bunsentagung 2016 - converia€¦ · and karriereforum 5 – 7 may 2016 · rostock · germany...
TRANSCRIPT

115.
Bu
nse
nta
gu
ng
201
6 · U
niv
ersi
tät
Ro
sto
ck
www.bunsentagung.de
Basic Mechanismsin Energy Conversion
BUNSENTAGUNG 2016115th General Assembly of the German Bunsen Society for Physical ChemistryFeaturing an industrial symposium with accompanying exhibition and Karriereforum
5 – 7 MAY 2016 · ROSTOCK · GERMANY
PR
OG
RA
MM
E

5 – 7 MAY 2016 · ROSTOCK · GERMANY
The organisers convey sincere thanks for supporting the Bunsentagung 2016 to the following companies and organizations:
BASF SE Ludwigshafen/D
Bruker BioSpin GmbH Rheinstetten/D
Clariant SESulzbach/Ts./D
DFG Deutsche Forschungsgemeinschaft e.V. Bonn/D
FCI Fonds der Chemischen Industrie Frankfurt am Main/D
HCS Group GmbH Frankfurt am Main/D
PCCP Physical Chemistry Chemical Physics Cambridge/UK
Sanofi-Aventis Deutschland GmbH Frankfurt am Main/D
SFB 652 Rostock/D
Universität Rostock Rostock/D
YARA Rostock Rostock/D
SPONSORS AND SUPPORTERS

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K · B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
PROGRAMME AT A GLANCE
Friday, 6 May 2016Chair Beller
08:30-09:15 Audimax PL01 PLENARY LECTURE: Xu Audimax Arno-Esch I Arno-Esch II Haus 1 Hörsaal 224 Haus 1 Hörsaal 323
Main Topic Soft Matter and Polymers
Industrial Applications Liquids and Solvation Chemical Dynamics and Kinetics
Chair Strunk Wagner Plass Urbanski Lauth09:30-09:50 V01.01 Litke V08.01 Gradzielski V13.01 Kohnke V02.01 Rathke V03.01 Moshammer09:50-10:10 V01.02 Walenta V08.02 Pacholski V13.02 Egger V02.02 Kerlé V03.02 Wezisla10:10-10:30 V01.03 Bredol V08.03 Hansen V13.03 Vargas-Barbosa V02.03 Knorr V03.03 Oum10:30-10:50 V01.04 Borchert V08.04 Plamper V13.04 Burggraf V02.04 Kiefer V03.04 Hüter10:50-11:10 COFFEE BREAK
Main Topic Soft Matter and Polymers
Solids and Nano-sized Matter
Liquids and Solvation Chemical Dynamics and Kinetics
Chair Ludwig Porada Bigall Bande Gerhards11:10-11:30 FL01.01 FULL LECTURE
KornyshevV08.05 Elsing* V07.01 Dorfs V02.05 Imoto V03.05 Van der Linde
11:30-11:50 V08.06 Nack V07.02 Kartouzian V02.06 Knake V03.06 Van Wilderen11:50-12:10 V01.05 Wark* V08.07 Herold V07.03 Schaumberg V02.07 Heyden V03.07 Torres-Alacan12:10-12:30 V01.06 Hausbrand V08.08 Urbanski* V07.04 Niedermaier V02.08 Gleim V03.08 Kahnt12:30-13:30 LUNCH BREAK (12:30 – 13:30 Women‘s Networking Lunch, Haus 1, Raum SR021)
Chair Wagner13:30-14:15 Audimax PL02 PLENARY LECTURE: Durrant
Audimax Arno-Esch I Arno-Esch II Haus 1 Hörsaal 224 Haus 1 Hörsaal 323Main Topic Electrochemistry
and EnergySolids and
Nano-sized MatterIndustrial Applications Hot Topics
Chair Kornyshev Hofmann Kartouzian Kragl Nickel14:30-14:50 V01.07 Chen V12.01 Senocrate FL07.01 FULL LECTURE
BigallV13.05 Hartmann V14.01 Starr
14:50-15:10 V01.08 Bickel V12.02 Tranca V13.06 Barsch V14.02 Hemberger15:10-15:30 V01.09 Neff V12.03 dos Santos
SardinhaV07.05 Scheele V13.07 Ondo V14.03 Pérez de Tudela
15:30-15:50 V01.10 Fischer V12.04 Chen V07.06 Boldt V13.08 Joseph V14.04 Usvyat15:50-16:10 COFFEE BREAK
Main Topic Experimental Techniques
Solids and Nano-sized Matter
Surfaces and Interfaces
Preisträgersession
Chair Kaiser Siefermann Niedner-Schatteburg Schmitt Sauer16:10-16:30 V01.11 Strunk V05.01 Waldt V07.07 Passow V09.01 Bünermann V15.01 Nernst-Haber-
Bodenstein Preis16:30-16:50 V01.12 Widmann V05.02 Kottke V07.08 Lauth V09.02 Paier V15.02 Paul-Bunge
Preis16:50-17:10 FL01.02 FULL LECTURE Papp
V05.03 Pramann V07.09 Nielsen V09.03 Yang17:10-17:30 V05.04 Hollmann V07.10 Kruss V09.04 Späth V15.03 Ewald-Wicke
Preis17:30-17:50 V01.13 Neumann V05.05 Zimina V07.11 Klinke V09.05 Kropp V15.04 Wilhelm-
Ostwald-Nach-wuchspreis
Tent18:00-19:30 POSTER SESSION (even numbers)19:30-21:00 POSTER SESSION (odd numbers)
Thursday, 5 May 2016
12:00-15:00 Arno-Esch II Karriereforum (in German only)
15:00-16:00 Arno-Esch I Ordentliche Mitgliederversammlung (only for DBG members, in German only)
Chair Sauer/Ludwig
16:30-18:30
Audimax
OPENING CEREMONY
18:30-19:15 OPENING LECTURE: Grätzel
19:30-22:00 Welcome Reception

· B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
PROGRAMME AT A GLANCE
Saturday, 7 May 2016Chair Kühn
08:30-09:15 Audimax PL03 PLENARY LECTURE: Lubitz Audimax Arno-Esch I Arno-Esch II Haus 1 Hörsaal 224 Haus 1 Hörsaal 323
Main Topic Electrochemistry and Energy
Biophysical Chemistry Surfaces and Interfaces
Molecular Structure
Chair Marschall Peuckert Kottke Backus Schnell09:30-09:50 V01.14 Lambert V12.05 Lück V10.01 Khadem V09.06 Schmid V04.01 Pitzer09:50-10:10 V01.15 Müh V12.06 Single V10.02 Luong V09.07 Antonsson V04.02 Wiese10:10-10:30 V01.16 Wächtler V12.07 Wu V10.03 Chmielewicz* V09.08 Schneider V04.03 Reusch10:30-10:50 V01.17 Greisch V12.08 Berger V10.04 Vogel V09.09 Siebels V04.04 Bumüller10:50-11:10 COFFEE BREAK
Main Topic Electrochemistry and Energy
Biophysical Chemistry Surfaces and Interfaces
Molecular Structure
Chair Papp Schuster Jung Wöll Kühn11:10-11:30 V01.18 Merschjann V12.09 Chanaewa V10.05 Bald FL09.01 FULL LECTURE
BackusV04.05 Poblotzki
11:30-11:50 V01.19 Dräger V12.10 Schedel-Niedrig V10.06 Roder V04.06 Bernhard11:50-12:10 V01.20 Feldhoff V12.11 Taffa V10.07 Langel V09.10 Munoz-Santiburcio V04.07 Schnell12:10-12:30 V01.21 Andrei V12.12 Pfeifer V10.08 Müller V09.11 Behler V04.08 Song12:30-13:30 LUNCH BREAK
Chair Kragl13:30-14:15 Audimax PL04 PLENARY LECTURE: DeBeer
Audimax Arno-Esch I Arno-Esch II Haus 1 Hörsaal 224 Haus 1 Hörsaal 323Main Topic Electrochemistry
and EnergyCatalysis Theoretical Techniques Hot Topics
Chair Lochbrunner Paschek Beller Paier Dick14:30-14:50 V01.22 Lenzer V12.13 Brummel V11.01 Kubis V06.01 Fabri V14.05 Heyda14:50-15:10 V01.23 Marschall V12.14 Oezaslan V11.02 Kunz V06.02 Ivanov V14.06 Gliemann15:10-15:30 V01.24 Schmitt FL12.01 FULL LECTURE
Schuster*V11.03 Wagner V06.03 Kandratsenka V14.07 Senske
15:30-15:50 V01.25 Kundu V11.04 Boubnov V06.04 Müller V14.08 Neugebohren16:00-16:30 Audimax CLOSING CEREMONY / POSTER AWARD CEREMONY
* different from the printed programme

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
TABLE OF CONTENTS
3
Page
PROGRAMME AT A GLANCE
COMMITTEE / ORGANISATION 4
EXHIBITORS 5
LECTURE PROGRAMME
Opening Lecture / Plenary Lectures / Full Lectures 6Opening Lecture 7Basic Mechanisms in Energy Conversion (Main Topic) 8, 9, 18, 19Soft Matter and Polymers 10Electrochemistry and Energy 11, 20, 21Experimental Techniques 11Industrial Applications 12, 15Solids and Nano-sized Matter 12, 13Liquids and Solvation 14Surfaces and Interfaces 15, 24Chemical Dynamics and Kinetics 16Hot Topics 17, 27Preisträgersession (Award session) 17Biophysical Chemistry 22Catalysis 23Theoretical Techniques 25Molecular Structure 26
POSTER PROGRAMME
Basic Mechanisms in Energy Conversion (Main Topic) 28Liquids and Solvation 31Chemical Dynamics and Kinetics 33Molecular Structure 35Experimental Techniques 37Theoretical Techniques 38Solids and Nano-sized Matter 38Soft Matter and Polymers 40Surfaces and Interfaces 41Biophysical Chemistry 43Catalysis 46Electrochemistry and Energy 46Industrial Applications 48
L IST OF AUTHORS 443
VENUE OVERVIEW 455
IMPRINT 455
As of 13 April 2016, subject to alterations. Abstracts, submission title and authors information as given by the submitter. No proof by DBG.

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
COMMITTEE / ORGANISATION
4
PROGRAMME COMMITTEE
Matthias Beller LIKAT, Rostock/DAngelika Brückner LIKAT, Rostock/DBernhard Dick Universität Regensburg/DAlexander Eychmüller TU Dresden/DFrank Gießelmann Universität Stuttgart/DAras Kartouzian TU München/DUdo Kragl Universität Rostock/DOliver Kühn Universität Rostock/DRalf Ludwig Universität Rostock/DChristian Ochsenfeld Universität München/D
Marcell Peuckert Hofheim/DJan Henryk Porada Universität Stuttgart/DJoachim Sauer Humboldt-Universität zu Berlin/DRolf Schäfer TU Darmstadt/DRobert Schlögl Fritz-Haber-Institut der MPG, Berlin/DMartin Suhm Universität Göttingen/DJoachim Wagner Universität Rostock/DRoland Winter TU Dortmund/D
SCIENTIFIC ORGANISATION
Ralf Ludwig Universität Rostock/DJoachim Wagner Universität Rostock/DAngelika Brückner LIKAT, Rostock/D
Udo Kragl Universität Rostock/DMatthias Beller LIKAT, Rostock/DOliver Kühn Universität Rostock/D
LOCAL ORGANISATION
Ralf Ludwig Universität Rostock/DJoachim Wagner Universität Rostock/DSergey Verevkin Universität Rostock/DDietmar Paschek Universität Rostock/DJochen Lehmann Universität Rostock/D
Gisela Boeck Universität Rostock/DAndreas Appelhagen Universität Rostock/DAlexander Wulf Universität Rostock/DAnika Wilhelms Universität Rostock/DAnne Knorr Universität Rostock/D
HOST
Deutsche Bunsen-Gesellschaft für physikalische Chemie e.V.German Bunsen Society for Physical ChemistryVarrentrappstraße 40-4260486 Frankfurt am Main/D
Phone: +49 (0)69 7917-363Fax: +49 (0)69 7917-1363E-Mail: [email protected]: www.bunsen.de
Organisational support by:
DECHEMA e.V. Universität Rostock/D Frankfurt am Main/D

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
5
6 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim/D
7 Bruker Optik GmbH, Ettlingen/D8 Helmholtz-Zentrum Berlin für Materialien
und Energie GmbH, Berlin/D9 Belltec – Ing. Büro Glocke, Wesel/D
Audimax Ground Floor
Arno Esch Ground Floor
1 PCCP Physical Chemistry Chemical Physics, Cambridge/UK
2 DFG Deutsche Forschungsgesellschaft, Bonn/D3 de Gruyter GmbH, Berlin/D4 SPECS Surface Nano Analysis GmbH, Berlin/D5 rhd instruments GmbH & Co. KG, Marburg/D
Catering
1 2
3
Catering
87
6
54
9
EXHIBITORS

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
6
OPENING LECTURE
Thursday, 5 May 2016
18:30OL01
Mesoscopic photosystems for the solar generation of electricity and fuels M. Grätzel1; 1 EPFL – École polytechnique fédérale de Lausanne/CH
51
PLENARY LECTURES
Friday, 6 May 2016
08:30PL01
Functional nanomaterials for energy conversion and storage Q. Xu, Advanced Industrial Science and Technology (AIST), Osaka/J
52
13:30PL02
Charge carrier dynamics for photoelectrochemical water splitting J. Durrant, Imperial College London/UK
60
Saturday, 7 May 2016
08:30PL03
Mechanism of light-induced water oxidation and oxygen release in photosynthesis W. Lubitz, MPI für Chemische Energiekonversion, Mülheim/D
137
13:30PL04
Making and breaking bonds: x-ray Spectroscopic studies of small molecule activation by biological catalysts S. DeBeer, MPI für Chemische Energiekonversion, Mülheim/D
146
FULL LECTURES
Friday, 6 May 2016
11:10FL01.01
Energy harvesting and storage with ionic liquids: the essential physics at the nanoscale A.A. Kornyshev1; 1 Imperial College London/UK
57
14:30FL07.01
Hydrogels and aerogels of nanocrystal building blocks N. Bigall1; 1 Leibniz Universität Hannover/D
95
16:50FL01.02
Dehydrogenation of liquid organic hydrogen carriers on Pt(111) C. Papp1; C. Gleichweit1; M. Amende1; S. Schernich1; W. Zhao1; M. Lorenz1; O. Höfert1; N. Brückner1; P. Wasserscheid1; J. Libuda1; H. Steinrück1; 1 Universität Erlangen-Nürnberg, Erlangen/D
67
Saturday, 7 May 2016
11:10FL09.01
Two types of water at the water−surfactant interface revealed by time-resolved vibrational spectroscopy R. Livingstone1; Y. Nagata1; M. Bonn1; E. Backus1; 1 Max Planck Institute for Polymer Research, Mainz/D
179
15:10FL12.01
Electrochemical microcalorimetry R. Schuster1; 1 Karlsruhe Institute of Technology (KIT)/D
162
Page
OPENING LECTURE / PLENARY LECTURES / FULL LECTURES

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
7
Thursday, 5 May 2016
12:00 KARRIEREFORUM* Arno-Esch II
15:00 ORDENTLICHE MITGLIEDERVERSAMMLUNG* Arno-Esch I(only for DBG members)
Chair: J. Sauer, Humboldt-Universität zu Berlin/D; R. Ludwig, Universität Rostock/D
16:30 OPENING CEREMONY (16:30 – 18:30) Audimax
18:30 –
19:15OL01
OPENING LECTURE: Audimax Mesoscopic photosystems for the solar generation of electricity and fuelsM. Grätzel1; 1 EPFL – École polytechnique fédérale de Lausanne/CH
51
19:30 WELCOME RECEPTION (19:30 – 22:00) Tent
* in German only
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
8
Friday, 6 May 2016
Chair: M. Beller, Leibniz-Institut für Katalyse e.V., Rostock/D Audimax
08:30 PL01
PLENARY LECTURE Functional nanomaterials for energy conversion and storageQ. Xu, Advanced Industrial Science and Technology (AIST), Osaka/J
52
Main Topic – Basic Mechanisms in Energy Conversion
Chair: J. Strunk, Max-Planck-Institut für chemische Energiekonversion, Mülheim/D Audimax
09:30 V01.01
TiO2 as a bottleneck for the photocatalytic water reduction on Cd0.5Zn0.5S@Pt-TiO2 composites A. Litke1; J. Hofmann1; T. Weber1; E. Hensen1; 1 Eindhoven University of Technology/NL
53
09:50 V01.02
Ethanol photocatalysis on rutile TiO2(110): unraveling the mechanistic role of defects and water C. Walenta1; S. Kollmannsberger1; M. Tschurl1; U. Heiz1; 1 TU München, Garching/D
54
10:10 V01.03
Electrocatalytic activity of sulfides M. Bredol1; V. Niemann2; J. Goossen1, 1 Fachhochschule Münster, Steinfurt/D; 2 Drexel University, Philadelphia, PA/USA
55
10:30 V01.04
Solar cells with a solution-processable heterojunction between CuInS2 and ZnO nanocrystals H. Borchert1; D. Scheunemann1; S. Wilken1; R. Miranti1; C. Krause1; J. Parisi1; 1 University of Oldenburg/D
56
10:50 COFFEE BREAK
Main Topic – Basic Mechanisms in Energy Conversion
Chair: R. Ludwig, Universität Rostock/D Audimax
11:10 FL01.01
FULL LECTURE Energy harvesting and storage with ionic liquids: the essential physics at the nanoscale A.A. Kornyshev1; 1 Imperial College London/UK
57
11:50 V01.05
High temperature proton conducting fuel cell membrane additives operating under anhydrous conditions M. Einemann1; C. Harms2; M. Wark1; 1 University of Oldenburg/D; 2 NEXT Energy Forschungszentrum, Oldenburg/D
58
12:10 V01.06
Electronic and ionic structure of Li-ion battery materials: concept and impact on interface properties R. Hausbrand1; M. Fingerle1; 1 TU Darmstadt/D
59
12:30 LUNCH BREAK
Chair: J. Wagner, Universität Rostock/D Audimax
13:30 PL02
PLENARY LECTURE Charge carrier dynamics for photoelectrochemical water splittingJ. Durrant, Imperial College London/UK
60
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
9
Friday, 6 May 2016
Basic Mechanisms in Energy Conversion
Chair: A.A. Kornyshev, Imperial College London/UK Audimax
14:30 V01.07
Structural insights into charged Li2-xMO2F by synchrotron X-ray PDF and XANES R. Chen1; R. Hempelmann2; E. Maawad3; M. Yavuz4; M. Knapp4; S. Zander5; 1 KIST Europe, Saarbrücken/D; 2 Saarland University, Saarbrücken/D; 3 Helmholtz-Zentrum Geesthacht (HZG), Hamburg/D; 4 Karlsruhe Institute of Technology (KIT)/D; 5 Helmholtz-Zentrum Berlin (HZB)/D
61
14:50 V01.08
Insights into the mechanism of the pyridine catalyzed electrochemical CO2 reduction K. Bickel1; A. Crespo-Yapur1; M. Daimer1; S. Filser1; Q. Li1; K. Schönleber1; K. Krischer1; 1 TU München, Garching/D
62
15:10 V01.09
Imaging ultrafast photo-induced dynamics in semiconducting polymer films with time-resolved photoemission electron microscopy A. Neff1; K. Siefermann1; 1 Leibniz Institute of Surface Modification, Leipzig/D
63
15:30 V01.10
Mechanistic Understanding of photocatalytic water reduction with iron carbonyl phosphines by means of in situ spectroscopy and time dependent – DFT calculations S. Fischer1; A. Lennox2; O. Bokareva1; A. Rösel1; N. Rockstroh2; M. Bauer3; O. Kühn1; M. Beller2; R. Ludwig1; 1 University of Rostock/D; 2 Leibniz-Institut für Katalyse e.V., Rostock/D; 3 Paderborn University /D
64
15:50 COFFEE BREAK
Main Topic – Basic Mechanisms in Energy Conversion
Chair: B. Kaiser TU Darmstadt/D Audimax
16:10 V01.11
Identification and exclusion of intermediates of photocatalytic CO2 reduction on TiO2 under conditions of highest purity A. Pougin1; M. Dilla2; J. Strunk2; 1 Ruhr-Universität Bochum/D; 2 Max-Planck-Institute for Chemical Energy Conversion, Mülheim a.d. Ruhr/D
65
16:30 V01.12
Methanol formation via CO2 hydrogenation on Au/ZnO catalysts – influence of CO and its role in the reaction mechanism D. Widmann1; 1 Ulm University/D
66
16:50 FL01.02
FULL LECTURE Dehydrogenation of liquid organic hydrogen carriers on Pt(111) C. Papp1; C. Gleichweit1; M. Amende1; S. Schernich1; W. Zhao1; M. Lorenz1; O. Höfert1; N. Brückner1; P. Wasserscheid1; J. Libuda1; H. Steinrück1; 1 Universität Erlangen-Nürnberg, Erlangen/D
67
17:30 V01.13
Mechanism of the aqueous-phase reforming of methanol – understanding the role of the base E. Alberico1; A. Lennox2; L. Neumann2; H. Jiao2; W. Baumann2; H. Drexler2; M. Nielson2; H. Junge2; M. Beller2; 1 Istituto di Chimica Biomolecolare, Sassari/I; 2 Leibniz-Institut für Katalyse e.V., Rostock/D
68
18:00 POSTER SESSION (even numbers) Tent
19:30 POSTER SESSION (odd numbers) (19:30 – 21:00) Tent
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
10
Friday, 6 May 2016
Chair: M. Beller, Leibniz-Institut für Katalyse e.V., Rostock/D Audimax
08:30 PL01
PLENARY LECTURE Functional nanomaterials for energy conversion and storageQ. Xu, Advanced Industrial Science and Technology (AIST), Osaka/J
52
Soft Matter and Polymers
Chair: J. Wagner, Universität Rostock/D Arno-Esch I
09:30 V08.01
Complexes of chitosan and alkylethoxycarboxylates: pH responsive systems of versatile structure and properties M. Gradzielski1; L. Chiappisi1; M. Simon1; 1 TU Berlin/D
69
09:50 V08.02
Benefits of using hydrogel microgels in colloidal lithography C. Pacholski1; M. Weiler2; 1 University of Potsdam/D; 2 Max Planck Institute for Intelligent Systems, Stuttgart/D
71
10:10 V08.03
Atomistic simulations of the formation of perylene-based supramolecular complexes in aqueous solution J. Baz1; N. Hansen1; 1 University of Stuttgart /D
72
10:30 V08.04
Interface-enforced complexation between copolymer blocks A. Steinschulte1; F. Plamper1; 1 RWTH Aachen University/D
73
10:50 COFFEE BREAK
Soft Matter and Polymers
Chair: J.H. Porada, Universität Stuttgart/D Arno-Esch I
11:10 V08.05
Monodisperse, ordered polystyrene foams and their mechanical properties J. Elsing1; M. Gilchrist2; W. Drenckhan3; C. Stubenrauch1; 1 University of Stuttgart/D; 2 University College Dublin/IRL; 3 Université Paris Sud/F
74
11:30 V08.06
Hindered nematic alignment of hematite spindles in viscoelastic matrices A. Nack1; J. Seifert1; C. Passow1; J. Wagner1; 1 University of Rostock/D
75
11:50 V08.07
Viral coefficients of anisotropic, hard solids of revolution: The influence of the geometry E. Herold1; R. Hellmann1; J. Wagner1; 1 University of Rostock/D
76
12:10 V08.08
New insights into nematic nanocomposites by dielectric spectroscopy: conductivity, polarization and relaxation M. Bourg1; M. Urbanski1; 1 University of Luxembourg /L
77
12:30 LUNCH BREAK
Chair: J. Wagner, Universität Rostock/D Audimax
13:30 PL02
PLENARY LECTURE Charge carrier dynamics for photoelectrochemical water splittingJ. Durrant, Imperial College London/UK
60
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
11
Friday, 6 May 2016
Electrochemistry and Energy
Chair: J.P. Hofmann, TU Eindhoven/NL Arno-Esch I
14:30 V12.01
Unraveling the nature of ionic conductivity in hybrid organic-inorganic halide perovskites A. Senocrate1; T. Yang1; G. Gregori1; N. Pellet2; M. Graetzel2; J. Maier1; 1 Max Planck Institute for Solid State Research, Stuttgart/D; 2 Ecole Polytechnique Fédérale de Lausanne/CH
78
14:50 V12.02
Codoped 2D conjugated metal-organic surface for efficient electrochemical hydrogen production: from molecules to solid catalysts D. Tranca1; R. Dong1; I. Tranca2; N. Chandrasekhar1; H. Liang1; J. Zhang1; S. Liu1; G. Seifert1; Z. Zheng1; X. Feng1; 1 TU Dresden/D; 2 Eindhoven University of Technology/NL
79
15:10 V12.03
Removal of the SiO2 native layer from silicon electrodes – scanning electrochemical microscopy and X-ray photoelectron spectroscopy E. dos Santos Sardinha1; H. Bülter1; G. Wittstock1; 1 University of Oldenburg/D
80
15:30 V12.04
Mass storage at heterojunctions C. Chen1; R. Usiskin1; L. Fu1; J. Maier1;1 Max Planck Institute for Solid State Research, Stuttgart/D
81
15:50 COFFEE BREAK
Experimental Techniques
Chair: K. Siefermann, Leibniz-Institut für Oberflächenmodifizierung e.V., Leipzig/D Arno-Esch I
16:10 V05.01
Gas phase studies of luminescent lanthanoid complexes by trapped ion laser induced fluorescence (TLIF) E. Waldt1; J. Greisch1; B. Schäfer1; M. Ruben1; M. Kappes2; D. Schooss1; 1 Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen/D; 2 Karlsruhe Institute of Technology (KIT)/D
82
16:30 V05.02
Indirect isotope effects need to be taken into account for the assignment of vibrational spectra of complex molecules T. Domratcheva1; E. Hartmann1; I. Schlichting1; C. Sommer2; M. Dietz2; T. Kottke2; 1 Max Planck Institute for Medical Research, Heidelberg/D; 2 Bielefeld University/D
83
16:50 V05.03
The role of HR-MC-ICP mass spectrometry in the redefinition of the SI unit kilogram A. Pramann1; O. Rienitz1; 1 Physikalisch-Technische Bundesanstalt (PTB), Braunschweig/D
84
17:10 V05.04
New spectroscopic in situ tools for monitoring electrocatalysts during water oxidation (WO) D. Hollmann1; K. Grabow1; J. Rabeah1; M. Polyakov1; A. Sukrus1; U. Bentrup1; S. Klink2; W. Schuhmann2; A. Brückner1; 1 Leibniz-Institut für Katalyse e.V., Rostock/D; 2 Ruhr-Universität Bochum/D
85
17:30 V05.05
The CAT-ACT beamline at ANKA: a new high energy X-ray spectroscopy facility for CATalysis and ACTinide research A. Zimina1; 1 Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen/D
86
18:00 POSTER SESSION (even numbers) Tent
19:30 POSTER SESSION (odd numbers) (19:30 – 21:00) Tent
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
12
Friday, 6 May 2016
Chair: M. Beller, Leibniz-Institut für Katalyse e.V., Rostock/D Audimax
08:30 PL01
PLENARY LECTURE Functional nanomaterials for energy conversion and storageQ. Xu, Advanced Industrial Science and Technology (AIST), Osaka/J
52
Industrial Applications
Chair: M. Plass, DOW Europe GmbH, Horgen/CH Arno-Esch II
09:30 V13.01
Electrochemical potential determination H. Kohnke1, 1 Gaskatel GmbH, Kassel/D
87
09:50 V13.02
La2NiO4 as anode material for high temperature steam electrolysis A. Egger1; N. Schrödl1; W. Sitte1; C. Gspan2; F. Hofer2; 1 Montanuniversität Leoben/A; 2 Graz University of Technology/A
88
10:10 V13.03
Investigating the viability of monopolar and bipolar membranes as separators in buffered water-splitting cells N. Vargas-Barbosa1; T. Mallouk2; 1 Universität Marburg/D; 2 The Pennsylvania State University, University Park, PA/USA
89
10:30 V13.04
Novel materials and components for Proton Exchange Membrane (PEM) water electrolysis: status and challenges F. Burggraf1; P. Lettenmeier1; A. Gago1; K. Friedrich1; 1 DLR – German Aerospace Center, Stuttgart/D
90
10:50 COFFEE BREAK
Solids and Nano-sized Matter
N.-C. Bigall, Universität Hannover/D Arno-Esch II
11:10 V07.01
Tuning the localized surface plasmon resonances in degenerately doped semiconductor nanocrystals A. Wolf1; D. Dorfs1; 1 Universität Hannover/D
91
11:30 V07.02
Plasmons in supported silver clusters T. Lünskens1; P. Heister1; M. Thämer1; A. Von Weber1; M. Jakob1; A. Kartouzian1; U. Heiz1; 1 TU München, Garching/D
92
11:50 V07.03
Laser irradiation of suspensions: mechanisms of nanoparticle generation C. Schaumberg1; 1 Humboldt-Universität zu Berlin/D
93
12:10 V07.04
Functionalization of oxide nanocrystals and composites with transition metal ions M. Niedermaier1; A. Gheisi2; J. Bernardi3; O. Diwald1; 1 Paris-Lodron Universität Salzburg/A; 2 Universität Erlangen-Nürnberg, Erlangen/D; 3 Vienna University of Technology/A
94
12:30 LUNCH BREAK
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
13
Friday, 6 May 2016
Chair: J. Wagner, Universität Rostock/D Audimax
13:30 PL02
PLENARY LECTURE Charge carrier dynamics for photoelectrochemical water splittingJ. Durrant, Imperial College London/UK
60
Solids and Nano-sized Matter
Chair: A. Kartouzian, TU München/D Arno-Esch II
14:30 FL07.01
FULL LECTURE Hydrogels and aerogels of nanocrystal building blocks N. Bigall1; 1 Leibniz Universität Hannover/D
95
15:10 V07.05
Conductive PbS mesocrystals A. André1; M. Samadi Khoshkhoo1; M. Weber1; M. Scheele1; 1 University of Tübingen/D
96
15:30 V07.06
Environmental effects on the stability and photo-chemistry of semiconductor nanocrystals K. Boldt1; N. Kirkwood2; S. Murphy2; J. Baldauf2; P. Mulvaney2; 1 University of Konstanz/D; 2 University of Melbourne & Bio21 Institute, Melbourne/AUS
97
15:50 COFFEE BREAK
Solids and Nano-sized Matter
Chair: G. Niedner-Schatteburg, TU Kaiserslautern/D Arno-Esch II
16:10 V07.07
Orientational distribution functions of suspended spindle-shaped hematite particles in external magnetic fields C. Passow1; A. Nack1; J. Wagner1; 1 University of Rostock/D
98
16:30 V07.08
Ultrafast dynamics of electron-hole pairs in 2D InSe nanosheets J. Lauth1; F. Spoor1; A. Kulkarni1; A. Houtepen1; J. Schins1; S. Kinge2; L. Siebbeles1; 1 Delft University of Technology/NL; 2 Toyota Motor Europe, Delft/NL
99
16:50 V07.09
Small-diameter semiconductor block nanowires synthesized by Solution-Liquid-Solid (SLS) technique and cation exchange A. Nielsen1; W. Buhro2; T. Kipp1; A. Mews1; 1 Uinversität Hamburg/D; 2 Washington University in St. Louis, MO/USA
100
17:10 V07.10
Impact of redox-active molecules on fluorescence and exciton dynamics in semiconducting carbon nanotubes S. Kruss1; 1 Universität Göttingen/D
101
17:30 V07.11
Electrical transport through colloidal PbS sheets C. Klinke1; 1 University of Hamburg/D
102
18:00 POSTER SESSION (even numbers) Tent
19:30 POSTER SESSION (odd numbers) (19:30 – 21:00) Tent
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
14
Friday, 6 May 2016
Chair: M. Beller, Leibniz-Institut für Katalyse e.V., Rostock/D Audimax
08:30 PL01
PLENARY LECTURE Functional nanomaterials for energy conversion and storageQ. Xu, Advanced Industrial Science and Technology (AIST), Osaka/J
52
Liquids and Solvation
Chair: M. Urbanski, University of Luxembourg/L Haus 1 – Hörsaal 224
09:30 V02.01
Protic ionic liquids: mesoscopic inhomogeneities in solutions of ethyl-ammonium nitrate and n-alkyl alcohols O. Russina1; A. Triolo2; W. Schröer3; B. Rathke3; 1 Università di Roma Sapienza, Rome/I; 2 Consiglio Nazionale delle Ricerche, Rome/I; 3 Universität Bremen/D
103
09:50 V02.02
A further step to the understanding of the solvation behavior of alcohols in ionic liquids A. Appelhagen1; D. Kerlé2; 1 University of Rostock/D; 2 Universität Bremen/D
104
10:10 V02.03
Spectroscopic evidence for clusters of like-charged ions in ionic liquids A. Knorr1; P. Stange1; R. Ludwig1; 1 University of Rostock/D
105
10:30 V02.04
Excess spectroscopy to study binary and ternary solutions J. Kiefer1; F. Zehentbauer1; K. Noack2; 1 Universität Bremen/D; 2 Universität Erlangen- Nürnberg, Erlangen/D
106
10:50 COFFEE BREAK
Liquids and Solvation
Chair: A. Bande, Helmholtz Zentrum Berlin (HZB)/D Haus 1 – Hörsaal 224
11:10 V02.05
Pressure effects on solvation structure and vibrational spectroscopy of aqueous TMAO solutions S. Imoto1; H. Forbert1; D. Marx1; 1 Ruhr-Universität Bochum/D
107
11:30 V02.06
The impact of TMAO and urea on the H-bond dynamics under high pressure L. Knake1; H. Vondracek1; M. Havenith1, 1 Ruhr-Universität Bochum/D
108
11:50 V02.07
Thermodynamic properties of water solvating biomolecular surfaces M. Heyden1; 1 Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr/D
109
12:10 V02.08
Ultrafast dynamics of vibrational relaxation of pseudohalides in liquid-to-supercritical water. a critical test of fermi’s golden rule J. Gleim1; D. Czurlok1; J. Lindner1; P. Vöhringer1; 1 University of Bonn/D
110
12:30 LUNCH BREAK
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
15
Friday, 6 May 2016
Chair: J. Wagner, Universität Rostock/D Audimax
13:30 PL02
PLENARY LECTURE Charge carrier dynamics for photoelectrochemical water splittingJ. Durrant, Imperial College London/UK
60
Industrial Applications
Chair: U. Kragl, Universität Rostock/D Haus 1 – Hörsaal 224
14:30 V13.05
FTIR spectroelectrochemistry – details of equipment and applications W. Hartmann1; M. Kessler1; 1 Bruker Optik GmbH, Ettlingen/D
111
14:50 V13.06
Application of in situ infrared spectroscopy to the production of a flavouring substance E. Barsch1; A. Petri2; P. Müller2; R. Ludwig1; 1 University of Rostock/D; 2 Miltitz Aromatics GmbH, Bitterfeld-Wolfen/D
112
15:10 V13.07
Heats of dilution of ionic liquids and their inorganic salt precursors in water D. Ondo1; V. Dohnal1; N. Ignatiev2; 1 University of Chemistry and Technology Prague/CZ; 2 Merck KGaA, Darmstadt/D
113
15:30 V13.08
Papierbasierte Chemiresistoren E. Wete1; R. Dittrich1; Y. Joseph1; 1 TU Bergakademie Freiberg/D
114
15:50 COFFEE BREAK
Surfaces and Interfaces
Chair: M. Schmitt, Universität des Saarlandes, Saarbrücken/D Haus 1 – Hörsaal 224
16:10 V09.01
Inelastic hydrogen atom scattering: role of electron-hole pair excitations O. Bünermann1; H. Jiang1; Y. Dorenkamp1; A. Wodtke1; 1 Universität Göttingen/D
115
16:30 V09.02
CO2 capture via adsorption on iron oxides: how a specific surface morphology enables host-guest-like chemistry X. Li1; J. Paier1; 1 Humboldt-Universität zu Berlin/D
116
16:50 V09.03
Structure and reactivity of ceria single crystal surfaces studied by IR spectroscopy C. Yang1; A. Nefedov1; Y. Wang1; C. Wöll1; 1 Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen/D
117
17:10 V09.04
Hydrogenation and dehydrogenation of hexagonal – Boron Nitride (h-BN) on Ni(111) F. Späth1; F. Düll1; C. Gleichweit1; U. Bauer1; P. Bachmann1; H. Steinrück1; C. Papp1; 1 Universität Erlangen-Nürnberg, Erlangen/D
118
17:30 V09.05
C–H bond activation at ceria-supported vanadia T. Kropp1; J. Paier1; J. Sauer1; 1 Humboldt-Universität zu Berlin/D
119
18:00 POSTER SESSION (even numbers) Tent
19:30 POSTER SESSION (odd numbers) (19:30 – 21:00) Tent
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
16
Friday, 6 May 2016
Chair: M. Beller, Leibniz-Institut für Katalyse e.V., Rostock/D Audimax
08:30 PL01
PLENARY LECTURE Functional nanomaterials for energy conversion and storageQ. Xu, Advanced Industrial Science and Technology (AIST), Osaka/J
52
Chemical Dynamics and Kinetics
Chair: J. Lauth, Delft University of Technology/NL Haus 1 - Hörsaal 323
09:30 V03.01
Advancing kinetic mechanisms for low-temperature combustion through the detection of highly elusive intermediates K. Moshammer1; A. Jasper1; N. Hansen1; 1 Sandia National Laboratories, Livermore, CA/USA
120
09:50 V03.02
The primary photochemical processes of a ferrocyclobutadiene in liquid solution studied by ultrafast mid-infrared spectroscopy B. Wezisla1; J. Lindner1; P. Vöhringer1; 1 University of Bonn/D
121
10:10 V03.03
A comprehensive kinetic model for the excited state dynamics of all-trans retinal O. Flender1; M. Scholz1; J. Hölzer1; T. Lenzer1; K. Oum1; 1 University of Siegen/D
122
10:30 V03.04
Vibronic coherence in the first excited pp* state of penta fluorobenzene by coupling to the pσ* state O. Hüter1; M. Sala1; H. Neumann1; S. Zhang1; H. Studzinski1; D. Egorova1; F. Temps1; 1 Kiel University/D
123
10:50 COFFEE BREAK
Chemical Dynamics and Kinetics
Chair: M. Gerhards, TU Kaiserslautern/D Haus 1 - Hörsaal 323
11:10 V03.05
Photochemistry of organic compounds adsorbed on salt clusters C. van der Linde1; N. Bersenkowitsch1; M. Beyer1; 1 University of Innsbruck/A
124
11:30 V03.06
Picosecond photorelease mechanism of coumarin-cages to trigger ultrafast bio/chemical processes L. van Wilderen1; C. Neumann1; D. Kern-Michler1; N. Seibert1; A. Rodrigues Correia1; M. Reinfelds1; A. Heckel1; J. Bredenbeck1; 1 University of Frankfurt/D
125
11:50 V03.07
o-Benzyne: An elusive intermediate investigated by time-resolved FTIR spectroscopy J. Torres-Alacan1; 1 University of Bonn/D
126
12:10 V03.08
Radiation chemical modification of graphene oxide A. Kahnt1; S. Eigler2; R. Flyunt3; W. Knolle3; S. Naumov3; B. Abel3; 1 Universität Erlangen-Nürnberg, Erlangen/D; 2 Chalmers University of Technology, Gothenburg/S; 3 Leibniz Institute of Surface Modification (IOM), Leipzig/D
128
12:30 LUNCH BREAK
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
17
Friday, 6 May 2016
Chair: J. Wagner, Universität Rostock/D Audimax
13:30 PL02
PLENARY LECTURE Charge carrier dynamics for photoelectrochemical water splittingJ. Durrant, Imperial College London/UK
60
Hot Topics
Chair: U. Nickel HCS GmbH, Frankfurt/D Haus 1 - Hörsaal 323
14:30 V14.01
Soft and hard X-ray ambient pressure X-ray photoelectron spectroscopic characterization of the BiVO4/potassium phosphate interface for water splitting applications D. Starr1; F. Abdi1; M. Favaro2; M. Kanis1; H. Bluhm2; E. Crumlin2; R. van de Krol1; 1 Helmholtz-Zentrum Berlin (HZB)/D; 2 Lawrence Berkeley National Laboratory, Berkeley, CA/USA
129
14:50 V14.02
Threshold photoelectron spectroscopy to trace chemistry in combustion, pyrolysis and catalysis P. Hemberger1; 1 Paul Scherrer Institut (PSI), Villigen PSI/D
130
15:10 V14.03
Dissociation of microsolvated acids R. Pérez de Tudela1; D. Marx1; 1 Ruhr-Universität Bochum/D
131
15:30 V14.04
Hierarchical quantum chemical treatment as a path to sub-kJ/mol accuracy in solid-state applications D. Usvyat1; 1 Universität Regensburg/D
132
15:50 COFFEE BREAK
Award Session
Chair: J. Sauer, Humboldt-Universität zu Berlin/D Haus 1 - Hörsaal 323
16:10 V15.01
NERNST-HABER-BODENSTEIN PREIS Measuring macromolecular properties in a “field-free” single molecule trapM. Krishnan1, 1 University of Zurich/CH
133
16:30 V15.02
PAUL-BUNGE PREISWhere has all the chemistry gone?R.G.W. Anderson1, 1 University of Cambridge/UK
134
17:10 V15.03
EWALD-WICKE PREISTowards stress imaging with diamond magnetometry SR. Hemelaar1; F. Perona Martinez1; M. Chipaux1; F. Oosterhof1; T. Hamoh1; A. Nagl1; R. Schirhagl1; 1 Groningen University/NL
135
17:30 V15.04
WILHELM-OSTWALD-NACHWUCHSPREISUnnatural amino acids as novel probes for ultrafast 2D-IR spectroscopy of proteinsH.M. Müller-Werkmeister1; 1 University of Frankfurt/D and University of Toronto/CAN
136
18:00 POSTER SESSION (even numbers) Tent
19:30 POSTER SESSION (odd numbers) (19:30 – 21:00) Tent
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
18
Saturday, 7 May 2016
Chair: O. Kühn, Universität Rostock/D Audimax
08:30 PL03
PLENARY LECTURE Mechanism of light-induced water oxidation and oxygen release in photosynthesisW. Lubitz, MPI für Chemische Energiekonversion, Mülheim/D
137
Main Topic – Basic Mechanisms in Energy Conversion
Chair: R. Marschall, Universität Gießen/D Audimax
09:30 V01.14
On the spin chemistry of donor-acceptor triads containing transition metal photosensitisers C. Lambert1; 1 University of Würzburg/D
138
09:50 V01.15
Linking structure and optical spectra of photosynthetic pigment-protein complexes F. Müh1; 1 University of Linz/A
139
10:10 V01.16
Multimetallic terpyridine complexes with highly conjugated ligands for spatially directed energy transport M. Wächtler1; J. Kübel1; K. Barthelmes2; A. Winter2; A. Schmiedel3; C. Lambert4; U. Schubert2; B. Dietzek1; 1 Leibniz Institute of Photonic Technology, Jena/D; 2 University of Jena/D; 3 Universität Würzburg, Jena/D; 4 University of Würzburg/D
140
10:30 V01.17
Gas-phase photoluminescence characterization of mass-selected lanthanoid complexes: energy transfer mechanisms J. Greisch1; M. Harding1; J. Chmela2; B. Schäfer1; M. Ruben1; W. Klopper2; D. Schooss1; M. Kappes2; 1 Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen/D; 2 Karlsruhe Institute of Technology (KIT)/D
141
10:50 COFFEE BREAK
Main Topic – Basic Mechanisms in Energy Conversion
Chair: C. Papp, Universität Erlangen-Nürnberg, Erlangen/D Audimax
11:10 V01.18
Graphitic carbon nitrides as graphene complements: new insights and challenges C. Merschjann1; 1 Freie Universität Berlin/D
142
11:30 V01.19
Titanium and Iron substituted LiCoTixMn1-xO4/ LiCo1-yFeyMnO4: energy storage via high-voltage spinels C. Dräger1; S. Indris2; H. Ehrenberg1; 1 Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen/D; 2 Karlsruhe Institute of Technology (KIT)/D
143
11:50 V01.20
Energy conversion in thermo-ionic-electronic materials and systems A. Feldhoff1; 1 Leibniz Universität Hannover/D
144
12:10 V01.21
Environmentally friendly composites for flexible thermoelectrics: concepts and applications V. Andrei1; K. Bethke1; K. Rademann1; 1 Humboldt-Universität zu Berlin/D
145
12:30 LUNCH BREAK
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
19
Saturday, 7 May 2016
Chair: U. Kragl, Universität Rostock/D Audimax
13:30 PL04
PLENARY LECTURE Making and breaking bonds: X-ray Spectroscopic studies of small molecule activation by biological catalystsS. DeBeer, MPI für Chemische Energiekonversion, Mülheim/D
146
Basic Mechanisms in Energy Conversion
Chair: S. Lochbrunner, Universität Rostock/D Audimax
14:30 V01.22
Charge carrier dynamics of methylammonium lead iodide: From PbI2-rich to low-dimensional broadly emitting perovskites J. Klein1; O. Flender1; M. Scholz1; K. Oum1; T. Lenzer1; 1 University of Siegen/D
147
14:50 V01.23
Improved charge carrier separation in barium tantalate composites investigated by laser flash photolysis J. Schneider1; K. Niktin2; D. Bahnemann2; R. Marschall3; 1 Leibniz Universität Hannover/D; 2 Saint-Petersburg State University/RU and Leibniz Universität Hannover/D; 3 Giessen University/D
148
15:10 V01.24
About the evaluation and simulation of solar energy M. Schmitt1; F. Heib1; 1 Saarland University, Saarbrücken/D
149
15:30 V01.25
Ab-initio prediction of pure and mixture gas adsorption of small gas molecules in CPO-27-Mg A. Kundu1; J. Sauer1; K. Sillar2; 1 Humboldt Universität zu Berlin/D; 2 University of Tartu/EST
150
16:00 CLOSING CEREMONY – POSTER AWARD CEREMONY Audimax
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
20
Saturday, 7 May 2016
Chair: O. Kühn, Universität Rostock/D Audimax
08:30 PL03
PLENARY LECTURE Mechanism of light-induced water oxidation and oxygen release in photosynthesisW. Lubitz, MPI für Chemische Energiekonversion, Mülheim/D
137
Electrochemistry and Energy
Chair: M. Peuckert, Hofheim/D Arno-Esch I
09:30 V12.05
Modeling of adsorption processes at nanostructured cathodes in lithium-sulfur batteries J. Lück1; A. Latz1; 1 German Aerospace Center (DLR), Ulm/D
151
09:50 V12.06
Modelling SEI growth: a novel description for porous layers F. Single1; B. Horstmann1; A. Latz1; 1 German Aerospace Center (DLR), Ulm/D
152
10:10 V12.07
Graphene-based nanocomposites with hierarchical structures as high-performance electrodes for lithium (sodium) storage C. Wu1; 1 Max Planck Institute for Solid State Research, Stuttgart/D
153
10:30 V12.08
Defects in porous networks of WO3 particle aggregates A. Márquez1; M. Rodríguez-Pérez2; J. Anta3; G. Rodríguez-Gattorno2; G. Bourret1; G. Oskam2; T. Berger1; 1 University of Salzburg/A; 2 CINVESTAV-IPN, Mérida, Yucatán/MEX; 3 Universidad Pablo de Olavide, Seville/E
154
10:50 COFFEE BREAK
Electrochemistry and Energy
Chair: R. Schuster, Karlsruhe Institute of Technology (KIT)/D Arno-Esch I
11:10 V12.09
Charge redistribution and extraction in photocatalytically synthesized Au−ZnO nanopyramids A. Chanaewa1; 1 VU University Amsterdam/NL
155
11:30 V12.10
Novel, efficient and stable composite photoelectrodes based on device-grade silicon and chalcopyrite for visible-light driven hydrogen and oxygen evolution T. Schedel-Niedrig1; 1 Helmholtz-Zentrum Berlin (HZB)/D
156
11:50 V12.11
Dye molecules induced porosity in electrodeposited zinc ferrite films for enhanced photoelectrochemical response D. Taffa1; M. Wark1; 1 University of Oldenburg/D
158
12:10 V12.12
The electronic structure of iridium oxides active in water splitting V. Pfeifer1; T. Jones1; J. Velasco-Vélez2; R. Arrigo2; M. Hävecker2; A. Knop-Gericke1; R. Schlögl1; 1 Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin/D; 2 Max-Planck-Institut für Chemische Energie konversion, Mülheim a. d. Ruhr/D
159
12:30 LUNCH BREAK
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
21
Saturday, 7 May 2016
Chair: U. Kragl, Universität Rostock/D Audimax
13:30 PL04
PLENARY LECTURE Making and breaking bonds: X-ray Spectroscopic studies of small molecule activation by biological catalystsS. DeBeer, MPI für Chemische Energiekonversion, Mülheim/D
146
Electrochemistry and Energy
Chair: D. Paschek, Universität Rostock/D Arno-Esch I
14:30 V12.13
Low platinum group metal fuel cell catalysts: from surface science to in-situ spectroelectrochemistry O. Brummel1; F. Waidhas1; F. Faisal1; R. Fiala2; M. Vorokhta2; I. Khalakhan2; G. Kovács3; S. Kozlov3; K. Neyman3; V. Matolín2; J. Libuda1; 1 Universität Erlangen-Nürnberg, Erlangen/D; 2 Charles University in Prague/CZ; 3 Universitat de Barcelona/E
160
14:50 V12.14
Electrochemical degradation mechanisms of shape-controlled platinum nanoparticles M. Oezaslan1; I. Safo1; J. Haverkamp1; 1 University of Oldenburg/D
161
15:10 FL12.01
FULL LECTURE Electrochemical microcalorimetry R. Schuster1; 1 Karlsruhe Institute of Technology (KIT)/D
162
16:00 CLOSING CEREMONY – POSTER AWARD CEREMONY Audimax
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
22
Saturday, 7 May 2016
Chair: O. Kühn, Universität Rostock/D Audimax
08:30 PL03
PLENARY LECTURE Mechanism of light-induced water oxidation and oxygen release in photosynthesisW. Lubitz, MPI für Chemische Energiekonversion, Mülheim/D
137
Biophysical Chemistry
Chair: T. Kottke, Universität Bielefeld/D Arno-Esch II
09:30 V10.01
Distinguishing different diffusion behaviors by novel analysis of FCS autocorrelation curves S. Jebreiil Khadem1; C. Hille2; H. Löhmannsröben2; I. Sokolov1; 1 Humboldt-Universität zu Berlin/D; 2 University of Potsdam/D
163
09:50 V10.02
Combined temperature, pressure, and cosolvent effects on enzyme activity T. Luong1; R. Winter1; 1 TU Dortmund/D
164
10:10 V10.03
Quantitative approaches in single-molecule micorscopy – can the stoichiometry of protein complexes be measured in cells? W. Chmielewicz1,2; K. Grußmayer1; S. Bierbaum1; P. Klein2; O.T. Fackler2; D.-P. Herten1; 1 Heidelberg University/D; 2 Universitätsklinikum Heidelberg/D
165
10:30 V10.04
Determination of the ionization threshold of oligonucleotides and sequence-specific VUV-induced DNA strand breaks using the DNA origami technique S. Vogel1; J. Rackwitz1; A. Milosavljevic2; A. Giuliani3; I. Bald1; 1 University of Potsdam/D; 2 University of Belgrade, Pregrevica/SRB; 3 Sychrotron SOLEIL, Paris/F
166
10:50 COFFEE BREAK
Biophysical Chemistry
Chair: G. Jung, Universität des Saarlandes, Saarbrücken/D Arno-Esch II
11:10 V10.05
DNA origami nanostructures as a platform for optical spectroscopy I. Bald1; L. Olejko1; J. Prinz1; C. Heck1; 1 University of Potsdam/D
167
11:30 V10.06
Local tissue manipulation using a force- and pressure-controlled AFM micropipette in a frontloading mode P. Roder1; C. Hille1; 1 University of Potsdam/D
168
11:50 V10.07
Networking of biopolymers – molecular dynamics on collagen and glycosaminoglycans W. Langel1; E. Peter1; N. Geist1; 1 University of Greifswald/D
169
12:10 V10.08
Unravelling an underrated noncovalent interaction in proteins through single- conformation IR spectroscopy of model peptides in the molecular beam C. Müller1; P. Walsh2; T. Zwier2; 1 Ruhr-Universität Bochum/D; 2 Purdue University, West Lafayette, IN/USA
170
12:30 LUNCH BREAK
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
23
Saturday, 7 May 2016
Chair: U. Kragl, Universität Rostock/D Audimax
13:30 PL04
PLENARY LECTURE Making and breaking bonds: X-ray Spectroscopic studies of small molecule activation by biological catalystsS. DeBeer, MPI für Chemische Energiekonversion, Mülheim/D
146
Catalysis
Chair: M. Beller, Leibniz-Institut für Katalyse e.V., Rostock/D Arno-Esch II
14:30 V11.01
In situ FTIR measurements and applied chemometrics for studying kinetic and mechanistic aspects of alkene hydroformylation C. Kubis1; M. König1; M. Sawall2; D. Selent1; R. Ludwig2; K. Neymeyr2; D. Hess3; R. Franke4; A. Börner1; 1 Leibniz-Institut für Katalyse, Rostock/D; 2 University of Rostock/D; 3 Evonik Performance Materials GmbH, Marl/D; 4 Evonik Performance Materials GmbH, Rostock/D
171
14:50 V11.02
Ligands on nanoparticles – combining the benefits of homogeneous and heterogeneous catalysis? S. Kunz1; 1 University of Bremen/D
172
15:10 V11.03
Synthesis and characterization of Ni-Ox and Ni-Nx catalysts for electrochemical water splitting S. Wagner1; J. Ziegler1; B. Kaiser1; W. Jaegermann1; 1 TU Darmstadt/D
173
15:30 V11.04
Uncovering deactivation mechanisms of Pd-Pt catalysts for methane oxidation by X-ray absorption and infrared spectroscopy A. Boubnov1; A. Gremminger1; M. Casapu1; O. Deutschmann1; J. Grunwaldt1; 1 Karlsruhe Institute of Technology (KIT)/D
174
16:00 CLOSING CEREMONY – POSTER AWARD CEREMONY Audimax
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
24
Saturday, 7 May 2016
Chair: O. Kühn, Universität Rostock/D Audimax
08:30 PL03
PLENARY LECTURE Mechanism of light-induced water oxidation and oxygen release in photosynthesisW. Lubitz, MPI für Chemische Energiekonversion, Mülheim/D
137
Surfaces and Interfaces
Chair: E. Backus, Max-Planck-Institut für Polymerforschung, Mainz/D Haus 1 - Hörsaal 224
09:30 V09.06
Using the molecular structure of a tetrapyrrole ligand to tune the oxidation state of metal ions: Co(III)- and Ni(III)-corroles on Ag(111) M. Schmid1; M. Zugermeier1; M. Chen1; F. Niefind1; J. Herritsch1; R. Tonner1; J. Gottfried1;
1 Universität Marburg/D
175
09:50 V09.07
Surface composition of free mixed nanoscale aerosols E. Antonsson1; B. Langer1; C. Raschpichler1; D. Marchenko1; E. Rühl1; 1 Freie Universität Berlin/D
176
10:10 V09.08
Porphyrin adsorption studies on metal oxide nanoparticles J. Schneider1; J. Bernardi2; T. Berger1; O. Diwald1; 1 Paris-Lodron University Salzburg/A; 2 Vienna University of Technology/A
177
10:30 V09.09
Coupling of quantum well emission to surface plasmon modes in rolled-up microtubes J. Siebels1; H. Vu2; A. Mews2; T. Kipp2; 1 Universität Hamburg, Krummhörn/D; 2 Universität Hamburg/D
178
10:50 COFFEE BREAK
Surfaces and Interfaces
Chair: C. Wöll, Karlsruhe Institute of Technology (KIT)/D Haus 1 - Hörsaal 224
11:10 FL09.01
FULL LECTURE Two types of water at the water−surfactant interface revealed by time-resolved vibrational spectroscopy R. Livingstone1; Y. Nagata1; M. Bonn1; E. Backus1; 1 Max Planck Institute for Polymer Research, Mainz/D
179
11:50 V09.10
Prebiotic chemistry in nanoconfined water D. Muñoz-Santiburcio1; D. Marx1; 1 Ruhr-Universität Bochum/D
180
12:10 V09.11
Molecular dynamics simulations of solid-liquid interfaces employing high- dimensional neural network potentials S. Kondati Natarajan1; V. Quaranta1; M. Hellström1; J. Behler1; 1 Ruhr-Universität Bochum/D
181
12:30 LUNCH BREAK
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
25
Saturday, 7 May 2016
Chair: U. Kragl, Universität Rostock/D Audimax
13:30 PL04
PLENARY LECTURE Making and breaking bonds: X-ray Spectroscopic studies of small molecule activation by biological catalystsS. DeBeer, MPI für Chemische Energiekonversion, Mülheim/D
146
Theoretical Techniques
Chair: J. Paier, Humboldt-Universität zu Berlin/D Haus 1 - Hörsaal 224
14:30 V06.01
The quantum dynamics of tunneling and electroweak parity violation in trisulfane (HSSSH) C. Fabri1; L. Horny1; M. Quack1; 1 ETH Zürich/CH
182
14:50 V06.02
Caldeira-leggett model description of condensed phase vibrational spectroscopy S. Ivanov1; F. Gottwald1; K. Oliver1; 1 University of Rostock/D
183
15:10 V06.03
Independent-electron surface hopping study of the nonadiabatic energy transfer at scattering NO molecule from Au(111) surface A. Kandratsenka1; B. Krüger1; T. Schäfer1; a. Wodtke1; 1 Universität Göttingen/D
184
15:30 V06.04
High energy resolution X-ray absorption spectroscopy and TD-DFT calculations on copper systems P. Müller1; M. Bauer1; 1 Paderborn University/D
185
16:00 CLOSING CEREMONY – POSTER AWARD CEREMONY Audimax
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
26
Saturday, 7 May 2016
Chair: O. Kühn, Universität Rostock/D Audimax
08:30 PL03
PLENARY LECTURE Mechanism of light-induced water oxidation and oxygen release in photosynthesisW. Lubitz, MPI für Chemische Energiekonversion, Mülheim/D
137
Molecular Structure
Chair: M. Schnell, Max-Planck-Institut für Struktur und Dynamik Haus 1 – Hörsaal 323 der Materie, Hamburg/D
09:30 V04.01
Direct determination of absolute stereochemical configuration by coulomb explosion imaging M. Pitzer1; M. Kunitski2; T. Jahnke2; H. Schmidt-Böcking2; R. Dörner2; M. Schöffler2; S. Marquardt3; S. Marquardt3; R. Berger3; 1 University of Kassel, /D; 2 University of Frankfurt/D; 3 Universität Marburg/D
186
09:50 V04.02
Molecular-frame photoelectron imaging of controlled complex molecules J. Wiese1; S. Trippel1; J. Küpper1; 1 Center for Free-Electron Laser Science, Hamburg/D
187
10:10 V04.03
Distinction of structural isomers by means of chirped femtosecond laser ionization N. Reusch1; V. Krein1; N. Wollscheid1; K. Weitzel1; 1 Universität Marburg/D
188
10:30 V04.04
Influence of hydrogen addition on the structures of Ruthenium cluster anions D. Bumüller1; E. Waldt1; A. Hehn1; R. Ahlrichs1; M. Kappes1; D. Schooss1; 1 Karlsruhe Institute of Technology (KIT)/D
189
10:50 COFFEE BREAK
Molecular Structure
Chair: O. Kühn, Universität Rostock/D Haus 1 – Hörsaal 323
11:10 V04.05
Reversed OH shifts in oxygen and p hydrogen bonded dimers of methanol with furan derivatives A. Poblotzki1; M. Suhm1; 1 Universität Göttingen/D
190
11:30 V04.06
The bonus of dispersion – the diphenyl ether-methanol complex investigated using a multi-spectroscopic and theoretical approach D. Bernhard1; A. Poblotzki2; C. Medcraft3; C. Holzer4; A. Stamm1; F. Dietrich1; J. Altnöder2; M. Heger2; S. Zinn3; M. Suhm2; M. Schnell3; W. Klopper4; M. Gerhards1; 1 TU Kaiserslautern/D; 2 Universität Göttingen/D; 3 Max Planck Institute for the Structure and Dynamics of Matter, Hamburg/D; 4 Karlsruhe Institute of Technology (KIT)/D
191
11:50 V04.07
Characterization of microsolvated polycyclic-aromatic hydrocarbons and crown ethers from broadband rotational spectroscopy A. Steber1; C. Perez1; J. Lopez2; A. Rijs3; M. Schnell1; 1 Max Planck Institute for the Structure and Dynamics of Matter, Hamburg/D; 2 Universidad de Valladolid, Valladolid/E; 3 Radboud Universiteit, Nijmegen/NL
192
Page
LECTURE PROGRAMME

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
27
Saturday, 7 May 2016
12:10 V04.08
Vibrational spectroscopy of titanium oxide anions: structure-reactivity relationship X. Song1; 1 Fritz-Haber-Institut der MPG, Berlin/D
193
12:30 LUNCH BREAK
Chair: U. Kragl, Universität Rostock/D Audimax
13:30 PL04
PLENARY LECTURE Making and breaking bonds: X-ray Spectroscopic studies of small molecule activation by biological catalystsS. DeBeer, MPI für Chemische Energiekonversion, Mülheim/D
146
Hot Topics
Chair: B. Dick, Universität Regensburg/D Haus 1 – Hörsaal 323
14:30 V14.05
Thermodynamic description of salt-specific effects in thermo responsive polymers and copolymers J. Heyda1; J. Dzubiella2; R. Chudoba2; B. Schulz2; 1 University of Chemistry and Technology, Prague/CZ; 2 Helmholtz-Zentrum Berlin (HZB)/D
194
14:50 V14.06
SURMOFs – highly porous materials with huge potential in sensor device application H. Dr. Gliemann1; C. Wöll2; 1 Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen/D; 2 Karlsruhe Institute of Technology (KIT)/D
195
15:10 V14.07
A thermodynamic study of different cosolute effects on the stability of RNase A M. Senske1; D. Constantinescu1; H. Weingärtner1; C. Herrmann1; S. Ebbinghaus1; M. Havenith11 Ruhr-Universität Bochum/D
197
15:30 V14.08
Ion imaging of reaction dynamics at surfaces: Eley-Rideal vs Langmuir-Hinshelwood J. Neugebohren1; D. Harding1; H. Hahn1; D. Auerbach1; T. Kitsopoulos1; A. Wodtke2; 1 Universität Göttingen/D
198
16:00 CLOSING CEREMONY – POSTER AWARD CEREMONY Audimax
Page
LECTURE PROGRAMME

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
28
Main Topic – Basic Mechanisms in Energy Conversion
P01-01 Rate and routes of electron transfer in an enzymatic fuel cell A. Petrenko1; M. Stein1; 1 Max Planck Institute for Dynamics of Complex Technical Systems, Magdeburg/D
201
P01-02 Viscosity dependence of hydrogen atom self-exchange: a CW-ESR line broadening study on N-hydroxyphtalimid/ phtalimide-N-oxyl J. Bächle1; 1 Graz University of Technology/A
202
P01-03 Electron transfer dynamics of indoline and triarylamine sensitizers on mesoporous ZnO, TiO2 and Al2O3 surfaces O. Flender1; J. Klein1; M. Scholz1; K. Oum1; T. Lenzer1; 1 University of Siegen/D
203
P01-04 In situ spectroelectrochemical study on the oxidation of a 4H-imidazole-ruthenium dye on TiO2 electrodes Y. Zhang1; S. Kupfer2; L. Zedler1; J. Schindler1; T. Bocklitz2; J. Guthmuller3; S. Rau4; B. Dietzek1; 1 Leibniz Institute of Photonic Technology Jena and University of Jena/D; 2 University of Jena/D; 3 Gdansk University of Technology/PL; 4 University of Ulm/D
204
P01-05 In-situ investigations on the norbornadiene/quadricyclane energy storage system on various metal surfaces by UPS and high-resolution-XPS U. Bauer1; 1 Universität Erlangen-Nürnberg, Erlangen/D
205
P01-06 Oxidation state control of MnOx thin films for water oxidation catalysis by pH value adjustment during the deposition process M. Shaker1; M. Khan1; M. Tesch1; J. Xiao1; L. Spiccia2; E. Aziz1; 1 Helmholtz-Zentrum Berlin (HZB)/D; 2 Monash University, Clayton/AUS
206
P01-07 Photocurrent of BiVO4 is limited by surface recombination, not surface catalysis C. Zachäus1; F. Abdi1; R. van de Krol1; 1 Helmholtz-Zentrum Berlin (HZB)/D
207
P01-08 Unraveling elementary steps of an Ir-based photocatalytic cycle for water splitting O. Bokareva1; G. Grell1; S. Bokarev1; O. Kühn1; 1 Universität Rostock/D
208
P01-09 Concentration effects on the entropy of electrochemical lithium deposition: implications for Li+ solvation S. Matthias1; J. Xu1; J. Lindner1; P. Novák2; R. Schuster1, ; 1 Karlsruhe Institute of Technology (KIT)/D; 2 Paul Scherrer Institut (PSI), Villigen PSI/CH
209
P01-10 Temperature programmed high-resolution X-ray photoelectron spectroscopy of formic acid on Ni(111) and Pt(111) P. Bachmann1; 1 Universität Erlangen-Nürnberg, Erlangen /D
210
P01-11 Towards understanding the mechanism of water splitting on TiO2 S. Hosseinpour1; 1 Max Planck Institute for Polymer Research, Mainz/D
211
P01-12 Recombination spectroscopy of organic photovoltaic devices M. Eckardt1; R. Wieczorek1; F. Laquai2; W. Harneit3; 1 Universität Mainz, Mainz/D; 2 King Abdullah University of Science and Technology (KAUST), Thuwal/SAR; 3 Universität Osnabrück/Dcancelled
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
29
P01-13 Physicochemical energy conversion to phase separation, conformational isomerisation or thermal energy change. G. Ahn-Ercan1; 1 ComputeChem und Universität Regensburg, Saal an der Donau/D
212
P01-14 Photon-stimulated processes on gallium nitride semiconductors S. Kollmannsberger1; C. Walenta1; M. Tschurl1; U. Heiz1; S. Weiszer1; A. Winnerl1; R. Pereira1; M. Stutzmann1; 1 TU München, Garching/D
213
P01-15 Plasma enhanced CVD preparation of CoOxOH2 and CoMOxOH2 (M=Ni,Fe) as catalysts for the oxygen evolution reaction N. Weidler1; 1 TU Darmstadt/D
214
P01-16 Structural investigations on a copper containing OLED complex by application of time-resolved step-scan FTIR spectroscopy and quantum chemical calculations M. Zimmer1; F. Dietrich1; F. Bäppler1; M. Wallesch2; D. Volz3; S. Bräse2; R. Diller1; M. Gerhards1; 1 TU Kaiserslautern /D; 2 Karlsruhe Institute of Technology (KIT)/D; 3 Cynora gmbH, Bruchsal/D
215
P01-17 Photoelectrochemical properties of CdS and CdSe/CdS nanorods J. Miethe1; J. Poppe1; N. Bigall1; 1 Leibniz Universität Hannover/D
216
P01-18 Electrochemical interphase formation at a buried sample/electrode interface M. Schäfer1; V. Wesp1; J. Martin1; K. Weitzel1; 1 Universität Marburg /D
217
P01-19 Electrodeposited nickel nanoparticles as catalysts for hydrogen evolution reaction S. Tao1; F. Yang1; B. Kaiser1; W. Jaegermann1; 1 TU Darmstadt/D
218
P01-20 Ultrafast photo-induced electron transfer in self-assembled donor-acceptor coordination cages J. Ahrens1; G. Clever2; M. Frank2; D. Schwarzer1; 1 Max-Planck-Institute for Biophysical Chemistry, Göttingen/D; 2 Universität Göttingen/D
219
P01-21 Inelasticity in Hydrogen atom scattering from surfaces: a probe for energy conversion Y. Dorenkamp1; H. Jiang1; A. Wodtke1; O. Bünermann1; 1 Universität Göttingen/D
220
P01-22 Monitoring reactive layers inside redox hydrogels by Raman microspectroscopy C. Klinkhammer1; S. Stapf1; N. Plumeré1; M. Havenith1; 1 Ruhr University Bochum/D
221
P01-23 Investigation of Pt- and Pt/Ni-nanoparticles as catalysts for the hydrogen evolution reaction in electrochemical cells J. Ziegler1; F. Tabary1; L. Wittern2; H. Weller2; W. Jaegermann1; B. Kaiser1; 1 TU Darmstadt/D; 2 Universität Hamburg/D
222
P01-24 Imaging photo-induced processes in organic-inorganic perovskites L. Martinez Uriarte1; A. Neff1; F. Meier2; C. Deibel2; K. Siefermann1; 1 Leibniz Institute of Surface Modification, Leipzig/D; 2 TU Dresden/D
223
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
30
P01-25 Understanding the effect of Ir(III/IV) aquo complexes on the photoelectrochemistry of IrOx•nH2O–based water-splitting cells N. Vargas-Barbosa1; T. Mallouk2; L. Jensen2; 1 Universität Marburg/D; 2 The Pennsylvania State University, University Park, PA/USA
224
P01-26 Group III-phosphide semiconductors for water splitting: surface preparation and water adsorption experiments A. Hajduk1; M. Lebedev2; B. Kaiser1; W. Jaegermann1; 1 TU Darmstadt, Darmstadt/D; 2 Ioffe Institute, St. Petersburg/RUS
225
P01-27 Atomistic modeling of light induced surface relief gratings M. Böckmann1; N. Doltsinis1; 1 Universität Münster, Münster/D
227
P01-28 (Self)assembling amphiphilic dyes and tuning of photonic properties via morphology control M. Presselt1; S. Das1; F. Herrmann1; M. Hupfer1; B. Dietzek1; 1 University Jena/D
228
P01-29 Ultrafast excited state dynamics of iridium(III) photosensitizers S. Tschierlei1; A. Neubauer2; N. Rockstroh3; M. Karnahl1; H. Junge3; M. Beller3; S. Lochbrunner2; 1 University of Stuttgart/D; 2 University of Rostock/D; 3 Leibniz-Institut für Katalyse, Rostock/D
229
P01-30 Real-time observation of Ir photosensitizer reductive quenching during light- activated molecular catalysis A. Britz1; T. Assefa1; A. Galler1; W. Gawelda1; E. Bajnoczi2; Z. Nemeth2; G. Vankó2; N. Rockstroh3; H. Junge3; M. Beller3; G. Doumy4; A. March4; S. Southworth4; S. Bokarev5; S. Lochbrunner5; O. Kühn5; C. Bressler1; 1 European XFEL, Hamburg/D; 2 Wigner Research Centre for Physics, Budapest/H; 3 Leibniz-Institut für Katalyse, Rostock/D; 4 Argonne National Laboratory, Argonne, IL/USA; 5 Universität Rostock/D
230
P01-31 Light-driven electron transfer in a photocatalytic model system based on a Copper photosensitizer A. Friedrich1; S. Luo2; H. Junge3; M. Beller3; S. Lochbrunner1; 1 Universität Rostock/D; 2 State Key Laboratory Breeding Base of Green Chemistry-Synthesis Technology, Zhejiang University of Technology, Hangzhou/CN; 3 Leibniz-Institut für Katalyse, Rostock/D
231
P01-32 In-operando atomic force microscopy for energy materials characterization F. Hausen1; 1 Forschungszentrum Jülich GmbH/D
232
P01-33 SURMOFs/CNCs as tuneable “designer solids” for light harnessing and energy applications E. Redel1; 1 Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen/D
P01-34 New reaction pathway on rutile B. Jasper1; M. Osmic2; S. Schmiedekind1; 1 University of Oldenburg/D; 2 University of Oldenburg, Wilhelmshaven/D
233
cancelled
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
31
P01-35 Enhanced stability of thin film silicon tandem solar cells based photocathodes with amorphous TiO2 layers coated with nanoparticle catalysts for solar hydrogen evolution F. Yang1; L. Movsesyan2; F. Urbain3; J. Becker3; M. Toimil-Molares2; B. Kaiser1; W. Jaegermann1; 1 TU Darmstadt/D; 2 GSI Helmholtzzentrum für Schwerionenforschung, Darmstadt/D; 3 Forschungszentrum Jülich GmbH/D
234
P01-36 Splittin water with novel dinuclear ruthenium complexes T. Stolper1, R. A. Mata1, 1 University of Göttingen/D
235
Liquids and Solvation
P02-01 Ionic dynamics in alkylammonium nitrates are decoupled from viscosity S. Roy1; M. Bonn1; J. Hunger1; 1 Max Planck Institute for Polymer Research, Mainz/D
236
P02-02 A molecular dynamics simulation study of direct cation-cation interactions in cholinium based ionic liquids J. Neumann1; R. Ludwig1; D. Paschek1; 1 Universität Rostock/D
237
P02-03 Structure and dynamics in mixtures of protic ionic liquids: a molecular dynamics simulation study B. Golub1; D. Paschek1; R. Ludwig1; 1 Universität Rostock/D
238
P02-04 Temperature dependence of computed osmotic second virial coeffients of apolar solutes in aqueous solutions E. Reiter1; 1 Universität Rostock/D
239
P02-05 Under pressure – amorphous ice – a MD simulation study M. Jehser1; M. Seidl2; T. Loerting2; G. Zifferer1; 1 University of Vienna/A; 2 University of Innsbruck/A
240
P02-06 Investigation of the formation of pharmaceutical nanoparticles within RESS by means of molecular dynamics simulation S. Braun1; T. Kraska1; 1 University of Cologne/D
241
P02-07 Nuclear quantum effects on charge transfer in the hydrogen bond of gas phase dimers and liquid water C. Schran1; O. Marsalek2; T. Markland2; 1 Ruhr-Universität Bochum/D; 2 Stanford University, CA/USA
242
P02-08 Terpene-based mixtures: molecular insights into the solid-liquid equilibria M. Alves da Rocha1; F. Zehentbauer1; B. Rathke1; J. Kiefer1; 1 Universität Bremen/D
243
P02-09 Imidazolium based ionic liquids: impact of cation symmetry on the liquid – liquid phase behavior of solutions with n-alkyl alcohols M. Alves da Rocha1; D. Kerlé1; W. Schröer1; B. Rathke1; 1 Universität Bremen/D
244
P02-10 Role of hydrogen bond donors in the formation of liquid mixtures M. Alves da Rocha1; F. Zehentbauer1; L. Zubeir2; B. Rathke1; M. Kroon2; J. Kiefer1; 1 Universität Bremen/D; 2 Eindhoven University of Technology/NL
245
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
32
P02-11 THz absorption spectroscopy of an insect lipoprotein ice nucleator reveals a long-range influence on the hydrogen bond dynamics A. Bäumer1; J. Duman2; M. Havenith1; 1 Ruhr-Universität Bochum/D; 2 University of Notre Dame, IN/USA
246
P02-12 Probing the acidity and solvent polarity of ionic liquids by determining the spectroscopic features of the proton sensitive dye N-methyl-6-oxyquinolone S. Schmode1; A. Petrosyan1; R. Ludwig1; 1 Universität Rostock/D
247
P02-13 Towards understanding glycan-assisted protein stabilization S. Schäfer1; H. Wirtz1; C. Hoberg1; J. Savolainen1; M. Havenith1; 1 Ruhr-Universität Bochum/D
248
P02-14 How does spatial confinement influence proton-transfer dynamics? E. Eisbein1; G. Seifert1; T. Lorenz1; J. Joswig1; 1 TU Dresden/D
249
P02-15 Investigation of ubiquitin using time-resolved kinetic thz absorption spectroscopy with temperature jumps H. Wirtz1; S. Schäfer1; C. Hoberg1; J. Savolainen1; M. Havenith1; 1 Ruhr-Universität Bochum/D
250
P02-16 Photoionization of sodium doped crystalline water clusters T. Zeuch1; C. Dierking1; 1 Universität Göttingen/D
251
P02-17 Determination of the electronic structure of aqueous urea and its derivatives: a combined soft x-ray – DFT approach M. Tesch1; R. Golnak1; F. Ehrhard1; R. Chuboda1; J. Dzubiella1; D. Schön1; J. Xiao1; A. Bande1; E. Aziz1; 1 Helmholtz-Zentrum Berlin (HZB)/D
252
P02-18 Solubility convergence and criticality in ionic liquids M. Namayandeh Jorabchi1; D. Kerlé2; R. Ludwig1; D. Paschek1; 1 University of Rostock/D; 2 University of Bremen/D
253
P02-19 Deuteron quadrupole coupling constants and reorientational correlation times in protic ionic liquids by means of NMR relaxation time experiments, DFT-calculations and molecular dynamics simulations M. Strauch1; V. Overbeck1; A. Bonsa1; B. Golub1; D. Michalik1; D. Paschek1; R. Ludwig1; 1 University of Rostock/D
254
P02-20 Critical radius of supercooled water droplets: on the transition towards dendritic freezing S. Bauerecker1; T. Buttersack1; 1 TU Braunschweig/D
255
P02-21 Quantifying dispersion forces in protic ionic liquids by means of low frequency spectroscopy P. Stange1; K. Fumino1; A. Knorr1; R. Ludwig1; 1 Universität Rostock/D
256
P02-22 Molecular topology and local dynamics govern the viscosity of imidazolium-based Ionic liquids Y. Zhang1; L. Xue2; F. Khabaz2; R. Doerfler1; E. Quitevis2; R. Khare2; E. Maginn1; 1 University of Notre Dame, IN/USA; 2 Texas Tech University, Lubbock, TX/USA
257
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
33
P02-23 Shear thinning of critical fluctuations in solutions of Ionic liquids W. Schröer1; 1 Universität Bremen/D
258
Chemical Dynamics and Kinetics
P03-01 Reactants induced dynamic responses of heterogeneous catalysts monitored by adsorption microcalorimetry S. Wrabetz1; T. Lunkenbein1; V. Pfeifer1; T. Jones1; J. Noack1; K. Mette1; A. Trunschke1; A. Knop-Gericke1; R. Schlögl1; 1 Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin/D
259
P03-02 Theoretical investigation of the thermolysis of urea D. Gratzfeld1; M. Olzmann1; 1 Karlsruher Institut für Technologie (KIT)/D
260
P03-03 Fuel-structure dependence of soot precursor formation processes in non-premixed laminar opposed-flow diffusion flames L. Ruwe1; K. Kohse-Höinghaus1; 1 Universität Bielefeld/D
261
P03-04 Thermal decomposition of propene – a shock-tube and modeling study L. Golka1; I. Weber1; M. Olzmann1; 1 Karlsruher Institut für Technologie (KIT)/D
262
P03-05 Experimental and detailed kinetic modeling study of ammonia combustion S. Vallabhuni1; L. Arnas1; F. Ravi1; 1 Physikalisch-Technische Bundesanstalt (PTB), Braunschweig/D
263
P03-06 Observation of b2 symmetry vibrational levels of the SO2 C~
1B2 state B. Park1; J. Jiang2; C. Saladrigas2; R. Field2; 1 Universität Göttingen/D; 2 Massachusetts Institute of Technology, Cambridge, MA/USA
264
P03-07 Ultrafast species-selective photochemistry in mixtures D. Kern-Michler1; C. Neumann1; N. Seibert1; L. van Wilderen1; M. Reinfelds1; J. von Cosel1; A. Heckel1; I. Burghardt1; J. Bredenbeck1; 1 Universität Frankfurt/D
265
P03-08 Stretching-libration dynamics in the methanol dimer M. Heger1; M. Suhm1; J. Andersen2; R. Wugt Larsen2; 1 Universität Göttingen/D; 2 Technical University of Denmark, Copenhagen/DK
266
P03-09 Ultrafast ionization and fragmentation dynamics of chloromethylsilanes E. Antonsson1; I. Halfpap1; C. von Randow1; C. Raschpichler1; E. Rühl1; 1 Freie Universität Berlin/D
267
P03-10 Femtosecond time-resolved dynamics of trans-methyl cinnamate by photoelectron imaging J. Kus1; O. Hüter1; F. Temps1; 1 Universität zu Kiel/D
268
P03-11 Geometric optimization of ICD electron emitter properties in quantum dots F. Weber1; E. Fasshauer2; A. Lode3; A. Bande1; 1 Helmholtz-Zentrum Berlin (HZB)/D; 2 University of Tromsoe/N; 3 University of Basel/CH
269
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
34
P03-12 Influence of the biofuel isomers diethyl ether and n-butanol on pollutant formation in premixed n-butane flames J. Pieper1; M. Zeng1; L. Tran1; I. Graf1; K. Kohse-Höinghaus1; 1 Bielefeld University/D
270
P03-13 Direct detection of CN radicals in the NCN reaction system behind shock waves S. Hesse1; N. Faßheber1; G. Friedrichs1; 1 Universität zu Kiel/D
271
P03-14 VIPER 2D-IR spectroscopy on coumarins: selectivity experiments and simulations C. Neumann1; L. van Wilderen1; D. Kern-Michler1; N. Seibert1; J. von Cosel1; R. Binder1; M. Reinfelds1; I. Burghardt1; A. Heckel1; J. Bredenbeck1; 1 Universität Frankfurt/D
272
P03-15 Strong field control of the interatomic coulombic decay process in quantum dots A. Haller1; Y. Chiang2; E. Aziz1; A. Bande1; 1 Helmholtz-Zentrum Berlin (HZB)/D; 2 The Chinese University of Hong Kong, Shatin, N.T./HK
273
P03-16 2-Butanone as an advanced biofuel – assessment of potential combustion emissions C. Hemken1; U. Burke1; K.A. Heufer2; I. Graf1; L. Ruwe1; K. Kohse-Höinghaus1; 1 Bielefeld University/D; 2 RWTH Aachen/D
274
P03-17 Measuring the kinetics of chemical reactions on metal surfaces in a molecular beam surface scattering experiment H. Hahn1; J. Neugebohren1; D. Harding1; D. Auerbach1; T. Kitsopoulos1; A. Wodtke1; 1 Universität Göttingen/D
276
P03-18 Experimental analysis of the passivation kinetics in aluminium M. Weimerskirch1; T. Nagy1; U. Pacher1; W. Kautek1; 1 University of Vienna/A
277
P03-19 High resolution analysis and quantum dynamics of fluoroform I. Bolotova1; O. Ulenikov2; E. Bekhtereva2; S. Albert1; H. Hollenstein1; M. Quack1; 1 ETH Zürich/CH; 2 Tomsk Polytechnic University, Tomsk/RUS
278
P03-20 Alignment- and orientation-dynamics of ground-state selected OCS S. Trippel1; J. Wiese1; 1 Deutsches Elektronen Synchrotron DESY, Hamburg/D
279
P03-21 Investigation of the methane number as measure for knock propensity of natural gas mixtures containing higher hydrocarbons L. Arnas1; S. Vallabhuni1; R. Fernandes1; 1 Physikalisch-Technische Bundesanstalt (PTB), Braunschweig/D
280
P03-22 Towards understanding ultrafast dynamics in complex systems like nanodiamonds or switchable polymer nanoparticles D. Hintzen1; 1 Leibniz-Institut für Oberflächenmodifizierung e. V., Leipzig/D
281
P03-23 The photodissociation of dimethylnitrosamine revisited – a 3d-REMPI and VMI study M. Schneider1; U. Kensy1; B. Dick1; 1 Universität Regensburg/D
282
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
35
Molecular Structure
P04-01 Vibrations of hemiporphycene in the S0 and S1 electronic states: a supersonic jet spectroscopy study S. Peukert1; M. Gil1; M. Kijak1; J. Ostapko1; J. Sepioł1; J. Waluk1; M. Broquier2; A. Zehnacker-Rentien2; 1 Polish Academy of Sciences, Warszawa/PL; 2 CNRS, Institut des Sciences Moléculaires d’Orsay (ISMO), Centre Laser Université Paris-Sud (CLUPS), University Paris-Sud, Orsay/F
283
P04-02 Excited state intramolecular proton transfer of 2,5-bis(5-ethyl-2-benzoxazolyl)-hydroquinone and its OH/OD-isotopomers studied in Supersonic jets S. Peukert1; M. Gil1; M. Kijak1; J. Sepioł1; 1 Polish Academy of Sciences, Warszawa/PL
284
P04-03 Nitrogen nanocoating effects on the conformational preferences of ethanol and its dimer detected by supersonic jet spectroscopy S. Oswald1; M. Suhm1; 1 Universität Göttingen/D
285
P04-04 Control over the hydrogen bond docking site in anisole by ring alkylation and halogenation H. Gottschalk1; M. Suhm1; 1 Universität Göttingen/D
286
P04-05 Three views on three xylene molecules: far-infrared spectroscopy, computational chemistry, and spectral modeling A. Bande1; I. Bravic1; C. Minnich2; L. Puskar1; E. Ritter3; U. Schade1; E. Aziz1; 1 Helmholtz-Zentrum Berlin (HZB)/D; 2 S-PACT GmbH, Aachen/D; 3 Humboldt-Universität zu Berlin/D
287
P04-06 Structural investigations on a linear depsipeptide by combined IR/UV spectroscopy in a molecular beam A. Stamm1; D. Bernhard1; M. Gerhards1; 1 TU Kaiserslautern/D
288
P04-07 Novel, heteroleptic copper(I)-4H-imidazolate complexes: synthesis and characterization of their structural, spectral and redox properties M. Schulz1; F. Dröge1; Y. Zhang2; C. Reichardt2; F. Herrmann-Westendorf1; J. Schindler2; H. Görls1; M. Presselt1; B. Dietzek1; 1 Universität Jena, Jena/D; 2 Leibniz-Institut für Photonische Technologien e.V., Jena/D
289
P04-08 Characterization of the cis- / trans-isomerization of peptide bond models in the gas phase T. Forsting1; K. Otto1; M. Suhm1; 1 Universität Göttingen/D
290
P04-09 Study of molecular interactions using broadband rotational spectroscopy M. Fatima1; 1 Max Planck Institute for Structure and Dynamics of Matter, Hamburg/D
291
P04-10 Millimeter-wavelength rotational spectroscopy for applications in astrochemistry B. Arenas1; 1 Max Planck Institute for the Structure and Dynamics of Matter, Hamburg/D
292
P04-11 Catching hydrides spectroscopically with hard X-rays L. Burkhardt1; 1 Universität Paderborn/D
293
cancelled
cancelled
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
36
P04-12 The role of dispersion forces for the stability of methane and carbon dioxide gas hydrates W. Polet1; R. Ludwig1; 1 Universität Rostock/D
294
P04-13 Gas phase vibrational spectroscopy of microhydrated sulfate dianions revisited H. Knorke1; 1 Universität Leipzig/D
295
P04-14 High-resolution spectrum of the asymmetric C-H stretch of CH2Br2: CRDS measurements and simulations I. Sadiek1; G. Friedrichs1; 1 Universität zu Kiel/D
296
P04-15 High resolution GHz and THz (FTIR) spectroscopy and theory of parity violation and tunneling for dithiine as a candidate for measuring the parity violating energy difference between enantiomers of chiral molecules I. Bolotova1; A. Sieghard1; Z. Chen1; C. Fábri1; L. Horny1; M. Quack1; G. Seyfang1; D. Zindel1; 1 ETH Zürich/CH
297
P04-16 Single-conformation IR spectroscopy of jet-cooled sulfanilamide and its homo- and heteromolecular 1:1 clusters T. Uhlemann1; S. Seidel1; C. Müller1; 1 Ruhr-Universität Bochum/D
298
P04-17 Spectral signatures of polarons and dynamics of charge separation in conjugated co-polymers C. Wiebeler1; S. Schumacher1; 1 Universität Paderborn/D
299
P04-18 Single-conformation spectroscopy, DFT, NBO, and charge density studies of the neuraminidase inhibitor analogue BANA 113 S. Seidel1; T. Uhlemann1; M. Seitz2; C. Müller1; 1 Ruhr-University Bochum/D; 2 University of Tübingen/D
300
P04-19 Influence of thermal fluctuations for the 2D-IR structure determination in the liquid phase K. Fehre1; D. Kern-Michler1; S. Combe2; P. Schreiner2; B. Kirchner3; J. Bredenbeck1; 1 Universität Frankfurt/D; 2 Universität Gießen/D; 3 Universität Bonn/D
301
P04-20 Photochemical dissociation of HOBr. a nonadiabatic dynamics study B. Allehyani1; R. Hilal1; S. Aziz1; 1 King Abdulaziz University, Jeddah/SAR
302
Experimental Techniques
P05-01 High energy resolution X-ray absorption and emission spectroscopy for the investigation of spin crossover processes R. Schepper1; M. Bauer1; 1 Universität Paderborn/D
303
P05-02 Construction of a surface plasmon spectrometer with enhanced sensitivity and time resolution for the study of electrochemical processes K. Schlag1; D. Nattland1; A. Timm1; R. Schuster1; 1 Karlsruhe Institute of Technology (KIT)/D
304
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
37
P05-03 Contactless microwave conductivity of oxide supported metal samples E. Wolf1; T. Risse2; R. Schlögl1; S. Cap1; 1 Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin/D; 2 Freie Universität Berlin/D
305
P05-04 Sensing of light induced magnetization by magnetic resonance force microscopy J. Deichmöller1; K. Kartaschew1; A. Rostas2; E. Schleicher2; S. Weber2; M. Havenith1; 1 Ruhr-Universität Bochum/D; 2 Universität Freiburg/D
306
P05-05 Hyperfine structure in the IR-spectrum of ammonia, NH3 G. Seyfang1; 1 ETH Zürich/CH
307
P05-06 Kinetics of carbon-dioxide hydrate formation S. Schulze1; J. Kiefer1; B. Rathke1; 1 Universität Bremen/D
308
P05-07 Vapor-liquid Equilibria (VLE) of binary imidazolium-based ionic liquid mixtures with volatile solvents M. Stuckenholz1; J. Kiefer1; B. Rathke1; W. Schröer1; 1 Universität Bremen/D
309
P05-08 Ionenmobilitäten: Bestimmung durch ESI/REMPI-IMS und Vergleich verschiedener Berechnungsmethoden D. Riebe1; A. Erler1; T. Beitz1; H. Löhmannsröben1; 1 Universität Potsdam/D
310
P05-09 Investigation of the thickness- and wavelength- dependence of laser stratigraphy on Cu and Ni using LIBS U. Pacher1; T. Nagy1; E. Khaybulkina1; W. Kautek1; 1 University of Vienna/A
311
P05-10 In situ studies of gold nanoparticle formation in microfluidic reactors G. Tofighi1; T. Sheppard1; G. Hofmann1; G. Rinke1; R. Dittmeyer1; J. Grunwaldt1; 1 Karlsruhe Institute of Technology (KIT)/D;
312
P05-11 On the polymerization efficiency of benzoin-type photoinitiator molecules by fs-TA spectroscopy, PLP-ESI-MS and TD-DFT C. Schweigert1; 1 Karlsruhe Institute of Technology (KIT)/D
313
P05-12 COMOTION - controlling the motion of large molecules and particles D. Horke1; S. Awel1; Z. Huang1; N. Roth1; N. Teschmit1; T. Ossenbrüggen1; D. Gusa1; J. Küpper1; 1 Deutsches Elektronen Synchrotron DESY, Hamburg/D
315
Theoretical Techniques
P06-01 Photoelectron spectra from optimally tuned TDDFT and Dyson orbital formalism T. Moehle1; S. Bokarev1; G. Grell1; O. Kühn1; 1 University of Rostock/D;
316
P06-02 Quantum-chemical calculations of metal-organic frameworks with open-shell adsorbates E. Dietze1; C. Herrmann1; 1 University of Hamburg/D
317
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
38
P06-03 Hydrogen atom scattering from M(111) surfaces: molecular dynamics simulations on a full-dimensional EMT Potential Energy Surface M. Kammler1; S. Janke1; D. Auerbach1; A. Wodtke1; A. Kandratsenka1; 1 Max-Planck-Institute for Biophysical Chemistry, Göttingen/D
318
P06-04 Computation and optimization of complex formation thermodynamics by a solute-solute integral equation theory F. Mrugalla1; S. Kast1; 1 TU Dortmund/D
319
P06-05 Transfer free energies between aqueous and nonaqueous phases from an integral equation-based quantum solvation model N. Tielker1; D. Tomazic1; S. Kast1; 1 TU Dortmund/D
320
P06-06 Isomerization reaction kinetics in 2-methyltetrahydrofuran oxidation P. Parab1; N. Sakade2; Y. Sakai2; R. Fernandes3; K. Heufer1; 1 RWTH Aachen University/D; 2 University of Fukui/J; 3 Physikalisch Technische Budesanstalt (PTB), Braunschweig/D
321
P06-07 Calculation of entropy-dependent thermodynamic properties of fluids at high pressure with Monte Carlo simulationS.Y. Nezhad1, U.K. Deiters1; 1 University of Cologne/D
322
Solids and Nano-sized Matter
P07-01 Imaging of gap plasmons and surface plasmon quenching in Au and Ag tip-sample-systems with cathodoluminescence M. Grüßer1; X. Ma1; R. Schuster1; 1 Karlsruhe Institute of Technology (KIT)/D
323
P07-02 Synthesis and control of emission properties of highly luminescent In(Zn)P/GaP/ZnS quantum dots C. Meerbach1; C. Rengers1; M. Adam1; N. Gaponik1; A. Eychmüller1; 1 TU Dresden/D
324
P07-03 Determination of the dimensions and extinction coeffiecient of CdSe seeded CdS nanorods through their UV/Vis spectra P. Adel1; J. Bloh1; D. Hinrichs1; T. Kodanek1; D. Dorfs1; 1 Universität Hannover/D
325
P07-04 The creation of highly emmissive green - to - red Cu - In - Zn - S/ZnS core/shell quantum dots J. Lox1; M. Adam1; T. Erdem2; H. Demir2; N. Gaponik1; A. Eychmüller1; 1 TU Dresden/D; 2 Bilkent University, Ankara/TR
326
P07-05 Modification of binary semiconductor nanorods by site-selective growth of a heterojunction F. Enders1; K. Boldt1; 1 Universität Konstanz/D
327
P07-06 Synthesis and spectroscopy of gold nanoclusters with different surface modifications T. Hadler1; T. Kipp1; A. Mews1; 1 University of Hamburg/D
328
P07-07 Optical and theoretical investigation of spectral diffusion of CdSe/CdS-Dotrods S. Lohmann1; C. Strelow1; T. Kipp1; A. Mews1; 1 University of Hamburg/D
329
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
39
P07-08 Studies on the twinning effects during the self-assembly of silver nanoparticles into supercrystals A. Reichhelm1; D. Haubold1; A. Eychmüller1; 1 TU Dresden/D
330
P07-09 Development of a new nanostructured i-Sb2S3 / p-NiO junction photoelectrode J. Schneider1; Y. Wu1; M. Barr3; L. Santinacci2; J. Bachmann1; 1 Universität Erlangen- Nürnberg, Erlangen/D; 3 Aix Marseille Université/F; 3 CNRS and Aix Marseille Université/F
331
P07-10 Influence of the modification on the initiation potential of nanoscale ZnO M. Schmitt1; 1 Saarland University, Saarbrücken/D
333
P07-11 The formation of alkali ion beams from MAlSi2O6 by thermionic emission S. Schuld1; M. Schäfer1; K. Weitzel1; 1 Universität Marburg/D
334
P07-12 Synthesis and characterization of CdSe/CdS nanorod decorated silica-coated plasmonic platinum nanoparticles F. Lübkemann1; N. Bigall1; D. Dorfs1; 1 Universität Hannover/D
335
P07-13 Mechanism of gold nanoparticle growth in organic solvents T. Redder1; P. Witthöft1; A. Mews1; 1 University of Hamburg/D
336
P07-14 Shape-controlled Synthesis of Photoactive Two Dimensional SnS Nanosheets M. Kobylinski1; A. Mews1; 1 University of Hamburg/D
337
P07-15 Simulations of alloying processes in nanoparticle assemblies D. Lenz1; T. Lorenz1; J. Joswig1; 1 TU Dresden/D
338
P07-16 Synthesis of tinsulfide-nanosheet networks via metal-organic chemical vapor deposition C. Ruhmlieb1; A. Mews1; 1 University of Hamburg/D
339
P07-17 Aerogel from CdSe and CdSe/CdS nanoplatelets S. Naskar1; S.S. Paradinas1; J.F. Miethe1; N. Bigall1; 1 Universität Hannover/D
340
P07-18 Combined local electrical and optical probes of individual Semiconductor nanostructures A. Kolditz1; 1 University of Hamburg/D
341
P07-19 Amine stabilized Au NP’s for catalytic applications L. Mohrhusen1; M. Osmic1; J. Kolny-Olesiak1; K. Al-Shamery1; 1 University of Oldenburg/D
342
P07-20 Versatile fabrication of voluminous nanocrystal aerogels A. Freytag1; F. Lübkemann1; N. Bigall1; 1 Universität Hannover/D
343
P07-21 Embedding nonpolar quantum Dots into ionic matrices C. Guhrenz1; A. Benad1; C. Bauer1; M. Adam1; J. Lox1; C. Ziegler1; N. Gaponik1; A. Eychmüller1; 1 TU Dresden/D
344
P07-22 Surface phenomena of metal nanowire networks as transparent electrodes for organic solar cells L. Sonntag1; 1 TU Dresden/D
345
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
40
P07-23 Impact of redox-active molecules on the fluorescence of carbon nanotubes E. Polo1; 1 Universität Göttingen/D
346
P07-24 Localized surface plasmon resonance of Au-ITO heterodimers prepared by colloid chemistry T. Mohamed1; D. Dorfs1; 1 Universität Hannover/D
347
P07-25 Influence of chloride ions on the shape and nucleation of CdS and CdSe on metal nanoparticles D. Hinrichs1; M. Galchenko1; T. Kodanek1; S. Naskar1; N. Bigall1; D. Dorfs1; 1 Universität Hannover/D
348
P07-26 Assembly of shape-controlled semiconductor nanocrystals L. Klepzig1; P. Jan1; N. Bigall1; 1 Universität Hannover/D
349
P07-27 Improvement of photostability of silica covered CdSe/CdS-Dot/Rods through surface attached AuNPs-Layer S. Schneider1; E. Kröger1; 1 University of Hamburg/D
350
Soft Matter and Polymers
P08-01 Internal complexation within starlike copolymers: influence of topology and composition P. Hebbeker1; A. Steinschulte1; F. Plamper1; S. Schneider1; 1 RWTH Aachen/D
351
P08-02 Gas permeation through carbon nanomembranes A. Beyer1; V. Chinaryan1; S. Shishatskiy2; J. Wind2; X. Zhang1; H. Vieker3; P. Angelova3; V. Abetz2; A. Gölzhäuser1; 1 University of Bielefeld/D; 2 Helmholtz-Zentrum Geesthacht (HZG)/D; 3 CNM Technologies GmbH, Bielefeld/D
352
P08-03 Polymer loops and tails attached to a surface, a dissipative particle dynamics study M. Jehser1; S. Eisenhaber1; G. Zifferer1; 1 Universität Wien/A
353
P08-04 Shielding effects in surface initiated polymerization studied by Dissipative Particle Dynamics (DPD) S. Eisenhaber1; M. Jehser1; G. Zifferer1; 1 University of Vienna/A
354
P08-05 Structure and time-dependent self-diffusion in binary mixtures of charge-stabilized colloidal suspensions F. Ziegert1; J. Wagner1; 1 Universität Rostock/D
355
P08-06 Detection of particle interaction in polydisperse highly concentrated polymer suspensions by Photon Density Wave spectroscopy L. Bressel1; S. Pfuhl2; O. Reich1; 1 University of Potsdam and innoFSPEC, Potsdam/D; 2 University of Potsdam/D
356
P08-07 Combination of fiber-optical methods for in-line multi-parameter analysis of the PNIPAm switching processes P. Werner1; 1 University of Potsdam and innoFSPEC, Potsdam/D
357
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
41
Surfaces and Interfaces
P09-01 Adsorption of CO2 on the Fe3O4(111) surface studied by density-functional theory X. Li1; J. Paier1; 1 Humboldt-Universität zu Berlin/D
358
P09-02 Formaldehyde adsorption on the rutile TiO2(110) surface probed by IR spectroscopy X. Yu1; C. Yang1; F. Bebensee1; A. Nefedov1; Z. Zhang2; Q. Ge3; Z. Dohnálek4; Y. Wang1; C. Wöll1; 1 Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen/D; 2 Baylor University, Waco, TX/USA; 3 Southern Illinois University, Carbondale, IL/USA; 4 Pacific Northwest National Laboratory, Richland, WA/USA
359
P09-03 Microcalorimetric measurement of double layer charging in ionic liquids J. Lindner1; S. Frittmann1; F. Endres2; R. Schuster1; 1 Karlsruhe Institute of Technology (KIT)/D; 2 TU Clausthal/D
360
P09-04 Structure dependent anchoring and thermal stability of organic molecules on atomically-defined oxide surfaces: phthalic acid on Co3O4(111), CoO(100) and CoO(111) T. Xu1; M. Schwarz1; K. Werner1; S. Mohr1; M. Amende1; J. Libuda1; 1 Universität Erlangen-Nürnberg, Erlangen/D
361
P09-05 LiCoO2-electrolyte interaction studied by surface science methods T. Späth1; R. Hausbrand1; W. Jaegermann1; 1 TU Darmstadt/D
362
P09-06 Relaxation dynamics and electron-transfer kinetics investigation on electrode|self-assembled monolayer|ionic liquid interfaces using electrochemical impedance spectroscopy N. Vargas-Barbosa1; B. Roling1; 1 Universität Marburg/D
363
P09-07 Investigation of the interface of immiscible liquids by fluorescence light under microfluidic conditions H. Fischer1; E. Antonsson1; E. Rühl1; 1 Freie Universität Berlin/D
364
P09-08 Scattering of highly vibrationally excited NO from Ge(111) S. Meyer1; B. Krüger1; A. Wodtke1; T. Schäfer1; 1 Universität Göttingen/D
365
P09-09 Vibrational energy pooling in multilayer CO on NaCl(100) studied with nanosecond time resolution using a superconducting nanowire single photon detector L. Chen1; D. Schwarzer1; A. Wodtke1; 1 Max-Planck-Institute for Biophysical Chemistry, Göttingen/D
366
P09-10 Multi-quantum vibrational relaxation in highly vibrationally excited NO/surface scattering: Influence of the surface B. Krüger1; S. Meyer1; A. Kandratsenka2; A. Wodtke1; T. Schäfer1; 1 Universität Göttingen/D; 2 Max-Planck-Institute for Biophysical Chemistry, Göttingen/D
367
P09-11 Energy transfer in collisions of highly vibrationally excited NO with vanadium dioxide surfaces A. Meling1; B. Krüger1; T. Schäfer1; I. Rahinov2; A. Wodtke1; 1 Universität Göttingen/D; 2 The Open University of Israel, Tel Aviv/IL
368
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
42
P09-12 Models systems for molecular energy storage: norbornadiene on Pt(111) and anchored to an atomically-defined cobalt oxide surface M. Schwarz1; S. Mohr1; T. Xu1; O. Brummel1; K. Werner1; K. Civale1; T. Döpper1; U. Bauer1; P. Bachmann1; D. Besold1; C. Papp1; H. Steinrück1; A. Görling1; J. Bachmann1; A. Hirsch1; J. Libuda1; 1 Universität Erlangen-Nürnberg, Erlangen/D
369
P09-13 Quenching electronically and vibrationally excited carbon monoxide at metal surfaces using velocity controlled molecular beams R. Wagner1; A. Meling1; T. Schäfer1; A. Wodtke1; 1 Universität Göttingen/D
370
P09-14 Fine structure in IR spectra of high quality CO2 ultrathin films J. Vogt1; 1 University of Magdeburg/D
371
P09-15 Direct scattering of formaldehyde from the Au(111) surface B. Park1; B. Krüger1; S. Meyer1; A. Wodtke1; T. Schäfer1; 1 Universität Göttingen/D
372
P09-16 Time-resolved IR spectroscopy during interfacial electrochemical reactions of organic corrosion inhibitors Y. Chen1; 1 Max-Planck-Institut für Eisenforschung GmbH, Düsseldorf/D
373
P09-17 In-situ IRAS studies of Tetraphenylporphyrins on Co3O4(111)/Ir(100) S. Mohr1; K. Werner1; M. Schwarz1; M. Amende1; T. Doepper1; A. Goerling1; J. Libuda1; 1 Universität Erlangen-Nürnberg, Erlangen/D
374
P09-18 The vanadium oxide monolayer on CeO2(111) studied by density functional theory C. Penschke1; J. Paier1; J. Sauer1; 1 Humboldt-Universität zu Berlin/D
375
P09-19 Direct growth of patterned graphene A. Turchanin1; 1 Universität Jena/D
376
P09-20 Scattering HCl from Au(111): vibrational excitation probabilities and dissociative adsorption J. Geweke1; P. Shirhatti1; C. Steinsiek2; C. Bartels3; I. Rahinov4; A. Wodtke1; 1 Max-Planck-Institute for Biophysical Chemistry, Göttingen/D; 2 Universität Göttingen/D; 3 Universität Freiburg/D; 4 Open University of Israel, Tel Aviv/D
377
P09-21 Electrostatic interactions and ligand effects at ionic-liquid modified interfaces: Co-adsorption of [EMIM][OTf] and CO on Pd(111) T. Bauer1; S. Mehl1; O. Brummel1; K. Pohako-Esko1; P. Wasserscheid1; J. Libuda1; 1 Universität Erlangen-Nürnberg, Erlangen/D
378
P09-22 Ab initio molecular dynamics simulations of CO/NO scattering from Au(111) J. Altschäffel1; A. Kandratsenka1; A. Wodtke1; 1 Universität Göttingen/D
379
P09-23 Monitoring oxide layer growth on manganese electrodes, by in-situ spectroscopic ellipsometry and raman spectroscopy M. Rabe1; A. Sarfraz1; A. Erbe1; 1 Max-Planck-Institut für Eisenforschung GmbH, Düsseldorf/D
380cancelled
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
43
P09-24 H at Au(111): an accurate full-dimensional potential energy surface S. Janke1; A. Kandratsenka1; Y. Dorenkamp1; H. Jiang1; O. Bünermann1; D. Auerbach1; A. Wodtke1; 1 University of Göttingen/D
381
P09-25 The effect of solvent and interfacial field affect on switching of a nitrospiropyran self-assembled monolayer at the solid/liquid interface T. Garling1; Y. Tong1; R. Campen1; 1 Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin/D
382
P09-26 Associative desorption of hydrogen from metal surfaces S. Kaufmann1; Q. Shuai1; D. Schwarzer1; D. Auerbach1; A. Wodtke1; 1 Max-Planck-Institute for Biophysical Chemistry, Göttingen/D
383
P09-27 CO adsorption on Pt3Ti(111) studied by IRRAS and XPS L. Schöttner1; M. Moors2; F. Bebensee3; A. Nefedov3; Y. Wang3; C. Wöll3; 1 Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen/D; 2 Forschungszentrum Jülich/D; 3 Karlsruhe Institute of Technology (KIT)/D
384
P09-28 Reactivity of CO on sulfur-passivated graphene-supported palladium nanocluster arrays F. Düll1; 1 Universität Erlangen-Nürnberg, Erlangen/D
385
P09-29 A new thin film fabrication technique by interconnecting nanoparticles via tetrazole-complexes A. Wolf1; C. Rengers1; S. Voitekhovich2; V. Lesnyak1; N. Gaponik1; A. Eychmüller1; 1 TU Dresden/D; 2 Belarusian State University, Minsk/BY
386
P09-30 Self-assembling amphiphilic fluorophores: in-situ monitoring and photonic characterization M. Hupfer1; 1 Universität Jena/D
387
P09-31 Photo-manipulation of the surface tension anisotropy at a liquid-crystal/ITO-glass interface A. Eremin1; H. Nadasi1; R. Stannarius1; 1 Universität Magdeburg/D
388
Biophysical Chemistry
P10-01 Molecular recognition in the organic corona around nanoparticles and applications to biosensing S. Kruss1; 1 Universität Göttingen/D
389
P10-03 Development of a label-free fiber optical biosensor based on etched fiber-Bragg-gratings S. Schulze1; C. Hille1; 1 Universität Potsdam/D
390
P10-04 High-resolution femtosecond FT2D-IR spectroelectrochemistry J. Schmidt-Engler1; Y. El Khoury1; L. van Wilderen1; J. Bredenbeck1; 1 Universität Frankfurt, Frankfurt/D
391
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
44
P10-05 Noninvasive, label-free analysis of stimulus-induced cellular activity using Raman spectroscopic imaging A. Post1; C. Hille1; 1 Universität Potsdam/D
392
P10-06 Liquid-liquid phase separation in therapeutic monoclonal antibody systems P. Garidel1; 1 Universität Halle-Wittenberg, Biberach/D
393
P10-07 Validation and comparison of biomolecular force fields regarding structural properties and binding affinities of cyclodextrins J. Gebhardt1; N. Hansen1; 1 University of Stuttgart/D
394
P10-08 Signal mechanism of integrin αVβ3 W. Langel1; M. Kulke1; 1 Universität Greifswald/D
395
P10-09 Probing the function of calmodulin adsorbed on and embedded in polyelectrolyte multilayers S. Cinar1; C. Czeslik1; 1 TU Dortmund/D
396
P10-10 Activation volumes of enzymes at polyelectrolyte brushes A. Levin1; C. Czeslik1; 1 TU Dortmund/D
397
P10-11 Cosolvent and crowding effects on the temperature and pressure stability of monomeric actin P. Schummel1; R. Winter1; 1 TU Dortmund/D
398
P10-12 Penetration of dexamethasone in human skin investigated by X-ray spectromicroscopy K. Yamamoto1; A. Klossek1; R. Flesch1; F. Rancan2; M. Weigand3; I. Bykova3; M. Bechtel3; P. Patoka1; G. Ulrich1; S. Ahlberg2; A. Vogt2; U. Blume-Peytavi2; P. Schrade2; S. Bachmann2; M. Schäfer-Korting2; E. Rühl1; 1 Freie Universität Berlin/D; 2 Charite Universitätsmedizin Berlin/D; 3 Max-Planck-Institut für Intelligente Systeme, Stuttgart/D
399
P10-13 CRMP2: switching structures D. Möller1; C. Lillig1; M. Gellert1; N. Geist1; W. Langel1; 1 Universität Greifswald/D
400
P10-14 Efficient energy transfer as switching mechanism for super-resolution microscopy A. Haderspeck1; D. Brox1; D. Herten1; 1 University of Heidelberg/D
401
P10-15 Allostery in PDZ3: azidohomoalanine as site-specific probe for IR spectroscopy K. Eberl1; P. Quoika1; H. Müller-Werkmeister1; J. Löffler1; M. Essig1; J. Bredenbeck1; 1 University of Frankfurt/D
402
P10-16 Shedding light on plasma-bacteria interaction using vibrational microspectroscopy K. Kartaschew1; S. Baldus1; F. Kogelheide1; M. Mischo1; J. Lackmann1; P. Awakowicz1; M. Havenith1; 1 Ruhr-Universität Bochum/D
403
P10-17 Understanding the primary steps of light-induced signal propagation of an artificial LOV2-Rac1 photoswitch I. Stambolic1; 1 University of Regensburg/D
404
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
45
P10-18 The flavin photochemistry in a LOV domain is tuned and controlled by the protein environment K. Magerl1; 1 Universität Regensburg/D
405
P10-19 Targeted delivery of aptamer- and antibody-conjugated AuNPs to Interleukin-6 receptor-carrying cells L. Prisner1; N. Bohn1; M. Bendig1; A. Mews1; 1 Universität Hamburg/D
406
P10-20 Effect of the N-terminal Helix and Nucleotide-loading on the membrane and effector binding of the GTPases Arl2 and Arl3 S. Kapoor1; E. Fansa2; S. Möbitz3; S. Ismail4; R. Winter3; A. Wittinghofer2; K. Weise3; 1 TU Dortmund and Max Planck Institute of Molecular Physiology, Dortmund/D; 2 Max Planck Institute of Molecular Physiology, Dortmund/D; 3 TU Dortmund/D; 4 Max Planck Institute of Molecular Physiology and CR-UK Beatson Institute, Glasgow/UK
407
P10-21 Kinetics of drug release and mobility in healthy and osteoporotic rat bone M. Rohnke1; 1 University of Giessen/D
408
P10-22 Calculation of relative folding free enthalpies of amide-to-ester mutants of protein Pin1 D. Markthaler1; 1 Universität Stuttgart/D
410
P10-23 Counting LAT and SLP-76 in the T-cell antigen receptor proximal signaling microcluster W. Chmielewicz1; S. Hänselmann1; K. Yserentant1; K. Grußmayer1; D. Herten1; 1 Heidelberg University/D
411
P10-24 Comparing azide and thiocyanate vibrational probes for infrared spectroscopy of proteins M. Mikorski1; D. Kern-Michler1; K. Eberl1; L. van Wilderen1; J. Bredenbeck1; 1 Universität Frankfurt/D
412
P10-25 Pressure-induced effects on self-cleavage reaction of ribozyme catalysis: a molecular dynamics study N. Kumar1; 1 Ruhr-Universität Bochum/D
413
P10-26 Cavity Enhanced Raman Spectroscopy (CERS) as an analytical tool in the biosciences: studying hydrogen production by microbes T.W. Smith1, M. Hippler1; 1 University of Sheffield/UK
414
P10-27 Scattering particles increase absorbance of dyes – a model study with relevance for sunscreens B. Herzog1, 1 BASF Grenzach GmbH, Grenzach-Wyhlen/D
415
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
POSTER PROGRAMME
46
Catalysis
P11-01 Oxygen chain structures and other forms of chemisorbed oxygen on gold-silver alloy surfaces: a DFT study L. Moskaleva1; T. Weiss1; M. Bäumer1; T. Klüner2; 1 Universität Bremen/D; 2 Universität Oldenburg/D
416
P11-02 Decomposition of formic acid on Pd-based surfaces H. Euchner1; 1 Ulm University/D
417
P11-03 Computational methods for pure component decompositions with application to the rhodium catalyzed hydroformylation K. Neymeyr1; M. Sawall1; A. Jürß1; H. Schröder1; 1 University of Rostock/D
418
P11-04 Formic acid dehydrogenation over Au/CeO2 catalysts: theoretical DFT study J. Kucera1; A. Groß1; 1 Ulm University/D
419
P11-05 Operando X-ray absorption and emission spectroscopy to understand reaction mechanisms in NOx-removal catalysts T. Günter1; D. Doronkin1; A. Boubnov1; M. Casapu1; J. Grunwaldt1; J. Rudolph2; C. Jacob2; G. Tofighi1; 1 Karlsruher Institut für Technologie (KIT)/D; 2 TU Braunschweig/D
420
P11-06 Nanoporous gold supported oxides as high-performance hydrogen production catalyst J. Shi1; A. Lackmann1; A. Wittstock1; M. Bäumer1; 1 University of Bremen/D
421
Electrochemistry and Energy
P12-01 Electroplating of polyurethane foams S. Keck1; A. Jung1; R. Hempelmann1; H. Natter1; 1 Saarland University, Saarbrücken/D
423
P12-02 New catalyst supports for energy conversion prepared by surface modification of Graphene- and CNT-structures E. Oh1; 1 Saarland University, Saarbrücken/D
424
P12-03 Characterization and improvement of the active materials in the vanadium redox flow battery K. Weißhaar1; H. Natter1; R. Hempelmann1; 1 Saarland University, Saarbrücken/D
425
P12-04 Development of an improved state – of – charge indicator for the all – vanadium redox flow battery J. Geiser1; H. Natter1; R. Hempelmann1; 1 Saarland University, Saarbrücken/D
426
P12-05 Perovskites containing protons, oxygen vacancies and holes: experiments and simulations for proton uptake and stoichiometry relaxation kinetics R. Zohourian Aboutorabi1; R. Merkle1; J. Maier1; 1 Max Planck Institute for Solid State Research, Stuttgart/D
427
Page

