~...,...,........j_
1,4 1,5 1,6 1,7 1,8 1,9 2,0 2,1 2,2 2,3 2,4 2,5
r(1,7) Distance [AI
a
20
25
35
40
45
50
55,-...,----------------------.
Fig,2, a. Determination of theequilibrium valueof the 1,7distanceof thehomotropylium cationbya) HF/6-31 Gtd) calculations(0 points) and ~) IOLO/6-310(d) 13C chemical shift calculations. In the latter case, the mean deviation of calculated andexperimental PCchemicalshiftsis plotted(0 points). Minimum valuesof rO,7) area) 2.28A(downward-directed arrow)and B)1.98 A(upward-directed arrow). b. MP2, MP3, and MP4(SDQ) potential energy surfaceof the homotropylium cation in thedirectionof the 1,7distance.The positionof the minimum is indicated in each case by an arrow.
do not oscillate. But the price one has to pay is to movefrom a one-step (MPn) to an iterative method that isneeded to calculate CC amplitudes. In the case of thehomotropylium cation, CC calculations would imply alarge amount ofcomputer time that is not available undernormal circumstances.
An alternative, less costly way of determining thecorrect structure on is provided by NMR chemical shiftcalculations for Ia, Ib, and Ie. Sincetwo ofthese structuresare just transient points on the HF potential energysurface (PES), a prerequisite for structure determinationby NMR chemical shift calculations is that the PES issystematically explored in the direction that connectspossible structures. In the case of I the three structuralalternatives Ia, Ib, and Ie correspond to three differentranges of the 1,7 distance: la implies a relatively shortdistance (r(1,7) < 1.7 A), Ie a relatively long distance(r(1,7) > 2.3 A), and Ib corresponds to intermediatevalues (2 A± 0.2 A). By fixing r(1,7) to a number of
discrete values ranging from 1.6 A to 2.4 A and byoptimizing the remaining geometrical parameters of I,the PES in the 1,7-direction is explored. Even though HFis not sufficient to give the correct value ofthe equilibriumI ,7distance, one can expect that HF describes the changesofthe other geometrical parametersofI in dependence onr(1,7) in a consistent way.
If for each of the optimized HF geometries, IGLO I3CNMR chemical shifts are calculated, the dependence ofthe o(l3C) shifts of Ion the 1,7 distance is obtained andcan also be investigated. One gets for all C atoms o(l3C)as a function of r(I,7), which for some 1,7 distancebecomes identical with the corresponding experimentalvalue. In Fig. 1,deviations from experimental'C chemicalshifts rather than calculated I3C shifts themselves areplotted against r(1,7). The five curves cross or approachthe zero line between 1.9 and 2 A, which means that thebest agreement with the experimental FCNMR chemicalshifts is achieved in this region. The minimum of the
Cremer et al. / NMR Chemical Shift Calculations

374
mean deviation between calculated and theoretical I3Cchemical shifts forI is calculated forr(I,7) = 1.98 A(seeFig. 2) which suggests that the equilibrium structure oncorresponds to Ib rather than Ia or Ic.42Cation I neitherpossesses a bicyclic structure with a long 1,7-bond nor aneight-membered ring with weak 1,7 through-spaceinteractions. At r(1,7)=2A, the through-space interactionsare sufficiently strong to influence rt-delocalization in theconjugated system: Bond equalization and uniform chargedistribution result, thus giving I the typical character ofahomoaromatic compound.P'"
Clearly, the determination of the equilibrium geometryof a compound such as I by IGLO/IHF NMR chemicalshift calculations cannot lead to high accuracy since thereare several approximations involved. But such a procedureis sufficiently accurate to characterize basic structuralfeatures. In addition, it can be improved by going toIGLO//MP2, IGLO//MP4, orGIAO-MP2//MP4.33,43 Thisis reflected by comparing the IGLO/6-31G(d)/IHF/63IG(d) value ofrO,7) =1.98 Awith the correspondingMP2/6-3IG(d), MP3/6-3IG(d)//MP2/6-3IG(d), andMP4(SDQ)/6-31G(d)//MP2/6-31G(d) values: 1.901,1.985,2.031 A(compare with Fig. 2b). The agreementwith the IGLO result is satisfactory in view of theapproximations involved. MP4(SDQ) leads to the mostreliable geometry on, which again can be checked withthe calculated 13C NMR chemical shift parameters. Whilethe smallest mean deviation between theoretical andexperimental 13C shifts at the HF level is 6 ppm, it isreduced to 4.9 ppm using the optimized MP4(SDQ)/63IG(d) geometry." Hence, MP4(SDQ)/6-3IG(d)provides a reasonably accurate prediction of theequilibrium geometry of I.
The procedure describedabove for thehomotropyliumcation is particularly useful if the molecule in questiondepends on just one or two critical geometry parametersthat can only be determined by expensive correlationcorrected ab initio calculations. Variation of theseparameters at the HF level in connection with NMR shiftcalculations helps to determine their equilibrium value.We have applied this approach in a number of cases, inparticular to compute interaction distances of potentiallyhomoaromatic compounds."
CALCULATION OF NMR CHEMICAL SHIFTSIN SOLUTION - BORANE
MONOAMMONIATEBoranemonoammoniate, BH3NH3,has been investigatedas a prototype of Lewis acid-base complexes.r'r" An r,geometry of the molecule has been reported by Thome etal.," which is largely reproduced by the MP2I6-31G(d.p)equilibrium geometry of BH
3NH3(Table 1). The HF/6
3IG(d,p) geometry gives a BN bond length that is 0.3 A
too large,which shows thatelectron correlation correctionsare important to correctly describe the charge transferfrom N to B and by this also the BN bond length.
In Table 1, IGLO/[5s4pld/3sIp]/IHF/6-3IG(d,p),IGLO/[5s4pld/3slp]/lMP2I6-3IG(d,p) liB,and lsNNMRchemical shifts" are compared with experimental valuesmeasured inan aqueous solutionofBH3NH3
.48 Theoreticalshifts differ as much as 10 and 20 ppm, respectively, fromexperimental shifts. This is surprising in view of the factthat" B IGLO chemical shifts are normally correct within5 ppm, provided the equilibrium geometry of the boroncompound is accurately reproduced by theory and used ino(IlB) calculations.2sh.3llb Since the latter is true for theMP2 geometry, something other than calculationaldeficiencies must be responsible for the discrepancybetween theoretical and experimental shift values."
A clue is given by the large dipole moment of 5.22debyes for BH
3NH3,46 which suggests that the molecule
will be strongly solvated in polar solvents because ofdipole-dipole interactions. Therefore, one can expectstrong solvent shifts for OCIlB) and o(lsN). To test thishypothesis, wehave simulated the influence of the mediumby the PISA solvent model" where the wave function ofthe solute is recalculated in a solvent cage under theinfluence of a polarizablecontinuum that is characterizedby the dielectric constant of the solvent. IGLO NMRchemical shifts are then obtained for a solvent-dependentSCF wave function. Such an approach may be denoted asPISA-IGLO.34
PISA-IGLO/[5s4pld/3s1p] shifts change by 2 and 5ppm to higher field when the dielectric constant of water(e = 80) is used. Changes in the shift values areaccompaniedby an increased charge transfer from NH3toB~ and anenlargementof the dipole moment. Obviously,the B and the N nuclei are more shielded, thus leading tochemical shift values closer to the experimental o-valuesthan the gas-phase O-values. However, the differencesbetween theoretical and experimental shifts are still toolarge to be acceptable.