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
POSTER PROGRAMME
47
P12-06 Investigation of co-adsorption and side processes during underpotential deposition of Cu and Ag on Au(111) by electrochemical microcalorimetry S. Frittmann1; V. Halka1; R. Schuster1; 1 Karlsruher Institut für Technologie (KIT)/D
428
P12-07 Designed architectures of electrode materials for advanced Li (Na)-ion batteries Y. Yu1; C. Wu1; 1 Max Planck Institute for Solid State Research, Stuttgart/D
429
P12-08 On the calculation of excess functions of electrolytes A. Maurer1; H. Krienke2; D. Horinek2; G. Schmeer2; 1 University of Regensburg, Tirschenreuth/D; 2 University of Regensburg/D
431
P12-09 Manganese oxides for the oxygen evolution reaction J. Doerfer1; J. Schuch1; B. Kaiser1; W. Jaegermann1; 1 TU Darmstadt/D
432
P12-10 In situ method for efficient catalyst layer production with very low catalyst loading R. Engstler1; D. Durneata1; H. Natter1; R. Hempelmann1; 1 Saarland University, Saarbrücken/D
433
P12-11 BaTiO3 modifications for photochemical hydrogen evolution Y. Xiong1; 1 Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr/D
434
P12-12 Long-term stability of silicon-poisoned La0.6Sr0.4Co0.2Fe0.8O3-δ SOFC-cathodes M. Perz1; E. Bucher1; A. Egger1; C. Gspan2; F. Hofer2; W. Sitte1; 1 Montanuniversitaet Leoben/A; 2 Graz University of Technology/A
435
P12-13 Development of a photocapacitor based on printed solar cells and super capacitors K. Anneser1; S. Braxmeier1; A. Baumann1; G. Reichenauer1; V. Dyakonov2; 1 Bavarian Center for Applied Energy Research, Würzburg/D; 2 University of Würzburg/D
436
P12-14 Electron bombardment induced cation transport in an ion conducting glass A. Hein1; J. Wiemer1; K. Weitzel1; 1 Universität Marburg/D
437
P12-15 Sulfide electrolytes based on Glyme-Lithium salt mixtures for new battery systems A. Bonsa1; A. Appelhagen1; J. Lehmann1; R. Ludwig1; 1 University of Rostock/D
438
P12-16 Electrochemical study of Si-nanodroplets deposited on graphite using a LPCVD process K. Nimmrich1; D. Klein1; T. Bartoldus2; S. Cap1; R. Schlögl1; 1 Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin/D; 2 Max-Planck-Institut für Chemische Energiekonversion, Mülheim an der Ruhr/D
439
P12-17 Development of electrocatalysts for alkaline water electrolysis A. Surkus1; M. Polyakov1; N. Rockstroh1; H. Junge1; A. Martin1; M. Beller1; 1 Leibniz-Institute for Catalysis, Rostock/D
440
P12-18 Preparation of titanium dioxide thin films by atomic layer deposition applied on tandem solar cells for photoelectrochemical water splitting T. Cottre1; J. Ziegler1; B. Kaiser1; W. Jaegermann1; 1 TU Darmstadt/D
441
Page

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
48
Industrial Applications
P13-01 Metrological support for LNG custody transfer and transport fuel applications J. Rauch1; K. Arrhenius2; M. Downey3; B. Gieseking3; P. Eilts4; H. Ent5; O. Kerkhof5; J. Li5; P. Albo6; A. Lucassen1; G. Nieuwenkamp5; M. Richter7; A. van der Veen5; 1 Physikalisch-Technische Bundesanstalt (PTB), Braunschweig/D; 2 SP Sveriges Tekniska Forskningsinstitut AB (SP), Borås/S; 3 National Physical Laboratory (NPL), Teddington/UK; 4 TU Braunschweig/D; 5 VSL Dutch Metrology Institute, Delft/NL; 6 Instituto Nazionale di Ricerca Metrologica (INRiM), Turin/D; 7 Ruhr-Universität Bochum/D
442
Page

49
LECTURES

50

Mesoscopic photosystems for artificial photosynthesisMichael Graetzel , Ecole Polytechnique Fédérale de Lausanne,
Lausanne,Switzerland, Mesoscopic photovoltaics have emerged as credible contenders to conventional p-njunction photovoltaics [1-3]. Mimicking light harvesting and charge carrier generation in natural photosynthesis, dye sensitized solar cells (DSCs) were the first to use three-dimensional nanocrystalline junctions for solar electricity production, reaching currently a power conversion efficiency (PCE) of over 14% in standard air mass 1.5 sunlight. By now, large-scale DSC production and commercial sales have been launched on the multi-megawatt scale. Recently, the DSC has engendered the meteoric rise of perovskite solar cells (PSCs) [4,5]. Todayʼs state of the art devices employ metal halide perovskite of the general composition ABX3 as light harvesters, where A stands for methylammonium, formamidinium or caesium, B denotes lead or tin and X iodide or bromide. Carrier diffusion lengths in the 100 nm - micron range have been measured for solution-processed perovskites and certified power conversion efficiencies (PCEs) attain 22 %, exceeding the PCE of polycrystalline silicon solar cells. These photovoltaics show intense electro-luminesence matchingthe external quantum efficiency of silicon solar cells. and Voc values close to 1.2 Vfor a 1.55 eV band gap material. This renders perovskite-based photosystem very attractive for applications in tandem cells and for the generation of fuels from sunlightmimicking natural photosynthesis [6,7].
1. B.OʼRegan and M. Grätzel. “A Low Cost, High Efficiency Solar Cell based on the Sensitization of Colloidal Titanium Dioxide,” Nature 353 (1991) pp 7377-7381.
2. M. Grätzel, “Photoelectrochemical Cells,” Nature 353 (2001) pp 7377-7381. 3. A.Yella, H.-W. Lee, H. N. Tsao, C. Yi, A.Kumar Chandiran, Md.K. Nazeeruddin, EW-G
.Diau,,C.-Y Yeh, S. M. Zakeeruddin and M. Grätzel,“Porphyrin-based Solar Cell with Co(II/III) Redox Electrolyte Exceed 12% Efficiency,“ Science 629 (2011) pp 334-341.
4. M. Grätzel, Light and Shade of Perovskite Solar Cells, Nature Mat. 13 (2014) pp 838-842.
5. J. Burschka, N. Pellet, S.-J. Moon, R.Humphry-Baker, P. Gao1, M K. Nazeeruddin and M. Grätzel, „Sequential deposition as a route to high-performance perovskite-sensitized solar cells“ Nature 499, (2013),pp 316-3199.
6. J. Luo, J.-H. Im, M.T. Mayer, M. Schreier, Md.K. Nazeeruddin, N.-G. Park, S.D.Tilley, H.J. Fan, M. Grätzel, Water photolysis at 12.3% efficiency via perovskite photovoltaics and Earth abundant catalysts Science, 345, (2014), pp 1593-1596.
7. M.Schreier L. Curvat, F. Giordano, L. Steier, A. Abate, S.M. Zakeeruddin, J. Luo, M. Mayer and M:Grätzel, "Efficient photosynthesis of carbon monoxide from CO2 using perovskite photovoltaics" Nature Commun. 6, (2015) , pp 7326-7332.
51

Functional nanomaterials for energy conversion and storageQiang Xu, National Institute of Advanced Industrial Science and Technology (AIST)
and Kobe University, Ikeda, Osaka 563-8577, JapanThis talk includes following two topics:
1) Porous metal-organic frameworks (MOFs) for energy applicationsWe have developed new applications of porous MOFs, especially for energy
conversion and storage.1 Novel porous metal-organic frameworks have been synthesized, which present stable catalytic activities for the oxidation of CO to CO2.Metal nanoparticles (NPs) have been immobilized to MOFs by the solid-grinding method, impregnation method and double-solvents approach in combination with the H2 reduction, liquid-phase concentration-controlled reduction and the CO-directed reduction at the solid-gas interface, which exhibit excellent catalytic performances for various reactions, including hydrogen generation from chemical hydrides. Porous carbons have been synthesized by using MOFs as templates/precursors and the resultant carbons display high specific surface areas and excellent electrochemical properties as electrode materials for electric double-layered capacitor (EDLC) and as catalysts for oxygen reduction reaction (ORR).
2) Metal nanoparticle-catalyzed hydrogen generationWe have reported excellent liquid-phase hydrogen generation systems, which are
based on metal nanoparticle-catalyzed hydrolysis of ammonia borane (NH3BH3),complete decomposition of hydrous hydrazine (H2NNH2) and decomposition of formic acid. The metal nanoparticles catalysts immobilized by the double-solvents method (DSM) inside the pores of MOFs, by the non-noble metal sacrificial approach(NNMSA) to reduced graphene oxide and by the weakly-capping growth approach(WCGA) to carbon nanospheres will be discussed. The use of soluble porous organic cages as a stabilizer and homogenizer toward the homogenization of heterogeneous metal nanoparticle catalysts with enhanced catalytic performance will also be discussed.References1. For recent reviews, see: (a) S.-L. Li, Q. Xu, Energy Environ. Sci., 2013, 6, 1656-1683. (b)
Q. L. Zhu, Q. Xu, Chem. Soc. Rev., 2014, 43, 5468-5512. (c) J.-K. Sun, Q. Xu, Energy Environ. Sci., 2014, 7, 2071-2100. (d) W. Xia, A. Mahmood, R.-Q.. Zou, Q. Xu, Energy Environ. Sci., 2015, 8, 1837-1866.
2. For recent reviews, see: (a) Yadav, M.; Xu, Q. Energy Environ. Sci. 2012, 5, 9698. (b) Zhu, Q.-L.; Xu, Q. Energy Environ. Sci. 2015, 8, 478.
52

TiO2 as a bottleneck for the photocatalytic water reduction on Cd0.5Zn0.5S@Pt-TiO2 composites
Anton Litke, Jan P. Hofmann, Thomas Weber, Emiel J.M. HensenEindhoven University of Technology, The Netherlands
Solar light is the most abundant renewable energy source on the Earth, but its intermittentnature requires an efficient technology for the conversion of photons into storable and transportable chemical fuels. In this regard photocatalytic water splitting is considered as an attractive and environmentally benign solution.Thus, development of an active photocatalytic systems is of high importance. Mixed Cd-Zn sulphides are considered as a promising class of inorganic photocatalysts because they are active under visible light for water reduction and theirproperties can be adjusted in a wide range by changing metal ratio and/or chalcogenide. Besides this, other strategies such as formation of a heterojunction with wide-bandgap metal oxides (e.g. TiO2 or ZnO) can be used to improve charge carrier separation and overall photocatalytic efficiency of sulphide-bases systems.
In this work we studied composites of Cd-Zn sulphides with photoplatinized TiO2.This type of materials was reported to have superior photocatalytic activity comparedto bulk CdS due to a vectorial electron transfer from sulphide to TiO2 and further to Pt. This approach was confirmed to work for sulphides with low intrinsic activity (i.e. precipitated CdS) but had a strong deteriorating effect on highly active hydrothermally prepared Cd-Zn sulphide (Cd0.5Zn0.5S). The combination of FTIR and XPS showed that the surface of Pt nanoparticles got poisoned during hydrothermal treatment. However poisoning of Pt co-catalyst had only minor effect on the activity of the composite with hydrothermal Cd0.5Zn0.5S. We found that electron transfer from sulphide to oxide in hydrothermally prepared composites was more efficient than in co-precipitated samples, but overall photocatalytic activities of all composites were similar and almost independent from the properties of sulphides. Hence, the mobility of the conduction band electrons in TiO2 was proposed as the limiting factor.
53

Ethanol Photocataylsis on Rutile TiO2(110): Unravelling the Mechanistic Role of Defects and Water
Constantin A. Walenta, Sebastian L. Kollmannsberger, Martin Tschurl and Ueli Heiz, Chair of Physical Chemistry & Catalysis Research Center, Technische Universität
München, Lichtenbergstr. 4, 85748 Garching b. München, Germany
Metal cluster-semiconductor systems are promising new hybrid materials for combined light harvesting and conversion of photon energy into chemical energy. As promising materials Au/TiO2 systems attracted considerable attention over the lastyears [1,2]. The most explored model reaction is photochemical water splitting, but the H2-production is still limited. Another renewable feedstock of interest is ethanol, because alcohols are the main product from biofermentation and as such are ideal precursors for biomass in general [3].In order to design ideal metal cluster-semiconductor hybrid systems and to maximize the hydrogen production yield, the understanding of the semiconductorʼs surface photochemistry is of paramount importance. To ensure very well defined conditions, rutile TiO2(110) is studied under UHV conditions. Exemplified on titania, we demonstrate how the mechanisms of photochemical reactions can be elucidated via the judicious choice of different experiments. It is shown, that ethanol is photooxidized to acetaldehyde. Although formally two hydrogen atoms remain from the stoichiometric reaction equation, no molecular hydrogen production is observed[4]. The hydrogen forms water on the surface which poisons the photocatalyst by site-blocking on a defect-rich surface. This mechanism enables even the interpretation of ambient photocatalytic systems.
References:[1] Murdoch et al., Nat Chem, 2011, 3, 6[2] Yoshida et al., J. Am. Chem. Soc., 2009, 131, 37[3] Navarro et al., Energy Environ. Sci., 2009, 2, 1[4] Walenta et al., Phys. Chem. Chem. Phys., 2015, 17, 35
54

Electrocatalytic Activity of Sulfides
Michael Bredol1, Valerie Niemann1,2 and Jan-David Goossen1
1 Münster University of Applied Sciences, Department of Chemical Engineering, Stegerwaldstraße 39, 48565 Steinfurt
2 Drexel University, Chemical and Biological Engineering, Philadelphia, PA
For electrochemical energy conversion at low and moderate temperatures, electrocatalysts are needed on both electrodes. State of the art catalysts are fabricated from noble metals, typically platinum. Such an approach is not sustainable for large scale applications due to the scarcity of these elements and the consequently high prices. In order to overcome this limitation, alternatives are sought, in most cases in the form of transition metals and their composites. In this contribution, the focus is shifted from metals to the electrocatalytic potential of sulfides on the reducing electrode. Sulfides offer a broad range of redox states for interaction with electrolytes, opening electrochemical windows for chemistries like thehydrogen evolution reaction (HER) from water or CO2 reduction to various target compounds.
Based on the experience with the electrochemical oxidation of ethanol in the presence of sulfides [1,2,3], compounds like nanoparticulate ZnS, ZnSe, CuInS2 or FeS2 have been synthesized and deposited on electrodes and tested with respect to their electrocatalytic activity in the HER and in CO2-containing electrolytes. First results in terms of electrochemical windows available, overvoltages observed and products achieved will be reported and discussed on the background of efficiency and kinetics.
[1] M.Bredol, M.Kaczmarek, H.-D.Wiemhöfer, J. Power Sources 255, 260-265 (2014)[2] M.Kaczmarek, M.Bredol, J. Mat. Sci. 46, 5400-5405 (2011)[3] M.Bredol, M.Kaczmarek, J. Phys. Chem. A 114, 3950-3955 (2010)
55

Solar cells with a solution-processable heterojunction between CuInS2 and ZnO nanocrystals
H. Borchert, D. Scheunemann, S. Wilken, R. Miranti, C. Krause, and J. Parisi,University of Oldenburg, Department of Physics, Energy and Semiconductor
Research Laboratory, Carl-von-Ossietzky Str. 9-11, 26129 Oldenburg, Germany
Colloidal quantum dot solar cells with a heterojunction between lead chalcogenide nanocrystals and titanium dioxide have made rapid progress in recent years. Their power conversion efficiency was reported to reach up to ~10%. A drawback remains the toxicity of the materials used. A more environmentally friendly material is copper indium disulfide (CuInS2, “CIS”). In principle, colloidal CIS and ZnO nanocrystals are also suitable to create depleted heterojunction solar cells [1,2], but their power conversion efficiency reached only up to 0.6%, so far. With the help of quantum efficiency measurements and optical simulations, we examined the collection probability for photo-generated charge carriers as dependent on the spatial region where they are generated. It was found that only a narrow zone in the absorber layer adjacent to the hetero-contact contributes to the photo-current [2]. This provides an explanation for the limited device performance. One possible reason for the narrow width of the collection zone might be the charge carrier mobility in the CIS nanocrystal layer. Therefore, we fabricated single carrier diodes to analyze the charge transport in CIS nanocrystal films. Strategies for improving the carrier mobility in the nanocrystal films as well as for improving the efficiency of corresponding nanocrystal solar cells are discussed.
[1] D. Scheunemann, S. Wilken, J. Parisi, and H. Borchert; Towards depleted heterojunction solar cells with CuInS2 and ZnO nanocrystals, Appl. Phys. Lett.103, 133902 (2013).
[2] D. Scheunemann, S. Wilken, J. Parisi, and H. Borchert; Investigation of the Spatially Dependent Charge Collection Probability in CuInS2/ZnO Colloidal Nanocrystal Solar Cells, ACS Photonics 2, 864-875 (2015).
56

Energy harvesting and storage with ionic liquids:the essential physics at the nanoscale
Alexei A. KornyshevImperial College London, SW7 2AZ UK
The renewal of interest to fundamental mechanisms of energy storage in electrochemical supercapacitors was boosted by the progress in development of novel materials (mainly carbon based) for nanostructured electrodes. It was also affected by the booming research in room temperature ionic liquids that offers virtually unlimited number of new electrolyte combinations. Some of them have large electrochemical windows that allow to charging capacitors to higher voltages and thereby store more energy. This material-science based progress had to be matched with detailed investigation of performance of such systems. In particular, exploiting ultrananoporous electrodes with pores just about to accommodate one layer or one row of ions, can increase the interfacial area and thereby the stored energy, but what will be the capacitance-voltage dependence and the dynamics of charging of such electrodes? What are generally the laws of population of such pores with ions subject to applied potential? What are the conditions for maximizing energy storage and power?
There was considerable progress in this area over the last five years. Basic mechanisms of charge storage equilibria in ultrananoprous electrodes have been understood based on statistical theory of ion-ion and ion interactions in nano-confinement. The patterns of charging dynamics has also been revealed based on kinetic theory and simulations. The lecture will overview these advances, with a focus on the effects that theory explains or predicts, as well as highlight pressing questions for future experiments and theory.
Sister systems to supercapacitors are electroactuators (that can be and a being used for the conversion of time dependent applied voltage into mechanical motion, or other way around for the conversion of the applied force into the AC current. The second class of gadgets is known as harvesters of mechanical work, and draw currently great attention in particular for various scenarios of current generation from walking. Advances in this class of systems will also be reviewed in this lecture.
Tending to give a broad overview of this research area, the lecture will be to a high degree based on a series of joint works [few example publications are cited below] of the Imperial College team and our partners at University of Strathclyde (UK), University of Drexel, Virginia Tech, ORNL and PennState (USA), FZ-Juelich (Germany), University of Tel Aviv, LPTL CNRS (France), and other groups, highlighted in the talk.
1. S.Kondrat, A.Kornyshev, Charging dynamics and optimization of nanoporous supercapacitors, J.Phys.Chem.C 117, 12399-12406 (2013).
2. S.Kondrat, P.Wu, R.Qiao, A.A.Kornyshev, Accelerating charging dynamics in subnanometer pores, Nature Materials 13, 387-393(2014).3. Y.He, J. Huang,B.G. Sumpter, A.A.Kornyshev, R.Qiao, Dynamic charge ctorage in Ionic liquids-filled nanopores: insight from a
computational cyclic voltammetry study, J. Phys. Chem. Lett. 2015, 6, 22−30 (2015).4. A.A.Lee, S.Kondrat, G.Oshanin, A. A.Kornyshev, Charging dynamics of a supercapacitor with narrow cylindrical nanopores,
Nanotechnology 25, #315401, 1-8 (2014).5. S. Kondrat, A.A.Kornyshev, Pressing a spring: what does it take to maximize the energy storage in nanoporous supercapacitors?
Nanoscale Horizons, B1, 45-52 (2016).6. A.A.Kornyshev, The simplest model of charge storage in single file metallic nanopores, Faraday Disc. 164 , 117-133 (2013).7. C.C. Rochester, G.Pruessner,and A.A.Kornyshev, Statistical mechanics of `unwanted electroactuation' in nanoporous supercapacitors,
Electrochim.Acta 174, 978-984 (2015).8. G.Feng, X.Jiang, R.Qiao, A.A.Kornyshev, Water in ionic liquids at electrified interfaces: the anatomy of electrosorption, ACS Nano 8,
11685–11694 (2014).9. A.A.Lee, S.Kondrat, A.A.Kornyshev, Single-file charge storage in conducting nanopores, Phys.Rev.Lett. 113, #048701, 1-5 (2014).10. M.V.Fedorov and A.A.Kornyshev, Ionic liquids at electrified interfaces, Chem.Rev. 114, 2978−3036 (2014).11. O.Y.Fajardo, F.Bresme, A.A.Kornyshev, M.Urbakh, Electrotunable lubricity with ionic liquid nanoscale films, Sci.Rep. 5, 2045-2322 (2015).12. O.Y. Fajardo, F. Bresme, A.A. Kornyshev, M. Urbakh, , Electrotunable friction with Ionic liquid lubricants: how important is the molecular
structure of the ions?, J.Phys.Chem.Lett. 6, 3998-4004 (2015).
57

High Temperature Proton Conducting Fuel Cell Membrane Additives Operating under Anhydrous Conditions
Madita Einemann1, Corinna Harms2, Michael Wark1
1 Institute of Chemistry, Carl von Ossietzky University Oldenburg, Oldenburg, Germany
2 -Forschungszentrum für Energietechnologie, Oldenburg, Germany
The application of inorganic (solid) additives has attracted attention as possible wayto reduce the humidity loss in PEM fuel cell membranes at elevated temperatures [1]. Here, the synthesis and characterization of proton conducting composites based on ordered mesoporous silica (OMS) of the Si-MCM-41 type and the protic ionic liquid (PIL) triethylammmonium trifluorosulfonate are described. The proton conductivity of composites can be tuned by: (i) the content of PIL in the composites (25 and 50 wt-%), (ii) the functionalization of the pores of OMS with sulfonic acid groups (-SO3H+)[2] or (iii) with trimethylamine-neutralized sulfonic acid groups (-SO3NH+Et3), and (iv) the porosity of non-functionalized and functionalized OMS. Proton conductivities as high as 2.6 and 16.3 mS cm-1 at 40 and 160 °C, resp., were obtained under anhydrous conditions for the composite with the lowest porosity, loaded with 50 wt % of PIL and -SO3NH+Et3 as the functional groups. Higher proton conductivities and lower activation energies are found as: (i) the content of PIL in the composite increases and (ii) the porosity of OMS used for the preparation of composites decreases, irrespective of the functionalization of OMS. This indicates that the mechanism of proton conductivity is mainly determined by the porous structure of the OMS [3]. The stability of the composites is determined by the nature of surface functionality: that is OMS>OMS-SO3NH+Et3>OMS-SO3H+.
Reference 1. S.P. Jiang, J Mater Chem A 2014, 2, 7637.2. R. Marschall, P. Toelle, W.L. Cavalcanti, M. Wilhelm, C. Koehler, T. Frauenheim,
M. Wark,. J Phys Chem C 2009, 113, 19218.3. M. Sharifi, C. Köhler, P. Tölle, T. Frauenheim, M. Wark, Small 2011, 8, 1086.
58

Electronic and ionic structure of Li-ion battery materials: concept and impact on interface properties
René Hausbrand and Mathias FingerleInstitute of Material Science, Darmstadt University of Technology,
Jovanka-Bontschits-Str. 2, 64287 Darmstadt, Germany
Lithium ion battery materials are ionic compounds with pure ionic or mixed conductivity. The electronic structure of electrode materials has been given considerable attention in the past due to its significance for the electrode potential. Despite its relevance for charge transfer, however, the ionic structure for Li-ions, i.e. the energy levels of Li-ions and of related defects, was hardly addressed directly in the past. In fact, the concept of energy levels, although successfully applied in semiconductor physics for electronic states, is not yet fully established for ions in ionic materials.This contribution gives an introduction to the concept of ion energy levels and points to its relevance for charge transfer. While ionic energy levels cannot be measured directly, they can be evaluated indirectly by spectroscopic methods and by quantum-mechanical simulation. Principally, such evaluation offers the possibility to establish the ionic structure of different materials on a common reference scale. At interfaces, differences in ionic energy levels between the materials manifest themselves as barriers for charge transfer. The relevance of relaxation effects and interlayer formation is discussed.
59

Charge carrier dynamics for photoelectrochemical water splitting
James R Durrant, Department of Chemistry, Imperial College London, London SW7 2AZ, U.K. and SPECIFIC IKC, College of Engineering, University of Swansea,
Swansea, U.K. Email: [email protected]
There is increasing interest in the development of artificial photosynthetic systems for solar driven fuel synthesis. My talk will focus upon the role of charge carrier dynamics in determining the efficiency of such systems, including both photoelectrodes for water oxidation and reduction and hybrid molecular / inorganic systems for solar driven proton and CO2 reduction. I will particularly focus on the charge carrier dynamics in metal oxide photoelectrodes for water oxidation. I will address the kinetics of charge separation and recombination and water oxidation. Experimentally, these studies will be based around transient absorption spectroscopy on timescales from femtoseconds to seconds, which will be correlated with the results of photoelectrochemical analyses of device efficiency. These techniques are employed to address the function of hematite, titania and bismuth vandate photoanodes, and the effect of deposition of cobalt phosphate on these electrodes, as well as studies of cuprous oxide photoelectrodes for proton reduction. I will focus mainly on the parameters limiting the efficiency of water oxidation on such electrodes. In particular I will present a rate law analysis of water oxidation and proton reduction on oxide (photo)electrodes, addressing the dependence of the reaction upon the density of surface localized holes and electrons. These studies will address in particular the extent to which such oxide surfaces are catalytic for the multi-hole water oxidation reaction, and the implications of these rate law dependencies for the mechanisms of water oxidation and proton reduction.
60

Structural insights into charged Li2-xMO2F by synchrotron X-ray PDF and XANES
Ruiyong Chen, Joint Electrochemistry Lab, KIST Europe, Saarbrücken/Germany;Rolf Hempelmann, Physical Chemistry, Saarland University, Saarbrücken/Germany;
Emad Maawad, Helmholtz-Zentrum Geesthacht, Hamburg/Germany;Murat Yavuz, Michael Knapp, Karlsruhe Institute of Technology, Karlsruhe/Germany;
Stefan Zander, Helmholtz-Zentrum Berlin, Berlin/Germany
A new class of Li2MO2F (M = V, Cr) oxyfluoride intercalation cathode materials with disordered rock-salt structure (space group Fm-3m) has been recently developed.1-3
High intercalation capacity of about 420 mAh g-1 has been achieved, compared to only 170 mAh g-1 for LiFePO4. Improved voltage and cycling has been obtained by using Cr-substituted oxyfluorides. Attractively, high specific energy of 960 Wh kg-1 is accessible at 1 C rate. These findings are expected to inspire new activities in high energy/power disordered rock-salt intercalation cathode materials and open up a new avenue to the search for advanced energy storage materials. In this contribution, we study the structural evolution of the materials upon charging by means of synchrotron X-ray pair distribution function (PDF) analysis and absorption spectroscopy.4
References1. R. Chen, S. Ren, M. Knapp, D. Wang, R. Witter, M. Fichtner, H. Hahn, Adv.
Energy Mater. 5 (2015) 1401814.2. S. Ren, R. Chen, E. Maawad, O. Dolotko, A. Guda, V. Shapovalov, D. Wang, H.
Hahn, M. Fichtner, Adv. Sci. 2 (2015) 1500128.3. R. Chen, S. Ren, M. Yavuz, A. Guda, V. Shapovalov, R. Witter, M. Fichtner, H.
Hahn, Phys. Chem. Chem. Phys. 17 (2015) 17288.4. R. Chen, S. Ren, M. Yavuz, M. Knapp, E. Maawad, S. Zander, P. Beran, R. Witter,
R. Hempelmann, H. Hahn, in preparation
61

Insights into the mechanism of the pyridine catalyzed electrochemical CO2 reduction
Katrin R. Bickel, Alfonso Crespo-Yapur, Michael Daimer, Simon Filser, Qi Li, Konrad Schönleber, Katharina Krischer; Nonequilibrium Chemical Physics, Technical
University of Munich; James-Franck Str. 1, 85748 Garching; [email protected]
The direct electrochemical reduction of CO2 to hydrocarbons usually requires large overpotentials rendering such processes energetically inefficient. It was recently reported that pyridine can catalyze the reduction of CO2 at palladium and platinumelectrodes at moderate overpotentials [1, 2]. The reduction product is methanol, which is an ideal fuel and predicted to be a key base chemical in the future [3].
It is well known that pyridine strongly adsorbs at platinum electrodes in a wide potential window (see e.g. [4-6]), and we believe that a surface process involving an adsorbed pyridine species plays an important role in the CO2 reduction reaction. Therefore, we have focused our study on the interface between platinum and the aqueous electrolyte containing pyridine. Electrochemical measurements have confirmed the presence of adsorbed pyridine, for example since the hydrogen UPD-peaks in cyclic voltammograms are being suppressed. Attenuated total reflection Fourier transform infrared spectroscopic (ATR-FTIR) experiments have been carried out to further investigate the adsorbed pyridine species. The results on platinum electrodes are compared to the well investigated pyridine adsorption on gold electrodes (see e.g. [7, 8]), where the CO2 reduction does not take place.
1. Seshadri, G., C. Lin, and A.B. Bocarsly, Journal of Electroanalytical Chemistry, 1994. 372(1–2), 145.
2. Barton Cole, E., et al., Journal of the American Chemical Society, 2010. 132(33), 11539.
3. Bertau, M., et al., eds. Methanol: The Basic Chemical and Energy Feedstock of the Future - Asinger's Vision Today. 2014, Springer: Berlin Heidelberg.
4. Krauskopf, E.K., L.M. Rice-Jackson, and A. Wieckowski, Langmuir, 1990. 6(5), 970.5. Schmiemann, U., Z. Jusys, and H. Baltruschat, Electrochimica Acta, 1994. 39(4),
561.6. Cai, W.-B., et al., Journal of the Chemical Society, Faraday Transactions, 1998.
94(20), 3127.7. Hoon-Khosla, M., et al., Electrochimica Acta, 1999. 45(4–5), 611.8. Li, N., et al., Journal of Electroanalytical Chemistry, 2002. 524–525, 43.
62

Imaging ultrafast photo-induced dynamics in semiconductingpolymer films with time-resolved photoemission electron
microscopyAndreas Neff, Katrin Siefermann, Leibniz Institute of Surface Modification, Leipzig,
GermanyOrganic semiconductors have great potential for applications in optoelectronicdevices. By now, it is well established that the performance of devices critically depends on the detailed morphology of the organic semiconducting films [1]. However, a profound understanding of the correlation between morphology and (photo)physical properties is still missing, and this remains a key challenge to overcome technological hurdles in the field.We address this challenge with a combination of a photoemission electron microscope (PEEM) and a femtosecond laser system. With this setup, we image the morphology of films of organic semiconductors, in particular the size and orientation of crystallites, with a lateral resolution of 100 nm. Ultrafast pump-probe experiments allow us to image photo-induced dynamics with a temporal resolution of 150 fs and a lateral resolution of 100 nm. Here, we present results from the ultrafast exciton decay dynamics in P3HT (Poly(3-hexylthiophen-2,5- diyl)) films. We find that these decay dynamics are - as expected but not previously detectable - not the same for all locations on the sample.These results demonstrate the potential of time-resolved PEEM to address key questions with regard to the relationship between nanoscale morphology of organic semiconductors and photo-physical properties.
[1] Y. Diao, Y. Zhou, T. Kurosawa, L. Shaw, C. Wang, S. Park, Y. Guo, J. a. Reinspach, K. Gu, X. Gu, B. C. K. Tee, C. Pang, H. Yan, D. Zhao, M. F. Toney, S. C. B. Mannsfeld, and Z. Bao, Nat. Commun. 6, 7955 (2015).
63

Mechanistic Understanding of Photocatalytic Water Reduction withIron Carbonyl Phosphines by means of in situ Spectroscopy and
Time Dependent - DFT Calculations.Steffen Fischer,a Alastair J. J. Lennox,b Olga S. Bokareva,c Arend Rösel,a Nils
Rockstroh,b Matthias Bauer,d Oliver Kühn,c Matthias Beller,b Ralf Ludwiga
a Institut für Chemie, Universität Rostockb Leibniz-Institut für Katalyse an der Universität Rostock e.V.c Institut für Physik, Universität Rostockd Institut für Chemie, Universität Paderborn
Gärtner et al. reported an efficient water reduction system using an iridium Photosensitizer (PS), simple iron carbonyls together with Phosphines as Water Reduction Catalyst (WRC) and Triethylamine as Sacrificial Reagent (SR).[1] While the system already shows a significant Turnover Number (TON) of 400 for the WRCFe3(CO)12 in the absence of phosphine, it is increased by more than 50 % when phosphines like P(-C6H4-4-CF3)3 are present. In this work a variety of spectroscopic methods were applied to investigate the exact reason for this finding: Stopped-Flow Rapid-Scan FTIR was used to examine quantitatively the preformation reactions of the catalyst on a short time scale, whereas operando FTIR, in situ XAS and ex situNMR were applied in qualitative long-term investigations. Experiments revealed the formation of the dimer [Fe2(CO)6(μ-CO)(μ-PR2)] as active WRC, being more stable than its phosphine deficient analogs and representing hydrogenase mimics.The formation of the same complex was also observed in a noble metal free system with a copper PS and Fe3(CO)12 as WRC.[2] In this case, the bridging PR2 unit was found to originate from the loosly bonded Xantphos ligand on the PS.Taking this findings into account, the WRC [NEt4][Fe2(CO)6(μ-CO)(μ-PR2)] has not only been analyzed and evaluated by electrochemistry and DFT calculations, but was also applied in catalysis along with different PS. Furthermore a convenient in situforming copper-iron based photocatalytic water reduction system had been developed.[3] This leads to optimized reaction conditions and shows the high potential of mechanistic investigations.[1] F. Gärtner, A. Boddien, E. Barsch, K. Fumino, S. Losse, H. Junge, D. Hollmann, A.
Brückner, R. Ludwig, M. Beller, Chem. Eur. J. 2011, 17, 6245 6436.[2] S. Fischer, D. Hollmann, S. Tschierlei, M. Karnahl, N. Rockstroh, E. Barsch, P.
Schwarzbach, S.-P- Luo, H. Junge, M. Beller, S. Lochbrunner, R. Ludwig, A. Brückner, ACS Catal. 2014, 4, 1845 1849.
[3] A. Lennox, S. Fischer, M. Jurrat, S.-P. Luo, N. Rockstroh, H. Junge, R. Ludwig, M. Beller et al., Chem. Eur. J. 2015, accepted.
64

Identification and Exclusion of Intermediates of Photocatalytic CO2 Reduction on TiO2 under Conditions of Highest Purity
Anna Pougin1, Martin Dilla2, and Jennifer Strunk2,1Ruhr-University Bochum, Bochum/Germany,
2Max-Planck-Institute for Chemical Energy Conversion, Mülheim a.d. Ruhr/Germany
It would be the simplest and most desirable way to convert solar energy directly into energy carriers such as methane over semiconductor powders. However, in spite of more than thirty years of intense research, no system has been developed yet withyields sufficiently high for the industrial scale [1]. The slow progress might potentially be attributed to little understanding of the reaction mechanism and the elementary processes, even in case of pure TiO2 [2]. In this contribution, significant new insight into the reaction mechanism of photocatalytic CO2 reduction on TiO2 will be presented. Experiments were performed in a high-purity gas-phase photoreactor with a highly-sensitive GC trace gas analysis, allowing to detect and to quantify C1 to C5
hydrocarbons, CO, H2, O2, CH3OH, and CO2 conversions. The setup and photo-catalyst pretreatment entirely excludes formation of products from impurities [3].Under those conditions of highest purity, CH4 and CO are found as main products of photocatalytic CO2 reduction with a ratio of 4:1. Under identical reaction conditions, CO was not converted at all within a time course of six hours. So, we can clearly rule out CO as a reaction intermediate to CH4 or any other product. To test the reactivity of other previously suggested intermediates, HCOOH, H2CO and CH3OH were added to the photocatalyst, but none of them led to methane formation. All C1
species were exclusively oxidized or degraded. Considering these observations, we strongly support a mechanism involving C-C bond formation and C2 intermediates as suggested previously [4]. We tested our hypothesis by reacting numerous C2
intermediates on the photocatalyst. The reactions of acetic acid and acetaldehyde yielded product distributions closely resembling those in photocatalytic CO2
reduction, thus providing significant evidence in favor of the C2 mechanism [5].
References [1] E.V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal, J. Pérez-Ramírez, Energy Environ. Sci. 6 (2013) 3112-3135.[2] S. N. Habisreutinger, L. Schmidt-Mende, J. K. Stolarczyk, Angew. Chem. Int. Ed. 52 (2013) 7372–7408.[3] B. Mei, A. Pougin, J. Strunk, J. Catal. 306 (2013) 184–189.[4] I. A. Shkrob, T. W. Marin, H. He, P. Zapol, J. Phys. Chem. C 116 (2012) 9450–9460.[5] A. Pougin, M. Dilla, J. Strunk, Phys. Chem. Chem. Phys., SI, submitted.
65

Methanol Formation via CO2 Hydrogenation on Au/ZnO Catalysts –Influence of CO and its Role in the Reaction Mechanism
Y. Hartadi, D. Widmann, R.J. BehmInstitute of Surface Chemistry and Catalysis, Ulm University, 89081 Ulm / Germany
Methanol (MeOH) synthesis via hydrogenation of CO2 with H2 produced by renewable energies represents a promising concept for chemical storage of excess electrical energy from renewable sources.1 By using CO2 from CO2 capture and sequestration, this can contribute also to a diminished overall CO2 emission. Previously we demonstrated that oxide supported Au catalysts, in particular Au/ZnO,have activities for MeOH formation from CO2 and H2 comparable to that of commercial Cu/ZnO/Al2O3 catalysts.2,3 The hydrogenation of CO2 to MeOH is, however, always accompanied by the formation of CO via the undesirable reverse water-gas shift (RWGS) reaction. Thus, selectivity towards MeOH formation is another crucial factor that has to be considered. Here supported Au/ZnO catalysts are even superior compared to commercial Cu catalysts.2,3 At 240 °C and 50 bar, for example, it is about 70% on Au/ZnO compared to 42% on Cu/ZnO/Al2O3.3
Since there will nevertheless always be significant amounts of CO present in the reaction atmosphere, we also studied the influence of CO on the MeOH formation on Au/ZnO catalysts. Here a detailed understanding is essential in order to minimize detrimental effects of CO on the main reaction. By adding various amounts of CO to the CO2-H2 reaction mixture, in combination with isotope labelling techniques (13CO2), we will provide evidence that the presence of up to 1% CO in the reaction atmosphere has almost no influence on the overall MeOH formation under present reaction conditions (5-50 bar, 240 °C). Besides, also insights into the reaction pathway of the CO2 and CO hydrogenation can be obtained from these studies. It will be demonstrated i) that CO2 is the preferred carbon source for MeOH, similar to previous proposals for Cu catalysts,4 and ii) that CO2 is directly hydrogenated to MeOH on Au/ZnO catalysts, and not reduced to CO in a first step (via the RWGS reaction), which is subsequently hydrogenated to MeOH CO hydrogenation.
References[1] G. Olah, Angew.Chem.,Int.Ed. 52 (2013), 104.[2] Y. Hartadi, D. Widmann, R.J. Behm, ChemSusChem 8 (2015) 456.[3] Y. Hartadi, D. Widmann, R.J. Behm, J Catal. 333, (2016), 238.[4] Y. Yang, C.A. Mims, D.H. Mei, C.H.F. Peden, C.T. Campbell, J.Catal. 298 (2013) 10.
66

Dehydrogenation of dodecahydro-N-Ethylcarbazol on Pt(111)C. Papp 1, C. Gleichweit1, M. Amende1, S. Schernich1, W. Zhao1, M. P. A. Lorenz1,
O. Höfert1, N. Brückner2, P. Wasserscheid2, J. Libuda1, H.-P. Steinrück1
(1) Lehrstuhl für Physikalische Chemie II, Friedrich-Alexander Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen, Germany
(2) Lehrstuhl für Chemische Reaktionstechnik, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen (Germany)
e-mail: [email protected]
The dehydrogenation of the liquid organic hydrogen carrier dodecahydro-N-ethylcarbazole (H12-NEC) on Pt(111) was studied by high-resolution X-ray photoelectron spectroscopy in order to elucidate its dehydrogenation reaction to N-Ethylcarbazol (NEC). The pair H12-NEC/NEC is a potential candidate for chemical hydrogen storage in mobile applications: The material is a high boiling organic molecule that can be reversibly hydrogenated and dehydrogenated in catalytic processes.
H12-NEC was adsorbed by physical vapor deposition, and subsequently the reaction was monitored during heating. Although the molecules are quite complex, identifiedthe reaction steps after analysis of the C 1s and N 1s data. During adsorption at low temperatures the development of monolayer and multilayer was observed. When heating the sample continuously, the multilayer desorbs up to temperatures of about 285 K while the signature of the monolayer increases due to decreased damping. Subsequently, the dehydrogenation of the H12-NEC to NEC follows successively in the range of 280 to 500 K. Above 430 K, the dealkylation reaction of the NEC to carbazole is observed. Upon further heating to 1000 K decomposition to adsorbed carbon takes place.
We acknowledge the Cluster of Excellence “Engineering of Advanced Materials” and the Helmholtz-Zentrum Berlin for the allocation of synchrotron beamtime.
67

Mechanism of the aqueous-phase reforming of methanol –Understanding the role of the base
Elisabetta Alberico, Istituto di Chimica Biomolecolare, CNR, Sassari/Italy; Alastair Lennox; Lydia Neumann; Haijun Jaio; Wolfgang Baumann; Hans-Joachim Drexler;
Martin Nielson; Henrik Junge; Matthias Beller; Leibniz Institute for Catalysis, Rostock/Germany
A promising concept for addressing the challenge of large-scale energy storage is the use of methanol as a secondary energy carrier. For the low-temperature reforming of aqueous methanol under basic conditions employing the ruthenium-based PNP-pincer complex (1a) an outer-sphere mechanism was tentatively proposed.[1] The reason for the dramatic increase in activity brought about by the high base content of 8 M has been unclear, as the current mechanistic proposal suggests that only catalytic quantities are required.
In order to elucidate the complete reaction pathway for the methanol reforming, a range of experimental, spectroscopic and theoretical tools were employed in our recent work. In addition to labelling experiments, the reaction was examined spectroscopically under the catalytic conditions, catalytic intermediates were prepared and their reactivity investigated, and the influence of ligand backbone manipulation was assessed. To support our observations, comprehensive DFT calculations were made. In conclusion, an overall mechanistic picture for methanol reforming is proposed using the bifunctional pincer complex (1a) and the corresponding N-methylated complex (Me-1a). It was found that a high base concentration significantly increases the activity by providing a lower energy pathway through the catalytic cycle. Hereby the formation of deprotonated intermediates and the sequestration of by-products are both essential factors.[2]
[1] M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge, S. Gladiali, M. Beller, Nature 2013,495, 85-89.
[2] E. Alberico, A. Lennox, L. Neumann, W. Baumann, H. Drexler, M. Nielson, H. Junge, H. Jaio, M. Beller, submitted.
68

Complexes of Chitosan and Alkylethoxycarboxylates: pH-Responsive Systems of Versatile Structure and Properties
Leonardo Chiappisi, Miriam Simon, Michael Gradzielski, Stranski-Laboratorium für Physikalische Chemie, Technische Universität Berlin, Berlin, Germany
Ionic co-assembly of polyelectrolytes and surfactants leads to a rich variety of structures, depending on the molecular architecture of the components. Parameters such as persistence length and location of the charges have a strong effect on the mixtures of polyelectrolytes and oppositely charged surfactants [1]. The co-assembly of positively charged biopolymer chitosan with weakly anionic alkyl ethoxycarboxylate yields a fully biocompatible system [2-3]. These complex mixtures are very interesting due to their high pH-responsiveness arising from both components. A detailed investigation of the resulting structures as a function of pH allows to correlate it with the phase behaviour and with the constituting molecules(see Figure 1), e.g., rigidity of the polymer or packing parameter of the surfactant.
The structural evolution of these complexes in aqueous solutions as a function of the mixing ratio Z of chitosan/surfactant and pH has been determined by a detailed SANS study combined with light scattering experiments. SANS shows that, depending on Z or pH one may observe a weak attachment of micelles to the chitosan network, a linear arrangement of micelles facilitated by the rather stiff chitosan, or densely packed micelles “glued together” by the chitosan. For bilayerforming surfactants also the formation of multilamellar vesicles can be induced where
Fig. 1: Self-assembled structures formed in chitosan / alkyl ethoxycarboxylate mixtures.
69

the degree of multilamellarity is controlled by the addition of oppositely charged chitosan. Such systems allow for the formulation of highly responsive systems, which can be employed for a large spectrum of applications. This was been demonstrated by some examples in which chitosan/alkylethoxycarboxylate mixtures are employed for the selective removal or release of more or less hydrophobic target molecules,showing that they can be separated effectively. In addition, it is shown to be able to induce pH-dependent sequestration of multivalent cations [4]. In summary, these are highly biocompatible systems with a large tuneable structural versatility, which can easily be employed for a multitude of interesting applications in aqueous dispersion.
References:1. Chiappisi, L.; Hoffmann, I.; Gradzielski, M. Soft Matter 2013, 9, 3896–3909. 2. Chiappisi, L.; Prévost, S.; Grillo, I.; Gradzielski, M. 2014, 30, 1778–1787.3. Chiappisi, L.; Prévost, S.; Grillo, I.; Gradzielski, M. Langmuir 2014, 30, 10608–10616. 4. Chiappisi, L.; Simon, M.; Gradzielski, M. ACS Appl. Mater. Interfaces 2015, 7, 6139-6145.
70

Benefits of using hydrogel microgels in colloidal lithographyMarkus Weilera and Claudia Pacholskia, b
aMax Planck Institute for Intelligent Systems, Stuttgart, GermanybPresent address: University of Potsdam, Potsdam, Germany
Colloidal lithography is an attractive method for creating nanostructures. Based on its cost-efficiency, simplicity, rapidity and applicability to structuring large areas colloidal lithography has extensively been investigated for years. Here, most often hard spheres such as polystyrene or silica colloids have been employed for generating highly ordered 2D arrays via self-assembly (reviewed in e.g. [1]). These arrays were mainly used as masks in subsequent etching/deposition processes.Only recently hydrogel microgels were exploited in colloidal lithography. Their special properties - abrupt volume changes caused by certain stimuli, such as temperature, ionic strength, pH, etc. - facilitate the formation of 2D nanostructures with remarkable properties (reviewed in e.g. [2]). In this contribution, we show that stimuli-responsive hydrogel microspheres are ideal candidates for colloidal lithography. By using colloids composed of poly-N-isopropylacrylamide loosely packed, hexagonally ordered arrays with exceptional long range order were prepared. Moreover, the lattice constant of the formed array and the diameter of the microspheres were controlled independently and the hydrogel matrix was successfully used as nano-reactor for generating nanoparticles.
[1] N. Vogel, C. K. Weiss, K. Landfester, Soft Matter, 2012, 8, 4044[2] C. Pacholski, Zeitschrift für Physikalische Chemie, 2015, 229 (1-2), 283
71

Atomistic Simulations of the Formation of Perylene-based Supramolecular Complexes in Aqueous Solution
Jörg Baz and Niels Hansen, Institute of Thermodynamics and Thermal Process Engineering, University of Stuttgart, Stuttgart, Germany
The design and synthesis of complex functional dye-based materials from components held together by intermolecular noncovalent forces is of increasing importance for many high technology applications. As a result there is growing interest in understanding dye-dye interactions and how these interactions impact the properties of nano- or bulk solid state materials. Such understanding can be derived from investigations on supramolecular dye assemblies that constitute the intermediate state of matter between monomeric dyes and supramolecular materials. During the last decade, perylene bisimide dyes (PBI) emerged as an archetype class of colorants for the elucidation of the transition from monomeric to bulk materials via the supramolecular state [1]. The self-assembly of amphiphilic molecules into complex structures is determined by size and shape of the monomeric unit, system composition and thermodynamic boundary conditions [2]. Given this diversity of factors for self-assembly, a molecular-level understanding of the various driving forces is a promising approach for the rational bottom-up material design. In the present work the influence of solvent composition on the thermodynamics of self-assembly of PBI is investigated by molecular dynamics free-energy calculations in aqueous solution.
References[1] F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat, D. Schmidt, Chem. Rev., DOI: 10.1021/acs.chemrev.5b00188.[2] P. Calandra, D. Caschera, V. T. Liveri, D. Lombardo, Colloids and Surfaces A: Physicochem. Eng. Aspects, 2015, 484: 164-183.
72

Interface-Enforced Complexation between Copolymer BlocksA. A. Steinschulte, F. A. Plamper,
Institute of Physical Chemistry, RWTH Aachen University, Germany
Amphiphilic copolymers are well known and understood for their self-assembly in bulk solution. At the same time, many amphiphilic block copolymers exhibit surface activity, i.e. the polymers can be anchored at water-air or water-oil interfaces. One important tool to investigate the properties of interfaces is the Langmuir-trough experiment.
Scheme 1: Interface as “catalyst” for complex formation between the two weakly interacting polymers in PPO-b-PDMAEMA; complexation is aided by the proximity of PPO and PDMAEMA within interfacial pancakes (right) while hardly any complexation is seen for PPO-b-PDMAEMA in bulk water (left).
Here, the compression isotherms of binary diblock copolymers and corresponding ternary miktoarm stars are compared at oil-water interfaces. All polymers contain oil-soluble poly(propylene oxide) PPO, water-soluble poly(dimethylaminoethyl methacrylate) PDMAEMA and/or poly(ethylene oxide) PEO. The features of their Langmuir compression isotherms are well related to the ones of the corresponding homopolymers. Unexpectedly, of all PPO containing copolymers only PEO-b-PPO is an effective amphiphile. In contrast, the compression isotherms show a complexation of PPO and PDMAEMA for PPO-b-PDMAEMA and the star, reducing their overall amphiphilicity. In bulk water, such complex was clearly observed for the star but not for PPO-b-PDMAEMA.1 However at the interface, the blocks of PPO-b-PDMAEMA are forced to form a complex already in the dilute pancake conformation due to their enhanced proximity in such a pseudo 2D-environment (see Scheme 1). Hence, the interface acts like a “catalyst” for complexation.2
[1] A. Steinschulte, B. Schulte, S. Rütten, T. Eckert, J. Okuda, M. Möller, S. Schneider, O. V. Borisov, F. A. Plamper, PCCP 2014, 16, 4917-4932[2] A. Steinschulte, W. Xu, F. Draber, P. Hebbeker, A. Jung, D. Bogdanovski, S. Schneider, V. V. Tsukruk, F. A. Plamper, Soft Matter 2015, 11, 3559-3565.
73

Monodisperse, ordered polystyrene foams and their mechanical properties
Jonas Elsing, Universität Stuttgart, Stuttgart, Germany; Michael Gilchrist, University College Dublin, Dublin, Ireland; Wiebke Drenckhan, Université Paris Sud, Paris,
France; Cosima Stubenrauch, Universität Stuttgart, Stuttgart, Germany
Polymer foams have a wide range of applications. They may serve as impact protection, insulation or packaging materials [1]. One of the most frequently used polymer foams is expanded polystyrene. The high complexity of traditional manufacturing processes makes it difficult to control the structure and porosity of the material. Here we show a route for the synthesis of monodisperse ordered polystyrene foams via a microfluidic device. Pure styrene cannot be foamed, even in the presence of surfactants. This problem is solved by foaming a styrene-in-water emulsion with a high styrene content of 64.5 vol% [2] where the surfactant stabilizes both the styrene droplets as well as the gas bubbles. Foaming is carried out by means of a microfluidic device with flow focusing architecture which allows the generation of a monodisperse foam. The foamed emulsion remains stable during the polymerization and the structure of the liquid foam is retained. We here show that this technique allows controlling the pore size of the polymer foam by adjusting the bubble size in the liquid foam. Moreover, we show first results on the mechanical behavior of monodisperse ordered polystyrene foams under compression and shear.
Figure: Concept of foaming an emulsion by means of a flow-focussing microfluidic device (left). Obtained liquid foam (middle), and resulting polymer foam (right) [3].
[1] Mills, N., Polymer Foams Handbook, 2007.[2] Schüler et al., Angew. Chem. Int. Ed., 51, 2213-2217, 2012.[3] Quell, A., Elsing, J., Drenckhan, W., Stubenrauch, C., Adv. Eng. Mat., 17, 604-
609, 2015.
74

Hindered nematic alignment of hematite spindles in viscoelasticmatrices
Annemarie Nack, Julian Seifert, Christopher Passow, Joachim WagnerUniversität Rostock, Institut für Chemie, Albert-Einstein-Straße 3a, 18051 Rostock
Due to a magnetic moment perpendicular to their long axis, hematite particles insuspension exhibit a field induced isotropic-nematic phase transition. Theorientational distribution functions of, both aqueous suspensions and particles indifferent hydrogel matrices are investigated by means of Small Angle X-rayScattering in dependence of an external magnetic field.The viscoelasticity of the hydrogel matrices hinders the field-induced alignment of thespindle-shaped particles as visible by the comparison of nematic order parametersdetermined for aqueous suspensions and hydrogel composites at the same numberdensity of magnetic particles. The stronger the elastic modulus, tuned by thecrosslinking density of the polymer, is, the more hindered is the field-inducedalignment.The field-induced change of the mesostructure influences the macroscopic,rheological properties of composites as investigated by means of oscillatory shearexperiments in dependence on an external magnetic field. With increasing fluxdensity, due to an orientational arrest of the particles, the elastic modulus rises, too.The magnetic particles act as additional field-dependent crosslinkers in thecomposite.
AcknowledgementsWe acknowledge the ESRF for providing beamtime at the beamlines ID02 and ID10as well as the DFG for financial support within the priority program SPP1681.
75

Virial coefficients of anisotropic, hard solids of revolution: The influence of the geometry
Elisabeth Herold, Robert Hellmann, Joachim WagnerInstitut für Chemie, Universität Rostock, Albert-Einstein-Straße 3a, 18051 Rostock
The phase behaviour of dense suspensions of hard solids is governed by their excluded volume. Many-particle interactions can be expanded in the virial series,which is not only important for the equation of state but also for the determination of the phase diagrams employing classical density functional theory.As a result of the orientation as an additional degree of freedom for anisotropic particles, the complexity of their phase diagrams is enhanced compared to spherical ones. Already Onsager predicted an isotropic-nematic phase transition for prolate particles approximated as hard cylinders. Due to the existence of nematic and smectic structures, such systems are mesoscale analogs of liquid crystals and interesting model systems for investigating the self-organization of soft matter.The second and third virial coefficients for anisotropic, convex bodies of revolution are calculated for different aspect ratios and particle geometries. The second virial coefficient is calculated analytically from the geometric quantities volume, surface, and, mean radius of curvature employing the Isihara-Hadwiger relation. For double cones, spindles, uniaxial ellipsoids, spherocylinders and cylinders, analytical expressions for the excluded volume and second virial coefficients are derived. Third virial coefficients of spindles and ellipsoids are calculated in dependence on the aspect ratio by means of the Mayer-sampling Monte Carlo approach. The virial coefficients depend beyond the particles' aspect ratios on their detailed shape.
76

New insights into nematic nanocomposites by dielectric spectroscopy: Conductivity, polarization and relaxation
Mathias Bourg and Martin Urbanski, University of Luxembourg, LuxembourgComposites based on liquid crystals doped with nanoparticles have emerged into a topical field of research due to promising opportunities for both basic science and innovative display applications [1]. Our recent results show that doping nematic liquid crystals with organically functionalized gold nanoparticles significantly increases the conductivity of the nematic host by several orders of magnitude [2].
We found that, consequently, the electro-optic performance of such nanocomposites suffers from ion drift and interfacial polarization, phenomena which have been rarely associated with nematic nanocomposites so far. We model the electrode polarization process using a sophisticated theoretical approach and succeed to reproduce the experimentally observed dielectric response with high accuracy. Based on this model, we derive new meaningful explanations for frequently observed doping-induced phenomena like changes of switching voltages, decrease of dielectric anisotropy or slower switching times [3, 4].
Beyond probing the electro-optical performance of nanocomposites, we utilizedielectric spectroscopy to get deeper insights into the dopant-host interactions. The nematic potential, responsible for the orientational order of the nematic state, hindersthe molecular rotation and therefore affects the kHz relaxation time accessible viadielectric measurements. We investigate the impact of differently shaped nanoparticles (spheres, rods) on this low-frequency dispersion of permittivity to see how doping influences the liquid crystalline state itself.
AcknowledgmentThe present project is supported by the Fonds National de la RechercheLuxembourg.
References[1] H. Qi and T. Hegmann, J. Mater. Chem. 16, 4197-4205 (2006).[2] M. Urbanski and J. Lagerwall, Talk 21-1, IMID 2015 conference proceedings.[3] M. Urbanski et al., ChemPhysChem. 15, 7, 1395-1404 (2014).[4] M. Urbanski, Liq. Cryst. Today 24, 4, 102-115 (2015).
77

Unraveling the nature of ionic conductivity in hybridorganic-inorganic halide perovskites
Alessandro Senocrate‡,†, Tae-Youl Yang‡, Giuliano Gregori‡, Norman Pellet‡,†,Michael Grätzel‡,† and Joachim Maier‡
‡Max-Planck-Institut für Festkörperforschung, Stuttgart, Germany†École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland
In recent years, hybrid organic-inorganic halide perovskites have attracted much attention for their potential use as light absorbers in solar cells[1]. Typically, in these compounds, an organic A cation (e.g. CH3NH3
+, HC(NH2)2+) and an inorganic B
cation (e.g. Pb2+, Sn2+) bond to a halide anion (Cl-, Br-, I-) to form a perovskite lattice. These materials present many outstanding properties, such as high absorptioncoefficients, panchromaticity, long electron-hole recombination lengths and high charge carrier mobilities[1]–[3]. In less than two years, efficiency of hybrid-perovskite-containing devices has rapidly increased, already exceeding 20%[4]. Nevertheless,there is still uncertainty about the physico-chemical properties underlying such anoutstanding performance. Furthermore, anomalous behaviors have been reported for these materials, such as high dielectric constants at low AC frequencies or photocurrent hysteresis of solar cell devices during operation[5]. In this contribution[6]
we discuss the relevance of mixed conductivity in CH3NH3PbI3 and, on the basis of DC galvanostatic polarization measurements, we show that the ionic conductivity can substantially exceed the electronic conductivity. The ionic transport can explain the above mentioned anomalies. Moreover, the nature of the moving ion was assessed (Iodine ions), and the conductivity response to different atmospheres (I2,O2) was measured. As a natural follow-up to better understand the defect chemistry of such materials, acceptor doping has been successfully applied.
Bibliography[1] S. D. Stranks, G. E. Eperon, G. Grancini et al., Science, 342, 6156, 341–344, 2013.[2] C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis Inorg. Chem., 52, 15, 9019–9038, 2013.[3] D. Shi, V. Adinolfi, R. Comin et al., Science, 347, 6221, 519–522, 2015.[4] NREL National Center for Photovoltaics, Research cell efficiency records, www.nrel.gov/ncpv/.[5] Z. Xiao, Y. Yuan, Y. Shao et al., Nat Mater, 14, 2, 193–198, 2015.[6] T.-Y. Yang, G. Gregori, N. Pellet et al., Angew. Chemie Int. Ed., 54, 27, 7905–7910, 2015.
78

Codoped 2D Conjugated Metal-Organic Surface for Efficient
Electrochemical Hydrogen Production: from Molecules to Solid Catalysts
Renhao Dong1, Diana Tranca1, Ionut Tranca2, Naisa Chandrasekhar1, Haiwei Liang1, Jian Zhang1,
Shaohua Liu1, Gotthard Seifert1, Zhikun Zheng1, Xinliang Feng1
1Dresden University of Technology, Department of Teoretical Chemistry and Molecular Functional Materials, Dresden,
Germany2Laboratory of Inorganic Materials Chemistry, Schuit Institute of Catalysis, Eindhoven University of Technology,
Eindhoven, The Netherlands
Two dimensional materials are considered interesting due to their possible applications as
semiconductors. Efforts have been increased in searching for 2D-materials which have a non-zero
band gap. In this sense, two different classes of materials have been identified : layered metal
chalcogenides (e.g. MoS2, WSe2) and 2D covalent organic frameworks (COFs). Our work consists
of experimentally and theoretically investigations of new materials for hydrogen evolution reaction
(HER).
Material Design :
N, S-codoped two-dimensional conjugated metal-organic flakes, nanosheets and thin films were
flexibly constructed via hydrothermal synthesis, interfacial self-assembly and Langmuir Blodgett
strategy, respectively. The highly efficient electrochemical hydrogen production was achieved
trough the immobilization of homogeneous molecular functional catalysts into heterogeneous 2D
conjugated solid surface. The HER performance of Co/N/S codoped 2D metal-organic hybrids is
better than both of Co/N doped and Co/S-doped systems. Regarding the N, S-codoped metal-
organic thin films, the different metal centers play key role. The order of HER activity is
Co>Fe>>Ni.
79

Removal of the SiO2 Native Layer from Silicon Electrodes -Scanning Electrochemical Microscopy and X-ray Photoelectron
SpectroscopyEduardo dos Santos Sardinha, Heinz Bülter, Gunther Wittstock
Carl von Ossietzky Universität Oldenburg, Oldenburg
The development of lithium-ion batteries involves many aspects, one of them is the use of alternative materials for the negative electrodes (typically made of graphite).Silicon is one of the promising materials that has advantages due to its wide abundance, low voltage and high theoretical gravimetric capacity of 3579 mAh g-1.1
The formation of a native SiO2 layer on the surface of silicon electrodes can passivate the desired electrochemical properties. The layer can be removed/damaged locally by gentle abrasion with a microelectrode probe of a scanning electrochemical microscope (SECM) produced form glass or by immersion in hydrofluoric acid (HF). The mechanical abrasion in the presence of the electrolyte did not lead to appreciable topographic changes (as detected by scanning force microscopy) despite quite striking changes in the passivating properties.
X-ray photoelectron (XP) spectroscopy and SECM allowed to follow the structural and functional changes introduced into the passive layer.2 SECM investigations have the additional advantage that they provide true in-situ information and have been used successfully on Li metal and lithiated graphite electrodes.3,4 By comparing a sample without any treatment, an etched sample and an abraded sample provided the information about the thickness and lateral heterogeneity of the passive layer and the damage to the passive layer.
The study will be expanded by study similar electrodes after cycling in Li+-containing electrolytes.
___________________________________________________________________1 M. N. Obrovac, L. Christensen. Electrochem. Solid State Lett., 7, A93 (2004).2 H. Bülter, M. Sternad, E. d. S. Sardinha, M. Wilkening, Gunther Wittstock. J. Electrochem. Soc.,Submitted Manuscript (2015). 3 H. Bülter, F. Peters, J. Schwenzel, G. Wittstock. Angew. Chem. Int. Ed., 53, 10531-10535 (2014).4 H. Bülter, F. Peters, J. Schwenzel, G. Wittstock. J. Electrochem. Soc., 162, A7024-A7036 (2015).
80

Mass Storage at Heterojunctions
Chia-Chin Chen, Robert Usiskin, Lijun Fu, and Joachim MaierMax Planck Institute for Solid State Research, Stuttgart, Germany
Anomalies of ion transports at solid-solid interfaces have been extensively studied in the field of nano-ionics [1]. In this contribution, we concentrate on mass storageanomalies and present a general thermodynamic model for interfacial non-stoichiometry, including both excess [2] and deficiency. Different from non-stoichiometry in the bulk, interfacial storage can occur in a heterogeneous way: Although neither of the constituent phases may be able to tolerate an excess or a deficiency of a component M, M (= Li, Ag, H) can be dissociatively stored at their junctions. We calculate the equilibrium voltage-charge curves for various phase combinations and also show that the related chemical diffusion coefficients are unprecedentedly fast. The thermodynamic and kinetic predictions are found to be consistent with the experiments.
References:[1] J. Maier, Nature Mater. 4, 805-815 (2005).[2] L.J. Fu, C.-C. Chen, D. Samuelis, J. Maier, Phys. Rev. Lett. 112, 208301 (2014)
81

Gas phase studies of luminescent lanthanoid complexes by trapped ion laser induced fluorescence (TLIF)
Eugen Waldt1 , Jean-Francois Greisch1,Bernhard Schäfer1, Mario Ruben1, Manfred M. Kappes1,2, Detlef Schooss1,2
1) Institut für Nanotechnologie, Karlsruher Institut für Technologie (KIT), Postfach 3640, 76021 Karlsruhe, Germany
2) Institut für Physikalische Chemie, Karlsruher Institut für Technologie (KIT), Kaiserstraße 12, 76128, Karlsruhe, Germany
E-mail: [email protected]
Luminescence spectroscopy in condensed phase is a well-established method to investigate the optical properties of different kind of chromophores. However, photophysical properties of chromophores can be strongly affected by their chemical environment. Additionally, interpretation of luminescence spectra is sometimes difficult due to the unknown stoichiometry of the emitting species. Here we present a gas phase experiment for luminescence spectroscopy of trapped ions, which combines laser induced fluorescence and mass spectrometry. The advantage of mass selective gas phase measurements is that the composition of the investigated chromophore is very well defined. With this technique we were able to acquire dispersed gas phase luminescence spectra and the intrinsic lifetime of excited states of organic dyes like Rhodamine 6G+ [1] and lanthanoid complexes [2].
Here we present our studies of ligand (1-Benzoyl-3,3,3-trifluoroacetone) sensitized Ln3+ complexes of Tb3+, Sm3+ and Pr3+. We discuss the role of the lanthanide nuclearity and the ligand shell on luminescence spectra, lifetimes and energy transfer processes. In combination with Yb3+ the presented Ln3+ complexes are potential candidates for a molecular near-infrared-downconverter. Mixed complexes are currently under investigation.
References[1] M. Kordel et al., J. Phys. Chem. A. 114 (2010) 5509-5514.[2] J.F. Greisch, et al., J. Phys. Chem. Lett. 5 (2014) 1727-1731.
82

Indirect Isotope Effects Need to be Taken into Account for theAssignment of Vibrational Spectra of Complex Molecules
Tatiana Domratcheva, Elisabeth Hartmann, Ilme Schlichting, Max Planck Institute for Medical Research, Heidelberg, Germany
Constanze Sommer, Marina Dietz, Tilman Kottke,Bielefeld University, Bielefeld, Germany
Specific isotope labeling is considered to be the most reliable method for an experimental assignment of vibrational bands to specific functional groups in complex molecules. Here, we present evidence from infrared difference spectra of labeled photoreceptors and quantum chemical calculations that the spectra cannot be interpreted satisfactorily without including indirect effects.First, all amino acids of a cryptochrome photoreceptor were globally labeled with 13C, whereas the chromophore flavin was non-covalently bound at natural abundance.Thereby, contributions from structural changes of the chromophore to the light-induced difference spectra should be strictly separated from those of the protein moiety, complementing previous assignments [1,2]. However, we observed some isotope shift also of bands that have been assigned unambiguously to the unlabeled chromophore.Second, all glutamines of a BLUF (blue light sensor using flavin) domain were labeled specifically with 15N. Time-resolved infrared double difference spectra and extensive quantum chemical calculations revealed the formation of a glutamine tautomer and an indirect isotope effect on the flavin C4=O stretch that originates from a coupling via hydrogen bonds. Therefore, additional information is provided by the spreading of the isotope-induced shifts, which is crucial to identify the molecular structure and the strength of interaction of the hydrogen-bonding network.
[1] D. Immeln, R. Pokorny, E. Herman, J. Moldt, A. Batschauer, and T. Kottke (2010)J. Phys. Chem. B 114, 17155-17161.[2] C. Thöing, S. Oldemeyer, and T. Kottke (2015) J. Am. Chem. Soc. 137, 5990-5999.
83

The Role of HR-MC-ICP Mass Spectrometry in the Redefinition of the SI Unit Kilogram
Axel Pramann, Olaf Rienitz Physikalisch-Technische Bundesanstalt (PTB), 38116 Braunschweig/Germany
In an interdiscipliniary approach of Physics, Chemistry and Metrology, the redefinition of the SI units is awaited for 2018. The presented work is focused on the determination of the Avogadro constant NA with lowest associated uncertainty urel
aiming at the redefinition of the SI unit of the mass, the kilogram. In the XRCD (X-ray-crystal-density)-approach, known as the “AVOGADRO-Project”, atoms are “counted” in a macroscopic sphere of high purity silicon, highly enriched in the 28Si isotope (amount-of-substance fraction x(28Si) 1 To further strongly reduce urel(NA), new silicon crystal material has been produced in a cooperation between PTB and institutes in Russia (with (x(28Si) ranging between 0.999985 mol/mol and 0.999995 mol/mol). The general mass spectrometric (MS) principles andbasic theory, the improvements and latest results of the MS determination of the isotopic composition of that material is presented with a complete measurement uncertainty budget.2,3 Recently, the new material enabled the measurment of the molar mass M(Si) with urel(M(Si)) < 1 x 10-9 which is a reduction in uncertainty by more than two orders of magnitude in 10 years and denotes a milestone of metrology in chemistry.
1Y. Azuma et al., Metrologia 52 (2015) 360.2O. Rienitz, A. Pramann, D. Schiel, Int. J. Mass Spectrom. 289 (2010) 47.3A. Pramann, K.-S. Lee, J. Noordmann, O. Rienitz, Metrologia 52 (2015) 800.
84

New spectroscopic in situ tools for monitoring electrocatalysts during water oxidation (WO)
D. Hollmann,a K. Grabow, a J. Rabeah, a M. Polyakov, a A.-E. Surkus, a U. Bentrup, a
S. Klink, b W. Schuhmann, b A. Brückner, a
aLeibniz-Institut für Katalyse e. V. Rostock, Germany; bAnalytical Chemistry and Center for Electrochemical Sciences, Ruhr-University Bochum, Germany
Rational development of efficient WO catalysts, driving one step of overall electrocatalytic water splitting, requires detailed information on structure-reactivity relationships, preferentially obtained under true reaction conditions. However, spectroscopic in situ studies in aqueous electrolytes under applied potentials are very challenging. That is the reason why suitable methods able to monitor working catalysts are hardly available so far. In this work, we present new setups for spectroelectrocatalytic in situ studies of working WO catalysts by EPR and Raman spectroscopy (Figure 1). The cells (directly implemented in the spectrometers) are equipped with a carbon working electrode covered with the catalyst layer, a counter (Pt) and a reference electrode (Ag/AgCl) and operate at potentials between 0 and 2V(vs RHE) and in the presence of aqueous KOH.
A) B)
Figure 1: Raman A) and EPR B) electrochemical in situ cells.
First in situ Raman studies with mixed oxide cobalt and/or nickel containing WO catalysts revealed the formation of NiOOH under reaction conditions in active catalysts while segregation of Co3O4 and/or spinel phases was observed for poorly active catalysts. Electron transfer processes as well as changes of the magnetic behavior (ferromagnetic/antiferromagnetic), reflecting reaction-induced structural changes of working catalysts, could be detected by in situ EPR spectroscopy.Currently, comprehensive investigations in dependence on catalyst modification with second metal components are under way to derive more detailed insights in the mechanism of water oxidation.
85

The CAT-ACT Beamline at ANKA: A new high energy X-ray spectroscopy facility for CATalysis and ACTinide research
A. Zimina, H. Lichtenberg, T. Pruessmann, J.-D. Grunwaldt, Institute of Catalysis Research and Technology, Institute for Chemical Technology and Polymer
Chemistry, Karlsruhe Institute of Technology, Karlsruhe, GermanyK. Dardenne, J. Rothe, T. Vitova, Institute for Nuclear Waste Disposal, Karlsruhe
Institute of Technology, Karlsruhe, GermanyE. Huttel, S. Mangold, R. Steininger, ANKA Synchrotron Radiation Source, Karlsruhe
Institute of Technology, Karlsruhe, Germany
To strengthen the capabilities for radionuclide and catalysis research at KIT a hard X-ray beamline for CATalysis and ACTinide research at the ANKA synchrotronradiation source has been recently built up and will be operated together with ANKA staff by the two KIT communities having essential expertise in these two application fields. The beamline design with a superconducting wiggler source places emphasis on X-ray spectroscopic techniques, including ʻflux hungryʼ photon-in / photon-out techniques and special infrastructure for catalysis and radionuclide research,including correlative spectroscopy. The CATalysis experimental station is operated by KIT-ITCP/IKFT and is presently equipped with a special infrastructure for in-situ and in-operando catalytic studies, including ventilation systems, supply of reactive gases, specially designed sample environments for catalytic experiments under realistic reaction conditions (e.g. high pressure, high temperature or liquid phase) and set-ups for combining X-ray absorption spectroscopy with other characterization techniques (e.g. infrared spectroscopy and X-ray diffraction). These set-ups will allow to analyse local molecular environments and long range order structures of catalytically active materials and to study simultaneously the adsorbed reaction species. The first commission phase has been successful , and first X-ray absorption spectra of realistic samples relevant for both catalysis and actinide research will be presented.
86

The electrode potential – a challenging variable in measurementsHans-Joachim Kohnke, Gaskatel GmbH, Kassel/Deutschland; Dr. Hans-Joachim
KohnkeElectrochemical potentials of electrode reactions are given in literature compared to the standard hydrogen electrode (SHE). But that value is not practicable. Even the IUPAC declares that the values towards SHE are defined for a hypothetical cell.Therefore in practice different potential measurements established with time like:
Corrosion Anodic Index (Gold pseudo reference)Measurement in neutral, chloric solutions (Calomel or Silver Chloride Electrode)
Measurement in neutral sulfate solutions (Copper or Mercury Sulfate Electrode)
Even though the AgCl and Hg2Cl2 potentials are well established, the accuracy of a measurement in general is often around +/- 15mV. But also aberrations of some hundred Millivolts are possible. This is due to temperature effects, concentrations effects, diffusion potentials and the interface and electrokinetic effects at the interface.It is a pitty not to be able to detect potentials with a higher precision. Otherwise one would easily differentiate between for example different crystal structure (ZnO amorphous or crystalin) or precipitation in metallic alloys (Phosphor and Oxygen in bronze).
We would like to present that the so called reversible hydrogen electrode (RHE) is not affected by diffusion or electrokinetic effects. The difference between the theoretical SHE and the practical RHE is due to Nernst coefficients of the different ion concentrations. And that is usually the answer of the most electrochemical questions.
Like the UPAC we would strongly point out to use a universal hydrogen electrode. We would like to present the benefits and the challenges of that concept in detail: For water based solutions and for other protic solutions as well.
87

La2NiO4+ as Anode Material for High TemperatureSteam Electrolysis
Andreas Eggera, Nina Schrödla, Werner Sittea, Christian Gspanb, Ferdinand Hoferb
a Montanuniversitaet Leoben, Chair of Physical Chemistry, Austriab Institute for Electron Microscopy and Nanoanalysis (FELMI), Graz University of
Technology & Graz Centre for Electron Microscopy (ZFE), Austria
Increasing supply of electrical energy from intermittent power sources such as wind or photovoltaics requires large-scale storage capacities to bridge the gap between energy production and consumption. Hydro pumping provides an effective method of storing excess power but is not everywhere available due to regional differences in topography. A promising alternative is the conversion of electrical to chemical energy by high-temperature steam electrolysis (HTSE), which offers a way for highly efficient water splitting, especially if thermal coupling to external heat sources is available. HTSE technology is based on solid oxide electrolyzer cells (SOECs) which are operated at temperatures between 600 and 1000°C. In this work the promising ceramic compound La2NiO is characterized as anode material for high temperature electrolyzer cells. Special emphasis is put on the Cr-tolerance of the material, which is an important feature for electrodes when applied in stacks containing metallic interconnects. Degradation processes are monitored by electrochemical techniques such as conductivity relaxation measurements, current voltage analysis and impedance spectroscopy. In-depth analytical investigations of surface-near regions by SEM and TEM were performed at several stages in the degradation process and are correlated with results from electrochemical measurements in order to gain insight into degradation mechanisms occurring under SOEC operating conditions.
88

Investigating the viability of monopolar and bipolar membranes as separators in buffered water-splitting cells
Nella M. Vargas-Barbosa* and Thomas E. MalloukDepartment of Chemistry, The Pennsylvania State University
University Park PA, USA
Abundant research has been dedicated to the discovery and optimization of light-absorbers and catalysts that can be utilized for water-splitting in photoelectrochemical cells (PECs). Indeed, immense strides have been done in identifying efficient materials composed of Earth-abundant elements. However, the long-term stability of some of these materials is contingent to a stable buffered electrolyte. Furthermore, robust separators will likely be a necessity to ensure maximum collection of gaseous products and overall electroneutrality. Here, a systematic study of the potential losses associated with employing a PEC with buffered electrolytes and commercial ion exchange membranes is presented. In particular, the ohmic drops caused by membrane resistivities, solution conductivity and the formation of a pH gradient were measured in a current density range up to 25 mA/cm2. We identified that buffer concentration polarization is the most detrimental potential loss in monopolar ion exchange membranes, which can be greater than 300 mV.1 However, this loss is somewhat mitigated by back-diffusion of neutral species when the cell is turned off. Furthermore, implementing bipolar membranes in a pH-polarized cell appears to provide an opportunity to de-couple the optimization and solution requirements of the materials for the anodic and cathodic reactions.2
References:(1) Hernandez-Pagan, E. A.; Vargas-Barbosa, N. M.; Wang, T.; Zhao, Y.; Smotkin,
E. S.; Mallouk, T. E. Energy Environ. Sci. 2012, 5 (6), 7582.(2) Vargas-Barbosa, N. M.; Geise, G. M.; Hickner, M. A.; Mallouk, T. E.
ChemSusChem 2014, 7 (11), 3017.
*Current institution: AG Prof. Dr. Bernhard RolingDepartment of Physical Chemistry, Philipps–Universität MarburgMarburg, Germany
89