In a second step, we have reoptimized the equilibriumgeometry ofBH3NH3under the influence of the solvent.A similar investigation has been carried out by Schleyerand coworkers," but since this investigation differs fromours in several ways, we find it desirable to present alsothe results of our PISA geometry optimizations. ThePISA/6-3IG(d,p) geometry shown in Table I reveals asignificant decrease in the BN bond length (1.610 A) ofas much as 0.05 A, which, according to the calculatedcharge distribution, is the result of an increase in chargetransfer from N to B. The calculated PISA geometry gainscredibility by the fact that X-ray structural analysis ofcrystalline BH3NH3soand various crown ether complexesofBH3NHt have led to BN bond lengths between 1.564
Israel Journal ofChemistry 33 1993

375
Table I. Geometry, dipole moment, and NMR chemical shifts of BH3NH3a
Parameter HF MP2 PISA MW b X-ray c
r(BN) 1.687 1.657 1.610 1.658 1.564 - 1.605r(BH) 1.209 1.202 1.213 1.216r(NH) 1.003 1.014 1.005 1.014<HBN 104.4 104.5 106.8 104.7<HNB 110.7 II I.1 111.6 110.3/..l 5.54 5.60 6.61 5.22
IGLO IGLO IGLO PISA-IGLO PISA-IGLO expo d
IIHF 11MP2 IIPISA 11MP2 IIPISA
~I1B) -13.2 -15.0 -17.8 -16.8 -19.5 -23.8l)(15N) 2.7 8.0 4.9 3.0 -0.8 -13.0
abond lengths in A, angles in deg, dipole moment /..l in debyes, ~I1B) in ppm relative to BF3·0Et2, ~15N) in ppm relative to NH3.IGLO l)(llB) values have first been calibrated with regard to BzH6and, then, adjusted with the experimental value ofB2H6 (16.6ppm) to the BF3·0Et2 scale. Geometry otimizations have been carried out with the 6-3IG(d,p) basis, IOLO calculations with a[5s4pld/3s1p] basis set denoted by Kutzelnigg and Schindler as basis II in their IGLO calculations,25h The notation method 1//method 2 indicates that method 1 calculations have been carried out at method 2 geometries.b rs-geometry from ref 46.c X-ray geometries from refs 50 and 51.d NMR chemical shifts measured in water, ref 48.
Hb-H
Dipole rnernentu
and 1.604 A. According to these investigations, BH3NH3is embedded in the crown ether and interacts with it by Hbonds in a similar way as it does in water solution.
When recalculating chemical shifts with either thePISN6-310(d,p) or the X-ray geometry," both a(IlB)and a(ISN) are shifted upfield by about 3 ppm. Adding thegeometry effect (3 ppm) and the direct solvent effect onthe shift calculation (2 ppm), a a(IlB) value is obtained(-19.6 ppm, Table 1) that is within 5 ppm of theexperimental 8(llB) value obtained in an aqueoussolution." For ISN, geometry and direct solvent effectalso improve the IOLO a(ISN) value considerably, but thedifference to the experimental 8(lsN) value is 13 ppm,still large. This, however, simply reflects the fact thatlarger basis sets are needed to reach the same accuracy for8(lsN) as that obtained for IGLOIlB NMR chemicalshifts. JOb
Schleyerand coworkers, in their study onB~NH3 andrelated borane adducts, obtained an even larger reductionofthe BN bond length when using the PISA method withdifferent physical constants for the solvent." As aconsequence, these authors calculated an upfield shift of5 ppm for 8(1 'B), thus leading to -20.5 ppm at IGLOIDZI/PISA/6-31G(d), in reasonable agreement withexperiment. They concluded that "medium effects onNMR chemical shifts may be indirect," caused by asolvent-dependent change in molecular geometry ratherthan by a direct influenceofthe solventon NMR chemicalshifts."
Our calculated a(llB) data seem to confirm thisconclusion (2.8 ppm geometry effect, 1.8 ppm directsolvent effect), but the computed 8{1SN) values, whichwere not reported by Schleyer and coworkers, suggest alarger direct influence ofthe solvent on the chemical shift(3.1 ppm geometry effect, 5 ppm direct solvent effect,Table 1). We conclude that neither of the two effects canbe neglected, and taken together they lead to a gooddescription of the total solvation effect (4.5 ppm for8(IlB) and 8.8 for a(ISN), Table 1). PISA-IGLO/IPISAcalculations can provide a reasonable approximation ofNMR chemical shifts of solvated molecules, while thecombination of IGLO/JMP2, IGLO/IPISA, and PISAIGLO/JMP2 calculations leads to the determination ofgeometry and direct solvent effect. Accordingly, IGLOchemical shift and PISA solvent calculations provide abasis for the analysis of solvent-solute interactions andhelp us to understand electronic structure changes underthe impact of the surrounding medium."
Cremer et al. / NMR Chemical Shift Calculations

376
INVESTIGATION OF SOLUTE-SOLVENTCOMPLEXES BY NMR CHEMICAL SHIFTS -
ORGANOTIN IONS IN SOLUTIONIn recent years, there has been much dispute on thequestion whether triorganosilyl cations can be generatedin solution and whether some of the carbocation chemistrycan also be found for silicon." Since this question has notbeen clearly answered so far because of experimentaldifficulties, it is interesting to extend the question to tinand ask whether triorganostannyl cations in solutionshow some similarities to carbocations. Indeed, evidencefor the formation of organotin cations was reported asearly as 192353 and the detection of SnH3+ in aqueousfluorosulfonic acid by 119Sn NMR spectroscopy waspublished in 1971.54Nevertheless, little is known aboutthe electronic structure of tin cations in solution and itremains to be proven whether they are comparable tocarbocations.
Again, NMR spectroscopy in combination with abinitio calculations seems to be an appealing method fordescribing the electronicstructure of organotincompoundsin solution. Some NMR investigations have focused onthe detection of stannyl cations54- 56 while relatively littlehas been done on the ab initio calculation of tincompounds.57 Most of the theoretical work has beenperformed with pseudopotential methods that do notprovide a good starting point for NMR chemical shiftcalculations."There are no ab initio investigations on tincompounds that include the calculation of both energy,geometry, and NMR chemical shiftparameters. Therefore,wehave taken up theproblem of calculating 119Snchemicalshifts for organotin compounds pursuing two objectives;first, to learn more about the electronic nature of stannylcations in solution and, second, to test the potential ofNMR chemical shift calculations for heavy elements.
As a suitable basis set, we have taken for Sn the(15s 11p6d) basis of Stromberg et al." that is between DZandTZ quality and have combined itwith variousDunningTZand DZ basis sets for first- and second-rowelements."Since our IGLO program does not contain f-functions wehad to perform shift calculations without polarizationfunctions at Sn. Nevertheless, we have also tested theeffect of p-type and d-type polarization functions addedto the Dunning basis set. A reasonable compromisebetween accuracy and costs was already obtained with amixed basis set of close to TZ quality for Sn and DZ+Pquality for all other elements, which we will denote asDZ+(P) in the following, where the parentheses indicatethat f-type polarization functions for Sn are missing."
In Figs. 3,4, and 5,calculated geometries of compounds1-12are shown. There are some microwave and electrondiffraction investigations on methylstannanes that can beused for comparison.62 They reveal that both the SnH and
the SnC bonds calculated at the HFIDZ level are 0.Ql0.02 Aand at the MP2IDZ level 0.02- 0.03 Alonger thanexperimental values. Including polarization functions atall atoms but the Sn atom leads to a significant decreaseof SnH and SnC bond lengths so that HFIDZ+(P)geometries show good agreement with experimentalvalues. Inclusion of f-type polarization functions at theSn atom leads to another small reduction of SnX (X =H,C, 0, CI, etc.) bond lengths but leaves other geometricalparameters unchanged. For example, the SnH bond isdecreased to 1.707 Aat the HFIDZ+P level and to 1.689Aat theMP2IDZ+Plevel. Hence, MP2IDZ+Pcalculationsprovide the best agreement with experimentalgeometries." But since f-functions could not be used inthe NMR shift calculations, we used the DZ and theDZ+(P) basis sets for the geometry calculations.
When replacing the H atoms of 1 consecutively bymethyl groups, both the SnH and the SnC bond lengthsincrease steadily from 1.709 to 1.722 Aand from 2.153to 2.167 A(HFIDZ+(P». At the same time the positivecharge at the Sn atom becomes larger (Table 2) becauseof electron withdrawal by the methyl groups. Obviously,the SnC and SnH bond lengths increase with increasingbond polarity. The experimental bond lengths do notshow any clear trends (Fig. 3). In view ofthe difficultiesand approximations involved in the experimentalmeasurements," the calculated geometries probablyprovide a more accurate description of general trendsthan the few experimental data available.
Alky1groups lead to somedestabilization of stannanes.From isodesmic reactions
(CH3)nSnH4.n + (n - 1) SnH4 ~ n CH3SnH3
SnC bond, bond interaction energies of-1.1 (n = 2), -2.8(n = 3), and -4.8 kcal/mol (n = 4) are calculated with theenergies listed in Table 2. This is different from CC bond,bond interaction energies which are stabilizing, as can beseen when replacing H atoms in CH4by methyl groups.Since the methyl group functions in CH3SnH3 as a (J
acceptor, a second methyl group could lead to stabilizationprovided it acts as a 1t-donor, thus giving the SnC bondsome double bond character. However, 2p1t(C)-5p1t(Sn)overlap is rather poor and, therefore, the 1t-donor abilityof CH3is strongly reduced. As a consequence, bothmethyl groups become negatively charged (see Table 2and Fig. 6), thus repelling each other. The more methylgroups are attached to Sn, the larger the electrostaticrepulsion between them and the greater the stannanemolecule destabilization.
In Table 3, calculated 119Sn NMR chemical shifts areshown. When comparing calculated 119Sn chemical shiftswith experimental ones,63--65 one has to keep in mind thatthe latter can change very much in dependence on the
Israel Journal ofChemistry 33 1993

377
1.7327.737
(1.680) ua«
110.8
111 .1111.4.1.lL2.
108.4108.9.l.QB..&
(2.143)
111.0777.3
1..1.1.J.
3, Me2SnH2' e2v
(2.144)
Fig.3. Calculatedgeometriesof methylstannanes (CH3)nSnH4_n 1-5. Numbersin normalprint:HFIDZvalues;in italics:MP2IDZvalues; in underlined italics: HFIDZ+(P) values; in parentheses: experimental values.
Cremer et al. / NMR Chemical Shift Calculations
378
180.4180.0us»
1.6941.702MJM
1.698J...QB.S.
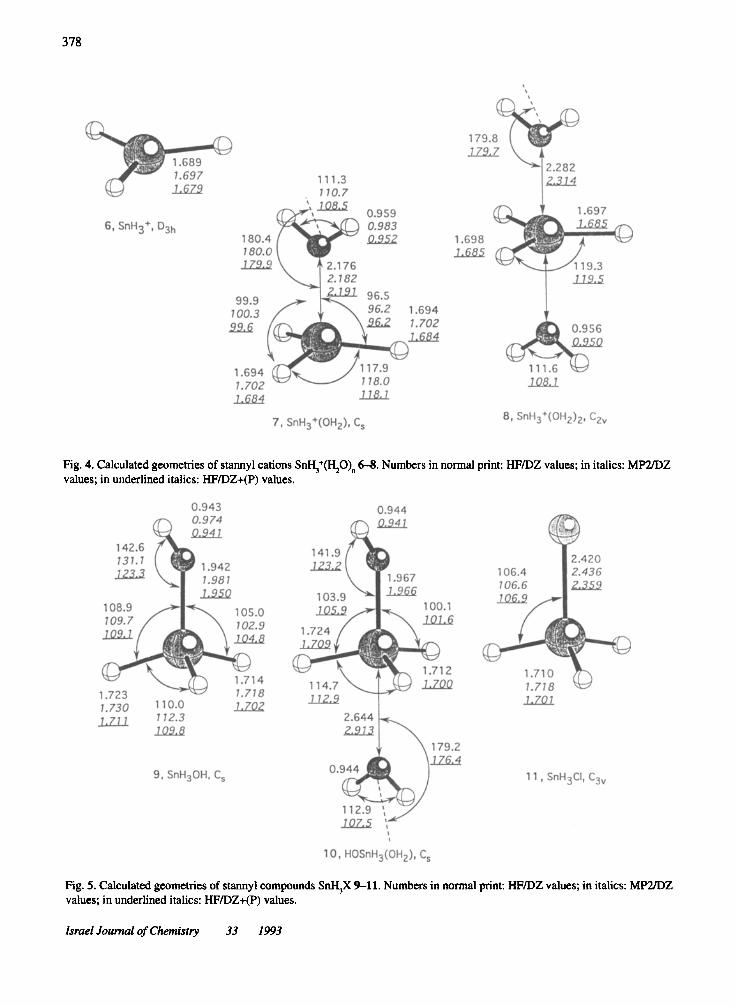
Fig. 4. Calculated geometries of stannyl cations SnH3+(Hp)n
6-8. Numbers in normal print: HFIDZ values; in italics: MP2IDZvalues; in underlined italics: HFIDZ+(P) values.
2.4202.436zss»
1.7141.718LZJl2.110.0
112.3us»
Fig. 5. Calculated geometries of stannyl compounds SnH3X
9-11. Numbers in normal print: HFIDZ values; in italics: MP2IDZvalues; in underlined italics: HFIDZ+(P) values.