Novel materials and components for Proton Exchange Membrane (PEM) water electrolysis: Status and challenges
F. Burggraf, P. Lettenmeier, S. Kolb, A.S. Gago, K.A. FriedrichInstitut für Technische Thermodynamik,
Deutsches Zentrum für Luft- und Raumfahrt, Stuttgart
Hydrogen generation by water electrolysis is expected to play an important role as a crosslinking technology between power generation on one hand and transport and industry on the other hand. When produced by water electrolysis from renewable energy sources - such as solar or wind - hydrogen can directly replace fossil fuels in transport and industry, thereby helping in the integration of renewable energies in other energy sectors. The relevant technologies are either the mature alkaline water electrolysis or the newer proton exchange membrane (PEM) water electrolysis, which exhibits higher potentials for performance enhancement and cost reduction. The major advantages of this new technology compared to the conventional electrolytic processes are its flexibility, its high loading capacities and its very compact system design. Nevertheless, there are still drawbacks to overcome: The acidic and oxidativeenvironment requires the utilization of precious metals such as platinum (Pt), titanium (Ti) or iridium (Ir). The availability of these materials represents a huge drawback in terms of cost with a massive deployment perspective of this technology in a hydrogen economic society[1]. Additionally, durability has to be investigated and enhanced for the use of PEM electrolyser systems in the future energy system.At the Institute of Engineering Thermodynamics of the German Aerospace Center we try to overcome these drawbacks by means of (electro)chemistry, chemical engineering and system modeling. Corrosion protective coatings that allow replacing cost intensive titanium plates by cheaper titanium-coated stainless-steel compoundshave been demonstrated[2]. Furthermore, the development of high performance electrocatalysts with reduced precious metal content allows for further cost reduction[3]. In parallel to experimental investigations, a dynamic PEM electrolyser cell and system model is developed to support the identification and the explanation of degrading mechanisms that limit the lifetime of current PEM electrolyser systems.[1] E. A. Paoli, F. Masini, R. Frydendal, D. Deiana, C. Schlaup, M. Malizia, T. W. Hansen, S. Horch, I. E. L. Stephens, I. Chorkendorff, Chem. Sci. 2014, 00, 1–7[2] Patent No.: WO2015000925 A1[3] P. Lettenmeier , L. Wang, U. Golla-Schindler, P. Gazdzicki, N. Cañas, M. Handl, R. Hiesgen, S. Hosseiny, A. Gago, K.A. Friedrich, Angewandte Chemie International Edition 2015, 54, 1 – 6
90

Tuning the Localized Surface Plasmon Resonances in Degenerately Doped Semiconductor Nanocrystals
A. Wolf, D. Dorfs, Leibniz Universität Hannover, Hannover, DeutschlandLocalized surface plasmon resonances (LSPRs) in nanoparticles are currently a research area with a lot of attention from many fields of science. The most well-known structures that exhibit this kind of resonant light absorption effect are metallic nanoparticles and more specific noble metal nanoparticles. One necessity for an LSPR to occur is a sufficiently high density of free charge carriers. Therefore, the large material class of degenerately doped semiconductors can in principle also show LSPRs. Due to the lower charge carrier densities compared to metals, nanoparticles from degenerately doped semiconductors show LSPRs in the near infrared spectral region rather than in the visible region (like for many metals). While the lower charge carriers density compared to metals might beseen as a disadvantage, these materials also have the huge advantage that the density of free charge carriers can be almost freely tuned by the amount of dopant. This effect was in depth studied for strongly self-doped Cu2-xSe nanocrystals. (1,2)In this presentation we will briefly summarize some of the recently developed procedures to synthesize monodisperse copper chalcogenide nanoparticles (Cu2-xSe and Cu1.1S) and the role of copper vacancies for their plasmonic behavior will be highlighted. The plasmonic properties of degenerately self-doped Cu2-xSe nanoparticles will be discussed in depth and the influence of dopant concentration on the LSPR is shown. Recent results of various other chemical approaches to influence the plasmonic behavior of this type of nanoparticles (e.g. shell growth, ion exchange etc.) will be presented, which greatly increase their application potential (e.g. enhanced stability under ambient conditions). (3-5)References:(1) Dorfs, D.; Hartling, T.; Miszta, K. et al.; J. Am. Chem. Soc., 2011, 133, 11175.(2) Scotognella, F.; Della Valle, G.; Kandada, A. R. S.; Dorfs, D. et al.; Nano Lett.,2011, 11, 4711.(3) Dilena, E.; Dorfs, D.; George, C.; et al.; J. Mater. Chem. 2012, 22, 13023.(4) Wolf, A.; Kodanek, T. and Dorfs, D.; Nanoscale, 2015, 7, 19519. (5) Wolf, A.; Härtling, T., Hinrichs. D. and Dorfs, D.; ChemPhysChem, 2015,accepted for publication, DOI: 10.1002/cphc.201500907.
91

Plasmons in supported silver clusters
T. Lünskens, P. Heister, M. Thämer, A. Von Weber, M. Jakob, A. Kartouzian,and U. Heiz, Technische Universität München, Lehrstuhl für Physikalische Chemie,
Catalysis Research Center, Garching, Germany
The plasmonic behavior of size-selected supported silver clusters is studied by surface second harmonic generation spectroscopy for the first time. A blue shift of ~0.2 eV in the plasmon resonance is observed with decreasing cluster size from Ag55
to Ag9. In addition to the general blue shift, also a nonscalable size-dependence is observed in plasmonic behavior of Ag nanoclusters that is attributed to varying structural properties of the clusters. The results are in quantitative agreement with a hybrid theoretical model based on Mie theory and existing DFT calculations.
92

Laser Irradiation of Suspensions: Formation Mechanisms of Surfactant-Free Nanoparticles
Christian Schaumberg, Department of Chemistry, Humboldt-Universität zu Berlin, Berlin, Germany; Markus Wollgarten, Helmholtz-Zentrum Berlin für Materialien und Energie, Berlin, Germany; Klaus Rademann, Department of Chemistry, Humboldt-
Universität zu Berlin, Berlin, Germany
Pulsed laser ablation in liquids has proved to be an elegant method to synthesize colloidal solutions of nanoparticles.[1] A stable colloid is formed by focusing a laser beam on a bulk target immersed in a liquid. An advantage of this approach is the avoidance of reducing or stabilizing agents. A further development is the use of suspended μm-sized powders as targets resulting in higher productivities.
While the experimental set up is rather simple, the underlying mechanisms appear to be quite complex. Short laser pulses introduce stress to crystal lattices leading to a fragmentation into smaller pieces while the chemical composition is preserved.[2] This mechanism can therefor regarded as fragmentation. In addition, the laser beam may also evaporate the material. The ablated atoms and clusters then nucleate to small primary nanoparticles. As the laser irradiation continues these particles may coalesceand form larger secondary nanoparticles. During these processes the chemical composition changes (e.g. Cu3N to Cu). Thus, this can be described as a reductive ablation mechanism.[3] In most cases, only one of these two mechanisms is observed for a certain precursor. As the formation mechanism determines the properties of the generated nanoparticles, it becomes vital to understand which experimental parameters trigger the one or the other mechanism.
Our work reveals the influence of the chemical composition of the precursor powder on the formation mechanism of copper colloids in organic liquids. Particle size distributions obtained from transmission electron microscopy studies are discussed together with the analysis of the chemical composition determined by electron energy
loss spectrosc
opy.[1] S. Barcikowski, G. Compagn
ini, PCCP 2013, 15, 3022-3026.[2] C. A. Schaumberg, M. Wollgarten, K. Rademann, PCCP 2015, 17, 17934-17938.[3] C. A. Schaumberg, M. Wollgarten, K. Rademann, J. Phys. Chem. A 2013, 118, 8329-8337.
93

Functionalization of oxide nanocrystals and composites with transition metal ions
Matthias Niedermaier a, Amir Gheisi b, Johannes Bernardi c, Oliver Diwald a
a Paris Lodron University of Salzburg, Hellbrunnerstrasse 34/III, A-5020 Salzburg, Austriab Friedrich Alexander University, Cauerstr. 4, D-91052 Erlangen, Germany
c Vienna University of Technology, Wiedner Hauptstrasse 8-10, A-1040 Vienna, Austria
Structure and functional properties of composite metal oxide nanoparticle systems are subject to stability and composition of related surfaces and interfaces. For vapor phase grown non-equilibrium solids, annealing induced ion diffusion provides efficient means to adjust the surface composition and, thus, the functional properties of the material. Insights into the underlying transformation processes, which can be controlled by annealing in different gas atmospheres, are essential for both nanomaterial design and material applications at elevated temperatures.
In the present contribution we will discuss properties and transformation behavior of metal oxide composites, such as Fe-Mg-O or Co-Mg-O nanoparticles, which were prepared by a hybrid chemical vapor synthesis approach using organic transition metal compounds as precursors. In addition to their combustion in the MgO formation flame, we also explored the feasibility of adsorption of transition metal oxides onMgO nanocubes in liquid particle dispersions followed by subsequent annealing in different chemical environments.
We used an integrated characterization approach involving electron microscopy (SEM, TEM), X-ray diffraction (XRD), electron dispersive X-ray spectroscopy (EDX) and Mößbauer spectroscopy to study the ensemble properties of nanoparticle composites, on the one hand, and structure and morphological features of characteristic local structures, on the other. The complex evolution of the composite nanostructures as a result of annealing induced segregation and phase separationwas characterized for samples with different thermal processing histories. These can give rise to a variety of material types in the range between nanostructures with bulk and surface admixed transition metal oxides to phase separated systems such as Fe-Mg-O - magnesioferrite MgFe2O4 particles. Advantages and experimental challenges related to the two types of functionalization approaches will be discussed.
94

Hydrogels and Aerogels of Nanocrystal Building Blocks
Nadja C. Bigall*
Institute of Physical Chemistry and Electrochemistry, Leibniz University Hannover, Callinstr. 3A, D-30167 Hannover
In this contribution, recent results from the development of gel structures from colloidal metallic, metal oxide and semiconductor nanoparticles will be presented and discussed.[1-3] One main focus will be hydrogels and aerogels from CdSe/CdS seeded nanorods, which exhibit extremely long fluorescence decay times and at the same time high photoluminescence quantum yields.[4] The spectroscopic characterization of such macroscopic monoliths is performed by means of an integrating sphere which helps determining the absorption and emission spectra as well as the photoluminescence quantum yield. By electron microscopy, the microscopic mesoporous morphology is investigated. It is furthermore revealed thatthe crystal-type interparticle linkage preferably takes place at the tip-sites of the nanorods. A model explaining the observations is derived. Further developments in the field of aerogels of nanocrystals will also be presented, such as the newly developed cryogelation method.[5] Possible applications will be discussed.
[1] A. Eychmüller, ANGEWANDTE CHEMIE INT. ED., 2005, 44, 4839
[2] I.U. Arachchige, S.L. Brock, ACCOUNTS OF CHEMICAL RESEARCH, 2007, 40, 801
[3] N. C. Bigall, et al., ANGEWANDTE CHEMIE INT. ED., 2009, 48, 9731
[4] Sánchez-Paradinas, S.; et al. ADVANCED MATERIALS, 2015, 27, 6152–6156
[5] Freytag, A.; et al. ANGEWANDTE CHEMIE INT. ED., 2016, 55, 1200–1203
95

Conductive PbS MesocrystalsAlexander André1, Mahdi Samadi Khoshkhoo1, Michelle Weber1, Marcus Scheele1,2
1 Institute of Physical and Theoretical Chemistry, University of Tübingen, 72076 Tübingen,
Germany.
2 Center for Light-Matter Interaction, Sensors & Analytics LISA+, University of Tübingen,
72076 Tübingen, Germany.
Mesocrystalline materials are “three-dimensional arrays of iso-oriented single-crystalline
particles with an individual size between 1 – 1000 nm”.1 Significant progress has been made
in the field of colloidal semiconductor nanocrystals towards mesocrystalline assemblies over
macroscopic domains by exploiting ligand-ligand interactions.2,3 Typically, the utilized
ligands consist of wide-gap, bulky hydrocarbons which render the mesocrystals insulating. On
the other hand, numerous ligand exchange strategies have been developed to promote efficient
charge carrier transport through large nanocrystal assemblies, but such post-assembly
treatments usually destroy the mesocrystalline structure.4
We will show how organic semiconductor small molecules can be utilized to guide the self-
assembly of PbS nanocrystals into 3D arrays which are conductive and mesocrystalline at the
same time. Our analysis is carried out in real space by high-resolution TEM, HAADF-STEM
and electron diffraction as well as in reciprocal space by GIXD, GISAXS and simultaneous
SAXS/WAXS utilizing a nano-beam set-up.
We will discuss how the structure of the 3D array can be modulated between body-centered
tetragonal, honeycomb and cubic as well as how the organic semiconductor small molecule
controls the lattice constant and directs the oriented attachment of the nanocrystals’ atomic
lattices.
References:
[1] J. Seto, Y. Ma, S.A. Davis, F. Meldrum, A. Gourrier, Y.-Y. Kim, U. Schilde, M. Sztucki,
M. Burghammer, S. Maltsev, C. Jäger, and H. Cölfen, PNAS 109, 3699 (2012).
[2] K. Bian, J.J. Choi, A. Kaushik, P. Clancy, D.-M. Smilgies, and T. Hanrath, ACS Nano 5,
2815 (2011).
[3] C.B. Murray, C.R. Kagan, and M.G. Bawendi, Science 270, 1335 (1995).
[4] J. Luther, M. Law, Q. Song, C. Perkins, M. Beard, A. Nozik, ACS Nano 2, 271 (2008).
96

Environmental Effects on the Stability and Photo-Chemistry of Semiconductor Nanocrystals
Klaus Boldt, Universität Konstanz, Konstanz/Germany; Nicholas Kirkwood, Sean Murphy, Julia Baldauf, and Paul Mulvaney, University of
Melbourne & Bio21 Institute, Melbourne/Australia
Experimental conditions for the synthesis of II-VI semiconductor quantum dots (QDs)such as CdSe have been improved continuously since the first published hot injection synthesis in 1993.1 Recent publications report core/shell particles that appear photo-chemically stable and exhibit photoluminescence quantum yields close to unity.2,3
Yet, many parameters that govern the result and reproducibility of a synthetic routeare not well understood. Here we present our findings concerning environmental effects on nanocrystal synthesis and long-term behaviour.Many routes towards Cd-based nanocrystals employ CdO, which releases water upon formation of the reactive precursor. The water is usually removed under reduced pressure before particle formation. We show that traces of water have a strong impact on the kinetics of QD nucleation by influencing the size and reactivity of magic size clusters that have been found to form during nucleation.4 This allows additional control over particle size and size distribution.Water as well as oxygen continue to play a significant role after the particles have been formed: exposure to ambient conditions irreversibly changes the photo-stability even of well-passivated core/shell QDs compared to samples that have beenmeticulously shielded from air and moisture. This history effect allows to explain the some-times conflicting reports in the literature on QD photo-enhancement and degradation.5,6
(1) C. B. Murray et al. J. Am. Chem. Soc. 1993, 115, 8706–8715.(2) O. Chen et al. Nat. Mater. 2013, 12, 445–451.(3) K. Boldt et al. Chem. Mater. 2013, 25, 4731–4738.(4) Z.-J. Jiang et al. ACS Nano 2010, 4, 1561–1572.(5) J. Müller et al. Appl. Phys. Lett. 2004, 85, 381–383.(6) K. Pechstedt et al. J. Phys. Chem. C 2010, 114, 12069–12077.
97

Orientational distribution functions of suspended spindle-shaped hematite particles in external magnetic fields
C.Passow, A. Nack, J. Wagner
Universität Rostock, Dr.-Lorenz-Weg 1, 18059 Rostock Germany
Colloidal hematite spindles are a well defined colloidal model system with tunable aspect ratio. Due to the interaction with an external field, an isotropic-nematic phase transition can be induced in the presence of moderate magnetic fields, whereby spindle-shaped hematite particles align perpendicular to the direction of an external field. To determine the magnetic properties of hematite spindles, their orientational distribution functions (odf) in dependence on the flux density of an external field are investigated by means of small angle X-ray scattering employing synchrotron radiation as a probe.
The energy of a hematite spindle in a magnetic field depends on a permanent magnetic moment as well as on an induced magnetic moment which results from the anisotropy of its magnetic susceptibility.
With the odf, in absence of particle-particle interactions, the magnetic parameters are determined from a simultaneous fit of the small angle scattering in dependence on the modulus, the direction of the external field and the magnetic flux density. The alignment of hematite particles in presence of an external field results both, from a negative magnetic anisotropy, and a permanent magnetic field. The main contribution to the interaction with an external field, however, results from the Zeeman energy of a permanent magnetic field nearly orthogonal to the long particle axis.
98

Ultrafast Dynamics of Electron-Hole Pairs in 2D InSe NanosheetsJannika Lauth, Delft University of Technology, Delft/The Netherlands,
Frank C. M. Spoor, Delft University of Technology, Delft/The Netherlands Aditya Kulkarni, Delft University of Technology, Delft/The Netherlands
Arjan J. Houtepen, Delft University of Technology, Delft/The Netherlands,Juleon M. Schins, Delft University of Technology, Delft/The Netherlands,
Sachin Kinge, Toyota Motor Europe, Zaventem/BelgiumLaurens D. A. Siebbeles, Delft University of Technology, Delft/The Netherlands
Graphene-like two-dimensional (2D) crystals with non-zero band gaps exhibit highly interesting anisotropic dimensionality-dependent properties and bear high potential for ultrathin electronics. Amongst the most investigated 2D semiconductors, one material recently moved into focus: Exfoliated InSe, a van-der-Waals layered III-VI metal chalcogenide with an atomically smooth surface. It has proven its highly promising (opto-)electronic properties as high mobility field-effect transistor[1] and even outperformed other 2D semiconductors like MoS2 and GaSe as high responsivity (visible to NIR) photodetector[2,3]. We have synthesized atomically thin2D InSe nanosheets (inorganic InSe layer thickness 1.7 nm, total thicknes <5 nm with organic ligands) by a straightforward ligand templated colloidal method and have fully characterized the 2D single crystals with electron and atomic force microscopy, X-ray photoelectron spectroscopy and scattering methods[4]. By applying ultrafast transient absorption spectroscopy, we evaluate the charge carrier and recombination dynamics in InSe nanosheets and extract (intrinsic) mobilities of ~30 cm2/Vs by using time-resolved terahertz spectroscopy. In photoexcited coupled InSe nanosheet thin films we detect free charges, whereas excitons are formed in InSe nanosheet solutions. The combination of colloidal synthesis and pump-probe spectroscopy allows us to assess the potential of wet-chemically processed ultrathin InSe nanosheets for next generation electronics.
[1] Sucharitakul, S. et al., Nano Lett. 2015, 15, 3815.[2] Tamalampudi, S. R. et al. Nano Lett. 2014, 14, 2800.[3] Lei, S. et al. ACS Nano 2014, 8, 1263.[4] Lauth, J. et al., submitted.
99

Small-Diameter Semiconductor Block Nanowires Synthesized bySolution-Liquid-Solid (SLS) Technique and Cation Exchange
Andreas Nielsen*, W. E. Buhro , Tobias Kipp*, Alf Mews**Institute of Physical Chemistry, University of Hamburg, GermanyDepartment of Chemistry, Washington University in St. Louis, USA
Small-diameter semiconductor nanowires (NW) are interesting for usage in nanoscale electronics, photonics and energy conversion. Due to their one-dimensional morphology and radii below their respective exciton-Bohr-radius, quantum-sized nanowires exhibitunique diameter-dependent optical and electronic properties. By introducing an axial-hetero-interface, the electronic band structure can be tuned. We prepared blockwires by subsequently growing CdSe and CdTe segments in a SLS synthesis (Fig. 1a). Scanning-force techniques and confocal microscopy proved this model system to exhibita type-II (staggered gap) band alignment at the interface, which enables exciton charge separation[1]. In consequence photo generated charges can be drained separately by electrodes. A second method to yield heterostructured NWs is cation exchange (CE). Applied on our model CdTe NW system, using silver ions, we produced Ag2Te/CdTe blockwires with atomically sharp Ag2Te-CdTe interfaces (Fig. 1b). The newly formed Ag2Te segments are highly susceptible to further CE, opening a window to a variety of new material combinations and hence modifications in the electronic properties. Investigation by high resolution transmissions electron microscopy (HRTEM) provides information on the nanowire crystal structure and therefore on the growth and exchange process.
Fig. 1: HRTEM images of heterointerfaces in block NWs. (a) CdTe(left)/CdSe(right) NW. (b) CdTe(left)/Ag2Te(right)NW with atomically sharp interface.[1] S. Schäfer, A. Reich, Z. Wang, T. Kipp, A. Mews, Appl. Phys. Lett. 2012, 100, 022110.
100

Impact of redox-active molecules on fluorescence and exciton dynamics in semiconducting carbon nanotubes
Sebastian Kruss, Universität Göttingen, Institut für Physikalische Chemie, Göttingen/Deutschland
Semiconducting single-walled carbon nanotubes (SWCNTs) are hollow cylinders of one-atom-thick sheets of carbon. They provide an intrinsic bandgap, which results in near infrared (nIR) fluorescence based on exciton formation. Interestingly, the fluorescence of polymer-wrapped SWCNTs is very sensitive to the local chemical environment. It has been shown that certain reducing molecules increase the fluorescence quantum yield of SWCNTs. So far the role of the polymer around the SWCNT, the structure of the reducing molecule as well as the mechanism is not understood. Weinvestigated how reducing and oxidizing small molecules affect exciton dynamics and quantum yields of polymer-wrapped SWCNTs. Our results show that the polymer plays an essential role. Reducing molecules such as ascorbic acid, epinephrine and trolox increased the nIR fluorescence by more than 400 % but only if suspended in negatively charged polymers such as DNA or poly-acrylic acid (PAA). In comparison, phospholipid-polyethylenglycole wrapped SWCNTs did not respond at all while positively charged polyallylamine-wrapped SWCNTs were quenched. In general, polymer wrapped SWCNTs that responded to reducing agents (fluorescence increase) also responded to oxidizing agents (fluorescence decrease). Furthermore we show that neither changes of absorption cross-sections, scavenging of reactive oxygen species nor calculated free surface areas on SWCNTs can explain the observed patterns. However, our data is in agreement either with a redox-reaction or conformational changes of the polymer that change fluorescence decay routesand create radiative exciton traps. In summary, we show that the polymer around SWCNTs governs if reducing molecules increase nIR-fluorescence (quantum yield) of SWCNTs and how they affect exciton diffusion.
Impact of redox-active molecules on fluorescence and exciton dynamics in semiconducting carbon nanotubes
Sebastian Kruss, Universität Göttingen, Institut für Physikalische Chemie, Göttingen/Deutschland
Semiconducting single-walled carbon nanotubes (SWCNTs) are hollow cylinders of one-atom-thick sheets of carbon. They provide an intrinsic bandgap, which results in near infrared (nIR) fluorescence based on exciton formation. Interestingly, the fluorescence of polymer-wrapped SWCNTs is very sensitive to the local chemical environment. It has been shown that certain reducing molecules increase the fluorescence quantum yield of SWCNTs. So far the role of the polymer around the SWCNT, the structure of the reducing molecule as well as the mechanism is not understood. Weinvestigated how reducing and oxidizing small molecules affect exciton dynamics and quantum yields of polymer-wrapped SWCNTs. Our results show that the polymer plays an essential role. Reducing molecules such as ascorbic acid, epinephrine and trolox increased the nIR fluorescence by more than 400 % but only if suspended in negatively charged polymers such as DNA or poly-acrylic acid (PAA). In comparison, phospholipid-polyethylenglycole wrapped SWCNTs did not respond at all while positively charged polyallylamine-wrapped SWCNTs were quenched. In general, polymer wrapped SWCNTs that responded to reducing agents (fluorescence increase) also responded to oxidizing agents (fluorescence decrease). Furthermore we show that neither changes of absorption cross-sections, scavenging of reactive oxygen species nor calculated free surface areas on SWCNTs can explain the observed patterns. However, our data is in agreement either with a redox-reaction or conformational changes of the polymer that change fluorescence decay routesand create radiative exciton traps. In summary, we show that the polymer around SWCNTs governs if reducing molecules increase nIR-fluorescence (quantum yield) of SWCNTs and how they affect exciton diffusion.
101

Electrical transport through colloidal PbS sheets
Christian Klinke
Institute of Physical Chemistry, University of Hamburg
I will show that the formation of ordered and densely packed ligand surface layers of oleic acid on {100} PbS surfaces can drive the normally isotropic growth of colloidal crystals into a two-dimensional oriented attachment of nanocrystals. Hereby, the presence of chlorine containing co-solvents plays a prominent role during the initial nucleation and growth process of the nanocrystals. The formation mechanism will be discussed, as well as its transfer to other two-dimensional material systems. Furthermore, the electrical transport through these materials will be analyzed in devices like transistors and solar cells.
102

Protic Ionic Liquids:Mesoscopic Inhomogeneities in Solutions of Ethyl-Ammonium
Nitrate and n-Alkyl AlcoholsOlga Russina1, Alessandro Triolo2, Wolffram Schröer3, and Bernd Rathke4,#
1Dipartimento di Chimica, Università di Roma Sapienza, Rome, Italy2Instituto Struttura della Materia, Consiglio Nazionale delle Ricerche, Rome, Italy
3Institut für Anorganische und Physikalische Chemie, Universität Bremen, Germany4Technische Thermodynamik, Universität Bremen, 28359 Bremen, Germany
(#) e-mail: [email protected]
To gain a more generalized understanding of the behavior of protic Ionic Liquids (ILs) in mixtures, we have been performed an interconnected study of both, the characterization of liquid-liquid phase behavior ethyl-ammonium nitrate (EAN) with n-alkyl alcohols (heptan-1-ol and octan-1-ol) and the characterization of inhomogeneities investigated by small angle x-ray scattering (SAXS). The systems studied so far in a temperature range of T = (240 – 313) K show limited miscibility with upper critical solution temperatures (UCST) at T = (260 and 315) K. The UCST decreases with decreasing length of the alkyl-chains of the alcohols. Both components are amphiphilic and characterized by an extended hydrogen bonding network. Although they show on a macroscopic scale an apparent homogeneous behavior, they are heterogeneous on the mesoscopic level. Especially in the vicinity of the critical point a superposition of critical fluctuations and inhomogeneities due to the separation into ionic and non-ionic domains is observed. In contrast to prior studies, which have been focused on shorter chained n-alkyl alcohols, mixtures with heptan-1-ol allow to study both contributions within the liquid region of the phase diagram. Both contributions are evaluated and discussed.
103

A further Step to the Understanding of the Solvation Behavior of Alcohols in Ionic Liquids.
Andreas Appelhagen 1 and Daniela Kerlé 2
1 Universität Rostock, Physikalische Chemie, Dr.-Lorenz-Weg 1, 18059 Rostock2 Universität Bremen, Technische Thermodynamik, Badgasteiner Str. 1, 28359 Bremen
Mixtures of Ionic Liquids (ILs) and molecular solvents show complex behavior and are of high interest for technological applications [1]. Alcohols are often used with ILs because they have similar solvation properties; moreover many ILs are totally miscible with alcohols, but others show miscibility gaps. Mixtures of alcohols with ILs are quite complex due to hydrogen bonding and van-der-Waals interactions.To get a more detailed understanding, atomistic simulations are a great help. By using molecular dynamics and Monte Carlo simulations it is possible to analyze the local dynamics and structures in detail.In this work we focus on vapor-liquid equilibria of a series of 1-n-alkyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)amide ([CnMIm][NTf2]) ILs with different alcohols and additionally the influence of contaminations of water. These systems were studied with the Monte Carlo simulations code Cassandra [2]. With the Gibbs ensemble Monte Carlo method we are able to calculate the entire isotherms for the vapor-liquid equilibria in excellent agreement with experimental results without any need of parameter refitting. Other properties we studied include activity coefficients, excess enthalpies, mixing volumes, cluster sizes and percolation rates. By the comparison to experimental data these results are validated. A detailed analysis of the driving forces, hydrogen bonding and preferred structures will complete this work and help to get a detailed understanding of these complex mixtures.
References[1] V.R. Vale, S. Will, W. Schröer, B. Rathke, ChemPhysChem 13, 2012, 1860.[2] J.K. Shah, E.J. Maginn, J. Chem. Phys. 135, 2011, 134121.
Fig. 1: Preferred locations of functional groups of the ions around a methanol molecule.
104

Spectroscopic evidence for clusters of like-charged ions in ionic liquids
Anne Knorr, Peter Stange and Ralf LudwigInstitute of Chemistry, Physical and Theoretical Chemistry, University of Rostock,
Dr.-Lorenz-Weg 1, D-18059 Rostock, Germany
The concept of attractive like-charge interaction usually requires the mediation of solvent molecules. In this work this counter-intuitive phenomenon can be shown the first time for pure ionic liquids. [1]Direct Spectroscopic evidence for hydrogen-bonded clusters of like-charged ions is reported for ionic liquids, based on the 1-(2-hydroxyethyl)-3-methylimidazolium cation. The measured infrared O-H vibrational bands of the hydroxyethyl groups can be assigned to the dispersion-corrected DFT calculated frequencies of linear and cyclic clusters. Compensating the like-charge Coulomb repulsion, these cationic clusters can range up to cyclic tetramers resembling molecular clusters of water and alcohols. These ionic clusters stabilized via anti-electrostatic hydrogen bonds(AEHBs) show strong cooperative effects at low temperature. [2,3]Additionally we are able to discuss the attractive like-charge interaction between cations for differently strong interacting anions, namely tetrafluoroborate, bis(trifluoromethylsulfonyl)imide, triflate and methylsulfonate as well as a function of OH-density in mixtures with the corresponding 1-methyl-3-propylimidazolium based ionic liquid. [4]
[1] A. Knorr, K. Fumino, A.-M. Bonsa, R. Ludwig, Phys.Chem.Chem.Phys., 2015, 17, 30978-30982.
[2] A. Knorr, R. Ludwig, Sci. Rep., 2015, 5, 17505.[3] F. Weinhold, R. A. Klein, Angew. Chem., 2014, 126, 11396 –11399
(Angew. Chem. Int. Ed., 2014, 53, 11214 –11217).[4] A. Knorr, P. Stange, K. Fumino, F. Weinhold, R. Ludwig, ChemPhysChem, 2015,
submitted.
105

Excess Spectroscopy to Study Binary and Ternary SolutionsJohannes Kiefer, Florian M. Zehentbauer, Universität Bremen, Germany;
Kristina Noack, Universität Erlangen-Nürnberg, Germany
Solutions comprising of different molecular liquids are of great practical relevance. Not only do they exhibit interesting macroscopic properties that may deviate significantly from those of a thermodynamically ideal mixture, but also their behavior at the molecular level can be exciting and unexpected. A prominent example for such a solution is the binary system of dimethyl sulfoxide (DMSO) and water, in particular at the eutectic composition. A complicated interplay of molecular interactions including hydrogen bonding, polar interactions and dispersion forces governs the observed behavior.Vibrational spectroscopy is frequently employed to study molecular interactions in solutions. In this context, excess spectroscopy, which compares an experimental mixture spectrum with that of the corresponding spectroscopic/thermodynamic ideal solution, experiences an ever-increasing interest in the analysis of vibrational spectra. In this work, we demonstrate how excess spectroscopy can be utilized to enhance the interpretation of vibrational spectra of binary and ternary solutions. This includes the identification and quantification of peak shifts as well as changes in the dipole moment. We also show what kind of artifacts can arise from an improper treatment of the spectra. As practical examples, results from mixtures of imidazolium ionic liquids and organic solvents are shown.
106

Pressure Effects on Solvation Structure and Vibrational Spectroscopy of Aqueous TMAO Solutions
Sho Imoto, Harald Forbert and Dominik Marx, Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, D-44780 Bochum, Germany
Although the effects of pressure perturbations on aqueous biomolecules and proteins are not well understood, pressure is an essential thermodynamic variable like temperature and concentration. Recent experiments revealed that the local solvation shell structure around proteins is strongly altered upon compressing to high hydrostatic pressures (HHP) in the kilobar regime. Especially the properties of trimethylamine N-oxide (TMAO) solutions at HHP conditions are of great interest because the molecule stabilizes proteins against pressure denaturation. The TMAO molecule has strongly hydrophilic and hydrophobic groups at its opposite ends and, thus, water molecules around the hydrophilic and hydrophobic groups are supposed to show different responses against pressure perturbations. We analyzed the structure [1] as well as the intermolecular (THz) and intramolecular (mid-IR) vibrational spectra of TMAO in water at 10 kbar compared to ambient pressure by using ab initio molecular dynamics simulations.
Reference:[1] Sho Imoto, Harald Forbert and Dominik Marx, Phys. Chem. Chem. Phys. 17(2015) 24224.
107

The Impact of TMAO and Urea on the H-Bond Dynamics under High Pressure
Lukas Knake, Hendrik Vondracek, Martina Havenith Physical Chemistry II, Ruhr-University Bochum, D-44801 Bochum, Germany
It is well known that life can withstand extreme conditions regarding pressure and temperature. Organic osmolytes such as amino acids, sugars and trimethylamine-N-oxide (TMAO) have been found to be accumulated under pressure and thermal stress [1]. TMAO has been found to be enriched in deep sea animals living under high pressure (up to 1 kbar) conditions, counteracting the denaturing effect of high pressure [2]. Previous studies suggest that the stabilizing mechanism of TMAO is based on a solvent-mediated effect, e.g. a modification of the hydrogen-bond network and an enhancement of the water structure [3]. Although at ambient conditions the influence of TMAO on the water network is known, the impact of high pressure on the solvent dynamics is still an open question.
In our study, we use broadband Terahertz (THz) absorption spectroscopy to study the molecular details of changes in the fast (sub-ps) hydrogen bond network dynamics around TMAO and Urea up to 14 kbar. Based upon a detailed analysis we reveal new insights into the solvent dynamics of these solvated biomolecules when changing from ambient to high pressure conditions.
References
[1] P.H. Yancey, J. Exp. Biol. 208 (2005), 2819-2830[2] P. Yancey, et al., Proc. Natl. Acad. Sci. U.S.A. 111 (2014), 4461–4465[3] F. Meersman, D. Bowron, A. Soper, M. Koch, Biophys. 97 (2009), 2559-2566
The Impact of TMAO and Urea on the H-Bond Dynamics under High Pressure
Lukas Knake, Hendrik Vondracek, Martina Havenith Physical Chemistry II, Ruhr-University Bochum, D-44801 Bochum, Germany
It is well known that life can withstand extreme conditions regarding pressure and temperature. Organic osmolytes such as amino acids, sugars and trimethylamine-N-oxide (TMAO) have been found to be accumulated under pressure and thermal stress [1]. TMAO has been found to be enriched in deep sea animals living under high pressure (up to 1 kbar) conditions, counteracting the denaturing effect of high pressure [2]. Previous studies suggest that the stabilizing mechanism of TMAO is based on a solvent-mediated effect, e.g. a modification of the hydrogen-bond network and an enhancement of the water structure [3]. Although at ambient conditions the influence of TMAO on the water network is known, the impact of high pressure on the solvent dynamics is still an open question.
In our study, we use broadband Terahertz (THz) absorption spectroscopy to study the molecular details of changes in the fast (sub-ps) hydrogen bond network dynamics around TMAO and Urea up to 14 kbar. Based upon a detailed analysis we reveal new insights into the solvent dynamics of these solvated biomolecules when changing from ambient to high pressure conditions.
References
[1] P.H. Yancey, J. Exp. Biol. 208 (2005), 2819-2830[2] P. Yancey, et al., Proc. Natl. Acad. Sci. U.S.A. 111 (2014), 4461–4465[3] F. Meersman, D. Bowron, A. Soper, M. Koch, Biophys. 97 (2009), 2559-2566
108

Thermodynamic properties of water solvating biomolecular surfaces
Matthias Heyden, Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, Germany
Changes in the potential energy and entropy of water molecules hydrating biomolecular interfaces play a significant role for biomolecular solubility and association. Free energy perturbation and thermodynamic integration methods allow calculations of free energy differences between two states from simulations. However, these methods are computationally demanding and do not provide insights into individual thermodynamic contributions, i.e. changes in the solvent energy or entropy. Here, we employ methods to spatially resolve distributions of hydration water thermodynamic properties in the vicinity of biomolecular surfaces from molecular dynamics trajectories. This allows direct insights into thermodynamic signatures of the hydration of 1) hydrophobic and hydrophilic solvent accessible sites of proteins; 2) small molecules and 3) comparisons to ideal model surfaces. Based on our results, we analyze correlations between dynamic properties of hydration water molecules, i.e. translational and rotational mobility, to their thermodynamics. The latter can be used as a guide to extract thermodynamic information from experimental measurements of site-resolved water dynamics. Further, we study energy-entropy compensations of water at different hydration sites of biomolecular surfaces.
109

Ultrafast Dynamics of Vibrational Relaxation of Pseudohalides in Liquid-to-Supercritical Water. A Critical Test of Fermiʼs Golden Rule
Jeannine Gleim, Denis Czurlok, Jörg Lindner and Peter Vöhringer
Institut für Physikalische und Theoretische Chemie, Universität Bonn
In recent years, the vibrational spectroscopy of pseudohalide anions in condensed media has attracted much attention. They were studied in the context of dynamical solute-solvent interactions and vibrational energy relaxation (VER) in hydrogen-bonded liquids like water [1]. In particular, the distinction between the various relaxation pathways arising from intramolecular vibrational redistribution (IVR) and intermolecular energy transfer (VET) has become a challenging issue for all triatomicpseudohalide anions in aqueous solutions.
Here, the stretching vibration, 3, of 15N-labelled thiocyanate (SC15N-), selenocyanate
(SeCN-) and azide (N3-) anions in aqueous solution were studied with Fourier-
Transform IR-spectroscopy (FTIR) as well as femtosecond IR-pump-probe spectroscopy (fs-IR-PP). All experiments were carried out under isobaric conditions at a pressure of 500 bar over a wide temperature range ( ) covering the liquid and supercritical phases of water.
Relaxation rate constants were determined from the decay of the v=1 excited state absorption. In all studied cases, the temperature dependencies of the relaxation rate constants are similar; namely, with increasing temperature, the rate constants decrease. However, the relaxation pathways differ. While the relaxation of 15N-labelled thiocyanate and selenocyanate follow the VET pathway that of azide follows a combination of both, the IVR and VET pathways. The results are interpreted in
terms of Fermiʼs Golden Rule and a resonant VET from the soluteʼs 3-mode to the
combination tone of the bending and librational modes of water [2].
References:
[1] M. Olschewski, S. Knop, J. Lindner, P. Vöhringer, J. Chem. Phys. 134, 214504 (2011)
[2] D. Czurlok, J. Gleim, J. Lindner, P. Vöhringer, J. Phys. Chem. Lett. 5, 3373 (2014)
110

FTIR Spectroelectrochemistry – Details of Equipment and Applications
Wilfried Hartmann and Mathias Keßler, Bruker Optik GmbH, Ettlingen / Germany
Modern technological developments in the field of mobility, traffic and energy are driven by the research in the field of batteries and energy storage. A deepunderstanding of the processes occurring during electrochemical reactions allows for the constant improvement of existing techniques and the control of reaction pathways.1 The combination of the experimental techniques electrochemistry and FTIR spectroscopy provides the possibility to study electrolytes and electrode surfaces directly during the application of an electric potential. Classical transmission cells for spectroelectrochemistry are often limited to the use of metal mesh electrodes. Alternatively the utilization of a reflection unit offers a way to conduct reflection measurements directly on an electrode surface and to investigate electrolyte solutions with ATR (attenuated total reflection) experiments. The VERTEXFTIR spectrometer in combination with a reflection accessory and an electrochemical cell is a powerful tool for this coupled experimental technique.2 In particular under vacuum measurement conditions residual spectral disturbances are avoided which might be caused by residual moisture absorption of room air. The set-up enables
scientists to conduct in situstudies of their samples with achanging applied potential.3
Fig. 1: Application example:Spectra of the oxidation of ferrocyanide.
1 R. v. Noorden, Nature 2014, 507, 26–28.2 Bruker Optics Application Note 125 (2015).3 A. Kellenberger, E. Dmitrieva and Lothar Dunsch, J. Phys. Chem. B 2012, 116 .
111

Application of in situ Infrared Spectroscopy to the Production of a Flavouring Substance
Enrico Barscha, Andreas Petrib, Peter Müllerb, Ralf Ludwiga
a) Universität Rostock, Physikalische und Theoretische Chemie, Dr.-Lorenz-weg 1, 18059 Rostockb) Miltitz Aromatics GmbH, Riechstoffstrasse, D-06766 Bitterfeld-Wolfen
The significance of process analytical technologies (PAT) in the chemical and pharmaceutical industry has increased over the last decade. Modern in situ spectroscopies allow insight into mechanisms and provide a deep understanding of chemical reactions. Optimization prospects can be developed and an improved process control in manufacturing usually ensures the return of investment.Here we present an application of in situ infrared spectroscopy to the trimerization of isoprene. Out-of-plane C-H wagging modes enabled the real-time identification of educts, products and the main by-products. Kinetic experiments gave mechanistic information and offered ideas for an optimal reactor design. Faults in manufacturing, caused by an incomplete catalyst preformation, could be predicted by infrared spectroscopy. Through online analysis the time of reaction stop could be determined with only a one-minutetime delay instead of one hour using conventional GC analysis.The performed investigations suggest, that a permanent installation of an in situ infrared spectroscopy will accelerate process analysis, increase feedstock efficiency and will significantly contribute to the final product quality.
112

Heats of dilution of ionic liquids an their inorganic salt precursors in water
Daniel Ondo, Vladimír Dohnal, University of Chemistry and Technology, Technická 5, 166 28 Prague 6, Czech
Republic, [email protected] Ignatiev,
PC-RL Ils, Merck KgaA, Frankfurterstr. 250, D-64293 Darmstadt, Germany
Heats of dilution of ionic liquids (ILs) and their inorganic salts precursors in water at 298.15 K were measured using the isothermal titration calorimetry. The ionic liquids involved EMIM+ and BMIM+ cations combined with sulphate, sulfonate, thiocyanate anions, the inorganic salts involved lithium, sodium and potassium salts with corresponding anions widely used in ILs synthesis (TfN2
-, B(CN)4-, FAP-, PF6
-) and used previously [1,2]. Measured heats of dilution below 0.2 mol/kg were correlated by 2-3 parameter Pitzer equation for apparent molar enthalpies. The data are shortly discussed in terms of ion size, charge allocation and aqueous solubility.
[1] D. Ondo, M. TkadlecoA. Heintz, N. Ignatiev: J. Phys. Chem. B 115 (2011) 10285-10297
[2] D. Ondo, V. Dohnal: J. Chem. Thermodyn. 75 (2014) 86-95
113

Papierbasierte ChemiresistorenEvariste Pasky Wete, Rosemarie Dittrich, Yvonne Joseph
Institute for Electronic and Sensor Materials/Technische Universität Bergakademie Freiberg, Freiberg/ Germany
Motivation
Die Anwendung chemischer Sensoren in den Lebenswissenschaften und im medizinischen Bereich zeichnet sich durch den schnellen Informationsgewinn, die nicht-invasive Anwendung und den Einmalgebrauch aus. Die Sensoren müssen daher kostengünstig hergestellt werden können und dabei hohe Empfindlichkeiten sowie anwendungsbezogene Selektivitäten aufweisen. Insbesondere die Aufbau-und Verbindungstechnik (Substrate, Chip Carrier etc.) ist bisher sehr teuer.
Ergebnisse
Zur Realisierung kostengünstiger Sensoren wurden die bisher verwendeten einkristallinen oxidierten Si-Wafer durch Papiersubstrate ersetzt. Diese wurden vor der Beschichtung bezüglich ihrer thermischen und chemischen Stabilität hinsichtlich unterschiedlicher Lösungsmittel geprüft und ihre Oberflächenmorphologie auf Vorder- und Rückseite rasterelektronenmikroskopisch untersucht. Auf ausgewählte Papiersubstrate sind dann selbstorganisierende Nanokomposite auf Basis von Gold-Nanopartikeln und verschiedenen organischen Molekülen in Form von ultradünnen Schichten aufgebracht worden (vergl. Abb.1).[1] Die großflächige Abscheidung der Au-Nanopartikel wurde durch automatisierte lagenweise Abscheidung mittels Selbstassemblierung realisiert. Die abgeschiedenen Schichten wurden licht- und
rasterelektronenmikroskopisch untersucht sowie mittels Photoelektronenspektroskopie (XPS) und elektrischer Messungen zur Ermittlung der sensorischen Eigenschaften charakterisiert.
Abbildung 1: Goldnanopartikel-Nonandithiolkomposit auf Papiersubstrat
Literatur
[1] Joseph Y, et al. ; J. Phys. Chem. B 107, 7406-7413 (2003).
114

Inelastic Hydrogen Atom Scattering: Role of Electron-Hole Pair Excitations
Oliver Bünermann, Hongyan Jiang, Yvonne Dorenkamp, and Alec WodtkeGeorg-August-Universität Göttingen, Institut für Physikalische Chemie,
Tammannstr. 6, 37077 Göttingen, Germany
Obtaining an atomic-level understanding of the dynamics of energy conversion at surfaces remains a complex and challenging area of modern research in physical chemistry. A general strategy to this field follows the lessons of gas-phase bimolecular chemical dynamics, where simple model systems are studied experimentally with great care while theoretical simulations are developed. One of the simplest systems to think of is Hydrogen atom scattering from a single crystalline surface. We built a new apparatus to experimentally investigate this model system with extraordinary precision. Laser photolysis is employed to produce a monochromatic H-atom beam. The H-atom beam strikes a single crystalline surface held in UHV. The kinetic energy and angular distributions of the scattered H-atoms are measured with extraordinary resolution employing Rydberg Atom Tagging.
Experimental results for scattering H-atoms from metal and insulator surfaces will be presented. In case of an insulator the scattering is nearly elastic while in case of metals strongly inelastic scattering is observed. Our results reveal that electron-hole-pair excitation is the dominate relaxation channel for H-atom translation in case of a metal surface, an interpretation supported by theoretical calculations [1].
[1] Buenermann, O et al., Science 350, 1346-1349 (2015)
115

CO2 capture via adsorption on iron oxides: How a specific surface morphology enables host-guest-like chemistry
Xiaoke Li and Joachim PaierHumboldt-Universität zu Berlin, Unter den Linden 6, 10099 Berlin, Germany
Control and reduction of global warming induced by so-called greenhouse gases like carbon dioxide (CO2) represents one of the biggest and most urgent challenges for mankind to date. However, CO2 removal from the atmosphere is technologically difficult.Motivated by that, we study CO2 adsorption on the magnetite Fe3O4(111) surface.
CO2 is known to be a strong electron acceptor and forms stable CO2 species on various metal oxide surfaces, as demonstrated, e.g., for Cr2O3(0001) [1]. These activated molecules may serve in further reactions and could contribute to the chemistry required for so-called “climate engineering” or “enhanced weathering”.We employ DFT corrected by a Hubbard-type U parameter, which was shown to perform accurately for water adsorption on Fe3O4(111) [2]. Two adsorption modes for CO2 were found. The first one involves molecularly adsorbed CO2 (see Figure 1). Wavenumbers as well as isotopic shifts (13C and 18O) of an upright standing CO2
agree excellently with observation [3]. In a redox step, CO2 forms an iron oxalate involving electron transfer from the surface to the molecule. The oxalate binds strongly by 150 kJ/mol and calculated IR wavenumbers agree well with experiment proving both adsorption modes. Induced by the surface morphology, the Fe-oct2 and Fe-tet1 sublattices form a coordinative host-environment facilitating the interaction with CO2 in a way similar to supramolecular host-guest chemistry.
Figure 1. Molecular CO2/Fe3O4(111) [left]; chemisorbed CO2 (= C2O42-)/Fe3O4(111) [right].
[1] H. Kuhlenbeck et al., Phys. Chem. Chem. Phys. 96, 15 (1992).[2] P. Dementyev et al., Angew. Chem. Int. Ed. 54, 13942 (2015).[3] F. Mirabella, S. Schauermann, H.-J. Freund 2016, in preparation.
116

Structure and reactivity of ceria single crystal surfaces studied by IR spectroscopy
Chengwu Yang, Alexei Nefedov, Yuemin Wang, and Christof WöllInstitute of Functional Interfaces, Karlsruhe Institute of Technology, 76344
Eggenstein-Leopoldshafen, Germany
Ceria (CeO2), one of the most easily reducible metal oxides, exhibits extraordinary reactivity in diverse catalytic processes. The importance of this material has triggered numerous experimental and theoretical studies, in particular aiming at elucidating the properties of oxygen vacancies. The studies on bulk single crystal surfaces, however, represent still a major challenge. On a novel apparatus combining a state-of-the-art FT-IR spectrometer with a dedicated UHV-chamber, we used carbon monoxide (CO) to probe regular sites and oxygen vacancies at stoichiometric and reduced surfaces of CeO2(111), CeO2(110), and CeO2(100) single crystals. It was found that the vibrational frequency of C-O stretching mode strongly depends on facet orientations as well as on the reduction of ceria surfaces. In addition, the coverage-induced frequency shift was also observed. The assignment of the CO stretch frequency as determined by IR-spectroscopy was supported by ab-initio electronic structure calculations using density functional theory. More importantly, our IR data provide direct spectroscopic evidence for the high reactivity of reduced ceria surfaces towards activation of O2 which occurs at oxygen vacancy sites. The obtained results clearly demonstrate that the application of CO adsorption as an IR-probe is quite suitable for the determination of facet orientations as well as for probing surface oxygen vacancies and allows us to clarify the ambiguous assignments derived from previous powder and thin film data.
117

Hydrogenation and dehydrogenation of hexagonal - Boron Nitride (h-BN) on Ni(111)
F. Späth, F. Düll, C. Gleichweit, U. Bauer, P. Bachmann, H.-P Steinrück and C. Papp
Lehrstuhl für Physikalische Chemie II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Germany
Hexagonal-Boron Nitride, a single atom thick sheet of hexagonally arranged boron and nitrogen atoms, is an interesting two-dimensional material. Here, we study its potential a suitable candidate for future hydrogen storage applications. Weinvestigate its reactivity towards atomic hydrogen, i.e. the hydrogenation, and subsequently the thermally activated dehydrogenation, with high-resolution tempe-rature programmed X-ray photoelectron spectroscopy and temperature programmed desorption. h-BN is prepared via chemical vapor deposition of borazine (B3N3H6) on a Ni(111) single crystal. After exposure to atomic hydrogen at low temperatures, we find distinct chemical shifts in the relevant core level spectra. In the temperature programmed investigation, different dehydrogenation steps are identified at different temperatures, depending on the initial hydrogen coverage. Furthermore, the hydrogen-exposed h-BN is investigated with NEXAFS. At high exposures, the spectra at both the nitrogen and the boron edge show distinctive featurescharacteristic to quasi free standing h-BN, similar to those previously observed forgold-intercalated h-BN on Ni(111). The results are compared to the hydrogenation of graphene on Ni(111).We acknowledge the support from SFB 953 ʻSynthetic Carbon allotropes” and HZB for the allocation of synchrotron beamtime.
118

C–H bond activation at ceria-supported vanadia
Thomas Kropp, Joachim Paier, and Joachim Sauer
Humboldt-Universität zu Berlin, Unter den Linden 6, 10099 Berlin, Germany
Monomeric and oligomeric vanadia species deposited on ceria as a support represent very active catalysts for the selective oxidation of methanol to formaldehyde.1 By virtue of temperature-programmed desorption spectroscopy (TPD) as well as scanning tunneling microscopy, vanadia monomers were found to be more active than aggregations of higher nuclearity.2
Based on a Mars-van Krevelen mechanism, different reaction pathways exploiting the cooperative effect between catalyst and support are presentedincluding transition structures for the rate-determining hydrogen atom transfers. The comparison of different vanadia species provides insight into the nature of the support effect, which affects both methanol adsorption and the hydrogen atom transfer. This will be shown for the kinetically favored pathway (see below), which is based on a structural feature specific to monomeric and dimeric vanadium dioxide.This explains the higher reactivity of catalysts with lower vanadia loadings observed in TPD experiments.3
(1) M. Baron, H. L. Abbott, O. Bondarchuk, D. Stacchiola, A. Uhl, S. Shaikhutdinov, H.-J. Freund, C. Popa, M. V. Ganduglia-Pirovano, J. Sauer; Angew. Chem.-Int. Edit. 2009, 48, 8006-8009.
(2) M. V. Ganduglia-Pirovano, C. Popa, J. Sauer, H. L. Abbott, A. Uhl, M. Baron, D.Stacchiola, O. Bondarchuk, S. Shaikhutdinov, H.-J. Freund; J. Am. Chem. Soc.2010, 132, 2345-2349.
(3) T. Kropp, J. Paier, J. Sauer; J. Am. Chem. Soc. 2014, 136, 14616-14625.
119

Advancing kinetic mechanisms for low-temperature combustion through the detection of highly elusive intermediates
Kai Moshammer, Ahren Jasper, Nils Hansen
Combustion Research Facility, Sandia National Laboratories, Livermore, CA 94551, USA
Understanding combustion mechanisms on a chemically detailed level is ultimately expected to contribute to the development of cleaner engines. In this context, low-temperature (400-700 K) combustion strategies are a promising alternative to conventional high-temperature processes, offering better efficiency and reduced CO2
emissions, while simultaneously decreasing NOx and soot formation. However, in the low-temperature regime, partially oxidized species, such as keto-hydroperoxides or other peroxy species, are of particular importance but have also experimentally been hard to detect.
In this study, we report about the detection, identification, and quantification of such highly elusive species enabled by coupling a jet-stirred reactor with molecular-beam sampling capabilities to a mass spectrometer that employs synchrotron-generated vacuum-ultraviolet radiation for single-photon ionization. The obtained experimental results support the generally accepted classical low-temperature oxidation scheme;in particular, this was shown for the proposed diesel alternative dimethyl ether. But simultaneously, the detection of unexpected species revealed new opportunities for refining the mechanism. Furthermore, highly oxygenated species that are not part of the classical scheme were detected in the experiments when using more-carbonized fuels. These new intermediates are found to be additional radical chain-branching intermediates, suggesting the presence of an extended set of auto-oxidation reactions.
120

The primary photochemical processes of a ferrocyclobutadiene in liquid solution studied by ultrafast mid-infrared spectroscopy
Boris Wezisla, Jörg Lindner, and Peter VöhringerInstitut für Physikalische und Theoretische Chemie,
Rheinische Friedrich-Wilhelms-Universität, Wegelerstraße 12, 53115 Bonn, Germany
Metallacyclobutadienes belong to a class of molecules, which are of particular interest in modern catalysis, since they have been shown to be key intermediates in alkyne metathesis reactions. These reactions are important tools in the synthesis of complex molecules, drugs and polymers.
Our group recently communicated laser flash photolysis experiments combined with time-resolved Fourier-transform infrared spectroscopy of the pseudo-square-pyramidal tricarbonyl ferracyclobutadiene, 2-C3(NEt2)3}(CO)3]+, in an effort to explore possible ways to generate yet unknown ferracycles with light. This study focused on the timescale of nanoseconds to milliseconds, and the signals were attributed to a primary cleavage of an equatorial carbonyl ligand within the time resolution, followed by a reoccupation of the formed coordinative vacancy by a solvent molecule or counter-ion.
Here, we expand this study by exploring the primary processes leading to the formation of this solvent species after excitation of metal-to-carbonyl charge-transfer states with 355 nm or 266 nm laser pulses. The ensuing photochemical processes are detected in a time- and frequency-resolved manner using femtosecond mid-infrared (fs-MIR) spectroscopy in the carbonyl stretching region (1900 – 2100 cm–1).
The spectro-temporal evolution of the transient signal reveals that the primary events are complete within 300 ps. The primary quantum yield for the formation of a FeCBD featuring a solvent molecule in the equatorial binding site is found to be less than 20%. The results are discussed in terms of TD-DFT calculations and extensive modeling of the time-dependent MIR response.
LiteratureJ. Torres-Alacan, U. Das, B. Wezisla, M. Straßmann, A. C. Filippou, P. Vöhringer Chem. Eur. J. 2015,21, 17184.
121

A comprehensive kinetic model for the excited state dynamics ofall-trans retinal
Oliver Flender, Mirko Scholz, Jonas Hölzer, Thomas Lenzer and Kawon OumUniversität Siegen, Physikalische Chemie, Adolf-Reichwein-Str. 2, 57076 Siegen
Derivatives of retinal, the aldehyde of vitamin A, play a key role as chromophores in rhodopsin photoreceptor proteins and fulfil diverse functions, such as energy conversion, cellular signalling, vision and photopigment regeneration. We present our very recent investigation on the ultrafast excited state dynamics of all-trans retinal in solvents of different polarity using UV-Vis broadband transient absorption spectroscopy in the 260-660 nm range. After photoexcitation of all-trans retinal at 400 nm, we observed excited state absorption of the short-lived (ca. 60 fs) 1Bu
+ state, and subsequent formation of spectral features of the 1Ag
- and 1n * states on the
subpicosecond time scale. The 1n * state mediated the formation of the long-lived T1
triplet state. The 1Ag- state exhibited considerable intramolecular charge transfer
(ICT) character which was particularly evident in highly polar solvents. Photoisomerization of 1Ag
-/ICT resulted in the formation of cis isomers with a solvent-dependent quantum yield. We will present a kinetic mechanism which provides a unified picture of the complex excited-state dynamics of this seemingly simple polyene aldehyde system, with the aid of accurate absolute steady-state absorption spectra of the ground-state isomer species.
122

Vibronic coherence in the first excited * state of
pentafluorobenzene by coupling to the O. Hüter,1 M. Sala,1 H. Neumann,1 Song Zhang,1 H. Studzinski,1
D. Egorova,1 F. Temps1
1Institute of Physical Chemistry, Christian-Albrechts-University Kiel,Olshausenstr. 40, D-24098 Kiel, Germany
Pronounced coherent oscillations observed after femtosecond laser excitation of
pentafluorobenzene to its optically bright * first excited electronic state have been
assigned to vibronic interactions with the energetically close optically dark * state and analyzed quantitatively using three- and six-mode vibronic coupling models. The ensuing dynamics have been monitored using femtosecond time-resolved time-of-flight mass spectrometry and femtosecond time-resolved photoelectron imaging spectroscopy following photoexcitation at wavelengths between = 273 and 240 nm. The observed temporal profiles exhibit a bi-exponential decay behavior with a superimposed, long-lived, large-amplitude oscillation corresponding to a vibrational
frequency of osc = 78 74 cm-1 with a damping time of D = 5 2 ps. The
photoelectron spectra show the population of the * state in the maxima of the oscillation, while the corresponding bands vanish in the minima.
The results of ab initio quantum chemical calculations and time-resolved quantum dynamics simulations show that the excited-state potential energy surface acquires apronounced double-well character along several out-of-plane modes of b1 symmetry. The excited wavepacket oscillates in the anharmonic potential for several picoseconds. In the out-of-plane distorted molecular configuration near the outer
turning points, the excited state gains substantial * character, thus modulating the ionization probability. The anisotropy of the photoelectron angular distribution confirms the periodically changing electronic character. The ionizing probe laser pulse directly maps the wavepacket motion into the observed signal oscillations.
123

Photochemistry of Organic Compounds Adsorbed on Salt ClustersChristian van der Linde, Nina K. Bersenkowitsch, Martin K. Beyer, Universität
Innsbruck, Institut für Ionenphysik und Angewandte Physik, Technikerstraße 25/3, 6020 Innsbruck, Austria
Marine aerosols consist of a large variety of different compounds. They play animportant role for atmospheric processes. Small salt clusters can act as model systems to understand the chemistry and photochemistry of organic compounds adsorbed on these particles. To study these clusters a Fourier Transform-Ion-Cyclotron-Resonance Mass Spectrometer (FT-ICR MS) is coupled to an UV/VIS OPO Laser, tunable from 225nm to 2600nm. Small salt clusters are produced via electrospray ionization. The organic substance of interest is added to the electrospray solution, and the source conditions are modified to optimize adduct formation. To validate the approach, first experiments were performed with three bromo-alcanoic acids with different chain length adsorbed on sodium iodide clusters. Their UV-VIS spectra are compared with the spectra of bare salt clusters. Work on atmospherically more relevant molecules is in progress. UV-VIS spectra are obtained via action spectroscopy. Analysis of the photofragments as a function of wavelengthgives insight into photochemical and photophysical processes.
124

Picosecond photorelease mechanism of coumarin-cages to trigger ultrafast bio/chemical processes
Luuk J.G.W. van Wilderen1, Carsten Neumann1, Daniela Kern-Michler1, Nicole Seibert1, Alexandre Rodrigues Correia2, Matiss Reinfelds2, Alexander Heckel2, Jens
Bredenbeck1
1 Institut für Biophysik, Goethe-Universität, Frankfurt am Main, Germany2 Institut für Organische Chemie und Chemische Biologie, Goethe-Universität, Frankfurt am Main, Germany
In this contribution we present the uncaging mechanism of one of the fastest available photocages. Depending on the leaving group (LG) that is attached to the DEACM coumarin cage ([7-(diethylamino)coumarin-4-yl]methyl-LG), the cargo is released only picoseconds after light absorption. Such light-triggerable molecules allow spatial and temporal triggering of biological and chemical processes if the LG's activity is switched on when unbound. The cleavage process is tracked by ultrafast infrared spectroscopy from 100 fs to 3 ns and shows the release of the thiocyanate and azide LGs. Currently only >ns time scale mechanistic details have been reported [1] which could not uncover the cleavage process itself. In the case of azide, the release kinetics decellerates with increasing water content (used to mimic a morebiological context). For the latter LG we also confirm previously observed minor side reactions that lead to photo-induced dimer formation [2] and even decarboxylation of the ring system of the coumarin cage [3]. The quantum yield of release is about 10-20% for azide as LG, but about a factor two higher for thiocyanate. Understanding the full mechanism may assist in the design of photocages that improve the yield of desired reaction products and suppress that of side products.
[1] B. Schade, V. Hagen, R. Schmidt, R. Herbrich, E. Krause, T. Eckardt, J. Bendig, J. Org. Chem. 1999, 64, 9109–9117[2] G. Ciamician, P. Silber, Ber. Dtsch. Chem. Ges. 1902, 35, 4128–4131[3] N. Kus, S. Breda, I. Reva, E. Tasal, C. Ogretir, R. Fausto, Photochem. Photobiol.2007, 83, 1541–1542
125

o-Benzyne: An elusive reactive intermediate investigated by time-resolved FTIR spectroscopy
J. Torres-Alacan,Abteilung für Molekulare Physikalische Chemie, Institut für Physikalische und
Theoretische Chemie, Rheinische Friedrich-Wilhelms-Universität, Bonn/Germany
Abstract:o-Benzyne (1) was discovered in 1927 accidentally by Bachmann and Clarke during the investigation of the reaction between metallic sodium and boiling chlorobenzene.[1] The formation of triphenylene as a side product could only be rationalized by invoking a bivalent reactive intermediate of molecular formula C6H4. A second benchmark experiment was conducted by Roberts and co-workers in 1953 by reacting 14C labelled chlorobenzene with potassium amide in liquid ammonia at -33 °C.[2] Roberts and co-workers observed that [1-14C]aminobenzene and [2-14C]aminobenzene formed in nearly 1:1 ratio, lending further credibility to the existence of 1 in polar liquids.
* ** *NH3
KNH2
1
NH 2
NH 2Cl+
Although 1 can be represented by several resonance structures, the one corresponding to a strained alkyne explains its reactivity better. Notably, there are more than 75 natural products, among them antitumor agents, which are synthesized using 1 as reactive intermediate.Because 1 is very reactive a large body of its spectroscopic data has been collected in the gas[3] or in the solid phase at cryogenic temperatures.[4] To date only one experiment in liquid [D8]-THF reports the NMR spectrum of 1 generated in the cavity of a hemicarcerand.[5]Here I describe the generation of 1 from phtaloyl peroxide by photolysis in liquid acetonitrile at room temperature. The photochemistry and the ensuing thermal reactions are studied using nanosecond time-resolved FTIR spectroscopy. The experimental data are compared to simulations conducted employing density functional theory calculations for better understanding.[6]
126

[1] Bachmann, W. E.; Clarke, H. T. J. Am. Chem. Soc. 1927, 49, 2089.[2] Roberts, J. D.; Simmons, H. E.; Carlsmith, L. A.; Vaughan, C. W. J. Am. Chem. Soc.1953, 75, 3290.[3] Diau, E. W. G.; Casanova, J.; Roberts, J. D.; Zewail, A. H. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 1376.[4] Radziszewski, J. G.; Hess, B. A.; Zahradnik, R. J. Am. Chem. Soc. 1992, 114, 52.[5] Warmuth, R. Angew. Chem. Int. Ed. Engl. 1997, 36, 1347.[6] Torres-Alacan, J. J. Org. Chem. 2015 Submitted
127

Radiation Chemical Modification of Graphene OxideAxel Kahnt, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen/Germany
Siegfried Eigler, Chalmers University of Technology, Göteborg/Sweden, Roman Flyunt, Leibniz Institute of Surface Modification (IOM), Leipzig/Germany,
Wolfgang Knolle, IOM, Leipzig/Germany, Sergej Naumov, IOM, Leipzig / Germany Bernd Abel; IOM, Leipzig/Germany
In our presentation we like to show our results in the field of reduction and modification of graphene oxide and reduced graphene oxide. Graphene oxide and reduced graphene oxide are heterogeneous materials which are very difficult to reduce or functionalize efficiently. Recently we established the benefit of utilizing reductive transients for the reduction of graphene oxide to reduced graphene oxide (see fig. 1 left)1,2. Here we observed only for e(aq)
- a direct electron transfer2, whereas for the other tested reducing transients the reduction occurred in a concerted electron transfer / reductive elimination mechanism.The observation that non-thermal activated chemical approaches seems to be feasible modifying graphene oxide and related material we investigated the radical addition reactions of alkyl radicals with reduced graphene oxide (fig. 1 right) which are assumed as model reactions for further radiation chemical modification / functionalization of unreactive carbon based nanomaterial. Mechanistic and kinetic aspects for these reactions will be presented.
Figure 1. Left: Scheme of the reduction of graphene oxide to reduced graphene oxide utilizing reductive transients. Right: Modification of reduced graphene oxide. References:[1] A. Kahnt, R. Flyunt, C. Laube, W. Knolle, S. Eigler, R. Hermann, S. Naumov, B. Abel, Nanoscale 2015, 7, 19432–19437.[2] R. Flyunt, W. Knolle, A. Kahnt, A. Prager, A. Lotnyk, J. Malig, D. Guldi, B. Abel, Int. J. Rad. Biol. 2014, 90, 486-494.
128

Soft and hard X-ray ambient pressure X-ray photoelectron spectroscopic characterization of the BiVO4/potassium phosphate
interface for water splitting applicationsDavid E. Starr,1 Fatwa F. Abdi,1 Marco Favaro,2,3 Michael Kanis,1 Hendrik Bluhm,4
Ethan Crumlin,2 Roel van de Krol11Institute for Solar Fuels, Helmholtz-Zentrum Berlin für Materialien und Energie,
Berlin, Germany2Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
3Joint Center for Artificial Photosynthesis (JCAP), Lawrence Berkeley National Laboratory, Berkeley, CA, USA
4Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
Ambient pressure X-ray photoelectron spectroscopy has proven itself a useful technique for in-situ characterization of solid/electrolyte interfaces. With soft X-rays (AP-XPS) water adsorption from the gas phase at pressures up to a few Torr can be studied providing information about the early stages of solid/electrolyte interface formation. The tender X-ray form (AP-HAXPES) can be used to directly interrogate a solid surface under an electrolyte film that is tens of nanometers thick. We have used both photon energy regimes to study the BiVO4/potassium phosphate buffer solution interface (BiVO4/K-Pi), among the most successful semiconducting metal oxide water splitting photoanode systems to date. Our AP-XPS measurements on BiVO4(010) single crystal surfaces indicate that the surface is fully hydroxylated by ~0.5 Torr in direct contrast to density functional theory calculations. We have also investigated the open-circuit behaviour of the thin-film BiVO4/bulk-like K-Pi electrolyte interface when illuminated with a solar-simulator (1 sun, ~900 W/m2). Upon illumination we observe changes in the BiVO4 consistent with the production of a small amount of H+
and significant restructuring of the electrolyte near the interface. These results provide fundamental information about the general behaviour of water splitting photoelectrodes under illumination.
129

Threshold Photoelectron Spectroscopy to trace Chemistry in Combustion, Pyrolysis and Catalysis
Patrick HembergerMolecular Dynamics Group, Paul Scherrer Institute, Villigen, Switzerland
In the last two decades soft photoionization with tunable vacuum ultraviolet (VUV) synchrotron radiation has evolved into a versatile workhorse for the identification and quantification of reactive intermediates and stable molecules in processes of energy conversion such as combustion. Photoionization mass spectra (PIMS), which can be obtained by scanning the photon energy and recording the ion signal, can be useful to distinguish between constitutional isomers, when compared with calculated or experimental data. However, this approach is limited if the ionization energies of different isomers are close, since PI spectra do not show sharp peaks and vibrational fine structure as conventional photoelectron spectra do. Photoelectron photoion coincidence (PEPICO) spectroscopy is an excellent extension to mass spectrometry, since it allows measuring (threshold) photoelectron spectra mass-selectively. These spectra show selective vibrational transitions into ion states, which mostly follow the Franck-Condon principle and thus serve as vibrational fingerprints to identify complex composition of isomers. We will show several examples, which profited greatly from this additional analytical dimension:Hydroxyphenylradicals (HPR) produced selectively in a microtubular reactor decompose to form soot precursor molecules such as propargyl and cyclopentadienyl radicals. Although HPRs were selectively synthesized, they are shown to rearrange to phenoxy radicals, the global minimum on the potential energy surface, prior to decomposition.In an m- entify up to four different isomers of the composition C9H10 and C10H10, which are important findings in terms of soot formation chemistry.Most recently we have carried out experiments in the field of catalytic pyrolysis of lignin model compounds. Zeolites like HUSY were tested towards their capabilities to form reactive molecules when exposed to guaiacol. Compared to bulk phase catalytic pyrolysis fulvene and methyl-cyclopentadienyl was identified, which might provide insight of the mechanisms taking place on the catalyst surface.
130

Dissociation of microsolvated acids.
Ricardo Pérez de Tudela and Dominik Marx,
Lehrstuhl für Theoretische Chemie,
Ruhr Universität Bochum, D-44780 Bochum, Germany
The fundamental question of how many water molecules are required to dissociate
an acid molecule in an aqueous microsolvation environment is both long-standing
and controversial. For HCl interacting with water molecules – one added after the
other – there is convincing evidence that an ion pair, and thus the dissociated acid
molecule, can be stabilized using a minimum number of only four water molecules[1].
Nevertheless, a very recently experiment appeared in the literature[2] which
suggested a revision on this topic. In this experiment the dipole moment of HCl(H2O)n
clusters was measured as a function of the number of water molecules. The key
result of those measurements was a noticeable rise of the total dipole moment of
these clusters when n=6. A tempting explanation was to assign this sudden rise in
the dipole moment to the dissociation of the HCl molecule. However, ab initio path
integral simulations performed in our group disprove this possibility (see Fig. 1). Our
calculations show that the value of the dipole
moment of HCl(H2O)n clusters is completely
uncorrelated to the dissociation state of the acid
molecule (see Fig. 2).
References
[1] A. Gutberlet, G. Schwaab, O. Birer, M. Masia, A. Kaczmarek, H. Forbert, M. Havenith and D. Marx, Science
324, 1545 (2009)
[2] N. Guggemos, P. Slavek and V. V. Kresin, Phys. Rev. Lett. 114, 043401 (2015)
Fig. 1: Ab initio path integral averages of the dipole
moment for the HCl(H2O)n clusters
Fig. 2: Correlation plot between the values of the
dipole moment for more than 2500 HCl(H2O)n
optimized structures and the minimum H-Cl
distance.
131

Hierarchical quantum chemical treatment as a path to sub-kJ/mol accuracy in solid-state applications
Denis Usvyat, Universität Regensburg, Regensburg/Germany
In solids or on surfaces van der Waals interactions play an important role and in many cases are even the dominant binding force. Accurate first principle description of van der Waals dispersion is difficult, especially given the weakness of the interaction and the associated shallowness of the potential surfaces. The latter sets an additional demand onthe accuracy of the underlying theoretical treatment.
Quantum chemical methodological hierarchy, which systematically approaches the exact result, offers a powerful technique to describe van der Waals interaction as well as other binding components in a correct and balanced way. We demonstrate that by combining a low-order periodic treatment and higher-order corrections, evaluated with finite clusters, one can reach sub-kJ/mol accuracy for energy differences in periodic systems [1,2,3,4]. The local correlation scheme defeats the unfavorable scaling wall of the quantum chemical methods, and makes them applicable to relatively complex periodic systems.
We apply the combined periodic/finite-cluster quantum chemical treatment to study physisorption of molecular hydrogen on graphane [4], and helium atoms on MgO surface [3]. In the latter case, the achieved accuracy of the ab initio predictions allowed us to resolve a long standing experimental controversy concerning the energy of the lowest bound state of He on MgO.
[1] J. Yang, W. Hu, D. Usvyat, D. Matthews, M. Schütz, and G. K.-L. Chan, Science 345, 640 (2014)
[2] C. Müller, D. Usvyat , J. Chem. Theory Comput. 9, 5590 (2013)
[3] R. Martinez-Casado, D. Usvyat, L. Maschio, G. Mallia, S. Casassa, J. Ellis, M. Schütz, and N. M. Harrison, Phys. Rev. B 89, 205138 (2014)
[4] D. Usvyat, J. Chem. Phys. 143, 104704 (2015)
132

Measuring macromolecular properties ina “field-free” single molecule trap
Madhavi Krishnan Departments of Chemistry & Physics, Winterthurerstrasse 190, University of Zurich,
CH 8057 Zurich, Switzerland
Mass and electrical charge are fundamental properties of biological macromolecules. While molecular mass has long been determined with atomic precision, we have achieved for the first time a high-precision (< 1 e), absolute measurement of the electrical charge of molecules such as nucleic acid oligomers, and globular and intrinsically disordered proteins in solution. The method is based on parallel, external field-free trapping of nanometer-scale entities in the fluid phase (1,2), requires one molecule, is rapid, and also facilitates an estimate of a dielectric coefficient of the molecular interior. We have further demonstrated direct detection of chemical modifications and single point mutations in proteins. Since net macromolecular charge strongly depends on 3D conformation, high precision electrometry will serve as a new approach to probe molecular structure, dynamics and interactions in solution, down to the level of the single molecule and in real time. The ability to experimentally link electrical charge and molecular structure will likely open up an unexplored physical dimension in biology.
References (1) Krishnan, M., Mojarad, N., Kukura, P. & Sandoghdar, V. Geometry-induced
electrostatic trapping of nanometric objects in a fluid. Nature 467, 692-695 (2010).
(2) Mojarad, N. & Krishnan, M. Measuring the size and charge of single nanoscale objects in solution using an electrostatic fluidic trap. Nature Nanotechnology 7,448-452 (2012).
133

Where has all the Chemistry Gone?
Robert G.W. AndersonClare Hall, University of Cambridge
CambridgeUnited Kingdom
Our knowledge and understanding of the past is helped by the survival and preservation of its material culture. Collecting by museums is an important part of this process, enabling historical research to be carried out, while also allowing for public education to take place through exhibitions. This has been well established for disciplines such as archaeology, natural history, ethnography and the history of art but it is less well appreciated for the history of science, and in particular, chemistry. The Bunge-Prize talk will consider why in general there is so little public presentation of chemistry and why today collecting of instruments and apparatus operates at such a low level. The few recent success stories will also be highlighted.
134

Towards stress imaging with diamond magnetometry Simon R. Hemelaar, Felipe Perona Martinez, Mayeul Chipaux, Femke Oosterhof,
Thamir Hamoh, Andreas Nagl, Romana Schirhagl,Groningen University, Department of Biomedical Engineering, 9713 AW Groningen
Free radicals (some of them are also know under the term reactive oxygen species or ROS) are reactive molecules that usually appear in cells under stress conditions. They are suspected to play a crucial role in numerous pathogenic conditions including the diseases responsible for most deaths worldwide (e.g. arteriosclerosis, cancer, immune responses to pathogens). Despite their relevance relatively little is known about how they work, where they are generated and which ones exactly play a role. Due to their reactivity free radicals are difficult to distinguish from each other by specific assays. Our aim is to provide a solution to this problem. To this end we want to apply diamond magnetometry which is based on a fluorescent defect in diamond which changes its brightness based on its magnetic surrounding. Thus it allows magnetic resonance imaging by reading out an optical signal (no magnetic resonance machine is required). This method so accurate that one can detect the magnetic field caused by a single electron spin or even a few nuclear spins. During this talk I will show the first steps we have achieved and current challenges on the way to this goal. I will talk about bringing diamond into cells, identifying where they reside, controling the position and their potential impact on the cell biology. Finally, I will show our current progress with intracellular diamond magnetometry.
Figure 1: Nanodiamonds ingested in HeLa cells for intracellular stress measurements
135

Unnatural Amino Acids as Novel Probes for Ultrafast 2D-IR Spectroscopy of Proteins
Henrike M. Müller-Werkmeister, Goethe-University, Frankfurt, Germany,
now at: University of Toronto, Toronto, Canada
Two-dimensional IR spectroscopy bears great promise to study structural dynamics in a wide range of biological systems. Using different unnatural amino acids (UAAs) with azide- or nitrile groups [1] as vibrational probes in 2D-IR spectroscopy of proteins, site-specific information on processes such as vibrational energy transfer and local conformational dynamics in proteins with ultrafast time-resolution can be accessed. Results from a comparative experimental study using FTIR and 2D-IR spectroscopy of single unnatural amino acids, suggest azidohomoalanine (Aha) as preferred label for further applications. Two applications are discussed to demonstrate the gain in information of combining 2D-IR with spatial resolution. The first application is the investigation of the local microenvironment in a small globular protein using Aha as probe. The azide moiety shows a shift of its absorption frequency depending on the hydrophobicity of its surrounding. This is used to monitor ultrafast conformational fluctuations on the picosecond time scale by spectral diffusion. Different subensembles with more or less hydrophobic environment around the vibrational probe can be distinguished for the model protein investigated. The second application shown for site-specific labels is the study of vibrational energy flow processes (VET), which are discussed to be relevant for allosteric communication in protein domains [2]. VET can be tracked in great detail by time-resolved IR spectroscopy with high spatial resolution by exciting a localized vibrational mode and probing separate modes in a two-colour 2D-IR experiment [3]. To extend this kind of experiment to proteins, a specific donor-acceptor pair of two UAAs [4] has been designed; with an azulene moiety as donor that can be excited in the visible range but deposits the excess energy by internal conversion into the vibrational modes of the ground state and Aha as acceptor to monitor VET arrival with spatial resolution. In small peptides this VET pair was applied successfully, showing a distant-dependent energy transfer induced signal for VET through covalent bonds, bearing great promise for the direct observation of vibrational energy flow in proteins in real-time [5].
References [1] H. Kim, M. Cho, "Infrared probes for studying the structure and dynamics of biomolecules" Chem. Rev., 113, 5817 (2013) [2] D. M. Leitner, J. E. Straub, “Proteins: Energy, Heat and Signal Flow”, CRC Press, (2009) [3] H. M. Müller-Werkmeister, Y.-L. Li, E.-B. W. Lerch, D. Bigourd, J. Bredenbeck, “Ultrafast Hopping from band to band: Assigning Infrared spectra based on Vibrational Energy Transfer” Angew Chem. Int. Ed. 52, 6214-6217 (2013) [4] H. M. Müller-Werkmeister, J. Bredenbeck, “A donor-acceptor pair for the real-time study of vibrational energy transfer in proteins”, Phys. Chem. Chem. Phys. 16, 3261-3266 (2014) [5] H. M Müller-Werkmeister, M Essig, P Durkin, N Budisa, J. Bredenbeck, “Towards direct measurement of ultrafast vibrational energy flow in proteins”, Ultrafast Phenomena XIX, 535-538 (2015)
136

Mechanism of light-induced water oxidation and oxygen release in photosynthesis
Wolfgang Lubitz,Max-Planck-Institut für Chemische Energiekonversion, Mülheim/Ruhr, Germany
The invention of water splitting by early photosynthetic organisms led to the oxygen-rich atmosphere of our planet and the protective ozone layer in the stratosphere, which were essential for the development of higher life on earth. Photosynthesis stores the sun´s energy via CO2 reduction in form of energy-rich organic compounds(sugars). It is the only basic source of food and biomaterials on earth and has created all fossil fuels (coal, gas, oil). The catalytic water oxidation reaction is performed by a protein-bound Mn4OxCa cluster located in photosystem II (PSII). [1]The cofactor´s reaction cycle is comprised of 5 distinct redox intermediates Sn, where the subscript indicates the number of stored oxidizing equivalents in the Mn cluster (n = 0 to 4) required to split two water molecule and release one O2. A redox-active tyrosine residue couples the fast light-induced single-electron charge separation to the slow catalytic four-electron water oxidation process. The S-states are trapped by laser flash/freeze techniques and their electronic structure is studied by advanced EPR techniques (ENDOR, ESEEM, ELDOR-detected NMR) [2,3]. These data are corroborated by DFT calculations. The results give information on the spin and oxidation states of the manganese ions and the spin coupling in the cluster [4], the function of the Ca2+ [5], the effect of the amino acid surrounding, and the binding, location and reaction dynamics of the 2 substrate water molecules, the O-O bond formation and the release of molecular oxygen [6]. A robust model for the water oxidation mechanism is derived. This information can be used for the design of bioinspired catalysts for water oxidation.
1. a) Umena Y. et al. Nature 473 (2011) 55; b) Suga M. et al. Nature 51 (2015) 99.2. Cox N., Pantazis D., Neese F., Lubitz W., Acc. Chem. Res. 46 (2013) 1588.3. Cox N., Retegan M., Neese F., Pantazis D., Boussac A., Lubitz W. Science 345 (2014) 804.4. Krewald V. et al. Chem. Sci. 6 (2015) 1676; Retegan M. et al. Chem. Sci. 7 (2016) 725. a) Lohmiller T. et al. J. Biol. Chem. 287 (2012) 24721; b) Cox N. et al. J. Am. Chem. Soc. 133 (2011) 3635.6. a) Krewald, V. et al. Inorg. Chem. 55 (2016) 488; b) Peréz Navarro M. et al. Curr. Opin. Chem. Biol.31 (2016), DOI: 10.1016/j.cbpa.2016.02.007
137
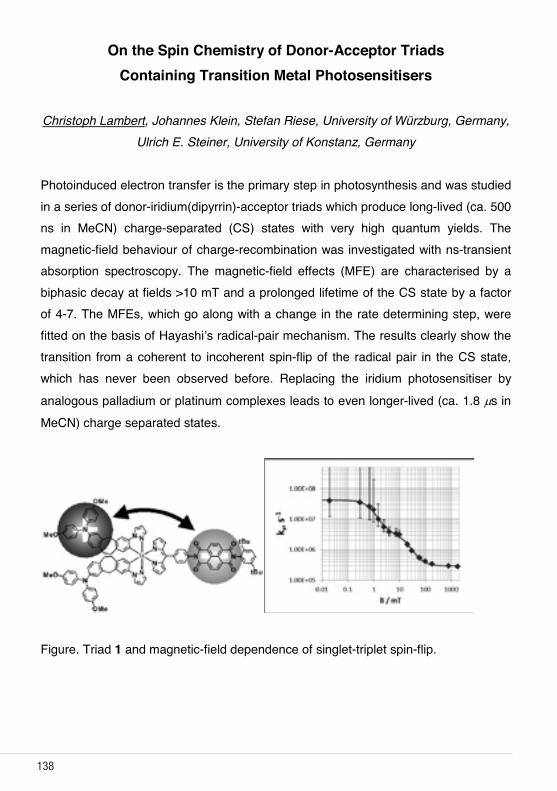
On the Spin Chemistry of Donor-Acceptor Triads Containing Transition Metal Photosensitisers
Christoph Lambert, Johannes Klein, Stefan Riese, University of Würzburg, Germany, Ulrich E. Steiner, University of Konstanz, Germany
Photoinduced electron transfer is the primary step in photosynthesis and was studied in a series of donor-iridium(dipyrrin)-acceptor triads which produce long-lived (ca. 500 ns in MeCN) charge-separated (CS) states with very high quantum yields. The magnetic-field behaviour of charge-recombination was investigated with ns-transient absorption spectroscopy. The magnetic-field effects (MFE) are characterised by a biphasic decay at fields >10 mT and a prolonged lifetime of the CS state by a factor of 4-7. The MFEs, which go along with a change in the rate determining step, were fitted on the basis of Hayashiʼs radical-pair mechanism. The results clearly show the transition from a coherent to incoherent spin-flip of the radical pair in the CS state, which has never been observed before. Replacing the iridium photosensitiser by analogous palladium or platinum complexes leads to even longer-lived (ca. 1.8 s in MeCN) charge separated states.
Figure. Triad 1 and magnetic-field dependence of singlet-triplet spin-flip.
138

Linking Structure and Optical Spectra of Photosynthetic
Pigment-Protein Complexes
Frank Müh
Johannes Kepler Universität Linz, Institut für Theoretische Physik, Altenberger Str. 69, 4040 Linz, Austria
Conversion of solar energy in natural photosynthesis involves two major steps: (i) Light-harvesting, where specialized pigment-protein-complexes (PPCs) absorb light and transfer the excitation energy (excitons) to distinct sites and (ii) the actual photochemical reaction that takes place in distinct PPCs at these sites. In order to understand the excitation energy flow and its efficiency in the light-harvesting systems, we combine quantum chemistry, classical electrostatics and the theory of open quantum systems [1, 2] to calculate optical spectra of the involved PPCs.Progress will be reported in the application of these methods to light-harvesting complexes of higher plants [3] and cyanobacteria [4] with emphasis on the identification of low-energy excited states of chlorophylls as well as to the influence of protonation states on the optical spectra of bacterial light-harvesting complex II [5].
[1] T. Renger, F. Müh (2013) Phys. Chem. Chem. Phys. 15, 3348 – 3371.[2] F. Müh, T. Renger (2014) in J. Golbeck, A. van der Est (eds.) The Biophysics
of Photosynthesis, Springer, pp. 3 – 44.[3] F. Müh, D. Lindorfer, M. Schmidt am Busch, T. Renger (2014) Phys. Chem.
Chem. Phys. 16, 11848 – 11863.[4] F. Müh, M. Plöckinger, H. Ortmayer, M. Schmidt am Busch, D. Lindorfer, J.
Adolphs, T. Renger (2015) J. Photochem. Photobiol. B: Biol. 152, 286 – 300.[5] F. Müh, M. Plöckinger, J. Adolphs, T. Renger, A.T. Gardiner, A.W. Roszak,
R.J. Cogdell (2015) unpublished.
139

Multimetallic terpyridine complexes with highly conjugated ligands for spatially directed energy transport
Maria Wächtler,1Joachim Kübel,1,2 Kevin Barthelmes,3,4 Andreas Winter,3,4 Alexander Schmiedel,5 Christoph Lambert,5 Ulrich S. Schubert,3,4 Benjamin Dietzek1,2,4
1Leibniz Institute of Photonic Technology (IPHT), Jena/Germany2Institute of Physical Chemistry and Abbe Center of Photonics, Friedrich Schiller
University, Jena/Germany 3Laboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller
University, Jena/Germany4 Jena Center for Soft Matter, Friedrich Schiller University Jena, Jena/Germany
5 Institut für Organische Chemie, Universität Würzburg, Wilhelm Conrad Röntgen Research Center for Complex Material Systems, Center for Nanosystems Chemistry,
Würzburg/Germany
Fundamental challenges in designing functional photoactive molecules and molecular materials, e.g., for organic photovoltaics or molecular photocatalysis, include: (i) achieving high absorbance in a broad spectral range and (ii) generating long-lived charge separated states. In this respect photoinduced energy transfer processes in polynuclear complexes presenting building blocks for hierarchically structured coordination polymers are investigated by time-resolved spectroscopy with respect to their ability to act as light-harvesting antennae. The investigated species combine Ru(II) (excitation energy donor) and Fe(II) or Os(II) (acceptor) centers by coordination to highly conjugated bis-4ʼ-terpyridine ligands. Due to delocalization and energetic stabilization of the 3MLCT states and additional formation of equilibria with low lying 3LC states these systems show exceptional long 3MLCT lifetimes.Excitation-intensity dependent effects are discussed in relation to the possibility of multiple excitation at high excitation intensities.
Acknowledgements Deutsche Forschungsgemeinschaft (DFG, Grant Nos. SCHU1229-16/1 and DI1517-3/1), the Fonds der Chemischen Industrie, COST Action CM1202 Perspect-H2O, LaserLab Europe (LLC001917), SolTech Initiative of the Bavarian State Ministry of Science, Research and the Arts
References[1] R. Siebert et al., Journal of Physical Chemistry C, 2011, 115, 12677-12688[2] K. Barthelmes et al., Inorganic Chemistry, 2015, 54, 3159-3171[3] M. Wächtler, et al., Phys. Chem. Chem. Phys., 2016, DOI: 10.1039/c5cp04447b
140

Gas-phase photoluminescence characterization of mass-selected lanthanoid complexes: energy transfer mechanisms
Jean-François Greisch1, Michael E. Harding1 2, Bernhard Schäfer1, Wim Klopper1,2, Mario Ruben1,3, Detlef Schooss1,2, Manfred M. Kappes1,2
1 Institute of Nanotechnology, Karlsruhe Institute of Technology (KIT), D.2 Institute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), D.
3 Institut de Physique et Chimie des Matériaux de Strasbourg (IPCMS),CNRS-Université de Strasbourg, F.
The potential application of charged lanthanoid-antenna complexes ranges from photovoltaics and light-emitting diodes to quantum memories. Rationalization of their design requires a thorough understanding of the intramolecular processes such as energy transfer, charge transfer, and non-radiative decay involving their subunits. Characterization of the complexesʼ excited state properties considerably benefits from mass spectrometric methods since their luminescence is strongly affected by stoichiometry, symmetry, and overall charge state. Furthermore, gas-phase measurements enable an almost direct comparison with computations.The photoluminescence of a series of electrodynamically trapped ions (Paul trap) has been both time- and energy-resolved with trends rationalized in terms of structural and composition changes. The role of the ligand shell and the induced ligand field on the lanthanoidʼs energy levels and related processes, as well as the differences in the vibrationally resolved spectra, are interpreted with the support of density functional theory as well as ligand-field and Franck-Condon computations.Cooperative effects between lanthanoid emitters are investigated.
141

Graphitic carbon nitrides as graphene complements:new insights and challenges
Christoph Merschjann, Freie Universität Berlin – Fachbereich Physik,Arnimallee 14, 14195 Berlin, Germany
"Graphitic" carbon nitride (CN) polymers are currently studied all over the world, mainly for photocatalytic processes like metal-free solar water-splitting [1], but also for solid-state lighting [2], hybrid photovoltaics [3], and organic electronics in general.Bearing its name due to a two-dimensional polymeric structure, the CN material class shows several fascinating properties, such as tunable optical band gap in the UV-VIS-IR range, comparably easy synthesis processes, and high chemical and thermal stability [4].Just recently we could deduce the charge carrier mobility from transient fluorescence measurements, showing that the CN material class comprises reasonable candidates for optoelectronic applications [5]. Moreover, we found that charge transport is mainly confined to channels perpendicular to the two-dimensional structure. The materials thus complement graphene, possibly opening the way for novel applications [5].The talk will give an overview of the latest opto-electronic physical findings in different CN materials. We will also address the challenges of optical and electronicmeasurements in highly scattering samples. Consequences of the unique charge-transport characteristics for energy-conversion applications will be discussed.
[1] X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen, M. Antonietti, Nat. Mater. 2009, 8, 76[2] Y. Zhang, Q. Pan, G. Chai, M. Liang, G. Dong, Q. Zhang, J. Qiu, Sci. Rep. 2013,3, 1943[3] J. Xu, T. J. K. Brenner, L. Chabanne, D. Neher, M. Antonietti, M. Shalom, J. Am. Chem. Soc. 2014, 136, 13486[4] A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl, J. M. Carlsson, J. Mater. Chem. 2008, 18, 4893[5] C. Merschjann, S. Tschierlei, T. Tyborski, K. Kailasam,.S. Orthmann, D. Hollmann, Th. Schedel-Niedrig, A. Thomas, S. Lochbrunner, Adv. Mater. 2015
142

Titanium and Iron substituted LiCoTixMn1-xO4/ LiCo1-yFeyMnO4:energy storage via high-voltage spinels
Christoph Dräger*, Sylvio Indris*, Helmut Ehrenberg*
(*)Karlsruhe Institute of Technology (KIT), Institute for Applied Materials - Energy Storage Systems (IAM-ESS), Eggenstein-Leopoldshafen
Mn-based spinel materials are highly promising high-voltage cathode materials due to their structural peculiarity, resulting in their high rate capability and excellent room temperature cycling stability. The energy density of these Mn spinel based materials can be further increased by substituting Mn with electrochemically active 3d transition metals like nickel, iron or cobalt, so that the operating potential of the cathode is further increased1,2. In the present work, the high-voltage spinel LiCoMnO4 and its titanium or iron substituted derivatives LiCoTixMn1-xO4 (0.02 x0.16) / LiCo1-yFeyMnO4 (0.1 .2) are investigated.
Besides the synthesis, our research is focused on the structural and electrochemical characteristics and the comparison of these materials. Therefore, several characterization techniques such as thermogravimetry in different states-of-charge, NMR spectroscopy, and in situ powder synchrotron diffraction during electrochemical cycling and also during heating are applied.
Galvanostatic cycling reveals that about 70% of the lithium can be extracted and reinserted electrochemically in the voltage window from 3.5 to 5.3 V against Li from/into LiCoMnO4, LiCoTi0,04Mn0,96O4 or LiCo0,9Fe0,1MnO4. In situ synchrotron powder diffraction results show that Li extraction occurs via a single-phase mechanism over the whole range of Li contents for Ti substituted and unsubstituted LiCoMnO4 spinels.
Financial support from the Federal Ministry of Education and Research (BMBF) within the DESIREE project, grant no. 03SF0477B, is gratefully acknowledged.
(1) Alcántara, R.; Jaraba, M.; Lavela, P.; Tirado, J. L. Chem. Mater. 2003, 15 (5), 1210–
1216.
(2) Bhaskar, A.; Mikhailova, D.; Kiziltas-Yavuz, N.; Nikolowski, K.; Oswald, S.;
Bramnik, N. N.; Ehrenberg, H. Prog. Solid State Chem. 2014, 42 (4), 128–148.
143

Energy Conversion in Thermo-Ionic-ElectronicMaterials and Systems
Armin Feldhoff, Institute of Physical Chemistry and Electrochemistry,Leibniz Universität Hannover, Hannover/German y
Recently, thermogalvanic systems known as thermocells have become of interest for thermal energy harvesting, which essentially make use of the Soret effect [1].Similarities with solid-state thermoelectric converters are obvious but have not been discussed in depth so far. It is proposed to look at these energy converters from the point of view of thermo-ionic-electronic (TIE) materials or systems. In addition to ionic and/or electric charge carriers, entropy is considered as further basic quantity being transported through the TIE material or system [2]. Conversion of energy is easily understood as the loading of energy from entropy current (thermal energy) to ionic current or electronic current (both electrochemical energy). Analogies between the Soret coefficient, the Seebeck coefficient and the ionic transfer number become evident. The latter plays an important role in the context of the mixed ionic-electronic conductor (MIEC) [3], which can be considered as TIE under isothermal conditions. If the TIE is simultaneously placed in gradients of temperature and electrochemical potential (ionic and/or electronic), currents of entropy, ionic charge carriers, andelectronic charge carriers are observed. In the basic transport equation, the TIE appears as tensor, which is a major advantage over the concept of the so-called thermodynamics of irreversible processes [4]. The role of energy and its conversion is easily understood by the flux of entropy, ionic charge carriers, and electronic charge carriers at their respective local potentials, which are the temperature, the ionic electrochemical potential, and the electronic electrochemical potential.
References[1] M.S. Romano et al., Carbon nanotube – reduced graphene oxide composites
for thermal energy harvesting applications, Adv. Mater. 25 (2013) 6602[2] A. Feldhoff, Entropy counts, Energy Harvesting and Systems 2 (2015) 3[3] W. Fang, F. Liang, Z. Cao, F. Steinbach, A. Feldhoff, A mixed ionic and
electronic conducting dual-phase membrane with high oxygen permeability,Angew. Chem. (Int. Ed.) 54 (2015) 4847
[4] A. Feldhoff, Thermoelectric material tensor derived from the Onsager – de Groot – Callen model, Energy Harvesting and Systems 2 (2015) 5
144

Environmentally friendly composites for flexible thermoelectrics: concepts and applications
V. Andrei, K. Bethke, K. Rademann, Humboldt-Universität zu Berlin,Berlin/Deutschland
Nowadays, thermoelectricity emerges ever more as an exciting alternative to common renewable energy sources, converting heat directly to electricity.Nevertheless, the large scale implementation of this technology is hindered by the need for rare and toxic elements, such as lead and tellurium, and the material brittleness. Recent work in this field has led to various unconventional substitutes (doped polymers,1 carbon based composites,2 or pastes,3 to name a few), which present a large potential for practical applications. In this presentation, several concepts from the field of thermoelectricity are briefly introduced, including the material optimization for a better energy conversion. After giving an overview of existing thermoelectric composites, we set out to present the benefits and challenges of our investigated oxide-polymer pastes (Fig. 1).3 For this purpose, the sample characterization by a custom made measurement cell is also discussed. Their potential is revealed by the high Seebeck coefficients recorded, reaching 606±5 V/K for a copper(I) oxide iron(III) oxide polychlorotrifluoroethene mixture. The suitability of the thermoelectric pastes for practical applications is also presented, by constructing a flexible temperature sensor. Further composite optimization could significantly increase their applicability, from sensing to electricity generators.
Figure 1. The development of thermoelectric pastes: from characterization and material optimization, to flexible devices.
1 Q. Zhang, Y. Sun, W. Xu, D. Zhu, Adv. Mater. 2014, 26, 6829–6851.2 C. Yu, K. Choi, L. Yin, J. C. Grunlan, ACS Nano 2011, 5, 7885-7892.3 V. Andrei, K. Bethke, K. Rademann, Appl. Phys. Lett. 2014, 105, 233902.
145

X-ray spectroscopic studies of nitrogenase and hydrogenase active sites
Serena DeBeer*
Max Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany; Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York, 14850 USA
e-mail: [email protected]
The efficient storage and release of energy in chemical bonds is a fundamental goal of molecular energy research. It is here that Nature provides an ideal paradigm in that earth abundant metals are utilized to enable bond formation and cleavage. Recent research in our laboratory has focused on understanding the fundamental chemistry of dintritrogen reduction and hydrogen production by the nitrogenase and hydrogenase families of enzymes, respectively. The development and application of advanced X-ray spectroscopic methods to detect the presence of single ligated atoms (e.g. carbide or hydride) will be highlighted. The ability to use valence-to-core X-ray emission spectroscopy as a measure of small molecule activation, as well as two-dimensional X-ray spectroscopic approaches for enhanced selectivity, will be discussed.1,2 Recent advances in our understanding of the electronic structure of the nitrogenase and hydrogenase actives sites will be emphasized.2-9
(1) Hall, E. R.; Pollock, C. J.; Bendix, J.; Collins, T. J.; Glatzel, P.; DeBeer, S. Journal of the American Chemical Society 2014, 136,10076.
(2) Pollock, C. J.; Grubel, K.; Holland, P. L.; DeBeer, S. Journal of the American Chemical Society 2013, 135, 11803.(3) Bjornsson, R.; Lima, F. A.; Spatzal, T.; Weyhermueller, T.; Glatzel, P.; Bill, E.; Einsle, O.; Neese, F.; DeBeer, S. Chemical
Science 2014, 5, 3096.(4) Bjornsson, R.; Neese, F.; Schrock, R. R.; Einsle, O.; DeBeer, S. Journal of Biological Inorganic Chemistry 2015, 20, 447.(5) Lancaster, K. M.; Hu, Y.; Bergmann, U.; Ribbe, M. W.; DeBeer, S. Journal of the American Chemical Society 2013, 135, 610.(6) Lancaster, K. M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.; Ribbe, M. W.; Neese, F.; Bergmann, U.; DeBeer, S. Science 2011, 334,
974.(7) Pollock, C. J.; DeBeer, S. Accounts of Chemical Research 2015, 48, 2967.(8) Rees, J. A.; Bjornsson, R.; Schlesier, J.; Sippel, D.; Einsle, O.; DeBeer, S. Angewandte Chemie International Edition 2015, 54,
13249.(9) Hugenbruch, S.; Shafaat, H. S.; Kramer, T.; Delgado-Jaime, M. U.; Weber, K.; Neese, F.; Lubitz, W.; DeBeer, S., Phys. Chem.
Chem. Phys., 2016, DOI: 10.1039/C5CP07293J.
146

Charge carrier dynamics of methylammonium lead iodide: From PbI2-rich to low-dimensional broadly emitting perovskites
Johannes R. Klein, Oliver Flender, Mirko Scholz, Kawon Oum and Thomas LenzerUniversität Siegen, Physikalische Chemie, Adolf-Reichwein-Str. 2, 57076 Siegen
Organic-inorganic hybrid perovskites hold great promise as efficient materials for solar energy conversion and optoelectronics applications, yet many aspects of their dynamics after photoexcitation are still not well understood. We investigated the charge carrier dynamics of methylammonium lead iodide (MAPbI3) synthesized using different mole fractions of methylammonium iodide (MAI, xMAI = 0.32-0.90).1 The dynamics were studied using ultrafast pump - supercontinuum probe spectroscopy over a wide range of pump laser fluences, allowing for an accurate variation of the initial carrier density. At high MAI excess (xMAI = 0.90), low-dimensional perovskites(LDPs) were formed, and their luminescence spectra were significantly blue-shifted and broadened compared to 3D perovskites. The shift is due to quantum confinement, and the inhomogeneous broadening arises from different low-dimensional MAPbI3 structures. Accurate carrier temperatures were extracted from the transient absorption spectra, and different carrier relaxation regimes were clearly distinguished. Perovskites with xMAI 0.71 exhibited extremely fast carrier cooling (300 fs) at low fluence (2 μJ cm-2), however cooling slowed down significantly at high fluence (50 μJ cm-2) due to the “hot phonon effect” (2.8 ps). A kinetic analysis of the electron-hole recombination dynamics provided second-order recombination rate constants in the range (1.8-6.0) x 10-9 cm3 s-1 for xMAI 0.71. Recombination in the LDPs was more than one order of magnitude faster, 6.4 x 10-8 cm3 s-1 (Fig. 1), which is related to the confined perovskite structure. This should be still slow enough for their application as broadband emitters or solar light harvesting materials.
Fig. 1 Broadband transient absorption spectroscopy reveals an increased carrier recombination rate constant of low-dimensional perovskites.
(1) J. R. Klein, O. Flender, M. Scholz, K. Oum* and T. Lenzer*, PCCP, submitted.
147

Improved charge carrier separation in barium tantalate composites investigated by laser flash photolysis
Jenny Schneidera, Konstantin Nikitina,b, Detlef W. Bahnemanna,b, Roland MarschallcaLeibniz University Hannover, Hannover/Germany
bSaint-Petersburg State University, Saint-Petersburg/RussiacJustus-Liebig-University Giessen, Giessen/Germany
Charge carrier dynamics in phase pure Ba5Ta4O15 and in a Ba5Ta4O15-Ba3Ta5O15
composite have been studied by means of diffuse reflectance laser flash photolysis spectroscopy in presence and absence of methanol, in order to reveal the reason for the improved photocatalytic performance of the composite.[1] For the first time the transient absorption of trapped electrons with a maximum around 650 nm and of trapped holes with a transient absorption maximum around 310 nm is reported for tantalates. The decay kinetics of the photogenerated charge carriers could be fitted by second order reaction kinetics, and the direct recombination of the trapped electrons with the trapped holes was proven. In the absence of an electron donor, no difference in the decay behavior between the phase pure and the composite material is found. In the presence of methanol, for the pure phase Ba5Ta4O15 the recombination of the charge carriers could not be prevented and the trapped electrons also recombine with the CH2OH radical formed via the methanol oxidation by the trapped holes. However, in the composite, the electron can be stored in the system, the CH2OH radical injects an electron into the conduction band of the second component of the composite, i.e., Ba3Ta5O15. Thus, the electrons are available for an extended period to induce reduction reactions.
[1] R. Marschall, J. Soldat, G. W. Busser, M. Wark, Photochem. Photobiol. Sci., 2013, 12, 671.
148

About the evaluation and simulation of solar energyMichael Schmitt, Florian Heib, Saarland University, Germany
It is well-know that solar energy is the by far largest exploitable renewable energy resource on earth. Therefore, understanding the basic mechanisms in energy conversion process is of uttermost importance and multiple scientific studies are under investigation. For all processes the knowledge of the input and output is essential e.g. to compute efficiency and so on. Unfortunately in a huge number of studies the input is often not easily/comprehensively obtained or clearly defined or even measured. This contribution summarizes a detailed study[1] containing meteorological input/verification dealing with the sun as references and the possibility to perform an absolute and spectral calibration of an easily accessible fiber optical detection unit, see left Figure. The procedure based on the data of a pyranometer from the DWD (Deutscher Wetterdienst) calibrated by official sources and areference spectrum (here ASTM G173-03[2]). Both of them are easily accessible/comparable. All of the influencing parameters/aspects of the fiber optical receiver can be calibrated by a developed statistical procedure so that for example a 100 W/m² xenon flash light have been calibrated[3][4], see right Figure. This study additionally underlines that using the sun as reference/illumination light is only possible if the photon flux is continuously monitored.
Figure right: Absolute und spectral calibrating of a fibre optical receiver; left:computed irradiation of a xenon flash light (solar simulated if using a high pass filter).Also the number of photons can be calculated.[1] M. Schmitt, F. Heib, RSC Advances, 2014, 4, 17639-17647.[2] ASTM G173-03, ASTM International, 2012, DOI: 10.1520/G0173-03E01.[3] M. Schmitt, RSC Advances, 2015, 5, 67284 - 67298.[4] M. Schmitt, Nanoscale, 2015, 7, 9532-9544.
149

Ab-initio prediction of pure and mixture gas adsorption of small gas molecules in CPO-27-Mg
Arpan Kundu, Joachim Sauer, Humboldt Universität zu Berlin, Institut für Chemie, Unter den Linden 6, 10099, Berlin, Germany
Kaido Sillar, University of Tartu, Institute of Chemistry, Ravila 14a, 50411, Tartu,Tartu, Estonia
Separation of gas mixture using selective adsorption in porous material is a challenging problem in CCS. The metal organic framework CPO-27-Mg can selectively adsorb CO2 at low partial pressure (below 1 bar) which is attributed to high concentrations of accessible under coordinated metal sites. Improved description of single and multi-component gas adsorption are necessary to design better material and to optimize the condition for gas purification and storage.
The basis of our method [1,2] is calculation of adsorption energies with chemical accuracy using a combination of density functional theory for periodic structures and wave function-based electron correlation methods for finite-size models of adsorption sites. Adsorption isotherms are then calculated analytically with the multisite
for lateral interactions, and with Grand Canonical Monte Carlo (GCMC) simulations on a lattice model with exact treatment of adsorbate-adsorbate interactions.
For adsorption of pure gases both of these methods predict identical adsorption isotherms which shows that mean field approximation ( ) for lateral interactions works very well in case of single component adsorption. We demonstrate that selectivity coefficients calculated from pure gas adsorption data wrongly predict gas mixture adsorption and adsorbate-adsorbate interactions have significant effect on selectivity and hence gas separating abilities.
[1] K. Sillar, J. Sauer J. Am. Chem. Soc. 2012, 18354.[2] L. Valenzano, B. Civalleri, K.Sillar, J. Sauer J. Phys. Chem. C. 2011, 21777.
150

Modeling of adsorption processes at nanostructured cathodes in lithium-sulfur batteries
Jessica Lück1,2,3 and Arnulf Latz1,2,3
1German Aerospace Center (DLR), Institute of Engineering Thermodynamics, Stuttgart , Germany
2Helmholtz Institute Ulm (HIU), Ulm , Germany3University of Ulm, Institute of Electrochemistry, Ulm , Germany
Due to its high theoretical energy density lithium-sulfur batteries are one of the most promising candidates for future energy storage. This position is further based on low costs, an intrinsic overload protection, non-toxicity and the natural abundance of itscompounds [1]. Nevertheless even after 5 decades of research lithium-sulfur cells are still far away from a commercialization. All occurring challenges in the operation of lithium-sulfur batteries are associated with intermediate species, which are produced when lithium reacts with sulfur [2]. These lithium polysufides differ in their chain length and their solubility. Soluble polysulfides generate a redox shuttle between cathode and anode, while insoluble polysulfides precipitate on the anode. This leads to a reduction of active sulfur and to a corrosion and polarization of the anode. One strategy to overcome this is a structuring of the cathode to retain soluble lithium polysulfides [3]. We present a thermodynamically consistent continuum model for nanostructured sulfur carbon composite cathodes. All polysulfides are assumed to be contained in carbon particles. Particular emphasis is put to the adsorption of Li ions on the carbon surface and covers the influence of the electrochemical double-layer. The surface effects are modeled from electrostatic considerations only, without assuming structural properties of the double-layer. Therefore, the model allows us insight into the chemical and physical processes at the electrode-electrolyte interface as well as their influence on the operation of a Li-S battery.
[1] L.F. Nazar et al., J. Mater. Chem., 20, 9821-9826, (2010).[2] S.S. Zhang, J. Power Sources 231, 153-162, (2013).[3] G. Zheng, Nano Lett. 11, 4462-4467, (2011).
151

Modelling SEI Growth: A Novel Description for Porous LayersFabian Single1,2, Birger Horstmann1,2, Arnulf Latz1,2,3
1 Helmholtz Institute Ulm, Helmholtzstraße 11, 89081 Ulm, Germany2 Institute of Engineering Thermodynamics, German Aerospace Center,
Pfaffenwaldring 38-40, 70569 Stuttgart, Germany3 University of Ulm, Albert-Einstein Alee 11, 89081 Ulm, Germany
During the first charge of a fresh lithium ion battery, lithium is intercalated into the negative graphite electrode. Simultaneously, the electrode potential drops and eventually leaves the electrochemical stability window of the electrolyte. Thus, the electrolyte is reduced. The solid reduction products form an electronicallypassivating, but ionically conducting layer on the graphite. This so called solid electrolyte interphase (SEI) prevents further electrolyte reduction and enables large cycle numbers of current lithium batteries [1,2].We simulate SEI growth in a 1D continuum model. Porous electrode theory is employed to describe transport through the porous SEI/electrolyte phase. This enables the simulation and study of porosity fluctuations in the direction of film growth.Similar to previous studies [3,4], we predict SEI thickness evolution. However, our novel approach also results in the prediction of SEI morphology. First, weunderstand that the porosity of the porous SEI layer is nearly constant and predict its value from transport parameters. Second, we predict the ratio between the thickness of the dense and the porous SEI layer depending on electrolyte reductionparameters. Both predictions are experimentally observable and allow model validation.
References:[1] E. Peled, Journal of The Electrochemical Society 126, 2047 (1979).[2] D. Aurbach, Journal of Power Sources 89, 206 (2000).[3] M. B. Pinson and M. Z. Bazant, Journal of the Electrochemical Society 160, A243 (2012).[4] J. Christensen and J. Newman, Journal of The Electrochemical Society 151,A1977 (2004).
152

Graphene-Based Nanocomposites with hierarchical structures as High-Performance Electrodes for Lithium (Sodium) Storage
Chao Wu ‡ Yuan-li Ding‡ Joachim Maier, Yan Yu,*,††Department of Materials Science and Engineering, University of Science and
Technology of China, 230026, Hefei, Anhui, P. R. China.‡ Max Planck Institute for Solid State Research, Heisenbergstr. 1, Stuttgart, 70569,
GermanyGraphene, the one-atom-thick 2D single layer sp2-bonded carbon, exhibits many unique and amazing properties such as high electrical conductivity, high surface area, and excellent chemical stability. These properties make graphene a good carbon candidate to improve the electrochemical properties of lithium (sodium)-ion and sodium-ion electrode materials. However, graphene sheets are easy to agglomerate, leading to an unsatisfied improved effect on graphene-based electrode materials. On the other hand, the active electrode materials based on different storage mechanism further propose other requirements for conductive graphene networks. For example, Sn-based materials, metal oxides, and metal sulfides with high theoretical capacity undergo large volume changes during lithiation/delithiation, and thus destroy the graphene networks. Currently, how to integrate graphene and active electrode particles into advanced electrode materials is still an important and challenging study. In this talk, I will summarize recent research results on graphene-based composites, including structure design, synthesis method and electrochemical properties. By the hierarchical architecture design and 3D graphene-network constructing, the obtained graphene-based composites1-3 (i.e. NaTi2(PO4)3, Sn-Co alloy and metal sulfides) show excellent electrochemical performances in terms of reversibility, cycle-stability and rate-capability. This study provides further understanding and new strategies for developing advanced graphene-based electrode composites.References:(1) Wu, C.; Kopold, P.; Ding, Y.-L.; van Aken, P. A.; Maier, J.; Yu, Y. Acs Nano 2015, 9, 6610.(2) Wu, C.; Maier, J.; Yu, Y. Advanced Functional Materials 2015, 25, 3488.(3) Wu, C.; Maier, J.; Yu, Y. Advanced Materials 2015, DOI: 10.1002/adma.20150396.
153

Defects in porous networks of WO3 particle aggregates
Augusto Márquez,a,b Manuel J. Rodríguez-Pérez,c Juan A. Anta,b Geonel Rodríguez-Gattorno,c Gilles Bourret,a Gerko Oskamc and Thomas Bergera,b
a Paris Lodron University of Salzburg, Salzburg, Austria, b Universidad Pablo de Olavide, Sevilla, Spain, c CINVESTAV-IPN, Mérida, Yucatán, Mexico
The most frequently studied functionalities of porous WO3 particle systems - namely their electrochromic and photocatalytic properties - are intimately related with thewanted or unwanted action of defects in the material. Here we report on the generation and action of three types of defects in WO3 electrodes: solid/solid interfaces between adjacent particle aggregates, defects generated in situ upon photoinduced electron accumulation and defects resulting from tungsten bronze formation. Solid/solid interfaces between preformed WO3 particle aggregates were generated upon thermal annealing at T = 450 °C and 550 °C, respectively. Especially at low annealing temperatures these interfaces constitute bottlenecks for charge transport and/or charge separation as highlighted for the photooxidation of a 0.1 M HClO4 aqueous electrolyte. The detrimental effect of these bottlenecks can be mitigated by the photoinduced or electrochemical double injection of electrons and protons into the oxide structure [1,2] at potentials EAg/AgCl > -0.05 V giving rise to an almost 3-fold, transient increase of the photocurrent. Strong electrochemical doping at EAg/AgCl < -0.05 V, on the other hand, is associated with a major structural modification of the oxide at the solid/electrolyte interface and leads to tungsten bronze formation. Although charge accumulation shows complete reversibility with respect to anodic polarization, electrodes suffer a significant decrease in photoelectrocatalytic activity. Obviously, charge accumulation/extraction cycles lead to the irreversible formation of defects in the thin film, which may act as recombination centers. The dynamic change of thin film properties associated with the doping process is tracked by a combined ATR-IR- and DR-UV/Vis-spectroscopic and electrochemical approach, which allows resolving on the electrochemical potential scale the beneficial and detrimental effects of defects.[1] Zhao, J.; Olide, E.; Osterloh, F. E. J. Electrochem. Soc. 2015, 162, H65-H71.[2] Idígoras, J., Berger, T., Anta, J. A. J. Phys. Chem. C 2013, 117, 1561-1570.
154

Charge Redistribution and Extraction in Photocatalytically
Alina Chanaewa, VU University Amsterdam, 1081 HV Amsterdam, NetherlandsJulius Schmitt, Institute of Physics, University of Freiburg, 79104 Freiburg, Germany
Michaela Meyns, Catalonia Institute for Energy Research, 08930 Barcelona, Spain
Mirjam Volkmann, University of Hamburg, 20146 Hamburg, Germany
Christian Klinke, University of Hamburg, 20146 Hamburg, Germany
Elizabeth von Hauff, VU University Amsterdam, 1081 HV Amsterdam, Netherlands
In this work we present synthesis and characterization of Au-ZnO hybrid nanostructures for solar water splitting application. The photocatalytic activity of pure zinc oxide is limited to a low solar-to-hydrogen conversion because the electron extraction is inhibited by the barrier formed at the solid/liquid interface due to the n-type character of ZnO. Coupling plasmonic gold particles to the zinc oxide nanostructures can help to overcome this limitation. Direct contact of gold and zinc oxide leads to a charge redistribution in the hybrid and facilitated charge extraction.
We present a facile two-step synthesis of tailored nanosized gold-zinc oxide hybrids. In the first step zinc oxide nanopyramids are generated via a basic hydrolysis using oleic acid as a stabilizing agent. In the second step a photocatalytic reaction yieldsgold nanoparticle formation exclusively at the tip of the pyramid. With the help of DFT calculations we discuss the mechanism of surface controlled deposition of gold. Further, charge distribution in novel hybrid nanostructures is investigated usingoptical spectroscopy as well as electrochemical methods. The results demonstrate facilitated electron extraction and therefore the potential of these hybrid nanostructures for solar energy conversion.
155

Novel, Efficient and Stable Composite Photoelectrodes Based on Device-grade Silicon and Chalcopyrite for Visible-Light Driven
Hydrogen and Oxygen Evolution
A. Azarpira1, M. Lublow2, K. Olech1, F. Zepezauer1, A. Steigert1, Ch. Höhn,Ch. Kaufmann1, A. Fischer2, Th. Schedel-Niedrig1
1Helmholtz-Zentrum Berlin GmbH, Hahn-Meitner-Platz 1, D-14019 Berlin 2Albert-Ludwig-Universität Freiburg, Institut für Anorganische und Analytische
Chemie, Albertstraße 2, D-79104 Freiburg
Novel composite photo-cathodes and -anodes based on device-grade Cu(In,Ga)Se2
chalcopyrite and silicon are presented for an efficient and stable solar-driven hydrogen (HER) and oxygen evolution (OER) under acidic conditions.Thin films of phase-pure anatase TiO2 implemented with Pt are used as novel transparent conductive oxide (TCO) deposited on top onto Cu(In,Ga)Se2. The Pt-concentrations are varied in order to optimize simultaneously (i) the conductivity of the films, (ii) the electrocatalytic activity, and (iii) the light-guidance toward the chalcopyrite. Thereby, high incident-photon-to-current-efficiencies (IPCE) of more than 80% could be achieved over the full visible light range [1]. In acidic electrolyte (pH0.3), the most efficient Pt-implemented TiO2-Cu(In,Ga)Se2 composite photo-cathodes reveal (i) photocurrent densities up to 38 mAcm-2 in the saturation region (-0.4 V RHE), (ii) 15 mAcm-2 at the thermodynamic potential for H2-evolution (HER; 0V RHE), and, furthermore, (iii) an anodic onset potential shift for the hydrogen evolution (+0.23 V RHE). Optimized light-incoupling into the device-grade chalcopyrite light-absorber as well as electron conductance properties within the surface-layer are achieved while no degradation was observed over more than 24 hours of operation. Water oxidation catalysts in the form of pre-synthesized powders offer enhanced synthetic variability and scale-up potential compared to crystalline films. Herein, we show the use of RuO2 powder as efficient electrocatalyst and simultaneous synthesis catalyst of protection layers for photoelectrochemical OER device. It is demonstrated that the dual-electrocatalytic capacity of RuO2 for water oxidation and catalytic
156

polymerization enables fabrication of hybrid Si/polymeric layer/RuO2Schottky-junctions. The electrosynthesized interfacial organic protection layer allows sustained photo-assisted water splitting at unprecedented photocurrent densities (15 mAcm-2
during more than 24 hours) for a hybrid system [2]. Detailed analysis of the one-step electrochemical fabrication principles is presented to describe the dehydration, dehydrohalogenation, oxidation and electrochemical reduction reactions upon polymer formation. This understanding on the atomistic/molecular level is allowing thus generalization of the synthesis principles and facilitating future approaches to polymer-protected photoelectrodes.
[1] A. Azarpira, M. Lublow, A. Steigert, P. Bogdanoff, D. Greiner, Ch. A. Kaufmann, M. Krüger, U. Gernert, R. van de Krol, A. Fischer, Th. Schedel-Niedrig, Adv.Energy Mat. 2015, 1402148-1402157. [2] A. Azarpira, J. Pfrommer, K. Olech, Ch. Höhn, M. Driess, B. Stannowski, Th.Schedel-Niedrig, M. Lublow, J. Mat. Chem. A (accepted 6.12.2015); A. Azarpira, Th.Schedel-Niedrig, H.-J. Lewerenz, M. Lublow, Adv.Energy Mat. (submitted 11/2015).
157

Dye molecules induced porosity in electrodeposited zinc ferrite films for enhanced photoelectrochemical response
Dereje H. Taffa and Michael WarkInstitute of Chemistry, Technical Chemistry, Carl von Ossietzky University Oldenburg
Carl-von-Ossietzky-Str. 9-11, 26129 Oldenburg, Germany
dereje. hailu. [email protected]
Zinc ferrite (ZnxFe3-xO4) is a ternary transition metal oxide grouped under spinel ferrites which have attractive magnetic and catalytic properties.1 Recently, it has been investigated as photoanode material for photoelectrochemical water oxidation as standalone photoanode or coupled with other oxide semiconductors and proved to enhance photogenerated charge carrier separation.2
Here we present the electrochemical deposition of ZnxFe3-xO4 thin films usingprotocol reported by Switzer et.al 3 but in the presence of dye molecules (Coumarin 343, Rhodamine B) a strategy we recently applied for hematite.4 The dye molecules possess carboxylate functionality which facilitates the controlled nucleation and growth of the film during the electrodeposition. The influences of deposition time, potential and concentration of the dyes on the structure, composition and photoelectrochemical activity of the electrodes were investigated. The amount of zinc and thus the stoichiometry is controlled by fine tuning of the deposition potential andverified by XPS measurements. The I–V curves show that the ZnxFe3-xO4 electrodes exhibit n-type semiconducting behavior and the photocurrent was strongly dependent on the deposition parameters. Photoelectrodes prepared in the presence of dye molecules exhibit photocurrent density 3-5 times higher than electrodes prepared under similar conditions without dyes.Reference
1. R. Dillert, D. H. Taffa, M. Wark, T. Bredow, D. W. Bahnemann, APL Mater.2015, 3, 104001.
2. .a) A. A. Tahir, K.G. U. Wijayantha, J. Photochem. Photobio. A: Chem. 2010,216,119; b) K. J. McDonald, K-S.Choi. Chem. Mater. 2011, 23, 4863.
3. J. A. Switzer, R.V. Gudavarthy, E. A. Kulp, G. Mu, Z. He, A. J. Wessel, J. Am. Chem. Soc. 2010, 132, 1258.
4. D. H. Taffa, I. Hamm, C. Dunkel, I. Sinev, D. W. Bahnemann, M. Wark, RSC Adv. 2015, 5, 103512.
158

The Electronic Structure of Iridium Oxides Active in Water Splitting V. Pfeifer1,2, T. E. Jones1, J. J. Velasco Vélez1,3, R. Arrigo 1,3, M. Hävecker1,3,
A. Knop-Gericke1, R. Schlögl1,3
1 Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin/Germany2 Helmholtz-Zentrum Berlin für Materialien und Energie, Berlin/Germany
3 Max-Planck-Institut für Chemische Energiekonversion, Mülheim a.d. Ruhr/Germany
Iridium oxide based electrodes are promising candidates for electrocatalyzing the oxygen evolution reaction (OER) in acidic media. To gain a deeper understanding of their electronic structure, we combined X-ray photoemission spectroscopy (XPS) and the near-edge X-ray absorption fine structure (NEXAFS) of benchmark catalysts with theoretical calculations. Our investigations reveal a pre-edge feature in the O K-edge of the catalytically more active X-ray amorphous IrOx, along with additional intensity at higher binding energies in the Ir 4f spectrum when compared to rutile-type IrO2.We identify the former as O 2p hole states and the latter as formally IrIII species. These electronic defects in the near-surface region of the anionic and cationic framework are likely critical for the enhanced activity of X-ray amorphous IrOx.1 To follow the formation of such highly defective iridium oxide surfaces on metallic iridium during the OER and as a function of the applied potential, we have developed two electrochemical cells, based on the proton exchange membrane Nafion®2 as well as graphene-covered SiNx
3, compatible with the Near-Ambient-Pressure XPS setup of the ISISS beamline at the synchrotron BESSYII/HZB.
Figure 1: Nafion® cell Figure 2: SiNx cell
1 Pfeifer, V.; Jones, T. E.; Velasco Vélez, J. J.; Massué, C.; Arrigo, R.; Teschner, D.; Girgsdies, F.;Scherzer, M.; Greiner, M. T.; Allan, J.; Hashagen, M.; Weinberg, G.; Piccinin, S.; Hävecker, M.; Knop-Gericke, A.;Schlögl, R., Surf. Interface Anal., 2015, DOI 10.1002/sia.5895.2 Arrigo, R.; Hävecker, M.; Schuster, M. E.; Ranjan, C.; Stotz, E.; Knop-Gericke, A.; Schlögl, R., Angew. Chem. Int. Ed. 2013, 52 (44), 11660 - 11664.3 Velasco Vélez, J. J.; Pfeifer, V.; Hävecker, M.; Weatherup, R. S.; Arrigo, R.; Chuang, C.-H.; Stotz, E.; Weinberg, G.; Salmeron, M.; Schlögl, R.; Knop-Gericke, A., Angew. Chem. Int. Ed. 2015, 54, 14554-14558.
159

Low Platinum Group Metal Fuel Cell Catalysts: From Surface Science to In-Situ Spectroelectrochemistry
Olaf Brummela, Fabian Waidhasa, Firas Faisala, Roman Fialab, Mykhailo Vorokhtab,Ivan Khalakhanb, Gábor Kovács c, Sergey Kozlov c,
Konstantin Neymanc,d, Vladimir Matolínb, Jörg Libudaa,e
aLehrstuhl für Physikalische Chemie II, Friedrich-Alexander-Universität Erlangen-Nürnberg, GermanybDepartment of Surface and Plasma Science, Charles University Prague, Czech RepubliccDepartament de Quimica Fisica, Universitat de Barcelona, SpaindInstitucio Catalana de Recerca i Estudis Avancats (ICREA), Barcelona, SpaineErlangen Catalysis Resource Center, Friedrich-Alexander-Universität Erlangen-Nürnberg, Germany
Pt-doped CeOx films have been identified as promising anode materials in proton exchange membrane fuel cells (PEMFC) with a high stability and an exceptionally high noble-metal-efficiency. On these films, atomically dispersed ionic platinum is anchored at (100) nanofacets of the nanostructured ceria support. This phenomenon was predicted by density functional theory (DFT) and verified by synchrotron radiation photoelectron spectroscopies under ultra high vacuum (UHV) conditions.1
To explore the chemical state of the Pt species in the ceria matrix under reaction conditions we investigated Pt-doped CeOx films by in-situ electrochemical IR spectroscopy in combination withDFT. Specifically we studied the methanol oxidation at different pH and on different types of thin film samples prepared by magnetron sputtering with and without C spacer layers. We systematically varied the Pt concentration and characterized all samples by SEM, EDX and XPS. Before measurement, we cleaned the surfaces by potential cycling. At pH 6 the film showed good stability even upon prolonged potential cycling. Using the CO that is formed during the reaction weprobe the Pt surface sites on the supported Pt nanoparticles and study their stability. Equivalent experiments on Pt(111) electrodes served as a reference.On all samples, the IR spectra in the CO stretching frequency region are dominated by the on-top CO band between 2000 and 2100 cm-1, identifying Pt0 as the active species. At the highest Pt concentration we differentiate between different COt bands for the adsorption on facets, edges and corners. Upon potential cycling we observe rapid sintering of the nanoparticles. For lower Pt concentrations, we identify “ultrasmall” Pt aggregates, which do not expose crystalline facets.Surprisingly these particle show enhanced stability against sintering. We assume that theseaggregates are formed upon reduction from the atomically dispersed ionic Pt on the ceria support.
We assume that the stabilization effect is causedby the interaction with the ceria support. The results suggest that metal support interaction may help tostabilize very small aggregatesand prevent them from sintering under conditions of dynamically changing potentials.
1. A. Bruix et al., Angewandte Chemie International Edition, 2014, 53, 10525-10530.
160

Electrochemical Degradation Mechanismsof Shape-controlled Platinum Nanoparticles
Isaac Adjei Safo, Carl von Ossietzky Universität Oldenburg, Oldenburg/Germany; Jan-Steffen Haverkamp, Carl von Ossietzky Universität Oldenburg,
Oldenburg/Germany; Mehtap Oezaslan, Carl von Ossietzky Universität Oldenburg,Oldenburg/Germany
In the last 20 years many research groups have studied the relationships between the surface structure and electrocatalytic properties for the cathode fuel cell reaction, oxygen reduction reaction (ORR), by using UHV-prepared Pt-based single crystals.[1] However, the transfer of the results obtained from single crystals to industrial Pt nanoparticles supported on high surface area carbon is very restricted.[2-3] A promising strategy to close or to reduce the material as well asknowledge gap for the ORR is the utilization of shape-controlled nanoparticles.
Here, we present the structural and electrochemical durability of monodispersed cubic-shaped Pt nanoparticles (NPs) for the ORR in acidic media. The focus of this work is to monitor in-situ the changes of surface structure and particle size for the cubic NPs under different electrochemical conditions (scan rate, potential range, cycle number, temperature etc.) by using high resolution (scanning) transmission electron microscopy (HR-(S)TEM), X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS) and surface-sensitive cyclic voltammetry. We provide a degradation model for cubic NPs with the important critical transition states. Our goal is to extend and to develop an advanced electrochemical degradation model for other shape-controlled pure Pt and multi-metallic nanoparticles and to fill the knowledge gap between single crystal surfaces and nano-scaled polycrystalline nanoparticles for the ORR.
Reference[1] N. M. Markovic and P. N. Ross Jr, Surface Science Reports, 2002, 45, 117-229.[2] M. Oezaslan, F.Hasché, P. Strasser, J. Phys. Chem. Lett., 4 (19) (2013) p. 3273–3291.[3] F. Hasché, M.Oezaslan, P. Strasser, ChemCatChem, 3 (2011) 11, p. 1805-1813.
161

Electrochemical Microcalorimetry
Rolf Schuster, Institute of Physical Chemistry,
Karlsruhe Institute of Technology, Kaiserstraße 12, 76131 Karlsruhe, Germany
We are measuring heat effects of electrochemical reactions at single electrodes with conversions down to a few percent of a monolayer. Therefore, we are sensitive to the heat exchange upon surface electrochemical reactions like e.g., hydrogen adsorption, the deposition of the first fractions of a monolayer of Cu on a Au electrode, adsorption of molecules or even charging of the double layer. Also Li-intercalation can be studied at single electrodes.The heat, which is reversibly exchanged at a single electrode is linearly correlated to the entropy of the electrochemical reaction plus a constant offset resulting from electron and ion transport, analogously to the Peltier effect at a junction of solid state conductors. The entropy of the electrochemical (half-cell) reaction includes all side reactions like charge neutral coadsorption processes or ordering of the solvent at the interface. It hence provides complementary information to the current-potentialrelation, which is usually addressed by conventional electrochemical methods.In the current contribution we will briefly review the experimental approaches for electrochemical calorimetry. We then present examples ranging from bulk metal deposition to underpotentially deposited monolayers and first results on double layer charging. We will also compare electrochemical microcalorimetry with other approaches to determine the entropy of electrochemical reactions.
Cyclic voltammogram and molar Peltier heat, measured for Ag underpotential deposition on Au(111)
162

Distinguishing different diffusion behaviors by novel analysis of FCS autocorrelation curves
Seyed Mohsen Jebreiil Khadem1,3, Carsten Hille2, Hans-Gerd Löhmannsröben2,3,Igor Sokolov1,3
1 Humboldt-Universität zu Berlin, Department of Physics, Statistical Physics and Nonlinear Dynamics, Berlin, Germany
2 University of Potsdam, Institute of Chemistry, Physical Chemistry, Potsdam-Golm, Germany
3 Humboldt-Universität zu Berlin, School of Analytical Sciences Adlershof (SALSA), Berlin, Germany
Molecular crowding in living cells can significantly influence diffusion processes. This is of high interest in many innovative strategies such as targeted drug delivery. Fluorescence correlation spectroscopy (FCS) is an ideal analytical tool for studying the diffusive transport of fluorescently labeled particles in living cells. With FCS, the fluorescence intensity fluctuations of particles are detected and analyzed. The resulting autocorrelation curves can be fitted to an appropriate physical model providing information about the diffusion behavior. In the case of anomalous diffusion, the anomalous diffusion exponent is often introduced in the model.Here, we show that fitting to standard model may lead to unreliable determination of the anomalous diffusion exponent , because the precise form of the autocorrelation function strongly depends on the higher moments of particle displacement distribution, and thus is highly sensitive to deviations from Gaussianity. However, we present new theoretical framework for analyzing autocorrelation curves. Thus, the
anomality parameter can be reliably estimated from asymptotic fitting at short lag times. One also obtains several lower moments from this fitting, which makes it possible to test for the Gaussian nature of the diffusion process. In addition, the comparison of the autocorrelation curve behavior at short and long lag times allows for evaluation of the fractality of the diffusion media. We show that the suggested theoretical analyzing procedure can be successfully applied to real experimental FCS data by investigating the diffusion of the fluorescent dye Atto655 in different crowded substrates as well as in living cells.
163

Combined temperature, pressure, and cosolvent effects on enzyme activity
Trung Quan Luong, Roland Winter, Department of Chemistry and Chemical Biology, Biophysical Chemistry, TU Dortmund University, D-44221 Dortmund, Germany
We studied the combined effects of pressure (0.1-200 MPa), temperature (20-40 °C)and cosolvents on the enzyme activity of -chymotrypsin upon the hydrolysis of N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide using a high-pressure stopped-flow system.A kosmotropic osmolyte (TMAO) and a chaotropic agent (urea) and mixtures thereof were used as cosolvents. High pressure enhances the hydrolysis rate as a consequence of a negative activation volume for all solution conditions (-2 to -4mL mol-1). The enhancement is most significant at 20 °C and smaller at high temperatures. Kinetic constants, such as the rate constant of the catalysis (kcat) and the Michaelis constant (KM), were determined as a function of pressure. Compared to the pure buffer solution, addition of 1 M TMAO has minor effects on the kinetic constants, while upon addition of 2 M urea kcat increases by 35% and KM increases6-fold. In the TMAO:urea 1:2 mixture, the urea-effect on kcat and KM is compensated to some extent, and, remarkably, pressure is found to have no effect on the rate of the enzyme reaction. Our data clearly show that by a combination of temperature, pressure and cosolvents, enzyme activity can be effectively modulated and optimized according to changes in environmental conditions.
164

Quantitative approaches in single-molecule micorscopy – can the stoichiometry of protein complexes be measured in cells?Wioleta Chmielewicz, Kristin Grußmayer, Sebastian Bierbaum, Philipp Klein, Oliver
T. Fackler, Dirk-Peter HertenInstitut für Physikalische Chemie, Heidelberg University, Germany
Department für Infektiologie, Virologie, Universitätsklinikum Heidelberg
T-cell receptor clusters play a detrimental role in transmitting the innate immune response, e.g. on viral infections. However, the human immunodeficiency virus type 1 (HIV1) manipulates T-cell receptor (TCR) clusters in infected cells in order to suppress immune response and support viral proliferation (6). There is indication that the viral protein Nef reorganizes the TCR-clusters and changes their constitution. Thus, quantitative data on constitution and stoichiometry of the TCR-clusters can help understanding HIV1 infection. This is, however, not possible with regular optical microscopy as the size of the TCR-clusters is 30-300 nm which is just below the limit of optical resolution. It is well known that the photon-statistics in single-molecule fluorescence spectroscopy carries information about the number fluorophores in the sample (2, 3). Over the past years, we have developed CoPS (Counting by photon statistics) as a robust method for counting the number of fluorophores in a confocal observation volume and to measure their molecular brightness (4-6). Currently, we try to use CoPS for quantifying the number of fluorescently labelled proteins in living cells to determine the stoichiometry of the TCR-clusters and learn about the influence of Nef on their constitution.
References[1] O. T. Fackler, A. Alcover, O. Schwartz, Nat Rev Immunol 2007, 7, 310.[2] H. Ta et al. Laser Phys, 2010. 20, 119.[3] H. Ta et al. PCCP, 2010, 12, 10295.[4] A.Kurz et al. Small 2013, 9, 1461.[5] K.S. Grußmayer et al. ChemPhysChem 2014, 15, 734.[6] K.S. Grußmayer et al. ChemPhysChem 2015, 16, 3578-3583.
165

Determination of the ionization threshold of oligonucleotides and sequence-specific VUV-induced DNA strand breaks using the DNA
origami techniqueS. Vogel, University of Potsdam, Potsdam/Germany; School of Analytical Science
Adlershof, Berlin/Germany, J. Rackwitz, University of Potsdam, Potsdam/Germany,A. Milosavlj , University of Belgrade, Pregrevica/Serbia, A. Guiliani, Synchrotron SOLEIL, Gif-sur-Yvette/France; 5UAR 1008 CEPIA, INRA, Nantes/France, I. Bald,
University of Potsdam, Potsdam/Germany; BAM Federal Institute for Materials Research and Testing, Berlin/Germany
DNA is one of the most important biological molecules and its interaction withionizing radiation is of ongoing interest as the mechanisms of energy deposition, deactivation and accordingly of DNA destruction are still mostly unclear. In this research approach the sequence-specific DNA damage induced by VUV photons on the single-molecule level is studied. Specifically designed oligonucleotides can be attached to DNA nanostructures and adsorbed on a VUV transparent substrate to be irradiated in an Argon filled chamber by a well-defined VUV beam. VUV induced bond dissociations can be visualized by AFM, quantified and plotted against the photon fluence to determine a cross section for single strand breaks for a DNA sequence at a certain irradiation energy (see Fig. below) [1,2].
To investigate the detailed pathway of the photon-induced damage, the same DNA targets are irradiated under the same experimental conditions (photon energies and fluxes), but isolated in an ion trap, thus allowing to determine the ionization thresholds for a specific oligonucleotide. The experiment can be performed by coupling a linear ion trap, fitted with an electrospray ionization source (ESI), to the synchrotron beamline and applying tandem mass spectrometry [3].References:[1] A. Keller et al. 2014 Sci. Rep. 4, 7391.[2] S. Vogel et al. 2015 J. Phys. Chem. Lett. 6, 4589.[3] A. Giuliani et al. 2012 L. Nahon, Angew. Chem. Int. Ed. 51, 9552.
166

DNA origami nanostructures as a platform for optical spectroscopyI. Balda),b), L. Olejko a),b),c), J. Prinza), C. Heck a),b),c)
a)University of Potsdam, Potsdam/Germany; b)BAM Federal Institute for Materials Research and Testing, Berlin/Germany; c)School of Analytical Science Adlershof,
Berlin/GermanyDNA origami nanostructures allow for the arrangement of different functionalities such as specific DNA structures, nanoparticles, proteins and various chemical modifications with unprecedented precision [1]. Additionally they provide a versatile platform, which can be immobilized on various materials, thereby enabling a simple quantification of adsorbed analyte molecules. Recently, we have demonstrated the ion-selective folding of telomeric DNA into guanine (G) quadruplexes on DNA origami platforms [2]. We have used Förster resonant energy transfer (FRET) to demonstrate that the G-quadruplex folding by Na+ ions is suppressed on DNA origami structures (see figure below) [2]. The folding can be reversibly switched, which can be e.g. exploited for selective K+ sensing and the switching of FRET based photonic wires.
Furthermore, gold nanoparticles (AuNPs) can be bound to the DNA origami platforms to create specific and complex plasmonic nanostructures. These can be used for highly-sensitive surface-enhanced Raman scattering (SERS) down to the single-molecule level [3]. Single-nanostructure characterization is conducted based on correlated AFM-Raman imaging. In this way, the Raman enhancement is characterized for different specific nanoparticle aggregates. The aim is to create a versatile and sensitive SERS platform to study complex biomolecules.
References[1] I. Bald and A. Keller, Molecules 19, 9 (2014).[2] L. Olejko, P. J. Cywinski, and I. Bald, Angew. Chem. Int. Ed. 54 (2015).[3] J. Prinz et al., J. Phys. Chem. Lett. 4, 23 (2013).
167

Local tissue manipulation using a force- and pressure-controlled AFM micropipette in a frontloading mode
Phillip Roder, Carsten HilleUniversity of Potsdam, Institute of Chemistry, Physical Chemistry / Applied Laser Sensing in Complex Biosystems (ALSComBi), 14476 Potsdam-Golm, Germany
For the investigation of epithelial transport processes and signaling cascades on single cell or even subcellular level, a precise local manipulation of living cells is advantageous. The combination of a conventional atomic force microscope (AFM) with nanofluidics via a microchanneled cantilever can provide the precise and gentle manipulation of target sites under physiological conditions [1]. This FluidFM technology enables the local volume release of only few fL directly at complex and irregularly structured cellular membranes.Here, a new frontloading and cleaning procedure is described for an AFM micropipette functioning as a reusable force- and pressure-controlled microscale liquid dispenser [2]. The loading and cleaning procedure is evaluated with the organic fluorescent dye rhodamine 6G and the AlexaFluor647-labeled antibody goat anti-rat IgG. The rapid, versatile and cost-efficient frontloading and cleaning procedure of microchanneled AFM micropipettes seems to be beneficial for various biological applications using local manipulation. Possible applications for such micromanipulation of living cells in a complex insect salivary gland tissue are shown.These experiments include the loading of single cells with specific fluorescent dyes and the local stimulation of single cells with biogenic amines. In doing so, the cellular responses can be simultaneously recorded using fluorescence microscopy.
Literature:[1] A. Meister et al., Nano Letters (2009), 9, 6, 2501-2507[2] P. Roder, C. Hille, PLoS ONE (2015), 10(12), e0144157
168

Networking of Biopolymers -Molecular Dynamics on Collagen and Glycosaminoglycans.
Walter Langel, Enrico Peter, Norman Geist, Universität Greifswald
Linear molecules such as glycosaminoglycans and collagens form networks,which form biological structures [1] or are used for cell immobilization [2].Experiments reveal interaction between polysaccharides and collagen strands, and specific contacts were proposed [2, 3].
Our force field molecular dynamics study aims at the atomistic understanding of such point interactions. - Chondroitin sulfate forms ion bridges and mediates the adhesion of longer collagen fragments to titanium dioxide surfaces. - The pure collagen triple helix aggregates to parallel bundles, and this is suppressed bytelopeptides and/or heparin molecules. We now obtained reliable structures of the flexible telopeptide termination from REMD and find the telopeptides wrappingaround the collagen fibrils. Heparin molecules attach to collagen by electrostatics or hydrogen bonds, and sequence specificity probably is due to matching of adjacent side chain structures with the heparine units. Contact maps are calculated and evaluated by statistical methods.
[1] P.N. Lewis, C. Pinali, et al., Structure, 18 (2010) 239-245.[2] D.R. Stamov, T.A.K. Nguyen, et al., Biomaterials, 32 (2011) 7444-7453.[3] J.D. Sanantonio, A.D. Lander, et al. , Journal of Cell Biology, 125 (1994) 1179-1188.
169

RL
Unravelling an underrated noncovalent interaction in proteins through single-conformation IR spectroscopy of model peptides
in the molecular beamChristian W. Müller, Ruhr-Universität Bochum, 44780 Bochum, Germany
Patrick S. Walsh, Timothy S. Zwier, Purdue University, West Lafayette, IN, U.S.A.
The shapes of the allowed regions in the Ramachandran plot of proteins, particularly the diagonal shapes of the R- and L-regions, have prompted a variety of explanations concerning their origin, invoking, for instance, 1–4 steric repulsions,
dipole–dipole electrostatics, and crowded H|Hi+1
atoms.1 Recently, we studied the single-conformation infrared spectra in the amide I and amide II regions for a total of 34 conformations of three -peptides, three -peptides, four / -
peptides, and one -peptide.2 Hessian reconstruction analysis in both the amide I (C=O stretch) and amide II (N–H bend) region revealedthat both the sign and magnitude of the amide I/I
and amide II/II coupling constants are characteristic of the size of the H-bonded ring
linking two amide groups. Seeming exceptions to this rule are I-turn conformations that exhibit a high amide II/II coupling constant for the middle amide group; this amide group, however, turns out to be engaged in an N···HNi+1 H-bond. Attention has
recently been drawn to the potential significance of this “unusual N–H···N hydrogen
bond” for secondary structure formation in proteins.3 Here, we show through Natural Bond Orbital (NBO) and Interacting Quantum Atoms (IQA) analysis that this unusual N···HNi+1 backbone interaction a) explains the diagonal shapes of the R- and L-
regions exhaustively, b) is a short-range resonance interaction, c) has the amide II/II coupling constant and the amide II vibration of the acceptor amide group as spectroscopic reporters, and d) is stable against solvation in an (implicit) solvent.
[1] B.K. Ho, A. Thomas, R. Brasseur, Protein Sci., 2003, 12, 2508.[2] E.G. Buchanan et al., J. Chem. Phys., 2012, 137, 094301.[3] R. Adhikary et al., J. Am. Chem. Soc., 2014, 136, 13474.
170

In situ FTIR Measurements and Applied Chemometrics for Studying Kinetic and Mechanistic Aspects of Alkene Hydroformylation
Christoph Kubisa, Matthias Königa, Mathias Sawallc, Detlef Selentb, Ralf Ludwigab,Klaus Neymeyrac, Robert Franked, Dieter Hessd, Armin Börnerab
aLeibniz-Institut für Katalyse e.V. an der Univ. Rostock, 18059 Rostock, GermanybUniversität Rostock, Institut für Chemie, 18059 Rostock, GermanycUniversität Rostock, Institut für Mathematik, 18057 Rostock, GermanydEvonik Performance Materials GmbH, 45772 Marl, Germany
In order to study mechanistic aspects of catalytic reactions, heterogeneous as well as homogeneous, one approach is to couple kinetic investigations with spectroscopic techniques. Nowadays this is more often accompanied with a chemometric treatment of the data obtained [1]. We perform selected hydroformylation reactions based on homogeneous and heterogeneous catalysts in semi-batch reactor systems which arecoupled with a high pressure transmission IR cell or ATR cell and an automated sampling device for ex situ gas chromatographic analysis [2]. This allows us to studyorganometallic catalyst species time resolved under real reaction conditions by FTIR spectroscopy as well as the often complex educt/product mixture by GC. The spectroscopic data obtained contain information about the particular catalyst species in form of the absorbance spectra and the respective concentration or molar fractionat a definite time. The software tool PCD (pure component decomposition) which is based on matrix factorization performs a recovery of the pure component spectra and their associated profiles of the concentration or molar fraction [3].The interpretation of experimental IR-spectra of catalytic intermediates, which are in most cases non-isolable and sometimes present in low concentrations only, are supported beneficially by considering vibrational spectra calculated by DFT methods.
[1] M. Garland in Mechanisms in Homogeneous Catalysis: A Spectroscopic Approach (Ed.: B. Heaton), Wiley-VCH, Weinheim, 2005, pp. 151-193.[2] C. Kubis, D. Selent, M. Sawall, R. Ludwig, K. Neymeyr, W. Baumann, R. Franke, A. Börner, Chem. Eur. J. 2012, 18, 8780-8794.[3] K. Neymeyr, M. Sawall, D. Hess, J. Chemometrics 2010, 24, 67-74.
171

Ligands on Nanoparticles – Combining the Benefits of Homogene-ous and Heterogeneous Catalysis?
Imke Schrader, Sarah Neumann, Rieke Himstedt, Jonas Warneke, Sebastian Kunz,Institute for Applied an Physical Chemistry (IAPC), Faculty 2, UFT, Bremen,
University of Bremen, Germany
A considerable benefit of supported metal nanoparticles (NPs) in comparison to ho-mogeneous catalysts is their handling in terms of catalyst recovering and performing continuous processes. Homogeneous catalysts however exhibit significant ad-vantages in terms of selectivity control. Especially, chemo- and stereoselectivity, which are fundamental challenges in heterogeneous catalysis, can be achieved with homogeneous catalysts by application of appropriate ligands that guide the reactionthrough specific interactions with the reactant. In the last years it has been demonstrated that the use of ligands is not restricted to homogenous catalysis. Chemo- and even stereoselectivity can be achieved with supported NPs when using ligands (see Fig. 1).1, 2 Evidence was found that the ligandsdo not just act as spectatorsbut enable for alternativereaction pathways, in which the ligand supports reac-tant activation (see Fig. 1). This enables for enhancing selectivity and activity simul-taneous, a phenomenon known from homogenous catalysis as ligand-acceleration. Ligands serve hence as a novel approach for tuning the properties of heterogeneous catalysts.
References:1. I. Schrader, J. Warneke, J. Backenköhler and S. Kunz, J. Am. Chem. Soc.,
2015, 137, 905-912.2. I. Schrader, S. Neumann, R. Himstedt, A. Zana, J. Warneke and S. Kunz,
Chem. Commun., 2015, 51, 16221-16224.
Fig. 1 Recently it was demonstrated that by binding L-proline (yellow) toPt NPs (grey) it is possible to achieve full chemoselectivity toward thedesired product (green) with some stereoselectivity and a simultenousenhancement of activity.1 These findings could be related to a ligand-accelration known from homogeneous catalysis as the N-H effect.
172

Synthesis and Characterization of Ni-Ox and Ni-Nx catalysts for electrochemical water splitting
S.Wagner, J. Ziegler, B. Kaiser, W. Jaegermann, Technische Universität Darmstadt, Darmstadt/Deutschland
Using hydrogen as storage for the discontinuous by available renewable energies requires the development of cheap and efficient catalysts for the water splitting reaction. The most promising noble-metal free catalyst for electrochemical water splitting is nickel. Nickel based catalysts are versatile and suitable for both hydrogen evolution reaction (HER) and oxygen evolution reaction (OER). Nickel is successfully applied in alloys and compounds to produce a low priced, by high efficient water splitting catalyst with an activity almost like platinum.
In this work we investigate nickel nitride and nickel oxide as most promising nickel compounds for the HER. We use reactive dc sputter deposition to control the structure and composition of Ni-Ox and Ni-Nx films by varying the deposition parameters. Thin film formation is also possible at a low temperature, which offers the possibility to prepare also meta-stable or amorphous Ni-phases.
The analysis of the chemical composition by X-ray photoelectron spectroscopy (XPS) before and after the electrochemical measurements ensures a comprehensive picture of the chemical and electrochemical behavior and the stability of the produced catalyst layers. Additionally, using scanning electron microscopy (SEM) information about the composition distribution and the topography of the Ni-Ox and Ni-Nx films can be traced.
173

Uncovering deactivation mechanisms of Pd-Pt catalysts for methane oxidation by X-ray absorption and infrared spectroscopy
Alexey Boubnov, Andreas Gremminger, Maria Casapu, Olaf Deutschmann, Jan-Dierk Grunwaldt, Karlsruhe Institute of Technology, Institute for Chemical Technology and
Polymer Chemistry, Karlsruhe
The catalytic total oxidation of methane is essential for decreasing methane emissions from natural gas vehicles, ships and power plants [1] and this process can be optimised by using supported Pt-Pd catalysts [2]. Our recent work has shown that methane conversion decreases with time [3]. However, addition of NOx can avoid this deactivation keeping a high conversion. The present work addresses the mechanism of the deactivation process by studying systematically the reactivation as well as the effect of NO addition. For this purpose, the structure of the noble metalcatalyst was probed by X-ray absorption spectroscopy (Pd K- and the Pt L3-edge),enhancing sensitivity by modulation excitation spectroscopy (periodic gas-mixture modulations). The bond strength and coordination of hydroxyl, nitro and other surfacegroups and their dynamics during deactivation/reactivation was additionally studied by diffuse-reflectance infrared Fourier-transform spectroscopy.The results indicate that the inhibition of methane oxidation over Pt-Pd by water at T50 = 350°C (corresponding to 50% conversion) is primarily a surface effect caused by hydroxyl group formation at the adsorption sites of the noble metal. The effect of NO strongly depends on the applied temperature with a promoting effect at T > 450°C and inhibition at lower temperatures.
References[1] O. Deutschmann and J.-D. Grunwaldt, Chem. Ing. Technik, 85 (2013) 595.[2] G. Lapisardi, L. Urfels, P. Gelin, M. Primet, A. Kaddouri, E. Garbowski, S. Toppi
and E. Tena, Catal. Today, 117 (2006) 564.[3] A.T. Gremminger, H.W. Pereira de Carvalho, R. Popescu, J.-D. Grunwaldt and
O. Deutschmann, Catal. Today, 258, Part 2 (2015) 470.
174

Using the molecular structure of a tetrapyrrole ligand to tune the oxidation state of metal ions: Co(III)- and Ni(III)-corroles on Ag(111)
Martin Schmid, Malte Zugermeier, Min Chen, Falk Niefind, Jan Herritsch,Ralf Tonner, J. Michael Gottfried
Fachbereich Chemie, Philipps-Universität Marburg Hans-Meerwein-Str. 4, 35032 Marburg
Metal(II)tetrapyrrole complexes are an intensively studied group of molecules withapplications ranging from heterogeneous catalysis to molecular electronics. It is wellestablished that thin films of metal-free tetrapyrroles (e.g. 2H-porphyrins and 2H-phthalocyanines) may be converted to metal(II)tetrapyrroles during a surface confined reaction by incorporating co-adsorbed metal atoms such as Co, Ni, or Fe into the central pockets of the molecules. In order to overcome the limitation to +2 oxidation states while retaining the vacant coordination sites perpendicular to the molecular plane, we substituted the regular porphyrin/phthalocyanine ligands by 3H-corrole ligands. Thesetetrapyrroles have a similar structure than their well-studied counterparts, however, in the unmetalated state their central cavities contain three instead of two hydrogen atoms (connected to N atoms) and they miss one bridging methin (CH) group between two of the four pyrrole subunits. In theory, the tighter coordination environment in combination with the possibility of removing up to three hydrogen atoms during metalation, should favor a +3 oxidation state. Our experiments show that it is possible to metalate corroles in-situ under ultrahigh vacuum (UHV) conditions with Co and Ni atoms, and equally important, to obtain exclusively metal(III)tetrapyrroles with vacant coordination sites perpendicular to the molecular planes. Thus, our approach successfully utilized the structure of an organic ligand as a switch for the oxidation state of the metal ion. In our experiments, we applied X-ray and UV Photoelectron Spectroscopy (XPS, UPS), Scanning Tunneling Microscopy (STM), and Density Functional Theory (DFT) to elucidate the fundamental properties of multilayers and monolayers of in-situ prepared metal(III)corroles on Ag(111) and their interaction with the substrate material.
175

Surface Composition of Free Mixed Nanoscale AerosolsE. Antonsson, B. Langer, C. Raschpichler, D. Marchenko, and E. Rühl
Physical Chemistry, Freie Universität Berlin, Takustr. 3, D-14195 Berlin, Germany
Nanoscopic NaCl/Na2SO4 as a model for marine sea salt aerosols. The crystallization process of droplets of such binary salt solutions isstudied using synchrotron radiation from BESSY II by photoelectron spectroscopy, which is particularly surface sensitive. Intensities of the chlorine 2p and the sulfur 2plines in photoelectron spectra taken at a photon energy of 270 eV are compared for different mixing ratios of the salts. This allows us to determine the chemical surface composition of free, mixed NaCl/Na2SO4 aerosols grown by drying aqueous saline droplets. It turns out that the ratio of the surface constituents deviates significantly from the mixing ratio in the aqueous solution, whereby the minority species in droplets are increasingly found on the surface of the solid mixed aerosols. This result can be explained by the nucleation process during crystallization, in which each of the two salts produces its own pure crystal nuclei rather than crystallizing together. The variation of the surface ion concentration as a function of the mixing ratio in the droplets, as observed here for nanoscopic aerosols, is in contrast to earlier findings suggesting a core-shell structure of mixed salt aerosols, that are in the micron range [1].
[1] Ge et al. J. Colloid Interface Sci. 183, 68-77 (1996).
176

Porphyrin adsorption studies on metal oxide nanoparticles
Johannes Schneider1, Johannes Bernardi2, Thomas Berger1 and Oliver Diwald1
1 Department of Chemistry and Physics, Paris-Lodron University of Salzburg, Austria2 University Service Center for Transmission Electron Microscopy, Vienna University
of Technology, Austria
The technological exploitation of porphyrin functionalized oxide nanostructures and their successful implementation in macroscopic, functional devices still requires abetter understanding of the microscopic interaction between porphyrins and inorganic components [1, 2]. We prepared MgO nanocubes and spherical TiO2 anatase nanoparticles by chemical vapor synthesis and subsequently annealed and oxidized them under high vacuum conditions to achieve clean and dehydroxylated surfaces. The functionalization of these well-defined particle systems with free-base porphyrin (2H-tetraphenylporphyrin, 2HTPP) was studied by DR-UV/Vis and photo-luminescence emission spectroscopy. We followed two different functionalization protocols such as a) porphyrin adsorption on nanoparticle powders at high vacuum conditions and b) porphyrin adsorption out of colloidal toluene based nanoparticle dispersions. For both adsorption protocols we observed in the case of MgO nanocubes ion exchange reactions between the two aminic protons of the porphyrin molecule and Mg2+ ions from the surface and the persistent adsorption of MgTPP at the oxide surface [3, 4]. From studies at high vacuum conditions (protocol a) we learned that low-coordinated surface cations like corners, edges or step-edges serve as reactive sites for porphyrin metalation. The impact of interface type, composition and temperature on the reactive adsorption of porphyrins will be discussed.
____[1] Urbani, M., Grätzel, M., Nazeeruddin, M.K., Torres, T. Chemical Reviews 2014, 114, 12330.[2] Ardo, S., Meyer, G.J. Chemical Society Reviews, 2009, 38, 115.[3] Schneider, J.; Kollhoff, F.; Bernardi, J.; Kaftan, A.; Libuda, J.; Berger, T.; Laurin, M.; Diwald, O. ACS Applied. Materials and Interfaces 2015, 7 (41), 22962–22969.[4] Schneider, J.; Franke, M.; Gurrath, M.; Röckert, M.; Berger, T.; Bernardi, J.; Meyer, B.; Steinrück, H.-P.; Lytken, O.; Diwald, O. Chemistry - A European Journal 2015, DOI: 10.1002/chem.201503661
177

Coupling of quantum well emission to surface plasmon modes in rolled-up microtubes
Jan Siebels, Hoan Vu, Alf Mews, and Tobias Kipp — Institute of Physical Chemistry,University of Hamburg, Grindelallee 117, 20146 Hamburg, Germany
It has recently been shown that dielectric grating structures with gradually varying eff-ective refractive indices in the direction perpendicular to the periodicity can be utilized for the excitation of surface plasmons in a wide spectral range [1]. This general idea of broad band surface plasmon excitation can be transferred to simple metallic gratings with gradually varying filling-factors. Here, we present the fabrication and optical characterization of silver grating structures positioned in rolled-up microtubes with embedded quantum well heterostructures. Time-resolved photoluminescence spectroscopy shows spectral shifts and changes of the decay lifetime exclusively in those areas of these structures in which calculations indicate an overlap between surface plasmon dispersion and emission energy of the quantumwell. Based on these observations we propose and discuss possible coupling mechanisms.
[1] J. Ehlermann et al., Appl. Phys. Lett. 106, 101106 (2015).
178

Time-Resolved Vibrational Spectroscopy
Ruth A. Livingstone, Yuki Nagata, Mischa Bonn, and Ellen H. G. Backus,Max Planck Institute for Polymer Research, Ackermannweg 10,
55128 Mainz, Germany
The surfactant sodium dodecyl sulfate (SDS) is widely used as a detergent for both domestic and industrial applications. It forms a self-assembled monolayer on thesurface of water. In a sum frequency generation process by combining a visible and an infrared laser beam the vibrational spectrum of solely the molecules at the interface can be obtained. An expansion of this method into time resolved two-dimensional sum frequency generation spectroscopy could provide information about the interaction between the surfactant and water and between water molecules at the interface on the microscopic scale. In this way two distinct sub-ensembles of water in the presence of this negatively charged SDS surfactant have been identified. One ensemble consists of water molecules close to the SDS headgroup having fairly
. The other group behaves more bulk like and consists of those whosei.e., shared between with subpicosecond energy transfer occurring between them. This is markedly different
interface, which are less heterogeneous, and indicates that the water molecules that interact with the surfactant headgroups havehydrogen-bonding properties different from those of water molecules interacting with the other water molecules.
179

Prebiotic Chemistry in Nanoconfined WaterDaniel Muñoz-Santiburcio and Dominik Marx,
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, Germany
Nanoconfined liquids can present very different properties with respect to the bulk phase. Especially nanoconfined water [1,2] is being extensively investigated nowadays, due to its deep implications in natural processes and technological applications.We present extensive ab initio MD simulations in which we compare several chemical reactions happening in a nanoconfined water lamella at high T and p with the same reactions in bulk water under ambient and high T and p conditions [3,4]. This set of reactions comprises a possible prebiotic peptide cycle [5] and the conclusions may shed some light on some of the many open questions about the origin of life and in the prebiotic chemistry field. On the other hand, our observations can be generalized to any chemical reaction happening in nanoconfined water.We will show that chemistry in nanoconfined water is conditioned by a subtle interplay between the statistical and sterical factors arising from the confinement in itself and the different dielectric behavior of nanoconfined water compared to bulk water. This interplay leads to dramatic changes in the reaction mechanisms and energetics when changing from the bulk regime to the nanoconfined regime.
[1] Wittekindt, C. & Marx, D. J. Chem. Phys. 137, 054710 (2012).[2] Muñoz-Santiburcio, D., Wittekindt, C. & Marx, D. Nat. Commun. 4, 2349 (2013).[3] Nair, N. N., Schreiner, E. & Marx, D. J. Am. Chem. Soc. 130, 14148–14160 (2008).[4] Schreiner, E., Nair, N. N. & Marx, D. J. Am. Chem. Soc. 131, 13668–13675 (2009).[5] Schreiner, E., Nair, N. N. & Marx, D. J. Am. Chem. Soc. 130, 2768–2770 (2008).
180

Molecular Dynamics Simulations of Solid-Liquid Interfaces Employing High-Dimensional Neural Network Potentials
Suresh Kondati Natarajan, Vanessa Quaranta, Matti Hellström, Jörg Behler,Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum,
D-44780 Bochum, Germany
Understanding the structural and dynamical properties of water at solid-liquid interfaces is essential for unraveling the atomistic details of key steps inelectrochemistry, heterogeneous catalysis and corrosion. In recent years, inparticular ab initio molecular dynamics simulations (MD) based on density-functional theory (DFT) have contributed significantly to the understanding of these processes.Still, due to their high computational costs, ab initio MD simulations of water interacting with solid surfaces are restricted to small systems and short simulation times. This limitation can be overcome by employing high-dimensional neural network potentials (NNPs), which are constructed from a set of electronic structure data and enable carrying out large-scale simulations with close to first-principles accuracy for a variety of systems [1,2].Here, we present NNPs for liquid water interacting with metal and oxide surfaces using copper and zinc oxide as benchmark systems. First, the convergence of various properties as a function of the supercell size and the thickness of the water layer in typical slab approaches will be investigated allowing to assess the reliability of conventional ab initio MD simulations. Then, the influence of copper and zinc oxidesurfaces on the properties of water will be discussed addressing in particular the local water structure, its density profile, changes in the mobility and hydrogen bond lifetimes as well as possible dissociation processes resulting in chemicalmodifications of the surfaces. Finally, some perspectives for the extension to general electrolyte solutions will be given.
[1] J. Behler, M. Parrinello, Phys. Rev. Lett. 98 (2007) 146401.[2] J. Behler, J. Phys.: Condens. Matter 26 (2014) 183001.
181

The Quantum Dynamics of Tunneling and Electroweak Parity Violation in Trisulfane (HSSSH)
Csaba Fábri, , and Martin Quack,
Physical Chemistry, ETH Zürich, 8093 Zürich, Switzerland
Measuring the parity- pvE between enantiomers of chiral molecules is one of the frontiers of highest resolution molecular spectroscopy and a considerable challenge, which has not been successful so far [1-4]. Over the last decade, considerable progress has been made in the accurate theoretical description of molecular parity violation [5-7], its possible implications for the origin of molecular chirality and biomolecular homochirality, and its role in the stereomutation dynamics of chiral molecules [1-4]. Accurate theoretical predictions of molecular parity violation importantly assist in a search for the most suitable molecular system. Moreover,tunneling must be studied for candidate molecules pvE E±
(the tunneling splitting). We present calculations of the parity-violating potential and the ground-state tunneling splitting for HSSSH. Our computations utilized the quasi-adiabatic channel reaction path Hamiltonian method, the rovibrational GENIUSH program and our coupled-cluster singles and doubles linear response (CCSD-LR) approach [7] to electroweak quantum chemistry. We demonstrate that the dynamics of chirality in HSSSH is dominated by de lege symmetry breaking [8] and HSSSH is
pvE, for which the basic experimental capabilities have been demonstrated in our laboratory [9]. If time permits we shall discuss also isotopic chirality and parity violation in HSSSH isotopomers and other prototypical molecules such as NHDT.
[1] M. Quack, Fundamental Symmetries and Symmetry Violations from High Resolution Spectroscopy, in Handbook of High-resolution Spectroscopy, edited by F. Merkt and M. Quack, (John Wiley & Sons, Ltd., Chichester, New York, 2011), pp. 659-772.[2] M. Quack, Chem. Phys. Lett. 132, 147 (1986).[3] M. Quack, Angew. Chem. Intl. Ed. (Engl.) 28, 571 (1989) and 41, 4618 (2002).[4] M. Quack, J. Stohner and M. Willeke, Ann. Rev. Phys. Chem. 59, 741 (2008).[5] A. Bakasov, T. K. Ha and M. Quack, J. Chem. Phys. 109, 7263 (1998).[6] R. Berger and M. Quack, J. Chem. Phys. 112, 3148 (2000).[7] and M. Quack, Mol. Phys. 113, 1768 (2015).[8] C. Fábri, and M. Quack, Chem. Phys Chem. 16, 3584 (2015).[9] P. Dietiker, E. Miloglyadov, M. Quack, A. Schneider and G. Seyfang, Proc. of the 49th SASP, 152 (2014) and J. Chem. Phys. 143 (2015) in press.
182

Caldeira-Leggett Model Description of Condensed Phase Vibrational Spectroscopy
Sergei D. Ivanov, Fabian Gottwald, Oliver KühnInstitute of Physics, Rostock University, Albert Einstein Straße 23-24, 18059 Rostock
Formulating a rigorous system-bath partitioning approach remains an open issue. In this context the famous Caldeira-Leggett (CL) model that enables a simple modeling of system-bath interactions via so-called spectral density functions has enjoyed popularity. Thus, parametrizing these spectral densities, e.g. from explicit molecular dynamics data, is important for using the model. We discuss that this task is naturally achieved in frequency domain and propose a Fourier-based method that outperforms existing time-domain techniques [1]. Moreover, the widely used rigid bond method turns out to be inappropriate in general and leads to a systematic overestimation of relaxation times, unless the system under study is indeed of the CL form. Although the CL model is a useful theoretical tool, its validity for describing anharmonic dynamics of real systems is often taken for granted. It is shown that such a use does not pass the self-consistency check for a broad class of solute-solvent systems, unless the system part of the potential is effectively harmonic, due to the "invertibility problem" [2]. The check is performed by comparing the spectra resulting from the corresponding generalized Langevin dynamics with their counterparts from explicit classical molecular dynamics. Despite the aforementioned failure there exist systems for which the model performs sufficiently well. Although the ultimate verdict is provided by the self-consistency check only, we discuss possible criteria based directly on molecular dynamics data.
[1] F. Gottwald, S. Karsten, S.D. Ivanov, O. Kühn,J. Chem. Phys.,142 (2015) 244110[2] F. Gottwald, S. D. Ivanov, O. Kühn, J. Phys. Chem. Lett., 6 (2015) 2722–2727
183

Independent-electron Surface Hopping Study of the Nonadiabatic Energy Transfer at Scattering NO molecule from Au(111) surface Alexander Kandratsenka, Bastian C. Krüger, Tim Schäfer, and Alec M. Wodtke
Max-Planck-Institut für biophysikalische Chemie, Göttingen, GermanyInstitut für physikalische Chemie, Georg-August-Universität Göttingen, Germany
Vibrationally excited NO molecules colliding with a Au(111) surface lose a large fraction of their vibrational energy into electronic excitation of the metal. Thisnonadiabatic process represents a major challenge to theories of molecule-surface interaction. We report a comparison of recently performed experiments characterizing the multiquantum vibrational relaxation of NO on Au(111) for a wide range of incidence translational and vibrational energies to two theoretical approaches most frequently used to model nonadiabatic e ects: Independent-Electron Surface Hopping (IESH) and molecular dynamics with electronic friction (MDEF) [1]. We consider the incidence conditions for which both IESH and MDEF fail to reproduce the experimental data and perform a detailed analysis aiming to reveal the de ciences of the theoretical models [2].
References[1] Shenvi, Roy, Tully JCP, 130, 174107 (2009)[2] Krüger et al, J. Phys. Chem. C 119, 3268 (2015)
184

High energy resolution X-ray absorption spectroscopy and TD-DFTcalculations on copper systems
Patrick Müller, Matthias Bauer, University of Paderborn, Paderborn/Germany;
Copper containing enzymes are a prominent example for charge transfer based bio catalysts that enable electron transfer. Modelling complexes like these is of great interest for catalytic applications. A very important factor is the geometry which strongly depends on the oxidation state of the metal. Exact knowledge of involved mechanisms is essential for understanding these compounds. HERFD-XAS provides information about local geometry and the local spin state, by reflecting the details of the lowest unoccupied states[1]. We therefore studied Copper I/II complexes by means of HERFD-XAS spectroscopy. These experimental spectra were compared to TD-DFT calculated spectra to check for the viability of both methods in elucidating geometrical and electronic parameters. One pair of copper complexes with a hybrid guanidine-pyridine ligand (Figure 1, left) only differing in oxidation state, forming a so called entatic state, was of great interest and value due to its biomimetic properties[2].Structures derived from gas phase optimization, crystal structure and relaxed crystal structure were used. In addition to that two extreme structures in tetrahedral and square planar geometry were studied to explore the potential of HERFD-XAS studies for such type of complexes. The experimental spectra of the Cu(I) and Cu(II) compound (Figure 1, right) show clearly distinguishable features in the edge and a clearly separated pre-peak for the copper(II) species.
[1] A. J. Atkins, C. R. Jacob, M. Bauer, Chemistry - A European Journal 2012, 18, 7021–7025.[2] A. Hoffmann et al., Angewandte Chemie (International ed. in English) 2014, 53, 299–304.
Figure 1: DMEGqu (N-(1,3-dimethylimidazolidin-2-ylidene)quinolin-8-amine) Cu complex (left) and experimental HERFD-XAS spectra of the Cu(I) (black) and Cu(II) (grey) compound with a zoom in on the pre-edge region
185

Direct Determination of Absolute Stereochemical Configuration by Coulomb Explosion Imaging
Martin Pitzer, Maksim Kunitski, Till Jahnke, Horst Schmidt-Böcking, Reinhard Dörner, Markus Schöffler, Goethe-Universität Frankfurt am Main, Deutschland
Sabrina Marquardt, Sebastian Marquardt, Robert Berger, Philipps-Universität Marburg, Deutschland
A multitude of techniques is currently in use to distinguish between the two enantiomers of a chiral species. However, the determination of the absolute configuration, i.e. the microscopic structure of the enantiomers, usually involves theory input or semi-empirical rules. The only technique to directly determine absolute configuration has so far been anomalous X-ray diffraction, introduced by Bijvoet in 1951.Recently, we have shown that direct determination of absolute configuration is possible with the technique of Coulomb Explosion Imaging [1]. In this method, several electrons are instantaneously removed from the molecule, e.g. by a femtosecond laser pulse, leading to repulsion between the positively charged cores. The momentum vectors of the ionic fragments after the “explosion” of the molecule can carry information about the initial structure. Coincidence measurements of the fragment momenta with COLTRIMS (Cold Target Recoil Ion Momentum Spectroscopy) allow thus the determination of absolute configuration of simple chiral species.In this contribution, we explain the technical implementation of Coulomb Explosion Imaging and present our results on the chiral prototype CHBrClF. We discuss the extension of the method to larger molecules and give an outlook on further topics in stereochemistry for which Coulomb Explosion Imaging can constitute a benefit.[1] M. Pitzer, et al., Science 341, 1096 (2013)
186

Molecular-Frame Photoelectron Imaging of Controlled ComplexMolecules
Joss Wiese1, Sebastian Trippel1, Jochen Küpper1,2,3
1Center for Free-Electron Laser Science, DESY, Hamburg2The Hamburg Center for Ultrafast Imaging, Hamburg
3Department of Physics, University of Hamburg
Since chemical function arises from the interplay amongst valence electrons, a viewat the evolution of the highest occupied molecular orbitals (HOMOs) during a chemi-cal reaction promises direct insight into the fundamentals of chemistry. Therefore, weemploy tomographic molecular-frame photoelectron imaging of spatially controlledcomplex molecules [1] strong-field ionized by intense near-infrared laser pulses [2].Reconstructed static three-dimensional photoelectron distributions in the molecular-frame velocity space of indole and its water complex will be presented. These allowfor the observation of the moleculesʼ electron density distributions, photoelectron ki-netic energies, and out-of-molecular-plane emission angles in 3D. The experimental-ly retrieved observables are discussed employing an extended strong-field approxi-mation model. Those three observables provide a close glimpse at the laser-distortedHOMO potential surfaces of the investigated molecules in the gas phase and yieldaccess to their changes in polarizability and dipole moment upon ionization. Further-more, the direct comparison of indole and its 1:1 water complex allows inspection ofthe nature of hydrogen bonding in heteroaromatic biomolecules.
[1] Chang, Horke, Trippel, Küpper, Int. Rev. Phys. Chem. 34(4), 557-590 (2015)[2] Holmegaard, Hansen, Kalhøj, Kragh, Stapelfeldt, Nat. Phys. 6(6), 428-432 (2010)
187

Distinction of structural isomers by means of chirpedfemtosecond laser ionization
Nicola Reusch, Viola Krein, Nikolaus Wollscheid, Karl-Michael Weitzel, Philipps-Universität Marburg, Germany
Structural isomers of disubstituted benzenes are difficult to distinguish with most mass spectrometric methods, in particular when both substituents are identical. In those cases, a distinction is possible by coupling mass spectrometry with a chromatographic method. An alternative approach is the combination of femtosecond laser ionization with time-of-flight mass spectrometry (fs-LIMS). Here, the variation of several laser pulse characteristics opens access to a multidimensional analytical technique.For a prototypical example, the xylenes, it has been shown that the ortho- and thepara- isomer can be distinguished in fs-LIMS either by binary phase pulse shaping [1]or by simply imprinting a linear chirp of about -1000 fs2 [2] onto fs-pulses at 800nm wavelength. In the current work we present a systematic chirped fs-LIMS investigation of two different ortho-, meta- and para-disubstituted benzenes, i. e. difluorobenzene and benzenediamine. In both cases the mass spectra for the three structural isomers look similar for transform limited laser pulses. By systematic variation of linear and quadratic chirp we are able to enhance small differences between the ortho-, meta-and para-isomers for specific fragments. Ultimately, we demonstrate, that we are able to distinguish the three structural isomers. In this context we will address i)differences in intensity-driven vs. non-intensity-driven fragmentation channels; ii) the mechanism for the formation of specific fragments; and iii) the relevance of a ring opening as start of the fragmentation process, as it is known for difluorobenzene [3].
[1] J. M. Dela Cruz, V. V. Lozovoy, M. Dantus, J. Phys. Chem. A, 109, 8447 –8450, (2005)[2] G. Urbasch, H. G. Breunig, K.-M. Weitzel, ChemPhysChem, 8, 2185-2188, (2007)[3] A.-M. Boulangera, D. M. P. Holland, D. A. Shaw, P. M. Mayer, J. Am. Soc. Mass Spectrom., 20,
20–24, (2009)
188

Influence of hydrogen addition on the structures ofRuthenium cluster anions
Dennis Bumüller1,2, Eugen Waldt1, Anna-Sophia Hehn2, Reinhart Ahlrichs2,Manfred M. Kappes1,2 and Detlef Schooss1,2;
1Institut für Nanotechnologie, KIT, Postfach 3640, 76021 Karlsruhe, Germany;2Institut für Physikalische Chemie, KIT, Kaiserstr. 12, 76128 Karlsruhe, Germany
Ruthenium nanoparticles are known to play an important role as heterogeneous catalyst for processes like methanation reactions, Fischer-Tropsch and ammonia synthesis. Gas phase studies on model catalysts have shown to be an useful tool to unravel underlying physical and chemical mechanisms in such systems. However, a precondition to a deeper understanding is the knowledge of the geometrical structures, ideally under process conditions. As first step into this direction we investigate the structural changes induced by adsorption of hydrogen for Ru14
- andRu19
- using trapped ion electron diffraction. Hydrogen adducts were generated by adding H2 or D2 to the seed gas in a magnetron sputter cluster source. Depending on the source condition varying hydrogen loads were observed. The response of the Ruthenium structures depends on the core structure type and the number of hydrogen atoms attached: Under high loading conditions adsorption of hydrogen(Ru14D35
- and Ru19D51-) results in a fundamental change of the Ru core structural
motif: for both cluster systems icosahedral structures were found. However, in the case of Ru14
- the hexagonal structure of the bare cluster [3] is preserved for lower H2-partial pressures (Ru14D16
-), while for Ru19- even the lowest experimentally achieved
H2 concentration led to an icosahedral core structure.
References
[1] Eugen Waldt, Anna-Sophia Hehn, Reinhart Ahlrichs, Manfred M. Kappes, and Detlef Schooss. Structural evolution of small ruthenium cluster anions.The Journal of chemical physics, 142(2):024319, 2015.
189

Reversed OH shifts in oxygen and dimers of methanol with furan derivatives
Anja Poblotzki; Martin A. Suhm, Institut für Physikalische Chemie, Georg-August-Universität Göttingen, Tammannstr. 6, 37077 Göttingen, Germany
Furan derivatives offer two sites for hydrogen bonding: The oxygen atom as a classical electrostatic acceptor and the -driven binding partner.coordination to the latter competitive. DFT calculations in combination with FTIR-analyzed supersonic jet expansions reveal the stability sequence of the isomers. For 2,5-dimethylfuran the OH stretching band of the -coordinated dimer is atypically further downshifted from the methanol monomer band than in the oxygen-boundcounterpart. A similar case for the coordination of 2,3-benzofuran has been reported by Sasaki et al.[1] and is revisited.The small energy difference between the conformers of such systems offers a testing ground for quantum chemical calculations. Methods with an insufficient or imbalanced description of dispersion energy are unlikely to predict the correct energy and frequency order.[2]
[1] H. Sasaki, S. Daicho, Y. Yamada and Y. Nibu, J. Phys. Chem. A, 2013, 117, 3183.[2] M. Heger, J. Altnöder, A. Poblotzki, M. A. Suhm, Phys. Chem. Chem. Phys., 2015, 17, 13045.
190

The bonus of dispersion – the diphenyl ether-methanol complex investigated using a multi-spectroscopic and theoretical approach
Dominic Bernhard1, Anja Poblotzki2, Chris Medcraft3, Christof Holzer4,Anke Stamm1, Fabian Dietrich1, Jonas Altnöder2, Matthias Heger2, Sabrina Zinn3,
Martin A. Suhm2, Melanie Schnell3, Willem Klopper4, Markus Gerhards1
1TU Kaiserslautern, Fachbereich Chemie, Erwin-Schrödinger-Str. 52, 67663 Kaiserslautern
2Universität Göttingen, Institut für Physikalische Chemie, Tammannstr. 6, 37077 Göttingen
3MPI für Struktur und Dynamik der Materie, Luruper Chaussee 149, 22761 Hamburg4KIT, Institut für Physikalische Chemie, Fritz-Haber-Weg 2, 76131 Karlsruhe
Dispersion interactions are ubiquitous regarding intra- and intermolecular interactions. Their relative contributions strongly vary depending on the investigated system and the theoretical prediction of these contributions is challenging. Diphenyl ether represents a system with competing docking sites for methanol. Both an OH Ohydrogen bond with the ether oxygen atom as a hydrogen bond acceptor and anOH binding to one of the two phenyl rings resulting from dispersion interactionshave to be discussed. Interestingly, both binding motifs are theoretically predicted to be almost equally stable, calling for experimental analyses to decide which structure is dominating. Therefore, we investigated the diphenyl ether-methanol complex indifferent molecular beam experiments using FTIR, IR/UV, IR/IR/UV and microwave spectroscopy in combination with a theoretical analysis by applying DFT and SCS-RICC2 methods. The spectroscopic and theoretical analyses are performed in the electronic ground state (S0), the electronically excited state (S1), and the ionic ground state (D0).A comparison of the experimental spectra with DFT calculations in the S0 state clearly shows, that the OH type structure is dominating, whereas the slightly less stable OH O isomer is far less abundant. In the S1 state different isomers are discussed and compared with theory. A structural rearrangement takes place in the D0 state due to electrostatic effects.
191

Characterization of Microsolvated Polycyclic-Aromatic Hydrocarbons and Crown Ethers
from Broadband Rotational Spectroscopy
Amanda Steber1,2, Cristóbal Pérez1, Juan C. López3, Anouk M. Rijs4 and Melanie Schnell1,2
1Max Planck Institute for the Structure and Dynamics of Matter, Hamburg, Germany.2The Hamburg Centre for Ultrafast Imaging, Hamburg, Germany.
3Departamento de Quimica Fisica y Quimica Inorganica,Universidad de Valladolid, Valladolid, Spain.
4Radboud Universiteit Nijmegen, Institute for Molecules and Materials, The Netherlands.
Polycyclic aromatic hydrocarbons (PAHs) are omnipresent in interstellar space and assumed to be at the basis of a complex chemistry, where they can, for example, act as catalysts. Their complexes with other molecules are therefore of great interest. We report here the broadband rotational spectroscopy study of several PAHs, such as acenapthene and corannulene and their complexes with water, which is of fundamental interest with respect to ice formation on grain surfaces. Complexes with up to three water molecules were observed so far. Using isotopically enriched H2
18O,the positions of the oxygen atoms can be precisely determined. Interestingly, water-water interactions are found to be dominant. By exploiting 13C isotopic substitution in natural abundance, we could furthermore precisely determine the carbon backbone structures. For the corannulene monomer, C20H10, the bond lengths very nicely resemble the bond characters predicted using Kekuleʼs resonance structures.In a related project, we investigate cluster formation of crown ethers. They areextensively used in organometallic chemistry due to their unparalleled binding selectivity with alkali metal cations. From a structural point of view, crown ethers are cyclic ethers containing oxygen and/or other heteroatoms, although the most common ones are formed from ethylene oxide units. Given their particular structural features, crown ethers are extremely flexible and possess both hydrophobic and hydrophilic binding sites. Previous studies identified microsolvated crown ethers but in all cases with a chromophore group attached to the structure. Here we present a broadband rotational spectroscopy study of microsolvated crown ethers produced in a pulsed molecular jet expansion. Several 1:1 and 1:2 crown ether-water aggregates are presented for 12-crown-4 and 15-crown-5. The changes induced in the structures of the crown ether monomer upon complexation and the hydrogen-bonding network that holds them together will be also presented and discussed.
192

Vibrational Spectroscopy of Titanium Oxide Anions: Structure-Reactivity Relationship
Xiaowei Song,1,2 Matias R. Fagiani,1,2 Marissa L.Weichman,3 Min Gao,4 Tetsuya Taketsugu,4,5 Andrey Lyalin,5 Daniel M. Neumark,3 and Knut R. Asmis2
1Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany.2Wilhelm-Ostwald-Institut, Universität Leipzig, Linnéstr. 2, 04103 Leipzig, Germany
3Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National Laboratory,Berkeley,California 94720, United States 4Department of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan5Global Research Center for Environment and Energy based on Nanomaterials Science (GREEN), National Institute for Materials Science (NIMS), Namiki 1-1, Tsukuba 305-0044, Japan
Titanium oxides are one of the most promising materials in solar energy conversion and photocatalysis. Their interactions with H2 and CO2 play important roles in water splitting, photocatalytic oxidation and CO2 chemistry. Spectroscopic studies on size-selected gas phase clusters, combined with high level electronic structure calculations, may aid in unraveling the underlying concepts governing such processes. Here, we use cryogenic ion trap vibrational spectroscopy equipped with the widely-tunable free electron laser (FHI-FEL) IR radiation to study the gas phase structure and reactivity of (TiO2)2-8¯ anions as well as their complexes with D2 and CO2 formed in a cryogenically-cooled ion trap. The bare anions exhibit structureswith 3- and 4-fold coordinated Ti-centers, substantially lower than the 6-fold coordination level found in bulk TiO2, as well as 2- to 4-fold coordinated O atoms.Molecular deuterium adsorbs dissociatively onto Ti2O4¯ already at cryogenic temperatures. Electronic structure calculations show the free energy reaction path way is essentially barrierless and involves migration of a D atom from one to the other Ti-center. Ti3O6¯ reacts with CO2 yielding both carbonate and carboxylate species. The structural characterization allows for new insights into the relationship between the degree of electron delocalization and the reactivity of these radical anions.
193

Thermodynamic Description of Salt-specific Effects in Thermoresponsive Polymers and Copolymers
Jan Heyda, University of Chemistry and Technology, Prague, Czech Republick; Joachim Dzubiella, Richard Chudoba, Bernhard Schulz, Helmholtz-Zentrum Berlin &
Humboldt-University Berlin, Germany
Salty solutions are prevalent in nature, and life and functionality of many species have evolutionary developed so to co-exist in solutions with salts thanks to well-balanced combination of hydration and ion-specific effects. Proteins are one important examples, however, soft materials, such as thermoresponsive polymers [1] are equally, or even more sensitive to the ion-specific interactions. In praxis, when these smart materials are synthesized, they are often characterized solely by simple cloud-point temperature (i.e., LCST) measurement.In our contribution we present a recently developed thermodynamic model, which allows for thermodynamic interpretation of cloud point data [2,3], and in particular to decipher the salt-specific contributions. These properties; transition enthalpy, m-value, and their derivatives, are found to be fairly transferable between biomolecules. Recently, our model was extended to account for electrostatic effects on LCST due to weak charging [3] as well as for the effect of copolymerization [4].Our theoretical findings are supplemented by systematic calorimetric study and microscopic view gained in all-atom MD simulations.
[1] Zhang, Y.; Furyk, S.; Sagle, L. B.; Cho, Y.; Bergbreiter, D. E.; Cremer, P. S. The Journal of Physical Chemistry C 2007, 111, 8916-8924.[2] Heyda, J.; Dzubiella, J. The Journal of Physical Chemistry B 2014, 118(37), 10979-10988[3] Heyda, J.; Soll, S.; Yuan, J.; Dzubiella, J. Macromolecules 2014, 47 (6), 2096-2102[4] Schulz, B.; Chudoba, R.; Heyda, J.; Dzubiella, J. The Journal of Chemical Physics, 2015, 143, 243119
194

SURMOFs – Highly Porous Materials With Huge Potential in Sensor Device ApplicationH. Gliemann, C. Wöll
Institute of Functional Interfaces (IFG), Karlsruhe Institute of Technology (KIT),Karlsruhe/Germany
Metal-organic frameworks MOFs [1] are highly porous, crystalline materials with a huge active surface area (up to 6,000 m2/g) and are usually prepared as bulk materials which might be well suited e.g. as gas storage materials and can be obtained in ton-quantities meanwhile. Due to these outstanding properties as host structures for molecules, MOFs have also high application potential in other fields such as sensor technology, where e.g. the changes in optical properties (refractive index, color) as a result of the guest molecule adsorption can be used as a readout parameter. For many of these applications it is necessary to coat substrates with MOF materials or even to prepare membranes out of it. However, the application of powder MOF material for surface coatings usually results in a polycrystalline, heterogeneous, granular layer of randomly arranged non-oriented MOF crystals with a huge number of defects and diffusion barriers which might hinder or even avoid the diffusion of molecules in or through this layer. Therefore, a layer-by-layer (LBL) deposition technique of MOFs has been developed where the components –inorganic building blocks and organic linkers – are deposited subsequently and iterative on a solid substrate [2]. By this method a homogenous highly transparent layer of surface-anchored metal-organic frameworks (SURMOFs) is obtained [3],whose thickness can be controlled exactly by the number of deposition cycles while the properties of the pores (size, shape, chemical properties) can be controlled by the used linkers. Even the preparation of hetero-layered SURMOFs is possible by the LBL method [4].In this paper the application of SURMOFs as highly porous host materials for molecules in the field of nanoplasmonic sensing (NPS) [5] is reported. SURMOFs are grown by a LBL procedure on the surface of the NPS sensors, which consist of a quartz glass with SiO2 coated gold islands. As the spectral position of the gold plasmonic bands depend on the refractive index of the surrounding medium, and as the loading of the SURMOF pores with guest molecules results in a change of the
195

refractive index of the SURMOF [6] the plasmon bands show a significant and measurable spectral shift as soon as the guest molecules adsorb in the pores (Figure 1) [7]. The combination of SURMOFs that change their optical properties with loading and the method of nanoplasmonic sensing open new fields of application in sensor technology. Here we show the proof of principle with HKUST-1 as the SURMOF material and with cyclohexane and ethanol as guest molecules from the gasphase.
Figure 1: When white light passes through the SURMOF coated sensor chip the plasmonic band of the gold nanodisks can be detected in transmission (a). After loading the SURMOF with guest molecules, the effective refractive index around the gold nanodisks changes, resulting in a spectral shift of the plasmonic band (b).
[1] O.M. Yaghi et al., Nature 378 (1995), 703.[2] O. Shekhah et al., J. Am. Chem. Soc. 129 (2007), 15118.[3] Z.-G. Gu et al., Microporous and Mesoporous Materials 211 (2015), 82.[4] Z. Wang et al., Nano Letters 14 (2014), 1526.[5] E. M. Larsson et al., Science 326 (2009), 1091.[6] E. Redel et al., Appl. Phys. Lett. 103 (2013), 091903/1-5.[7] H. Gulati et al., Proceedings of the 12. Dresdner Sensor-Symposium 2015,
ISBN: 978-3-9813484-9-1, DOI: 10.5162/12dss2015/P7.6 (2015), 234.
196

A thermodynamic study of different cosolute effectson the stability of RNase A
Michael Senske1, Diana Constantinescu2, Hermann Weingärtner1, Christian Herrmann2, Simon Ebbinghaus1, Martina Havenith1
1 Department of Physical Chemistry II, Ruhr-University Bochum, Germany2 Department of Physical Chemistry I, Ruhr-University Bochum, Germany
Understanding the structure and stability of biomolecules in the cellular environment requires knowledge of their interplay with high concentrations of macromolecules,osmolytes, salts and membranes. Since Hofmeister ranked salts according to their potential to precipitate hen egg white proteins, researchers aim to rationalize this Hofmeister series by studying salt effects on protein stability, kinetics, and aggregation [1]. Here, we describe the cosolute effect on the thermal stability of RNase A comparing inorganic salts, ionic liquids and uncharged cosolutes. Dissecting the free energy changes into enthalpy and entropy contributions led to a classification of the cosolutes into specific regimes of the enthalpy-entropy compensation plot [2]. While inorganic salts and ionic liquids caused a protein destabilization at moderate concentrations, their thermodynamic signatures are largely different. Inorganic salts cause an enthalpic destabilization, while all ionic liquids are entropically destabilizing and enthalpically stabilizing. Thereby, the enthalpic stabilization correlated with an increasing hydrophobicity of the cation. The transition from salts to uncharged cosolutes can be understood in the same way. The thermodynamic dissection reveals that Hofmeister series are by no means unique –even for properties of the same system: While the Gibbs free energy and entropy of the ionic liquids follow the same series as constructed based on the changes of the melting temperature, the series for the enthalpy is inverted.
References:[1] Ion Specific Hofmeister Effects, Faraday Disc., 2013, Vol. 160[2] M. Senske et al., J. Am. Chem. Soc., 2014, 136, 9036-9041
197

Ion Imaging of Reaction Dynamics at Surfaces: Eley-Rideal vs Langmuir-Hinshelwood
Jannis Neugebohren1, Dan J. Harding1, Hinrich W. Hahn1, Daniel J. Auerbach1,Theofanis N. Kitsopoulos1,2, Alec M. Wodtke1
1Institute for Physical Chemistry, Georg-August University of Göttingenand Max Planck Institute for Biophysical Chemistry, 37077 Göttingen, Germany
2Department of Chemistry, University of Crete and Institute of Electronic Structure and Laser, Foundation for Research and Technology-Hellas, 71003 Heraklion,
Greece
AbstractA UHV surface scattering apparatus is combined with a spatial imaging setup to obtain a complete picture of the dynamics involved in direct scattering, trapping-desorption and reaction processes at surfaces. Using an imaging plane parallel to the surface normal, laser slicing and delayed extraction allows us to extract the speed and angular distributions of molecules coming from the surface in a single image.In the current experiment we investigate the dynamics of the Eley-Rideal and the Langmuir-Hinshelwood reaction channels when scattering CO from an oxygen covered Pt(111) surface. The image shows both oxidation products, which are separated spatially due to their different velocities.
CO2 product
CO beam
Laser
Pt surface
ER
L
198