Israel Journal ofChemistry 33 1993

379
Table 2. Calculated energies and Sn charges q a
Molecule Sym Energy q(Sn) q(SnHn) q(Sn) q(SnHn)HFIDZ HFIDZ+(P) HFIDZ HFIDZ+(p)
I. SnH4 Td --6024.51685 --6024.53845 0.926 0.000 1.018 0.0002. CH3SnH3 C3v --6063.55801 --6063.59331 1.139 0.430 1.209 0.4333. (CH3hSnH2 C2v --6102.59735 --6102.64636 1.356 0.877 1.403 0.8804. (CH3hSnH C3v --6141.63583 --6141.69863 1.585 1.344 1.607 1.3445. (CH3)4Sn Td --6180.67384 --6180.67384 1.825 1.825 1.823 1.8236. SnH3+ D3h --6023.66346 --6023.68194 1.330 1.000 1.409 1.0007. SnH3+ H2O Cs --6099.75755 --6099.79827 1.362 0.899 1.438 0.9048. SnH3+ 2 H2O C2v --6175.82247 --6175.88849 1.417 0.843 1.487 0.8559. HOSnH3 Cs --6099.41327 --6099.45191 1.417 0.676 1.463 0.657
10. HOSnH3H2O Cs --6175.43729 --6175.50605 1.466 0.663 1.501 0.65111. C1SnH3 C3v --6483.46006 --6483.49166 1.199 0.552 1.317 0.575
aEnergiesin hartrees,Mullikenchargesq in electron.q(Sn) is thechargeat the Sn atom,q(SnHn) is thechargeof the SnHn group.The H20 reference energies are: -76.01100 (HFIDZ), -76.06163 (HFIDZ+P).
Fig.6. DependenceoflGLO ~1I9Sn)valueson thechargeat theSn atom (Mulliken valuescalculated with the DZ basis).
100....------------------.
I -150
~ -zoo:gii -250
Iu ·300
1,6 1,7 I,' 1,11,0 ',1 l,Z 1.1 1,4 '.5
Sn chugl [lllctron]
IGLO/DZCalculations
0.' 0,1
o
so
-se
-400
-450
-sse
-100
s
medium. Polar coordinating solvents can produce shiftchanges of 200 ppm and more if five-coordinated or sixcoordinated adducts with trigonal bipyramidal oroctahedral configuration are formed between the Sncompound and the solvent molecule (S):63.64
Also, dimerization of Sn compounds in solution ispossible." Therefore, comparison of calculated andexperimental 119Sn chemical shifts requires a carefulconsideration of environmental and structural effects.
The IGLO 119Sn chemical shifts show a cleardependence on the partial charge at Sn, as is shown in Fig.6. Since the electronegativity of a C atom is larger than
c:that of a H atom, the positive charge at the Sn atom :t.'increases withincreasing methylsubstitution (see Table ..2). Accordingly, the Sn nucleus becomes more and moredeshielded and ~119Sn) is shifted downfield, which is inline with experimental shift values (Table 3). However,
R R
R.....S1-R R-"S1 ..... R."",.- n ~ no,."",R I R""""- I S
I I
Sthe latter are between 120 and 30 ppm more negative thanthe IGLO shift values. Media effects can be partiallyresponsible for these differences, but since experimentalmeasurements have been carried out in weakly ornoncoordinating solvents, solvent corrections should besmaller than 10 ppm. On the other hand, inspection of thecalculated charge distributions in dependence on basis setand correlation corrections reveals that the major part ofthe differences between theoretical and experimental119Sn NMR shifts results from calculational deficiencies:1. The use of f-type polarization functions at Sn leads to
a redistribution of0.1 electron from the H atoms to theSn atom in 1, thus shielding the Sn nucleus. Utilizingthe linear relationship between 119Sn chemical shiftsand the charge at the Sn atom (Fig. 6), a a-value of-440 ppm is calculated for 1.
2. Response density calculations at the MP2IDZ+P levelof theory lead to further screening of the Sn nucleus(q(Sn) =0.790 e). This is because MP2 emphasizesbiradical states and, therefore, decreases the polarityof SnH and SnC bonds. We expect that correlationcorrected NMR chemical shift calculations lead to a
Cremer et al. / NMR Chemical Shift Calculations

380
Table 3. Calculated 119Sn NMR chemical shiftsa
Molecule Sym DZ DZ+(p) expo value
1. SnH4 Td -374 -380 -493 b2. CH3SnH3 C3v -275 -273 -346b3. (CH3hSnH2 C2v -169 -162 -225b4. (CH3hSnH C3v -76 -104b5. (CH3)4Sn Td 0 0 Oa6. SnH3+ D3h 777 7747. SnH3+H2O Cs 54 848. SnH3+2 H2O C2v -238 -181 _186 c9. HOSnH3 Cs -215 -181
10. HOSnH3H20 Cs -310 -22111. CISnH3 C3v -138 -17912. Sn(CH3h+ (1075)13. Sn(CH3h+ H2O (352)14. Sn(CH3h+ 2 H2O (60) 54 d15. HOSn(CH3h (83) 77b16. HOSn(CH3h H2O (-12) -47 e17. CISn(CH3h (160) 153 b
aShiftsrelative to that of5. Numbers in parenthesesare estimated frommethyl group increments.See text.
bFrom refs 63 and 64.cSnH3+/FS03H, ref 54.dSnBu3+12 CH3CN, ref 56.eRef65.
0(119Sn) value close to -450 ppm in the case of SnH4
•
3. For 0(1I9Sn) also relativistic corrections have to beconsidered. Judging from NMR shift calculations ofother heavy elements, these can be very large.66
However, there are no data for 0(1I9Sn) available thatsuggest a reasonable correction.
To get better oC 19Sn) values, calculations have to beperformed with a correlation-corrected method and basissets including f-type polarization functions." In this paper,however, we are more interested in trends than in accuratevalues and the former are satisfactorily reproduced at theIGLOIDZ and IGLOIDZ+(P) levels of theory.
In Fig. 4, three different SnH3+ cations 6, 7, and 8 are
shown. The naked cation 6 is planar and has a SnH bondlength that is clearly shorter (1.679 A) than that of 1(1.709 A). Because ofthe deshielding of the Sn nucleus(q(Sn) =1.33 e, Table 2), 0(1I9Sn) is shifted to 774 ppm(Table 3), which is ca. 960 ppm lowfield from theexperimental value of -186 ppm measured in FS03H
acid." Clearly, 6 does not exist in solution, but formsdonor-acceptor complexes with solvent molecules.Depending on the donor strength of the solvent, thenegative charge is transferred to cation 6, thus leading toshielding of the Sn nucleus. To model this process wehave calculated the mono- and di-water complex ofSnH
3+, 7, and 8 (Fig. 4).
Complex 7 is characterized by a rather short Sn,O
distance of 2.19 Aand a significant pyrarnidalization ofthe SnH
3group «OSnH =96.2 and 99.6°, Fig. 4). The
OH bonds arrange in a staggered conformation withregard to the SnH bonds, but move from a bent position(start of the geometry optimization: flipping angleSnO(HH) < 180°) into a lineararrangementwith SnO(HH)close to 180°. In this way, 2-electron stabilizationinteractions between the 0'(0) lone pair orbital and theempty p7t(Sn) orbital become possible while interactionsbetween the p7t(0) lone pair orbital and the pseudo7t*(SnH
3) orbital are probably negligible because of
insufficient 2p7t-5p7t overlap.This is confirmed by a complexation energy of 54
kcal/mol (HFIDZ+(P); 52 kcal/mol, HFIDZ), a chargetransfer of 0.10 electrons from water to SnH3+, a
• '1'c:JD •. lone ••"o lone pair orbital - orbital
CI,,_s~pseudo-lt* • 0
orbital
Israel Journal ofChemistry 33 1993

lengthening of the SnH bonds by 0.005 Ato 1.684 A, anda widening of the HOH angle to 108.5° (HFIDZ+(P».The value of()(1I9Sn) is strongly shifted upfield from 774ppm to 84 ppm (IGLOIDZ+(P); 54 ppm, IGLOIDZ,Table 3).