199
POSTER

200

Rate and Routes of Electron Transfer in an Enzymatic Fuel CellAlexander Petrenko, Max-Planck-Institut für Dynamik Komplexer Technischer
Systems, Magdeburg/Germany; Matthias Stein, Max-Planck-Institut für Dynamik Komplexer Technischer Systeme, Magdeburg/Germany
Hydrogen converting enzymes, hydrogenases, are versatile biological catalysts with a high turnover number and selectivity towards hydrogen only. Uptake [NiFe]-hydrogenases can be used in enzymatic fuel cells and generate protons and electrons from a heterolytic splitting of molecular hydrogen (H2).
From the buried active site, a relay of three iron-sulfur clusters transports the released electrons to the electron acceptor, which may also be a graphite electrode in fuel cells. The distal iron-sulfur cluster (FeSd) is the most critical redox center of the electron transport chain since it connects the enzyme with the electrode. In [NiFe]-hydrogenases, the FeSd is a [4Fe4S]-cluster with three cysteines and one histidine coordinating the iron atom. From Brownian dynamics simulations, the protein diffusion to the graphite anode surface was simulated and interaction spots of the protein were identified. Different possible routes of electron transfer from the distal FeS-cluster to the electron acceptor were investigated by calculating the rates of electron transfer from the Marcus equation. Only one electron transfer pathway leads to a rate of electron transfer which is in excellent agreement with experimental data from cyclovoltammetry [1].
[1] A. Petrenko, M. Stein, J. Phys. Chem. B (2015), 119, 13870–13882.
201
Basic Mechanisms in Energy Conversion P01-01

Viscosity dependence of hydrogen atom self-exchange:a CW-ESR line broadening study on
N-hydroxyphtalimid/ phtalimide-N-oxyl
Josua Bächle, Freskida Goni, Günter GramppGraz University of Technology, Institute of Physical and Theoretical Chemistry,
Stremayrgasse 9/I, 8010 Graz
The process of hydrogen atom transfer (HAT) is the simplest kind of proton coupled electron transfer (PCET) which plays an important role in a wide range of chemical, biological and industrial processes. These include reactions inthe interconversion of chemical and electrical energy, water splitting and in fuel cells. [1] All state of the art theories predict strong medium effects on the kinetics of such processes [2] and propose an analogy to the Marcus Cross Relation. [3] In our current research we successfully performed CW-ESR line-broadening experiments – displayed in Figure 1 - to determine the kinetics of hydrogen atom self-exchange. The system N-hydroxyphtalimid/ phtalimide-N-oxyl (NHPI/PINO) was investigated at room temperature in different organic solvents with a range of one order of magnitude in dynamic viscosity. [4] These first results could be interpreted by a theoretical description from Alexandrov and Gold´anskii. They predict a linear dependence of the rate k on the
exp(- – 2.8 Pa s. [5]
[1] M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004.[2] S. Hammes-Schiffer, Acc. Chem. Res., 2001, 34, 273.[3] J. M. Mayer, J. Phys. Chem. Lett., 2011, 2, 1481.[4] J. Bächle, F. Goni and G. Grampp, Phys. Chem. Chem. Phys., 2015, 17, 27204.[5] I. V. Aleksandrov and V. I. Golʼdanskii, Chem. Phys., 1984, 87, 455.
Figure 1. CW-ESR spectra (black) of (a)0.5 mM PINO in MeCN containing 0.1 MNaClO4 (b) with 5 mM NHPI and (c) with10 mM NHPI and correspondingsimulations (red).
202
Basic Mechanisms in Energy ConversionP01-02

Electron transfer dynamics of indoline and triarylamine sensitizerson mesoporous ZnO, TiO2 and Al2O3 surfaces
Oliver Flender, Johannes R. Klein, Mirko Scholz, Kawon Oum and Thomas LenzerUniversität Siegen, Physikalische Chemie, Adolf-Reichwein-Str. 2, 57076 Siegen
Photoinduced charge carrier separation and relaxation steps of indoline1 andtriarylamine solar cell dyes2 were investigated on mesoporous thin films using pump -supercontinuum probe (PSCP) transient absorption spectroscopy in the range 370-770 nm. Different types of mesoporous oxides were compared: Electrodeposited ZnO thin films prepared by using the structure-directing agents Eosin Y (EY) andCoumarin 343 (C343), which are known to produce ZnO layers of different morphology, as well as mesoporous ZnO and TiO2 thin films consisting of sintered nanoparticles. “Non-injecting” mesoporous thin films with large band-gap, consisting of Al2O3 nanoparticles, served as reference systems. On all ZnO and TiO2 thin films, the dyes efficiently injected electrons. We observed two components, a dominant one
with 70fs (time-resolution-limited) and a slower one with a time constant in the several hundred femtosecond range. Fast initial charge separation was consistentwith the immediate appearance of oscillatory structure due to a change in refractive index of the ZnO films upon electron injection. For ZnO and TiO2 thin films, we observed a transient spectral shift with a time constant in the picosecond regime which was assigned to a transient Stark effect (= electrochromism). The absorption band of the unexcited dye molecules was shifted due to the build-up of a local electric field between dye radical cations and conduction band electrons. On longer timescales, step-scan FTIR spectroscopy revealed a slightly faster cation-electron recombination on ZnO/C343 than on ZnO/EY thin films. On Al2O3, metastable cation-electron pairs were formed initially which recombined, and no transient Stark effect was observed. Complementary PSCP studies for the dyes in solution showed that they are all ideally suited for solar cell applications because of their long S1 lifetime.
(1) O. Flender, P. W. Lohse, J. Du, T. Oekermann, M. Scholz, K. Oum and T. Lenzer, Z. Phys. Chem., 229, 1907 (2015).(2) K. Oum, O. Flender, P. W. Lohse, M. Scholz, M., A. Hagfeldt, G. Boschloo and T. Lenzer, Phys. Chem. Chem. Phys., 16, 8019 (2014).
203
Basic Mechanisms in Energy Conversion P01-03

In Situ Spectroelectrochemical Study on the Oxidation of a 4H-Imidazole-Ruthenium Dye on TiO2 Electrodes
Ying Zhang†1,2, Stephan Kupfer†2, Linda Zedler1,2, Julian Schindler1,2, Thomas Bocklitz2, Julien Guthmuller3, Sven Rau4 and Benjamin Dietzek1,2
1 Leibniz Institute of Photonic Technology Jena (IPHT), Jena/Germany2 Friedrich Schiller University Jena, Jena/Germany3 Gdansk University of Technology, Gdansk/Poland4 University Ulm, Ulm/Germany† Both authors contributed equally to this work.Terpyridine 4H-imidazole-Ru(II) complexes are candidates for sensitizer in dye sensitized solar cells (DSSCs) by displaying broad absorption in the visible range, where the dominant absorption features are due to metal-to-ligand charge transfer (MLCT) transitions. The Ru(III) intermediates resulting electron transfer from the photoexcited complexes into the conduction band of a metal-oxide semiconductor are essential intermediates in the photoredox-cycle of the DSSC. However, the excited-state properties of these Ru(III) species are much less studied compared to the Ru(II) parent systems. To this end, weinvestigated the photophysical and structural alterations accompanying one-electron oxidation of the 4H-imidazole-Ru dye via in situ experimental and theoretical UV-Vis absorption and resonance Raman spectroelectrochemistry. Apronounced wavelength dependence with respect to the charge transfer character of the optical transition has been observed: Excitation at long wavelengths leads to the population of ligand-to-metal charge transfer states, i.e., photoreduction of the central Ru(III) ion, while high-energy excitation features an intra-ligand charge transfer state localized on the 4H-imidazole moiety. Therefore, the 4H-imidazole-Ru complexes investigated here are potential multi-photoelectron donors: One electron is donated from MLCTstates, and additionally, the 4H-imidazole ligand of the ruthenium(III) complexmight contribute to the excited states and reveal electron-donating characterupon blue-light irradiation.Reference:Y. Zhang, S. Kupfer, L. Zedler, J. Schindler, T. Bocklitz, J. Guthmuller, S. Rau,B. Dietzek, Physical Chemistry Chemical Physics, 2015, 17(44), 29637-29646.
204
Basic Mechanisms in Energy ConversionP01-04

In-situ investigations on the norbornadiene/quadricyclane energy storage system on various metal surfaces by UPS and high-
resolution-XPSUdo Bauer, Philipp Bachmann, Florian Späth, Fabian Düll, Christian Papp, Hans-
Peter SteinrückFAU Erlangen-Nürnberg, Germany
Fossil fuel-based energy technologies lack a long-term perspective. The further development of existing renewable energy concepts is needed regarding energy distribution and storage. One possible scenario is the storage of energy by chemical means in the form of strained organic molecules, e.g. the multi-cyclic hydrocarbon quadricyclane (QC) and its strain-released counterpart norbornadiene (NBD). By absorption of light, NBD is transformed to the energy-rich QC, followed by a catalytic energy release to reform NBD. We investigated the adsorption of QC and itsconversion to NBD; in addition, we also followed the decomposition of both over different catalytically active metal surfaces. Ultraviolet photoelectron spectroscopy provides characteristic spectra of NBD/QC. By applying heating ramps, we observe the conversion of QC to NBD at certain temperatures on Ni(111) and Au/Ni(111), but not for Pt(111) surface, where the reaction occurs already below 120 K. High-resolution X-ray photoelectron spectroscopy of the adsorption at 120 K and during the heating ramps reflects the different interaction strength between molecules and surface, and reveal a fundamentally changed decomposition behavior of NBD at higher temperatures for each surface. The work was supported by the Cluster of Excellence Engineering of Advanced Materials (EAM).
205
Basic Mechanisms in Energy Conversion P01-05

Oxidation state control of MnOx thin films for water oxidation catalysis by pH value adjustment during the deposition process
Maryam Shaker1,2, Munirah Khan1,2, Marc F. Tesch1, Jie Xiao1, Leone Spiccia3,Emad F. Aziz1,2
1 Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany,
2 Department of Physics, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany,
3 School of Chemistry and ARC Centre of Excellence for Electromaterials Science, Monash University, Clayton, VIC, 3800, Australia
Abstract
Manganese Oxide (MnOx) materials have been widely studied as electro-catalysts for the water oxidation reaction. Recently, a novel method for the deposition of MnOx
thin films in ionic liquids was reported [1]. Based on this method, it has been proven that heat treatment during the MnOx deposition process is an effective method to improve the catalytic performance [2]. Here we will show that the pH value of the electrolyte during the depositions process also has a significant effect on the catalytic activity by altering the film composition that consists of several MnOx species. The influence of the pH value - reaching from highly acidic to highly basic - on the oxidation state of the Mn is revealed systematically by x-ray absorption spectroscopy at the Mn L-edges and oxygen K-edge. The thin films are identified to contain multiple MnOx species. It turned out that films deposited in near neutral environmentconsisting of birnessite and Mn2O3 showed the best catalytic performance. Thecombination of different MnOx species by a fine tuning of the pH value might be one key to achieve a high water splitting efficiency for future devices.
[1] Zhou, F., Izgorodin, A., Hocking, R. K., Spiccia, L. and MacFarlane, D. R., Adv. Energy Mater. 2, 1013-1021 (2012), doi:10.1002/aenm.201100783
[2] Khan, M., Xiao, J., Zhou, F., Yablonskikh, M., MacFarlane, D. R., Spiccia, L. and Aziz, E. F., ChemSusChem 8, 1980–1985, (2015), doi:10.1002/cssc.201500330
206
Basic Mechanisms in Energy ConversionP01-06

Photocurrent of BiVO4 is limited by surface recombination, not surface catalysis
Carolin Zachäus, Fatwa F. Abdi, Roel van de Krol, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Berlin/Deutschland
To enhance the performance of metal oxide photoanodes for water splitting, their surfaces are usually functionalized with water oxidation catalysts (e.g. CoOx,NiFeOx). These catalysts greatly enhance the photocurrent by addressing the slow oxygen evolution kinetics of metal oxides. However, the true nature of semiconductor-catalyst interactions is still unclear.We use intensity modulated photocurrent spectroscopy (IMPS) to examine the surface charge carrier dynamics of spray-deposited bismuth vanadate (BiVO4)photoanodes and the effect of cobalt phosphate (Co-Pi) catalysts on its surface. Using a model developed by Peter et al.1, we can distinguish the rate constants for surface recombination and charge injection into the electrolyte. We find that Co-Pisignificantly reduces (>20-fold) the surface recombination, whereas the charge injection kinetics are much less affected. This suggests that the main role of Co-Pi on our BiVO4 is that of a surface passivation layer, as opposed to its usual role as an electrocatalyst. Based on this we predict that modification of the BiVO4 surface with RuOx, a well-known non-passivating oxygen evolution catalyst, does not improve the performance; our experiments show that this is indeed the case.
(1) Ponomarev, E. A.; Peter, L. M. J. Electroanal. Chem. 1995, 396 (1-2), 219.
207
Basic Mechanisms in Energy Conversion P01-07

Unraveling Elementary Steps of an Ir-based Photocatalytic Cycle for Water Splitting
O. S. Bokareva, G. Grell, S. I. Bokarev, O. KühnInstitut für Physik, Universität Rostock, Albert-Einstein-Str. 23-24, D-18059 Rostock
A theoretical perspective is given on the photophysical and photochemical processes which are operative in a photocatalytic system for water splitting, comprised of an Ir(III) heteroleptic photosensitizer (IrPS), triethylamine (TEA) as sacrificial reductant, and a series of iron carbonyls as water-reduction catalyst (see figure) [1-5].
This includes optical absorption of IrPS (I), its reduced species (II) as well as of a hybrid complex with a silver cluster (III), the electron transfer from the sacrificial reductant (IV) and to the iron carbonyl catalys (V). In order to have a reliable description of charge transfer excitations within density functional theory, the LC-BLYP functional with optimally-tuned range-separation parameter is used. Its perfomance is discussed and contrasted to wavefunction based multi-reference methods.
[1] S.I. Bokarev, O.S. Bokareva, O. Kühn, Coord. Chem. Rev. 304-305, 133 (2015).[2] A. Neubauer, G. Grell, A. Friedrich, et al, J. Phys. Chem. Lett. 5, 1355 (2014).[3] S.I. Bokarev, D. Hollmann, A. Pazidis et al., PhysChemChemPhys 16, 4789
(2014).[4] O.S. Bokareva, G. Grell, S.I. Bokarev et al., J. Chem. Theory Comput. 11 1700,
(2015).[5] S. Fischer, O. S. Bokareva, E. Barsch et al., ChemCatChem (2015);
doi:10.1002/cctc.201500872
208
Basic Mechanisms in Energy ConversionP01-08

Concentration Effects on the Entropy of Electrochemical Lithium Deposition: Implications for Li+ Solvation
Matthias J. Schmid,† Junjie Xu,† Jeannette Lindner,† Petr Novák,‡ and Rolf Schuster*,†
†Institut für Physikalische Chemie, Karlsruhe Institute of Technology, Fritz-Haber-Weg 2, D-76131 Karlsruhe, Germany
‡Electrochemistry Laboratory, Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland
LiPF6 is a widely used electrolyte for energy storage, for which the understanding of the behaviour of the lithium ions is a major concern. In this work, we used electrochemical microcalorimetry to study the solvation of Li+ in a solution of LiPF6 inethylene carbonate and dimethyl carbonate. Heat effects were measured upon electrochemical Li deposition and dissolution on bulk Li at different Li+ concentrationsbetween 0.01 M and 1 M. From the data the reversibly exchanged heat, i.e., the Peltier heat for the Li deposition at the different concentrations was determined. The concentration dependence of the Peltier heat reflects the changes of the entropy of solvated Li+ at the different concentrations. It was found that the entropy of Li+
increased by about 5 KJ/mol when the Li+ concentration was lowered by a factor of ten. This can be fully explained by the entropy change expected for the dilution of the Li+ solution. Hence we conclude that the inner coordination shell of Li+ does notsignificantly change between 0.01 M and 1M Li+ concentration. Furthermore, ion contact pairs, which are probably present in our solution, have to be similarly solvated as Li+ in the investigated concentration interval.
209
Basic Mechanisms in Energy Conversion P01-09

Temperature programmed high-resolution X-ray photoelectron spectroscopy of formic acid on Ni(111) and Pt(111)
Philipp Bachmann, Udo Bauer, Florian Späth, Fabian Düll, Christian Papp, Hans-Peter Steinrück, Lehrstuhl für Physikalische Chemie II, Universität Erlangen-
Nürnberg, Germany
Renewable energy sources will replace fossil fuels in the long term. Therefore, new energy storage systems and energy-storage materials have to be found. One promising candidate as energy storage material is hydrogen. Elemental hydrogen needs to be stored under unfavorable conditions such as high pressures or cryogenic temperatures, due to its low volumetric storage density. To circumvent these unfavorable conditions, the concept of chemical hydrogen storage is brought forward:Here, hydrogen is covalently bound in molecules, which can be dehydrogenated catalytically when the energy is needed.Formic acid is nontoxic and has favorable properties to be a safe hydrogen carrier. In order to further understand its interactions with dehydrogenation catalysts, we investigated the adsorption and temperature-dependent hydrogen release of formic acid on the model catalysts Ni(111) and Pt(111). High-resolution X-ray photoelectron spectroscopy during adsorption at 130 K and subsequent heating reveals differences in the decomposition behavior, such as different desorption temperatures and theformation of CO on Ni(111) in contrast to the Pt(111) surface.We acknowledge funding from the Cluster of Excellence ʻEngineering of Advanced Materialsʼ. We thank Helmholtz Zentrum Berlin for the allocation of synchrotron beamtime.
210
Basic Mechanisms in Energy ConversionP01-10

Towards understanding the mechanism of water splitting on TiO2
Saman Hosseinpour, Simon J. Schmitt, Ellen H.G. Backus, Max Planck Institute for Polymer Research, Mainz/Germany
Finding a clean and renewable energy source to replace fossil fuels has attracted much attention, the past few decades, as a requirement for the sustainable development of societies. Direct hydrogen generation on TiO2 by photocatalytic dissociation of water using sunlight was already proposed more than 40 years ago. However, despite extensive work in this area, the fundamentals of the process remain ill-understood, mainly due to the lack of a proper tool to specifically explore the interface between water and TiO2. Sum frequency generation spectroscopy (SFG), is an inherently surface sensitive tool, allowing the study of specifically the water molecules at the TiO2 surface. We present data on water in contact with TiO2
thin films at various pH values and isotopic dilution and correlate the spectral observations with surface species. Moreover, we show first UV-pump—SFG-probe data, illustrating the first steps towards following the photo-induced dissociation of water at the TiO2 interface in real-time.
211
Basic Mechanisms in Energy Conversion P01-11

Physicochemical energy conversion to phase separation, conformational isomerisation or thermal energy change.
Gudrun Ahn-Ercan, University of Regensburg, D-93040 Regensburg, Germany
The solvation energy of a solute depends on the different components in the solution as well as on temperature and on pressure. It is a physicochemical energy, which mainly comes from the energetical interaction of all solution particles with the concerned solute. In the framework of thermodynamics, the molar inner energy of solvation is solvU. Considering the thermodynamic relationships in closed systems, with the energy U of the particles of the system, then there is in a k-component system H=U+pV, –TS, G=H–TS and G=μ1N1+...+μkNk with known meanings. Thus U=TS–pV+μ1N1+...+μkNk and further dU=TdS+(–pdV+μ1dN1+...+μkdNk) and dG=–SdT+Vdp+μ1dN1+...+μkdNk. So it follows from this dU=dH–pdV–Vdp or in the case of ideal mixtures dU=dH–pdV. So the molar inner energy of solvation reads solvU= solvH–p solvV.The investigated systems are liquid electrolyte solutions at standard conditions as for example sodium chloride solutions in 1,4-dioxane-water mixtures [1]. Other systems contain N-methylformamide, N,N-dimethylformamide, water and tetraalkylammonium cations and halides or amino acid anions. The different solutions are simulated at constant N,V,T conditions by utilising Monte Carlo calculations of interacting potential models of molecules, ions and molecular ions. Thus inner energies of solvation are accessible and here it is focused on the solvation energies of the ionic solutes. According to Fischer et al. [2] the average ion-solvent solvation energy is estimated by an expression considering the solute-solvent pair distribution function and the angle averaged pair energy at distance R.The physicochemical energy of individual solvation of specific solute components in specific solvents may change with concentration and may correlate with changes in the whole liquid system, as for example phase separation. Also conformational isomerisation or thermal energy change may correlate with solute solvation energy changes in suitable systems.[1]Krienke H,Ahn-Ercan G,Barthel J. Alkali metal halide solutions in 1,4-dioxane-water mixtures. A Monte Carlo simulation study. J. Mol. Liq. 2002; 109: 115-124.[2]FischerR,RichardiJ,FriesPH,KrienkeH.The solvation of ions in acetonitrile and acetone. II. MonteCarlo simulations using polarizable solvent models.J.Chem.Phys.2002;117:8467-8478.
212
Basic Mechanisms in Energy ConversionP01-13

Photon-Stimulated Processes on Gallium Nitride SemiconductorsSebastian L. Kollmannsberger, Constantin A. Walenta, Martin Tschurl and Ueli Heiz,
Lehrstuhl für Physikalische Chemie & Catalysis Research Center, Chemistry Department, Technische Universität München, Garching bei München, GermanySaskia Weiszer, Andrea Winnerl, Rui Pereira and Martin Stutzmann, Lehrstuhl für Experimentelle Halbleiter Physik II, Physics Department, Technische Universität
München, Garching bei München, Germany
Understanding the influence of particular properties of semiconductors (e.g their doping) on the photocatalytic activity is of major importance for metal-cluster semiconductor hybrid materials [2]. One of the most stable systems is gallium nitride decorated with metal nanoparticles as a co-catalyst [1]. Furthermore, GaN is a semiconductor (3.4 eV band gap), which can either be p- or n-doped. Moreover, its band gap can be tuned by alloying.In this work we demonstrate the elucidation of reaction pathways on clean extended GaN surfaces in the UHV with a chemical probe molecule. For this purpose CO is themolecule of choice, as it exhibits photon-stimulated desorption (PSD) upon UV irradiation on the bare semiconductor surfaces. Further, it is shown that the CO adsorption properties are dependent on semiconductor doping, while the PSD kinetics remain the same.
References: [1] Ohno et al., J. Am. Chem. Soc., 2012, 134, 8254-8259[2] Kim et al., Nano Lett., 2013, 13, 1352-1358
213
Basic Mechanisms in Energy Conversion P01-14

Plasma Enhanced CVD Preparation of CoOxOH2 and CoMOxOH2
(M=Ni, Fe) as Catalysts for the Oxygen Evolution Reaction N. Weidler, S. Paulus, J. Schuch, B. Kaiser, W. Jaegermann
Technical University Darmstadt, Germany
One of the major obstacles, which make the hydrogen production via water-splittinguneconomical at this time, is the overvoltage loss for the oxygen evolution reaction (OER). First-row transition metal oxides and hydroxides could be identified as promising catalysts alternatively to the highly priced RuO2. Combining surface science techniques like XPS with the electrochemical characterization of the catalysts by measuring XPS from the initial state and the electrochemical treated one, we can get information of changes in the oxidation states and the effect on the electrochemical activity. These results contribute to the development of a better mechanistic understanding of the catalytically active sites. We observed, that PECVD deposited Co(II)OxOH2/Ti transform almost completely to the highly active Co(III)OOH surface (Co(III)max= 95%) under operating with an overpotential of only 0.37 V at 10 mA/cm2.[1] These results identify Co(III) as the active state, which is in good agreement with the in literature proposed active redox coupled Co(III)/Co(IV).[2]
Further optimization will be done by depositing mixed metal oxides/hydroxides.
[1] N. Weidler, J. Schuch, J. Klett, P. Stenner, P. Borowski, S. Hoch, A. Maljusch, J. Brötz, C. Wittich, B. Kaiser, W. Jaegermann, PCCP.
[2] Y. Surendranath, M. W. Kanan, D. G. Nocera, Journal of the American Chemical Society 2010, 132, 16501-16509.
214
Basic Mechanisms in Energy ConversionP01-15

Structural investigations on a copper containing OLED complex by application of time-resolved step-scan FTIR spectroscopy and
quantum chemical calculations
Manuel Zimmer1, Fabian Dietrich1, Florian Bäppler2, Manuela Wallesch3,Daniel Volz4, Stefan Bräse3, Rolf Diller2, Markus Gerhards1
1TU Kaiserslautern, Fachbereich Chemie, Erwin-Schrödinger-Str. 52, 67663 Kaiserslautern
2 TU Kaiserslautern, Fachbereich Physik, Erwin-Schrödinger-Str. 46, 67663 Kaiserslautern
3Karlsruher Institut für Technologie, Institut für Organische Chemie, Fritz-Haber-Weg 6, 76131 Karlsruhe
4Cynora GmbH, Werner-von-Siemens-Straße 2-6, 76646 Bruchsal
Time-resolved (TR) infrared experiments enable the investigation of chemical reactions, photochemical/-physical processes and their kinetic traces. With the step-scan technique we are able to identify electronically excited states and excited state structures by comparison with theoretical results from quantum chemical calculations. Here we present the first TR step-scan FTIR measurements on a dimetallic Cu(I)-NHETPHOS-complex in a KBr matrix. These complexes which are developed for emitter material in OLEDs are a very promising substitute for currently used Ir-complexes. We show the capabilities of step-scan FTIR measurements as well as femtosecond transient absorption and reflectivity measurements in a KBr matrix and thin films. Time-resolved IR spectra of electronically excited states have been obtained. Furthermore, wavelength and temperature dependent measurements have been performed to gain detailed information about the excited state processes and deactivation mechanisms.The interpretation of measured spectra succeeded in combination with quantum chemical calculations (DFT and TD-DFT). Different electronically excited states are discussed and the electronic state s could be assigned to the T1 state by comparing the calculated vibrational spectra with the experimentally observed one.
215
Basic Mechanisms in Energy Conversion P01-16

Photoelectrochemical Properties of CdS and CdSe/CdS Nanorods
Jan F. Miethe, Jan Poppe, Nadja C. Bigall,Leibniz Universität Hannover, Hannover/Germany
In the last decades, nanochemistry developed manifold of ways to synthesize homo-and heterogeneous semiconductor nanoparticles, which are based on a wide rangeof materials and show broad diversity in shape, size and attributes.1
Photoelectrochemical properties of these semiconducting materials are only in a few cases investigated.2 In the present work a possibility to prepare monolayers of anisotropic CdS and CdSe/CdS nanocrystals on FTO is shown. These systems have been applied for specific experiments and the investigation of intrinsic properties like external and internal photocurrent efficiencies.2 It is possible to determine the position of the conduction band with potential modulated absorption spectroscopy studies.2 Intensity modulated photocurrent spectroscopy experiments show the influence of the excitation frequency onto the photocurrent response and thereforeinformation about the kinetics of the charge transfer process.
1. Carbone, L. et al.; Nano Lett. 7, 2942–2950 (2007).2. Hickey, S.G. et al.; J. Phys. Chem. B 104, 7623–7626 (2000).
216
Basic Mechanisms in Energy ConversionP01-17

Electrochemical Interphase Formation at a Buried Sample/Electrode Interface
M. Schäfer, V. Wesp, J. Martin, K.-M. Weitzel, Philipps-Universität Marburg, Marburg
The electrochemical interphase formation at the former interface between a sample and a single electrode has been investigated within the Bombardment induced ion transport approach [1]. This technique is based on inducing ion transport in a wide range of materials by attaching slow ions to a sample in contact with a single electrode. Eventually, the ion transport leads to electrodeposition at the interface.In the case of an ion-conducting solid electrolyte we are able to selectively generate a Na interphase from a glass containing Na and K as mobile species [2]. The implications for the electrochemical potentials at the former interface will be discussed.As an example of a polymer the interphase formation of cesium between a PPX film and a Pt electrode has been investigated. 3D-imaging of this interphase formation allows to reconstruct structural properties of the polymer film connected to its porosity[3].In the case of a polyelectrolyte membrane we have been able to measure the conductivity of (PAH/PSS)n multi layers as a function of the number of bilayers from n=16 down to a single bilayer. The variation of the conductance is non-monotonic going through a maximum for n=8. A model for rationalization is proposed.
[1] M. Schäfer, K.-M. Weitzel, PCCP, 13, 20112–20122, (2011)[2] J. Martin, S. Mehrwald, M. Schäfer, T. Kramer, C. Jooss, K.-M. Weitzel, to be submitted[3] V. Wesp, J. Zakel, M. Schäfer, I. Paulus, A. Greiner, K.-M. Weitzel, Electrochimica Acta
170, 122–130, (2015)[4] V. Wesp, M. Hermann, M. Schäfer, J. Hühn, W.J. Parak, K.-M. Weitzel, PCCP, in press
217
Basic Mechanisms in Energy Conversion P01-18

Electrodeposited Nickel Nanoparticles as Catalysts for Hydrogen
Evolution Reaction
S. Tao, F. Yang, B. Kaiser, W. Jaegermann
Institut für Materialwissenschaft, Technische Universität Darmstadt, Jovanka-Bontschits-Straße 2, 64287, Darmstadt,
Germany
E-mail: [email protected]
Abstract
Hydrogen production by electrochemical reduction of water is an important component of several
developing clean-energy technologies. So far, platinum has shown the highest activity for facilitating
the hydrogen evolution reaction (HER). However, the concerns over the practical usage of water
splitting cells have motivated the search for a low cost catalyst. Nickel (Ni) or Ni based compounds,
typically in the form of Nanoparticles (NPs) are often used due to the high abundance, high activity
and low cost.
In this work, we deposited Ni NPs on titanium substrates by electrodeposition using cyclic
voltammetry and solution of 10 mM NiCl2 in 1 mM H2SO4. High-resolution electron microscopy
(HREM), X-ray photoelectron spectroscopy (XPS) and electrochemical measurements are employed
to figure out the influence of cycle ranges, cycles and scan rates on the surface morphology and
chemical composition as well as on the catalytic performance of the Ni NPs.
XPS results and SEM images indicate that it is possible to prepare nearly mono-
dispersed Ni/Ni(OH)2 or Ni/Ni(OH)2/NiO NPs by changing the cycle range. With the increase of
the scan rate from 10 mV s-1 to 100 mV s-1, the average particle size decreases from 130 nm to 60 nm.
Furthermore, electrochemical measurements show that under alkaline conditions the overpotential of
the most active samples for the HER at a the current density of 10 mA cm-2 is around -0.08 V, but
after two hours of galvanostatic measurements, it decreases to around -0.15 V. Therefore, in a
further step the stability of the Ni NPs should be improved.
218
Basic Mechanisms in Energy ConversionP01-19

Ultrafast photo-induced electron transfer in self-assembled donor-acceptor coordination cages
Jennifer Ahrens1, Guido H. Clever2, Marina Frank2, Dirk Schwarzer1
1Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany2Institute for Inorganic Chemistry, University Göttingen, Germany
Electron donor-acceptor complexes built from self-assembled palladium coordinatedphenothiazine (D) and 9, 10-anthraquinone (A) based ligands, Pd4DmA8-m, where investigated by femtosecond spectroscopy with UV excitation and Vis/mid-IR detection to explore their potential use for photovoltaics. Coordination of the acceptor ligand to palladium in the complex Pd4A8 had no influence on the excited state dynamics of the anthraquinone chromophore which is determined by fast intersystem crossing to a long lived triplet state. On the other hand, excitation of the pure donor cage Pd4D8 leads to efficient ligand-to-metal charge transfer. For mixed complexes Pd4DmA8-m clearly ligand-to-ligand charge transfer within 200 fs is detected regardless of whether donor or acceptor ligands are photo-excited. This is evident from the appearance of a carbonyl stretching band in the mid-IR assigned to the anthraquinone radical anion. The lifetime of the charge separated state appears to be very inhomogeneous ranging from 3.0-2000 ps. The reason for this observation could be the broad distribution of donor-acceptor cage configurations, different spin states of the charge-separated diradical ion pairs or a combination of both.
219
Basic Mechanisms in Energy Conversion P01-20

Inelasticity in Hydrogen atom scattering from surfaces: A probe for energy conversion
Yvonne J. Dorenkamp, Hongyan Jiang, Alec Wodtke, and Oliver BünermannGeorg-August-Universität Göttingen, Institut für Physikalische Chemie,
Tammannstr. 6, 37077 Göttingen, Germany
Developing an understanding of energy conversion at surfaces is of great fundamental and practical interest and the subject of extensive investigation over many years. Measuring the change in momentum and quantum state in collisions of atoms and molecules with surfaces are fundamental tools to evolve an understanding of how atomic and molecular degrees of freedom couple to surfaces.Despite this extensive effort, we do not have an understanding of even the simple problem of how Hydrogen atoms lose kinetic energy and become adsorbed on surfaces. Recently, we have developed an apparatus to measure energy conversion in H-atom surface collisions: A mono energetic H-atom beam is pointed towards asurface at defined incidence angles. Employing Rydberg Atom Tagging the final scattering angular and kinetic energy distributions are measured. A first experiment showed that drastic differences are observed when Hydrogen atoms are scattered from a metal versus an insulator surface. Comparison of theexperimental results to resent theoretical work indicate that electron-hole-pair excitation plays an important role in scattering processes with metals.[1] In further studies the influence of various experimental parameters were investigated: Kinetic energy, incidence angles, isotope of the incident beam and temperature and structure of the surface.
[1] O. Bünermann et al., Science, 2015, 350, 1346-1349.
220
Basic Mechanisms in Energy ConversionP01-21

Monitoring reactive layers inside redox hydrogels by Raman microspectroscopy
Christina Klinkhammer1, Stefanie Stapf2, Nicolas Plumeré2, Martina Havenith1,1Physical Chemistry II, 2Center for Electrochemical Sciences - Molecular
Nanostructures, Ruhr-University Bochum, Germany
Hydrogenases are efficient catalysts for H2 conversion. However, their sensitivity towards high potentials and O2 impede the application in fuel cells.Plumeré et al. recently developed a viologen-based redox polymer that prevents high-potential inactivation and O2 damage of the polymer-bound [NiFe] hydrogenase.This effect is achieved by formation of protective layers inside the polymer ensuringthe functionality of the hydrogenase in the active site of the film. [1] However, little detail is known about the dimensions of these layers. We employ confocal Raman microspectroscopy in order to obtain spatial information about the hydrogel film. For this purpose a special measuring cell was designed which allows the combination of the electrochemical and the spectroscopic experiment. Preliminary results suggest that layers with different oxidation states canbe discriminated by tracing the oxidation state of viologen.
[1] Plumeré et al. Nature Chemistry 6, 822-827 (2014).
221
Basic Mechanisms in Energy Conversion P01-22

Investigation of Pt- and Pt/Ni-nanoparticles as Catalysts for the Hydrogen Evolution Reaction in Electrochemical Cells
J. Ziegler1, F. Tabary1, L. Wittern2, H. Weller2, W. Jaegermann1 and B. Kaiser1*
1*Institut für Materialwissenschaften, Technische Universität Darmstadt, [email protected]
2Institut für Physikalische Chemie, Universität Hamburg, Germany
A major drawback for using hydrogen as an efficient solar fuel are its high production costs from renewable energy and electrolysis compared to the classic synthesis route. This is due to the large overpotentials associated with the electrochemical water splitting process. In order to reduce these, precious metal electrodes from Pt and Pd are very efficient, but at the same time rare and very expensive. They can be replaced either by using these metals in the form of nanoparticles on a nonreactive carrier or by other more abundant and cheaper materials. Furthermore, it is an important task to elucidate the working mechanism of these compounds.Pt- and Pt/Ni-particles are generated by magnetron sputtering and by spin coating on TiO2 substrates. The samples are then characterized by cyclovoltammetry to test for their catalytic activity. Before and after each electrochemical experiment the surface structure and the surface composition are investigated by scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS) to observe changes induced by the electrochemical treatment. We observe, that sputter deposited nanoparticles are more stable during electrochemical testing for up to several hours, whereas spin coated nanoparticles will have to be optimized to achieve a more stable catalyst layer.
222
Basic Mechanisms in Energy ConversionP01-23

Imaging photo-induced processes in organic-inorganic perovskites
Lucas M. Uriarte1, Andreas Neff1, Fabian Meier2, Carsten Deibel2, Katrin Siefermann1, 1Leibniz Institute of Surface Modification, 04318 Leipzig,
Germany, 2Institut für Physik, Technische Universität Chemnitz, 09126 Chemnitz, Germany.
Lead iodide perovskite (CH3NH3-PbI3) solar cells have been one of the most exciting developments in photovoltaics in the past decade [1]. The promise of high conversion efficiencies and easy production processes has triggered numerous research efforts with the goal to understand and improve material properties. Despite significant advances, many aspects related to the photo-physics of these materials remain unclear, and impede a transfer from the laboratory to industry application.
Here, we present a microscopic experimental approach in which we combine a photoemission electron microscope (PEEM) with a femtosecond laser system. This setup allows us to image photo-induced processes in organic-inorganic perovskites with a lateral resolution of 100 nm. In first measurements, we have monitored photo-induced charging of individual perovskite crystallites, which ultimately results in photo-damage of these crystallites. We find that further photo-induced degradation of the material spreads from initially photo-damaged crystallites, and is thus not a homogeneous process.
In future experiments, we will perform ultrafast (pump-probe) photoemission-electron microscopy to directly image fundamental photo-induced processes in organic-inorganic perovskites.
[1] Tze Chien Sum and Nripan Mathews. Energy Environ. Sci., 2014, 7, 2518
223
Basic Mechanisms in Energy Conversion P01-24

Understanding the effect of Ir(III/IV) aquo complexes on the photoelectrochemistry of IrOx nH2O–based water-splitting cells
Nella M. Vargas-Barbosa*, Lasse Jensen and Thomas E. MalloukDepartment of Chemistry, The Pennsylvania State University
University Park PA, USA
The synthesis of IrOx 2O colloids by alkaline hydrolysis of Ir(III) or Ir(IV) salts proceeds through monomeric intermediates that were characterized using electrochemical and spectroscopic methods and modeled in TDDFT calculations. In air-saturated solutions the monomers exist in a mixture of Ir(III) and Ir(IV) oxidation states, where the most likely formulations at pH 13 are [Ir(OH)5(H2O)]2- and[Ir(OH)6]2, respectively. The results from this study address a very important and current dilemma in IrOx nH2O–based PECs: how does the chemistry of the catalyst and its interface with the semiconductor influence the photoresponse of the cell? The careful preparation, purification and characterization of both IrOx nH2O catalysts and photoanodes in this work provide reasonable explanations to previously observed discrepancies in PECs that utilize IrOx nH2O colloids as co-catalysts on the photoanode electrode. The monomeric anions strongly adsorb onto TiO2, and they promote the adsorption of ligand-free IrOx 2O colloids onto mesoporous titania photoanodes. However, the reversible adsorption/desorption of electroactive monomers effectively short-circuits the photoanode redox cycle and thus dramatically degrades the photoelectrochemical performance of the cell. and identified the iridium aquo intermediates as possible recombination sites.
*Current institution: AG Prof. Dr. Bernhard RolingDepartment of Physical Chemistry, Philipps–Universität MarburgMarburg, Germany
224
Basic Mechanisms in Energy ConversionP01-25

Group III-phosphide semiconductors for water splitting: Surface preparation and water adsorption experiments
Andreas Hajduk1, Mikhail Lebedev2, Bernhard Kaiser1, Wolfram Jaegermann1
1Technische Universität Darmstadt, Germany2Ioffe Institute, St. Petersburg, Russia
Photoelectrochemical water splitting offers the possibility to convert solar energy di-rectly into a chemical fuel and therefore is a promising candidate for a sustainable energy solution in the future.
In this study we focus on group III-phosphide semiconductors i.e. GaInP2 and GaP,which possess a sufficiently wide bandgap for water splitting and show a favorable alignment of their band edges in respect to the redox potentials of water. In order to better understand the photoelectrochemical processes at the interface between these semiconductors and the aqueous solutions, we performed water adsorption experi-ments on clean semiconductor surfaces. To prepare clean surfaces, the samples were etched with HCl in 2-propanol solution and annealed afterwards. Surface prepa-ration and water adsorption were carried out under inert gas atmosphere and subse-quently investigated by X-ray photoelectron spectroscopy. As shown by the reduction of the O 1s photoemission intensity by a factor of 20, we were able to remove the native oxide layer on p-GaInP2. Contact to liquid water resulted in the formation of Ga–O bonds on the p-GaInP2 surface and in an increase of its ionization energy by 0.45 eV, as well as in a reduction of the surface band bending from 0.55 to 0.4 eV.
225
Basic Mechanisms in Energy Conversion P01-26

Atomistic Modeling of Light Induced Surface Relief Gratings
Marcus Böckmann and Nikos L. Doltsinis,Institut für Festkörpertheorie, Westfälische Wilhelms-Universität Münster,
Münster, Germany
An increasing number of photoresponsive smart materials is made from polymers or smart materials that contain azobenzene (AB) as their photochromic unit [1,2]. While typical applications range from, e.g., conversion of photonic into mechanic energy to light control in thin film formation [3,4], a special focus nowadays is on the phenomenon of photofluidity [5,6]. Here, energy conversion leads to effective mass transport in thin film layers that eventually results in characteristic surface relief gratings (SRGs) [7, 8]. In this contribution, we report on the photoinduced behaviour of such an active layer based on the PDO3M azo-polymer that we have investigated by means of multiscale molecular dynamics simulation techniques [9] making use of a molecular mechanics switch for AB photoisomerisation that we designed recently[10]. In particular, we will show how multiple E Z activation cycles account for the mass transport and yield different results as compared to simple thermal heating [11].
This work is a joint project with the groups of Prof. C. Denz (Nonlinear Photonics, WWU) and Prof. Z. Wang (Key Lab of Organic Solids, Chinese Academy of Science) in the framework of the TRR61 (DFG).
[1] Y. Zhao and T. Ikeda, eds., Smart Light-Responsive Materials (Wiley-VCH, Weinheim, 2009).[2] H. Yu and T. Ikeda, Adv. Mater. 23, 2149 (2011).[3] D. A. Doshi, N. K. Huesing, M. Lu, H. Fan, Y. Lu et al., Science 290, 107 (2000).[4] L. Pithan, C. Cocchi, H. Zschiesche, C. Weber, A. Zykov, S. Bommel, S. J. Leake, P. Schäfer,
C. Draxl, and S. Kowarik, Cryst. Growth Des. 15, 1319 (2015).[5] P. Karageorgiev, D. Neher, B. Schulz, B. Stiller, U. Pietsch, M. Giersig, and L. Brehmer,
Nature Materials 4, 699(2005).[6] Y. Kim and N. Tamaoki, J. Mater. Chem. C 2, 9258 (2014).[7] N. C. R. Holme, L. Nikolova, S. Hvilsted, P. H. Rasmussen et al., Appl. Phys. Lett. 74, 519 (1999).[8] N. S. Yadavalli, F. Linde, A. Kopyshev, and S. Santer, ACS Appl. Mater. Int. 5, 77437747 (2013).[9] M. Böckmann, D. Marx, C. Peter, L. Delle Site, K. Kremer, and N. Doltsinis, PCCP 10, 1039 (2011).[10] M. Böckmann, S. Braun, N. L. Doltsinis, and D. Marx, J. Chem.Phys. 139, 084108 (2013).[11] M. Böckmann, T. Schemme, C. Denz, and N. L. Doltsinis (in preparation).
226
Basic Mechanisms in Energy ConversionP01-27

(Self)Assembling Amphiphilic Dyes and Tuning of Photonic Properties via Morphology Control Martin Presselt, Saunak Das, Felix Herrmann, Maximilian Hupfer, Benjamin Dietzek
Thin organic chromophore layers are the essential functional elements in devices for light to electric
energy conversion and vice versa, i.e. in organic solar cells (OSCs) and organic light emitting diodes
(OLEDs). In OLEDs the morphology of the active layer needs to be engineered for optimal directional
characteristics of light emission while in OSCs the morphology of the active layer needs to be
elaborately tuned for optimal power conversion efficiency. However, the morphology is generally
challenging to target starting from molecular chromophore design. This is due to the fact that
predictions of supra-molecular structures are still computationally very demanding at a high accuracy
level and the formation of particular morphologies depends on various external physical, chemical
and processing parameters. Therefore, various approaches have been developed to control the
morphology. One promising approach to design defined supra-molecular structures that are
thermodynamically stable is self-assembly. A prominent strategy to induce self-assembly is to
introduce amphiphilicity to functional molecules. Beyond self-assembly, amphiphilicity additionally
enables utilization of the Langmuir-Blodgett (LB) technique for superior control of molecular ordering
and phase formation.
One research focus in our group is therefore on self-assembly, morphological and photonic
properties of novel donor and novel acceptor molecules that were made amphiphilic. Using the LB-
technique we are able to produce thin films of both donor and acceptor molecules with controlled
morphology. The surface sensitive photothermal deflection spectroscopy[1-3] is not just applied to
determine the UV-vis absorption spectra of even molecular monolayers, but also to monitor self-
assembly on surfaces from solutions in situ. For further characterization of the essential processes
happening after photoexcitation a variety of advanced spectroscopic techniques is applied, ranging
from highly sensitive to time-resolved techniques to characterize ultra-fast dynamics. Within the
proposed PhD-project, thin films with well-defined morphology will be produced using the above
mentioned methods. Thus, stack architectures with tunable complexity will be produced and the
photo- or electrically induced processes happening at the interfaces will be analyzed with the above
mentioned advanced techniques. The obtained results will provide the basis for novel devices with
superior properties.
1. Presselt, M., et al., Influence of Phonon Scattering on Exciton and Charge Diffusion in Polymer-Fullerene Solar Cells. Advanced Energy Materials, 2012. 2(8): p. 999-1003.
2. Presselt, M., et al., Sub-bandgap absorption in polymer-fullerene solar cells studied by temperature-dependent external quantum efficiency and absorption spectroscopy. Chemical Physics Letters, 2012. 542: p. 70-73.
227
Basic Mechanisms in Energy Conversion P01-28

3. Herrmann, F., et al., Influence of Interface Doping on Charge-Carrier Mobilities and Sub-Bandgap Absorption in Organic Solar Cells. The Journal of Physical Chemistry C, 2015. 119(17): p. 9036-9040.
The authors are grateful for financial support (BMBF FKZ 03EK3507).
228
Basic Mechanisms in Energy ConversionP01-28

Ultrafast Excited State Dynamics of Iridium(III) PhotosensitizersS. Tschierleia,b; A. Neubauera; N. Rockstrohc; M. Karnahlb,c; H. Jungec; M. Bellerc;
S. Lochbrunnera
a Universität Rostock, Rostock; b Universität Stuttgart, Stuttgart; c Leibniz-Institut fürKatalyse e.V., Rostock
Subject of this contribution is the investigation of the photophysical properties of a series of heteroleptic iridium(III) photosensitisers bearing 2-phenyl-pyridine and different 2,2ʼ-bipyridine (bpy) ligands (Fig. 1a). These complexes have been already successfully applied in proton reducing systems for the photocatalytic production of hydrogen. In this report, the initial light-induced electron transfer and intramolecular relaxation steps are elucidated by means of time-resolved optical spectroscopy.An substitution at the bpy ligand influences the absorption and emission features as well as the excited state absorption properties of the respective complexes. We could assign these processes after light excitation (Fig. 1b) to vibrational relaxation (sub
1 ps) and an interligand charge transfer ( 30 ps). The interligand charge transfer leads to absorption changes which depend on the polarisation of the probe light.Surprisingly, the interligand charge transfer, from the 2-phenyl-pyridine to the bpy ligand, occurs only for the unsubstituted bpy ligand and not for the ligand substiuted with carboxyl anchor groups.
a) b)
Fig. 1: a) General structure of the investigated Ir(III) complexes 1 and 2.b) Schematic representation of the underlying excited state processes.
References: [1] F. Gärtner, B. Sundararaju, A.-E. Surkus, A. Boddien, B. Loges, H. Junge, P. Dixneuf and M. Beller, Angew. Chem. Int. Ed., 2009, 48, 9962. [2] A. Neubauer, G. Grell, A. Friedrich, S. I. Bokarev, P.Schwarzbach, F. Gärtner, A.-E. Surkus, H. Junge, M. Beller, O. Kühn and S. Lochbrunner, J. Phys. Chem. Lett., 2014, 5, 1355. [3] S. Fischer, O. S. Bokareva, E. Barsch, S. I. Bokarev, O. Kühn and R. Ludwig, ChemCatChem, doi: 10.1002/cctc.201500872
229
Basic Mechanisms in Energy Conversion P01-29

Real-time observation of Ir photosensitizer reductive quenching during light-activated molecular catalysis
Alexander Britz1,2, Tadesse Assefa1, Andreas Galler1, Wojciech Gawelda1, Eva Bajnoczi3, Zoltan Nemeth3, György Vankó3, Nils Rockstroh4, Henrik Junge4, Matthias Beller4, Gilles Doumy5, Anne Marie March5, Steve Southworth5, Sergey Bokarev6,Stefan Lochbrunner6, Oliver Kühn6, and Christian Bressler1,2
1European XFEL, 22761 Hamburg, Germany2Hamburg Centre for Ultrafast Imaging, 22761 Hamburg, Germany
3Wigner Research Centre for Physics, Hung. Acad. Sci., H-1525 Budapest, Hungary4Leibniz-Institut für Katalyse an der Universität Rostock, 18059 Rostock, Germany
5Argonne National Laboratory, Argonne, Illinois 60439, USA6Universität Rostock, Institut für Physik, 18055 Rostock, Germany
We will present how employing ultrafast combined optical pump/X-ray probespectroscopy allows visualizing in “real-time” transient electronic and geometric changes accompanying a sequence of redox reactions taking place during molecular catalysis. Their coupling governs both the light harvesting and redox functions, and therefore determines the efficiency of the conversion of the visible light-inducedelectronic excitation into energy-rich electrons used in the subsequent water splitting reactions. A deeper understanding of the structure-function relationship is critical for the development and optimization of efficient hydrogen production in molecularphotocatalysis.We have studied a photoexcited Ir-based photosensitizer (IrPS) complex in a homogenous solution using a suite of complementary time-resolved hard X-ray spectroscopic tools with 100 ps temporal resolution. Our study delivers a more complete picture about how transient geometric and/or electronic structures of long-lived (>1 ns) charge-separated excited states of those molecules determine the efficiency of the subsequent reductive quenching in the presence of sacrificial reductant molecules and the intermolecular electron transfer to the water splitting catalyst. We have used a fully functional photocatalytic system, which includes triethylamine, acting as a reductive quencher of the photoexcited IrPS site and Fe3(CO)12 as the water reduction catalyst.
230
Basic Mechanisms in Energy ConversionP01-30

Light-Driven Electron Transfer in a Photocatalytic Model SystemBased on a Copper Photosensitizer
Aleksej Friedrich1, Shu-Ping Luo2, Henrik Junge3, Matthias Beller3, and Stefan Lochbrunner1
1Institute of Physics, University of Rostock,Albert-Einstein-Straße 23, 18059 Rostock, Germany
2State Key Laboratory Breeding Base of Green Chemistry-Synthesis Technology,Zhejiang University of Technology, 310014 Hangzhou, China3Leibniz-Institut für Katalyse an der Universität Rostock e.V.,
Albert-Einstein-Straße 29a, 18059 Rostock, Germany
Photocatalysis is a promising route to generate solar fuels with the help of sun light. In this work time resolved spectroscopy is applied to understand the behavior of aphotocatalytic model system for hydrogen generation. It consists of a copper complex as light absorbing sensitizer, an iron complex as catalyst, triethylamine (TEA) as electron donor, and water as hydrogen source [1,2].Directly after the absorption event ultrafast intersystem crossing occurs in the copper sensitizer [3]. The subsequent interaction between the copper complex and the electron donor as well as the catalyst is characterized by photoluminescence quenching using a streak camera as well as time resolved absorption measurements on the nano- and microsecond timescale. It is found that the iron catalyst quenches the triplet lifetime of the copper photosensitizer efficiently from 280 ns to 110 ns and that this oxidative quenching path way dominates over reductive quenching by TEA.Long living products of the quenching events are observed by the transient absorption measurements. By comparing these data to reference measurements with methyl viologen the electron transfer efficiency from the sensitizer to the catalyst is determined to 30%.
[1] S. Luo et al., Angew. Chem., 2013, 125, 437.[2] E. Mejía et al., Chem. Eur. J., 2013, 19, 15972.[3] S. Tschierlei et al., ChemPhysChem, 2014, 15, 3709.
231
Basic Mechanisms in Energy Conversion P01-31

In-operando Atomic Force Microscopyfor Energy Materials Characterization
Florian HausenForschungszentrum Jülich, Institute of Energy and Climate Research, IEK-9, Jülich;
RWTH Aachen University, Institute of Physical Chemistry, Aachen, Germany
Turning away from conventional energy sources and towards a sustainable energy landscape is closely related to the ability to store energy and release it on demand. Independently of the exact route how this task is performed, for numerous energy materials it is based on the understanding of processes occurring at the interface between an electrode and an electrolyte. In order to develop optimized device properties such as increased capacity, better durability and faster charging rates in the case of batteries or with enhanced catalytic properties for chemical energy conversion, analysis and characterization of mechanisms taking place at thenanometer scale are of utmost importance. Combining classical electrochemical methods like cyclic voltammetry with atomic force microscopy allow for precise measurements with extremely high spatial resolution down to the sub-nanometer level. Within this contribution various techniques based on atomic force microscopy (AFM) for the analysis of energy storage principles at small scales are presented and discussed. This includes in-operando experiments of degradation mechanisms as well as a thorough analysis of double layer properties in ionic liquids as a function of applied potential. Additionally, a method to distinguish between the formation of the so-called Solid Electrolyte Interface (SEI)-Layer or an expansion of the material under electrochemical load is introduced for Li-battery materials. Conductive AFM as well as AFM - Scanning Electrochemical Microscopy experiments are used to differentiate locally between electrochemically active and less-active parts of the surface. Furthermore, the recently developed Electrochemical Strain Microscopy which is based on the relationship between ion density and molar volume, is discussed.
232
Basic Mechanisms in Energy ConversionP01-32

New Reaction Pathway on Rutile
Boerchert Jasper, Milena Osmi , Simon Schmiedekind, Katharina Al-Shamery
Carl von Ossietzky University of Oldenburg, Institute of Chemistry, Physical Chemistry 1, D-26129 Oldenburg, Germany
Due to its high occurrence in nature, its high chemical stability and toxicological safety, titanium dioxide is used in a huge variety of applications. As it has a band gapof 3,02 eV (rutile), it is able to absorb UV-A-light and hence can use roughly 4 % of the sunlight for solar energy conversion, e.g. photocatalytic reduction of CO2 to methane by water [1].Habisreutinger et al. suggested a pathway for photocatalytic CO2 reaction by stepwise reduction via formaldehyde and methanol to form methane [2].
Using temperature programmed desorption (TPD) and Fourier transformed infrared reflection absorption spectroscopy (FT-IRRAS) we investigate the (photo-)oxidation and (photo-)reduction of methanol under ultra high vacuum conditions and present anew reaction pathway via a methoxy intermediate. We shall show that the defect density is a key factor for titania chemistry in accordance with our ealier findings on the reduction of benzaldehyde to stilbene [3].
[1] A. Linsebigler, G. Lu, J. Yates, Photocatalysis on TiO2 Surfaces, Chem. Rev. 1995, 95, 735-758.[2] Habisreutinger et al., Photocatalytic Reduction of CO2 on TiO2 and Other Semiconducters, Angew. Chem. Int. Ed., 2013, 52, 7372-7408.[3] P. Clawin, C. Friend, K. Al-Shamery, Defects in Surface Chemistry – Reductive coupling of Benzaldehyde on Rutile TiO2(110), Chemistry: A European Journal, 2014, 90, 7665-7669.
233
Basic Mechanisms in Energy Conversion P01-34

Enhanced Stability of Thin Film Silicon Tandem Solar Cells based Photocathodes with Amorphous TiO2 Layers Coated with
Nanoparticle Catalysts for Solar Hydrogen Evolution
Florent Yang1*, Liana Movsesyan2, Félix Urbain3, Jan-Philipp Becker3, Maria Eugenia Toimil-Molares2, Friedhelm Finger3, Bernhard Kaiser1, and Wolfram Jaegermann1
1 Technische Universität Darmstadt, Germany2 GSI Helmholtzzentrum für Schwerionenforschung, Germany
3 Forschungszentrum Jülich, Germany
Photoelectrochemical (PEC) water splitting is an attractive approach to directly convert solar energy into solar fuels, e.g. hydrogen as a storable, clean and future important energy carrier. Thin film silicon based tandem solar cells as photocathodes are very promising semiconductor materials for PEC integrated systems, because they can provide the necessary photovoltage required to split water into hydrogen without any additional bias. Moreover, these tandem cells are made from abundant and low-cost materials, and they have already shown photovoltaic conversion efficiencies of However, their stability in aqueous media is a critical issue.Therefore, a passivation layer, here TiO2, is either deposited by magnetron sputtering or atomic layer deposition on top of the tandem cell to protect the Si-based absorber/electrolyte interface against photocorrosion and/or degradation under operating conditions. Moreover, Pt or Ni-Mo nanoparticle catalysts werephotoelectrodeposited onto the tandem cell to efficiently improve its PEC performance by reducing the kinetic barrier for the water reduction half-reaction. The electronic properties of the samples were characterized using X-ray photoelectron spectroscopy before and after PEC measurements, and the morphology changes were determined by scanning electron microscopy. First results reveal that the TiO2
buffer layer shifts the onset potential to higher anodic potential by about +160 mV in comparison to the bare substrate, which is further increased by +110 mV with the presence of Pt nanoparticles. Moreover, these Pt nanoparticles on TiO2 efficiently stabilize the tandem cells for more than 10 hours against photocorrosion in acidic solution.
234
Basic Mechanisms in Energy Conversion P01-35

������������������������������ ����������������������
�����������������������������
���������������������������� ��������������������������������� ���
����������������������������������������������� ������������������������������ �������� ���
������ ����������������������������� ������������������������������ ����������������������
������������������������������������������������������������������������������� �����������
����������������������������� ����� �����
�� ���������������������� ���������� �����������
� ���������������������������� ������� ����������������������� ������������������ ���������
������ � ��������� � ������ � ��� � ���� � �������� � ��� � �������� � � ��� � ��������� � ��� ����
�� ������� � ��� � ���� � �������� � ��� � ����� � �� � �� � � ��� � �� � ������ � ��������� � ���
��������� ���������������������������������������� ������������� �������� ����� ������������
���� ��������� ������������������������������������������������������ � ���������������
����������� ������� �� ��������
������������ ������� �����������������������������������������������
�� � ���� ���������� ���������� �������� ����������� �� ����������������������� ���� ����������
������ ����������� ������������������������� �� �������������������� ������������ ����
��� ������������������������������������������ ���������������������� ����������������� ����
������� ���� ���������������� �� �� ����� ������������������ ����������������� ������������ ��
����������������������� ����������������� ��������������������������� ������������� ����
������������� ������������ ������������������������������������� ��������������������� �����
������������������������������������� �� ������������������������ ���������������������
������
��������������������������� ����� �����������������������
������������������������¡���������¢������������£�������£��� �����������������£�¤�
£���������¥������������������������������ ��������������������������
���������� �������¦������§������������������������������������� ����� ����������������
�����
�����������¨�¥���������������� ����������� ������������
����¨��� ��������� ����������������������
����¤������������¤�¨������£��£�������������¤����� ������������� ����������������
�����¢������������������ �����������������
����¥�������¤�������������� ����¢����������� �����������������������
Ru
Ru
O
O
Ru
Ru
OO
Ru
Ru
OO+ H2O
Ru
RuOO
OHH Ru
Ru OHO
OH + H2O- O2
Ru
Ru
OH2
OH2
Ru
Ru
O
O
+ H2ORu
Ru
O
OOH
H
235
Basic Mechanisms in Energy Conversion P01-36

Ionic dynamics in alkylammonium nitrates are decoupled from viscosity
Soham Roy1, 2, Mischa Bonn1, Johannes Hunger1
1Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz2Graduate School Materials Science in Mainz, Staudinger Weg 9, 55128 Mainz
Despite the heterogeneous structure of ionic liquids (ILs) having been firmly established, the consequences of the nanoscopic segregation on their dynamics has remained elusive. We use femtosecond infrared pump-probe spectroscopy [1] to probe the local dynamics of the ammonium group for a series of alkylammonium nitrates, with the alkyl chain length increasing from ethyl to pentyl. Vibrational relaxation of the N-D stretching vibration is fast (~ 200 fs) and followed by slow relaxation dynamics (~ 15-25 ps). The slow dynamics are assigned to thermal equilibration of the three-dimensional structure of the ILs [2]. The thermal equilibration and also the rotational dynamics, as extracted from the excitation anisotropy, are decoupled from viscosity [3]. Interestingly, our results suggest that increasing hydrophobic domain size goes along with an acceleration of the restricted rotational motion of the ammonium group, which is indicative of a more fragile inter-ionic interaction. Thus, our study underlines that the structural heterogeneity of ILs results in heterogeneous local dynamics.
[1] J. Hunger, T. Sonnleitner, L. Liu, R. Buchner, M. Bonn, H. J. Bakker, J. Phys. Chem. Lett., 2012, 3, 3034.
[2] Z.-P. Zheng, W.-H. Fan, S. Roy, K. Mazur, A. Nazet, R. Buchner, M. Bonn, J. Hunger, Angew. Chem. Int. Ed. Engl., 2015, 54, 687.
[3] S. Bouzón Capelo, T. Méndez-Morales, J. Carrete, E. López Lago, J. Vila, O. Cabeza, J. R. Rodríguez, M. Turmine, L. M. Varela, J. Phys. Chem. B, 2012,116, 11302.
236
Lipids and SolvationP02-01

A Molecular Dynamics Simulation Study of Direct Cation-Cation Interactions in Cholinium Based Ionic Liquids
Jan Neumann, Ralf Ludwig, and Dietmar Paschek
Institut für Chemie, Physikalische und Theoretische Chemie, Universität Rostock,Albert-Einstein-Str. 21, D-18059 Rostock, Germany
In previous studies our group was able to provide spectroscopic evidence for direct cation-cation interactions in the ionic liquid (2-hydroxyethyl)-trimethylammonium bis(trifluoro-methylsulfonyl)amide ([Ch][NTf2]) via hydrogen bonding between adjacent hydroxyl groups [1]. Here we present an accompanying molecular dynamics simulation study in search of these hydrogen bond interactions. For this purpose we parametrized a cation-forcefield employing ab initio calculations.To validate our forcefield we compare our simulation results with available thermodynamic data. Utilizing various geometric criteria for the hydrogen bond we report evidence of the existence of these specific intermolecular interactions. However, the observed linear hydrogen bonds are characterised by an unusually large oxygen-oxygen distance. In our study we provide a detailed comparison of the cation-cation interaction with the cation-anion hydrogen bond regarding quantity and geometry.
[1] A. Knorr, K. Fumino, A.-M. Bonsa, R. Ludwig, Phys. Chem. Chem. Phys. 17,30978-30982 (2015).
237
Lipids and Solvation P02-02

Structure and Dynamics in Mixtures of Protic Ionic Liquids: A Molecular Dynamics Simulation Study
Benjamin Golub, Dietmar Paschek and Ralf Ludwig
Institute of Chemistry, Physical Chemistry, University of Rostock,Dr.-Lorenz-Weg 1, D-18059 Rostock, Germany
Hydrogen bonding plays an important role for the local structure in mixtures of protic ionic liquids [1-2]. In previous studies we were able to describe and understand the hydrogen bonding network in binary mixtures of Triethylammonium [(C2H5)3NH]+ andTrihexylammonium [(C6H13)3NH]+ based ionic liquids, using Methylsulfonate [CH3SO3]-, Trifluoromethylsulfonate [CF3SO3]- and Bis(trifluorome-thylsulfonyl)imide [(CF3SO2)2N]- as anions. In addition, we developed a simple lattice model to describe the hydrogen bonded cluster distributions in these mixtures [2]. Now, we would like to show the relation between the structure and the dynamics of these systems. Therefore we present the diffusion coefficients for the ions and molecular reorientation times and their relation to structural properties such as cluster size distributions. The existence of large hydrogen bonded clusters, where an anion interacts with up to three cations at the same time, leads to a significantly reduced local mobility, sometimes even smaller than what is observed for the pure ionic liquids. Finally, also the behaviour of Triethylammonium based ternary mixtures of [(C6H13)3NH]+[CH3SO3]-, [(C6H13)3NH]+[CF3SO3]- and [(C6H13)3NH]+[(CF3SO2)2N]-,are discussed with respect to their structure and dynamics.
[1] K Fumino, A.-M. Bonsa, B. Golub, D. Paschek, R. Ludwig, Chem. Phys. Chem.16, 299-304 (2015).
[2] D. Paschek, B. Golub, R. Ludwig, Phys. Chem. Chem. Phys. 17, 8431-8440 (2015).
238
Lipids and SolvationP02-03

Temperature Dependence of Computed Osmotic Second Virial Coeffients of Apolar Solutes in Aqueous Solutions
Elias Reiter, Dietmar Paschek, and Ralf Ludwig
Institut für Chemie, Physikalische und Theoretische Chemie, UniversitätRostock,
Dr.-Lorenz-Weg 1, D-18051 Rostock, Germany
The hydrophobic interaction of solvated particles in aqueous solutions is described by their second osmotic virial-coefficient. However, it is hard to quantify this property, since it is systematically affected by concentration- andsystem-size-effects. In a previous contribution K. Koga [1] has shown, that a proper representation of the second virial-coefficient also requires an accurate computation of the asymptotic long-range behaviour of the pair distribution function. Here we present computed pair distribution functions and related second virial-coefficients for selected Lennard-Jones solutes, such as methane, xenon and mercury in a realistic water environment represented by several models, for example the TIP3P, TIP4P2005, Tip4PEwald and the TIP5P water model. We show that the necessary asymptotic behaviour of the pair-distribution functions can be (under certain conditions) obtained accurately from fitting computational data to an asymptotic decay function, described by Evans et al. [2] From this data we can examine the second virial-coefficient in dependence of temperature.
[1] K. Koga, J. Phys. Chem. B 2014, 117, 12619-12624[2] R. Evans, J. Chem. Phys. 1994 100, 591-603
239
Lipids and Solvation P02-04

Under Pressure – Amorphous Ice – A MD Simulation Study
Martin Jehsera, Markus Seidlb, Thomas Loertingb and Gerhard Zifferera
aUniversity of Vienna, Department of Physical Chemistry, Währinger Straße 42, 1090 WienbUniversity of Innsbruck, Institute of Physical Chemistry, Innrain 80-82 ,6020 Innsbruck
There are at least sixteen different crystalline modifications of ice. Solid water also exists in several supercooled amorphous forms ranging from LDA (low-density amorphous ice) over HDA (high-density amorphous ice) to VHDA (very high-density amorphous ice) [1].Around the glass transition temperature Tg the metastable glassy and liquid states often undergo spontaneous crystallisation, thus interfering in experiments aiming at observing and characterizing the metastable liquid. Although a lot of experimental effort is put on shrinking the so called waterʼs no manʼs land [2], atomistic molecular dynamic simulations provide highly efficient alternative routes for studying amorphous to liquid transitions [3].In the present contribution the effects of pressure on the amorphous structure as well as the method of pressure induced amorphisation are studied in detail. Making use of the open source package GROMACS [4] and the Tip4P2005 force field [5] the focus lies on closely resembling the experimental protocols for a broad range of external pressures.
____[1] T. Loerting, K. Winkel, M. Seidl, M. Bauer, C. Mitterdorfer, P. H. Handle, C. G. Salzmann, E. Mayer, J. L. Finney, and D. T. Bowron, Phys. Chem. Chem. Phys. 13, 8783 (2011)[2] M. Seidl, A. Fayter, J.N. Stern, G. Zifferer, T. Loerting, Phys. Rev. B 91, 144201 (2015)[3] M. Jehser, M. Seidl, C. Rauer, T. Loerting, G. Zifferer, J. Chem.Phys. 140, 134504 (2014)[4] http://www.gromacs.org[5] J. L. Abascal, C. Vega, J Chem Phys, 123 (23), 234505, (2005)
240
Lipids and SolvationP02-05

Investigation of the formation of pharmaceutical nanoparticles within RESS by means of molecular dynamics simulation
Stephan Braun, Thomas Kraska, Institute of Physical Chemistry, Universität zu Köln
Within the formulation of pharmaceuticals it is desired to improve the availability of pharmaceutical substances in the human body as well as to steer the structure of the crystals by means of polymorphic behavior. The formation of such particles from the vapor phase is restricted by the chemical stability of the substances at high temperature. In order to achieve fine disperse particles of a pharmaceutical substance, the rapid expansion of a supercritical solution process (RESS) is suitable. Due to the swift change in pressure and the resolution change in solubility, high values of supersaturation can be obtained. While experimental results indicate a dependence of the particle properties on the state conditions of the expansion process, the molecular process for the formation of different structures remains unclear.
Here we investigate the particle formation of such systems on the molecular level by means of molecular dynamics simulations. Potential models for two pharmaceuticals (carbamazepine and naproxen) were developed and solutions of these model-substances in CO2 at supercritical conditions are set up and equilibrated. The expansion process is modeled in the simulation method by stepwise increasing the size of the simulation box, providing a method to model the adiabatic expansion of a supercritical fluid. The expansion of such a solution leads to a highly supersaturated system that starts to form particles.From this formation process the mechanism can be deduced.
241
Lipids and Solvation P02-06

Nuclear Quantum Effects on Charge Transfer in the Hydrogen Bond of Gas Phase Dimers and Liquid Water
Christoph Schran, Ruhr-Universität Bochum, GermanyOndrej Marsalek, Thomas E. Markland, Stanford University, USA
The hydrogen bond is stabilized by partial charge transfer (CT) from the donor to the acceptor molecule, besides effects as electrostatics, polarization and dispersion.Since atomic charges are not uniquely defined, many different methods exist to study CT in simulations and the absolute amount of transferred charge and the resulting stabilization is under discussion and dependent on the specific method. Studies of CT so far have been performed using the minimum energy structure or a classical description of the nuclei of the system of interest, ignoring nuclear quantum effects (NQEs) as zero point motion, quantum delocalization and tunneling. Here we are looking at CT including the interplay of nuclear and electronic quantum effects employing ab initio path integral molecular dynamics (AI-PIMD) for various gas phase dimers and liquid water to compare with classical ab initio molecular dynamics (AIMD) for Bader, Hirshfeld and Natural population analyses. Due to competing NQEs, CT is influenced in different directions dependent on the system which isconnected to the change of the proton transfer coordinate. CT in liquid water is shown to be a 2-body effect and a method of predicting the molecular charge of the molecules in liquid water is presented.
242
Lipids and SolvationP02-07

Terpene-based Mixtures: Molecular Insights into the Solid-Liquid Equilibria
Marisa A. A. Rocha, Florian M. Zehentbauer, Bernd Rathke, and Johannes KieferTechnische Thermodynamik, Universität Bremen, Badgasteiner Str. 1, 28359 Bremen, Germany
Ionic liquids (ILs) and deep eutectic solvents (DESs) have emerged as green and designable solvents showing properties desirable for applications in numerous fields such as, synthesis, separation processes, and material science [1-3]. DESs can be prepared by simply combining a hydrogen bond donor (HBD) and a hydrogen bond acceptor (HBA) with moderate melting points to form a liquid at room temperature. Alike to ILs, DESs also offer the opportunity of modifying their physicochemical properties by an appropriate selection of the components in terms of their molecular structure and chemical nature, as well as the compositions and water content of their mixtures [2,3]. Further, DESs present the advantage of solvent free preparation without the need for purification steps. In this work, binary mixtures containing terpenes and long chain alcohols are explored on both the macroscopic as well as the microscopic level. Solid-liquid phase equilibria (SLE) of the binary mixtures are investigated to determine the concentration range where a liquid mixture is formed at room temperature and to characterize the eutectic composition of the mixture. This study is complemented by a comprehensive investigation of the molecular interactions and bulk structural organization of the binary mixtures by vibrational spectroscopy (FTIR). On the basis of the obtained results, the effects of the chemical nature and concentration of each component in the binary mixture on the formation of a liquid mixture at room temperature are analyzed.
References[1] F. Pena-extraction processes” ChemSusChem, 7, 1784-1800 (2014).[2] E. L. Smith, A. P. Abbott, K. S. Ryder “Deep eutectic solvents (DESs) and their applications” Chemical Reviews, 114, 11060-11082 (2014).[3] M. Francisco, A. van den Bruinhorst, M. C. Kroon “Low-transition-temperature mixtures (LTTMs): a new generation of designer solvents” Angewandte Chemie International Edition, 52, 3074-3085(2013).
AcknowledgementsThe authors gratefully acknowledge financial support by the Institutional Strategy funded by the Federal Government's and the Federal States' Excellence Initiative.
243
Lipids and Solvation P02-08

Imidazolium Based Ionic Liquids:Impact of Cation Symmetry on the Liquid – Liquid Phase
Behavior of Solutions with n-Alkyl AlcoholsMarisa A. A. Rocha1, Daniela Kerlé1, Wolffram Schröer2, and Bernd Rathke1
1 Technische Thermodynamik, Universität Bremen, Badgasteiner Str. 1, 28359 Bremen, Germany2 Institut für Anorganische und Physikalische Chemie, Universität Bremen, Leobener Str. NW II,
28359 Bremen, Germany [email protected]
To gain a more generalized understanding of the behavior of Ionic Liquids (ILs) in mixtures, we studied the liquid-liquid phase diagrams of binary mixtures of thesymmetric 1,3-diethylimidazolium bis(trifluoromethylsulfonyl)amide ionic liquid ([C2C2im][NTf2]) with n-alkyl alcohols (propan-1-ol, butan-1-ol, hexan-1-ol, and octan-1-ol). The phase diagrams were determined at atmospheric pressure and over a temperature range of T = (258 – 386) K. Partial miscibility with an upper critical solution temperature (UCST) between T = (284 – 386) K, was observed for the studied systems. The UCST decreases with decreasing alkyl chain of the alcohols. By comparing the data obtained with the asymmetric 1-methyl-3-propylimidazolium bis(trifluoromethylsulfonyl)amide IL ([C3C1im][NTf2]) [1], it was possible to evaluate the effect of symmetry of the IL cation on the liquid-liquid behavior.The experimental study is supported by molecular-dynamics simulations (MD), in order to attain a better understanding on a molecular level. The local dynamics and structures are analyzed in detail and reveal the effect of the symmetry on different physico-chemical properties.
References[1] X. Shao, W. Schröer, and B. Rathke, J. Chem. Eng. Data (2014)
244
Lipids and SolvationP02-09

Role of Hydrogen Bond Donors in the Formation of Liquid Mixtures
Marisa A. A. Rocha1, Florian M. Zehentbauer1, Lawien F. Zubeir2, Bernd Rathke1,Maaike C. Kroon2,3, and Johannes Kiefer1
1 Technische Thermodynamik, Universität Bremen, Badgasteiner Str. 1, 28359 Bremen, Germany2 Separation Technology, Eindhoven University of Technology, Den Dolech 2, 5612 AZ Eindhoven, The
Netherlands3 The Petroleum Institute, Dept. Chemical Engineering, P.O. Box 2533, Abu Dhabi, United Arab Emirates
Deep eutectic solvents (DESs) or eutectic mixtures can be defined as the combination of two or more solid compounds, which form a liquid upon mixing with a depression in the melting point compared to the pure substances. A hydrogen bond donor (HBD) and a hydrogen bond acceptor (HBA) are required, since the DESs associate via hydrogen bonding [1-3]. Most of the DESs studied until now are based on ammonium salts as HBA, and several HBDs such as carboxylic acids, amides, sugars, or (poly-)alcohols.Nowadays, these new solvents are also prepared by combining of only uncharged molecules which can contribute to a thermal lower stability.Binary mixtures composed of menthol as HBA and one of the three HBDs, acetic acid, lactic acid, and decanoic acid are explored in order to understand the role of thehydrogen bond donor in the formation of liquid mixtures at room temperature. In an initialpart of this work, different preparation and drying methodologies are evaluated. Usually DESs are considered as low volatile mixtures, but when it comes to mixtures composed of uncharged volatile compounds, several precautions should be taken into account in their preparation. Thermogravimetric analysis is used to evaluate the thermal stability as well as to give information about the volatility of the prepared mixtures. The energetics of the phase transitions of the mixtures and the strength of the intermolecular interactions are investigated using differential scanning calorimetry and Fourier transform infrared spectroscopy, respectively. References[1] F. Pena- p eutectic mixtures: sustainable solvents for extraction processes” ChemSusChem, 7, 1784-1800 (2014).[2] E. L. Smith, A. P. Abbott, K. S. Ryder “Deep eutectic solvents (DESs) and their applications” Chemical Reviews, 114, 11060-11082 (2014).[3] M. Francisco, A. van den Bruinhorst, M. C. Kroon “Low-transition-temperature mixtures (LTTMs): a new generation of designer solvents” Angewandte Chemie International Edition, 52, 3074-3085(2013).
AcknowledgementsThe authors gratefully acknowledge financial support by the Institutional Strategy funded by the Federal Government's and the Federal States' Excellence Initiative.
245
Lipids and Solvation P02-10

THz absorption spectroscopy of an insect lipoprotein ice nucleator reveals a long-range influence on the hydrogen bond dynamics
Alexander Bäumer, Martina Havenith, Ruhr-Universität Bochum, Bochum/D;John G. Duman, University of Notre Dame, Notre Dame/USA
Very little is known about the mechanism of action of ice nucleation proteins, although their special ability to trigger ice nucleation at high sub-zero temperatures could be used in a broad variety of applications. We study an insect lipoprotein ice nucleator (LPIN) by circular dichroism spectroscopy and Terahertz absorption spectroscopy. myo-Inositol is the functional group of phosphatidylinositol and is crucial for the activity of LPIN. Interestingly, it is not present in other insect lipoproteins, which lack ice nucleation activity. myo-Inositol shows a significant interaction with the hydration water compared to other inositol isomers. We observe that this is conserved in LPIN. The ice nucleation inhibitor sodium borate perturbs the influence in both cases. Our CD measurements show that the lipoproteins of LPIN consist of a high amount of -structures (35 %) revealing a potential structural similarity with bacterial INPs and insect antifreeze proteins (AFPs). We propose asimilar mechanism for LPIN as that found for AFP activity: A necessary local ice-binding mechanism is supported by a long-range retardation of the hydrogen bond network dynamics. Both conditions are mandatory for ice nucleation activity in LPIN.
Fig 1. Left: Concentration-dependent THz absorption of LPIN with and without addition of sodium borate. Right: Inhibition scheme of LPIN by sodium borate.
246
Lipids and SolvationP02-11

Probing the acidity and solvent polarity of ionic liquids by determining the spectroscopic features of the proton sensitive dye
N-methyl-6-oxyquinolone
Stella Schmode, Andranik Petrosyan, Ralf LudwigInstitute of Chemistry, Physical and Theoretical Chemistry, University of Rostock,
Dr.-Lorenz-Weg 1, D-18051 Rostock, Germany
N-Methyl-6-Oxyquinolinium (6MQz) is a potent dye in optical spectroscopy. The native zwitterionic molecule shows a strong negative solvatochromism. The S1
absorption band in DMSO compared to that in water shifts by 100 nm. The solvatochromic effect can also be followed by stationary fluorescence spectroscopy (stokes shift 169 nm / 3931 cm-1). Due to excitation the dipole moment decreases from 10 D to 6 D.[1,2]Among solvent polarity the acidity of various media can be investigated using 6MQz. By enlarging the solvent proton donor capability the probe is protonated easily. In 6MQc the intramolecular donor/acceptor system is abolished. Therefore, the optical properties of the dye change drastically. In one sample both forms can be detected quantitatively by absorption as well as fluorescence spectroscopy at the same time. Thus 6MQz is exerted as a molecular pH meter in ILs. For characterizing the molecular interactions IR techniques are adopted. The distinguishing C-O stretching mode classifies intermolecular attraction and repulsive forces of 6MQz and its chemical surrounding. Experimental results are supported by Density Functional Theory (DFT) calculations on the B3LYP level of theory. We also focus on the synthesis of RTILs formed by 6MQc cations. With this chemical tool we want to probe mixtures of two or more (protic) ILs and common solvents in the full range of concentration. A very exciting feature is to observe the possibility to transfer the proton from the cation to the environment by photo excitation as can be similarly observed for 6MQc.
References[1] C. Allolio, D. Sebastiani, Phys. Chem. Chem. Phys. 2011, 13, 16395.[2] J.L. Pérez Lustres, N.P. Ernsting, Angew. Chem. 2005, 44, 5635.
247
Lipids and Solvation P02-12

Non-PEGylated
PEGylated
a) b)
Towards Understanding Glycan-Assisted Protein Stabilization
Sarah Schäfer , Hanna Wirtz, Claudius Hoberg, Janne Savolainen and Martina Havenith, Physical Chemistry II, Ruhr-University Bochum, Germany
Posttranslational attachment of glycans to asparagin amino acid side chains, namely N-glycosylation, is a frequent decoration of proteins involved in signaling and recognition. Furthermore, addition of polyethylene glycol (PEG), i.e. PEGylation, to proteins is a widely used carbohydrate mimic to improve stability and therefore delivery in pharmaceutical applications. Microscopic understanding of these effects is,however, still lacking. Recent studies have shown that PEG can stabilize a protein by interacting with the surface and/or by extending into the solvent (Fig. 1a).[1]
Our approach towards better understanding of glycan-assisted protein stabilization is a Terahertz-based real time view on the rearrangement of the hydrogen bond network surrounding the non-equilibrated PEGylated hPin1 WW domain. A strong nanosecond IR laser pulse (T-jump) excites an OH stretching overtone, thereby heating the water and inducing the protein folding and unfolding kinetics. Terahertz radiation captures the relaxation of bulk water along with the solvation shell around PEGylated WW domain. Our kinetic THz absorption experiments will provide a unique insight into cooperative motions of water and PEGylated WW domain. Preliminary results show a distinct change in the water network relaxation in the presence of WW-PEG45(Fig.1b). Further experiments with (non-)PEGylated WW domain will shed light on their intermolecular interplay with water and the physical basis of glycan-assisted protein stabilization.
[1] S. Chao et al., J. Phys. Chem. B 118, 8388–8395 (2014).
Figure 1. a) CD melting curves for PEGylated (gray curve) and non-PEGylated (black curve)WW-domain variants. The stabilizing effect of PEG4 is nearly 8 °C, assuming two-state folding.[1]b) T-jump traces for WW-PEG45 (gray curve) and corresponding buffer solution (black curve).
248
Lipids and SolvationP02-13

How does spatial confinement influence proton-transfer dynamics?Emanuel Eisbein, Tommy Lorenz, Gotthard Seifert, and Jan-Ole Joswig
Physikalische Chemie, TU Dresden, 01062 Dresden, Germany
Controlling the hydrogen-bond network is a crucial requirement for designing applicable proton-conducting soft-matter materials. On the one hand, the network has to be flexible enough to react on spontaneous occurrences; on the other hand, it has to be stable in such a way that the proton-conduction is permanent. In this contribution, we present results from molecular-dynamics simulations of different proton-conducting materials. Special attention has been paid to the influence of spatial confinement on the hydrogen-bond network and the proton-transfer events. Additionally, the hydrogen-bond lifetime and the occurrence of Zundel-like complexes during the transfer have been analyzed.Proton-conducting liquids and polymers have been studied, as well as imidazole systems that are inspired from applications, such as arrays of anchored alkyl-imidazole molecules [1] and imidazole confined in a MIL53AL metal-organic framework [2,3]. Most of these systems have direct applications in fuel-cell membranes.
1. W. L. Cavalcanti, D. F. Portaluppi, J.-O. Joswig, J. Chem. Phys. 133 (2010),104703.
2. E. Eisbein, J.-O. Joswig, G. Seifert, J. Phys. Chem. C 118 (2014), 13035.3. E. Eisbein, J.-O. Joswig, G. Seifert, Micropor. Mesopor. Mater. 216 (2015), 36.
249
Lipids and Solvation P02-14

Investigation of Ubiquitin Using Time-Resolved Kinetic THzAbsorption Spectroscopy with Temperature Jumps
Hanna Wirtz, Sarah Schäfer, Claudius Hoberg, Janne Savolainen, Martina HavenithRuhr-Universität Bochum, Bochum/Germany
Terahertz (THz) spectroscopy is a powerful technique to study solvation dynamics around solutes like proteins, sugars and salts.[1] With frequencies ranging from 0.1 THz to 10 THz, intermolecular and collective motions in the water network can be investigated.We use kinetic THz absorption spectroscopy in order to probe solvation dynamics during the (un)folding of Ubiquitin (UBQ). Equilibrium conditions are perturbed by fast temperature jumps (T-jumps) heating the solution by up to 10 K in 5 ns with 1542 nm laser pulses. The response of the H-bond network following the induced structural changes of UBQ is then monitored with microsecond time resolution using time-domain THz spectroscopy.UBQ unfolds upon cold denaturation at 275 K as determined by Circular Dichroism(Fig. 1a). First T-jump measurements from 270 K show different response signals for UBQ compared to the pure buffer solution (Fig. 1b). Based on these preliminary results, we propose that these differences stem from structural rearrangements of UBQ coupled to the dynamics of the surrounding water molecules.
[1] B. Born, M. Havenith, J Infrared Milli Terahz Waves 2009, 30, 1245-1254.[2] Ibarra-Molero et al., Biochemistry 1999, 38, 8138-8149.
Fig. 1a) Cold denaturation melting curve for 0.2 mM UBQ (in 10 mM glycine, 3.55 M GuHCl, pH 2 [2])determined using Circular Dichroism Spectroscopy. The region of the T-jump is shaded.Fig. 1b) T-jump traces for buffer and 0.6 mM UBQ from a starting temperature of 270 K. Inset: zoom-in on the microsecond timescale.
b)a)
250
Lipids and SolvationP02-15

Photoionization of Sodium Doped Crystalline Water Clusters Thomas Zeuch and Christoph W. Dierking, Institut für Physikalische Chemie,
Georg-August-Universität Göttingen, Germany
A special interest in the photoionization properties of sodium doped water clusters originates from the formation of solvated electrons in these microsolutions. In this work the high-energy (>3.5 eV) part of the photoionization spectrum is examined for clusters with different physical states. The interaction of sodium atoms with neutral water clusters was studied in a molecular beam experiment by means of single photon ionization mass spectrometry with a tunable UV light source and ReTOF-MS.Whereas former studies[1,2] focused on the appearance ionization energies (aIE) of Na(H2O)n clusters, the aim of our work is to determine the ionization energy at which a saturation of the ion signal occurs (sIE) and how the physical state of the clusters (amorphous/crystalline) affects the photoionization spectrum.The surprising breadth of the photoionization spectrum lead to the assignment of two isomer classes with aIEs of 2.8 eV for 10-15[1] and 3.2 eV for 4[2]. The sIE for n > 9 is constantly around 4 eV for clusters with up to 100 molecules. The occurrence of a the isomer class with an aIE of 2.8 eV has been explained by ab initio molecular dynamics simulations to result from clusters featuring solvent separated ion pairs in the clusters[1]. The high abundance of clusters with IEs higher than 3.4 eV indicatesthe presence of a third isomer class where the sodium atom and the water cluster interact weakly and thus the 3s electron is likely unseparated from the sodium atom.Since the sodium pickup technique is believed to yield reliable cluster size distributions it is of particular interest to determine if the physical state of the clusters(amorphous/crystalline), which can be probed by IR-UV action spectroscopy[3], has an influence on the photoionization spectrum and thus on the cluster detection.[1] R. M. Forck, I. Dauster, Y. Schieweck, T. Zeuch, U. Buck, Milan On ák, Petr
Slaví ek, Communications: Observation of two classes of isomers of hydratedelectrons in sodium-water clusters, J. Chem. Phys., 132, 221102 (2010).
[2] I. V. Hertel, C. Hüglin, C. Nitsch, C. P. Schulz, Photoionization of Na(NH3)nand Na(H2O)n Clusters: A Step Towards the Liquid Phase?, Phys. Rev. Lett.,67(13), 1767 (1991).
[3] C. C. Pradzynski, R. M. Forck, T. Zeuch, P. Slavícek, U. Buck, A Fully Size-Resolved Perspective on the Crystallization of Water Clusters, Science, 337,1529 (2012).
251
Lipids and Solvation P02-16

Determination of the Electronic Structure of Aqueous Urea and its Derivatives: A combined Soft X-Ray – DFT Approach
Marc Tesch1, Ronny Golnak1, 2, Felix Ehrhard1, Richard Chudoba1, Joachim Dzubiella1, Daniela Schön1, 3, Jie Xiao1, Annika Bande1, Emad F. Aziz1, 3
1 Institute of Methods for Material Development, Helmholtz-Zentrum Berlin für Materialien und Energie, Berlin, Germany
2 Department of Chemistry, Freie Universität Berlin, Berlin, Germany3 Department of Physics, Freie Universität Berlin, Germany
We present first x-ray absorption and emission measurements on the nitrogen K-edges of the biomolecules urea and its derivatives acetamide, thiourea, biuret and dimethylurea dissolved in water. A combination of absorption (XAS) and emission (XES, RIXS) soft x-ray spectroscopy and DFT (BP86-D3/def2-TZVP, COSMO solvent treatment) calculations reveals the electronic structure of the investigated molecules. On this poster, we put special focus on the theoretical aspects of this investigation. Especially in the more extended pi electron system of biuret, conformational variation of structure via bond rotation revealed a strong variation of the spectrum and led to the assumption, that the minimum energy geometry spectrum differs considerably from the experimental spectrum. To examine this case beyond zero Kelvin vacuum conditions, MD simulations followed by DFT calculations were taken into account for an explicit solvent treatment.
252
Lipids and SolvationP02-17

Solubility Convergence and Criticality in Ionic Liquids
Majid Namayandeh Jorabchi1, Daniela Kerlé2, Ralf Ludwig1, Dietmar Paschek1
1Institute of Chemistry, Physical and Theoretical Chemistry, University of Rostock, Dr.-Lorenz-Weg 1, D-18059 Rostock, Germany
2FB 4 Department of Production Engineering, University of Bremen, Badgasteiner Str. 1, D-28359 Bremen, Germany
With the aim of exploring the solvation behavior of gases in Ionic Liquids (ILs), we domolecular dynamics simulations of a large collection of important light gases in imidazolium-based Ionic Liquids of type 1-n-alkyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([Cnmim][NTf2], with n=2,4,6,8) over a broad
temperature range from 300 K to 800 K. We are able to determine the temperature dependent solvation free energies for those selected light gases in simulated imidazolium based ILs with high statistical accuracy. Our simulations demonstrate that the magnitude of the solvation free energy of a gas molecule at a chosen reference temperature and its temperature-derivatives are intimately related with respect to one-another. Solvation entropy-enthalpy compensation effect is the consequence of a funnel-like free energy landscape of the solvated small gas molecules, which we have coined “solvation funnel” model. A direct consequence of the enthalpy-entropy relation for gases with varying interaction-strength is that the infinite dilution solubility data converge with increasing temperatures and finally meet at a single temperature, given that the associated heat capacities of solvation are similar. We examine the relation of this convergence temperature with the location of the liquid-gas critical point of the Ionic Liquid.
253
Lipids and Solvation P02-18

Deuteron Quadrupole Coupling Constants and Reorientational Correlation Times in Protic Ionic Liquids by Means of NMR Relaxation Time Experiments,
DFT-Calculations and Molecular Dynamics Simulations
Matthias Strauch1, Viviane Overbeck1, Anne-Marie-Bonsa1, Benjamin Golub1, Dirk Michalik2, Dietmar Paschek1 and Ralf Ludwig1
1Physikalische und Theoretische Chemie, Universität Rostock,Dr.-Lorenz-Weg 1, D-18059 Rostock, Germany
2Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Str. 29a, D-18059 Rostock, Germany
We describe a method for the accurate determination of deuteron quadrupole
coupling constants D for N-D bonds in triethylammonium-based protic ionic liquids (PILs). This approach has been first introduced by Wendt and Farrar for molecularliquids [1], and is based on the linear relationship between the deuteron quadrupole
coupling constants D, and the proton chemical shifts 1H, as obtained from DFT calculated properties in differently sized clusters of the compounds. Thus the
measurement of 1H provides an accurate estimate for D, which can then be used
for deriving reorientational correlation-times ND, by means of NMR deuteron quadrupole relaxation time measurements. The method is applied to pure PILs
including differently strong interacting anions. The obtained D values vary between 152 and 204 kHz, depending on the cation-anion interaction strength, intensified by H-bonding. We find that considering dispersion corrections in the DFT-calculations
leads to only slightly decreasing D values. The determined reorientational correlation times indicate that the extreme narrowing condition is fulfilled for these
PILs. The c values along with the measured viscosities provide an estimate for the
volume/size of the clusters present in solution. In addition, the correlation times c,and the H-bonded aggregates were also characterized by molecular dynamics (MD) simulations.[2]
[1] M. A. Wendt, T. C. Farrar, Mol. Phys. 1998, 95, 1077-1081.[2] D. Paschek, B. Golub, R. Ludwig, Phys. Chem. Chem. Phys. 2015, 17, 8431-
8440.
254
Lipids and SolvationP02-19

Critical radius of supercooled water droplets: on the transition towards dendritic freezing
Tillmann Buttersack and Sigurd BauereckerInstitut für Physikalische und Theoretische Chemie TU Braunschweig
The freezing of freely suspended supercooled water droplets with a diameter of bigger than a few micrometers splits into two rather different freezing stages. Within the first very fast dendritic freezing stage a spongy network ice with an ice portion of less than one third forms and more than two thirds of liquid water remain [1-3]. In the present work the distribution of the ice portion in the droplet directly after the dendritic freezing phase as well as the evolution of the ice and temperature distribution has been investigated in dependence of the most relevant parameters as droplet diameter, dendritic freezing velocity (which correlates with the supercooling) and heat transfer coefficient to the surroundings (which correlates with the relative droplet velocity compared to the ambient air). For this purpose on the experimental side acoustically levitated droplets in climate chambers have been investigated in combination with high-speed cameras. The obtained results have been used for finite element method (FEM) simulations of the dendritic freezing phase under consideration of the beginning second, much slower heat-transfer dominated freezing phase. A theoretical model covering 30 layers and 5 shells of the droplet has been developed which allows to describe the evolution of both freezing phases at the same time. The simulated results are in well agreement with experimental as well as with calculated results exploiting the heat balance equation. The most striking result of this work is the critical radius of the droplet which describes the transition of one-stage freezing of the supercooled water droplet towards the thermodynamically forced dendritical two-stage freezing where the droplet cannot sufficiently get rid of the formation heat anymore. Depending on the parameters named above this critical radius was found to be in the range of 0.1 to 1 micrometer by FEM simulation [4].
[1] H.R. Pruppacher, J.D. Klett, Microphysics of Clouds and Precipitation, 2nd ed.(1997).[2] S. Bauerecker, P. Ulbig, V. Buch, V., L. Vrbka, P. Jungwirth, J. Phys. Chem. C,112, 7631-7636, (2008).[3] S. Bauerecker, T. Buttersack, J. Phys. Chem. B, 118, 13629-13635 (2014).[4] T. Buttersack, S. Bauerecker, J. Phys. Chem. B, (2016), in press.
255
Lipids and Solvation P02-20

Quantifying dispersion forces in protic ionic liquids by means of low frequency spectroscopy
Peter Stange1, Koichi Fumino1, Anne Knorr1, Ralf Ludwig1,2
1Physical & Theoretical Chemistry, University of Rostock,18059 Rostock, Germany2Leibniz Institute for Catalysis, 18059 Rostock, Germany
The properties of ionic liquids are determined by a subtle balance of forces, including Coulomb-interaction, hydrogen bonds and dispersion forces. Although Coulomb-interactions are the dominant force in ionic liquids, both hydrogen bonds and dispersion forces can significantly influence be behavior of these coulomb-fluids.[1] The investigation of the role of London dispersion interaction in ionic liquids is usually limited to density functional theory (DFT) calculations using coupled-cluster-methods as benchmarks.[2] However, few experimental data analyzing the importance of dispersion in Coulomb-dominated liquids exist.By investigating a carefully chosen set of protic ionic liquids (PILs), we can tune thedispersion component of the interaction by varying the alkyl-chain length of the cationand observe the effects on low frequency vibrations characterizing the anion-cation interaction. We can additionally vary the strength of the anion-cation-interaction via hydrogen bonds by choosing different anions.Investigations of PILs consisting of trialkylammonium cations and methylsulfonate and trifluoromethylsulfonate (triflate) anions using far-infrared spectroscopy showed characteristic vibrational features indicating the transition from hydrogen-bond-dominated to dispersion-dominated ion pairs-interactions, provided that the dispersion component of the interaction is large enough and the hydrogen bond between cation and anion is sufficiently weak.[3] This approach, in conjunction with dispersion-corrected DFT-calculations, enables us to determine the relative strength of hydrogen bonds and dispersion interactions in Coulomb-dominated systems.[4]
REFERENCES[1] K. Fumino, S. Reimann, R. Ludwig, Phys. Chem. Chem. Phys., 40, 21903-21929 (2014)[2] E. Izgorodina, D. Golze, R. Maganti, V. Armel, M. Taige, T. J. S. Schubert, D. R. MacFarlane,
Phys.Chem.Chem.Phys., 16, 7209- 7222 (2014)[3] K. Fumino, V. Fossog, P. Stange, D. Paschek, R. Hempelmann, R. Ludwig, Angew. Chem. Int.
Ed., 54, 2792-2795, (2015), Angew. Chem., 127, 2834–2837 (2015)[4] R. Ludwig, Phys. Chem. Chem. Phys., 17, 13790-13794 (2015)
256
Lipids and SolvationP02-21

Molecular topology and local dynamics govern the viscosity of imidazolium-based Ionic liquids
Yong Zhang,1 L. Xue,2 Fardin Khabaz,3 Rose Doerfler,1 Edward L. Quitevis,2 RajeshKhare,3 Edward J. Maginn1
1Department of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, Indiana 46556, United States
2Department of Chemistry & Biochemistry and3Department of Chemical Engineering, Texas Tech University, Lubbock, Texas
79409, United States
A series of branched ionic liquids (ILs) based on the 1-(iso-alkyl)-3-methylimidazoliumcation from 1-(1-methylethyl)-3-methylimidazolium bistriflimide to 1-(5-methylhexyl)-3-methylimidazolium bistriflimide and linear ILs based on the 1-(n-alkyl)-3-imidazoliumcation from 1-propyl-3-methylimidazolium bistriflimide to 1-heptyl-3-methylimidazolumbistriflimide were synthesized and their physical properties characterized.[1] For ILs with the same number of carbons in the alkyl chain, the branched IL was found to have the same density but higher viscosity than the linear one. In addition, the branched IL 1-(2-methylpropyl)-3-methylimidazolium bistriflimide ([2mC3C1Im][NTf2])was found to have an abnormally high viscosity. In order to obtain a molecular level understanding of these trends, the same ILs were studied using molecular dynamics (MD) simulations.[2] The viscosities of each IL were calculated using the equilibrium MD method at 400 K and the non-equilibrium MD method at 298 K. The results agree with the experimental trends. A quantitative correlation between the liquid structure and the viscosity was observed. Analysis shows that the higher viscosities in the branched ILs are due to the relatively more stable packing between the cations and anions. The abnormal viscosity of [2mC3C1Im][NTf2] was found to be the result of the specific side chain length and molecular structure.
[1] L. Xue, E. Gurang, G. Tamas, Y. Koh, M. Shadeck, S. L. Simon, M. Maroncelli, and E. L. Quitevis, J. Chem. Eng. Data 2016 (submitted).[2] Y. Zhang, L. Xue, F. Khabaz, R. Doerfler, E. L. Quitevis, R. Khare, E. J. Maginn, J. Phys. Chem. B 2015, 119, 14934-14944.
257
Lipids and Solvation P02-22

Shear thinning of critical fluctuations in solutions of Ionic liquidsW. Schröer, A. Butka, V.R. Vale, A. Elshwishin, D. Saracsan
Fb2 Biologie-Chemie, Universität Bremen, 28359 Bremen, Germany
It is well known that viscosity measurements in the vicinity of the liquid-gas phase-transition are influenced by gravity in a manner that causes apparent leveling off the critical enhancement. When approaching the critical temperature, the viscosity may reach an apparent plateau because the divergence of the viscosity is suppressed. Even if the gravity effect is not clearly seen it may lead to false values of the experimental critical exponent. In order to reduce the gravity influence experiments in space have been carried out [1].
For the liquid-liquid phase transition of mixtures the plateau of the temperature dependence of the viscosity is commonly not observed. However, in solutions of ionic liquids, that show an upper critical solution point, such plateau is reached if the measurements are carried out in a capillary viscosimeter with a temperature control with mK accuracy. The shear in the capillary limits the size of the cuts of the critical fluctuations that in ionic are larger than in non-ionic mixtures. The plateau is determined by the mentioned cutting effect, described by mode coupling theory [2] and a shift of the critical temperature [3] in the sheared system. With this corrections the temperature dependence of the viscosity is well represented by the crossover theory [4] that describes the crossover from regular to critical Ising behavior. In this work we report measurements on seven solutions (C18mim NTF2/Cyclohexane, C18mimNTF2/MethylCyclohexane, C12mim-chlorid/Benzene, P6,6,6,14Cl/n-Nonan, C6mimTriflat/ Chlorobenzene, C8mimTriflat-4 Chloro toluen, C5 mim-triflat/Wasser) of ionic liquids in non-polar and polar solvents and also in water.
References[1]R. F. Berg and M. R. Moldover, J. Chem. Phys, 93 (1990) 1926.[2]D.W. Oxtoby and W.M.Gelbart, J. Chem. Phys., 61(1974) 2957.[3] A. Onuki, K. Yamazaki and K. Kawasaki, Ann. Phys. 131 (1981) 217.[4] J. K. Bhattacharjee, R. A. Ferrell, R. S. Basu and J. V. Sengers,Phys. Rev. A, 24( 1981) 1469.
258
Lipids and SolvationP02-23

Reactants induced dynamic responses of heterogeneous catalysts monitored by adsorption microcalorimetry
S. Wrabetz, Th. Lunkenbein, V. Pfeifer, T. Jones, J. Noack, K. Mette, A. Trunschke, A. Knop-Gericke, R. SchlöglFritz-Haber-Institut der MPG, Dept. of Inorg. Chem., Berlin, Germany
In the present work, adsorption microcalorimetry1 is applied to investigate the adsorption-desorption cycles under near-reaction conditions which gives new insights into the dynamic behavior of the catalyst surface during adsorption /desorption steps. In our studies we find a dynamic response of the working catalyst to the reactants. The observed chemical dynamicscan be considered as a key process in the field of creating active sites. The focus is placed on three impressive projects: 1) CO chemisorption on Ni/MgAl oxide catalyst for dry reforming reaction of methane3. Figure 1 shows that the most active 50%Ni/MgAl oxide catalyst (XCH4,10h=73%) exhibits abnormally high differential heats of ~270kJmol-1 attributable to the Ni catalyzed CO oxidation with the MgAl oxide matrix. Further adsorption-desorption cycles cause drastic change of the heat profiles. It can be concluded, that metallic Ni sites are regenerated and more Ni aggregates are formed by the interaction with CO, verified by FTIR spectroscopy. This behavior evidences a highly dynamic surface. In contrast, the less active 5%Ni/MgAl oxide catalyst (XCH4,10h=50%) shows a strong reduced dynamic character. Thus, the dynamic character of the Ni-based adsorption sites of the most active surface is apparently favorable for the catalytic performance due to a continuous generation and/or regeneration of the used active sites.2) Adsorption of propane on MoV oxide, which is a versatile oxidation catalyst in liquid-phase oxidation reactions or in the oxidative dehydrogenation of ethane, but shows low selectivity in the oxidation of propane. Thislow selectivity may be attributed to the dynamic generation of strong adsorption sites provoked by the reactant itself (Figure 2). Adsorption of propane at thesesites can lead to over-oxidation.3) CO chemisorption at Treaction on Ir based catalysts for oxygen evolution reaction.2
1. S. Wrabetz, X. Yang, G. Tzolova-Müller, R. Schlögl, F. C. Jentoft, J. Catal. 269 (2010) 351.; Amakawa, K., Wrabetz, S., Kröhnert, J., Tzolova-Müller, G., Schlögl, R., Trunschke, A.; J. Am. Chem. Soc., 134 (28) (2012) 11462-11473;
2. V. Pfeifer,S. Wrabetz, R. Schlögl, A. Knop-Gericke et. al., „The electronic structure of iridium and its oxides” in progress.3. K. Mette et.al. “Development of HT stable Ni nano-particles for the dry reforming of methane at 900°C” in progress.4. R. S. Bordoli, J. C. Vickerman, J. Wolstenholme, Surf. Sci. 1979, 85, 244-262.5. A. Tanksale, J.N. Beltramini, J.A. Dumesic, G.Q. Lu, J. of Catalysis 258 (2008) 366–377; J. T. Stuckless, N. AI-Sarraf, C.
Wartnaby, D. A. King, J. Chem. Phys. 1993, 99, 2202-2212.6. M. Cerro-Alarcó, B. Bachiller-Baeza, A. Guerrero-Ruiz, I. Rodríguez-Ramos, J. Mol. Catal. Chem. 2006, 258, 221-230.
Figure 1. Differential heat of CO adsorbed on 50mol% Ni/MgAl oxide. Adsorption-desorption cycles at 30°C.
Figure 2. Differential heats of C3H8 adsorption on fresh MoV oxide via the amount of ads. Adsorption-desorption cycles at 40°C.
259
Chemical Dynamics and Kinetics P03-01

Theoretical investigation of the thermolysis of ureaDennis Gratzfeld and Matthias Olzmann
Institut für Physikalische Chemie, Karlsruher Institut für Technologie (KIT),Karlsruhe, Germany
Selective catalytic reduction (SCR) of nitrogen oxides (NOx) with ammonia is currently the most promising technology for reducing the NOx emissions of lean-burn combustion devices [1]. The need for more compact and mobile systems, due to increasing legislative restrictions on NOx emissions from automobiles, lead to the useof urea-water solutions to produce the gaseous reducing agent ammonia directly in the exhaust line. Although this technique is widely applied, the detailed reaction mechanism of ammonia production from urea-water mixtures is still unknown. Also, experimental studies [2,3,4,5] identified several byproducts whose formation has not been examined any further.We investigated elementary gas-phase reactions of the thermolysis of urea in presence of water with quantum chemical calculations and methods based on statistical rate theory. Stationary points on relevant potential energy surfaces were characterized with explicitly-correlated CCSD(T) calculations, and rate constants were determined with transition state theory and a simplified version of the statistical adiabatic channel model.In our contribution we energetically characterize reaction pathways for the production of ammonia and the formation of byproducts and discuss implications of our findings.
[1] Granger, P.; Parvulescu, V. I., Chem. Rev. 2011, 111, 3155–3207.[2] Stradella, L.; Argentero, M., Thermochim. Acta 1993, 219, 315–323.[3] Schaber, P. M. et al., Thermochim. Acta 2004, 424, 131–142.[4] Eichelbaum, M. et al., Appl. Catal., B 2010, 97, 90–97.[5] Brack, W. et al., Chem. Eng. Sci. 2014, 106, 1–8.
260
Chemical Dynamics and KineticsP03-02

Fuel-structure dependence of soot precursor formation processes in non-premixed laminar opposed-flow diffusion flames
L. Ruwe, K. Kohse-HöinghausDepartment of Chemistry, Bielefeld University, 33615 Bielefeld
Soot emissions from combustion processes are associated with harmful influences on human health and climate [1,2]. The relationship between the fuel molecule, the combustion process and the chemical structure of soot particles is not yet well understood. It is assumed that information on the formation pathways of soot will assist in predicting their nature and consequently some of their effects. Uncertainties remain in the mechanism of soot formation, especially in the regime between gas-phase chemistry and soot nucleation. Because most industrial combustion applications are non-premixed, non-premixed laminar opposed-flow diffusion flames are a suitable laboratory test bed and thus of great interest.
This study deals with the fuel-structure dependence of soot precursor formation processes in non-premixed laminar opposed-flow diffusion flames fueled by 1-pentene and n-pentane. The chemical compositions were determined by flame-sampling molecular-beam mass spectrometry employing electron ionization and single-photon ionization by synchrotron-generated vacuum-ultraviolet radiation. Molefraction profiles and isomer-resolving photoionization efficiency curves provide information on the gas-phase chemistry. As a result, a significant fuel-structure dependence of the reaction pathways was observed, resulting in substantial differences in soot formation tendency. For the 1-pentene flame reactions mainly proceed via allylic radicals, leading to an increased soot formation compared to the n-pentane flame.
[1] B. Frank, M.E. Schuster, R. Schlögl, D.S. Su, Angew. Chem. Int. Ed. 52 (2013) 2673-2677.
[2] J. Quaas, Nature 471 (2011), 456-457.
261
Chemical Dynamics and Kinetics P03-03

Thermal decomposition of propene – A shock-tube and modeling study
Leonie Golka, Isabelle Weber, Matthias OlzmannInstitut für Physikalische Chemie, Karlsruher Institut für Technologie (KIT),
Karlsruhe, Germany
Propene plays an important role as an intermediate species in many combustion and pyrolytic processes of higher hydrocarbons. A profound knowledge of the kinetics of the thermal decomposition of propene is therefore important for the understanding and modeling of these processes. In our contribution, we present results of anexperimental and theoretical study on the kinetics of the initial decomposition steps of propene and set up a mechanism describing not only the decomposition itself but also taking into account consecutive reactions.We performed experiments behind reflected shock waves at temperatures between 1340-1910 K and pressures in the range of 0.3 to 4.6 bar, using atomic resonance absorption spectroscopy for the detection of hydrogen atoms. The rate constant for the initial step was inferred from the measured concentration-time profiles and its temperature and pressure dependence was parameterized. A mechanism containing 29 reactions involving 15 species is proposed that is able to reproduce the measured profiles over the whole pressure and temperature range with reasonable accuracy.To analyze the observed pressure dependence, a SACM /Master-equation modelingwas performed.
262
Chemical Dynamics and KineticsP03-04

Experimental and Detailed Kinetic Modeling study of Ammonia Combustion
Sonal Kumar Vallabhuni, Arnas Lucassen, Ravi Fernandes, Physikalisch-Technische Bundesanstalt (PTB), Braunschweig, Germany.
Ammonia has been discussed as a potential alternative energy carrier for transportation and stationary engines, due to the fact that it can be easily produced from renewable sources using concepts such as power-to-gas.
It is an important potential clean energy source (no soot) for transportation. However,knowledge of NOx formation and krocking pollutants in gas based engines for high compression ratios is lacking. For its use in conventional and advanced engines, a detailed understanding of the combustion characteristics of ammonia over a wide range of conditions is desired for fuel-engine optimization.
The present study focuses on shock tube and Rapid Compression Machine (RCM) experiments for ignition delays and detailed kinetic modeling of ammonia oxidation. We report results in the intermediate temperature regime (700 – 1500 K) over a wide pressure range of (20 – 60 atm) and equivalence ratios (0.5, 1.0, 2.0). The experimental results are used for detailed kinetic model validation.
263
Chemical Dynamics and Kinetics P03-05

Observation of b2 symmetry vibrational levels of the SO21B2
state: OS— —SO asymmetry staggering and the vibronic coupling mechanism
G. Barratt Park, Jun Jiang, Catherine A. Saladrigas, and Robert W. Field, Massachusetts Institute of Technology, Cambridge, MA, USA
1B2 state of SO2 has a double-minimum potential in the antisymmetric stretch coordinate, such that the minimum energy geometry has nonequivalent SO bond lengths. We use IR-UV double resonance to observe the (one-photon-dark) b2
vibrational levels below 1600 cm 1 , which
3
the asymmetry in the potential energy surface near equilibrium. Our results are consistent with a vibronic coupling model for the double-minimum, which involves direct coupling to the 2 1A1 state and indirect coupling to the repulsive 3 1A1 state.Because the interactions among these three levels also cause the state to correlate adiabatically to the ground state SO(3 ) + O(3P) dissociation products, high-resolution characterization of the vibronic interactions near equilibrium provides mechanistic information about photodissociation dynamics that occur at much higher energy.
264
Chemical Dynamics and KineticsP03-06

Ultrafast species-selective photochemistry in mixturesDaniela Kern-Michlera, Carsten Neumanna, Nicole Seiberta, Luuk J.G.W. van
Wilderena, Matiss Reinfeldsb, Jan von Coselc, Alexander Heckelb, Irene Burghardtc,Jens Bredenbecka
a Institut für Biophysik, b Institut für Organische Chemie und Chemische Biologie, c Institut für Physikalische und Theoretische Chemie, Goethe-Universität, Frankfurt am Main, Germany
In a mixture of two spectroscopically very similar molecules we show that photochemistry can be induced and monitored species-selectively. As extreme test case of spectroscopically similar molecules we use a mixture of isotopologues. Our system consists of two isotope-labelled species of the common photolabile protection group [7-(diethylamino)coumarin-4-yl]methyl (DEACM) carrying azide as a leaving group. Their UV-Vis spectra are indistinguishable, but their infrared spectra exhibit clearly distinct bands.The selectivity in the infrared is exploited by using the VIPER 2D-IR pulse sequence[1,2], which uses a narrowband IR-pump pulse to select one species by exciting it to the first vibrationally excited state. The following visible pump pulse promotes the marked molecules to undergo a photoreaction. Tuning the IR-pump frequency results in signals specific for the photochemistry of each species in the mixture. Thepresented data will be discussed in terms of species and mode selectivity.
[1] van Wilderen, Luuk J.G.W.; Messmer, A. T.; Bredenbeck, J. Angew. Chem. Int. Ed. 2014; 53 (10), 2667-2672[2] van Wilderen, Luuk J.G.W.; Bredenbeck, J. Angew. Chem. Int. Ed. 2015; 54 (40), 11624-11640
265
Chemical Dynamics and Kinetics P03-07

Stretching-Libration Dynamics in the Methanol Dimer Matthias Heger, Martin A. Suhm
Georg-August-Universität Göttingen, 37077 Göttingen, GermanyJonas Andersen, René Wugt Larsen
Technical University of Denmark, 2800 Kgs. Lyngby, Denmark
The methanol dimer has seen intense experimental and theoretical characterization due to its model character for the simplest organic hydrogen bond between two aliphatic alcohol units. [1] Particular focus has been placed on the vibrational dynamics of the OH stretching vibration and its well-characterized ~110 cm–1
bathochromic dimerization shift in the donor molecule. We have undertaken efforts to dissect this observable into its different harmonic and anharmonic contributions, revealing two important core findings: first, that the former are notoriously misjudged by most popular quantum chemical methods, and high levels of electron correlation are mandatory to assess this quantity [2]; and second, that there is an intricate balance of intra-mode anharmonic weakening and inter-mode coupling effectscontained in the dimerization shift. Among the latter, the coupling of the stretching motion to the libration – i.e., the hindered OH torsion – particularly stands out. [3]
We present the first experimental characterization of these important inter-mode dynamics by matrix-FTIR spectroscopy. The results further provide an important benchmark for standard anharmonic quantum chemical approaches, which are still needed to fill in the remaining experimental gaps in the interpretation of the overall dimerization shift.
[1] F. Kollipost, K. Papendorf, Y.-F. Lee, Y.-P. Lee, M. A. Suhm, Phys. Chem. Chem. Phys. 16, 15948 (2014).[2] M. Heger, R. A. Mata, M. A. Suhm, J. Chem. Phys. 141, 101105 (2014).[3] F. Kollipost, J. Andersen, D. W. Mahler, J. Heimdal, M. Heger, M. A. Suhm, R. W. Larsen, J. Chem. Phys. 141, 174314 (2014).
266
Chemical Dynamics and KineticsP03-08

Ultrafast Ionization and Fragmentation Dynamics of Chloromethylsilanes
E. Antonsson, I. Halfpap, C. von Randow, C. Raschpichler, and E. RühlPhysical Chemistry, Freie Universität Berlin, Takustr. 3, D-14195 Berlin, Germany
The femtosecond pump-probe technique is known to be a suitable approach to study molecular strong-field ionization and fragmentation processes in the time domain. To follow specific fragmentation pathways, mass spectrometry is the most suitable approach. The fragmentation dynamics of highly excited molecular neutrals and ions can be affected by dynamic resonances, where the wavepacket motion in ionic potential energy surfaces plays a crucial role.
We present recent results on the ionization and fragmentation dynamics of trichloromethylsilane (SiCl3CH3), dichlorodimethylsilane (SiCl2(CH3)2), and chloro-trimethylsilane (SiCl(CH3)3) investigated by a one-color 804 nm femtosecond pump-probe experiment recording the ion yields of fragment ions. The intense pump pulse (250 μJ/pulse) is used to ionize the gas phase molecules which it is followed by a time delayed less intense probe pulse (70 μJ/pulse). This leads to additional fragmentation of the parent and fragment ions. The yields of parent and fragment ions show broad dynamic resonances with maxima in a range of t = 0.5 ps. The experimental findings are explained in terms of dynamic resonances leading to an enhanced dissociation of the parent ions as well as some fragment ions.
267
Chemical Dynamics and Kinetics P03-09

Femtosecond time-resolved dynamics of trans-methyl cinnamate by photoelectron imaging
Jonas A. Kus, Ole Hüter, Friedrich TempsInstitute of Physical Chemistry, Christian-Albrechts-University Kiel,
Olshausenstr. 40, D-24098 Kiel, Germany
Cinnamic acid and its derivates can be found in the UV photoprotection cycle of plants and are also commonly used as UV filter agents in sunscreens because of their broad absorption in the UV range and the ultrafast relaxation of the excited states. The deactivation of the initially excited states occurs within picoseconds [1].Despite numerous studies, however, the electronic structure of the molecules and the corresponding relaxation pathways are not well understood.
Figure 1: Structure of trans-methyl cinnamate.
We used femtosecond time-resolved time-of-flight mass spectrometry and photoelectron imaging to obtain deeper insight into the relaxation dynamics of jet-cooled trans-methyl cinnamate. We excited the molecule at = 295 nm and
= 260 nm and ionized the molecules by a time-delayed probe pulse at = 402 nm.
After excitation at = 295 nm, we observe bands in the photoelectron images which
we assign to a *-state, while we see no conclusive evidence for a population of the
n *-state. The complementary mass signal of the parent ion decays within 6 ps. At
= 260 nm, we observe no major changes in the photoelectron band structure,although we found shorter lifetimes of below 2 ps of the parent ion signal, hinting at amore efficient relaxation pathway at this wavelength.
[1] M. Vengris, D. S. Larsen, M. A. van der Horst, O. F. A. Larsen, K. J.Hellingwerf, R. van Grondelle, J. Phys. Chem. B 2005, 109, 4197–4208.
268
Chemical Dynamics and KineticsP03-10

Geometric optimization of ICD electron emitter propertiesin quantum dots
F. Weber1), E. Faßhauer2), A. Lode3), A. Bande1)
1) Helmholtz-Zentrum-Berlin, Berlin / Germany; 2) University of Tromsø,Tromsø / Norway; 3) University of Basel, Basel / Switzerland
In previous theoretical studies on electron dynamics[1], the interatomic Coulombic decay (ICD) process was predicted to take place in charged double quantum dot (QD) systems. ICD represents an ultrafast energy transfer process[2], that is triggered by photoexcitation of a conduction band (CB) electron in a photon-absorbing QD. Itsdeexcitation leads to the release of a CB electron in a neighbouring electron-emittingQD. As ICD heavily depends on the electronic properties[3], which in turn are defined by the QD geometry and the used material[4], the process can be optimized to yield bestmost efficiency. The facile tunability of QD materials therefore make them the best canditate for applied sciences.So far, however, the optimization of the system only focused on the photon-absorber QD[5] while keeping the properties of the electron-emitter QD constant. The present work therefore focuses on maximizing the ICD rate in a double QD model system by additional optimization of the emitter QD by studying the electron dynamics with the multiconfiguration time-dependent Hartree method in two different implementations[6,7]. Using both the optimization of absorber and emitter, we conclude that a nanowire of longitudinally arranged cigar-shaped QDs is suitable for experimental studies.
Literature[1] A. Bande, K. Gokhberg, L. S. Cederbaum, J. Chem. Phys. 135, 144112 (2013)[2] L.S. Cederbaum, J. Zobeley, F. Tarantelli, Phys. Rev. Lett. 79, 4778 (1997)[3] U. Hergenhahn, J. Electron Spectrosc. Relat. Phenom. 184, 78 (2011)[4] A. Zunger, MRS Bulletin 23, 32 (1998)[5] P. Dolbundalchok, D. Peláez, A. Bande, E.F. Aziz, in preparation[6] H.-D. Meyer, U. Manthe, L.S. Cederbaum, Chem. Phys. Lett. 165, 73 (1990)[7] A. Lode, E. Faßhauer, arXiv:1510.02984 (2015)
269
Chemical Dynamics and Kinetics P03-11

Influence of the biofuel isomers diethyl ether and n-butanol on pollutant formation in premixed n-butane flames
Julia Pieper, Meirong Zeng, Luc-Sy Tran, Isabelle Graf, Katharina Kohse-Höinghaus,Bielefeld University, Bielefeld, Germany
Increasing energy demand especially in the transport sector, coupled with the need to reduce greenhouse gas emissions, continues to motivate research directed towards renewable fuels. Biofuels such as ether, esters, and alcohols are being discussed as additives or replacement fuels.
In this study, effects of the addition of isomeric oxygenated fuel additives to a hydrocarbon fuel have been analyzed for premixed low-pressure flame conditions by electron ionization molecular-beam mass spectrometry (EI-MBMS). Fuel mixtures of the LPG representative n-butane with either diethyl ether (DEE) or its isomer n-butanol, an advanced biofuel, have been investigated. The analysis of the flame structure has revealed fuel-specific differences, especially in the formation ofintermediate species. For example, soot precursor formation is reduced by adding DEE to n-butane, while n-butanol surprisingly shows no significant reduction effects or even higher formation of soot precursor species. Also, the formation of toxic carbonyls upon oxygenated fuel addition is of special concern. The increase in aldehyde mole fractions, including formaldehyde and acetaldehyde, was observed to be significantly lower for DEE than for n-butanol addition to n-butane. The observed tendencies that show the superior properties of DEE as a fuel additive will be discussed in terms of the respective kinetic mechanisms.
270
Chemical Dynamics and KineticsP03-12

Direct Detection of CN Radicalsin the NCN Reaction System Behind Shock Waves
Sebastian Hesse, Nancy Faßheber, Gernot FriedrichsInstitut für Physikalische Chemie, Christian-Albrechts-Universität zu Kiel
The mechanistic understanding of NOx formation in combustion processes relies on detailed investigations of elementary reactions. The NCN radical plays a key role for prompt NO formation in flames, which – according to the theoretical study of Moskaleva et al. [1] – is initiated by the reaction CH + N2 NCN + H. In recent years, both experimental and theoretical studies have been performed to develop a reliable NCN submechanism such as the NOMecha2.0 [2]. As a product of bimolecular NCN reactions, the formation and secondary chemistry of CN radicals are crucial for modeling overall NO formation. In this work, CN radical concentration-time profiles have been measured behind shock waves in order to validate recent reaction kinetic data in the NCN reaction system. The combination of the shock tube method and the sensitive frequency modulation (FM) spectroscopy enables detection of the CN radical on the (4 – 0)
Q1(10) transition of the red A21 – X2 system at a wavelength of = 622.2733 nm.
The thermal decomposition of BrCN at 2200 K < T < 3200 K and of NCN3 at 700 K < T < 2500 K served as a quantitative source for the CN and NCN radical, respectively. The CN profiles have been analyzed with the current version of the GDF-Kin®3.0/NOMecha2.0 mechanism [2] setting a special focus on the roles of the
reactions NCN + NCN 2 CN + N2, CN + NCN C2N2 + N, CN + CN + M C2N2
+ M, and BrCN + M Br + CN + M.
[1] L. V. Moskaleva, M. C. Lin, Proc. Combust. Inst. 2000, 28, 2393-2401.[2] N. Lamoureux, H. E. Merhubi, L. Pillier, S. de Persis, P. Desgroux, Combust.
Flame 2015, doi: 10.1016/j.combustflame.2015.11.007, in press.
271
Chemical Dynamics and Kinetics P03-13

VIPER 2D-IR Spectroscopy on Coumarins: Selectivity Experimentsand Simulations
Carsten Neumann1, Luuk J.G.W. van Wilderen1, Daniela Kern-Michler1, Nicole Seibert1, Jan von Cosel2, Robert Binder2, Matiss Reinfelds3, Irene Burghardt2,
Alexander Heckel3, Jens Bredenbeck1
1Institut für Biophysik, 2Institut für Physikalische und Theoretische Chemie, 3Institut für Organische Chemie und Chemische Biologie, Goethe Universität Frankfurt am
Main, 60438 Frankfurt am Main
Here, we demonstrate the potential for subensemble-selective photo control by using the ultrafast VIPER 2D-IR pulse sequence. This mixed IR/non-IR pulse sequence has been used previously to study chemical exchange of coumarin 6 beyond the limit of vibrational lifetime [1]. In recent experiments, the chemical and bond selectivity is applied to a coumarin photocage, 7-[(diethylamino)coumarin-4-yl]methyl (DEACM),that carries an azide which is released upon photo-excitation. In DEACM, the VIPER-pulse sequence is used to trigger photochemistry by a combination of an IR- and a Vis-pump pulse. The narrow-band IR-pump pulse is set to be resonant with differentvibrational modes and followed by a Vis-pump pulse. The Vis-pulse is tuned to provide only sufficient energy to bring the vibrationally-excited molecules to the electronically excited state. The subsequent cargo release then takes place and is monitored in the infrared spectral region.In this work, the influences of tuning the IR- and Vis-pump pulse frequencies and of the relative timing between the pump pulses on the signal size are compared for the two coumarins, DEACM-N3 and coumarin 6. For both molecules, quantum chemical calculations were performed to predict the effect of pre-exciting different vibrational modes on the Vis-absorption spectrum, identifying the lower frequency ring mode to be the most promising mode in terms of VIPER efficiency. These theoretical predictions agree with our experimental findings.
[1] L.J.G.W van Wilderen, A. T. Messmer, J. Bredenbeck; Angew. Chem. Int. Ed.2014; 53 (10), 2667-2672
272
Chemical Dynamics and KineticsP03-14

Strong Field Control of the Interatomic Coulombic Decay Process in Quantum Dots
Anika Haller1), Ying-Chih Chiang2,3), Emad F. Aziz1), Annika Bande1,2)
1)Helmholtz-Zentrum Berlin, Berlin/Germany
2)Universität Heidelberg, Heidelberg/Germany
3)The Chinese University of Hong Kong, Shatin, N.T., Hong Kong
The interatomic Coulombic decay (ICD) is an ultrafast energy transfer process between atoms and molecules induced by long-range electron correlation. ICD is also possible for systems of electrons confined in general, non-infinite binding potentials as they are commonly used to model quantum dots (QD) [1,2].
We consider a system of two electrons in a double well potential with descrete and continuous energies. The ground state is resonantly excited and the excited state then decays via ICD, where one electron is transferred to the continuum.
We theoretically investigate the electron dynamics for the described system exposed to a time-dependent laser field and show how ICD can be controlled via laser initiation. We use the ansatz as it had been used for the resonant Auger decay in [3,4] to obtain energy resolved spectra for the quantum states. Further, an antisymmetrized multi-configuration time-dependent Hartree (MCDTH) method [5] is used to solve the time-dependent Schödinger equation. This gives, beyond the
-pulse had been studied [2]. This is now extended to a whole range of pulse strengths from extremely weak to extremely strong fields.
Literature[1] A. Bande, K. Gokhberg, and L. S. Cederbaum. J. Chem. Phys., 135 (2011), 144112.
[2] A. Bande. J. Chem. Phys., 138 (2013), 214104.
273
Chemical Dynamics and Kinetics P03-15

[3] Y.-C. Chiang, P. V. Demekhin, A. I. Kuleff, S. Scheit, and L. S. Cederbaum. Phys. Rev. A, 81 (2010), 032511.
[4] P. V. Demekhin and L. S. Cederbaum. Phys. Rev. A, 83 (2011), 023422.
[5] H.-D. Meyer, F. Gatti, and G. A. Worth. Multidimensional Quantum Dynamics: MCTDH Theory and Applications. Wiley-VCH, Weinheim, 2009.
274
Chemical Dynamics and KineticsP03-15

2-Butanone as an advanced biofuel - assessment of potential combustion emissions
Christian Hemken, Ultan Burke, K. Alexander Heufer, Isabelle Graf, Lena Ruwe,Katharina Kohse-Höinghaus
Bielefeld University, Bielefeld, GermanyRWTH Aachen University, Aachen, Germany
Regarding future transportation strategies, biofuels will play an increasingly important role. 2-Butanone has recently been identified as a next-generation biofuel candidate by a model-based design strategy from about 10.000 molecular structures by the Excellence Cluster Tailor-Made Fuels from Biomass, RWTH Aachen University.The detailed combustion of 2-butanone under both high- and low-temperature conditions was studied here for the first time, using a laminar flame and a plug flow reactor, analyzed by molecular-beam mass spectrometry; both using electron ionization and synchrotron-based photoionization in two independent experiments. Quantitative mole fraction profiles for about 45 species were obtained. Among those, toxic oxygenated compounds were identified, including specifically methyl vinyl ketone, acetaldehyde and formaldehyde. Soot precursors were seen to be notably low in concentration.The measurements are considered useful targets for the development of detailed reaction mechanisms, and first simulation results with an existing comprehensive reaction model will be shown.
275
Chemical Dynamics and Kinetics P03-16

Measuring the kinetics of chemical reactions on metal surfaces in a molecular beam surface scattering experiment
Hinrich W. Hahn1, Jannis Neugebohren1, Dan J. Harding1, Daniel J. Auerbach1,Theofanis N. Kitsopoulos1,2, Alec M. Wodtke1
1Institute for Physical Chemistry, Georg-August University Göttingen, 37077 Göttingen, Germany
2Department of Chemistry, University of Crete, 71003 Heraklion, Greece and Institute of Electronic Structure and Laser, Foundation for Research and Technology-Hellas,
71003 Heraklion, Greece
AbstractWe use spatial ion imaging and slicing in combination with intense femtosecond laser pulses for strong field ionization to examine the kinetics of reactions at surfacesoccurring after molecular beam metal surface collisions. The universal ionization method together with spatial imaging make a versatile detection scheme that enables us to measure speed and angular distributions of the mass resolved reaction products. Ions created in the detection chamber of our UHV machine are extracted by means of a pulsed electric field and accelerated towards a gated MCP/phosphor screen assembly and a CCD camera.The reaction particularly featured in this work is the oxidation of CO on an oxygen pre-covered Pt(111) surface. While a constant O2 background pressure in themachine maintains certain oxygen coverage, a pulsed molecular beam of CO is shot at the surface with an incidence angle of 45°. Coverage dependent CO2 desorption rates and angular distributions are measured at the surface normal (i.e. far off the specular angle) for the products of two reaction pathways that significantly differ in velocity and angular distribution. Known CO dose, CO sticking probability and CO desorption rates (residence time) result in a reaction probability for the CO oxidation.
276
Chemical Dynamics and KineticsP03-17

Experimental Analysis of the Passivation Kinetics in Aluminium
Morris Weimerskirch, Tristan O. Nagy, Ulrich Pacher, Wolfgang Kautek;
Universität Wien, Wien/Österreich
Hints to the unification of the Point Defect Model and the High Field Model
Following our work on the repassivation mechanism[1] as a time-dependent linear combination of a high-field model of oxide growth (HFM)[2,3] and the point defect model (PDM)[4] we present our findings on the repassivation process as two concurrent charge consumption channels. For that, in situ laser depassivation[5,6] of plasma electrolytically oxididized (PEO) coatings on aluminium was performed with nanosecond laser pulses at 266 nm under potentiostatic control and the repassivation current transients were recorded as a function of pulse number.There are hints to a material reminiscence to laser treatment as the charge contribution of the PDM channel grows with the number of laser pulses. Furthermore, we determined a point tswitch where, according to our combined model, the contribution of the HFM supersedes the one of the PDM. Interestingly, the ratio of tswitch and the overall passivation time seems to remain constant over any number of pulses, providing hints for a possible unified theory describing the passivationprocess from the blank metal to the steady state. [7]
Keywords: aluminium, current-transients, laser-depassivation, repassivation, high field model, point defect model
[1] T. O. Nagy, U. Pacher, A. Giesriegl, L. Soyka, G. Trettenhahn, W. Kautek, Laser-induced electrochemical de- and repassivation investigations on plasma-oxidized aluminium alloys, Applied Surface Science, 302 (2014), 184-188.
[2] T.P. Hoar, N.F. Mott, A mechanism for the formation of porous anodic oxide films on aluminium, Journal of Physics and Chemistry of Solids, 9 (1959) 97-99.
[3] G.S. Frankel, C.V. Jahnes, V. Brusic, A.J. Davenport, Repassivation transients measured with the breaking-electrode technique on aluminum thin-film samples, Journal of the Electrochemical Society, 142 (1995) 2290-2295.
[4] D.D. Macdonald, The point defect model for the passive state, Journal of the Electrochemical Society, 139 (1992) 3434-3449.
[5] A. Cortona, W. Kautek, Microcorrosion and shock-affected zone investigation at anodic films on aluminium alloys by pulse laser depassivation, Physical Chemistry Chemical Physics, 3 (2001) 5283-5289.
[6] W. Kautek, G. Daminelli, Electrochemical reactivity of laser-machined microcavities on anodised aluminium alloys, Electrochimica Acta, 48 (2002) 3249-3255.
[7] T. O. Nagy, M. Weimerskirch, U. Pacher, W. Kautek, Repassivation Investigations on Aluminium: Physical Chemistry of the Passive State, to be published.
277
Chemical Dynamics and Kinetics P03-18

High resolution analysis and quantum dynamics of fluoroformI. Bolotova, Physical Chemistry, ETH-Zürich, CH-8093 Zürich, Switzerland;
O. Ulenikov, E. Bekhtereva, Institute of Physics and Technology, National Research Tomsk Polytechnic University, 634050 Tomsk, Russia; S. Albert, H. Hollenstein,
M. Quack, Physical Chemistry, ETH-Zürich, CH-8093 Zürich, Switzerland
The high resolution spectroscopy of CHF3 has been the basis of the study of time independent and time dependent quantum dynamics for a long time [1-9]. There have also been substantial efforts concerning the ab initio potential hypersurface [10, 11]. We shall present here a survey of some of our recent analyses ranging from the Terahertz (Far infrared) spectral range to about 3000 cm 1, with particular emphasis on the pure rotational (FIR) spectra measured at the infrared beamline of the Swiss synchrotron Light Source (SLS), v3 fundamental (700 cm 1 range), the v2, v5, v3+v6
polyad (1200 cm 1 range), the v4/2v3 dyad (1400 cm 1), the 2v4 (A1 and E) dyad and extensions as available at the time of the Bunsentagung. The implications for the study of intramolecular vibrational energy redistribution (IVR) will be outlined.
References[1] a) S. Albert, K. Keppler Albert, H. Hollenstein, C. Manca-Tanner, and M. Quack Fundamentals of Rotation-Vibration Spectra, Vol. 1, pp. 117-173; b) S. Albert, K.Keppler Albert, and M. Quack High-Resolution Fourier Transform InfraredSpectroscopy, Vol. 2, pp. 965-1019; c) M. Quack Fundamental Symmetries and Symmetry Violations from High-Resolution Spectroscopy, Vol. 1, pp. 659-722 in Handbook of High Resolution Spectroscopy, M. Quack and F. Merkt eds., Wiley Chichester 2011.[2] H. R. Dübal, and M. Quack, Chem. Phys. Lett. 80, 439-444 (1981).[3] H. R. Dübal, and M. Quack, J. Chem. Phys. 81, 3779-3791 (1984).[4] R. Marquardt, M. Quack, J. Stohner and E. Sutcliffe, J. Chem. Soc., Far. Tr. 82,1173-1187 (1986).[5] A. S. Pine, and J. M. Pliva, J. Mol. Spectrosc. 130, 431-444 (1988).[6] J. Segall, R. N. Zare, H. R. Dübal, M. Lewerenz, and M. Quack, J. Chem. Phys.86, 634-646 (1986).[7] A. Amrein, M. Quack, and U. Schmitt, Mol. Phys. 60, 237-248 (1987).[8] A. Amrein, M. Quack, and U. Schmitt, J. Phys. Chem. 92, 5455-5466 (1988).[9] M. Quack, Annu. Rev. Phys. Chem. 41, 839-874 (1990).[10] T. K. Ha, M. Lewerenz, R. Marquardt, and M. Quack, J. Chem. Phys. 93, 7097 7109 (1990).[11] J. Breidung, J. Coslou, J. Demaison, K. Sarka, and W. Thiel, Mol. Phys. 102, 1827-1841 (2004).
278
Chemical Dynamics and KineticsP03-19

Alignment- and orientation-dynamics of ground-state selected OCS
S. Trippel,1 T. Mullins,1 J. Kienitz,1, 2 N. Müller,1 J. Wiese,1 J. Küpper,1,2,3
1 Center for Free-Electron Laser Science, DESY, 22607 Hamburg, Germany2 Department of Physics, University of Hamburg, 22761 Hamburg, Germany
3 Center for Ultrafast Imaging, University of Hamburg, 22761 Hamburg, Germany
State-selected, strongly aligned and oriented molecular ensembles serve as ideal samples to study ultrafast chemistry directly in the molecular frame [1]. Possible probing mechanisms include the investigation of molecular-frame photoelectron angular distributions [2] or the detection of structural changes via X-ray or electron diffraction [3].We have developed techniques to manipulate the motion of molecules in cold supersonic beams using strong inhomogeneous electric and laser fields [4]. State-selected molecules are aligned by strong laser fields or oriented in combination of laser fields and static electric fields.Here, we will present our work on the alignment and orientation carbonyl of sulfide(OCS). We have created a coherent superposition of pendular states in the strong field of the alignment laser and follow its propagation as a function of time [5].Furthermore we have obtained strong laser field free orientation by controlling the coupling between the lowest field free rotational states [6]. In addition we have accomplished adiabatic orientation by making use of strong DC electric fields.Furthermore, molecular frame photoelectron angular distributions of OCS will be presented.
References: [1] J. Hansen, H. Stapelfeldt, D. Dimitrovski, M. Abu-samha, C. Martiny and L. Madsen, Phys. Rev. Lett., 106,
073001 (2011).[2] L. Holmegaard, J. L. Hansen, L. Kalhøj, S. L. Kragh, H. Stapelfeldt, F. Filsinger, J. Küpper, G. Meijer, D.
Dimitrovski, M. Abu-samha, C. P. J. Martiny, and L. B. Madsen, Nat. Phys. 6, 428 (2010). [3] J. Küpper, S. Stern, L. Holmegaard, F. Filsinger et al, Phys. Rev. Lett. 112, 083002 (2014)[4] Y.-P. Chang, D. A. Horke, S. Trippel, and J. Küpper, Int. Rev. Phys. Chem. 34, 557 (2015).[5] S. Trippel, T. Mullins, N. Müller, J. Kienitz, J. Omiste, H. Stapelfeldt, R. González Férez, J. Küpper, Phys.
Rev. A, 89, 051401(2014)[6] S. Trippel, T. Mullins, N. Müller, J. Kienitz, R. Gonzalez-Ferez, J. Küpper, Phys. Rev. Lett., 114, 103003
(2015)
279
Chemical Dynamics and Kinetics P03-20

Investigation of the Methane number as measure for knock propensity of natural gas mixtures containing higher hydrocarbons
Arnas Lucassen, Sonal Kumar Vallabhuni, Ravi Fernandes,Physikalisch- Technische Bundesanstalt (PTB), Braunschweig, Germany.
The methane number (MN) is a parameter that is representative for the knocking behavior natural gas mixtures. In analogy to the octane number and the cetane number the values are based on a binary model mixture in this case of Methane(pure methane has a MN of 100) and Hydrogen (pure hydrogen has an MN of 0). The currently used comprehensive approach to the development of a correlation between the natural gas composition and the MN dates back to the early 1970s [1]. One of the shortcomings of this work is that the number of components considered is quite limited. This becomes crucial with the increased use of LNG as transport fuel ,because LNG composition differs from that of other natural gas by increased content of higher hydrocarbons. These hydrocarbons have a suppressing effect on the MN.
This study aims to link the MN and the knocking behavior to the composition of a mixture by detailed understanding of the combustion kinetics and use of kinetic models. To achieve this we studied a wide range of prototypical LNG model compositions in a Rapid Compression Machine, compared the results to simulated ignition delay times and validate newly derived Methane Number calculation method against the dataset. The results are furthermore compared to tests with the same mixtures in two different spark ignition engines.
[1] "AVL method" Leiker, M., Cartillieri, W., Christoph, K., Pfeifer, U., Rankl, M.: Evaluation of Antiknocking Property of Gaseous Fuels by Means of Methane Number and its Practical Application to Gas Engines. ASME-Paper 72-DGP-4.
280
Chemical Dynamics and KineticsP03-21

Towards understanding ultrafast dynamics in complex systems like nanodiamonds or switchable polymer nanoparticles
Daniel Hintzen, Leibniz-Institute of Surface Modification, Leipzig, Germany, Bernd Abel, Leibniz-Institute of Surface Modification, Leipzig, Germany
Our group is concerned with the fundamental understanding of ultrafast processes in complex systems like Nanodiamonds or photoswitchable polymer nanoparticles.[1]
To get insights in the dynamic processes in these complex systems we use innovative and unique techniques from the field of ultrafast spectroscopy, with a particular focus on the Ultra-Broadband Femtosecond Transient Absorption Spectroscopy. Our goal is to understand the associated ultrafast dynamics of the switching process in order to optimize the molecular processes which are the key to the unique properties of these polymer nanoparticles. With this we take a first step towards rational and smart design of photon controlled devices.
[1] Anwar, N., et al., Light-Switchable and Monodisperse Conjugated Polymer Particles. ACS MacroLetters, 2013. 2(9): p. 766-769.
281
Chemical Dynamics and Kinetics P03-22

The photodissociation of dimethylnitrosamine revisited –a 3d-REMPI and VMI study
Manuel Schneider, Uwe Kensy, Bernhard Dick, Universität Regensburg, Germany;
Nitrosamines are a pervasive group of molecules with dimethylnitrosamine (DMN) as a highly toxic, cancerogenic member, that is formed as an industrial by-product and can also be found in foodstuffs, drinks and tobacco1. The ground state chemistry of nitrosamines has been studied for more than three decades and is mostly well understood. Much less is known on their photochemistry, and in particular on the dynamics of photodissociation. In this study we employed velocity map imaging (VMI) and the 3d-REMPI technique to obtain a detailed insight into the photodissociation dynamics of DMN following the excitation into either its S1 or S2 state.Our results indicate a direct dissociation from the S1 level on a purely repulsive potential energy surface, which is in good agreement with previous studies on this molecule2,3,4. We investigated the influence of additional vibrational excitation within the S1 state, which provided us with a complete overview of the dissociation process from these levels in terms of energy and angular distribution of the photofragments. We present evidence that this extra vibrational energy remains in the N(CH3)2-fragment. The corresponding vibration must hence be assigned to an internal vibration of the aminyl fragment rather than the NN or NO stretching modes as suggested in earlier studies4.In the case of dissociation from the S2 level, the observed data also indicate a fast and direct dissociation. Apparently two distinct dissociation channels exist, which differ in the amount of kinectic energy released. One channel produces the aminyl radical in the electroncally excited state, the other produces the electronic ground state. This second channel was theoretically proposed by Peláez et al.5 and already confirmed for N-nitrosopyrrolidine (NNPy) by Wenge et al.6 in our group.
1 Lü J.-M., Wittbrodt J. M., Wang K., Wen Z., Schlegel H. B., Wang P. G. & Cheng J.-P.; J. A. C. S., 2001, 1232Lavi R. & Rosenwaks S.; J. Chem. Phys., 1988, 893Brühlmann U., Dubs M. & Huber J. R.; J. Chem. Phys., 1987, 864Dubs M. & Huber J. R.; Chemical Physics Letters, 1984, 108, 123-1275Peláez D., Arenas J. F., Otero J. C. & Soto J.; J. Org. Chem., 2007, 726Wenge A. M., Schmaunz A., Kensy U. & Dick, B.; Phys. Chem. Chem. Phys., 2010, 12
282
Chemical Dynamics and KineticsP03-23

Vibrations of hemiporphycene in the S0 and S1 electronic states:A supersonic jet spectroscopy study
S. PeukertM. Broquier,§ A. Zehnacker-Rentien§
Institute of Physical Chemistry, Polish Academy of Sciences, Warsaw, Poland§CNRS, Institut des Sciences Moléculaires dʼOrsay (ISMO), Centre Laser Université
Paris-Sud (CLUPS), University Paris-Sud, Orsay, France
Previously, much effort has been devoted to spectroscopic and photophysical studies of porphyrin (Pr) and porphycene (Pc), since both molecules represent model systems for tautomerization processes. They are characterized by symmetric double minimum potential energy surfaces and proton tunneling splitting can occur. In these compounds the extent of proton tunneling correlates with the properties of intramolecular NH…N hydrogen bonds. They are stronger in Pc and this is regarded as a reason why – amongst others - tautomerization rates are orders of magnitude larger in Pc than in Pr1 and why the fluorescence excitation spectrum of Pc isolated in supersonic jets consists of doublets separated by 4-5 cm-1, whereas the spectrum of supersonic jet isolated Pr does not show any tunnelling splitting.2,3
Parent hemiporphycene (HPc) is another constitutional isomer of of Pr and it has been recently synthesized. To the best of our knowledge there is no spectroscopic characterization of this molecule so far. In HPc the geometry of the inner cavity is formally similar, but its dimensions are larger than those of Pc and Pr. Moreover, HPc is of lower symmetry (Cs) than Pc (C2h). The intention of this work is to probe if there is any evidence for proton tunneling in HPc and to characterize the vibrational structure of HPc in both, its electronic ground (S0) and excited state (S1). Therefore we have conducted experiments applying the supersonic jet technique in combination with laser-induced fluorescence excitation and dispersed fluorescence as well as hole burning spectroscopy. The experiments are complemented by electronic structure calculations at the (T)DFT/B3LYP/6-31+G(d,p) level of theory.
References1 - Eur. J. 15 (2009) 4851.
Phys. Lett. 296 (1998) 549.
174201.
283
Molecular Structure P04-01

Excited State Intramolecular Proton Transfer of 2,5-bis(5-ethyl-2-benzoxazolyl)-hydroquinone and its OH/OD-Isotopomers studied in
Supersonic Jets
S. PeukertInstitute of Physical Chemistry, Polish Academy of Sciences, Warsaw, Poland
Excited-State Intramolecular Proton Transfer (ESIPT) is a photochemical process creating tautomers with different fluorescence properties. Among numerous ESIPT systems,1 some emit dual fluorescence. One group of such compounds includes 2,5-bis(2-benzoxazolyl)-hydroquinone (BBHQ) and its derivatives.2,3
In the present work, ESIPT reactions of dually fluorescent 2,5-bis(5-ethyl-2-benzoxazolyl)-hydroquinone (DE-BBHQ) and its isotopomers have been studied in the supersonic jet applying laser induced fluorescence (LIF) and hole burningspectroscopy.4 LIF-spectra measured at photo-tautomeric (red) fluorescence exhibit a characteristic triplet pattern of vibronic bands, which gradually collapses upon successive deuteration. In comparison to BBHQ, clustering bands show a decreased splitting. Experimental and theoretical results indicate that the torsional motions of the ethyl-substituents couple with excited state proton tunneling, which can contribute to a decreased splitting of clustering bands in DE-BBHQ. Complementary (T)DFT calculations at the B3LYP/6-31+G(d,p) level of theoryindicate also the possibility of two consecutive ESIPT reactions yielding an excited state diketo-tautomer. However, concerning this matter the present experimental results are not unambiguous and could be also rationalized without assuming the formation of an additional photo-tautomer.
References1. T. Elsaesser, H.J. Bakker, Ultrafast Hydrogen Bonding Dynamics and Proton Transfer Processes in the Condensed Phase; Springer: Heidelberg, Germany, 2002.2. N.P. Ernsting, J. Phys. Chem. 89 (1985) 4932.3. N.P. Ernsting, T. Arthen-Engeland, M.A. Rodriguez, W. Thiel, J. Chem. Phys. 97 (1992) 3914.4. S. Peukert, M. Gil, M. Kijak
284
Molecular StructureP04-02

Nitrogen nanocoating effects on the conformational preferences of ethanol and its dimer detected by supersonic jet spectroscopy
Sönke Oswald, Martin A. SuhmInstitut für Physikalische Chemie, Georg-August-Universität Göttingen, Germany
Ethanol is particularly suitable to study the interplay between hydrogen bonding and molecular conformation. The isolated monomer favours the trans state over the gauche conformer, whereas the global minimum structure of the dimer consists of two homochiral gauche monomers [1]. Three out of five theoretically predicted metastable dimer conformations have already been detected experimentally [1,2] and we discuss the search for the missing two. Furthermore, we present spectra of ethanol monomers and dimers with single and multiple attached nitrogen molecules, because the ethanol conformational preference is inverted in solid nitrogen matrices [3]. A combination of direct absorption FTIR spectroscopy and spontaneous Raman scattering [1,2] is helpful in interpreting the spectral fingerprints of conformational change and nitrogen nanocoating.
[1] Emmeluth, C., Dyczmons, V., Kinzel, T., Botschwina, P., Suhm, M. A., Yáñez, M., Combined jet relaxation and quantum chemical study of the pairing preferences of ethanol, Phys. Chem. Chem. Phys., 2005, 7, 991-997.[2] Wassermann, T. N., Suhm, M. A., Ethanol Monomers and Dimers Revisited: A Raman Study of Conformational Preferences and Argon Nanocoating Effects, J. Phys. Chem. A, 2010, 114, 8223-8233.[3] Coussan, S. Bouteiller, Y., Perchard, J. P., Rotational Isomerism of Ethanol and Matrix Isolation Infrared Spectroscopy, J. Phys. Chem. A, 1998, 102, 5789-5793.
285
Molecular Structure P04-03

Control over the hydrogen bond docking site in anisole by ring
alkylation and halogenationHannes C. Gottschalk, Martin A. Suhm,
Georg-August-Universität Göttingen, Tammannstr. 6, 37077 Göttingen, Germany Supramolecular docking of methanol to anisole may occur via an OH O hydrogen bond or via an OH contact.[1] The subtle balance between these two structures can be varied in supersonic jets over one order of magnitude through aromatic ring methylation, tert-butyl substitution or halogenation as evidenced by infrared spectroscopy. This steep variation allows to benchmark relative quantum-chemical energy predictions on a kJ mol-1 level, promising insights into inductive, mesomeric and dispersive effects. For ring alkylation, the zero point corrected B3LYP-D3/aVTZ level is shown to provide an accurate relative description of the two very different hydrogen bonds, similar to a wavefunction-based protocol including CCSD(T) corrections applied to the same structures. M06-2X alone systematically overestimates coordination.[2]
References: [1] M. Heger, J. Altnöder, A. Poblotzki and M. A. Suhm, To or not to – how does methanol dock onto anisole? Phys. Chem. Chem. Phys.,2015, 17, 13045-13052. [2] H. C. Gottschalk, J. Altnöder, M. Heger and M. A. Suhm, Control over the hydrogen bond docking site in anisole by ring methylation, Angew. Chem., accepted.
286
Molecular StructureP04-04

Three views on three xylene molecules: far-infrared spectroscopy, computational chemistry, and spectral modeling
Annika Bande1, Ivona Bravic1, Clemens B. Minnich2, Ljiljana Puskar1, Eglof Ritter3,Ulrich Schade1, Emad F. Aziz1,4
1Helmholtz-Zentrum Berlin, Berlin/Germany; 2S-PACT GmbH, Aachen/Germany; 3Humboldt-University Berlin, Berlin/Germany; 4Free University Berlin, Berlin/Germany
In the characterization of chemical compounds far-infrared (FIR) spectroscopy in the THz region has not been considered over decades. Now it had an advent in the detection of hazardous liquids in plastic bottles useful for airport safety controls.1
Among the characterized liquids, ortho-, meta-, and para-xylene have been re-investigated for the first time after 50 years,2 however, without a clear assignment of spectral peaks to molecular vibrations.
Here we present exactly this detailed analysis of xylene vibrations in the FIR based on density-functional theory (DFT) calculations in neat solution. As theoretical works on FIR spectra are as scarce as experiments, critical focus lies on the applicability of computational concepts used as a standard in calculations of near-and mid-IR spectra. These are namely the choice of functionals and basis sets and the treatment of solvent effects as e.g. through polarizable continuum models3 or scaling factors.4 With the DFT protocol developed we are able to reproduce and explain experimental spectra and discuss trends among the three isomers.
Alongside a new analysis procedure for optically appealing and meaningful comparisons of spectral data at all wave lengths has been developed based on spectral Hard Modeling.5 Its core piece is the fit of calculated line spectra to experimental FIR spectra with a quantitative track record on the modifications.
References 1. J. Beckmann et al., submitted.2. J. H. Green, W. Kynaston, H. A. Gebbie, Nature 159 (1962) 595.3. S. Sinnecker et al., J. Phys. Chem. A 110 (2006) 2235.4. M. K. Kesharwani, B. Brauer, J. M. L. Martin, J. Phys. Chem. A 119 (2015) 1701.5. F. Alsmeyer, H.-J. Koß, W. Marquardt, Appl. Spectrosc. 58 (2004) 957.
287
Molecular Structure P04-05

Structural investigations on a linear depsipeptide by combined IR/UV spectroscopy in a molecular beam
Anke Stamm, Dominic Bernhard, Markus Gerhards
TU Kaiserslautern, Erwin-Schrödinger-Str. 52, 67663 Kaiserslautern
Physikalische und Theoretische Chemie
Depsipeptides are an important class of natural products showing a diverse biological activity. As a special subclass of peptides they contain at least one ester bond replacing a peptide bond which gives rise to a different folding behavior compared to an ordinary peptide and probably leads to a different biological activity. They are often found to be natural antibiotics, such as the cyclic depsipeptides beauvericin and valinomycin. For a better understanding of their biological activity on a molecular level, knowledge on the structure of the isolated molecules is of importance. As firstlinear depsipeptide investigated in a molecular beam experiment we have chosen cyclohexylcarbonyl-glycine-lactate-2-anisidine (CyCO-Gly-Lac-NH-PhOMe; abbrev.MOC). MOC represents a model system whose structural investigation should give basic insights into preferred conformations of isolated depsipeptides. Furthermore the system contains both an aromatic phenyl ring and an aliphatic cyclohexyl ring. These two groups interact viadispersion is taken into account. Dispersion corrections are of significant importance
An experimental analysis is performed by application of mass- and isomer-selective combined double and triple resonance IR/UV methods in a molecular beam. In order to assign structures, DFT calculations including dispersion corrections were performed and compared with the experimental IR spectra.
288
Molecular StructureP04-06

Novel, heteroleptic copper(I)-4H-imidazolate complexes: synthesis and characterization of their structural, spectral and redox
propertiesM. Schulz,*a,b F. Drögea, Y. Zhanga,b, C. Reichardta,b, F. Herrmann-Westendorfa, J.
Schindlera,b,, H. Görlsc, M. Presselta, Benjamin Dietzeka,b
aInstitute for Physical Chemistry and Abbe Center of Photonics, Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena,
bLeibniz Institute of Photonic Technology (IPHT) Jena e. V., Albert-Einstein-Str. 9, 07745 Jena/Germany.
cInstitute for Inorganic and Analytical Chemistry, Friedrich-Schiller University Jena, Humboldtstr. 8, 07743 Jena/Germany
Photosensitizers for solar energy conversion should ideally exhibit a large molar extinction coefficient over as much of the solar irradiance spectrum as possible ahigh quantum yield of the charge separated state and tunable redox properties. Over the past decades, the enormous potential of Cu(I) photosensitizers has been realized and lead to intensive research activities in this field. Here we present the facile synthetic access to novel, neutral, heteroleptic copper(I)-complexes, incorporating 4H-imidazolates as well as the bisphosphane ligands XantPhos and DPEPhos. The complexes were characterized in the solid state as well as in solution and exhibit abroad intense absorption that spans almost the entire visible range. The spectral properties were further investigated by means of UV-Vis absorption and emission spectroscopy, ultrafast transient absorption spectroscopy, spectroelectrochemical methods as well TD-DFT calculations. NMR as well as electrochemical investigations and UV-Vis absorption suggest electronic communication of the phosphane and 4H-imidazolate ligands as well as the possibility to control the complex properties by choice of both the phosphane and 4H-imidazolate ligands. The photophysical and redox properties will be discussed as well as the potential of these complexes to serve as photosensitizer in molecular catalysis.
289
Molecular Structure P04-07

Characterization of the cis- / trans-isomerization of peptide bond models in the gas phase
Thomas Forsting, Katharina E. Otto, Martin A. SuhmInstitut für Physikalische Chemie, Georg-August Universität Göttingen, Germany
One of the simplest and yet realistic model systems for peptide bonds is N-methylformamide. Like true peptide bonds, it allows for cis/trans isomerism with a high interconversion barrier. In order to benchmark quantum chemical calculations, it would be valuable to have accurate experimental values for the isomerization enthalpy in the isolated gas phase. Previous estimates based on quenching the hot gas phase equilibrium in an Ar matrix yielded isomerization enthalpies of 5.4 kJ/mol1
and 7.4(7) kJ/mol.2
By quenching the hot gas phase equilibrium in a supersonic jet expansion and probing the cooled isomers in a spontaneous Raman scattering setup3 as a function of nozzle temperature, the enthalpy of isomerization is obtained in an independent way and compared to quantum chemical results. We also report on the Raman spectra of singly hydrogen-bonded trans-trans dimers4 and the search for cis-cisdimers, which are predicted to be much more stable, but kinetically hindered due to the high isomerization barrier. Finally, we give an outlook on N-methylacetamide.
1. S. Ataka, H. Takeuchi, M. Tasumi, J. Mol. Struct. 1984, 113, 147.2. S. Shin, A. Kurawaki, Y. Hamada, K. Shinya, K. Ohno, A. Tohara, M. Sato, J. Mol. Struct. 2006, 791, 30.3. K. E. Otto, Z. Xue, P. Zielke, M. A. Suhm, Phys. Chem. Chem. Phys. 2014, 16, 9849.4. M. Albrecht, C. A. Rice, M. A. Suhm, J. Phys Chem A. 2008, 112, 7530.
290
Molecular StructureP04-08

Study of Molecular Interactionsusing Broadband Rotational SpectroscopyMariyam Fatima1, Chris Medcraft1,*, Melanie Schnell1,2
1Max Planck Institute for the Structure and Dynamics of Matter at the Center for Free Electron Laser Science, Hamburg, Germany, 2The Hamburg Centre for Ultrafast
Imaging (CUI) at the University Hamburg, Hamburg, Germany, and, *PresentAddress: University of Newcastle upon Tyne, England
Intermolecular interactions play an important role in our life, from the folding of proteins and stability of DNA. Molecular processes such as (bio) molecular aggregation and recognition are driven by the interplay between different interactions, e.g. hydrogen bonding and dispersion. Therefore, the quantitative understanding of the various types of intermolecular interactions and their interplay in deciding the molecular structure is an interesting topic.In our group, we are studying these interactions using high-resolution molecular spectroscopy (1) under the isolated conditions of a molecular jet in the gas phase.This technique provides precise molecular parameters like bond length, bond angle etc. for developing a broad understanding of the underlying processes at the molecular level. With this we are interested in understanding the molecular recognition process. In order to do this, we are investigating the interactions of diphenylether ((C6H5)2O) and camphor (C10H16O) (2) with different alcohol systems, using rotational spectroscopy in the region of 2-8 GHz. It is expected that for these molecules as the alcohol will get larger the propensity towards hydrogen bonding should decrease whilst dispersion interactions become dominant.
(1) M.Schnell “Chirped-pulse rotational spectroscopy for molecular structure and dynamics studies” Zeitschrift für Physikalische Chemie, 227 (2013) 1-21.
(2) C.Pérez, et al “Wetting Camphor: Multi isotopic Substitution Identifies the Complementary Roles of Hydrogen Bonding and Dispersive Forces”, Journal of Physical Chemistry Letters, under revision.
291
Molecular Structure P04-09

Millimeter-wavelength Rotational Spectroscopy for Applications in Astrochemistry
Benjamin E. Arenas1, Amanda L. Steber1, 2, Sébastien Gruet1 and Melanie Schnell1, 2,1Max Planck Institute for the Structure and Dynamics of Matter at the Centre for Free Electron Laser Science, Hamburg, Germany and 2The Hamburg Centre for Ultrafast
Imaging (CUI) at Universität Hamburg, Hamburg, Germany.
With the availability of state-of-the-art observational technologies at the Atacama Large Millimeter/submillimeter Array (ALMA)[1], the extended Very Large Array (eVLA)[2] and others, scientists are beginning to explore some long-standing astronomical questions about chemical evolution. The rapid increase in data from these sources necessitates a concurrent increase in high-quality laboratory spectroscopy. Here, we present the use of a commercial[3] segmented chirped-pulse Fourier-transform millimeter-wave spectrometer, that operates from 75-110 GHz, to study astrochemically relevant complex organic molecules including alcohols and aldehydes. Our spectral region falls within the range of ALMAʼs Band 3 regime, allowing for us to directly compare the laboratory data with that from radio astronomy observatories[4]. Modification of the laboratory set-up, including the use of discharge and laser ablation apparatus, will grant us the ability to probe the reactive mechanisms of these interstellar molecules.
References[1] ALMA: http://www.eso.org/sci/facilities/alma.html, accessed 11 December 2015.[2] eVLA: https://science.nrao.edu/facilities/vla, accessed 11 December 2015.[3] BrightSpec: http://brightspec.com/, accessed 11 December 2015.[4] ALMA and ESO: https://almascience.eso.org/proposing/sensitivity-calculator,accessed 11 December 2015.
292
Molecular StructureP04-10

Catching hydrides spectroscopically with hard X-rays
Lukas Burkhardt, Matthias Bauer, Universität Paderborn, Fakultät für Naturwissenschaften, Department Chemie, Warburger Straße 100, 33098 Paderborn
Transition metal hydrides are of great importance in biological and chemical catalysis. They are known for many chemical transformations, hydrogen storage[1] or in renewable energy[2] applications like water reduction[3]. To understand the characteristics of hydride binding intermediates of hydrogenases, hydrogenasemodel complexes and other transition metal complexes which catalyze hydrogen production, spectroscopic methods are required. Normally metal hydride compounds are characterized via IR-/Raman-, NMR-, EPR-spectroscopy or neutron/X-ray scattering. In case of paramagnetic complexes, NMR is no longer possible. Furthermore infrared and Raman frequencies can be very weak or overlaid by other vibrations. Scattering techniques are also challenging or it is almost impossible to detect hydrogen.VtC-XES (Valence to Core X-ray Emission Spectroscopy) and HERFD-XANES (high-energy resolution fluorescence detected X-Ray absorption near edge structure) in combination with ground state DFT and TD-DFT calculations could overcome this limitations. VtC-XES probes the HOMO-structure of a given compound, it is highly sensitive to the ligand sphere, because valence orbitals change notable for different types of coordination[4,5]. The XANES region contains a lot of electronic and structural information, particularly the pre-edge signal is a probe for the LUMO-structure.It has been shown previously, that VTC-XES signals, their shape and energy positions are sensitive to ligand identity and coordination[6]. With a series of related iron hydride and non-hydride complexes, which are known to catalyze hydrogen formation[7], we will present a probe for metal-hydride-bonds via VtC-XES and HERFD-XANES.
[1] N. Armaroli, V. Balzani, ChemSusChem 2011, 4, 21–36.[2] T. Abbasi, S. A. Abbasi, Renewable and Sustainable Energy Reviews 2011, 15, 3034–3040.[3] D. Hollmann, F. Gärtner, R. Ludwig, E. Barsch, H. Junge, M. Blug, S. Hoch, M. Beller, A.
Brückner, Angew. Chem. Int. Ed. 2011, 50, 10246–10250.[4] N. Lee, T. Petrenko, U. Bergmann, F. Neese, S. DeBeer, J. Am. Chem. Soc. 2010, 132, 9715–
9727.[5] C. J. Pollock, S. DeBeer, Acc. Chem. Res. 2015, 48, 2967-2975.[6] M. Bauer, Phys. Chem. Chem. Phys. 2014, 16, 13827–13837.[7] S. Rommel, L. Hettmanczyk, J. Klein, B. Plietker, Chemistry, an Asian journal 2014, 9, 2140–2147.
293
Molecular Structure P04-11

The role of dispersion forces for the stability of methane and carbon dioxide gas hydrates
Wigbert S.K. Polet1 and Ralf Ludwig1,2,1Institute of Chemistry, Physical Chemistry, University of Rostock, Dr.-Lorenz-Weg 1, D-
18059 Rostock, Germany, [email protected] Institute for Catalysis, D-18059 Rostock, Germany, [email protected]
Natural gas hydrates can be found under sediments on the ocean floors of the earth as well as in Siberia and Alaska in sandstone and siltstone beds. Gas hydrates have been a subject of intense research for many years. The potential technical exploitation of these inclusion compounds have been mainly discussed in terms of a new carbon based energy source [1]. A thorough theoretical and thermodynamical description is of paramount importance for a fully understanding the stability of these compounds. Two main factors determine the stability of gas hydrates: hydrogen bonding between the water molecules forming the cage and dispersion interaction of the guest molecules with the water cage. Recent studies have shown that a very accurate thermodynamical description can only be achieved if the guest-guest interaction via dispersion forces is included as well [2][3].This study reports the calculation of interaction energies between CO2 and CH4 guest molecules in gas hydrates, comparing density functional theory (DFT) calculations includingdispersion correction D3 and Møller-Plesset perturbation theory (MP2) [4]. We investigated clathrate hydrate structures of type sI, which forms unit cells consisting of small 512- andlarger 51262-cages. The guest molecules are located at the centre of each cage. In the completely filled gas hydrate structures, each guest molecule is surrounded by 12 or 14 other species and coordinated by 20 or 24 water molecules, depending on the type of cage.To obtain the full guest-guest interaction energy, each cage was fully surrounded by 12/14 guest molecules. All guest molecules were placed at measured geometries. We can conclude that the guest-guest interaction cannot be neglected for the stability of gas hydrates.
References[1] E.D. Sloan, Nature 426, 353-363 (2003)[2] J.B. Klauda and S.I. Sandler, Chem. Eng. Sci. 58, 27-41 (2003)[3] S. Ravipati and N. Punnathanam, J. Phys. Chem. C, 18549-18555 (2013)[4] S. Grimme; J. Antony, S. Ehrlich and H. Krieg J. Phys. Chem. 132 (2010)
294
Molecular StructureP04-12

Gas Phase Vibrational Spectroscopy of Microhydrated Sulfate Dianions Revisited
Harald Knorke1, Martin Mayer1, Marcel Jorewitz1, Tim Esser1, Knut R. Asmis1
1Wilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstr. 2, 04103 Leipzig (Germany)
Hydrated sulfate dianion (SO42-) play an important role in the environment as well as
in several biological contexts. However, how these prototypical multiply charged anions are microhydrated in finite-size clusters remains heatedly debated. Here, wepresent gas phase infrared photodissociation spectra of the D2-tagged microhydrated sulfate dianions SO4
2-(H2O)n with n = 3–9. The spectra are recorded at a temperature of 13 K and in the spectral range from 650 to 3800 cm-1, covering the intramolecular modes of the solvent (O-H stretches and H2O bends) at higher energies, those of the solute (sulfate stretches) at intermediate energies and the intermolecular solute librational modes at the lowest energies. The IRPD spectra of the messenger-tagged complexes are considerably narrower than the previously published IRMPD spectraA,B and shows more details. Comparison to the results of electronic structure calculations leads to new insights into the nature of the solute-solvent and solvent-solvent interactions and the formation of the first hydration shell around the sulfate dianion core.
A J. Zhou, G. Santambrogio, M. Brümmer, D. T. Moore, L. Wöste, G. Meijer, D. M. Neumark and K. R. Asmis, J. Chem. Phys. 2006, 125, 111102.B M. F. Bush, R. J. Saykally, and E. R. Williams, J. Am. Chem. Soc. 2007, 129, 2220
295
Molecular Structure P04-13

High-Resolution Spectrum of the Asymmetric C-H Stretch of CH2Br2: CRDS Measurements and Simulations
Ibrahim Sadiek, Gernot FriedrichsInstitut für Physikalische Chemie, Christian-Albrechts-Universität zu Kiel
Photochemically active very short-lived brominated compounds are emitted from the ocean and reach the upper tropical tropopause layer by strong convective transport in equatorial regions. They exhibit high ozone destruction potential and affect the oxidation capacity of the atmosphere. High-resolution cavity enhanced absorption schemes such as cavity ringdown spectroscopy (CRDS) allow for sensitive and selective detection of environmental trace gases. However, potential applications for halocarbon detection in the IR spectral range, which requires careful line selection and hence relies on the availability of high resolution spectra, have not been fully exploited as yet. In this work, fundamental CRDS measurements and spectral simulations are presented. A single-frequency, continuous-wave (cw) optical parametric oscillator(cw-OPO) has been used as a light source for recording the Doppler-limited high-
resolution spectrum of the 6 band of CH2Br2 for the first time. The recorded spectra have a noise equivalent absorption of 3.4 × 10-9 cm-1 Hz-1/2. In addition, anharmonic frequency calculations have been performed using different density functionals as well as the MP2 approach. Vibrational ground state constants were taken from microwave measurements [1, 2]. The capabilities of the different theoretical methods to predict a reliable change in the state-dependent rotational constants upon vibrational excitation were investigated. Allowing for an additional explicit Coriolis perturbation due to the nearby symmetric C-H vibration, the main transitions of all three isotopologues of CH2Br2 could be assigned and hence the spectroscopic constants could be determined. A fit of the effective asymmetric rotor Hamiltonian to the spectrum resulted in a standard deviation of ~0.001 cm-1, which is in the order of the spectrometer resolution.
[1] R. Davis, M. Gerry, J. Mol. Spectrosc. 1985, 109, 269-282.[2] Y. Niide, H. Tanaka, I. Ohkoshi, J. Mol. Spectrosc. 1990, 139, 11-29.
296
Molecular StructureP04-14

High resolution GHz and THz (FTIR) spectroscopy and theory of parity violation and tunneling for dithiine as a candidate for measuring the parity violating energy difference between
enantiomers of chiral moleculesS. Albert, I. Bolotova, Z. Chen, C. Fabri, L. Horny, M. Quack, G. Seyfang and
D. Zindel, Physical Chemistry, ETH-Zürich, CH-8093 Zürich, SwitzerlandIn the framework of ordinary “electromagnetic” quantum theory the ground states of the enantiomers of chiral molecules are energetically equivalent. However, with the electroweak quantum chemistry and parity violation, one predicts a small “parity violating” difference PVE on the order of 100 aeV, typically, depending on the molecule, corresponding to a reaction enthalpy of stereomutation of about 10-11 J mol-1
[1,2]. So far, this effect has never been observed experimentally. In our paper, we report exploratory spectroscopy and theory in view of a possible use of the chiral C2-symmetric molecule dithiine (C4H4S2) for detecting molecular parity violation using a current experimental setup in our laboratory [3]. Using high resolution FTIRspectroscopy [4] we were able to provide a first rovibrational analysis of two bands, one centered at 623.3121 cm-1 consisting of c-type transitions and the second centered at 1308.8724 cm-1 consisting of a-type transitions. We also report GHz spectra. The assignments have been verified by combination differences. In parallel we calculated parity violating potentials using our recent coupled cluster approach [5]and tunneling using our quasiadiabatic channel reaction path Hamiltonian approach [6]. The implications of our results for the study of molecular parity violation will be discussed (see also refs. [7] and [8]).
References[1] M. Quack, Fundamental symmetries and symmetry violations from high resolutionspectroscopy, in Handbook of High-Resolution Spectroscopy, Vol. 1, (Eds. M. Quack and F. Merkt), Wiley, Chichester, 659-722 (2011).[2] M. Quack, Angew. Chem.-Int. Edit. Engl. 28, 571-586 (1989).[3] P. Dietiker, E. Milogyadov, M. Quack, A. Schneider and G. Seyfang, J. Chem. Phys. 143,(2015) in press.[4] S. Albert, K. K. Albert and M. Quack, High Resolution Fourier Transform Infrared Spectroscopy, in Handbook of High-Resolution Spectroscopy, Vol. 2, (Eds. M. Quack and F. Merkt), Wiley, Chichester, 965-1021 (2011). S. Albert and M. Quack, ChemPhysChem., 8,1271–1281 (2007).[5] L. Horny and M. Quack, Mol. Phys. 113, 1768-1779, (2015).[6] B. Fehrensen, D. Luckhaus, M. Quack, Chemical Physics 338, 90-105 (2007).[7] C. Fabri, L. Horny and M. Quack, Chem. Phys. Chem. 16, 3584-3589, (2015).[8] R. Prentner, M. Quack, J. Stohner and M. Willeke, J. Phys. Chem. A (2015).DOI:10.1021/acs.jpca.5b08958
297
Molecular Structure P04-15

Single-Conformation IR Spectroscopy of Jet-Cooled Sulfanilamide and its Homo- and Heteromolecular 1:1 Clusters
T. Uhlemann; S. Seidel; C.W. Müller, Ruhr-Universität Bochum/D
Here, we present our results from vibrationally state-selective, mass-resolved, and conformer-specific UV and IR spectroscopy of sulfanilamide, the sulfanilamide dimer and its monohydrated cluster in a cold molecular beam. The conformer-specific UV and IR spectra revealed that only one conformer of the monomer is present in the molecular beam.Additionally, we could show that the dimer as well as the monohydrated cluster of sulfanilamide also adopt only one isomeric form in the molecular beam. Unequivocal assignments could be made both for the dimer and the monohydrated cluster based on harmonic frequency calculations at the B3LYP-D3(BJ)/6-311++G(3df,3pd) level of theory. The dimer structure is of particular interest, since it provides, as smallest homomolecular cluster, some insight into crystal formation.We compare our findings with those of Borba and coworkers who recently conducted a matrix isolation FTIR study on the sulfanilamide conformers.1
[1] A. Borba; A. Gómez-Zavaglia; R. Fausto, J. Phys. Chem. A, 2013, 117, 704.
Chemical structure of sulfanilamide.
298
Molecular StructureP04-16

Spectral Signatures of Polarons and Dynamics of Charge Separation in Conjugated Co-Polymers
C. Wiebeler and S. Schumacher, Department of Physics and Center for Optoelectronics and Photonics Paderborn, University Paderborn, 33098 Paderborn
In organic photovoltaics molecules carrying a net charge emerge as a desired product after successful charge separation. We study the electronic and optical properties of ions of the low-bandgap co-polymer PCPDT-BT and compare it with the corresponding homopolymer using (time-dependent) Density Functional Theory. One of our main findings is that the ions of the homopolymer show a polaron absorption that is symmetric between anion and cation whereas for polaron excitations in the co-polymer this symmetry is absent [1] (c.f. left panel of the figure below).
In addition, it is found in experiment that a higher polaron pair formation yield is observed at shorter excitation wavelengths [2]. We explain this observation with the more pronounced initial charge transfer character of the higher lying excited states[2]. However, to fully understand the ultrafast photophysical process that is observed in neat films of conjugated co-polymers, the dynamics after photoexcitation have to be investigated. To model the relaxation through the excited state manifold, nonadia-batic ab initio molecular dynamics based on Trajectory Surface Hopping is used (c.f. right panel of the figure for a sample trajectory).
References
[1] C. Wiebeler, R. Tautz, J. Feldmann, E. von Hauff, E. Da Como, and S. Schumacher., J. Phys. Chem. B 2013, 117, 4454.[2] R. Tautz, E. Da Como, C. Wiebeler, G. Soavi, I. Dumsch, N. Fröhlich, S. Allard, U. Scherf, G. Cerullo, S. Schumacher, and J. Feldmann, J. Am. Chem. Soc. 2013, 135, 4282.
299
Molecular Structure P04-17

Single-Conformation Spectroscopy, DFT, NBO, and Charge Density Studies of the Neuraminidase Inhibitor Analogue BANA 113
Sebastian Seidel1, Thomas Uhlemann1, Michael Seitz2 and Christian W. Müller1,1Department of Physical Chemistry II, Ruhr-University Bochum,
Universitätsstraße 150, 44780 Bochum, Germany2Institue of Inorganic Chemistry, Eberhard Karls University Tübingen, Auf der
Morgenstelle 18, 72074 Tübingen, Germany
Due to numerous degrees of freedom the neuraminidase inhibitor analogue 4-(N-acetylamino)-3-guanidino benzoic acid (BANA 113) adopts a large number of conformations. These conformations are stabilised by different intramolecular interactions depending on the individual structure of the conformer and stereoelectronic effects. Here, we present conformer-specific UV and IR spectra of BANA 113 in the N–H and O–H stretching region. The experiments are supported by calculations of harmonic frequency spectra at DFT level of theory (B3LYP-D3(BJ)/6-311+G(d,p)). In order to gain deeper insight into intramolecular stabilisation, we explored the potential energy surface (PES) by systematically scanning two dihedral angles with respect to the aromatic core for all three substituents. Stabilising effects have been quantified by means of Quantum Theory of Atoms in Molecules (QTAIM) and Natural Bond Orbital (NBO) analysis. We compare our findings with the computational results of Yang and co-workers who studied the conformational preferences of BANA 113 and its analogues at B3LYP/6-31G(d) and B3LYP/6-311++G(d,p) levels of theory.[1,2]
[1] Z. Yang, G. Yang, Y. Zu, Y. Fu, L. Zhou, Phys. Chem. Chem. Phys., 2009,11, 10035 – 10041.[2] Z. Yang, Y. Nie, G. Yang, Y. Zu, L. Zhou, J. Theo. Bio., 2010, 267, 363 –374.
300
Molecular StructureP04-18

Influence of thermal fluctuations for the 2D-IR structure determination in the liquid phase
Kilian Fehre,a Daniela Kern-Michler,a Sascha Combe,b Peter R. Schreiner,b Barbara Kirchner,c and Jens Bredenbecka
a.Institute of Biophysics, Johann Wolfgang Goethe-University, Max-von-Laue-Str. 1, 60438 Frankfurt, Germany. b.Institute of Organic Chemistry, Justus-Liebig University,
Heinrich-Buff-Ring 58, 35392 Giessen, Germany. c.Mulliken Center for Theoretical Chemistry, Institut für Physikalische und Theoretische Chemie, Universität Bonn,
Beringstr. 4+6, 53115 Bonn, Germany.Ultrafast polarization-dependent two-dimensional infrared (P2D-IR) spectroscopydetermines molecular structure via the measurement of vibrational transition dipole angles (TDAs).1,2,3 P2D-IR has been used to identify the major conformer of N-crotonyl-oxazolidinone, a Diels-Alder dienophile equipped with a chiral auxiliary(Evans auxiliary).1,2 Computations at the density functional theory (DFT) levelsuggest a planar structure with the transition dipoles of the investigated marker modes lying exactly in a plane. Surprisingly, TDAs retrieved from temperature dependent P2D-IR spectroscopy in the temperature range between -70°C and 20°Creveal a non-planar structure. We show by potential energy surface scans on the DFT level and by ab initio molecular dynamics trajectory computations that thermal fluctuations lead to considerable deviations from the planar minimum energy structure and to according changes in the TDAs. Even at the low end of the accessible temperature range, which is limited by the freezing point of the solvent CH2Cl2 (-97°C), the thermal ensemble is sufficiently broad that the experimentally determined structure is expected to measurably deviate from the structure at the energy minimum. If very accurate structure determination in liquids is attempted andcomparison to theoretical results or crystal structures is made, the thermal distribution around the minimum energy structure therefore has to be taken into account.
1A. T. Messmer, K. M. Lippert, P. R. Schreiner, J. Bredenbeck, Phys. Chem. Chem. Phys. 2013, 15,1509–1517.2A. T. Messmer, K. M. Lippert, S. Steinwand, E.-B. W. Lerch, K. Hof, D. Ley, D. Gerbig, H. Hausmann,P. R. Schreiner, J. Bredenbeck, Chem. Eur. J. 2012, 12, 14989–14995.3H. Bian, J. Li, X. Wen, Z. Sun, J. Song, W. Zhuang, J. Zheng, J. Phys. Chem. A 2011, 115, 3357–3365.
301
Molecular Structure P04-19

Photochemical Dissociation of HOBr. A Nonadiabatic Dynamics Study
Saadullah G. Aziz1, Basmah H. Allehyani1 and Rifaat Hilal1,2
1Chemistry Department, Faculty of Science, King Abdulaziz University, Jeddah, SA2Chemistry Department, Faculty of Science, Cairo University, Giza, Egypt
The photochemical dissociation of hypobromous acid, HOBr, was investigated by computing excited-state properties with a number of single-reference methods, namely CIS-, SAC-CI, EOM-CCSD and time-dependent density functional theory(TDDFT). Excited states calculated with these methods, especially TDDFT, show good agreement with experimental results. UV photoabsortion spectrum and nonadiabatic dynamics simulations were carried out to understand the decay of the excited states of HOBr in the gas phase. These calculations have been performed at the TDDFT level (with the M06-2x functional) using NEWTON-X [1,2]. The spectrum was computed with the nuclear ensemble approximation [3] sampling a Wigner distribution with 500 points. 10 excited states were considered here. Dynamics simulations were started in two separate spectral windows, at the 4 ± 0.25 eV and 8 ±0.25 eV, to be compared to experimental UV spectral data. One hundred points were stochastically sampled from each of these windows according to their excitation probabilities. Dynamics simulations were done with the surface hopping method [4].Nonadiabatic effects between excited states were taken into account by the fewest switches algorithm with decoherenc The excited-state lifetime was determined. Photodissociation occurred through several different reaction pathways with different products. The main photochemical channel observed were H and BrO eliminations, representing 54% of all processes.
[1] Barbatti, M.; Ruckenbauer, M.; Plasser, F.; Pittner, J.; Granucci, G.; Persico, M.; Lischka, H., WIREs: Comp. Mol. Sci. 2014,[2] Barbatti, M.; Granucci, G.; Ruckenbauer, M.; Plasser, F.;Crespo-Otero, R.; Pittner, J.; Persico, M.; Lischka, H. NEWTON-X: A package for Newtonian Dynamics Close to the Crossing Seam. www.newtonx.org, 2013.[3] Crespo-Otero, R.; Barbatti, M., Theor. Chem. Acc. 2012, 131, 1237.[4] Tully, J. C.,[5] Barbatti, M.,
302
Molecular StructureP04-20

[1] P. Gütlich, H.A. Goodwin, Top. Curr. Chem. 2004, 233, 1-47. [2] J.-F. Letard, P. Guionneau, L. Goux-Capes, Top. Curr. Chem. 2004, 235, 221-249. [3] G. Vanko, F. de Groot et al., J. Phys. Chem. B2006, 110, 11647-11653. [4] H.-J. Krüger, Coord. Chem. Rev. 2009, 253, 2450-2459.
High energy resolution X-ray absorption and emission spectroscopy for the investigation of spin crossover processes
Rahel Schepper, Matthias Bauer, Universität Paderborn, Paderborn, Deutschland
Spin crossover (SCO) is an example of potential molecular bistability, in which the high-spin (HS) and low-spin (LS) states are interconvertible by physical perturbation,such as temperature.[1] Since its first discovery the phenomenon has attracted much attention from chemists and physicists, because it offers an important application of ligand field theory. But despite the tremendous insights that have been gained into the underlying electronic principles responsible for the occurrence and the specific behaviour of spin transition processes, the study of SCO complexes is still a highly attractive research field due to their high potential for electronic devices.[2] Most characterizations are based on either determining the structural changes (XRD, XAS) or the changes of the spin state (Mößbauer, SQUID). For further understanding of the molecular mechanism of SCO processes the simultaneous measurement of structural and spin state changes is desirable.
X-ray emission based methods offer new possibilities to record both parameters (quasi simultaneously) in one experiment by means of HERFD-XAS, K 1,3- 1,3-XES and HERFD-XAS provide information about the metal spin state, more exactly, about the effective number of unpaired 3d electrons[3], EXAFS allows to access the local structure around the metal centre.
We therefore studied the quasi octahedral SCO compound [Fe(L-N4Bz2)(NCS)2] [4], which shows a gradual spin transition from LS to HS, by temperature dependent HERFD-
1,3-XES and EXAFS. The experimental basics, data analysis approach and results are presented with this contribution.
References:
Figure 1: Structure of [Fe(L-N4Bz2)(NCS)2]
High energy resolution X-ray absorption and emission spectroscopy for the investigation of spin crossover processes
Rahel Schepper, Matthias Bauer, Universität Paderborn, Paderborn, Deutschland
Spin crossover (SCO) is an example of potential molecular bistability, in which the high-spin (HS) and low-spin (LS) states are interconvertible by physical perturbation,such as temperature.[1] Since its first discovery the phenomenon has attracted much attention from chemists and physicists, because it offers an important application of ligand field theory. But despite the tremendous insights that have been gained into the underlying electronic principles responsible for the occurrence and the specific behaviour of spin transition processes, the study of SCO complexes is still a highly attractive research field due to their high potential for electronic devices.[2] Most characterizations are based on either determining the structural changes (XRD, XAS) or the changes of the spin state (Mößbauer, SQUID). For further understanding of the molecular mechanism of SCO processes the simultaneous measurement of structural and spin state changes is desirable.
X-ray emission based methods offer new possibilities to record both parameters (quasi simultaneously) in one experiment by means of HERFD-XAS, K 1,3- 1,3-XES and HERFD-XAS provide information about the metal spin state, more exactly, about the effective number of unpaired 3d electrons[3], EXAFS allows to access the local structure around the metal centre.
We therefore studied the quasi octahedral SCO compound [Fe(L-N4Bz2)(NCS)2] [4], which shows a gradual spin transition from LS to HS, by temperature dependent HERFD-
1,3-XES and EXAFS. The experimental basics, data analysis approach and results are presented with this contribution.
References:
Figure 1: Structure of [Fe(L-N4Bz2)(NCS)2]
High energy resolution X-ray absorption and emission spectroscopy for the investigation of spin crossover processes
Rahel Schepper, Matthias Bauer, Universität Paderborn, Paderborn, Deutschland
Spin crossover (SCO) is an example of potential molecular bistability, in which the high-spin (HS) and low-spin (LS) states are interconvertible by physical perturbation,such as temperature.[1] Since its first discovery the phenomenon has attracted much attention from chemists and physicists, because it offers an important application of ligand field theory. But despite the tremendous insights that have been gained into the underlying electronic principles responsible for the occurrence and the specific behaviour of spin transition processes, the study of SCO complexes is still a highly attractive research field due to their high potential for electronic devices.[2] Most characterizations are based on either determining the structural changes (XRD, XAS) or the changes of the spin state (Mößbauer, SQUID). For further understanding of the molecular mechanism of SCO processes the simultaneous measurement of structural and spin state changes is desirable.
X-ray emission based methods offer new possibilities to record both parameters (quasi simultaneously) in one experiment by means of HERFD-XAS, K 1,3- 1,3-XES and HERFD-XAS provide information about the metal spin state, more exactly, about the effective number of unpaired 3d electrons[3], EXAFS allows to access the local structure around the metal centre.
We therefore studied the quasi octahedral SCO compound [Fe(L-N4Bz2)(NCS)2] [4], which shows a gradual spin transition from LS to HS, by temperature dependent HERFD-
1,3-XES and EXAFS. The experimental basics, data analysis approach and results are presented with this contribution.
References:
Figure 1: Structure of [Fe(L-N4Bz2)(NCS)2]
303
Experimental Techniques P05-01

Construction of a surface plasmon spectrometer with enhanced sensitivity and time resolution for the study of electrochemical
processes
Karin Schlag, Detlef Nattland, Axel E. Timm, Rolf SchusterInstitute of Physical Chemistry, Karlsruhe Institute of Technology
Kaiserstr. 12, D-76131 Karlsruhe, Germany
We combine electrochemical methods like cyclic voltammetry and pulsepotentiometry with time-resolved surface plasmon resonance (SPR) utilizingKretschmannʼs configuration for attenuated total reflection (ATR) to study adsorption and deposition phenomena at thin gold film working electrodes. Plasmon angle and width are highly sensitive to compositional changes at the electrode-electrolyte interface. We strive for a significant improvement with respect to the detection of the surface plasmon angle as well as to the response time in comparison to a SPR spectrometer previously built in our lab. Our new apparatus can be easily and reproducibly aligned and has a flexible range of the angle of incident.Plasmons are excited by a laser diode (635 nm) at the 50 nm working electrode gold film. The full SPR profile is simultaneously recorded by an array photodiode detector(512 Pixel, which in our geometry spans 12°) in one millisecond. For interpretation we subsequently fit a multilayer model to the plasmon profiles. Each layer is characterized by its thickness and its complex refractive index.As a first test system we have chosen sulfate adsorption on Au(111). The noise of the plasmon curve while cycling through the adsorption/desorption region did not exceed about 0,0001°. This corresponds to a refractive index uncertainty of a six angstrom thick sulfate adlayer of about 0,0002.
304
Experimental TechniquesP05-02

Contactless Microwave Conductivity of Oxide Supported Metal Samples
Elisabeth Hannah Wolf1, Thomas Risse2, Robert Schlögl1, and Sebastien Cap1,1Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin/Deutschland,
2Freie Universität, Berlin/DeutschlandBased on the microwave cavity perturbation technique1, a contact-free method of measuring the conductivity of powdered catalysts under reaction conditions (changeable gas atmosphere at elevated temperatures), has been developed. Themethod utilises the perturbation of microwave radiation inside a resonant cavity by the dielectric properties of the sample. In particular, shifts of resonant frequency and the quality factor of the resonator are used to calculate the conductivity of the sample2.The conductivity of samples consisting of small copper particles supported on metal oxides (ZnO, MgO) was thus analysed under reducing and oxidising conditions (5% H2 and 5% O2 flow in N2 respectively). When changing the support of the metal from ZnO to MgO, the conductivity response varies drastically. Cu/ZnO and Cu/MgO showed a difference in conductivity of several orders of magnitude, as ZnO is a semiconductor, whereas MgO is an insulator. The conductivity increased when switching to reducing feed over the Cu/ZnO system (Fig. 1a) which could indicate a charge transfer from copper to zinc oxide, as previously proposed in the literature3.By contrast, over the Cu/MgO system, switching to reducing feed resulted in adecrease in conductivity (Fig. 1b). The observed behaviour was reversible and reproducible in both cases. Possible reasons for these observations will be discussed.
1. Eichelbaum, M. et al. Phys. Chem. Chem. Phys. 14, 1302–12 (2012).2. Slater, J. C. Rev. Mod. Phys. 18, 441–512 (1946).3. Liu, X.-Y. et al., Thin Solid Films 520, 5372–5377 (2012).
Figure 1: Conductivity changes of (a) Cu/ZnO (230°C) and (b) Cu/MgO (250°C)
100% N2
5% H2 in N2
5% O2 in N25% O2 in N2
5% H2 in N2
a) b)
305
Experimental Techniques P05-03

Sensing of Light induced Magnetization by Magnetic Resonance Force Microscopy
Jacqueline Deichmöller1, Konstantin Kartaschew1, Arpad Rostas2, Erik Schleicher2,Stefan Weber2 and Martina Havenith1
1 Ruhr-Universität Bochum, Physical Chemistry II, 44780 Bochum, Germany,2 Albert-Ludwigs-Universität Freiburg, Institute for Physical Chemistry,79104 Freiburg, Germany
Many functions of biological systems cannot be unraveled without the knowledge of the mechanisms on the nanometre-scale. Over the past decades an extensive effort has therefore been made pushing the limits of magnetic resonance force microscopy (MRFM) based methods to a resolution of single spins [1]. Temperature and magnetic field dependencies hamper this objective enormously. We present the implementation and first application of a novel approach called Light induced Magnetization sensed by Magnetic Resonance Force Microscopy (LIMRFM). Light induced magnetic forces, in the regime of attonewtons, are detected with the aid of a ferromagnetic nanometric cantilever. The application of light excitable paramagnetic chromophore spin samples in LIMRFM has the advantage of strong initial spin polarizations even under ambient conditions. The sample excitation into the first paramagnetic triplet state is conducted by laser irradiation from the diamagnetic ground state (S =0) into the first excited state (S =1). Inter system crossing of the electron into the first triplet state results in a sufficient net magnetization even at room temperature. The existence of an external magnetic field leads to a triplet sublevel splitting, while spin inversions within the triplet sublevels are initiated by amicrowave magnetic field which satisfies the resonance condition. We will show first results of LIMRFM measurements of the chromophore pentacene at room temperature. Our aim is to introduce LIMRFM as a promising tool for spin detection and to extend the existing MRFM detection schemes to new applications.
[1] D. Rugar, R. Budakian, H. J. Mamin, B. W. Chui, Nature 2004
306
Experimental TechniquesP05-04