Complexation by another water molecule leads to atrigonal bipyramidal geometry with a planarSnH
3+cation
in the center. The SnO interaction distance is significantlyincreased from 2.19 (7) to 2.31 A(8), indicating that thetwo water molecules hinder each other in their interactions with the cation. This is in line with the calculatedcharge transfer ofjust 0.079 e from each water molecule,leading to a reduction of the positive charge from +1 (6)and +0.899 (7) to +0.843 e (8). As a consequence, the Snnucleus is more shielded and ()(119Sn) becomes -181 ppm(IGLOIDZ+(P); -238 ppm, IGLOIDZ, Table 3) for 8.This value comes close to the -186 ppm measured inaqueous fluorosulfonic acid" and suggests that in thismedium a similar SnH
3+complex exists.
Clearly, electronic structure, geometry, NMR shiftvalues, and other properties of SnH3+ in solution willdepend on the donor capacity of the solventmolecule and,therefore, it may be erroneous to speak in this case ofstannyl cations. For example, some of the properties ofcomplexes 7 and 8 are very close to those of a covalentlybonded SnHpH (9) molecule (Fig. 5, Tables 2 and 3).The SnO bond length of 9 (1.950 A) is just 0.241 and0.364 Ashorter than the SnO distances calculated for thetwo water complexes. More important, the Sn atomcarries a positive charge that is comparable to that of thewater complex 8 (Table 2). These similarities between 7,8, and 9 become understandable if one no longer considers7 and 8 as SiH
3+-watercomplexes but as protonated
SnH,oH (7), or protonated SnHpH-water complex (8)with covalent SnO bonds. Protonation will lead to SnObond lengthening and to charge redistributions as foundfor 7 and 8. On the other hand, it is still true that for both7 and 8 the positi ve charge is predominantly located at theSn atom (see Table 2) and, therefore, one can consider 7and 8 as coordination complexes of SiH
3+.In any case,
relationship between 7, 8, and 9 will make it difficult todistinguish covalently bonded SnR;X compounds andcoordination complexes ofstannyl cations. For example,9 has a similar ()(1l9Sn) value (-181 and-215 ppm) to thatof8 (-238 and -181 ppm) (Table 3).
Of course, 8e 19Sn) of coordination complexes of tincations with solvent molecules should show a strongdependence on the donicity of the solvent. For example,NMR measurements of Bu
3SnCl,Bu
3SnCl04, and·Bu3SnBF4 in different solvents ranging from C~Cl2 tohexamethylphosphoramide have found upfield shifts of()(119Sn) by 200 to 260 ppm, with the less ionized startingcompound having the more positive and, hence, more
381
deshielded Sn nucleus.56This is inline withour calculationsthat suggest a relatively high positive charge at Sn in thecovalently-bonded compounds SnH
3X,with X being an
electronegative group or atom such as OH (9) or Cl (11)(See Table 3). Hence, the detection of the cation requiresa series of NMR measurements for a variety of solventswith increasing donicity.
There is, however, some ambiguity with this procedurefor detecting stannyl cations. Because of the relativelyhigh positive charge at Sn in a covalently-bonded tincompound, the latter can coordinate at least one solventmolecule without dissociating into cation and anion. InTables 2 and 3 and in Fig. 5, calculated energies, Sncharges, and geometry parameters of the mono-watercomplex of 9, 10, are shown. The properties of 10 aremuch more sensitive to changes in the basis set than hasbeen found for the other tin compounds listed in Table 2.The charge transfer from water to Sn (0.036 (HFIDZ) and0.024 e (HFIDZ+(P» is one-third smaller if calculatedwith the larger basis set, but in any case it is much smallerthan that found for complexes 7 and 8. This is due to thefact that the o lone pair orbital ofHP interacts in 10 withthe energetically higher-lying cr*(SnO) orbital and,therefore, interactions are weaker than in the case of astannyl cation.
As a consequence, complex 10has a binding energy ofjust 8.2 (HF/DZ) and 4.7 kcallmol (HFIDZ+(P»,respectively,much smaller than thatof7or 8.Experimentalcomplexation enthalpies measured by Torocheshnikov etal, for Me
3SnCland Et3SnCl in acetone, acetonitrile, and
dioxane range from 2.8 to 4.2 kcal/mol," Accordingly,HFIDZ+(P) seems to give a better description ofcomplex10 than HFIDZ, which is in line with the discussionabove. The relative weakness of the interactions betweenwater and 9 is also reflected by the calculated Sn,Ointeraction distance that with 2.644 (HFIDZ) and 2.913 A(HFIDZ+(P», respectively, is much larger than Sn,Odistances obtained forcations 7and8 (Fig.4). Nevertheless,complexation of 9 changes the electronic structure of theSnH
3group in the direction of that of a Sn~+cation. This
can be seen from the beginning planarization of the SnH3
group (reduction of the OSnH angles) and the increase inthe SnO bond length (0.015 A, Fig. 5) as a consequence ofthe population of the cr*(SnO)orbital.
The 8(119Sn) value of 10 is shifted to highfield becauseofcharge transfer from water to Sn and stronger shieldingof the Sn nucleus. However, for the DZ+(P) basis thisshift is moderate (8(119Sn) =-221 ppm), while it is muchlarger for the DZ basis «)(l 19Sn =-31 0 ppm, Table 3). Onthe basis of these results we cannot exclude that H
3SnX
compounds such as 9 and 11 show a relatively strongdependence on the coordinating capacity of the solvent.Definitely, careful NMR investigations and a broad
Cremer et al. / NMR Chemical Shift Calculations

382
variation of the solvent are required to distinguish R3Sn+cations from ~SnX compounds in solution. This isillustrated by some examples from the literature.
For HOSn(CH3)3' 15, and ClSn(CH3)3,17, 0(1I9Sn)values of 77 and 153 ppm have been measured.P-"Utilizing the shift values obtained for compounds 1-5,we can predict IGLOIDZ values of 83 (15) and 160 ppm(17), in good agreement with experiment. These 119Snchemical shifts have to be compared with an estimated0(119Sn) value of 60 ppm for Sn(CH3)3+ 2 ~O, (14). In acareful NMR investigation of'Bu.Snt ions using CH2CI2,acetonitrile, pyridine, N,N-dimethylpropylenurea,dimethylsulphoxide, and hexamethylphosphoramide assolvents, 0(1I9Sn) values between 130 and -47 ppmresulted when starting from Bu3SnCl, between 220 and-43 ppm for Bu
3SnCI04and between 160 and -44 ppm
for Bu3SnBF/6
When comparing our model solvent,water, with these solvents, we predict that with regard toits electron-donating capacity ~O should be locatedsomewhere between sulfolan (0(1I9Sn) = 139 ppm forBu
3SnCI04) and acetonitrile (O(\I9Sn) = 54 ppm forBu
3SnCI04) . Hence, an estimated 0(119Sn) value of 60ppm for 14 is in line with experimental results.