Hyperfine Structure in the IR-Spectrum of Ammonia, NH3
Eduard Miloglyadov, Martin Quack, Georg SeyfangPhysical Chemistry, ETH Zurich, Switzerland
Ammonia is a one of the simplest prototype molecules with a double minimum potential and its tunneling dynamics in the ground state and the lower vibrational states is well understood [1-3]. In accordance with theoretical calculations a larger quadrupole constant eQq0 was found in the symmetric tunneling states as compared to the anti-symmetric tunneling states for the vibrational ground state and for the tunneling mode ν2 [4-7].Using a high resolution, single mode OPO (Qioptiq) we have measured selected lines of the ν1, ν3
±1, 2ν40 and 2ν4
±2 vibrational modes with hyperfine resolution in a molecular beam and determined the quadrupole coupling constant eQq(νi) for the corresponding vibrational levels, also in relation to a proof-of-principle experiment as preparation to detect parity violation in chiral molecules [8]. To increase the accuracy and precision of the measured spectra the OPO was locked to a 250 MHz repetition rate frequency comb (Menlo Systems) and a spectral line with of 350 kHz was obtained using REMPI detection of the excited molecules in a doubly skimmed pulsed molecular beam. The hyperfine transitions were measured with an estimated uncertainty of ±100 kHz.References:
1. R. Marquardt, K. Sagui, J. Zheng and W. Thiel, D. Luckhaus,
S. Yurchenko, F. Mariotti and M. Quack, J. Phys. Chem. A, 117, 7502 (2013)
2. C.Fabri, R. Marquardt, M. Quack, Bunsentagung 2015, Bochum and to be published
3. M. Snels, H. Hollenstein and M. Quack, J. Chem. Phys. 125, 194319 (2006) and
M. Snels, V. Horká-Zelenková, H. Hollenstein, M. Quack in ‚Handbook of High
Resolutiom Spectroscopy’, M. Quack and E. Merkt (edt.), Vol. II, 1021 (2011)
4. S.G. Kukolich and S.C.Wofsy, J. Chem. Phys. 52, 5477 (1970)
5. J. T. Hougen, J. Chem. Phys. 57, 4207 (1972)
6. V. Špirko, Mol. Phys. 38, 1761 (1979)
7. P.W.Fowler and V. Špirko, J. Chem. Soc. Farad. Trans. 86, 1991, (1990)
8. P. Dietiker, E. Myloglyadov, M. Quack, A. Schneider and G. Seyfang, J. Chem. Phys.
143, (2015) in press
307
Experimental Techniques P05-05

Kinetics of Carbon-Dioxide Hydrate FormationSebastian Schulze, Johannes Kiefer, and Bernd Rathke#
Universität Bremen, Technische Thermodynamik, Badgasteiner Str. 1, 28359 Bremen, Germany
(#) e-mail: [email protected]
The formation of gas hydrates is a key issue in technical processes dealing with elevated pressures, humidity and low temperatures. From the applied point of view, the sequestration of carbon-dioxide, the basic understanding of processes in oil industry and natural gas processing are the most prominent applications, which require a profound knowledge of the physico-chemical aspects of the underlying mechanisms. Unfortunately, basic research on the kinetics of hydrate formation taking into account the different aspects of equilibrium thermodynamics and the kinetics of phase transitions are scarce. Within the present study we have been characterized onset-conditions of the formation of gas hydrates from carbon-dioxide saturated water.For this purpose a set of experiments has been performed using a high pressure apparatus suitable up to pressures of p = 700 bar. The set-up consists of two independent parts, which allow for a preparation of binary mixtures under defined conditions and rapid kinetic studies of phase transitions induced by fast pressure changes, respectively. This concept allows for an independent control of temperature and pressure without a change of the composition of a sample.First results indicate a strong variation of induction times of hydrate formation at different degrees of supersaturation and are discussed in terms of classical nucleation theories.
308
Experimental TechniquesP05-06

Vapor-liquid Equilibria (VLE) of Binary Imidazolium-Based IonicLiquid Mixtures with Volatile Solvents
Marcus Stuckenholz1,#, Johannes Kiefer1, Bernd Rathke1, and Wolffram Schröer2
(1) Universität Bremen, Technische Thermodynamik, Badgasteiner Str. 1,28359 Bremen, Germany
(2) Universität Bremen, Institut für Anorganische und Physikalische Chemie, Leobener Str. NW II , 28359 Bremen, Germany
(#) e-mail: [email protected]
The knowledge of the phase behavior is a prerequisite both for the application of Ionic Liquids (ILs) in different processes as well as for a physico-chemical understanding. However, up to now a large fraction of studies published during the last years has been focused on the characterization of liquid-liquid equilibria. Systematic investigations on the vapor-liquid equilibria (VLE) of binary mixtures of an Ionic Liquid (IL) and an organic solvent, complementing these data, are still scarce. Therefore, the present paper will contribute to this topic. First results of isobaric VLEs of mixtures of 1-hexyl-3-methylimidazolium trifluoromethanesulfonate (C6mimTfO) and water at temperatures in between T = 303- 353 K under conditions of a slightly reduced pressure (p < 1 bar) are presented. For that purpose a micro-ebulliometer is used. The pressure is monitored and the temperature of the boiling liquid phase is measured simultaneously. After achieving conditions of equilibrium, a sample of the liquid phase is analyzed by Karl-Fischer titration to characterize the composition of the sample. Results are discussed in terms of commonly used engineering approaches (e.g., the NRTL-model) in order to determine the activity coefficients of the investigated system.
309
Experimental Techniques P05-07

Ionenmobilitäten: Bestimmung durch ESI/REMPI-IMS und Vergleich verschiedener Berechnungsmethoden
Daniel Riebe, Alexander Erler, Toralf Beitz, Hans-Gerd LöhmannsröbenUniversität Potsdam
Eine zuverlässige Vorhersage von Ionenmobilitäten (IM) ist die Voraussetzung für ein besseres Verständnis der IM-Spektrometrie (IMS). Für sehr ähnliche Ionen (z.B. in homologen Reihen) können IM durch empirische Masse-Mobilitäts-Korrelationen berechnet werden. Modelle, die die Wechselwirkung zwischen Ionen und Driftgasmolekülen durch verschiedene Potenziale berücksichtigen, erlauben nach Anpassung an experimentelle Werte die Extraktion verschiedener molekularer Parameter.1
Eine andere Möglichkeit ist die Ermittlung der Geometrien von Molekülionen durch quantenchemische Rechnungen und die Bestimmung der Stoßquerschnitte in Trajektorien-Simulationen, z.B. mit der Software Mobcal.2 Diese ist allerdings ursprünglich nur für die Berechnung von Mobilitäten in Helium parametrisiert und muss modifiziert werden um die stärkeren Wechselwirkungen mit Stickstoff zu berücksichtigen. Eine entsprechend angepasste Variante wurde von Campuzano et al. entwickelt.3
In dieser Arbeit wurden verschiedene Modelle der Berechnung von IM miteinander verglichen und deren Vor- und Nachteile näher diskutiert. Zusätzlich wurde ein neuer Ansatz der Berechnung von IM entwickelt, der die o. g. Methoden miteinander verbindet. Außerdem wurden erste Versuche zur Bestimmung und Berechnung von Hochfeld-Mobilitäten unternommen. Dazu wurde ein IM-Spektrometer entworfen, bei dem der Druck bis 10 mbar reduziert werden kann. Dadurch wurden Werte des reduzierten elektrischen Feldes E/N von 80 Td erreicht.
Referenzen:[1] Mason, E. A.; OʼHara, H.; Smith, F. J., J. Phys. B At. Mol. Phys. 1972, 5, 169[2] Mesleh, M. F.; Hunter, J. M.; Shvartsburg, A. A.; Schatz, G. C.; Jarrold, M. F.,
J. Phys. Chem. 1996, 100, 16082[3] Campuzano, I.; Bush, M. F.; Robinson, C. V.; Beaumont, C.; Richardson, K.;
Kim, H.; Kim, H. I., Anal. Chem. 2012, 84, 1026
310
Experimental TechniquesP05-08

Investigation of the thickness- and wavelength- dependence of laser stratigraphy on Cu and Ni using LIBS
Ulrich Pacher, Tristan O. Nagy, Evgeniya Khaybulkina, Wolfgang KautekUniversity of Vienna, Vienna, Austria
The rapid qualitative and quantitative stratigraphic analysis of metal coatings is of substantial importance in modern industrial and medical applications. Laser-Induced Breakdown Spectroscopy (LIBS) is one of the most promising options to receive fast and reliable results in this field. Consequently, finding the optimal parameters for the analysis of given systems is a major task.In this Study, galvanic coatings of Cu and Ni at various thicknesses were applied to Al bulk material and subsequently subjected to stratigraphic analysis via LIBS, according to previously tested procedures1. Ablation experiments were carried out using the first four harmonic wavelengths of a Nd:YAG laser (1064, 532, 355 and 266 nm), and the resulting plasma spectra were analyzed using the correlation method2,3.For all wavelengths, the ablation behavior was observed in relation to the layer thickness and compared to corresponding theoretical values4.
[1] Pacher, U.; Dinu, M.; Nagy, T.O.; Radvan, R.; Kautek, W., Multiple Wavelength Stratigraphy by Laser-Induced Breakdown Spectroscopy of Ni-Co Alloy Coatings on Steel, Spectrochimica Acta B, in Publication
[2] Mateo, M. P.; Costa, G. N.; Piñon, V.; Yañez, A., Improvements in depth-profiling of thick samples by laser-induced breakdown spectroscopy using linear correlation. Surface and Interface Analysis 2006, 38, (5), 941-948.
[3] Nagy, T. O.; Pacher, U.; Pöhl, H.; Kautek, W., Atomic emission stratigraphy by laser-induced plasma spectroscopy: Quantitative depth profiling of metal thin film systems. Applied Surface Science 2014, 302, 189-193.
[4] Matthias, E.; Reichling, M.; Siegel, J.; Kaeding, O. W.; Petzoldt, S.; Skurk, H.; Bizenberger, P.; Neske, E., Influence of thermal diffusion on laser ablation of metal films. Applied Physics A: Solids and Surfaces 1994, 58, (2), 129-136.
311
Experimental Techniques P05-09

In Situ Studies Of Gold Nanoparticle Formation In Microfluidic Reactors
Ghazal Tofighi1, Thomas Sheppard1,2, Georg Hofmann1, Günter Rinke3,Roland Dittmeyer3, Jan-Dierk Grunwaldt1,2
1Institute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Karlsruhe/Germany
2Institute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology (KIT), Karlsruhe/Germany
3Institute for Micro Process Engineering (IMVT), Karlsruhe Institute of Technology (KIT), Karlsruhe/Germany
In order to design highly efficient catalysts, metal colloids are promising candidates that can be deposited on metal oxide supports. The size, distribution and shape of nanoparticles play an important role in tuning physical and chemical properties1.Moreover, the effects of different synthesis methods on nanomaterial properties resulting from structure, size, morphology, nanoparticle size distribution and interparticle interactions should be closely investigated. Understanding and controlling the intermediate processes occurring during synthesis is highly important. This highlights the necessity for in situ and operando studies and the combination of complementary characterization techniques2. Production of nanoparticles in a continuous flow microreactor is a promising method, offering several benefits such as homogenous mixing, easier process handling, higher mass and heat transport and process parameters within a wide range of operating conditions3-6.In this work, a microfluidic setup was built to investigate the early stages (down to milliseconds) of fast reduction reactions, i.e. to produce metallic gold nanoparticlesand to apply X-ray based techniques (EXAFS, SAXS) on them. The first reaction of interest was the reduction of HAuCl4 by tetrakis(hydroxylmethyl)phosphonium chloride (THPC) and NaBH4 yielding small Au nanoparticles (<2 nm). Good mixing and short residence times enable to study the reaction by X-ray absorption spectroscopy in the millisecond time regime.
312
Experimental TechniquesP05-10

1. J.-D. Grunwaldt, C. Kiener, C. Wögerbauer and A. Baiker, Journal of Catalysis, 1999, 181, 223-232.
2. J.-D. Grunwaldt and C. G. Schroer, Chemical Society Reviews, 2010, 39, 4741-4753.3. J. Wagner and J. M. Köhler, Nano Letters, 2005, 5, 685-691.4. M. Luty- Chemical Engineering
Journal, 2013, 226, 46-51.5. - Colloids
and Surfaces A: Physicochemical and Engineering Aspects, 2012, 413, 208-215.6. C. Fräulin, G. Rinke and R. Dittmeyer, Microfluidics and nanofluidics, 2014, 16, 149-
157.
313
Experimental Techniques P05-10

On the polymerization efficiency of benzoin-type photoinitiator molecules by fs-TA spectroscopy, PLP-ESI-MS and TD-DFT
Caroline Schweigert,a Elena Frick,b Benjamin B. Noble,c
Hanna A. Ernst,a Yu Liang,a Andrea Lauer,b
Michelle L. Coote,c Christopher Barner-Kowollikb and Andreas-Neil Unterreinera
a Karlsruhe Institute of Technology (KIT), Molecular Physical Chemistry, Institute of Physical Chemistry, Karlsruhe, Germany
b KIT, Preparative Macromolecular Chemistry, Institute of Chemical Technology and Polymer Chemistry and Institute of Biological Interfaces, Karlsruhe, Germany
c Australian National University, ARC Centre of Excellence for Electromaterials Science, Research School of Chemistry, Canberra, Australia
Radical photoinitiators and their initiation efficiency play a fundamental role in variousapplications.[1] Recent investigations[2] revealed that a trifold combination of femtosecond transient absorption (fs-TA) spectroscopy, pulsed laser polymerization followed by electrospray mass spectrometric analysis (PLP-ESI-MS) and theoretical calculations (TD-DFT) can be used to elucidate fundamental steps after photoexcitation. In efforts towards a higher degree of quantification, we present here investigations of a library of benzoin-type photoinitiators with varied substitution pattern highlighting the importance of taking into account of more than one parameter as selection criteria. Among these are i) the choice of molecules with relatively weak extinction coefficients, ii) the characterization of the nature of ground and excited states and iii) the description of the reactivity of the generated radical fragments with the monomer double bonds. It turned out that within this family of molecules efficient initiators are governed by an ultrafast decay of the fs-TA traces within fewpicoseconds whose origin is attributed to an intersystem crossing to an energetically close-lying triplet state.[3] Further details of the analysis will be presented.
[1.] Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245-6260.[2.] E. Frick, H. A. Ernst, D. Voll, T. J. A. Wolf, A.-N. Unterreiner and C. Barner-
Kowollik, Polymer Chemistry, 2014, 5, 5053-5068.[3.] E. Frick, C. Schweigert, B. B. Noble, H. A. Ernst, A. Lauer, Y. Liang, D. Voll,
M. L. Coote, A.-N. Unterreiner, and C. Barner-Kowollik, Macromolecules, DOI:10.1021/acs.macromol.5b02336 (2015).
314
Experimental TechniquesP05-11

COMOTION - Controlling the motion of large molecules and particles
D. A. Horke1,3, S. Awel1,3, Z. Huang1, N. Roth1, N. Teschmit1, T. Ossenbrüggen1,D. Gusa1, and J. Küpper1,2,3
1Center for Free-Electron Laser Science, DESY, Notkestr. 85, 22607 Hamburg2Dept. of Physics, University of Hamburg, 22761 Hamburg
3Center for Ultrafast Imaging, University of Hamburg, 22761 Hamburg
Recent years saw the development of several techniques to control small neutral molecules, and we can now routinely select single structural isomers, create beams of size-selected clusters and disperse rotational quantum states for the production of colder molecular ensembles, in certain cases even creating single-quantum-state samples [1]. Here we report on our efforts to extend these techniques to significantly larger molecules, nanoparticles and biological systems. We are developing new sources for these large systems based on soft vaporization techniques (aerodynamic lenses, laser desorption, acoustic desorption, …) to provide high molecular fluxes over long measurement times (> 12 h), as required in typical Free-Electron laser (FEL) based X-ray diffractive imaging experiments. Using novel characterizationtools, such as super-resolution particle localization [2] and plasma-formation imaging, new injectors are studied in detail and optimized. Coupling to efficient cooling schemes (supersonic expansions, buffer-gas cells) will provide internally cold molecules for further manipulation and control using static or dynamic electric fields. These will include the separation of structural isomers using Stark separation [1], alignment and orientation using laser-based strong-field techniques [3], as well as spatial control of particles using shaped quasi-Bessel laser beams [4]. This will allow the guiding of cold and controlled nanoparticles into a focused FEL beam, a major step towards single-particle diffractive imaging with atomic resolution.
[1] Chang, Horke, Trippel, Küpper, Int. Rev. Phys. Chem. 34, 557 (2015)[2] Awel, Kirian, Eckerskorn, Wiedorn, Horke, Rode, Küpper, Chapman, submitted (2015)
, Mol. Phys. 111, 1738 (2013)[4] Eckerskorn, Li, Kirian, Küpper, DePonte, Krolikowski, Lee, Chapman, Rode, Opt. Expr. 21, 30492 (2013)
315
Experimental Techniques P05-12

Photoelectron spectra from optimally tuned TDDFT and Dyson orbital formalism
Tobias Moehle, Sergey I. Bokarev, Gilbert Grell, Oliver KühnInstitut für Physik, Universität Rostock, Germany
Photo-electron spectroscopy is an important tool for studying the electronic structure of a wide range of materials. Often the photo-electron spectra (PES) are predicted theoretically within one-electron picture from the ground state orbital energies, neglecting the variation of cross-section for ionization from different levels. With this respect, optimally tuned range separation hybrid (OT-RSH) density functionals have been used to eliminate the self-interaction error what have led to an improvement of the Kohn-Sham orbital energies and agreement with experiment [1]. However, to further improve the agreement one needs to get the accurate intensities and to include many-electron combination transitions.In present communication, the PES calculations including many-electron effects were performed on the basis of time-dependent density functional theory with OT-RSH functionals. The Dyson orbital (DO) formalism was applied to obtain cross-sections depending on the kinetic energy of outgoing electron and interfaced to NWChem program suite [2]. The PES matrix elements involving the DOs and various types of free-electron wave functions were evaluated by means of numerical integration [3]. This protocol was applied to simple organic conjugated molecules whose PESs are experimentally well studied. Further, it was applied to sulphur clusters Sn (n=6–8) aiming at explaining the large differences in the integral cross sections observed experimentally.We acknowledge financial support by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, (grant No. D-003-435) and the Deutsche Forschungsgemeinschaft (grant No. KU952/10-1).
[1] Thomas Körzdörfer and Jean-Luc Brédas,Acc. Chem. Res. 2014 47 (11), 3284-3291
[2] M. Valiev, et. al., "NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations"
[3] C.M. Oana and A.I. Krylov, J. Chem. Phys. 127, 234106 (2007)
316
Theoretical TechniquesP06-01

Quantum-chemical calculations of metal-organic frameworks with open-shell adsorbates
Elisabeth Dietze, Carmen HerrmannDepartment of Chemistry, University of Hamburg, Germany.
HKUST-1 is a common example of a multi-functional metal-organic framework, containing metal dimers in the form of paddle-wheel structures, with a large field of applications such as gas storage and catalytic activity [1]. Jee et al. observed complex magnetic behaviour when introducing di-tert-butyl nitroxide (DTBN) as guest molecules which provides a promising avenue for radical heterogeneous catalyses or magnetic sensor applications [2].Our density functional theory (DFT) calculations concentrated on the coordination of DTBN at HKUST-1 to clarify the coordination structure and spin states of the radical with the Cu(II) metal centres of HKUST-1. EPR parameters and IR spectra were also calculated for comparison with experiment.
[1] C. H. Hendon, A. Walsh, Chem. Sci. 2015, 6, 3674.[2] B. Jee, K. Koch, L. Moschkowitz, D. Himsel, M. Hartman, A. Pöppl, J. Chem. Phys. Lett. 2011, 2, 357-361.
317
Theoretical Techniques P06-02

Hydrogen atom scattering from M(111) surfaces: Molecular dynamics simulations on a full-dimensional EMT
Potential Energy SurfaceMarvin Kammler, Svenja M. Janke, Daniel J. Auerbach, Alec M. Wodtke, and
Alexander KandratsenkaMax-Planck-Institut für biophysikalische Chemie, Göttingen, Germany
Institut für physikalische Chemie, Georg-August-Universität Göttingen, Germany
Potential Energy Surfaces (PES) have long been a useful means to simulate scattering experiments much faster than using “on-the-fly” ab initio molecular dynamics (AIMD). This work extends the successful theoretical considerations concerning molecular dynamics (MD) studies done in this group [1,2] for H at Au(111) to the case of some other transition metal surfaces.The H atom is placed in a grid-like fashion at several hundred different positions in and above the surface and the energy of these configurations is evaluated by the PBE functional. Additionally, about a dozen AIMD trajectories are run to also sample non-equilibrium configuration space of the system. Next, the energies of one AIMD trajectory as well as of the entire grid are fit to an analytical function resulting from Effective Medium Theory [3]. It contains seven physically meaningful parameters for each atomic species which can be used to derive structural properties, as well as to run classical MD simulations. The quality of the fit is defined by the RMS error regarding AIMD trajectories not included in the fit and by the reproduction of the metal elastic properties. This work extends the understanding of scattering processes of a hydrogen atom from various transition metals on the atomic level by employing a highly accurate PES. The dependence of H atom trapping probability and energy losses on the metal is analyzed and related tointrinsic metal properties.
References[1] S. M. Janke et al., J. Chem. Phys., 143, 124708, 2015.
[2] O. Bünermann et al., Science, 350, 1346, 2015
[3] K. W. Jacobsen et al., Surf. Sci., 366, 394, 1996.
318
Theoretical TechniquesP06-03

Computation and optimization of complex formation thermodynamics by a solute-solute integral equation theory
Florian Mrugalla, Stefan M. KastPhysikalische Chemie III, TU Dortmund, 44227 Dortmund, Germany
The development of novel drugs is an arduous procedure which can take more than a decade and can accumulate expenses in the triple digit million euro range [1]. These facts remain true in spite of recent advances in virtual screening and computer-aided drug design [2]. The need for reasonably accurate and fast methods to predict and optimize the binding affinity which can drive the design process of drug-like molecules is therefore very high.
We here present novel methodology for computing binding free energies and their analytical dependence on chemical properties based on the so-called solute-solute equation of the reference interaction site model (RISM) [3]. This integral equation theory is derived from the molecular Ornstein-Zernicke equation and enables us to calculate the potential of mean force (PMF) including physics-based solvation contributions with very high computational efficiency. The PMF is the key quantity for characterizing chemical and biological processes since it represents the free energy change along a given reaction coordinate from which the binding free energy can be computed.
We derive the conceptual framework and demonstrate various application areas of the model. PMF results from the solute-solute RISM approach are compared with explicit-solvent free energy perturbation molecular dynamics simulations in order to assess the approximations inherent to the theory for simple model systems. Optimal binding chemistry is defined by minimizing the free energy with respect to nonbonded complex interaction parameters. The results lead to suggestions for novel approaches to define protein-ligand scoring functions.
[1] A. Lavecchia, C. Di Giovanni, Curr. Med. Chem. Phys. 20, 2839 (2014).[2] J. Michel, Phys. Chem. Chem. Phys. 16, 4465 (2014).[3] T. Kloss, S. M. Kast, J. Chem. Phys. 128, 134505 (2008).
319
Theoretical Techniques P06-04

Transfer free energies between aqueous and nonaqueous phases from an integral equation-based quantum solvation model
Nicolas Tielker, Daniel Tomazic, Stefan M. KastPhysikalische Chemie III, TU Dortmund, 44227 Dortmund, Germany
Reliable yet fast prediction of free energies of solvation or of partition coefficients of molecules between immiscible or partly miscible phases such as water and n-octanol requires proper theories as for instance provided by the integral equation approach to fluid phase thermodynamics [1]. To accurately model the solvation of small molecules we here combine such a statistical-mechanical description of the solvent with a quantum level description of the solute in the form of the “embedded cluster reference interaction site model” (EC-RISM). This combination takes into account both the electronic relaxation and the excess chemical potential governing the solvation process for predicting the free energy of solvation [2].
To extend the scope of EC-RISM theory to complex solvents other than water we here examine several models for n-octanol, taking molecular flexibility into account. This is achieved by parameterizing suitable analytical expressions for the intramolecular distribution functions with respect to reference data from explicitmolecular dynamics simulations. One known drawback of the RISM formalism is anoverestimation of the free energy contribution accompanying the formation of the solute cavity, leading to significant errors in the absolute free energy of solvation.This error is highly correlated with the partial molar volume (PMV) of the solute [3].To address this issue we parametrize a PMV correction to increase the accuracy ofthe calculated free energies of solvation within the EC-RISM context. We discuss the application of this framework to the calculation of properties such as acid dissociation constants and octanol-water partition coefficients of unionizable (log P) and ionizable compounds (log D).
[1] E. L. Ratkova, D. S. Palmer, M. V. Fedorov, Chem. Rev. 115, 6312 (2015)[2] T. Kloss, J. Heil, S. M. Kast, J. Phys. Chem. B, 112, 4337 (2008).[3] D. S. Palmer, A. I. Frolov, E. L. Ratkova, M. V. Fedorov J. Phys.: Condens.
Matter 22, 492101 (2010).
320
Theoretical TechniquesP06-05

Isomerization Reaction Kinetics in 2-MethyltetrahydrofuranOxidation
Prajakta R. Parab1, Naoki Sakade2, Yasuyuki Sakai2, Ravi Fernandes3, K. Alexander Heufer1
1Physico Chemical Fundamentals of Combustion, RWTH Aachen University, Templergraben 55, 52056 Aachen, Germany
2University of Fukui, Bunkyo 3-9-1, Fukui, 9108507, Japan3Physikalish Technische Budesanstalt (PTB) Bundesallee 100, 38116 Braunschweig,
Germany
Abstract2-Methyltetrahydrofuran (2-MTHF) is proposed to be potential biofuel component by “Tailor Made Fuels from Biomass (TMFB)” at RWTH Aachen University, Germany [1]. Biosynthetic approach is also reported for 2-MTHF via selective conversion of levulinic acid. In this work, reaction kinetics for the isomerization (ROO to QOOH) reactions in 2-MTHF oxidation is theoretically investigated. All together eight ROO isomers of 2-MTHF has been considered as the starting reactants for the corresponding isomerization reactions. Formation of the QOOH product species from the ROO isomers of 2-MTHF are observed to proceed via formation of the cyclic transition states. CBS-QB3 composite method is employed for the computation of C-H Bond dissociation energies in 2-MTHF which is found to be lowest at the carbon sites neighboring ring oxygen atom. For the treatment of hindered rotors in reactants, transition states and products in the titled reactions, Pitzer-Gwinn like approximationhas been implemented. Thermodynamic properties (standard enthalpies of formation, standard entropies and heat capacities at constant pressure) of the reactants and products species involved in the ROO to QOOH reactions in 2-MTHF oxidation are presented. High pressure limit rate constants for all the isomerization reactions are determined from the three-parameter Arrhenius expression in the temperature range 500-2000 K. All theoretical calculations have been carried out at CBS-QB3 composite method. Reference [1] Janssen, A. J., Kremer, F.W., Baron Jan, H., Muether, M., Pischinger, S., Klankermayer, J. (2011). Tailor-Made Fuels from Biomass for Homogeneous Low-Temperature Diesel Combustion. Energy Fuels, 25, 4734.
321
Theoretical Techniques P06-06

Calculation of entropy-dependent thermodynamic properties offluids at high pressure with Monte Carlo simulation
Simin Yazdi Nezhad, Ulrich K. Deiters
Institute of Physical Chemistry, University of Cologne, Luxemburger Str. 116D-50939 Cologne, Germany
Entropy and entropy-dependent properties (e.g., the chemical potential) are difficult to obtain by computer simulation. The existing computation methods, require considerable effort and CPU time. A well-known example is Widom's test particle insertion method [1]. It has long been known that there is a connection between the entropy and the free volume of fluids [2]. More recently, a correlation between the entropy and the diffusion constant in liquid metals was observed. In this work, the relation between
the residual entropy and the maximum displacement parameter of Monte Carlo simulations (at constant acceptance ratio) is studied. This parameter is obtained during simulations without additional computational cost. Simulations were performed for single- and two-center molecules; the potential energy was obtained as a sum of site–site pair potentials. For these, Mie potentials (particularly Lennard-Jones potentials) and hard-sphere potentials were assumed. At middle and high densities, the relation between the logarithm of the residual entropy and the logarithm of the Monte Carlo displacement parameter can be represented by second-order polynomials; these polynomials depend on the molec-ular structure and pair potential, but practically not on the temperature. At low densities, the application of these polynomials is limited by a percolation transition.
[1] B. Widom, J. Chem. Phys. 39 (1963) 2808.
[2] D. F. Steyr, Am. J. Phys. 68 (2000) 1090.
[1] B. Widom, J. Chem. Phys. 39 (1963) 2808.[2] D. F. Steyr, Am. J. Phys. 68 (2000) 1090.322
Basic Mechanisms in Energy ConversionP06-07

Imaging of Gap Plasmons and Surface Plasmon Quenching in Au and Ag Tip-Sample-Systems with Cathodoluminescence
Martin Grüßer, Xinzhou Ma, Rolf Schuster
Institute of Physical Chemistry, Condensed Matter Division, Karlsruhe Institute of Technology (KIT), Germany
Sharp metal tips act as optical antennas and are accompanied by a locally enhanced electromagnetic field which can be further enhanced in the vicinity of a metal surface [1]. This allows, e.g., for tip-enhanced raman spectroscopy [2, 3] on the single molecule level. In addition to the lightning rod effect [3] and surface plasmonslocalized at the tip apex [5] at small distances also gap plasmons between a tip and asample surface [6] contribute to the field enhancement. At very small distance in the quantum regime a competing effect of surface plasmon quenching sets in [7].
We employ cathodoluminescence (CL), i.e., light emission upon e--irradiation in a scanning electron microscope (SEM), to investigate the local excitation of surface and gap plasmons in the tip-surface system. The distance between tip and surface is controlled by a scanning tunneling microscope (STM), mounted inside the SEM. We observed enhanced photon emission upon e--irradiation near the tip-surface gap.Spectra upon irradiation of tip or sample show strong differences in intensity and peak position. Both the evolution of additional peaks, which may depend on the formation of gap plasmons, and the vanishing of peaks, which may be due to quenching of radiative plasmon modes are observed.
[1] N. Behr, M. B. Raschke, J. Phys. Chem. C 2008, 112, 3766.[2] R. M. Stöckle, Y.D. Suh, V. Deckert, R. Zenobi, Chem. Phys. Lett. 2000, 318, 131.[3] B. Pettinger, P. Schambach, C. J. Villagómez, N. Scott, Annu. Rev. Phys. Chem.2012, 63, 379.[4] A. V. Zayats, Opt. Commun. 1999, 161, 156.[5] X. Ma, Ph.D thesis, Karlsruhe Institute of Technology, 2012.[6] Z. Yang, J. Aizpurua, H. Xu, J. Raman Spectrosc. 2009, 40, 1343.[7] V. Kravtsov, S. Berweger, J. M. Atkin, M. B. Raschke, Nano Lett. 2014, 14, 5270.
323
Solids and Nano-sized Matter P07-01

Synthesis and Control of Emission Properties of Highly Luminescent In(Zn)P/GaP/ZnS Quantum Dots.
Christian Meerbach, Christin Rengers, Marcus Adam, Nikolai Gaponik,
Alexander Eychmüller
Physical Chemistry, TU Dresden, Bergstraße 66b, Dresden
Since the beginning of the discovery of semiconductor nanoparticles 30 years ago,research is focusing on these materials owing to their unique optical properties. They are distinguished by their size-dependent, adjustable emission wavelength (quantum size effect), narrow emission signals and high emission quantum yields. Thesecharacteristics make quantum dots promising materials for various applications such as LEDs, screens or medical applications. The materials of the first choice are CdSe,CdTe, InP, CuInS2 or ZnO semiconductors. In the next few years especially cadmium-free quantum dots will be pushed in applied research because of their lower toxicity. Due tothe fact that there is a high demand for thismaterial a published synthesis of In(Zn)P/GaP/ZnS quantum dots[1] was modified to get a broad color range and to optimize thevisual characteristics.
The result is a series of quantum dots with emission maxima of 500-650 nm, a FWHM of less than 60 nm and quantum yields up to 64%. Furthermore, by replacing the long-chain hydrophobic ligands by short-chain hydrophilic ligands we succeeded in transferring these semiconductor nanoparticles into the aqueous medium. In thenext step the particles have been successfully embedded in a salt matrix to protect them from photooxidation.[2] The quantum dot salt composites were used as color conversion material in white LEDs with a long lifetime.
[1] Kim, S.-W., Kim, T., Kang, M. Journal of the American Chemical Society,2012, 134, 3804–3809.
[2] Adam, M., Wang, Z., Dubavik, A., Stachowski, G. M., Meerbach, C., Soran-Erdem, Z., Eychmüller, A. Advanced Functional Materials, 2015, 18(25), 2638–2645.
Overview of the QDs under UV lamp excitation.
324
Solids and Nano-sized MatterP07-02

Determination of the Dimensions and Extinction Coefficients of CdSe seeded CdS Nanorods through their UV/Vis Spectra
Patrick Adel, Julian Bloh, Dominik Hinrichs, Torben Kodanek and Dirk DorfsInstitute for Physical Chemistry and Electrochemistry, Leibniz Universität Hannover,
Germany
CdSe seeded CdS nanorods are broadly used in nanochemistry. Until now, to be able to apply further steps to these particles, different analytical methods to deter-mine the size parameters and the concentration of particles had to be utilized.
In this work, we will show that all the necessary size parameters are well accessible through the UV/Vis spectra of these particles in solution. Starting from the high wave-lengths region, first the size of the CdSe seed of the nanorod can be determined through the position of the first absorption maximum. Following this, the intercept of CdS band tangent with the wavelength axis gives access to the width of the rod. The extinction of the CdS peak relative to that of the CdSe peak correlates with the length of the nanorods. Finally, with the total volume of the nanorods now known, the extinc-tion coefficients can be calculated from the extinction at a low wavelength, e. g. at 350 nm. With this, the concentration of particles and all their dimensions can be cal-culated.
The accuracy as well as the boundaries of this method are carefully analyzed and discussed.
325
Solids and Nano-sized Matter P07-03

The creation of highly emmissive green - to - red Cu - In - Zn - S/ZnScore/shell quantum dots
Josephine F.L. Loxa, Marcus Adama, Talha Erdemb, Hilmi Volkan Demirb,c, Nikolai Gaponika, Alexander Eychmüllera
a Physical Chemistry, TU Dresden, Bergstr. 66b, 01062 Dresden, Germanyb Department of Physics, Department of Electrical and Electronics Engineering and
UNAM-Institute of Materials Science and Nanotechnology, Bilkent University, TR-06800, Ankara, Turkey
c School of Electrical and Electronic Engineering and School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 639798,
SingaporeWith the invention of the blue emitting InGaN-based LED in 1993 by Shuij Nakamura an important step for lighting was made. The possibility to replace Edisonʼs incandescent bulb by the LED was born and with it a new era in the lighting research has started.Inspired by Edmund Germer's gas discharge tube (1926), the idea was to use a blue primary LED and to create white light by blending color conversion layers onto it. So far, semiconductor quantum dots (QDs) with their spectral tunability and color purity have proven themselves as outstanding candidates. However, the cadmium - based quantum dots have been well studied and show, despite of their numerous advantages, a limitation because of their high toxicity.As a result cadmium - free quantum dots are getting more and more in the focus of research among which the quartenary Cu - In - Zn - S/ZnS quantum dots are serving asrepresentatives. Besides their lower toxicity, the possibility of tuning their bandgap andthe emission wavelength by varying the composition in the multicomponent systemsmakes them potentially even more attractive.The Cu - In - Zn - S/ZnS core/shell nanoparticles were synthesized in various colors via a surfactant induced nucleation process and resulted in untypical high quantum yields up to 47 %. By embedding those high quality Cu - In - Zn - S/ZnS QDs in lithium chloride, a highly stable and processable bulk material was built.
[1] Schmiechen, S.; Pust, P. et al. Nachrichten aus der Chemie 2014, 62, 847.
[2] Pan, H.; Lai, C. et al. Mater. Express 2012, 2, 224.
326
Solids and Nano-sized MatterP07-04

Modification of binary semiconductor nanorods by site-selective growth of a heterojunction
Florian Enders, Klaus Boldt, Universität Konstanz, Konstanz/Germany
The development and improvement of wet-chemical procedures for the synthesis of highly fluorescent semiconductor nanocrystals has attracted intense research interest in the last 20 years. The hot-injection and seeded growth syntheses made it possible to produce nanocrystals with high crystallinity, narrow size distributions, and good control over particle shape.[1,2] More recently, the introduction of fast cation exchange reactions made it possible to utilize even more materials in the synthesis of nanocrystals. In these reactions the sublattice of cations in ionic nanocrystals can be replaced by different cations while the anion sublattice remains in place.[3]
Heterostructures consisting of more than one semiconductor material can also be created using the cation exchange approach.[4]
The focus of our work is the modification of such heterostructures in a controlled way.We employ artificially introduced lattice faults in nanorods as nucleation sites for the growth of a semiconductor heterojunction.CdS nanorods were synthesized by a seeded growth approach with spherical CdSenanocrystals as seeds. Employing a partial cation exchange reaction, Cu2S regions were created on the rods. These areas were then used as nucleation sites for Zinc-chalcogenide nanocrystals. The modified crystals were analyzed with high-resolution transmission electron microscopy, UV/Vis absorption and photoluminescence emission spectroscopy. Control of this modification step paves the way to new, more advanced semiconductor nanomaterials with novel electronic properties. For example, with the use of the right materials, type II heterojunctions that lead to an efficient electron-hole separation can be created.[5] These types of nanocrystals are interesting for many possible applications in electronic devices.
[1] C. B. Murray, D. J. Norris, M. G. Bawendi, J. Am. Chem. Soc. 1993, 115, 8706 8715.[2] L. Carbone, C.Nobile, M. de Giorgi, F. D. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, E.; Fiore, I.
R. Franchini, et al., Nano Lett. 2007, 7, 2942–2950. [3] H. Li, M. Zanella, H. Zheng, A. Genovese, M. Povia, A. Falqui, C. Giannini, L. Manna,
Nano Lett. 2011, 11, 4964-4970[4] B. Sadtler, D.O. Demchenko, H. Zheng, S. M. Hughes, M. G. Merkle, U. Dahmen, L. W. Wang,
A. P. Alivisatos, J. Am. Chem. Soc. 2009, 131, 5285–5293.[5] K. Boldt, N. Kirkwood, G. A. Beane, P. Mulvaney, Chem. Mater. 2013, 25, 4731–4738.
327
Solids and Nano-sized Matter P07-05

Synthesis and Spectroscopy of Gold Nanoclusters with Different Surface Modifications
Tim Hadler, Tobias Kipp, Alf MewsInstitute of Physical Chemistry, University of Hamburg, Germany
Gold nanoparticles (AuNPs) show unique optical properties which make them suitable for labeling in biological applications. Besides the well-known light scattering properties, AuNPs also show fluorescence properties which are far less understood. We investigate AuNPs with diameters smaller than 2 nm, which are commonly calledgold nanoclusters (AuNCs). When the surface of these AuNCs is functionalized with thiol ligands they are able to exhibit strong photoluminescence (PL), similar to semi-conductor nanocrystals. The PL emission depends on several parameters including size, oxidation state of surface atoms and surface ligands, but the underlying processes are not yet completely understood. The PL of thiol-functionalized AuNCs is typically assigned to Au-S hybrid orbitals on the AuNCs surface. By using different ligands and reducing agents the emission energy of these AuNCs can be tuned over the visible and NIR spectrum.
Here, we present results on the PL of thiol-stabilized AuNCs that show how the surface composition with different ligands of the AuNCs affects the PL emission. PL spectroscopy combined with IR-spectroscopy (Fig. 1) proves that the phosphorus in the reducing agent THPC contributes to the AuNCs PL.
Fig. 1: (a) PL spectra of thiol stabilized AuNCs synthesized with tetrakis-(hydroxymethyl)phosphonium chloride (THPC) and sodium borohydride (NaBH4). (b) IR spectra of the purified AuNCs synthesized with THPC compared to free 11-mercaptoundecanoic acid (MUA).
328
Solids and Nano-sized MatterP07-06

Optical and theoretical investigation of fluorescence spectral diffusion of CdSe/CdS-Dotrods
Sven-Hendrik Lohmann, Christian Strelow, Tobias Kipp and Alf MewsUniversity of Hamburg, Institute of Physical Chemistry, Grindelallee 117, 20146
Hamburg, Germany
Semiconductor nanoparticles show many possible applications in the field of opto-electronics, such as LEDs or photovoltaic cells. For the development of these devices it is necessary to precisely control the optical properties. Core/shell heterostructures are very well suited because they allow adjusting of the emission wave length and band alignment via composition and size of the system. In this work we investigate the photoluminescence of single elongated CdSe/CdS core/shell dotrods, at low temperature. We observe a spectral diffusion of the narrow photoluminescence lines, which is a random shift of the emission wavelength over time. Commonly, this is explained by migrating of surface charges[1]. Hence we recorded a series of time- and energy-resolved spectra of single dotrods at cryogenic temperatures, which show a correlation between wavelength shifts and changes in the fluorescence lifetime. Here we developed a model based on the effective mass approximation, where we calculated the effect of surface charges on the overlap of the electron and hole wave functions and the photoluminescence energies and compared it to the experimental results. These studies corroborate the model after which the photo generated electrons are highly mobile across the dotrod, while the holes are strongly localizes within the CdSe cores.
[1] S. A. Empedocles, M. G. Bawendi, Science 1997, 278, 2114.
329
Solids and Nano-sized Matter P07-07

Studies on the twinning effects during the self-assemblyof silver nanoparticles into supercrystals
Annett Reichhelm, Danny Haubold, Alexander Eychmü[email protected]
Physical Chemistry, TU Dresden, Bergstraße 66b, 01062 Dresden
Similar to atoms in classic crystals, nanoparticles are able to self-assemble to highly ordered superstructures with symmetric shapes and defined edges and facets. In contrast to the atomistic crystallisation, the assembly of nanoparticles allows the direct investigation of the arrangement of the resulting supercrystals by electron microscopy. This promises a better understanding of crystallisation processes and the similarities and differences of the self-arrangement of atomic and nanoscale objects.
In this study, we prepared supercrystals by the self-assembly of 5 nm silver nano-particles via a gas phase destabilisation method and investigated their morphologiesby means of SEM. Beside octahedral crystals, which are typical for fcc assemblies, various twinned structures have been observed. The examination of these crystal shapes led to the geometric reduction of the structures to sections of octahedrons,which are connected by twin planes. The twinning of these structures occurs only at {111}-facets leading to the orientation of the twin planes parallel or in an angle of 70,5° and yielding complex symmetrical crystal shapes like five-armed stars (figure 1). The twinning incidents were found to occur partly simultaneously at the moment of the seed formation or in other cases independently during the growth of the supercrystal.
Fig. 1: Dissection of a typical star-like twinned supercrystal and completion of one part to a wholeoctahedron.
1. Murray, C. et al. Science 270, 1335-1338 (1995).
330
Solids and Nano-sized MatterP07-08

Development of a new nanostructuredi-Sb2S3 / p-NiO junction photoelectrode
Julia Schneider, Yanlin Wu, Maïssa Barr, Lionel Santinacci, Julien BachmannDepartment Chemie und Pharmazie, Friedrich-Alexander-Universität Erlangen-
Nürnberg; CINaM and Aix-Marseille Université
Nanostructured interfaces featuring ordered arrays of parallel, cylindrical rods or pores with accurately tunable geometry provide a platform for the systematic study of how chemical and physical properties vary with the geometric parameters. In photovoltaic and (photo)electrochemical energy conversion, the exchange of electrons at interfaces and the transport of charge carriers towards interfaces can be adjusted based on geometry. This work aims at realizing a ʻZ-schemeʼ water splitting based on separate i-p and i-n photoelectrodes with systematically optimizable geometry.
Starting with an Al foil we grow a porous Al2O3 membrane by a two-step anodization. The anodic alumina membranes (AAO) exhibit ordered pores ofdiameter from 180 nm to 400 nm and length adjustable between 1 μm and 100 μm.The functional thin layers of nickel oxid (NiO) and antimony sulfide (Sb2S3) are successively deposited into the porous matrix by atomic layer deposition (ALD). Asubsequent annealing step crystallizes the Sb2S3 layer. Finally, a thin gold layer is sputter-coated for the electrochemical testing.
331
Solids and Nano-sized Matter P07-09

Combination of microscopic and spectroscopic investigation techniques characterize the concentric organization of the hole-conducting NiO and light-absorbing Sb2S3
layers, their nearly perfect stoichiometry, as well as their clean surfaces. Preliminary experiments demonstrate a photoelectrochemical activity of the samples in buffered aqueous electrolyte.
.
332
Solids and Nano-sized MatterP07-09

Influence of the modification on the initiation potential of nanoscale ZnOMichael Schmitt, Saarland University, Germany
Semiconductors like ZnO are well-known for their charge transfer potential and their photo-catalytic properties. Recently we have proven that modified nanoscaled ZnO have the potential to be useful as photo-initiator for radical polymerisation of bulk materials[1][2]. NanoPI can be migration-less/free, “harmless” photo-initiators with tuneable, selective absorbance. Such initiators are of dire need for applications resulting to products like food packages. This contribution deals with the effect of modifier content (e.g. levulinic acid) during the synthesis. Levulinic acid is a small simple molecule which to not contain reactive or aromatic/ chromophore functionalities. The synthesis is a two-step procedure whereby the injection procedure leads to non-surface modified bare, well-defined nanoparticles (spherical, smaller than 10 nm). This masterbatch is used for different modifications so that the effect of the modifier can be analysed independent from the influence of the precipitation. Monitoring of the polymerisation of a multifunctional acrylic ester resin took place by using novel transition UV-curing equipment[3], compare to Figure. Thereby an absolute and spectral calibrated xenon flash light illuminates the sample and a diode array detector measures the reaction progress down to the time range of the flash distance (down to 30 ms).
11000
11500
12000
12500
13000
13500
14000
14500
15000
15500
0 5000 10000 15000 20000 25000 30000 35000
inte
nsity
number of flashes
counts
linear 1
linear 2
linear 3
linear 4
linear 5
linear 6
Figure: Example analysis for one wavelength point of the UV-curing experiment. The transmitted intensity results in a reaction dependent slope. [1] M. Schmitt; Macromol. Chem. Phys., 213, 2012, 1953-1962[2] M. Schmitt, Nanoscale, 2015, 7, 9532-9544.[3] M. Schmitt; Macromol. Chem. Phys., 212, 2011, 1276-1283
333
Solids and Nano-sized Matter P07-10

The formation of alkali ion beams from MAlSi2O6 by thermionic emission
Stephan Schuld, Martin Schäfer, Karl-Michael Weitzel, Philipps-Universität Marburg,Marburg/Germany
Thermionic emission has been a subject of interest for about 100 years, not only for the formation of electron beams but also for cation beams [1]. Today experiments based on alkali ion beams again receive considerable interest in part because of the relevance in several fields, e.g. energy storage (Li, Na) but also sensory perception (Na, K) and material profiling (Cs).In this contribution we present a systematic investigation of thermionic emission from silicates of the MAlSi2O6-type (M= Li, Na, K, Rb, Cs). The emission flux density has been measured as a function of the temperature and the electric field at the emitter surface. From a Richardson-Dushman analysis [2] we have derived the ionic work function. The data allow to correlate the work function with structural parameters of the silicates. Detailed analysis of the current-voltage characteristics show a transition from Child-Langmuir [3] to Schottky [4] characteristics significantly extending earlier work from our group [5].We also present a full numerical calculation based on Nernst-Planck-Poisson theoryof the current-voltage-curves taking into account the transport of ions in the emitter material, the potentials relevant for ionization at the surface and the consequences for the kinetic energy of the ion beam.
[1] O. W. Richardson, The Emission of Electricity from Hot Bodies, Longmans, Green and Co., London, 1916.
[2] S. Dushman, Rev. Mod. Phys., 2, 0381, (1930) [3] C.D. Child, Phys. Rev., 32, 492-511, (1911)[4] W. Schottky, Z. Phys., 14, 63-106, (1923)[5] T. Kolling, A. Schlemmer, B. Harbrecht, K.-M. Weitzel, J. Appl. Phys., 107, 014105, (2010)
334
Solids and Nano-sized MatterP07-11

Synthesis and Characterization of CdSe/CdS Nanorod Decorated Silica-coated Plasmonic Platinum Nanoparticles
Franziska Lübkemann, Nadja-Carola Bigall, Dirk DorfsInstitute of Physical Chemistry and Electrochemistry, Leibniz Universität Hannover
In this presentation we will show the synthesis and characterization from Pt@SiO2
nanoparticles with different silica shell thicknesses and the concepts to connect CdSe/CdS nanorods on the Pt@SiO2 surface. The synthesized hybrid materials are characterized by optical spectroscopy like absorption, emission and excitation and lifetime. Also dynamic light scattering as well as Transmission electron microscopywere done.
Pt nanoparticles are synthesized in a seeded growth method to create particles with an adjustable size.[1] To get a defined distance between the Pt nanoparticles and the CdSe/CdS nanorods, which are synthesized according to Carbone et al.,[2] a silica shell is grown around them. The silica shell is grown following the typical Stöber synthesis.[3] To achieve binding between the silica-coated Pt nanoparticles and the CdSe/CdS nanorods, the surface of the silica shell is modified with amine functional silanes.
[1] N. C. Bigall et al., Nano Lett. 2008, 8, 4588–4592.
[2] L. Carbone et al., Nano Lett. 2007, 7, 2942–2950.
[3] S. Liu et al., Adv. Funct. Mater. 2007, 17, 3147–3152.
335
Solids and Nano-sized Matter P07-12

Mechanism of gold nanoparticle growth in organic solvents
Tobias Redder, Phillip Witthöft and Alf Mews
University of Hamburg, Institute of Physical Chemistry, Grindelallee 117, 20146 Hamburg, Germany
Here we present a study of gold nanoparticle growth in organic solvents, were samples after different growth time were investigated directly within a free liquid jet of only 50 m in diameter. In particular we performed X-ray diffraction of dissolved particles and simulated the diffraction pattern based on atomistic structures and size distributions as taken from TEM data. In parallel we took UV-Vis spectra within the jet and determined the particle concentrations based on discrete dipole approximation calculations. In combination with several other analytical techniques we developed a comprehensive model of gold nanoparticle growth, which is based on the existence of three different species throughout the reaction. Here we propose that particle growth is induced by the reduction of Au3+ ions to Au0 precursor clusters, which in turn transform into gold nanoparticles and grow to several tens of nanometers.
336
Solids and Nano-sized MatterP07-13

Shape-controlled Synthesis of Photoactive Two Dimensional SnS Nanosheets
M. Kobylinski and A. Mews
University of Hamburg, Institute of Physical Chemistry, Grindelallee 117, 20146 Hamburg, Germany
Two dimensional semiconductors have a great potential for applications in field effect transistors, lithium ion batteries, or photovoltaic devices. Especially the IV-VI semiconducting layer structure Tin(II)sulfide is very attractive because of its natural abundance and low toxicity. In combination with promising electrical properties of like the barrow band gap of 1.3 eV, the excellent hole mobility, and the high absorption coefficient, this material is highly suitable for thin film photo voltaics.
Here we show how Tin(II)sulfide nanosheets with lateral sizes in the range of several micrometers and thicknesses of only tens on nanometers can be synthesized by using the hot injection method. The cold precursor S-oleylamine is injected into a hot solution of oleylamine, oleic acid, HMDS and tin chloride. We will show how the size and shape of the nanosheets can be controlled by adjusting the reaction parameters. Especially the influence of octadecene plays an important role to prepare planar and stacked cubic or hexagonal nanosheets. Further we characterize these nanostructures by several techniques such as TEM and SEM, UV/Vis/NIR and Raman spectroscopy and also EDX measurements. Based on the experimental we developed and growth model to explain the structure formation of these 2D-Tin(II)sulfide nanosheets in solution.
Fig.1: TEM images of (a) hexagonal and (b) cubic SnS nanosheets.(c) 3D growth model based on HRTEM and FFT analysis showing the growth direction of hexagonal (blue) and
cubic (green) orthorhombic nanosheets.
337
Solids and Nano-sized Matter P07-14

Simulations of Alloying Processes in Nanoparticle AssembliesDennis Lenz, Tommy Lorenz, and Jan-Ole Joswig
Theoretische Chemie, TU Dresden, 01062 Dresden, Germany
Formation of gels and aerogels from nanoparticles as well as the controlled handling of these processes has moved into focus of colloidal-particle research. The gelation products connect the nanoscopic with the macroscopic world, i.e. properties depend on both the building block and the final macro-structure. As they have an extremely low density and high porosity, aerogels provide access to the capacious inner surface of the interconnected nanoobjects they consist of. In this contribution, we concentrate on the alloying process that follows the gelationand monitor the long-term behaviour of the nanoparticle assembly. This process turned out to be element-dependent [1]. Therefore, classical molecular-dynamics simulations of noble-metal nanoparticle assemblies of different sizes and composition were employed. Particle sizes were varied between a few hundred and several thousand atoms, and the focus was laid on the noble metals gold, silver, platinum, and palladium. The alloying was studied as a function of time and showed indeed composition-dependent characteristics.
1. Multimetallic Aerogels by Template-Free Self-Assembly of Au, Ag, Pt, and Pd Nanoparticles, A.-K. Herrmann, P. Formanek, L. Borchardt, M. Klose, L. Giebeler, J. Eckert, S. Kaskel, N. Gaponik, and A. Eychmüller, Chem. Mater.
.
338
Solids and Nano-sized MatterP07-15

Synthesis of Tinsulfide-Nanosheet Networks via Metal-Organic Chemical Vapor Deposition
Charlotte Ruhmlieb and Alf MewsUniversity of Hamburg, Institute of Physical Chemistry, Hamburg, Germany
Tin(II)sulfide (SnS) is an inorganic p-type semiconductor and a very interesting material for optoelectronic devices, e.g. solar cells. Naturally it grows in layers ofnanoscopic thickness and is usually synthesized via chemical vapor deposition (CVD) or even by wet-chemical approach methods. Chemical vapor deposition of SnS is usually performed by evaporating SnS powder, or by evaporation of elemental tin under H2S supply. Both methods require high temperatures and low pressure, and typically result in massive SnS layers.Here we present a modified CVD synthesis which results in high-surface SnS-nanosheet networks with nanoscopic crystal thicknesses by using bis(N,Nʼ-diethyldithiocarbamato)tin(II) as single-source precursor. The non-toxic precursor melts at 250 °C and evaporates at 350 °C under normal pressure. Placed in a three-zone horizontal tube furnace, the generated vapor is transported via Argon flow to an activated substrate, where the crystalline SnS network grows. Here we found that in particular gold in the form of small gold nanoclusters is highly suited to activate the SnS growth. The resulting SnS nanosheets show photoluminescence at 473 nm and 640 nm, which might be a result of strong quantization effect.
Fig.1: (a) Schematic illustration of the experimental setup. The single-source precursor is placed in the Argon flow, while the substrate is placed in temperature zone 2. (b) SEM
picture of the SnS-nanosheet network synthesized via MOCVD.
(a) (b)
339
Solids and Nano-sized Matter P07-16

Aerogel from CdSe and CdSe/CdS NanoplateletsSuraj Naskar, Sara Sánchez Paradinas, Jan. F. Miethe, Nadja C. Bigall*
Institute of Physical Chemistry and Electrochemistry, Leibniz Universität Hannover Callinstraße 3A, 30167 Hannover, Germany.
Extremely porous, fluorescent aerogel monoliths are achieved for the first time from strongly quantum confined quasi 2D CdSe and CdSe/CdS nanoplatelets (NPL). The NPL are synthesized in organic medium, subsequently transferred to aqueous solution and converted to hydrogels over aerogels by supercritical drying. Controlled destabilization of the aqueous NPL solution is achieved following a technique reported earlier with variable concentration of H2O2.1 Microscopic analysis proves interconnected self-supported superstructure consists of a fractal type network with pore distribution ranging from micro to macro meter regime, where NPL are randomly oriented with (111) as the exposed crystal facets. The aerogels have significantly high BET specific surface area in the range of 219-189 m2·g-1 and very low densities ~ 0.038 g·cm-3 respectively. Luminescent aerogel monoliths have quantum yield upto 10.3 %. High porosity, low density and quantum confined properties of the here developed aerogels allow the possible reactant molecules to access the surface of the NPL making them promising as substrate for the future application in the field e.g. catalysis, optical sensing, photovoltaic, light emitting diodes.1. Sánchez-Paradinas, S. et.al. Adv. Mater. 2015, 27, 6152-6156.
340
Solids and Nano-sized MatterP07-17

Combined local electrical and optical probes of individualSemiconductor Nanostructures
Andreas Kolditz, Christian Strelow, Tobias Kipp and Alf MewsUniversity of Hamburg, Institute of Physical Chemistry, Grindelallee 117, 20146
Hamburg, Germany
Elongated 1D-nanowires and 2D-nanosheets are promising materials forphotovoltaics as well as for light emitting applications. In order to explore their optical and electrical characteristics we combine scanning probe techniques with optical and electrical measurements of individual contacted nanostructures. In particular we performed Electrostatic Force Microscopy (EFM) and Kelvin Probe Force Microscopy (KPFM) in combination with electrical transport and fluorescence measurements of the same individual nanostructures, which were contacted by electrodes. This setupallows us to investigate the effect of band bending on the photo current and also the effects of external charging on the fluorescence properties of individual structures.
Fig.1: schematic illustration of the experimental setup. The individual nanostructures are
placed on an ITO substrates coated with SiO2. While the scanning probe microscope (AFM,
EFM, KPFM) is on top the optical measurements (CLSM) were carry out beneath. The photo
current measurements (SPCM) are realized with the contacts and external amplifier.
341
Solids and Nano-sized Matter P07-18

Amine Stabilized Au NPʼs for Catalytic ApplicationsL. Mohrhusen1 1, J. Kolny-Olesiak2, K. Al-Shamery1
1Carl von Ossietzky University of Oldenburg, Institute of Chemistry,Physical Chemistry 1, D-26129 Oldenburg, Germany
2Carl von Ossietzky University of Oldenburg, Institute of Physics,Energy and Semiconductor Research Laboratory (EHF), Nanochemistry, D-26129 Oldenburg,
Germany
Nanostructured materials are important in applications such as heterogeneouscatalysis. Their catalytic properties depend on the particle size and shape. In more recent research colloids are tested for better control of these parameters. Furthermore the stabilizing ligands have the potential to be a set screw for selectivity and reaction control. For catalytic applications gold nanoparticles (Au NPʼs) with atight particle size distribution in a range of <5 nm are desired.[1,2] While common thiol ligands suppress any catalytic activity because of strong interactions more weakly bound amine ligands appear to be promising candidates for catalytic applications. We adopted a simple colloidal single phase synthesis protocol developed for Au colloid synthesis with DDA (dodecylamine) and thiol ligands to the synthesis of small gold nanoparticles with various amine ligands (e. g. oleylamine).[3] We shall discuss the strong temperature dependence during reaction and ripening on the particle sizes and distributions and present an optimized procedure (Figure 1a-d).
Fig. 1: Overview TEM images of Au nanocrystals: a, b Au dodecylamine NPʼs before and after cleanup. c, d Au oleylamine NPʼs before and after heat induced ripening.
[1] Y. Kuwauchi et al., Angew. Chemie, 124, 7849-7853 (2012) [2] T. Ishida, M. Haruta, Angew. Chemie 119, 7288–7290 (2007) [3] N. R. Jana, X. Peng, J. Am. Chem. Soc., 125, 14280-14281 (2003)
342
Solids and Nano-sized MatterP07-19

Versatile fabrication of voluminous nanocrystal aerogelsAxel Freytag[a], Nadja C. Bigall[a],*
[a] Leibniz University Hannover, Institute of Physical Chemistry and Electrochemistry, Callinstr. 3A, 30167 Hannover, GermanyE-Mail: [email protected]
The presented work will give an insight into a complete new and versatile fabrication process to assemble nanoparticles from an aqueous, colloidal solution into high voluminous, self-supported macroscopic aerogel monoliths with extremely low density[1]. With our method it is possible to directly transfer nanocrystals from colloidal solutions into dry aerogel-like superstructures and therefore overcoming problems caused by gelation, liquid exchange or supercritical drying as would be the case for conventional aerogel fabrication. As a result the fabrication of aerogels will become easier to produce, is environmentally friendly in fabrication and will be produced at lower cost. The morphology of the resulting superstructures can be described as thin platelets of assembled nanocrystals, which form a voluminous network responsible for the low macroscopic density of the resulting cryogel. Our fabrication process is applicable to many colloidal nanoparticle materials and was already successfully tested for (noble) metals, metal oxides (e.g. hematite) or semiconductor materials (e.g. CdSe/CdS quantum rods), while most of the physical properties of the nanoparticles could be retained. In addition this method allows the shaping of the resulting aerogel not only as monolith but also as i.e. thin films or more sophisticated geometries.
Figure 1. A) Thin film of CdSe/CdS Aerogel on glass substrate. B) Around 70 mg of Pt as “Smiley” aerogel (black bar represents 1cm)
[1] A. Freytag, S. Sánchez-Paradinas, S. Naskar, N. Wendt, M. Colombo, G. Pugliese, J. Poppe, C. Demirci, I. Kretschmer, D. W. Bahnemann, P. Behrens, N. C. Bigall, Angew Chem Int Ed Engl 2015, DOI: 10.1002/anie.201508972
343
Solids and Nano-sized Matter P07-20

Embedding Nonpolar Quantum Dots into Ionic Matrices
Chris Guhrenz, Albrecht Benad, Christoph Bauer, Marcus Adam, Josephine F. L. Lox, Christoph Ziegler, Nikolai Gaponik, Alexander Eychmüller
Physical Chemistry, TU Dresden, Bergstraße 66b, 01062 Dresden, Germany
The incorporation of colloidal quantum dots (QDs) into ionic crystals by using saturated salt solutions was first described by OTTO et al. in 2012.[1] In comparison to the commonly used incorporation into polymers, this approach results in an even higher increase of the photo- and chemical stability and processability while preserving the strong luminescence of the embedded QDs.[1,2] However, this method relies short, polar ligands on the QD-surface and requires a ligand exchange for organic-synthesized QDs.[3] Therefore, a seed-mediated liquid-liquid diffusion-assisted crystallization (LLDC) has been established for a direct encapsulation of organic-synthesized QDs into salt crystals.[4]
Herein, we present yet another new, facile approach to incorporate a wide range oforganic-synthesized QDs into ionic matrices (e. g. CsI, KBr). This method needs high pressure and does not require a ligand exchange procedure. The optical properties of the initially used QDs are retained in the ionic crystals making them attractive for optoelectronic devices including light-emitting diodes (LEDs) and lasers.
[1] T. Otto, M. Müller, P. Mundra, V. Lesnyak, H. V. Demir, N. Gaponik, A. Eychmüller, Nano Lett. 2012, 12, 5348–5354.
[2] M. Müller, M. Kaiser, G. M. Stachowski, U. Resch-Genger, N. Gaponik, A. Eychmüller, Chem. Mater. 2014, 26, 3231–3237.
[3] M. Adam, T. Erdem, G. M. Stachowski, Z. Soran-Erdem, J. F. L. Lox, C. Bauer, J. Poppe, H. V. Demir, N. Gaponik, A. Eychmüller, ACS Appl. Mater. Interfaces2015, 7, 23364–23371.
[4] M. Adam, Z. Wang, A. Dubavik, G. M. Stachowski, C. Meerbach, Z. Soran-Erdem, C. Rengers, H. V. Demir, N. Gaponik, A. Eychmüller, Adv. Funct. Mater. 2015, 25, 2638–2645.
344
Solids and Nano-sized MatterP07-21

Surface Phenomena of Metal Nanowire Networks as Transparent Electrodes for Organic Solar Cells
L. Sonntag,1 F.Eichler,1 N. Weiß,1 L. Bormann,2 N. Gaponik,1 K. Leo,2 A. Eychmüller1
1 Physical Chemistry and Cluster for Advancing Electronics Dresden (cfaed),Technische Universität Dresden
2 Institut für Angewandte Photophysik (IAPP), Technische Universität Dresden
The worldʼs growing market of organic optoelectronic devices requires low-cost and easy processable, transparent conductors with high transmittance and low sheet resistance. Networks assembled from silver nanowires (AgNWs) are promising candidates to substitute commonly used and expensive indium tin oxide (ITO). State-of-the-art AgNW electrodes show comparable performances to ITO while being distinctly cheaper. Nanowires with different favorable diameters and lengths can be obtained by means of the polyol synthesis utilizing Polyvinylpyrrolidone (PVP) as capping agent.However, the non-uniform surface of AgNW networks makes an incorporation into thin-film devices, for instance in organic solar cells (OSC), challenging. The corresponding devices suffer from short circuits, when NWs penetrate into the layer on top of the electrode, since typical NW diameters are in the same range as common thicknesses of organic layers. Herein, we report on the advances in the synthesis of AgNWs and the role of PVP in post-treatment of fabricated electrodes. Moreover, we demonstrate an opportunity to overcome the surface roughness of the deposited nanowire network. Our technique allows an incorporation of AgNWs into OSCs without additional pressing of the network or without a thick, planarizing polymer layer, which is usually deployed to smooth the rough NW surface. Our optimized electrodes can also be suitable for other thin film devices, such as (organic) light emitting diodes or transistors. Moreover, our approach is applicable to nanowire networks based on other promising metals, such as copper or gold.
345
Solids and Nano-sized Matter P07-22

Impact of redox-active molecules on the fluorescence of carbon nanotubes
Elena Polo, Dr, Sebastian Kruss, Georg-August-Universität Göttingen, Institut für Physikalische Chemie,Göttingen/Deutschland
Our research focus on optical biosensors based on carbon nanotubes. Carbon nanotubes are hollow cylinders of rolled-up one-atom-thick sheets of carbon. Depending on the roll-up vector of the nanotube, one can create useful optical and chemical properties. Semiconducting single-walled carbon nanotubes (SWCNTs) provide an intrinsic bandgap which results in near infrared (nIR) fluorescence. The nIR fluorescence of polymer-wrapped SWCNTs is very sensitive to the local chemical environment. It has been shown that certain small reducing molecules increase the fluorescence of SWCNTs. The exact mechanism of the interaction between the organic phase around the nanotube and the analyte molecule is still unknown.
Recently, we investigated the influence of reducing and oxidizing small molecules on the nIR fluorescence of polymer-wrapped SWCNTs. Our results show that the polymer plays an essential role. Reducing molecules such as ascorbic acid, epinephrine and trolox increased the nIR fluorescence up to 250%, but only if SWCNTs were suspended in negatively charged polymers such as DNA or poly-acrylic acid (PAA). In comparison, phospholipid-polyethylenglycole wrapped SWCNTs did not respond at all, while positively charged polyallylamine-wrapped SWCNTs showed reverse behavior (fluorescence decrease). Furthermore, we show that neither changes of absorption cross-sections, scavenging of reactive oxygen species nor calculated free surface areas on SWCNTs can explain the observed patterns. However, our data is in agreement either with a redox-reaction or conformational changes of the polymer around the nanotube. In summary, we showed that the polymer around SWCNTs governs how reducing molecules changenIR-fluorescence (quantum yield) of SWCNTs. Molecules with a low redox-potential are more likely to increase SWCNT fluorescence but it is not sufficient.
346
Solids and Nano-sized MatterP07-23

Localized Surface Plasmon Resonance of Au-ITO heterodimersPrepared by Colloid Chemistry
Tarek Mohamed, Dirk DorfsInstitute of Physical Chemistry and Electrochemistry
Leibniz Universität HannoverHannover/Germany
Highly doped semiconductors and oxide nanoparticles play an increasingly important role with promising applications in various fields such as photovoltaicʼs, sensors and catalysis. [1] With colloid chemistry, it is possible to produce nanoparticles of different sizes, shapes and compositions. By controlling these three parameters, the physical properties of the nanoparticles can be tuned. [2] Coupling different nanoparticles to create heterodimers offer the advantage of combining various characteristics of theses nanoparticles in one system. [3,4]The Au-ITO hetrodimer nanoparticles were prepared by a injection synthesis and show localized surface plasmon resonances (LSPRs) in the near infrared (NIR) region and an LSPR in the visible region. With the variation of the tin content of the ITO nanoparticles the LSPR can be set in the NIR region. In this work, the influence of different parameters on the properties of the Au-ITO hetrodimers is investigated.
[1] a) A.P. Alivisatos, Science 1996, 271, 933-937; b) H. Weller, Angew. Chem. Int. Ed. 1993, 32, 41-53.
[2] M. Kenehara, H.Koike, T.Yoshinaga, T. Teranishi, J. Am. Chem. Soc. 2009,131, 17736
[3] T.R. Gordon, R. Schaak, Chem. Mater. 2014, 26, 5900-5904[4] X. Ye, D. Refisnyder Hickey, J.Fei et al, J. Am. Chem. Soc. 2014, 136,
347
Solids and Nano-sized Matter P07-24

Influence of chloride ions on the shape and nucleation of CdS and CdSe on metal nanoparticles
Dominik Hinrichs, Torben Kodanek, Michael Galchenko, Suraj Naskar, Nadja Bigall, Dirk Dorfs,
Leibniz Universität, Hannover, Niedersachsen
Semiconductor-metal hybrid nanoparticles are of high interest in nowadays research, e.g. due to their combination of properties for the application in photocatalysis orsolar energy applications.[1] To obtain the hybrid nanoparticles often the metal is grown on a separately prepared semiconductor particle.[2] However, it is also interesting to pursue the vice versa approach.We present a synthetic route to prepare metal-semiconductor hybrid nanoparticles.Addition of chloride ions shows a high degree of control over the shape andnucleation behavior of the semiconductor material, which is grown on a separately prepared metal nanoparticle seed. In literature chloride ions were just investigated as shape influencing additive for pure CdS/ CdSe but not for their nucleation/growth on a metal nanoparticle.[3-6]
Gold, platinum and cobalt platinum nanoparticles are used as metal seeds, on which CdS or CdSe is grown. The chloride ions, present in the reaction mixture, show a significant influence on the primary and secondary nucleation as well as on the shape development of the semiconductor. Neither pure CdO nor pure CdCl2 as precursor for cadmium but only a mixture of both makes these hybrids obtainable.
[1] R. Costi, A. E. Saunders, U. Banin, Angewandte Chemie, 2010, 49, 4878-97.[2] L. Carbone, P. D. Cozzoli, Nano Today, 2010, 5, 449-493.[3] M. R. Kim, K. Miszta, M. Povia, R. Brescia, S. Christodoulou, M. Prato, S. Marras,L. Manna, ACS nano, 2012, 6, 11088-96.[4] J. Lim, W. K. Bae, K. U. Park, L. zur Borg, R. Zentel, S. Lee, K. Char, Chemistry of Materials 2013, 25, 1443-1449.[5] M. Meyns, F. Iacono, C. Palencia, J. Geweke, M. D. Coderch, U. E. A. Fittschen,J. M. Gallego, R. Otero, B. H. Juárez, C. Klinke, Chemistry of Materials, 2014, 26, 1813-1821.[6] Y. Zou, D. Li, D. Yang, Nanoscale research letters, 2011, 6, 374.
348
Solids and Nano-sized MatterP07-25

Assembly of shape-controlled semiconductor nanocrystals
Lars F. Klepzig, Jan Poppe, Nadja C. Bigall, Institute of Physical Chemistry and Electrochemistry, Leibniz Universität Hannover, Hannover/Germany
Self-supporting aerogels of semiconductor nanoparticles are a recently emerged class of solid materials facilitating low densities and high surface areas. These highly porous networks of connected nanocrystals provide the possibility to retain the nanoscopic characteristics enabling the macroscopic use of e.g. the size quantization effect or the high surface to volume ratio. The introduction of anisotropic crystals as building blocks offers further possibilities to alter the properties such as increasing the surface to volume ratio, thus leading to new applications in catalysis or sensing.
In this work the gel formation of shape-controlled anisotropic nanocrystals is investigated. The phase transfer to aqueous solution by ligand exchange and the gelation induced by destabilizing the particles by partially removing the surface ligands is accomplished for two different systems with various conditions. An investigation on the dynamics of the gelation process is performed as well as acomparison to the new method of freezing and subsequent freeze-drying [1]. The achieved hydrogels are supercritically dried and characterized by optical absorption, N2-physisorption and electron microscopy. The obtained aerogels show high porosities and surface areas of up to 85 m2g-1.
[1] A. Freytag; S. Sánchez-Paradinas; S. Naskar; N. Wendt; M. Colombo, G. Pugliese; J. Poppe; C. Demirci; I. Kretschmer; D. Bahnemann; P. Behrens; N. Bigall, Angew.Chem.Int.Ed. 2015, 54
349
Solids and Nano-sized Matter P07-26

Improvement of Photostability of silica covered CdSe/CdS-Dot/Rodsthrough surface attached AuNPs-Layer
Elvira Huber, Xiao Tang, Simon Schneider, Alf MewsInstitute for Physical Chemistry, University of Hamburg, Germany
Elongated semiconductor nanocrystals with a spherical CdSe-dot embedded into an elongated CdS-rod (DotRods, DRs) exhibit high photo stability and also a high fluorescence quantum yield of more than 50 %. In this contribution we show how heterostructures of DRs and gold metal particles (AuNPs) can be prepared to investigate the electronic interaction between the excitons generated inside the DRsand the plasmon of the AuNPs. This interaction depends on several parameters, forexample the distance between DR and AuNPs and the overlap/resonance of the fluorescence of the DRs and the plasmon of the AuNPs. To adjust the distance werealized a silica coating of the DRs with a shell thickness of only several nm by a micro emulsions method1. Then we prepared a layer if AuNPs on a glass substrate and attached the silica coated DRs to the AuNP-layer. It was observed that the fluorescence properties of individual DRs change considerably due to their proximity to the AuNPs. For example, we found a distinct change in fluorescence blinking where the on-time fraction of the silica covered DRs (SiDRs) increases dramatically, which was accompanied by an increase in photo stability.
Fig. 1: Sketch of the sample preparation. AuNPs on a glass substrate with coupled SiDR.
1. F. Pietra et al., Chem. Mater., 25, 3427-3434, 2013.
350
Solids and Nano-sized MatterP07-27

Internal Complexation within Starlike Copolymers: Influence of Topology and Composition
P. Hebbeker, A. A. Steinschulte, F. A. Plamper, S. Schneider, Institute of Physical Chemistry, RWTH Aachen University, 52074 Aachen, Germany
Experimentally, it was found that the local mutual segment density has an effect on the intramolecular complexing behavior of poly(propylene oxide) (PPO) and poly(dimethylaminoethyl methacrylate) (PDMAEMA).1 We investigated linear block-copolymers as well as miktoarm star polymers of the two components. A complexation of the PPO chain in the miktoarm stars was found for conditions under which the linear polymer is not complexed.To rationalize these observations the polymers were modeled using a coarse grained bead spring model. The polymers consist of two types of hard sphere beads (A and B), interacting by an attractive Lennard-Jones Potential. Using Monte Carlo Simulations the structure of a miktoarm starpolymer and a diblock copolymer was investigated as a function of the strength of the attractive potential.2
To deepen the insight into the complexation behavior different topologies like alternating and random copolymers as well as comb copolymers are simulated in addition to the miktoarm star and the diblock copolymer topologies (Fig. 1). Atintermediate interaction strengths the complexation is more pronounced in diblock copolymers and even more in miktoarm star polymers.Comparing different compositions of A and B are compared to shows the role of the composition on the complex formation. It is shown that in a miktoarm star polymer the same degree of complexation can be achieved for a much lower molecular weight compared to the linear diblock copolymer. This was also observed experimentally.
Fig. 1 Sketch of the simulated topologies
1 Steinschulte, A. A.; Schulte, B.; Erberich, M.; Borisov, O. V.; Plamper, F. A. ACS MacroLett. 2012, 1, 504–507.2 Hebbeker, P.; Plamper, F. A.; Schneider, S. Macromol. Theory Simul. 2015, 24, 110–116.
351
Soft Matter and Polymers P08-01

Gas permeation through carbon nanomembranes
André Beyer1, Vahe Chinaryan1, Sergey Shishatskiy2, Jan Wind2, Xianghui Zhang1,Henning Vieker3, Polina Angelova3, Volker Abetz2 and Armin Gölzhäuser1
1 University of Bielefeld, 33615 Bielefeld, Germany 2 Helmholtz-Zentrum Geesthacht, 21502 Geesthacht, Germany
3 CNM Technologies GmbH, 33609 Bielefeld, Germany
The gas permeation characteristics of carbon nanomembranes (CNMs) from self-assembled monolayers are reported. CNMs are 1 nm thick sheets, made by the cross-linking of molecular monolayers. CNMs were placed onto polydimethylsiloxane (PDMS) membranes to determine the gas permeation characteristics for single- andmulti-layers of CNMs. CNMs made from different molecules having thicknesses from 0.6 nm up to 1.2 nm were investigated with regard to the permeation of different gases (He, H2, CO2, O2, N2, …). The CNM-PDMS composites were also characterized by X-ray photoelectron spectroscopy and helium-ion microscopy. The permeation characteristic of CNMs indicates a molecular-sieve-like transport mechanism which can be attributed to molecular-sized channels in CNMs. Additionally, the permeance of CNMs is adjustable by varying the length of the precursor molecules, i.e. the thickness of single-layer CNMs. Multilayers of CNMs show a permeation that differs significantly from single-layer CNMs, possibly due to the diffusion of the permeating molecules in between the CNM layers.
352
Soft Matter and PolymersP08-02

Polymer Loops and Tails attached to a Surface A Dissipative Particle Dynamics Study
Martin Jehser, Stephan Eisenhaber and Gerhard Zifferer
University of Vienna, Department of Physical Chemistry, Währinger Straße 42, 1090 Wien
Inorganic particles can be modified with organic polymer layers leading to well defined hybrid nanoparticles with tuneable surface properties. Usually, simple linear chains (so called tails or trails) are anchored to the surface. A straightforward modification to this is the introduction of surface-confined loops opening a new class of hybrid material, with the benefit of no free chain ends. Recently, experimental methods have been developed allowing the production of such systems [1]. In the present contribution equivalent situations are studied by means of computer simulations based on Dissipative Particle Dynamics (DPD) [2,3].As a coarse-grained method for equivalent chains, consisting of beads connected via springs, DPD is highly efficient and able to smoothly cover the range of several nanometers up to the mesoscale region. The explicit treatment of solvent particles allows reproducing realistic hydrodynamic behaviour. Following Newtonʼs 3rd law allinteractions, namely conservative, dissipative and random forces, are strictly pairwise. The latter two represent the thermostat and the former the thermodynamic conditions, i.e., solvent quality, sort of the polymer bead, etc.In order to reproduce the behaviour of adsorbed polymers in addition to the conservative forces mentioned above an attractive interaction between one (or, in case of loops, both) chain end(s) and a surface – represented by a soft wall – are introduced as well.The aim of the study is the comparison of properties of loops to those of tails and those of unattached polymers as a function of surface coverage, solvent quality and the distance between anchoring segments of the loop.____[1] R. Rotzoll, P. Vana, Journal of Polymer Science Part A: Polymer Chemistry 46, 7656 (2008)[2] R.D. Groot, P.B. Warren, J. Chem. Phys. 107, 4423 (1997)[3] M.M. Nardai, G. Zifferer, J. Chem. Phys. 131, 124903 (2009)
353
Soft Matter and Polymers P08-03

Shielding Effects in Surface initiated Polymerization studied by Dissipative Particle Dynamics (DPD)
Stephan Eisenhaber, Martin Jehser, and Gerhard Zifferer
University of Vienna, Department of Physical Chemistry, Währinger Straße 42,1090 Vienna, Austria
Hybrid nanoparticles, inorganic surfaces modified by polymer layers, may be built either by attching the polymer to the particle (ʻgrafting toʼ approach) or growing the polymer from the surface (ʻgrafting fromʼ techniques). The latter usually allows higher grafting densities. Among others a most promising grafting from technique is the so-called Z-group approach of surface initiated RAFT (reversible addition fragmentation chain transfer) polymerization [1] where the RAFT agent is directly linked to the solid substrate. The propagating species in the solution, i.e., the polymer radicals are in equilibrium with the dormant polymeric RAFT agent leading to a narrow distribution of chain-lengths as all radicals have the same probability to grow. A critical factor is the contact-formation between the RAFT agent and the radical end of the approaching polymer, which may be hindered by both – the approaching as well as the leaving – polymers. If in addition at least one of the polymers is attached to a surface (as in the present case), the shielding of the reactive site is further enhanced. For not infinitely low concentrations of reactive sites – or here, surface coverage –,shielding effects may be calculated from the dynamical approach of polymers, e.g., making use of Dissipative Particle Dynamics. This method has already been succesfully introduced and applied to Z-RAFT star polymerization [2,3]. In the present investigation this technique is adapted to study shielding effects of reactive centers near to surfaces.____[1] S. Perrier, P. Takolpuckdee, C. A. Mars, Macromolecules 38, 6770 (2005)[2] M.G. Fröhlich, M.M. Nardai, N. Förster, P. Vana, G. Zifferer, Polymer 51, 5122-5134 (2010)[3] M.M. Nardai, G. Zifferer, Polymer 54, 4183-4193 (2013)
354
Soft Matter and PolymersP08-04

Structure and time-dependent self-diffusionin binary mixtures of charge-stabilized colloidal suspensions
Falko Ziegert, Joachim WagnerInstitut für Chemie, Universität Rostock, Albert-Einstein-Straße 3a, 18051 Rostock
Binary mixtures of highly charged colloids consisting of polystyrene- (PS) and poly-2,2,2-trifluoroethyl acrylate- (pTFEA) particles are investigated. Due to the low refractive index of fluorinated polymers, pTFEA can be index-matched using water-glycerol mixtures as a suspending medium. Under this condition in light scattering experiments the scattered light solely originates from the PS particles with significantly higher refractive index.The time-dependent self-diffusion of electrostatically interacting PS tracers is investigated in dependence of both, the total number density = PS + pTFEA of colloidal particles and the ratio of number densities PS / pTFEA by means of photon correlation spectroscopy (PCS). The temporal decay of the self-diffusion coefficient as well as the short and long time limits DS and DL are determined. From the partial structure factors of PS the number of effective charges and the Debye screening lengths are derived. Brownian Dynamics simulations using these interaction parameters predict a time-dependence of self-diffusion coefficients in good agreement to the experimental data obtained from PCS.
AcknowledgmentsFinancial support by the BMBF within the Verbundprojekt 605 is gratefully acknowledged.
355
Soft Matter and Polymers P08-05

Detection of particle interaction in polydisperse highly concentrated polymer suspensions by Photon Density Wave spectroscopy
L. Bressel, S. Pfuhl, O. Reich, University of Potsdam/Institute of Chemistry/Physical Chemistry - innoFSPEC, Potsdam/Germany
Nano-/Microstructured materials are present nearly everywhere in our world. From biological systems like cells to food to industrial products like paints, cosmetics and polymers, all of these systems consist of absorbers and particles or droplets in the nano-/micrometer regime dispersed in a liquid. Due to the typically high volume
fraction of particles ( = 1-60%) these samples therefore exhibit multiple light scattering in addition to absorption. Both phenomena, quantifiable by the absorption and reduced scattering coefficient μa and μs by the type, amount and size (in case of particles) of absorbers and particles. Hence, by a profound understanding of the interaction between absorption and scattering the desired optical properties can be adjusted. One important effect one here has to consider is the effect of dependent scattering, i.e. a nonlinear relationship between μs , resulting from interference of the scattered light due to particle interaction
and ordering at high . The interference can be accounted for mathematically via the so called static structure factor S applying an appropriate interaction model.In the past the case of spherical monodisperse particles exhibiting solely hard sphere interaction has been extensively studied applying the analytical structure factor from the Percus Yevick approximation [1]. In this study we will focus on the influence of polydispersity on dependent scattering. Measurements are carried out on mixtures of monodisperse polymer dispersions and compared to theoretical results [2]. Mie theory for spherical particles is used to account for single particle light scattering.Photon Density Wave spectroscopy [1] is used as the experimental method to determine independently and without calibration μa and μs at the present high volume fractions.
[1] L. Bressel, R. Hass, O. Reich, JQSRT, 126, 122-129 (2013).[2] A. Vrij, J. Chem. Phys., 69, 1742-1747 (1978). A. Vrij, J. Chem. Phys., 71, 3267-3270 (1979).
356
Soft Matter and PolymersP08-06

Combination of fiber-optical methods for in-line multi-parameter analysis of the PNIPAM switching processes
Peter Werner, Oliver Reich, Universität Potsdam, Institut für Chemie, Physikalische Chemie-innoFSPEC, Potsdam/Deutschland;
The chemical and physical sensitivity of smart polymers like poly(N-isopropylacrylamide) (PNIPAM) have great potential for diverse applications, thus it is of high interest to understand the complete mechanism behind these effects.In order to detect more details behind the switching process of thermo-sensitive polymers it is necessary to use different technologies to get as much information as possible. With the combination of different inline methods dynamic changes during the switching process are detected.In this contribution a combination of photon density wave (PDW) spectroscopy and focused beam reflectance measurement (FBRM) in a highly automated reactor is presented. With the combination of these two methods the dependence of the switching process of PNIPAM on different heating/cooling rates could be investigated. For both methods it could be figured out that a direct correlation between the heating/cooling rates and the broadening of the hysteresis exists, i.e.the switching temperature (lower critical solution temperature LCST) is coupled directly to the heating/cooling rates.
26 28 30 32 34 36 38 400.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
cool
ing
red.
sca
tterin
g co
effic
ient
μs'
/ mm
-1
temperature T / °C
heating/cooling rate 0.1 K/min heating/cooling rate 0.3 K/min heating/cooling rate 0.5 K/min heating/cooling rate 0.7 K/min
heat
ing
Fig. 1: reduced scattering coefficient μsʼ at different heating/cooling rates as function of the temperature
357
Soft Matter and Polymers P08-07

Adsorption of CO2 on the Fe3O4(111) surface studied by density-functional theory
Xiaoke Li and Joachim PaierHumboldt-Universität zu Berlin, Unter den Linden 6, 10099 Berlin, Germany
Atomic level understanding on the interaction of CO2 with oxide surfaces is of high importance in solving the problems as e.g. CO2 capture and storage, global warming and energy recycling. Fe3O4 (magnetite) represents an outstanding oxide materialdue to many industrial applications [1-3].This work reports results on periodic models of the Fe3O4(111) surface [p(1x1) unit cell; 12 atomic layers] adsorbed by a single CO2 molecule, as well as two molecules and an oxalate (C2O4) group. We study the adsorption thermodynamics and infrared spectra of the aforementioned aggregations. We employ density-functional theory using the Perdew, Burke, Ernzerhof [4] gradient-corrected exchange-correlation functional including a correction for the Fe 3d levels by an onsite Hubbard-type U parameter.Even though several studies involving CO2 adsorption and reduction on Fe3O4
combined materials have been reported [5-7], the adsorption behavior of CO2 on Fe3O4(111) clean surface is still missing. This work attempts to fill this knowledge gap.
[1] Xu, F., et al. CrystEngComm. 2013, 15, 4431.[2] Smit, E. de; Weckhuysen, B. Chem. Soc. Rev. 2008, 37, 2758.[3] Ratnasamy, C. Catal. Rev. 2009, 51, 325.[4] Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865.[5] Lv, B., et al. Int. J. Biol. Macromol. 2015, 79, 719.[6] Galvita, V. V et al. J. Power Sources 2015, 286, 362.[7] Furukawa, T; Rouillard, F. Prog. Nucl. Energ. 2015, 82, 136.
358
Surfaces and InterfacesP09-01

Formaldehyde Adsorption on the Rutile TiO2(110) Surface Probed by IR Spectroscopy
Xiaojuan Yua, Chengwu Yanga, Fabian Bebenseea, Alexei Nefedova,Zhenrong Zhangb, Qingfeng Gec, Zdenek Dohnálekd, Yuemin Wanga and
Christof Wölla
aInstitute of Functional Interfaces, Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, Germany.
bDepartment of Physics, Baylor University, Waco, Texas 76798, USA.cDepartment of Chemistry and Biochemistry, Southern Illinois University, Carbondale, Illinois 62901, USA.
dFundamental and Computational Sciences Directorate, Institute for InterfacialCatalysis, Pacific Northwest National Laboratory, Richland, Washington 99352, USA.
TiO2 is one of the most important metal oxides used in catalysis and
photocatalysis. The interaction of TiO2 with aldehydes is of particular interest
due to its promising applications in ethanol fuel cells, hydrogen production,
biomass and in the control of air pollution. Here, the adsorption of
formaldehyde (CH2O) on the rutile TiO2(110) surface was studied by infrared
reflection-absorption spectroscopy (IRRAS). The IR data reveal the presence
of different species depending on the temperature and coverage. The CH2O
monomer is identified after submonolayer adsorption at 70 K, in which CH2O is
weakly bound to the surface Ti5c sites. Further adsorption at 70 K leads to the
formation of multilayer CH2O, which desorbs completely upon heating to 120 K.
Simultaneously, paraformaldehyde is formed at Ti5C sites along [001] direction
and becomes the dominant surface species. In addition, dioxymethylene is
detected as a minority one formed via reaction of CH2O with neighboring Obr
along [1-10] direction.
359
Surfaces and Interfaces P09-02

Microcalorimetric measurement of double layer charging in ionic liquids
Jeannette Lindner1, S. Frittmann1, F. Endres2, R. Schuster1, 1Institute of Physical Chemistry, Karlsruhe Institute of Technology, Karlsruhe/Germany; 2Institute of
Electrochemistry, TU Clausthal, Clausthal/Germany
The reversible molar heats, i.e., the Peltier heats of processes in the electrified double layer at an IL/Au(111) interface were measured via electrochemicalmicrocalorimetry [1]. Peltier heat corresponds to entropy change of the electrochemical reaction including all side processes and ion transport in solution. From the entropy of the double layer charging processes we conclude on the ad-/desorption and reorientation of the IL at the metal interface.
1-Butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)amide was used as ionic liquid. Microcalorimetric measurements gave positive Peltier heats in the whole double layer region with a maximum of = 65 kJ/mol. Because simple ad-/desorption processes should lead to much smaller values, we assume the collective reorientation of ions in the compact and diffuse layers near the electrode as described by molecular dynamics simulations [2]. The cations in the innermost layer might play an important part because they are expected to change their orientation to the surface depending on electrode potential [3].
Furthermore, we found a strong, potential-dependent asymmetry of the time constants for positive and negative charging of the double layer. This indicates the presence of several processes with different characteristic timescales upon double layer charging as also found by electrochemical impedance spectroscopy [4] and surface plasmon resonance spectroscopy [5].
References:
[1] S. Frittmann et al. S. Rev. Sci. Instrum. 2015, 86, 064102.[2] C. Merlet et al. Phys. Chem. Chem. Phys. 2013, 15, 15781.[3] S. Baldelli. Acc. Chem. Res. 2008, 41, 3, 421-431.[4] J. Wallauer et al. Z. Naturforsch. 2013, 68b – 1153.[5] N. Nishi et al. Phys. Chem. Chem. Phys., 2013, 15, 11615.
360
Surfaces and InterfacesP09-03

Structure Dependent Anchoring and Thermal Stability of Organic Molecules on Atomically-Defined Oxide Surfaces:
Phthalic Acid on Co3O4(111), CoO(100) and CoO(111)Tao Xu, Matthias Schwarz, Kristin Werner, Susanne Mohr, Max Amende, Jörg
Libuda*
Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Erlangen, GermanyWe have performed a model study to explore the influence of the surface structure on the anchoring and thermal stability of organic molecules on oxide materials.Specifically, we investigated the adsorption and desorption of phthalic acid (PA) on atomically defined (i) Co3O4(111), (ii) CoO(111) and (iii) CoO(100) grown on Ir(100). Formation of the PA films and interfacial reactions were monitored in-situ during growth by isothermal time-resolved infrared reflection absorption spectroscopy (TR-IRAS) under ultrahigh vacuum (UHV) conditions. We observe pronounced structure dependencies on the three surfaces with different binding geometries and characteristic differences as a function of temperature and coverage: (i) PA/Co3O4(111): PA binds initially by formation of a chelating bis-carboxylate with the molecular plane oriented perpendicular to the surface. Similar species are observed at low temperature (130 K) and at room temperature (300 K). (ii) PA/CoO(100): At 130 K, PA binds in form of a bridging bis-carboxylate at low coverage which converts to bis-carboxylates at higher coverage. At 300 K, the bis-carboxylate layer is formed and shows no coverage dependent changes. (iii) PA/CoO(111): we observe a temperature-dependent change in the adsorption mechanism. While PA binds as a mono-carboxylate in a bridging bidentate fashion at low temperature (130 K), a strongly distorted bis-carboxylate is formed at 300 K, possibly due to a temperature-dependent restructuring of the surface.Desorption of PA from the oxides was monitored by recording temperature-programmed IRAS between 130 and 600 K. Pronounced differences in stability are observed. The most stable species was found on CoO(100) where a bridging bis-carboxylate is formed involving easily accessible Co surface ions. The chelating bis-carboxylate on Co3O4(111) is found to be less stable and the distorted bis-carboxylate on CoO(111) shows the lowest stability due to the limited accessibility of the surface Co ions.
361
Surfaces and Interfaces P09-04

LiCoO2-electrolyte interaction studied by surface science methods
Thomas Späth, René Hausbrand and Wolfram JaegermannInstitute of Material Science, Darmstadt University of Technology,
Jovanka-Bontschits-Str. 2, 64287 Darmstadt, Germany
Reactions and SEI formation at electrode-electrolyte interfaces play an important role for performance and lifetime of batteries. For a better understanding of the reactivityof solvents with Li-ion cathode materials, we perform adsorption experiments on thin film model electrodes.In these experiments we use High Resolution Electron Energy Loss Spectroscopy (HREELS) and Photoemission Spectroscopy (PES) to study both vibrational modes and composition close to the surface. With this information we can conclude on chemical reactions at the interfaces.In this contribution we report on the status of our investigations regarding LiCoO2-solvent interaction. We demonstrate that HREELS is a suitable technique for studying polycrystalline thin electrode films. First we discuss the assignment of thedifferent features in the spectrum for the pristine LiCoO2 samples. After that, we present HREELS and PES spectra of the electrolyte adsorbed samples. For both techniques adsorption leads to new spectral features, assigned to physicallyadsorbed and chemically reacted species.From this data we can conclude that HREELS is an appropriate technique for study adsorption at electrode-electrolyte interfaces. We believe that HREELS supports our PES measurement due to its high surface sensitivity and the complementary information.
362
Surfaces and InterfacesP09-05

Relaxation dynamics and electron-transfer kinetics investigation onelectrode|self-assembled monolayer|ionic liquid interfaces using
electrochemical impedance spectroscopy
Nella M. Vargas-Barbosa and Prof. Dr. Bernhard Roling,Philipps–Universität Marburg, Marburg, Germany
Ionic liquids (ILs) are compounds that are entirely made up of anions and cations andhave melting points below 100 °C. Among their many intensive properties, their large electrochemical stability and low vapor pressure are most attractive for applications in energy generation and storage devices, e.g. rechargeable batteries, supercapacitors, liquid-electrolyte solar cells, among others. Although ILs are actively researched as electrolytes and solvents for many electrochemical systems, a fundamental understanding of the phenomena governing heterogeneous electron-transfer at the electrode-electrolyte interface is lacking. In this study, self-assembled monolayers (SAMs) of thiol-modified alkane molecules are used to control the electronic and spatial coupling between the electrode and the redox-active species in the electrolyte. As model system, the electrochemistry of 1-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (HMImTFSI) is probed at various SAM-modified gold electrodes (1-hexanethiol, 1-dodecanethiol and 1-octadecanethiol). Moreover, ferrocene (Fc) is used as the redox-active probe to assess permeability of the SAMs,as well as solvent effects on electron-transfer kinetics. Cyclic voltammetry and potential-dependent impedance measurements are used to characterize and identify various interfacial processes, as well as SAM length-dependent electron-transfer rates for the Fc0/+ couple.
363
Surfaces and Interfaces P09-06

Investigation of the interface of immiscible liquids by fluorescence light under microfluidic conditions
H.Fischer, E.Antonsson, E.Rühl, Freie Universität BerlinThe emission properties of fluorescence dyes, e.g. quantum yields, lifetimes, and band position are sensitive to the local solvent surroundings. We use a microfluidic setup to study the photoexcitation- and relaxation dynamics of Coumarin 153 at the interface of the immiscible liquids methanol and cyclohexane. The experimental conditions are tuned such that the two solvents are prepared in a laminar flow without the occurrence turbulent mixing, where the interaction between both phases is characterized by diffusion only.
The solvated dye is excited by a 85 fs laser pulse at a central wavelength of 400 nm. The fluorescence is collected using an optical fiber and a spectrometer equipped with a CCD camera, which is used for the detection of the shape and position of the emission band. A Y-shaped microfluidic-chip is used to investigate dynamic properties of the fluorescence at the interface of the two liquids.
We present fluorescence spectra of Coumarin 153 at the cyclohexane/methanol interface. The deconvolution of the broad emission bands reveals three components, which are assigned to Coumarin 153 in methanol and in cyclohexane as well as Coumarin 153 located at the interface of the immiscible liquids. By exciting the sample at different positions in the microfluidic cell or by changing the flow velocity of the liquid samples, the dynamics of diffusion between both phases and at the interface are monitored. This yields specific information on the diffusive transport of the dye across the interface between both liquids.
364
Surfaces and InterfacesP09-07