In view of the results obtained here the followingconclusions can be drawn.
a. R3SnXcompounds with an appropriate leaving groupsuch as Cl, OH, etc. have 0(119Sn) values at lower fieldthan R3Sn+ cations in solution. Although one wouldexpect the cation to have a much more deshieldednucleus, this is not the case since only stronglycoordianted Sn cations exist in solution.
b. Increasing the donicity of the solvent leads to trigonalbipyramidal R
3SnX·S(S =solvence) complexes. The
0(119Sn) value appears at higher field becauseofchargetransfer from S to Sn and resultant higher shielding ofthe Sn nucleus.
c. Formation of the cation probably involves another Smolecule and the reaction steps (l) and (2) or (l)and (3).
d. For an electronegative X, ~Sn (S)2+ cations shouldposses o(l19Sn) values at higher field than eitherR3SnXor ~SnX·S. However, for any R-X-S combinationthis has to be tested.
In view of the high binding energies between ~Sn+ andsolvent molecules (in the case of~Omore than 50 kcallmol) it is certainly not justified to speakofstannyl cationsinsolution. Inreality, there exist differentpentacoordinated~Sn (S)/ cations with strong Sn,S interactions that leadto properties that differ considerably from those of a
+8 ..xI
R.....Sn-RR~'
1
S
(1)
x X sI I I
+8 1$
R--'Sn-R ~
R........S
__ R-xs- R.....Sn-R (2)
R~; R~ in"'IIII'S R~i1 1 1
S S S
XI
R.....Sn-RR""""" i1
S
Israel Journal ofChemistry 33 1993
RR, (f) R-S.n
II
S
+8
S.II(f)
R--'Sn-RR~i
I
S
(3)

naked stannyl cation. This is best reflected by a O(1l9Sn)value that, according to our calculations, appears ca. 1000ppm at higher field than that of an isolated cation in thegas phase.
CONCLUSIONSThe calculation of NMR chemical shifts by ab initiomethods means an important addition to the repertoire ofquantum chemistry. It has started the third phase ofcollaboration between experimentalists and quantumchemists, since theoretical NMR chemical shifts can beuseful to solve a variety of chemical problems. This hasbeen demonstrated in the presentpaperfor three examples,namely, the structuredeterminationofthe homotropyliumcation, the description of BH
3NH3in solution, and the
investigation oforganostannyl cation-sovent complexes.
Acknowledgments. This work was supported by theSwedish Natural Science Research Council (NFR). Allcalculations were done on the CRAY XMP/416 of theNationellt Superdatorcentrum(NSC), Linkoping, Sweden.The authors thank the NSC for a generous allotment ofcomputer time. Useful discussions with Professor UlfEdlund, Umea, Sweden, and Professor Paul von RagueSchleyer, Erlangen, Germany are acknowledged.
REFERENCES AND NOTES(1) Boys, S.F. Proc. R. Soc. London 1950, A200: 542.(2) Roothaan, C.C.J. Rev. Mod. Phys. 1951,23: 69. Hall,
G.G. Proc. R. Soc. London 1951, A205: 541.(3) Light, J.C. J. Phys. Chem. 1982,86: 2111.(4) Barnett, M.P. Rev. Mod. Phys. 1963, 35: 571.
POLYATOM, Version 2. Neumann, D.B.; Basch, H.;Kornegay, R.L.; Snyder, L.C.; Moskowitz, J.W.;Hornback, C.; Liebmann, S.P. Program 199, QCPE,Indiana Univ., Bloomington, Indiana.
(5) Clementi, E.; Davis, D.R. J. Comput. Phys. 1966,1: 223.(6) Rothenberg, S.; Kollman, P.; Schwartz, M.E.; Hayes,
E.F.; Allen, L.C. Int. J. Chem., Symp.• 1970,3: 715.(7) Bagus, P.S.; Liu, B.; McLean, A.D.; Yoshimine, M. In
Wave Mechanics: The First Fifty Years; Price, W.C.;Chissick, S.S.; Ravensdale, T., Eds.; Butterworth:London, 1973. McLean, A.D.; Yoshimine, M.;Lengsfield, B.H.; Bagus, P.S., Liu, B. In MOTECC.Modern Techniques in Computational Chemistry;Clementi, E., Ed.; ESCOM Science Publishers: Leiden,1990, p. 593.
(8) GAUSSIAN 70. Hehre, W.J.; Lathan, W.A.; Ditchfield,R.; Newton, M.D.; Pople, J.A. Program 236, QCPE,Indiana Univ., Bloomington, Indiana. See also,GAUSSIAN 90. Frisch,M.J.; Head-Gordon, M.;Trucks,G.W.; Foresman, 1.B.; Schlegel, H.B.; Raghavachari,K.; Robb, M.A.; Binkley, J.S.; Gonzalez, C.; DeFrees,DJ.; Fox, DJ.; Whiteside, RA; Seeger, R.; Melius, c.F.;
383
Baker, J.;Martin, R.L.; Kahn,L.R.; Stewart, U.P.;Topiol,S.; Pople, JA, Gaussian Inc., Pittsburgh, PA., 1990.
(9) Almlof', J. USIP Rep. 72-09, Univ. Stockholm, 1972.Almlof, J. USIP Rep. 74-29, Univ. Stockholm, 1974.Roos, B.D.; Karlstrom, G.; Malmqvist, p.A.; Sadlej,A.J.; Widmark, P.O. In MOTECC, Modem Techniquesin Computational Chemistry;Clementi,E., Ed.; ESCOMScience Publishers: Leiden, 1990, p. 533.
(10) Dupuis,M.; Rys,J.; King, H.F.J. Chern.Phys.1976,65:Ill. Dupuis, M.; Farazdel, A.; Kama, S.P.; Maluendes,S.A.InMOTECC, Modem Techniques inComputationalChemistry, Clementi, E., Ed.; ESCOM SciencePublishers: Leiden, 1990, p. 277.
(11) GAMESS. Guest, M.F.; Kendrick, J.; Pope, SA SERCDaresbury Laboratory, England, 1983. Dupuis, M.;Spangler, D.;Wendolowski, JJ. NRCC Software Catalog,Vol. I, Program QGOl, 1980.
(12) CADPAC: The Cambridge Analytic DerivativePackage,4.0 ed. Amos, R.D.; Rice, J.E., Cambridge, 1987.
(13) M0ller, C.; Plesset, M.S. Phys. Rev. 1934, 46: 618.Pople, JA; Binkley, J.S.; Seeger, R. Int. J. QuantumChem., Symp. 1976, 10: 1. Krishnan, R.; Pople, J.A. Int.J. Quantum Chern. 1978,14: 91. Krishnan, R.; Frisch,MJ.; Pople, J.A. J. Chem. Phys. 1980, 72: 4244.Raghavachari, K.; Pople, J.A.; Replogle, E.S.; HeadGordon, M. J. Phys. Chern. 1990,94: 5579.
(14) Kelly, H.P. Phys. Rev. 1963,131: 684.(15) For reviews on analytical derivative methods, see
Jergensen, P.; Simons, J., Eds.; Geometrical Derivativesof Energy Surfaces and Molecular Properties; Reidel:Dordrecht, 1986. Pulay, P. Adv. Chem. Phys. 1987,67:241. Schlegel, H.B. Adv. Chem. Phys. 1987,67: 249.Amos, R.D. Adv. Chern. Phys. 1987,67: 99. Gauss, J.;Cremer, D. Adv. Quantum Chern. 1990,27: 101.
(16) Pulay, P. Mol. Phys. 1969,17: 197.(17) Gerratt, J.; Mills, I.M. J. Chem. Phys. 1968,49: 1719.(18) Hameka, H. Mol. Phys. 1958,1: 203. Hameka, H. Rev.
Mod. Phys. 1962,34: 87. Zeroka, D.; Hameka, H.F. J.Chern. Phys. 1966, 45: 300.
(19) London, F. Naturwissenschaften 1937, 15: 187. London,F. J. Phys. Rad. 1937,8: 397.
(20) Pople, J.A. J. Chem. Phys. 1962,37: 53. Pople, J.A. J.
Chem.Phys.1962,37:60.(21) Ditchfield, R.J. Chem. Phys. 1972,56: 5688. Ditchfield,
R. 1. Chern. Phys. 1976,65: 3123. Ditchfield, R. Mol.Phys. 1974,27: 789. Ditchfield, R. In Topics in Carbon13 NMR Spectroscopy; Wiley: New York, 1974; Vol I.
(22) Galasso, V. Theor. Chim. Acta 1983,63: 35. Jaszunski,M.; Adamowicz, L. Chern. Phys. Lett. 1981,79: 133.Lazzeretti, P.; Zanasi, R. J. Chern. Phys. 1983,105: 12.
(23) Wolinski, K.; Hinton, J.F.; Pulay, P. J. Am. Chem. Soc.1990, 122: 8251.
(24) Kutzelnigg, W. lsr. J. Chern. 1980,19: 193. Schindler,M.; Kutzelnigg, W. J. Chern. Phys. 1982,76: 1919.
(25) (a) Schindler, M.; Kutzelnigg, W. J. Arn. Chem. Soc.1983,105: 1360. (b) Schindler, M.; Kutzelnigg, W. Mol.Phys. 1983,48: 781. (c) Schindler, M.; Kutzelnigg, W.J.
Cremer et al. / NMR Chemical Shift Calculations

384
Am. Chem. Soc. 1987,109: 1021. (d) Schindler, M. J.Am. Chem. Soc. 1987, 109: 5950. (e) Schindler, M.Magn. Reson. Chem. 1988, 26: 394. (t) Schindler, M. J.Am. Chem. Soc. 1988, 110: 6623. (g) Schindler, M. J.Chem. Phys. 1988, 88: 7638. (h) Kutzelnigg, W.;Schindler, M.; Fleischer, U. NMR, Basic Principles andProgress; Springer: Berlin, 1989; Vol. 23.
(26) Hansen, Aa.E.; Bouman, T.D. J. Chem. Phys. 1985,82:5035.
(27) Gauss, J. Chem. Phys. Lett. 1992,191: 614. Gauss, J. J.Chem. Phys. 1993,99: 3629.
(28) Kutzelnigg, W.; van WOllen,Ch.; Fleischer, U.; Franke,R. Proceedings ofthe NATO AdvancedWorkshop on TheCalculation ofNMR Shielding Constants and their Usein the Determination of the Geometric and ElectronicStructures ofMolecules and Solids, 1992.
(29) Bouman, T.D.; Hansen, Aa.E. Chem. Phys. Lett. 1990,175: 292. Geertsen, 1.; Oddershedde, 1. J. Chem. Phys.1984,90: 301. Geertsen, 1.; Jergensen, P.; Yaeger, DLComput. Phys. Rep. 1984,2: 33. Darbom, G.T.; Handy,N.C. Mol. Phys. 1983,49: 1277. Jaszunski, M.; Sadlej,A. Theoret. Chim. Acta 1975,40: 157.
(30) Summaries of the work on carbocations, boron, andorganolithium compounds can be found in (a) Buzek, P.;Schleyer, P.v.R.; Sieber, S. Chem. Unserer Zeit 1992,26: 116. (b) BUhl, M.; Schleyer, P.v.R. In ElectronDeficientBoron andCarbon Clusters; Olah, G.A.; Wade,K.; Williams, R.E., Eds.; Wiley: New York, 1991. (c)BUbI, M.; Hommes, NJ.R. v.E.; Schleyer, P.v.R.;Fleischer, U.; Kutzelnigg, W. J. Am. Chem. Soc. 1991,113: 2459.
(31) See, e.g., Hnyk, D.; Vajda, E.; Buehl, M.; Schleyer,P.v.R.lnorg.Chem.I992,31:2464. Buehl,M.;Schleyer,P.v.R. J. Am. Chem. Soc. 1992,114: 477. Buehl, M.;Schleyer, P.v.R.; McKee, M.L. Heteroat. Chem. 1991,2:499. Buehl, M.; Schleyer, P.v.R. Angew. Chem. 1990,102: 962. Schleyer, P.v.R.; Buehl, M.; Fleischer, D.;Koch, W.lnorg. Chem. 1990,29: 153. Schleyer, P.v.R.;Koch, W.; Liu, B.; Fleischer, U. J. Chem. Soc., Chem.Commun. 1989: 1098.Bremer, M.; Scheetz, K.; Schleyer,P.v.R.; Fleischer, U.; Schindler, M.; Kutzelnigg, W.;Koch, W.; Pulay, P. Angew. Chem. 1989,101: 1063.
(32) Onak, T.;Tseng,J.;Diaz,M.; Tran,D.; Arias, 1.;Herrera,S.; Brown, D. Inorg. Chem. 1993,32: 487.
(33) Cremer, D.; Reichel, F.; Kraka, E. J. Am. Chem. Soc.1991,113: 9459, and references therein.
(34) Reichel, F. Ph.D. Dissertation, Univ. of Keln, 1991.(35) Paquette, L.A.; Broadhurst, MJ.; Warner, P.; Olah,
G.A.; Liang, G. J. Am. Chem. Soc. 1973,105: 3386.(36) Childs, R.F.; Varadarajan, A.; Lock, CJ.L.; R. Faggiani,
R.; Fyfe, C.A.; Wasylishen, R.E. J. Am. Chem. Soc.1982, 104: 2452.
(37) Childs, R.F.; Faggiani, R.; Lock, CJ.L.; Mahendran, M.J. Am. Chem. Soc. 1986,108: 3613.
(38) Kraka, E.; Gauss, J.; Cremer, D. J. Mol. Struct.(Theochem) 1991,234: 95.
(39) Gauss, J.; Cremer, D. Adv. Quantum Chem. 1990,27:101.
(40) Zhi He; Cremer, D.lnt. J. Quantum Chem., Symp. 1991,25: 43. Zhi He; Cremer, D. Theor. Chim. Acta 1993,85:305.
(41) See, e.g., Bartlett, R.J. J. Phys. Chem. 1989,93: 1697.(42) Schleyer and coworkers have carried out an IGLOIDZI
IMP2I6-31G(d) investigation oflb, which led to similarresults. JOaHowever, the agreementbetweenexperimentaland IGLO '3(; chemical shifts obtained in this work islower because of the deficiencies of the MP2 method todescribe nonclassical carbocations such as lb.
(43) (a) Cremer, D.; Svensson, P.; Kraka, E.; Ahlberg, P. J.Am. Chem. Soc. 1993, 115: 7445. (b) Cremer, D.;Svensson, P.; Kraka, E.; Konkoli, Z.; Ahlberg, P. J.Am.Chem. Soc. 1993,115: 7457. (c) Svensson, P.; Reichel,F.; Ahlberg, P.; Cremer, D. J. Chem. Soc., Perkin Trans.1991, 2: 1463. (d) Sieber, S.; Schleyer, P.v.R.; Otto,A.H.; Gauss, 1.; Reichel, F.; Cremer, D. J. Phys. Org.Chem. 1993,6: 445.
(44) See, e.g., Redmon, L.T.; Purvis, G.D.; Bartlett, R.L. J.Am. Chem. Soc. 1974,101: 2856. Zirz, C.; Ahlrichs, R.J. Chem. Phys. 1981,75: 4980. Binkley, J.S.; Thome,L.R.J. Chem. Phys. 1983,79: 2932. Brint, P.; Sangchakr,B.; Fowler, P.W. J. Chem. Soc., Faraday Trans. 2, 1989,85: 29, and references therein.
(45) BUhl,M.;Steinke, T.; Schleyer,P.v.R.; Boese,R.Angew.Chem., Int. Ed. Engl. 1991,30: 1160.
(46) Thome, L.R.; Suenram, R.D.; Lovas, FJ. J. Chem. Phys.1983,78: 167.
(47) The [5s4pld/3s1p] basis is called basis II in ref 25.(48) Neth, H.; Wrackmeyer, B. Chem. Ber. 1974,107: 3070.(49) Miertus, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981,
55: 117. Bonaccorsi, R.; Cimiraglia, R.; Tomasi, J. J.Comput. Chem. 1983,4: 567. Bonaccorsi, R.; Pala, P.;Tomasi,J. J. Am. Chem. Soc. 1984, 106: 1945. PascualAhuir, JL; Silla, E.; Tomasi, J.; Bonaccorsi, R. J.Comput. Chem. 1987,8: 778.
(50) Shore, S.G.; Parry, R.W. J. Am. Chem. Soc. 1955,77:6084. Hughes, E.W. J. Am. Chem. Soc. 1956,78: 502.Lippert, E.L.; Lipscomb, W.N. J. Am. Chem. Soc. 1958,80: 503. Schleyerand coworkers, who have redeterminedthe X-ray structure of BH
3NH3, give for the B-N bond
length 1.564(6) A.45(51) See, e.g., Colquhoun, H.M.; Jones, G.; Maud, J.M.;
Stoddart, J.F.; Williams, DJ. J. Chem. Soc., DaltonTrans. 1984, 63. Alston, D.R.; Stoddart, J.F.;Wolstenholme, J.B.; Allwood, B.L.; Williams, DJ.Tetrahedron 1985, 41: 2923. Pears, D.A.; ShahriariZavareh, H.; Stoddart, J.F.; Crosby, J.; Allwood, B.L.;William, D.l.Acta Crystallogr. 1988, C44: nor.1106,11l2, ius.
(52) See, e.g., Olah, G.A.; Rasul, G.; Heiliger, L.; Bausch, 1.;Prakash, G.K.S. J. Am. Chem. Soc. 1992, 114: 7737, andreferences therein.
(53) Kraus, C.A.; Callis, C.C. 1. Am. Chem. Soc. 1923,45:2624.
(54) Webster, J.R.; Jolly, W.L.lnorg. Chem. 1971,10: 877.(55) Tobias, R.S. Organomet. Chem. Rev. 1966,1: 93.(56) Edlund, U.; Arshadi, M.; Johnels, D. J. Organomet.
Israel Journal ofChemistry 33 1993

Chem: 1993,456: 57.(57) See, e.g., Marquez, A.; Sanz, J.F. J. Am. Chem. Soc.
1992,114: 10019.Schleyer,P.v.R.; Kaupp, M.; Hampel,F.; Bremer,M.; Mislow, K.J.Am. Chem.Soc. 1992,114:6791. Nguyen, K.A.; Gordon,M.S.; Wang, G.; Lambert,lB. Organometallics 1991,10: 2798. Kudo, T.; Nagase,S. J. Phys. Chem. 1992,96: 9189. Nagase, S. Angew.Chern. 1989,101: 340.
(58) In principle, NMR chemical shift calculations should bepossible with pseudopotential methods. However, withthe existing ab initio programs for chemical shiftcalculations pseudopotentials cannot be used.
(59) Stromberg, A.; Gropen, 0.; Wahlgren, U. J. Comput.Chern. 1983,4: 181.
(60) Dunning,T.H.;Hay,P.J.InMethodsinElectronicStruetureTheory, Modem Theoretical Chemistry; Schaefer, H.F.,Ed.; Plenum: New York, 1977; Vol. 3, p. 1.
(61) For thecorresponding TZ+P data, seeOlsson, L.;Cremer,D. Inorg, Chem., in press.
(62) Vilkov, L.V.; Mastryukov, V.S.; Sadova, N.I.Determination of the Geometrical Structure of Free
385
Molecules; MirPublishers:Moscow, 1983,andreferencestherein. Wilkinson, G.R.; Wilson, M.K. J. Chem. Phys.1956,25: 784. Beagley, B.; McAloon, K.; Freeman, J.M.Acta Crystallogr. 1974, B30: 444. Clark, H.C.; Furnival,S.G.; Kwon,J.T. Can. J. Chem. 1963,41: 2889. Fujii, H.;Kimura, M. Bull. Chem. Soc. Jpn. 1970,44: 2643.
(63) Smith, P.I. Annu. Rep. NMR Spectrosc. 1978,8: 291.(64) Wrackmeyer, B. Annu. Rep. NMR Spectrosc. 1985,16:
73.(65) Holecek, J.; Nadvornik, M.; Handlir, K.; Lycka, A. J.
Organomet. Chem. 1983, 24: 177. Nadvornik, M.;Holecek, I.; Handlir, K.; Lycka, A. J. Organomet.Chern. 1984, 275: 43. AI-Allaf, T.A.K. J. Organomet.Chem. 1986,337: 177. Holecek,J.; Handlir, K.;Cerny,V.; Nadvornik, M.; Lycka, A. Polyhedron 1987,6:1037.
(66) See, e.g., Tossell, JA.; Lazzeretti, P. J. Magn. Reson.1988,80: 39.
(67) Torocheshnikov, V.N.; Tupciauskas, A.P.; Sergeyev,N.M.; Ustynyuk, Y.A. J. Organomet. Chem. 1972,35:C25.
Cremer et al. / NMR Chemical Shift Calculations