Scattering of highly vibrationally excited NO from Ge(111)Sven Meyer, Bastian C. Krüger, Alec M. Wodtke and Tim Schäfer, Georg-August-
Universität Göttingen, GermanyScattering small molecules from well-defined surfaces is an exceptional way to investigate energy transfer processes at the gas-solid interface. Experiments on highly vibrationally excited molecules are especially interesting, because nonadiabatic effects might be enhanced due to the stretched geometry of the molecule. In this work we have scattered highly vibrationally excited NO (v''=11) from a semiconducting Ge(111) surface. Similar to earlier experiments on Au(111) and LiF(001), final vibrational, rotational and translational energies have been measured. In contrast to metal surfaces, scattering from germanium shows very inefficient vibrational relaxation. We explain this difference by the bandgap of the semiconducting germanium crystal and so a less efficient coupling to electron hole pairs.
365
Surfaces and Interfaces P09-08

Vibrational energy pooling in multilayer CO on NaCl(100) studied with nanosecond time resolution using a superconducting
nanowire single photon detectorLi Chena, Dirk Schwarzera, and Alec M. Wodtkea,b
aMax-Planck-Institut für biophysikalische Chemie, Göttingen, bInstitut für Physikalische Chemie Georg-August-Universität, Göttingen
Laser induced infrared fluorescence from multilayer CO (1000-1600 layers) on NaCl(100) at 10-20 K is studied in both time and frequency domain using a superconducting nanowire single photon detector (SNSPD). Upon vibrational overtone excitation (v = ns laser pulse, infrared emission is detected in the range 1700 to 5200 cm-1 covering one-quantum ( v = -1), two-quanta ( v = -2), and three-quanta ( v = -3) emission lines of CO. The emission originates from states up to v = 29 implying efficient vibrational energy pooling within a few microsecondsafter the laser pulse. Due to the high time-resolution of the SNSPD the pooling dynamics in solid CO was resolved for the first time. For the v = 1 and v = 2 states, the emissions decay within about 1 s; while for the higher vibrational states, the signals are characterized by a fast rise followed by a multi-exponential decay on a millisecond timescale. With increasing quantum number the rise time increases from 0.2 s for v =10 to 50 s for v=26. Kinetic modelling of the experimental data provides detailed information about the rate constants involved in the vibrational energy pooling process.
366
Surfaces and InterfacesP09-09

Multi-quantum vibrational relaxation in highly vibrationally excited NO/surface scattering: Influence of the surface
Bastian C. Krüger1, Sven Meyer1, Alexander Kandratsenka2, Alec M. Wodtke1,2, Tim Schäfer1
1 Institute for Physical Chemistry, Georg-August-University of Göttingen, Tammannstr. 6, 37077 Göttingen, Germany
2 Department of Dynamics at Surfaces, Max-Planck-Institute for Biophysical Chemistry, Am Faßberg 11, 37077 Göttingen, Germany
Multi-quantum vibrational relaxation is a very efficient energy transfer process in NO/Au(111) surface scattering.[1] The most probable scattering channel for an initially highly vibrationally excited molecule (vibrational quantum number v = 16) involves the electron transfer mediated conversion of nearly 2 eV of vibrational energy into electronic excitation in the metal within a single collision. While it is precisely known how the molecule´s initial degrees of freedom play an important role during these scattering events,[2] the knowledge regarding the influence of the surface properties is limited to a comparison between an insulator (LiF, where multi-quantum vibrational relaxation is completely absent) and a metal (Au) surface.[1]
Here, we report a comparison of highly vibrationally excited NO (in this study v = 11) surface scattering between more similar systems (Au(111) and Ag(111)). For Au(111)surface scattering, we find low but nonzero survival probabilities for the initial state and identify the most probable scattering channel as the loss of 5 - 6 vibrational quanta (1.1 eV). In contrast to that, the survival probability of the incidence vibrational state scattered from Ag(111) is zero and the average vibrational energy loss (at least 8-9 quanta) is by far exceeding the observations for Au(111). The results are discussed by considering the following properties: character of the potential energy surface, work function, surface states, and surface plasmons.
[1] Y. Huang, C. T. Rettner, D. J. Auerbach, A. M. Wodtke, Science 2000, 290,111-114.[2] N. Bartels, B. C. Krüger, D. J. Auerbach, A. M. Wodtke, T. Schäfer, Angew. Chem. Int. Ed. 2014, 53, 13690-13694.
367
Surfaces and Interfaces P09-10

Energy Transfer in Collisions of highly vibrationally excited NO with Vanadium Dioxide Surfaces
Artur Meling1,2, Bastian C. Krüger1,2, Tim Schäfer1,2, Igor Rahinov3, Alec M. Wodtke1,2
1Universität Göttingen, Institut für physikalische Chemie, Göttingen, Germany2Max-Planck-Institut für biophysikalische Chemie, Göttingen, Germany
3The Open Universtity of Israel, Department of Natural Sciences, Tel Aviv, Israel
Scattering of highly vibrationally excited NO from both insulating and conducting surfaces has increased our understanding of energy exchange channels between molecules and surfaces. It turns out that non-adiabatic effects play an important role during the interaction of the molecule with metals due to coupling of vibrational degrees of freedom to electron hole pairs (EHP).[1,2] In this study, we present work on the interaction of highly vibrationally excited NO molecules with vanadium dioxide (VO2). VO2 has a Mott transition at 60°C, at which it changes its properties from insulating to conducting.[3] This allows for switching the band gap of the surface on and off by only slightly adjusting its temperature. In the future, we hope to observe corresponding temperature effects on the vibrational energy transfer in NO/VO2 surface collisions.
[1] Y. Huang, et. al., Vibrational Promotion of Electron Transfer, Science, 2000, 209,111-114.[2] B.C. Krüger, et. al., Vibrational Inelasticity of Highly Vibrationally Excited NO on Ag(111), Journal of Physical Chemistry Letters, 2016, in submission.[3] N. F. Mott, Metal-Insulator Transition, Review of Modern Physics, 1968, 40,677-683.
368
Surfaces and InterfacesP09-11

Models systems for molecular energy storage: norbornadiene on Pt(111) and anchored to an atomically-defined cobalt oxide surfaceM. Schwarz, S. Mohr, T. Xu, O. Brummel, K. Werner, K. Civale, T. Döpper, U. Bauer,
P. Bachmann, D. Besold, C. Papp, H.-P. Steinrück, A. Görling, J. Bachmann, A. Hirsch, J. Libuda; Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU),
Erlangen, GermanySolar energy can be stored in form of chemical energy in a monomolecular process by photochemical conversion of norbornadiene (NBD) to its strained and energy-richvalence isomer quadricyclane (QC).We investigated the catalytically triggered energy release from QC to NBD on Pt(111) by infrared reflection absorption spectroscopy (IRAS) under ultrahigh
vacuum (UHV) conditions. NBD adsorbs molecularly on Pt(111) binding in a 2: 1
geometry involving an agostic H-Pt bond via the methylene group. Upon adsorption at 130 K, QC forms the same intermediate as NBD, indicating facile and catalytically triggered cycloreversion on the clean Pt surface. On the other hand, adsorbed QC is stable on hydrocarbon-precovered Pt(111) surfaces and in the multilayer. Theagostic H-Pt bond is a precursor state for dehydrogenation, a reaction which is activated at a temperature of 215 K. Further dehydrogenation steps follow at higher temperatures.While the energy release from free molecular QC can be triggered either catalytically or electrochemically, an alternative strategy to better control the conversion processinvolves immobilization of the energy storage couple at an interface. Anchoringprovides additional control over the chemical processes and over the energy transfer associated with the cycloreversion reaction. To immobilize NBD, we have anchored5-( -norbornadienyl)pentanoic acid (NBDA), i.e. a derivative of NBD functionalized with an carboxylic acid group, to a well-ordered Co3O4(111) film grown on Ir(100).We follow the anchoring process in-situ by time-resolved IRAS and compare the film growth of NBDA to that of other carboxylate films, for instance of benzoic acid (BA). Both molecules bind to the surface by the formation of symmetric chelating carboxylates, which are stable up to temperatures of at least 400 K.
369
Surfaces and Interfaces P09-12

Quenching electronically and vibrationally excited carbon monoxide at metal surfaces
using velocity controlled molecular beamsRoman J. V. Wagner,1,2 Artur Meling,1,2 Tim Schäfer1,2 and Alec M. Wodtke1,2
1 Georg-August-Universität Göttingen, Institut für Physikalische Chemie, Tammannstraße 6, 37077 Göttingen, Germany
2 Max-Planck-Institut für Biophysikalische Chemie, Department of Dynamics at Surfaces, Am Fassberg 11, 37077 Göttingen, Germany
When scattering highly vibrationally excited NO(v = 18) from a low-work function surface, the molecules lose several quanta of vibrational energy during the collision.[1] The large-amplitude vibration is converted into electronic excitation in the metal leading to electron emission from the surface.[2] This non-adiabatic behavior of NO molecules on metal surfaces can be explained by a mechanism involving anelectron transfer from the metal to the NO molecule, forming a transient anionicspecies which subsequently decays by auto-detachment of an electron. Our work focuses on molecule-surface scattering events that involve electron emission. In order to compare the dynamics of CO and NO at metal surfaces, we scatter highly vibrationally excited CO(v = 17) from a low-work function surface. In addition, we investigate the de-excitation of electronically excited, metastable CO
(a31) at clean metal surfaces, a process which proceeds via a similar mechanism as
the quenching of NO(v = 18).[3] In particular, we want to determine the importance of the velocity of incident molecules on the electron emission probability, as an inverse velocity dependence of vibrationally promoted electron emission was observed for NO(v = 18) on metal surfaces.[4] For these velocity dependent measurements, a machine has been built that combines a Stark decelerator for accurate velocity control of molecular beams with a state-of-the-art beam-surface scattering chamber.
[1] Y. Huang et al., Science 2000, 290, 111.[2] J. D. White et al., Nature 2005, 433, 503.[3] F. Grätz et al., J. Chem. Phys. 2014, 141, 044712.[4] N. H. Nahler et al., Science 2008, 321, 1191.
370
Surfaces and InterfacesP09-13

Fine structure in IR spectra of high quality CO2 ultrathin filmsJochen Vogt, Uni Magdeburg, Magdeburg, Germany
The splitting of the 3 asymmetric stretch mode into a longitudinal optical (LO) and a transverse optical mode (TO) is a well-known feature of CO2 ice thin films [1]. In new experiments high quality ultrathin films were grown by exposure of a saturated monolayer CO2 p(2x1)/NaCl(100) to CO2 at 40 K. Films which are prepared in thisway are highly ordered and show additional fine structure between the TO and LO peaks. Some of these additional bands appear and disappear during film growth. In s- and p-polarized spectra a band at 2353 cm-1 is observed which shifts only weakly with increasing film thickness. An interpretation of these additional peaks is based on geometry optimizations using pair potentials and vibrational exciton theory. The band at 2353 cm-1 is assigned to the vibrational excitation of molecules in the first two layers at the film-substrate interface. The additional features with only temporary visibility are assigned to excitations of film domains of special thickness. Within the model, film domains with 5, 6, 7, and 8 layers can be identified. [1] O. Berg, R. Disselkamp, G. E. Ewing, Surf. Sci. 277 (1992), 8.
371
Surfaces and Interfaces P09-14

Direct scattering of formaldehyde from the Au(111) surfaceG. Barratt Park, Bastian Krüger, Sven Meyer, Alec Wodtke, Tim Schäfer, Georg-
August Universität, Göttingen, Germany
Molecule-surface interactions are of vital importance to chemical processes at surfaces. Experiments have demonstrated that orientation and mode-specific vibrational excitation play an important role in the chemical reactivity of polyatomic molecules at surfaces, which underscores the need for a fundamental understanding of the interaction of molecular vibrations with surfaces. A number of recent studies have demonstrated signatures for qualitatively different mechanisms of vibrational energy transfer in direct scattering of diatomic molecules from metal surfaces. In particular, the dependence of vibrational excitation on surface temperature and molecular kinetic energy can distinguish electronically non-adiabatic electron hole pair-vibration (eHP-V) coupling from mechanical kinetic energy-vibration (T-V) coupling. In direct scattering of formaldehyde from Au(111), these two mechanisms may occur simultaneously and mode-selectively. We will describe a new experimental set-up, in which the rovibrational state distribution of direct scattering products of formaldehyde from the Au(111) surface are detected. Initial results will be discussed.
372
Surfaces and InterfacesP09-15

Time-resolved IR spectroscopy during interfacial electrochemical reactions of organic corrosion inhibitors
Ying-Hsuan Chen and Andreas Erbe,Department of Interface Chemistry and Surface Engineering, Max-Planck-Institut für
Eisenforschung GmbH, Düsseldorf, Germany Preventing metals from corrosion is an important issue. Many organic molecules serve as corrosion inhibitors by forming a passivating layer on metal.1 It was also shown that organic molecules containing aromatic heterocyclic structures, such asbenzotriazole and mercaptobenzotriazole, are more effective in inhibition.2 Despite the importance in industry, it remains unclear why certain substances protect better than others. This work studies electron transfer reactions and especially degradation mechanisms by electrochemical processes of corrosion inhibitors. By a combination of electrochemical studies with operando optical spectroscopy, chemical transformations at electrode interfaces should be investigated. This contribution shows mainly initial results from infrared spectroelectrochemical experiments, using the cell design shown in Fig. 1. Final goal is to engineer corrosion inhibitors based on their reactivity, their tendency to adsorb on the metal surface and their tendency to passivate a surface.
Fig. 1: Principle setup for internal reflection IR spectroscopy used to study corrosion inhibitor electrochemistry.
Reference1. Finšgar, Milošev Corrosion Science, 2010, 52, 2737-2749.2. Finšgar, Jackson Corrosion Science, 2014, 86, 17-41.
373
Surfaces and Interfaces P09-16

In-situ IRAS Studies of Tetraphenylporphyrins on Co3O4(111)/Ir(100)S. Mohr1, K.Werner1, M. Schwarz1, M. Amende1, T. Döpper2
, A. Görling2, J. Libuda1
1Physikalische Chemie II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany2Theoretische Chemie, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
Thin layers of functional organic molecules, such as porphyrins, on oxide surfaces offer potential applications in solar cells or molecular electronics. While the growth and structure of such organic films are subject to many studies, little is known about the processes of molecule-oxide bond formation at the atomic scale. To obtain a deeper understanding of the chemical bonding of tetraphenylporphyrins to oxides we apply infrared reflection absorption spectroscopy (IRAS) under UHV conditions. Here, IRAS provides both, information on the molecular orientation and on the molecular interactions of surface-bound species [1,2].As a model oxide surface, we employ a well-defined Co3O4(111) film grown on a Ir(100) single crystal. Thin films of about 8 nm thickness were prepared by evaporationof Co onto Ir(100) in O2 atmosphere. On this surface we explored the interactions of two free base porphyrins, namely tetraphenylporphyrin (2HTPP) and its mono-carboxylated derivative mono-para-carboxyphenyltriphenylporphyrin (MCTPP).All growth processes were monitored in-situ by time-resolved IRAS. By comparison to calculated spectra (DFT) and transmission spectra, we are able to extract the molecular orientation and identify specific surface interactions. We show that 2HTPP adsorbs molecularly in a flat-lying orientation at 300 K.Molecular adsorption is also observed for MCTPP at 300 K. At higher temperatures the molecule binds by formation of a chelating bidentate carboxylate. Previously we had studied the bonding of simpler carboxylic acids to the same oxide film [3].During film growth the bonding geometry of MCTPP changes. This could be monitored by time-resolved IRAS (TR-IRAS). A procedure is proposed for the calculation of the tilt angle at each point in time during film growth. At low coverage the molecules are found to adopt flat-lying geometry (tilt angle =25±15°). At higher coverage the tilt angle is increasing and the porphyrins are standing nearly upright ( =77±5°). Above the monolayer region a disordered multilayer is formed ( =61±9°).
[1] S. Mohr et al.; Langmuir 31, 7806 (2015)[2] S. Mohr et al.; Surface Science DOI:10.1016/j.susc.2015.06.027[3] T. Xu et al.; J. Phys. Chem. C 119, 26968 (2015)
374
Surfaces and InterfacesP09-17

The vanadium oxide monolayer on CeO2(111) studied by density functional theory
Christopher Penschke, Joachim Paier, and Joachim SauerHumboldt-Universität zu Berlin, Unter den Linden 6, 10099 Berlin, Germany
A frequently studied model reaction for C-H bond activation is the oxidative dehydrogenation of methanol to formaldehyde at VOx/CeO2(111).[1,2] The activity for this reaction decreases with increasing vanadia loading up to monolayer coverage.[2-4] For small vanadium oxide species, reactivity descriptors obtained by virtue of density functional calculations under periodic boundary conditions showed the same trend.[5] Later mechanistic studies offered details about the support effect,proving that density functional theory is a valuable tool to gain insight into this catalyst system.[6]However, atomic level details on reactivity for the supported 2D vanadia monolayer is still missing. The average number of vanadium atoms per surface area in the monolayer system varies in the available experimental studies. In addition, differentcompositions, i.e. V:O ratios, are possible.Using a combination of educated guess and genetic algorithm,[7] we attempt to determine the global minimum energy structure for different compositions. The most stable VO2 monolayer resembles the structure of bulk V2O5. The evaluation of reactivity descriptors suggests that the monolayer is less reactive than the small ceria-supported vanadium oxide clusters, in agreement with experiment.
[1] B. M. Weckhuysen, D. E. Keller, Catal. Today 78, 25, (2003).[2] I. E. Wachs, Catal. Today 100, 79, (2005).[3] G. S. Wong, M. R. Concepcion, J. M. Vohs, J. Phys. Chem. B 106, 6451 (2002).[4] M. V. Ganduglia-Pirovano et al., J. Am. Chem. Soc. 132, 2345 (2010).[5] C. Penschke, J. Paier, J. Sauer, J. Phys. Chem. C 117, 5274 (2013).[6] T. Kropp, J. Paier, J. Sauer, J. Am. Chem. Soc. 136, 14616 (2014).[7] R. W odarczyk et al., J. Phys. Chem. C 115, 6764 (2011).
375
Surfaces and Interfaces P09-18

Direct growth of patterned graphene
Nils-Eike Weber1, Stefan Wundrack2, Rainer Stosch2, Andrey Turchanin3,4
1Faculty of Physics, University of Bielefeld, 33615 Bielefeld, Germany2Physikalisch-Technische Bundesanstalt, 38116 Braunschweig, Germany
3Institute of Physical Chemistry, Friedrich Schiller University Jena, 07743 Jena, Germany4Jena Center for Soft Matter (JCSM), 07743 Jena, Germany
We demonstrate direct growth of single-layer graphene patterns employing electron irradiation of aromatic self-assembled monolayers (SAM) and subsequent annealing. The process consists of the following technological steps: (i) formation of an aromatic SAM on a Cu substrate; (ii) electron-beam-irradiation of the SAM resulting in the locally crosslinked SAM areas; (iii) conversion of these areas into single-layer graphene via annealing. In this way graphene patterns of various architectures are directly defined in the SAM by electron beam lithography reducing several manufacturing steps, which are typically applied for the patterning of two-dimensional sheets including graphene (baking and developing electron-beam resist, plasma etching, resist stripping). The formed micro- and nanostructures were characterized by Raman spectroscopy, scanning electron, scanning Auger, optical and atomic force microscopy. We demonstrate their successful transfer from the original copper foils onto oxidized silicon wafers, where they can be directly employed for applications.
Weber N.-E., Wundrack S., Stosch R., Turchanin A. Small (2015) DOI: 10.1002/smll201502931.
Figure: Schematic representation of the graphene growth (left) and SEM images of the grown patterns
376
Surfaces and InterfacesP09-19

Scattering HCl from Au(111):Vibrational Excitation Probabilities and Dissociative Adsorption
Jan Gewekea,b, Pranav Shirhattib,c, Christoph Steinsiekb,c, Christof Bartelsd, I. Rahinove, Alec M. Wodtkeb,c,f
a École Polytechnique Fédérale de Lausanne, Lausanne, Switzerlandb Max Planck Institute for Biophysical Chemistry, Göttingen, Germany
c University of Göttingen, Göttingen, Germanyd University of Freiburg, Freiburg, Germanye Open University of Israel, Tel Aviv, Israel
f International Center for Advanced Studies of Energy Conversion, Göttingen, Germany
Gas phase dynamics as well as dynamics at surfaces deal with the most elementary steps of chemical reactions like energy transfer between molecular and surface degrees of freedom, electron transfer, and the making and breaking of chemical bonds. We report absolute vibrational excitation probabilities and dissociative adsorption of HCl molecules encountering a Au(111) surface.Employing molecular beam techniques in ultrahigh vacuum we are able to study the scattering of gas phase molecules like HCl under well-defined, collision free conditions. A narrow-linewidth, high-intensity pulsed IR laser in combination with UV laser based Resonance Enhanced Multiphoton Ionization (REMPI) enables us to quantum state selectively prepare and detect molecules before and after scattering from the single crystal surface, thus giving insight into the flow of energy between the aforementioned degrees of freedom.HCl exhibits vibrational excitation upon scattering, depending on incident translational and vibrational energy as well as surface temperature (TS). Arrhenius-like behaviour at elevated TS suggests electronically non-adiabatic processes involving surface electrons. At higher incident translational energies (> 1 eV), HCl undergoes dissociation as proven by detection of Cl atoms using Auger Electron Spectroscopy (AES). However, measured energetic dissociation barriers and vibrational efficacy appear to be much higher than theoretically predicted.
377
Surfaces and Interfaces P09-20

Electrostatic interactions and ligand effects at ionic-liquid modified interfaces: Co-adsorption of [EMIM][OTf] and CO on Pd(111)
T. Bauer, S. Mehl, O. Brummel, K. Pohako-Esko, P. Wasserscheid, J, Libuda, FAU Erlangen-Nürnberg, Germany
A well-known concept in catalysis relies on the modification of heterogeneous catalysts by thin films of ionic liquids (ILs) so-called solid catalysts with IL layer (SCILL). It has been shown in several cases that such coatings can have beneficial effects on the catalystʼs activity and stability.[1]
To explore the molecular interactions at the metal/IL interface, we investigate the co-adsorption of the room-temperature IL [EMIM][OTf] (1-ethyl-3-methylimidazolium trifluoromethanesulfonate) and CO on Pd(111).[2]
Deposition of approximately one monolayer (ML) [EMIM][OTf] onto a Pd(111) surface fully pre-saturated by CO leads to the formation of a dense and well-defined coadsorbate layer. Temperature programmed desorption (TPD) experiments showed that the CO adsorbate structure is stable and preserved upon IL-deposition. The [OTf]- anions of the IL adsorb specifically via the SO3 group to the Pd(111) surface in the first ML, as shown in previous studies,[3] and are located in between the CO molecules at the interface. Time-resolved infrared reflection adsorption spectroscopy (TR-IRAS) spectra recorded upon IL deposition show a strong red-shift of the CO stretching frequency in the order of 50 cm-1. This effect may in part be due to the electronic ligand effect in accordance with the Blyholder model but this effect is very short ranged. The second and more prominent contribution is due to the vibrational Stark effect.[4] The latter is caused by the interaction of the electric field generated by the IL with the dipole moment of the CO molecule and is also observed in electrochemical studies.
[1] H. P. Steinrück, J. Libuda, P. Wasserscheid, T. Cremer, C. Kolbeck, M. Laurin, F. Maier, M. Sobota, P. S. Schulz, M. Stark, Adv. Mater. 2011, 23, 2571-2587.
[2] T. Bauer, S. Mehl, O. Brummel, K. Pohako-Esko, P. Wasserscheid, J. Libuda, submitted.
[3] S. Schernich, D. Kostyshyn, V. Wagner, N. Taccardi, M. Laurin, P. Wasserscheid, J. Libuda, J. Phys. Chem. C. 2014, 118, 3188-3193.
[4] O. Brummel, F. Faisal, T. Bauer, K. Pohako-Esko, P. Wasserscheid, J. Libuda, Electrochimica Acta, 2015, in press.
378
Surfaces and InterfacesP09-21

Ab initio molecular dynamics simulations of CO/NO scattering from
Au(111)Jan Altschäffel, Alexander Kandratsenka, and Alec M. Wodtke
Max-Planck-Institut für biophysikalische Chemie, Göttingen, GermanyInstitut für physikalische Chemie, Georg-August-Universität Göttingen, Germany
Recent advances in the experimental studies of the diatomic molecule scattering from the metal surfaces provided the detailed information as to the final translational, rotational and vibrational energy distributions of the scattered molecules [1,2]. These results suggest the necessity to account for the nonadiabatic effects when considering the diatomic vibrational energy (de-)excitation. From the other hand, the translational energy losses as well as the final rotational energy spectra appear to be explainable on the level of an adiabatic model. To justify this, we carried out ab initiomolecular dynamics simulations for scattering CO and NO molecules from the gold surface for various incidence conditions. The translational energy inelasticity andvibrational energy loss (gain) channels are studied in detail. The dependence of the resulting energy distributions on the choice of DFT-GGA functional is investigated and comparison to the available experimental data is performed.
References[1] R. Cooper et al., Angew. Chemie Int. Edt. 51, 4954, 2012.[2] K. Golibrzuch et al., J. Chem. Phys. 140, 044701, 2014.
379
Surfaces and Interfaces P09-22

Monitoring Oxide Layer Growth on Manganese Electrodes, by in-situ Spectroscopic Ellipsometry and Raman Spectroscopy
Martin Rabe, Adnan Sarfraz, and Andreas Erbe. Interface Spectroscopy Group, Department of Interface Chemistry and Surface Engineering, Max-Planck-Institut für Eisenforschung GmbH, Max-Planck-Str. 1,
40237 Düsseldorf.
Manganese compounds are promising materials for catalysts of the oxygen evolution reaction (OER) in water electrolysers. We aim to study the oxides forming on the interfaces of Manganese containing electrode during the OER. In the present study metallic Mn electrodes are prepared by physical vapor deposition. The native oxide layer is reduced electrochemically under basic conditions, to yield a metallic interface as a reference state. Subsequent changes in the electrode potential are probed by spectroscopic ellipsometry and Raman spectroscopy. These techniques allow in situmonitoring of the changes in layer thickness and electronic structure of the oxides on the electrode interfaces upon polarization and in the course of electrochemical reactions. The spectroscopic data is complemented with ex-situ X-ray photoelectron spectroscopy measurements. Cyclic voltammetry and chronoamperometryexperiments at transpassive electrode potentials are performed. These experiments give a detailed insight on the electrode structure and will help to reveal the active species during the OER on the electrodes and assist the design of novel OER catalysts.
380
Surfaces and InterfacesP09-23

H at Au(111): An Accurate Full-dimensional Potential Energy Surface
Svenja M. Janke1,2, Alexander Kandratsenka1,2, Yvonne Dorenkamp1,2, Hongyan Jiang1,2, Oliver Bünermann1,2,3, Daniel J. Auerbach1,2, Alec M. Wodtke1,2,3
1Institute for Physical Chemistry, Göttingen University, 2Max Planck Institute for Biophysical Chemistry, Göttingen/Germany, 3International Center for Advanced
Studies of Energy Conversion, Göttingen University, Göttingen/Germany
We have constructed a full-dimensional potential energy surface (PES) for H-atoms interacting with Au(111) by fitting DFT energies to Nørskovʼs Effective Medium Theory[1] expression. Our procedure provides an accurate treatment for displacements of Au atoms and adiabatic molecular dynamics (MD) simulations on the PES reproduce the energy loss behavior obtained from ab initio molecular dynamics scattering calculations[2]. The embedding electron density can be easily extracted from the EMT and allows us to simulate nonadiabatic energy loss due to electron hole pair excitation in a self-consistent manner by means of local density friction approximation[3]. Inclusion of the nonadiabatic effects into MD simulations allows us to accurately reproduce the large energy loss and the isotope effect observed experimentally in a collision of H and D with a Au(111) surface[4]. Using the same PES, we also treated diffusion of H in Au.
[1] K. W. Jacobsen, Surface Science, 366, 394, 1996.[2] G.-J. Kroes, Journal of Chemical Physics, 141, 054705, 2014.[2] S. M. Janke, Journal of Chemical Physics, 143, 124708, 2015.[3] O. Bünermann, Science, 350, 1346, 2015.
381
Surfaces and Interfaces P09-24

The effect of solvent and interfacial field affect on switching of a nitrospiropyran self-assembled monolayer at the solid/liquid
interface T. Garling, Y. Tong, R. K. Campen, Fritz-Haber-Institut, Berlin/Germany;
On irradiation with UV-light nitrospiropyran undergoes a reversible ring-opening reaction to merocyanine. Differences in electronic structure between the molecules suggest numerous possible applications of nitrospiropyrans in devices. These devices require attaching nitrospiropyran to surfaces. While the switching mechanism in solution is well understood, how attachment to surfaces, particularly in the presence of solvent or interfacial fields, alter this mechanism is not. To address this question we attached nitrospiropyrans, functionalized as below, to gold surfaces and investigated their switching, using the symmetric NO2 and (CH2)-N stretches as local probes, as a function of surface potential and solution polarity. We find that both strongly affect the photostationary state under UV irradiation: polar solvents and negative potentials favor merocyanine while positive potentials have minimal effect. While all switching is slowed relative to solution, we observe increasing solution polarity speeds up, while shifting to either negative or positive surface potentials slows, ring-opening. This potential dependence suggests switching in monolayers proceeds by rapid ring-opening followed by slow intramolecular rearrangement whose extent is proportional to surface potential.
382
Surfaces and InterfacesP09-25

Associative Desorption of Hydrogen from Metal SurfacesSven Kaufmann a,b, Quan Shuai a, Dirk Schwarzer a, Daniel J. Auerbach a,b, Alec M.
Wodtke a,b
a)Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germanyb)Institute for Physical Chemistry, Georg-August-University, Göttingen, Germany
The interaction of hydrogen molecules with metal surfaces is one of the most fundamental reactions in surface chemistry. Dissociative adsorption is often the first step in heterogeneous catalysis and tightly related to its reverse process, the associative desorption. In this experiment we studied the product energy distribution of nascent hydrogen molecules desorbing from different metal surfaces after recombination of the atoms by measuring their flight time through a field-free drift region. Using the principle of detailed balance allows us to determine the translational threshold of dissociative adsorption, E0, for each ro-vibrational state.Our results show that E0 only weakly depends on the rotational state, whereas vibrational excitation results in a strong reduction of the threshold. We present data for H2, HD and D2 desorbing from Cu(111), Cu(211) and Au(111) surfaces and compare these with results from high-level ab initio quantum chemical calculations.
383
Surfaces and Interfaces P09-26

CO adsorption on Pt3Ti(111) studied by IRRAS and XPS Ludger Schöttnera, Marco Moorsb, Fabian Bebenseea, Alexei Nefedova, Yuemin
Wanga, and Christof WöllaaInstitute of Functional Interfaces, Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, GermanybPeter Grünberg Institute, Forschungszentrum Jülich, Wilhelm-Johnen-Straße, 52425 Jülich, Germany
The intermetallic alloy Pt3Ti is a promising representative for titania (TiO2) based catalysts. In this work we present a surface science study of CO adsorption on pure and oxidized Pt3Ti(111) single crystal surfaces by employing infrared reflection absorption spectroscopy (IRRAS) and X-ray photoelectron spectroscopy (XPS). After CO adsorption on the pure Pt3Ti(111) surface at 110 K and room temperature adominant peak at about 2080 cm-1 was observed, which is assigned to CO adsorbed terminally at the surface Pt sites. With increasing CO coverage, the frequency of the CO band exhibits a blue-shift to 2102 cm-1 at saturation (full monolayer), in excellent agreement with the results of CO adsorption on Pt(111). The coverage-dependent frequency shift is attributed to the lateral adsorbate-adsorbate interaction including dipole-dipole coupling and chemical shift. In addition to the on-top CO as majority species, the IR spectra show a weak peak at 1865 cm-1 originating from the CO bound to bridge Pt sites. The adsorbed CO species was further characterized by XPS at low temperatures. Our IR and XPS results did not provide evidence for the decomposition of CO at surface Ti sites. Finally, the oxidation of Pt3Ti(111) with oxygen at elevated temperatures was monitored by IRRAS using CO as a probe molecule.
384
Surfaces and InterfacesP09-27

Reactivity of CO on Sulfur-passivated Graphene-supported Palladium Nanocluster ArraysFabian Düll, Florian Späth, Udo Bauer, Philipp Bachmann, Hans-Peter Steinrück,Christian Papp, Lehrstuhl für Physikalische Chemie II, Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen
Heterogeneous catalysts are involved in most processes of chemical industry. Poisoning with sulfur is a main cause of their deactivation. Graphene-supported metal nanocluster arrays are a good model system for studying the changes in reactivity caused by poisoning. Palladium nanocluster arrays were grown on Moiré-patterned graphene on Rh(111), which forms due to the lattice mismatch of the two materials. SO2 was adsorbed on the Pd nanoclusters and subsequently heated to 500 K, which leads to the formation of sulfur on the surface. Subsequently, we studied the influence of sulfur on the adsorption and desorption behavior of CO atdifferent sulfur coverages. All experiments were conducted with in situ high resolution X-ray photoelectron spectroscopy at the synchrotron light source BESSY II. Sulfur reduces the overall amount of adsorbed CO by blocking of adsorption sites. Thereby, specific sites are selectively blocked, while on others strong adsorption is still possible. Interestingly, the CO desorption behavior is not influenced by poisoning of the Pd-particles with sulfur.We acknowledge support from SFB 953 ʻSynthetic Carbon Allotropesʼ, the Cluster of Excellence ʻEngineering of Advanced Materialsʼ, and HZB for the allocation of synchrotron beamtime.
385
Surfaces and Interfaces P09-28

A New Thin Film Fabrication Technique by Interconnecting Nanoparticles via Tetrazole-Complexes
André Wolf*, Christin Rengers*, Sergei Voitekhovich†, Vladimir Lesnyak*, Nikolai Gaponik*, Alexander Eychmüller*
*Physical Chemistry, TU Dresden, Bergstr. 66b, 01062 Dresden, Germany
†Research Institute for Physical Chemistry Problems, Belarusian State University, Lenigradskaya Str. 14, 220030 Minsk, Belarus
Colloidal nanoparticles with their wide range of size-tunable properties show great prospects for modern applications in catalysis, optoelectronics, and photonics. However, assembly of nanoparticles into multifunctional macroscopic solids is often demanded for these applications but still challenging.1 Many different approaches have been developed such as embedding in matrices, formation of ordered mesocrystals and non-ordered gels as well as Layer-by-Layer deposition.2,3 Latter was shown in the early 1990s and is one of the approaches showing a facile way to scale up.4,5 In this present work, the method is extended by transferring the concept of advanced gel formation into a layer by layer process.6 The characterization of these two dimensional micro porous assemblies was focused on their optical properties, energy relations between their components, and morphology. The new process reduces the amount of polyelectrolytes to a minimum, leads to shorter distances between the layers, and opens a wide range of nanomaterials to work with. These architectures are promising for the construction of transparent fluorescent solar concentrators and for sensing devices.
(1) Gaponik, N. J. Mater. Chem. 2010, 20, 5174–5181.
(2) Lesnyak, V.; Gaponik, N.; Eychmüller, A. Chem. Soc. Rev. 2013, 42, 2905–29.
(3) Bahrig, L.; Hickey, S. G.; Eychmüller, A. CrystEngComm 2014, 16, 9408–9424.
(4) Kotov, N. A.; Meldrum, F. C.; Fendler, J. H.; Tombacz, E.; Dekany, I. Langmuir 1994, 10, 3797–3804.
(5) Dabbousi, B. O.; Rodriguez-Viejo, J.; Mikulec, F. V; Heine, J. R.; Mattoussi, H.; Ober, R.; Jensen, K. F.; Bawendi, M. G. J. Phys. Chem. B 1997, 101, 9463–9475.
(6) Wolf, A.; Lesnyak, V.; Gaponik, N.; Eychmüller, A. J. Phys. Chem. Lett. 2012, 3, 2188–2193.
386
Surfaces and InterfacesP09-29

Self-Assembling Amphiphilic Fluorophores: In-Situ Monitoring and Photonic Characterization
Maximilian Hupfer1,3, Felix Herrmann1,3, Stefan Fischer1, Martin Kaufmann2, Rainer Beckert2, Benjamin Dietzek1,3, Martin Presselt1,3,*
1 Institute of Physical Chemistry, Friedrich Schiller University Jena, Lessingstr. 8, 07743 Jena, Germany,
2 Institute of Organic and Macromolecular Chemistry, Friedrich Schiller University Jena, Humboldtstr. 10, 07743 Jena, Germany,
3 Leibniz Institute of Photonic Technology (IPHT), Albert-Einstein-Str. 9, 07745 Jena, Germany
* corresponding author: [email protected]
Due to their high cost-efficiency and the possibility to produce large-scale systems, organic light emitting diodes (OLEDs) are intensively investigated for display and lightning applications. Extensive research has been devoted to the synthesis of new materials, the implementation of interlayers and the development of device-structures to improve the long-term-stability and efficiency of the device. The optimization of morphology and control of the supra-molecular geometry plays an important role to establish new active layers with defined directional photonic characteristics and is generally advantageous for derivation of morphology-photonic properties and morphology-electronic property relationships in organic electronics.
Here we introduce amphiphilicity to dyes to enable self-assembling of the dyes[1] and, additionally, to control the morphology of active layers produced from these dyes. If certain preconditions, like a balanced spatial demand of hydrophilic and lipophilic moieties within the dye, are fulfilled, flat mono- and bilayers with well-defined and stable morphologies and interfaces can be fabricated. As chromophores we use 4-hydroxy-1,3-thiazoles as they are photochemical stable and exhibit a fluorescence quantum yield up to 95%.[2-4] To study self-assembly of the dyes we utilize in-situ absorption spectroscopy, particularly the highly surface-sensitive photo thermal deflection spectroscopy, on layers of amphiphilic thiazoles. The influence of the morphology in thin solid films made from the thiazoles on their photonic properties is discussed in detail.
References:
[1] M. Hupfer, F. Herrmann, S. Fischer, M. Kaufmann, R. Beckert, B. Dietzek, M. Presselt, to be submitted (2015).
[2] R. Menzel, D. Ogermann, S. Kupfer, D. Weiß, H. Görls, K. Kleinermanns, L. González, R. Beckert, Dyes Pigm. 94 (2012) 512.
[3] E. Täuscher, D. Weiß, R. Beckert, H. Görls, Synthesis 2010 (2010) 1603. [4] L.K. Calderón-Ortiz, E. Täuscher, E. Leite Bastos, H. Görls, D. Weiß, R. Beckert, Eur. J. Org.
Chem. 2012 (2012) 2535.
Acknowledgements:
We thank the Deutsche Forschungsgemeinschaft (DFG Grant No. PR 1415/2-1) for the financial support.
387
Surfaces and Interfaces P09-30

Photo-manipulation of the surface tension anisotropy at a liquid-
crystal/ITO-glass interfaceH. Nadasi, A. Eremin, R. Stannarius, Otto-von-Guericke-Universität,
Magdeburg/Germany
We report on optical manipulation and direct measurements of the surface energy anisotropy
(anchoring energy) at a glass-nematic liquid crystal interface containing a photo-active azo-
dendrimeric surfactant using low-power UV irradiation. The photoisomerisation at the surface
layer drives an anchoring transition from the homogeneous to the planar sate of the liquid
crystal. The anchoring energy is measured using the Frederiksz transition as a function of the
intensities of the UV and VIS light and is compared with a theoretical model. We also
demonstrate optical manipulation of the nematic director field around microshperes and rods
dispersed in the liquid crystal. In case of rod-shaped particles, the photo-driven change of the
anchoring energy results in a reversible macroscopic rotation of the particles.
388
Surfaces and InterfacesP09-31

Molecular Recognition in the Organic Corona around Nanoparticles andApplications to Biosensing
Sebastian Kruss, Universität Göttingen, Institut für Physikalische Chemie, Göttingen/Deutschland
Nanomaterials are versatile building blocks for new applications in bioanalytical chemistry. Typically they are functionalized with organic molecules and this organic phase around them is governing how they interact with other molecules (molecular recognition). In the case of nanobiosensors a specific interaction is desired that also changes e.g. optical properties of the particle. So far it is not understood how molecular recognition in the organic corona phase and signal transduction through the corona phase to nanomaterials works. Our goal is to develop optical nanosensors for interesting biomolecules such as neurotransmitters and to understand the underlying physicochemical processes. For this purpose we used semiconducting single-walled carbon nanotubes (SWCNTs), which fluoresce in the near Infrared (nIR). The SWCNTs were non-covalently functionalized with polymers such as DNA in order to solubilize them in aqueous solution. This organic polymer phase (corona) around the SWCNTs governs how they interact with molecules in their environment and how these interactions affect optoelectronic properties. We systematically changed the polymer phase (e.g. DNA sequence) around SWCNTs and quantified how the structure of small molecules such as the neurotransmitter dopamine affects the fluorescence of SWCNTs. We found that the nucleotide/sequence determines if a SWCNT/DNA-complex responds to dopamine. Additionally, small changes in the structure of the analyte strongly affected the magnitude of the fluorescence response. By using molecular dynamics simulations and a one dimensional exciton diffusion model we furthermore show that dopamine interacts and displaces phosphate groups of the DNA-backbone, which is correlated to the removal of exciton quenching sites. In summary, we provide insights into conformational dynamics offunctionalized nanoparticles during molecular recognition and how this affects the nanoparticleʼs optical properties.
389
Biophysical Chemistry P10-01

Development of a label-free fiber optical biosensor based on etched fiber-Bragg-gratings
Sven Schulze, Carsten HilleUniversity of Potsdam, Institute of Chemistry, Physical Chemistry / Applied Laser Sensing in Complex Biosystems (ALSComBi), 14476 Potsdam-Golm, Germany
Optical biosensors are analytical devices that integrate a biological recognition element with a physical transducer, whereby the interaction between a target molecule and the recognition element is translated into a measurable optical signal.They are a powerful alternative to conventional analytical techniques due to their high specificity, sensitivity, multiplexing capability, small size and cost effectiveness. Thus, these devices offer manifold application fields such as biomedical diagnostics, food safety, drug discovery or environmental monitoring and process control. For fiber Bragg grating (FBG)-based optical biosensors, the sensing capability is directly encoded into the reflected Bragg wavelength. Interaction of the target molecule nearthe fiber surface changes the surrounding refractive index (SRI), resulting in an easily measurable shift in the reflected Bragg wavelength.Here, we present results of the development of an optical biosensor based on etched FBGs. Gratings with Bragg wavelengths in a spectral range of 1520-1570 nm could be created by inscribing periods of refractive index modulations into the core of a single-mode fiber using a fs-pulsed Ti:Sa laser system working at = 800 nm. While FBG-based fibers can intrinsically provide access to physical parameters such as temperature and strain, the fibers have to be chemically modified to create FBG-sensitivity to SRI changes. Thus, the fibers were etched with hydrofluoric acid, at which the influence of acid concentration and etching time on the Bragg wavelength shift was studied. Therefore, differently etched fibers could be analyzed for their sensitivity to changes in SRI by immersing them into well-known test solutionsexhibiting refractive indices from 1.33 to 1.48. Once a better understanding of thepreparation process of etched FBG-fibers is gained, their application for biochemical sensing is investigated by using fibers functionalized with antibodies or aptamers as recognition elements.
390
Biophysical ChemistryP10-03

High-resolution femtosecond FT2D-IR spectroelectrochemistry
Julian M. Schmidt-Engler, Youssef El Khoury, Luuk J. G. W. van Wilderen and Jens Bredenbeck
Institute of Biophysics, Johann Wolfgang Goethe-University, Max von Laue Straße 1, 60438 Frankfurt am Main, Germany
Recently we introduced ultrafast 2D-IR spectroelectrochemistry in the frequency domain and demonstrated its capabilities on a small inorganic molecule.1 This approach can be used to infer direct structural information of a system under controlled redox conditions. However, the achieved spectral resolution along the pump axis and time resolution were limited by the width (10-15 cm-1) and duration (approx. 1 ps) of the Lorentzian pump pulse. These methodologial limitations hinder insights into systems with congested spectra or very fast vibrational dynamics.2
To gain access to the structure and dynamics of complex molecules both key figures have been substantially improved by using a pump pulse interferometer and corresponding Fourier transform methods.3 The highest possible spectral resolution and a 10-fold improvement in the time resolution, limited by the pulse duration of the laser fundamental, have been achieved, enabling access to redox-dependent dynamics of molecules that suffer from spectral congestion and/or exhibit shortvibrational lifetimes. The developed spectroelectrochemical cell offers temperature control accompanied by adjustable redox conditions and high durability. Due to the reflection geometry thin samples are possible, allowing for rapid redox equilibration.1
Pointing out the technique's benefits in combination with electrochemistry we presenthigh-resolution redox-coupled FT2D-IR spectra of a selection of molecules that rangefrom small and simple to complex.
References1 El Khoury Y., van Wilderen LJGW, Vogt T., Winter E. and Bredenbeck J.,
Rev. Sci. Instrum. 2015, 86 (8), 083102-62 El Khoury Y., van Wilderen LJGW and Bredenbeck J., J. Chem. Phys. 2015,
142(21), 212416-73 Helbing J. and Hamm P., JOSA B 2011, 28, 171-178
391
Biophysical Chemistry P10-04

Noninvasive, label-free analysis of stimulus-induced cellular activity using Raman spectroscopic imaging
Andreas Post, Carsten HilleUniversity of Potsdam, Institute of Chemistry, Physical Chemistry / Applied Laser Sensing in Complex Biosystems (ALSComBi), 14476 Potsdam-Golm, Germany
In living cells, there are always a plethora of processes taking place at the same time, but for a comprehensive understanding of intracellular homeostasis we needadequate analytic methods. Among other, Raman spectroscopic imaging has emerged as a powerful tool for the analysis of biological samples. Intuitively, it seems to be unsuitable for live cell imaging, since Raman scattering efficiency is quite low leading to long acquisition times and high laser intensities. However, it can easily provide detailed information on the chemical composition of cells under physiological conditions in a noninvasive and label-free modality.Here, we present results of Raman spectroscopic imaging of living cells from insect salivary gland tissue to study metabolic activity. Salivary gland lobes are mounted in a perfusion chamber and excitation at 532 nm provides a good compromise between Raman signal and cellular autofluorescence. At first, Raman spectra extracted from acquired Raman images of different gland cell types and of cellular compartmentsare compared. Next, gland tissue is stimulated with the biogenic amines dopamine or serotonin to induce saliva production. Thus, higher metabolic activity can be correlated with changes in the Raman spectra regarding the occurrence of Raman peaks and/or their relative intensities. Once the stimulus-induced relevant differences in the Raman spectra are unraveled, cellular metabolic pathways can be investigated in pharmacological studies.
392
Biophysical ChemistryP10-05

Liquid-Liquid Phase Separation in Therapeutic Monoclonal Antibody Systems
Reiche, K., Blume, A., and Garidel, P.Martin-Luther-University Halle/Wittenberg
Institute of Chemistry - Physical Chemistry, 06120-HalleSaale, GermanyBoehringer Ingelheim Pharma GmbH & Co. KG, Protein Science, 88400-Biberach an
der Riss, Germany
Monoclonal antibodies are the main driving protein class in the field of new biological entities. The therapeutic doses applied to a patient are often high, reaching the gram level with protein concentrations up to 200 mg/ml. A challenge with these high protein concentrations is the colloidal stability, which i.e. can induce protein precipitation (solid-liquid) as well as liquid-liquid phase separation.The objective of this study is focussed on understanding and elucidation of causes that might induce liquid-liquid phase separation in concentrated protein solutions.Causes that have to be considered are for example, counter-ion-protein-complexformation, protein hydration, protein-protein interactions, charge neutralisation, specific excipient binding, etc. [1].Our working hypothesis is that protein ion interactions induce a neutralisation of protein net charge with concomitant reduction of protein hydration. [2] As a consequence a macroscopic liquid-liquid phase separation is induced.The study will describe solution conditions prone to favour phase separation. Furthermore, model(s) to describe the phase diagram are presented and discussed.
[1] M.J.Voorn, Recueil, 75 (1956), 317 – 330, Complex coacervation. I. General theoretical considerations.
[2] C.L. Cooper, P.L. Dubin, A.B.Kayitmazer, S.Turksen, Current Opinion in Colloid & Interface Science 10 (2005) 52 – 78, Polyelectrolyte-protein complexes
393
Biophysical Chemistry P10-06

Validation and comparison of biomolecular force fields regarding structural properties and binding affinities of cyclodextrins
Julia Gebhardt, Niels Hansen, Institute of Thermodynamics and Thermal Process Engineering, University of Stuttgart, D-70569 Stuttgart, Germany
Cyclodextrins, CyDs, are macrocyclic oligosugars most commonly composed of 6, 7, - - -CyD. Other, usually smaller,
molecules (called guests) can enter their cavity forming inclusion complexes with these hosts. As a result of this complexation CyDs are widely used in many industrial products, technologies and analytical methods. CyDs have also been extensively studied by means of molecular dynamics simulations, concentrating on structural
-CyD is studied with different biomolecular force fields in its pure form and in complexes with linear alcohols in aqueous solution. The evaluation is based on structural properties from crystallographic X-ray and solution NMR data as well as on experimental standard binding free enthalpies. Relative binding free enthalpies for pairs of alcohols differing by two carbon atoms were calculated from twin-system enveloping distribution sampling simulations. The standard binding free enthalpy of 1-hexanol was calculated via the double decoupling method. Based on this analysis recommendations of biomolecular force fields applicable for cyclodextrin are provided.
394
Biophysical ChemistryP10-07

Signal Mechanism of Integrin V 3Martin Kulke, Walter Langel, Universität Greifswald, Greifswald/Germany
The membrane protein integrin plays a major part in the adhesion process of cells. It is a heterodimer consisting of a highly structured bend ectodomain, a helical transmembrane domain and a cytoplastic random coil domain. Integrin transmitssignals through the membrane. Several mechanisms for the activation have been postulated. The protein talin may bind to the cytoplastic tail of integrin and forces the two helices of the transmembrane domain of the dimer apart, which triggers an unbending of the ectodomain. This unbending would make the integrins metal ion-dependent adhesion site (MIDAS) accessible. Another possibility is that small conformational changes only initiated by binding a ligand to the adjacent ADMIDASdomain are sufficient.We analyzed the conformational change of the whole integrin during its bending and unbending for the first time with constant velocity steered molecular dynamic simulations. The integrin model was build up from the experimental crystal structure of the ectodomain and the homology model of the transmembrane and cytoplastic domain with an NMR derived model of i as template. Still missing amino acids were assumed to have random coil conformation.
395
Biophysical Chemistry P10-08

Probing the function of calmodulin adsorbed on and embedded in polyelectrolyte multilayersSüleyman Cinar, Claus Czeslik
TU Dortmund, Fakultät für Chemie und Chemische Biologie, D-44221 Dortmund
Polyelectrolyte multilayers are known to provide a mild environment for proteins. There are several studies in the literature that have found a high degree of native protein structure and enzyme activity, when proteins and enzymes are adsorbed on or embedded in polyelectrolyte multilayers. Here, we have studied the build-up and the properties of those multilayers that contain calmodulin, a functional protein that binds four Ca2+ ions and a further ligand molecule in turn. We have performed X-ray and neutron reflectivity measurements on the density profile of calmodulin-polyelectrolyte multilayers under different solvent conditions and FTIR spectroscopy measurements in attenuated total reflection (ATR) mode to probe the secondary structure of the built-in calmodulin. In this way, the multilayer thickness and the secondary structure of the protein have been measured as a function of the solvent composition, such as Ca2+, ligand, and EGTA concentration. Based on these data, the preservation of the functional properties of calmodulin adsorbed on and embedded in polyelectrolyte multilayers is discussed.
396
Biophysical ChemistryP10-09

Activation volumes of enzymes at polyelectrolyte brushesArtem Levin, Claus Czeslik
TU Dortmund, Fakultät für Chemie und Chemische Biologie, D-44221 Dortmund
Polyelectrolyte brushes can provide a native-like environment for the immobilization of proteins at aqueous-solid interfaces. In particular, poly(acrylic acid) (PAA) brushes have been shown to preserve the secondary structure of proteins and maintain enzyme activity to a high degree [1,2]. Moreover, the degree of protein adsorption at a PAA brush can be controlled by the ionic strength of the protein solution [3]. In view
of these interesting properties, we have investigated the structure and activity of -
chymotrypsin ( -CT) at a PAA brush as a function of pressure using TIRF
spectroscopy, neutron reflectometry, and ATR-FTIR spectroscopy. -CT is hydrolyzing peptide bonds, and its activity can be enhanced by pressure [4,5]. We
have found that this pressure response is largely maintained, when -CT is adsorbed
at a PAA brush, i.e. pressure still increases the catalytic rate of -CT. From ATR-
FTIR measurements, no significant changes of the secondary structure of -CT can be observed upon adsorption confirming the benign properties of the brush.
Furthermore, the density profile of -CT at a PAA brush has been determined as a function of pressure from neutron reflectivities. The profiles indicate a strong
interaction between -CT and the PAA chains and little pressure effects on the interfacial structure. Overall, a PAA brush seems to be a favorable surfacemodification for the immobilization of enzymes that can even be activated by pressure.
References[1] C. Reichhart, C. Czeslik Langmuir 25 (2009) 1047-1053.[2] C. Reichhart, C. Czeslik Colloids and Surfaces B 75 (2010) 612-616.[3] O. Hollmann, C. Czeslik Langmuir 22 (2006) 3300-3305.[4] M. J. Eisenmenger, J. I. Reyes-De-Corcuera, Enzyme Microbial Technol. 45(2009) 331-347.[5] V. Schuabb, C. Czeslik, Langmuir 30 (2014) 15496-15503.
397
Biophysical Chemistry P10-10

Cosolvent and crowding effects on the temperature and pressure stability of monomeric actin
Paul Hendrik Schummel and Roland Winter, TU Dortmund University, D-44227 Dortmund, Germany
Actin can be found in nearly all eukaryotic cells and is responsible for many different cellular functions. The polymerization process of actin has been found to be among the most pressure sensitive processes in vivo. In this study, we explored the effects of chaotropic and kosmotropic cosolvents, such as urea and the compatible osmolyte trimethylamine-N-oxide (TMAO), as well as the crowding agent Ficoll® PM 70 on the temperature and pressure stability of global actin (G-actin). The temperature and pressure of unfolding as well as thermodynamic parameters upon unfolding, such as volume and enthalpy changes, have been determined by fluorescence spectroscopy over a wide range of temperatures and pressures, ranging from 10-80 °C and 1-3000 bar, respectively. Different from the chaotropic agent urea, TMAO increases both, the temperature and pressure stability for the protein most effectively. In mixtures of these osmolytes, urea counteracts the stabilizing effect of TMAO to some extent. To create a more cell-like environment, Ficoll® PM 70 was added as macromolecular crowding agent as well. Addition of the crowding agent increases the temperature and pressure stability even further, thereby allowing sufficient stability of the protein at the temperature and pressure conditions encountered under extreme environmental conditions on Earth.
398
Biophysical ChemistryP10-11

Penetration of Dexamethasone in Human SkinInvestigated by X-Ray Spectromicroscopy
K. Yamamoto*, A. Klossek*, R. Flesch*, F. Rancan#, M. Weigand§, I. Bykova§, M. Bechtel§, P. Patoka*, G. Ulrich*, S. Ahlberg#, A. Vogt#, U. Blume-Peytavi#, P.
Schrade , S. Bachmann , M. Schäfer-Korting¶ and E. Rühl*
*Physikalische Chemie, Institut für Chemie und Biochemie, Freie Universität Berlin, 14195 Berlin, Takustrasse 3, Germany
#Klinisches Forschungszentrum für Haut- und Haarforschung, Charité Universitätsklinikum, 10117 Berlin, Germany
§Max-Planck-Institut für Metallforschung, Heisenbergstraße 3, 70569 Stuttgart, Germany
¶Institut für Pharmazie, Freie Universität Berlin, 14195 Berlin, GermanyAbteilung für Elektronenmikroskopie at CVK, 13353 Berlin Germany
The transport of drugs into human skin is of interest for improving the efficacy of medical treatments. In this work we focus on topical treatment of human skin. Here,the stratum corneum, which acts as the main barrier of skin, plays a key role. Bygetting a deeper insight into the time dependent penetration of the anti-inflammatory drug dexamethasone (DXM) the topical treatment of inflammatory skin diseases can be improved. Therefore, measurements on the DXM distribution in human skin were conducted for different drug exposure times by using Scanning Transmission X-Ray Microscopy (STXM). We characterized the time-resolved penetration profiles of DXM that was contained in HEC-gel between 10 min and 1000 min exposure time. Tape-stripped skin was also investigated in order to determine the importance of the skin barrier. Significant differences in penetration profiles between taped and untapped skin were observed.We also conducted experiments in which core-multi-shell nanotransporters (CMS) were topically applied to human skin. The localization of CMS nanocarriers wasdetermined by X-ray microscopy after topical treatment. CMS nanocarriers only penetrated into the stratum corneum showing their potential to act as drug reservoirsas well as penetration enhancers for lipophilic drugs overcoming the stratum corneum barrier.
399
Biophysical Chemistry P10-12

CRMP2: Switching StructuresD. Möller1 , C.H. Lillig2, M. Gellert2, N. Geist1, W. Langel1, Institut für Biochemie1 and Institut für Medizinische Biochemie und Molekularbiologie2, Universität Greifswald,
Germany
The CRMP2 (Collapsin response mediator protein)homotetramer controls the attachment of the extracellular matrix protein plexin to the membrane protein semaphorin.Switching between two states of CRMP2 is triggered by reducing two disulfide bridges between Cys504 in adjacentmonomers only, but these states are considered to have significantly different structures [1]. X-ray experiments reproduced the scaffold of the protein but not the flexible control unit at the C-term with the respective cysteines and phosphorylation sites. By force field molecular dynamics simulation with Ca2+ and Mg2+, we followed up the effect of opening the cystin bonds on relaxing the overall structure of the protein with 2064 residues in discrete water. The reactive part of the C-terminus was modelled by REMD. The structures relaxed during the simulation times of 250-300 ns, but the protein responded to reduction only by moderate displacements. Bivalent ions attach to the charged surface side chains, resulting in shifts in the electrostatic surface potentials and an increase of the surface areas around the C-termini.
[1] C.H. Lillig, http://www.thiolswitches.de/projekte/140220_homepageLillig.pdf(accessed 14/12/2015).
400
Biophysical ChemistryP10-13

Efficient Energy Transfer as Switching Mechanism for Super-Resolution Microscopy
Andreas Haderspeck,1) Dominik Brox,1) Dirk-Peter Herten1)
1)Institute for Physical Chemistry, Heidelberg University, Germany
Single-molecule based super-resolution microscopy is meanwhile well established for investigating cellular structures below the Abbe limit. Current methods make use of light-driven on/off-switching to enable imaging and subsequent localization of individual fluorophores with nanometer accuracy. Previously, we reported that switching between bright and dark states can also be achieved by chemical reactions, e.g. reversible coordination of metal ions to a ligand [1]. Presence of the metal ion in close proximity to the excited fluorophore enables efficient energy transfer to the formed complex, resulting in a nonradiative decay. The stochastic nature of the association/dissociation process can be used in localization microscopy to decouple switching from excitation, making additional laser irradiation obsolete and highly reducing phototoxicity [2]. More recently, we could show that the same principles can be employed to establish a novel mode of multiplexing in fluorescence microscopy [3]. Therefore, we currently work on modular systems allowing facile combination of different dye and receptor moieties to establish fluorescent probes that can be switched by chemical reactions. Here, we present new fluorescent probes which we recently synthesized and characterized in respect of their photophysical properties in bulk solution as well as on a single molecule level. We furthermore demonstrate their potential as fluorescent probe in localization microscopy imaging and their application in chemical multiplexing.
References[1] A. Kiel et al., Angew. Chem. Int. Ed., 2007, 119, 3427.[2] M. Schwering et al., Angew. Chem. Int. Ed., 2011, 50, 2940.[3] D. Brox et al., PLoS ONE, 2013, 8, e58049.
Efficient Energy Transfer as Switching Mechanism for Super-Resolution Microscopy
Andreas Haderspeck,1) Dominik Brox,1) Dirk-Peter Herten1)
1)Institute for Physical Chemistry, Heidelberg University, Germany
Single-molecule based super-resolution microscopy is meanwhile well established for investigating cellular structures below the Abbe limit. Current methods make use of light-driven on/off-switching to enable imaging and subsequent localization of individual fluorophores with nanometer accuracy. Previously, we reported that switching between bright and dark states can also be achieved by chemical reactions, e.g. reversible coordination of metal ions to a ligand [1]. Presence of the metal ion in close proximity to the excited fluorophore enables efficient energy transfer to the formed complex, resulting in a nonradiative decay. The stochastic nature of the association/dissociation process can be used in localization microscopy to decouple switching from excitation, making additional laser irradiation obsolete and highly reducing phototoxicity [2]. More recently, we could show that the same principles can be employed to establish a novel mode of multiplexing in fluorescence microscopy [3]. Therefore, we currently work on modular systems allowing facile combination of different dye and receptor moieties to establish fluorescent probes that can be switched by chemical reactions. Here, we present new fluorescent probes which we recently synthesized and characterized in respect of their photophysical properties in bulk solution as well as on a single molecule level. We furthermore demonstrate their potential as fluorescent probe in localization microscopy imaging and their application in chemical multiplexing.
References[1] A. Kiel et al., Angew. Chem. Int. Ed., 2007, 119, 3427.[2] M. Schwering et al., Angew. Chem. Int. Ed., 2011, 50, 2940.[3] D. Brox et al., PLoS ONE, 2013, 8, e58049.
401
Biophysical Chemistry P10-14

Allostery in PDZ3: Azidohomoalanine as Site-Specific Probe for IR Spectroscopy
Katharina B. Eberl,Patrick Quoika, Henrike M. Müller-Werkmeister, Jan G.Löffler,Martin Essig, Jens Bredenbeck, Institute of Biophysics, Johann Wolfgang Goethe-
University, Max-von-Laue-Str. 1, 60438 Frankfurt, Germany.
The unnatural amino acid Azidohomoalanine (Aha) is an excellent label for IR spectroscopy that provides local information of the interior of a protein. Its absorption frequency is highly sensitive to the local microenvironment and probes hydrogen-bonding and electrostatic effects. By placing Aha site-specifically into a protein it is possible to monitor even subtle conformational changes at a specific site using IR spectroscopy1-2.Here it is used to investigate allosteric communication within the 3rd PDZ domain of the synaptic protein PSD-95, which is a well studied but controversially discussedmodel for allostery without major conformational changes. A wire-like network of coupled amino acids, which might be involved in intradomain information transfer, has been predicted by Lockless and Ranganathan in 19993. Many other theoretical and some experimental studies tried to proof or disproof the existence of a coupled network of amino acids involved in allosteric communication; however the presence and mechanism of intradomain information transfer remain still unclear4-7. Our approach allows measuring tiny conformational changes which have been invisible before and thus detecting weak influences of ligand binding, as a trigger for allosteric effects. Thereby we are able to get a deeper insight in allosteric communication of the PDZ3 domain.
1H. Taskent-Sezgin, J. Chung, P. S. Banerjee, S. Nagarajan, B. Dyer, I. Carrico and, D. P. Raleigh, Angew.Chem. Int. Ed. 2010, 49, 7473-74752M. M. Waegele, R. M. Culik, and, F. Gai , J. Phys. Chem. 2011, 2, 2158-21623S. W. Lockless and R. Ranganathan, Science 1999, 286, 294-2994N. Ota and D. A. Agard, J. Mol. Biol. 2005, 351, 345-3545C. N. Chi, S. R. Haq, S. Rinaldo, J. Dogan, F. Cutruzzolà, Å. Engström, S. Gianni, P. Lundström, and P. Jemth , Biochemistry 2012, 51, 8971-89796C. N. Chi, L. Elfström, Y. Shi, T. Snäll, Å. Engström and, P. Jemth, Proc. Natl. Acad. Sci. 2008, 105, 12, 4679-4684
7C. M. Petit, J. Zhang, P. J. Sapienza, E. J. Fuentes and, A. L. Lee, Proc. Natl. Acad. Sci. 2009, 106, 43, 18249-18254
Allostery in PDZ3: Azidohomoalanine as Site-Specific Probe for IR Spectroscopy
Katharina B. Eberl,Patrick Quoika, Henrike M. Müller-Werkmeister, Jan G.Löffler,Martin Essig, Jens Bredenbeck, Institute of Biophysics, Johann Wolfgang Goethe-
University, Max-von-Laue-Str. 1, 60438 Frankfurt, Germany.
The unnatural amino acid Azidohomoalanine (Aha) is an excellent label for IR spectroscopy that provides local information of the interior of a protein. Its absorption frequency is highly sensitive to the local microenvironment and probes hydrogen-bonding and electrostatic effects. By placing Aha site-specifically into a protein it is possible to monitor even subtle conformational changes at a specific site using IR spectroscopy1-2.Here it is used to investigate allosteric communication within the 3rd PDZ domain of the synaptic protein PSD-95, which is a well studied but controversially discussedmodel for allostery without major conformational changes. A wire-like network of coupled amino acids, which might be involved in intradomain information transfer, has been predicted by Lockless and Ranganathan in 19993. Many other theoretical and some experimental studies tried to proof or disproof the existence of a coupled network of amino acids involved in allosteric communication; however the presence and mechanism of intradomain information transfer remain still unclear4-7. Our approach allows measuring tiny conformational changes which have been invisible before and thus detecting weak influences of ligand binding, as a trigger for allosteric effects. Thereby we are able to get a deeper insight in allosteric communication of the PDZ3 domain.
1H. Taskent-Sezgin, J. Chung, P. S. Banerjee, S. Nagarajan, B. Dyer, I. Carrico and, D. P. Raleigh, Angew.Chem. Int. Ed. 2010, 49, 7473-74752M. M. Waegele, R. M. Culik, and, F. Gai , J. Phys. Chem. 2011, 2, 2158-21623S. W. Lockless and R. Ranganathan, Science 1999, 286, 294-2994N. Ota and D. A. Agard, J. Mol. Biol. 2005, 351, 345-3545C. N. Chi, S. R. Haq, S. Rinaldo, J. Dogan, F. Cutruzzolà, Å. Engström, S. Gianni, P. Lundström, and P. Jemth , Biochemistry 2012, 51, 8971-89796C. N. Chi, L. Elfström, Y. Shi, T. Snäll, Å. Engström and, P. Jemth, Proc. Natl. Acad. Sci. 2008, 105, 12, 4679-4684
402
Biophysical ChemistryP10-15

Shedding light on plasma-bacteria interaction using vibrational microspectroscopy
Konstantin Kartaschew1, Sabrina Baldus2, Friederike Kogelheide², Meike Mischo1,Jan-Wilm Lackmann², Peter Awakowicz2, Martina Havenith1
1Physical Chemistry II, Ruhr-Universität Bochum, Germany2Institute for Electrical Engineering and Plasma Technology, Ruhr-UniversitätBochum, Germany
Cold atmospheric-pressure plasma (CAP) is well known as a promising tool for the inactivation of bacteria [1], however a lack of distinct understanding of the chemical mechanism remains up to now. As the fourth state of matter, plasma consist of a broad mix of stable and reactive species, electrons, ions and UV light. This broadvariety is one of the reasons why the bacterial inactivation effect of plasma in detail remains unclear.We investigated the chemical impact of CAP on both, discrete molecules such as cysteine and bacteria by using vibrational microspectroscopy. Raman- and Fourier transform infrared-microspectroscopy as the two used label-free vibrational spectroscopic methods do not harm the sample and provide directly chemical information of the investigated material. Hence, these methods allow an immediateidentification of plasma-induced chemical modifications.We demonstrate how vibrational microspectroscopy can be used practically to shed light on the question how CAP influences not only distinct cell components, but full organisms. Thus, this approach contributes to a better understanding of plasma-cell interactions.
[1] Lackmann, J.-W. & Bandow, J. E. Appl. Microbiol. Biotechnol. 98, 6205–6213 (2014).
403
Biophysical Chemistry P10-16

Understanding the primary steps of light-induced signal propagation of an artificial LOV2-Rac1 photoswitch
I. Stambolic, B. Dick, S.A. Baeurle, University of Regensburg, Regensburg/Germany
LOV (light-, oxygen-, voltage – sensitive) domains are the photosensory units of various photoreceptor proteins, using flavin as a chromophore. In most cases they are coupled to an effector domain, through which the signals can be transmitted downstream and initiate a large number of vital processes, like e.g. phototropism, stomatal opening or chloroplast relocation. Recently, artificial photoswitches evolved as an interesting extension to naturally occurring light-regulated enzymes. However, our current level of understanding of the natural photoswitches is still not enough to allow for rational design of new optogenetic tools [1].Wu et al. [2] fused a LOV2 domain with Rac1 mutants, forming artificial photoswitches. They found that localized Rac1 activation or inactivation was sufficient to produce cell motility and control the direction of cell movement on atimescale of seconds and with a submicron precision. The effector binding site of Rac1 was sterically blocked due to an extensive interface formed between LOV and Rac1. Furthermore, they suggested that the interface is hydrophobic in nature due to the un-evolved interactions where most of the hydrogen bonds are satisfied by the buried waters instead of the inter-domain interactions. We chose this LOV2-Rac1 construct as a model system to understand the primary steps of light-induced signal propagation following the adduct formation using molecular dynamics simulations. Additionally, we investigated the types of interactions between these two domains. We found that the light- as well as the dark-state of LOV2 subunit is promoting the activation and inactivation of the Rac1 domain, respectively. The adduct formation in LOV2 promotes even further strengthening of the LOV2-Rac1 domain interactions. However, inducing an active state in Rac1 does not lead to a change in the domain interactions nor the LOV2-Rac1 contact surface.
[1] Winkler A. et al., ACS Chem Biol., 2015, 10(2):502-9, doi: 10.1021/cb500744m[2] Wu Y. et al., Nature, 2009, 461(7260): 104–108, doi: 10.1038/nature08241
404
Biophysical ChemistryP10-17

The flavin photochemistry in a LOV domain is tuned and controlled by the protein environment
K. Magerl, I. Stambolic, B. Dick, University of Regensburg, Regensburg/Germany
LOV (light-, oxygen- or voltage- sensitive) and BLUF (blue light sensor using FAD)domains are photoreceptor proteins sensitive to blue light due to a non-covalently bound flavin. They are found in a variety of organisms in which they control a large number of biological functions, from circadian clock to phototropism. Excitation of theflavin leads to redox and protonation state changes which induce structural rearrangements of the protein core and ultimately lead to changes in protein surface and interactions with binding partners. Flavin photochemistry differs in these two protein groups, where in LOV domains the photochemistry is characterized by an adduct formation via the excited flavin triplet state while in BLUF domains there is an electron transfer process occurring via the excited singlet state of the flavin. To understand the factors that control the differences in the photochemistry of these two protein groups, we chose the LOV1 domain of the phototropin from the green alga C. reinhardtii (CrLOV1) as a model system for the investigation of electron transfer processes within these domains. We produced a number of CrLOV1 mutants, in which tyrosine was introduced as an electron donor amino acid in the protein binding pocket nearby the flavin. The reactive cysteine was mutated to either a glycine or a serine. To identify spectral species we performed UV/VIS, emission and transient absorption measurements. Additionally, MD simulations were also performed in an effort to explain the differences in dynamics of amino acid residues surrounding the flavin cofactor. In most cases no efficient electron transfer could be observed, even though the tyrosine-flavin interactions and orientations seemed favorable. Of all the systems studied, only the CrLOV1 F41Y-C57S mutant showed an efficient electron transfer from the introduced tyrosine to the T1 state of the flavin.Apparently the reactivity of the electron donor/flavin pair depends not only on distance and orientation but is tuned by additional properties of the protein environment that will be discussed in the presentation.
405
Biophysical Chemistry P10-18

Targeted Delivery of Aptamer- and Antibody-Conjugated AuNPs to Interleukin-6 Receptor-Carrying Cells
Lisa Prisner, Nadine Bohn, Mareike Bendig, and Alf MewsUniversity of Hamburg, Institute of Physical Chemistry, Hamburg, Germany
Gold nanoparticles (AuNPs) are well suited for biological applications due to their small size, their good biocompatibility and low toxicity as well as the fact that they are easily soluble and stable in aqueous solution e.g. cell culture media. In addition, gold nanoparticles display size- and shape-dependent optical properties which make them very promising candidates for imaging applications.
The receptor-ligand mediated approach for specific gold nanoparticle delivery to cells strongly depends on the type of used affinity molecule, but also on the size of the AuNPs. We here present an Interleukin-6 receptor based approach for the targeteddelivery of gold nanoparticle to receptor-carrying cells. We show a high specificbinding of the biofunctionalized particles as well as uptake into the cells. We investigate the effect of particle size, ligand coverage, and incubation parameters on their delivery efficiency for this system. In addition to the investigation and optimization of the aptamer-mediated system, we compare their delivery efficiency and specificity to the targeted delivery of antibody-functionalized gold particles recognizing the same receptor to validate the performance of this approach.Moreover we test their potential for imaging applications using different microscopic methods.
Figure 1: amount of gold particles in cells determined by ICP-MS (left) as well as dark field images (middle) and SEM-images of gold particles incubated with cells (right).
406
Biophysical ChemistryP10-19

Effect of the N-terminal Helix and Nucleotide-Loading on the Membrane and Effector Binding of the GTPases Arl2 and Arl3
Shobhna Kapoor,1,2 Eyad K. Fansa,3 Simone Möbitz,1 Shehab A. Ismail,3,4 Roland Winter,1 Alfred Wittinghofer,3 and Katrin Weise1
1 Physical Chemistry I, TU Dortmund University, Dortmund, Germany; 2 Department of Chemical Biology and 3 Structural Biology Group, Max Planck Institute of
Molecular Physiology, Dortmund, Germany; 4 Structural Biology of Cilia, CR-UK Beatson Institute, Glasgow, United Kingdom
The small GTP-binding proteins Arl2 and Arl3, which are close homologs, share a number of interacting partners and act as displacement factors for prenylated and myristoylated cargo. Nevertheless, both proteins have distinct biological functions. Whereas Arl3 is considered a ciliary protein, Arl2 has been reported to be involved in tubulin folding, mitochondrial function, and Ras signaling. How these different roles are attained by the two homolog proteins is not fully understood. Recently, weshowed that the N-terminal amphipathic helix of Arl3, but not that of Arl2, regulates the release of myristoylated ciliary proteins from the GDI-like solubilizing factor UNC119a/b [1]. In this biophysical study, both proteins are shown to exhibit a prefer-ential localization and clustering in liquid-disordered domains of phase-separated membranes. However, the membrane interaction behavior differs significantly between both proteins with regard to their nucleotide loading. Whereas Arl3 and other Arf proteins with an N-terminal amphipathic helix require GTP-loading for the interaction with membranes, Arl2 binds to membranes in a nucleotide-independent manner. In contrast to Arl2, the N-terminal helix of Arl3 increases the binding affinity to UNC119a. Furthermore, UNC119a impedes membrane binding of Arl3, but not of Arl2. Taken together, these results suggest an interplay among the nucleotide status of Arl3, the location of the N-terminal helix, membrane fluidity and binding, and the release of lipid modified cargos from carriers such as UNC119a [2].
References:[1] Ismail SA, Chen YX, Miertzschke M, Vetter IR, Koerner C, Wittinghofer A (2012)
EMBO J. 31:4085–4094.[2] Kapoor S, Fansa EK, Möbitz S, Ismail SA, Winter R, Wittinghofer A, Weise K.
(2015) Biophys. J. 109:1619–1629.
407
Biophysical Chemistry P10-20

Kinetics of drug release and mobility in healthy and osteoporotic rat bone
Marcus Rohnke, Boris Mogwitz, Anja Henß, Seemun Ray, Volker Alt and Jürgen Janek, Justus-Liebig University Gießen, Gießen / Germany
Drug functionalization of biomaterials is an innovative and popular approach in biomaterials research. Amongst others this concept is used for the functionalization of bone implants to stimulate locally the osseointegration. Due to practical and technical reasons it is almost impossible to track the drug release kinetics, drug distribution and the degradation of the implant material in vivo. In consequence of low drug concentrations in bone it is also not easy to image the drug distribution by analytical methods in post mortem examinations. Based on the results of various analytical experiments we developed a two-phase model for the release and mobility of the pharmaceutical agents Sr2+ and Bortezomib. Sr2+ is used as drug in osteoporosis therapy to stimulate bone growth and was released locally by a strontium doped cement. Bortezomib is an anti-cancer drug administered in therapy of multiple myeloma and was released by a collagen scaffold. The presented simulation experiments allow the determination of appropriate functionalized biomaterials and might help to reduce the number of animal experiments in the future. The validity of the applied model is proven by animal experiments.
Materials and MethodsThe time and spatially resolved drug movement was calculated by a finite element calculation for the femur of healthy and osteoporotic rats. Therefore scanning electron microscopy (SEM) images were taken of cross sections of healthy and osteoporotic rat femur. The backscattering electron detector (BSE) was used to obtain materials contrast in the image. The dark regions of the images belong to mineralised areas and were defined as area 1. The bright areas correspond to bone marrow and were defined as area 2. By using the finite element software Comsol 5.1 the time dependent drug mobility was calculated in a 2D model. For the Sr2+ release from the cements we assumed a first order kinetics and defined two different diffusion coefficients for area 1 and 2 with D1 < D2. For the second model (Bortezomib mobility) we assumed a finite source with immediate drug availability 408
Biophysical ChemistryP10-21

and D1 = D2. The obtained results are compared to mass images that were obtained from sections of in vivo experiments by Time of Flight Secondary Ion Mass Spectrometry (ToF-SIMS).
Results and DiscussionIn contrast to healthy rat bone the osteoporotic bone shows a reduced trabecular network and the trabeculae itself are thinner. The first model focuses on the Sr2+
release from the doped calcium phosphate cement. For the simulation a virtual drill hole of 2 mm diameter, which is filled with the cement, is placed in the bone area. A fast distribution of Sr2+ in the bone marrow is calculated due to the reduced trabecular network in the osteoporotic bone. In contrast to healthy bone the single trabeculae act as diffusion barrier for the drug. The simulated distribution image fits quite well to the experimentally obtained results from ToF-SIMS imaging. In the second model the distribution of an organic drug molecule is simulated. In contrast to Sr2+ the mobility is definitely higher and it seems to be the same mobility in the bone marrow as in the mineralised regions. Here no significant difference between the healthy and osteoporotic bone is calculated that is again in line with the obtained results from the ToF-SIMS measurements.
ConclusionsThe presented simple models for drug release and mobility in bone fit quite well tothe obtained experimental data. It appears that drug removal via the vascular system is negligible. This is a good basis for predictions of drug mobility in bone and might help to reduce the number of animal studies. Certainly, it is necessary to determine more precise diffusion coefficients for the drug in cortical, trabecular bone and bone marrow in the future.
409
Biophysical Chemistry P10-21

Calculation of relative folding free enthalpies of amide-to-ester mutants of protein Pin1
Daniel Markthaler, Niels Hansen, Institute of Thermodynamics and Thermal Process Engineering, University of Stuttgart, D-70569 Stuttgart, Germany
Understanding thermal stability of proteins is crucial for protein engineering applications. The prediction of the effect induced by mutations, however, is highly challenging as thermal stability changes are caused by alterations in the free energy of folding. In recent work the effect of removing a hydrogen-bond donor from the backbone of the 34-residue WW domain of protein Pin1 was investigated for 20
-sheet fold of this protein using molecular dynamics free energy simulations [1].
Representation of the unfolded state
A proper representation of unfolded state conformations is essential for an adequate description of relative stabilities of protein mutants. In [1] the most promising approximation was to use capped tri-peptides according to the sequence from the protein with the mutated residue in the middle. The influence of the peptide chain length in the unfolded state approximation was investigated by using penta- and hepta-peptides for all 20 mutants.
Influence of co-solutes
In addition the effect of co-solutes on the stability is of importance for a meaningful comparison between simulated and experimental relative stabilities of protein mutants. Therefore a comparison of existing force fields for trimethylamine N-oxide (TMAO) and guanidinium hydrochloride (GdmCl) has been performed. The force fields were tested for their ability to describe density and volumetric properties of various binary aqueous mixtures at different temperatures and pressures. For the present work the influence of the co-solute concentration on the relative folding free energy differences was also investigated for several protein mutants.
References
[1] A. P. Eichenberger, W. F. van Gunsteren, S. Riniker, L. von Ziegler, N. Hansen, Biochim. Biophys. Acta 2015, 1850, 983–995
410
Biophysical ChemistryP10-22

Counting LAT and SLP-76 in T-cell antigen receptor proximal signaling microclusters
Wioleta Chmielewicz, Heidelberg University, GermanySiegfried Hänselmann, Heidelberg University, Germany
Nikolaos Tsopoulidis, Heidelberg University Hospital, GermanyKlaus Yserentant, Heidelberg University, GermanyKristin Grußmayer, Heidelberg University, Germany
Oliver T. Fackler, Heidelberg University Hospital, GermanyDirk-Peter Herten, Heidelberg University, Germany
External activation of the T-cell antigen receptor (TCR) complex is an essential trigger for intracellular signaling cascades that ultimately lead to the mounting of adaptive immune responses. Signal transduction via the TCR critically depends on the rapid formation of TCR proximal signaling platforms referred to as microclustersfrom which downstream signalling is elicited. TCR microcluster formation requires the orchestrated interaction of many proteins including the adaptor proteins LAT (linker for the activation of T-cells) and SLP-76 (Src homology 2 (SH2) domain-containing leukocyte phosphoprotein of 76 kD). This process is also hijacked by human immunodeficiency virus type 1 (HIV-1) to modulate T-cell activation and optzimize virus spread via the accessory protein Nef[1]. The exact composition and stochiometry of microclusters and how modulation of these parameters impacts on their signaling competence has remained unclear. To address these issues, we want to apply Counting by Photon Statistics (CoPS)[2, 3] to count the number of phosphorylated and unphosphorylated LAT and SLP-76 proteins within an activated TCR cluster in dependence on Nef. We currently explore different labeling strategies based on fluorescently labeled primary antibodies and the genetically encoded HaloTag and develop a calibrator to determine its degree of labeling.
[1] O. T. Fackler, A. Alcover, O. Schwartz, Nat Rev Immunol 2007, 7, 310.[2] A. Kurz, J. J. Schmied, K. S. Grußmayer, P. Holzmeister, P. Tinnefeld, D.-P. Herten, Small 2013, 9, 4061.[3] K. S. Grußmayer, A. Kurz, D.-P. Herten, ChemPhysChem 2014, 15, 734.
411
Biophysical Chemistry P10-23

Comparing azide and thiocyanate vibrational probes for infrared spectroscopy of proteins
Marcus Mikorski, Daniela Kern-Michler, Katharina Eberl, Luuk J. G. W. van Wilderen,Jens Bredenbeck, Institute of Biophysics, Johann Wolfgang Goethe-University, Max-
von-Laue-Str. 1, 60438 Frankfurt, Germany.The infrared spectral properties of two different artificial vibrational probes that can be site-specifically introduced in a protein are presented and compared. The thiocyanate (-SCN) functional group is introduced by cyanylation of cystein.1,2 The azide (-N3) group is introduced by inserting azidohomoalanine (Aha) as a methionine surrogate.3 -SCN and -N3 infrared absorptions are sensitive to hydrogen bonding and electrostatic fields in their local environment. Both the -N3 and -SCN probe are introduced at six different mutation sites of the PDZ3 domain of PSD95 and IR spectra of the protein with and without bound peptide ligand are measured.A discussion about the local environment at the mutation sites and its impact on the IR spectra of both vibrational probes will be presented. Both probes are sensitive to changes in H-bonding and electrostatics of their local environment. The shifts caused by the environment, however, are different for each of the two probes. Using both probes at the same mutation site thus can lead to a more detailed interpretation of the IR spectra regarding the local properties of the protein.
1A. T. Fafarman, L. J. Webb, J. I. Chuang, S. G. Boxer, J. Am. Chem. Soc. 2006, 128, 13356–133572L. J. G. W. van Wilderen, D. Kern-Michler, H. M. Müller-Werkmeister, J. Bredenbeck, Phys. Chem. Chem. Phys. 2014, 16, 19643–196533H. Taskent-Sezgin, J. Chung, P. S. Banerjee, S. Nagarajan, R. B. Dyer, I. Carrico, D. P. Raleigh, Angew. Chem. Int. Ed. 2010, 49, 7473-7475
412
Biophysical ChemistryP10-24

Pressure-induced Effects on Self-cleavage Reaction of Ribozyme Catalysis: A Molecular Dynamics Study
Narendra Kumar and Dominik MarxLehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, 44780 Bochum, Germany.
Email: [email protected]
Ribozymes, a very important class of non-coding RNA molecules, catalyze site specific self-cleavage of phosphodiester bonds and play prominent roles in several biological processes such as RNA splicing, the controlling of gene expression, and processing of tRNA. Ribozymes also serve as a potential drug target. Replica exchange molecular dynamics simulations including explicit solvent and ions were utilized to understand pressure induced effect on self-cleavage reaction of hairpin ribozyme. We find that pressure-induced enhanced hydrogen bonding network stabilizes the active conformation. The results also provide strong support for the involvement of water molecules trapped in active site as a base. This work provides very first molecular simulation study understanding the pressure-induced effects on site-specific catalytic reaction mechanism which can be a stepping stone in further understanding of ribozyme catalysis using quantum mechanical methods to exactly see how bonding breaking or making takes place under high pressure.
413
Biophysical Chemistry P10-25

Cavity Enhanced Raman Spectroscopy (CERS) as an Analytical Tool in the Biosciences: Studying Hydrogen Production by
MicrobesT.W. Smith and M. Hippler, Department of Chemistry, University of Sheffield,
Sheffield S3 7HF, England
With current concerns about curbing emissions of greenhouse gases and dwindling supplies of fossil fuels, H2 is a promising replacement. Biologically derived hydrogen (biohydrogen) is a promising alternative to other sources of the gas as it provides efficient H2 production without the need for precious metal catalysts and with minimal greenhouse gas emissions. However, optimising this biological process for commercial H2 production requires a reliable method for measuring the concentration of H2, ideally on-line and in situ. With existing analytical techniques, it is difficult to monitor hydrogen in situ reliably and quantitatively. Recently, we have introduced a highly sensitive technique for detecting Raman active gases, Cavity Enhanced Raman Spectroscopy (CERS). [1,2]
Here we present the application of CERS to the simultaneous on-line, in situ andquantitative detection of H2, N2, O2 and CO2 in the headspace of glucose and glycerol fed Escherichia coli. Adding D2, we are also able to observe the reverse process of hydrogen consumption simultaneously with hydrogen production, due to the different spectroscopic signature of the isotopomers. This allows us to separate the two processes and to obtain kinetic parameters for hydrogen production and consumption by Escherichia coli. [T.W. Smith, M. Hippler, in preparation]
[1] R. Salter, J. Chu, M. Hippler, Analyst, 137, 4669 (2012).[2] M. Hippler, Anal. Chem., 87, 7803 (2015).
414
Biophysical ChemistryP10-26

Scattering particles increase absorbance of dyes –a model study with relevance for sunscreens
Bernd Herzog
Sunscreens used for the protection of human skin against the harmful effects of solar radiation contain UV absorbers as key ingredients, which are either dissolved in one of the phases of the preparation or, when insoluble, suspended as particles. Although the UV protective effect of particulate UV filters, inorganic and organic, is mainly due to absorption, they scatter UV and visible light. The scattering can have an additional attenuating effect on the incoming radiation by increasing the pathlength of the photons, especially when soluble filters are also present. This is investigated with model systems of dyes and absorbing and non-absorbing particles. The presence of particles causes an increase of the dye absorbance without changing dye concentration or cuvette thickness. It is possible to relate this amplification of dye absorbance to the turbidity of the system. Plots are constructed which allow for a given particle type the representation of all data on one single curve, though measured at different turbidity and cuvette thickness. With that, extrapolations to practical applications of sunscreens are possible.
415
Biophysical Chemistry P10-27

Oxygen chain structures and other forms of chemisorbed oxygen on gold-silver alloy surfaces: a DFT study
Lyudmila V. Moskaleva,* Theodor Weiss, Marcus Bäumer, Institut für Angewandte und Physikalische Chemie und Zentrum für Umweltforschung und nachhaltige
Technologien, Universität Bremen, 28359 Bremen, Germany;Thorsten Klüner, Institut für Chemie, Carl von Ossietzky Universität Oldenburg,
26129 Oldenburg, Germany
In recent years, the research on nanostructured gold catalysts has been extended to gold alloys with less noble metals. Several recent studies reported high catalytic activity of Au-Ag nanoparticles for alkene epoxidation, low-temperature CO oxidation,and other reactions, in many cases exceeding that of monometallic Au or Ag catalysts. In this work, we investigate theoretically on the basis of DFT the adsorbed forms of oxygen on pure gold and on Au-Ag alloy surfaces. The stepped and kinked Au(321) surface has been chosen as a realistic model to represent gold surfaces with a high density of low-coordinated sites. The relative stabilities of on-surface, subsurface, and surface-oxide forms of O have been analyzed and compared on pure gold and on the surfaces containing silver atoms. We identify (–Au–O–) chain structures as especially energetically favorable forms of adsorbed oxygen on gold-rich surfaces even at relatively low O coverage. We address the reactivity of these chains with CO and we show that –O–Au–O–linear fragments can form as a result of O2 dissociation on the surface with pre-adsorbed O, significantly lowering the dissociation barrier of O2. Ag atoms replacing Au on the Au(321) surface are shown to stabilize the O-covered surface with respect to the clean surface. However, the stabilization of (–M–O–) chain structures is significantly less for M = Ag than for M = Au.
416
CatalysisP11-01

Decomposition of formic acid on Pd-based surfacesHolger Euchner, Jan Ku era, Axel Groß, Institute of Theoretical Chemistry,
Universität Ulm, Ulm/Germany;
Pd-based catalysts have shown promising activity with respect to heterogeneous [1] and electrochemical [2] dehydrogenation of formic acid (FA), thus offering high potential for future H2 energy technologies. The performance of current catalysts, however, so far has not met the technological demands and, unfortunately, rational strategies towards better catalysts are difficult to establish. Despite the apparent simplicity of the overall reaction, this is mainly owed to the complexity of FA decomposition. To gain insight into underlying mechanisms and potential for improvement, we have conducted a computational study of the decomposition of FA on Pd-based surfaces. Using periodic density functional theory, including dispersion correction (DFT-D3), the effect of replacing sub-surface Pd by different transition metals is studied. The impact of changing electronic structure on the decomposition pathway of FA, caused by the vertical ligand effect, is studied and discussed with respect to desired dehydrogenation and undesired dehydration reaction. A step towards more realistic modeling is achieved by investigating the effect of hydrogen as a co-adsorbent, being a reaction intermediate at the same time.
[1] Grasemann et al., Energy Environ. Sci. 5, 8171 (2012).[2] Jiang et al., Phys. Chem. Chem. Phys. 16, 20360 (2014).
417
Catalysis P11-02

Computational methods for pure component decompositionswith application to the rhodium catalyzed hydroformylation
Klaus Neymeyr, Mathias Sawall, Annekathrin Jürß, Henning Schröder, Universität Rostock, Germany
Multivariate curve resolution (MCR) methods are extremely useful tools for the extraction of pure component information from spectral mixture data. However, MCR methods suffer from the intrinsic non-uniqueness of the underlying nonnegative matrix factorization problem.The talk provides an overview on the spectral recovery problem and the ambiguity ofthe pure component factorizations. Recent results are presented on a systematic approach to obtain full control of the ambiguity by the Area of feasible solutions (AFS).We present the Matlab toolbox FACPACK for computing sets of feasible solutions and demonstrate its usefulness for FT-IR spectral mixture data from the rhodium catalyzed hydroformylation of alkenes. This work is part of a cooperation with the Leibniz Institute for Catalysis (LIKAT) in Rostock and Evonik Industries.
References:1. K. Neymeyr, M. Sawall,
On an SVD-free approach to the complementarity and coupling theory: A note on the elimination of unknowns in sums of dyadic products.Accepted for J. Chemometrics 2015
2.. M. .Sawall, C. Kubis, A. Börner, D. Selent, K. Neymeyr,
A multiresolution approach for the convergence acceleration of multivariate curve
resolution methods. Analytica Chimica Acta 891, 101-112 (2015).
3.. M. Sawall, C. Kubis, R. Franke, D. Hess, D. Selent, A. Börner, K. Neymeyr,
How to apply the complementarity and coupling theorems in MCR methods: Practical
implementation and application to the rhodium-catalyzed hydroformylation.ACS Catal. 4, 2836-2843 (2014)
4.. M. Sawall, C. Kubis, D. Selent, A. Börner, K. Neymeyr,
A fast polygon inflation algorithm to compute the area of feasible solutions for three-
component systems. I: Concepts and applications. J. Chemometrics 27 (2013), 106-116.
418
CatalysisP11-03

Formic acid dehydrogenation over Au/CeO2 catalysts: Theoretical DFT study
Jan Ku era and Axel Groß, University Ulm, Ulm/Germany
Heterogeneous catalysts based on nano-sized Au supported on various oxides (e.g., TiO2,CeO2, or SiO2) can selectively decompose formic acid (FA) to provide pure hydrogen already at low temperatures - a process of high technological relevance in the context of hydrogen technology as part of a sustainable energy concept. Despite of the activity ascribed to Au, the decomposition mechanism depends strongly on the nature of the oxide. Unfortunately, the mechanistic understanding of the function of the support remains unclear, which hampers to establish a route towards better catalysts.Our computational study focuses on the elucidation of elementary processes behind the FA decomposition over Au/CeO2. In particular, the reducible surface of CeO2
facilitates the dynamical formation of catalytically active subnanometer/atomic Au species detached from Au-nanoparticles. We have employed periodic DFT+U models together with a periodic embedded cluster method (PEECM) to investigate the dehydrogenation process of FA over single-Au species on CeO2. We will discuss the influence of the charge transfer between Au and the CeO2 support on the catalytic mechanism in the presence of various surface defects. Besides we will discuss the reliability of two approaches periodical DFT+U and PEECM with hybrid functional for the presented FA/Au/oxide catalytic system.
419
Catalysis P11-04

Operando X-ray absorption and emission spectroscopy to understand reaction mechanisms in NOx-removal catalysts
T. Günter, D. E. Doronkin, A. Boubnov, M. Casapu, J.-D. GrunwaldtInstitute for Chemical Technology and Polymer Chemistry and Institute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Karlsruhe, Germany
J. Rudolph, C. R. JacobInstitute of Physical and Theoretical Chemistry, TU Braunschweig, Germany
Meeting the strict legislations for NOx-emissions from cars has become a challenge in catalyst development. Fe- and Cu-exchanged zeolites are particularly interesting due to their high performance during the selective catalytic reduction (SCR) of NOx with NH3. Further improvement of the catalyst formulation and exhaust gas aftertreatment control algorithm demand for detailed understanding of the occurring reactions. X-ray absorption and emission spectroscopy (XAS / XES) enable a detailed view at the catalytically active sites under realistic reaction conditions.Here we report a comparison of a series of SCR-catalysts using spatially- and time-resolved operando XAS and additionally XES for Fe-ZSM-5 and Cu-SSZ-13 during NH3-SCR of NOx in a quartz capillary reactor. Strong gradients of the oxidation state and coordination number along the reactor show the reaction progress and the importance of spatially-resolved vs. integral measurements for all catalysts [1]. The studies have recently been extended by high-energy fluorescence detected (HEFRD) XAS and XES, which allowed the identification of adsorbed species combined with DFT-calculations [2,3]. This led to the proposal of more detailed SCR-mechanisms explaining the different catalytic behavior of Fe- and Cu-based catalysts.
[1] D.E. Doronkin, M. Casapu, T. Günter, O. Müller, R. Frahm, J.-D. Grunwaldt, J. Phys. Chem. C 2014, 118, 10204-10212.[2] A. Boubnov, H.W.P. Carvalho, D.E. Doronkin, T. Günter, E. Gallo, A.J. Atkins, C.R. Jacob, J.-D. Grunwaldt, J. Am. Chem. Soc. 2014, 136, 13006-13015.[3] T. Günter, H.W.P. Carvalho, D.E. Doronkin, T. Sheppard, P. Glatzel, A.J. Atkins, J. Rudolph, C.R. Jacob, M. Casapu, J.-D. Grunwaldt, Chem. Commun. 2015, 51,9227-9230.
420
CatalysisP11-05

Nanoporous gold supported oxides as high-performance hydrogenproduction catalyst
Junjie Shi1, Anastasia Lackmann1, Arne Wittstock1, Marcus Bäumer1
1 Institute of Applied and Physical Chemistry and Center for Environmental Research and Sustainable Technology, University Bremen, Leobener Str. NW 2, 28359 Bremen, Germany
Catalysis by nanoporous gold spurred considerable interest since the first discovery of its low temperature CO oxidation activity around 10 years ago.1 This unsupported skeletal gold catalyst was shown to be a very active and selective Au type catalyst for a variety of oxidation reactions in catalysis employing dioxygen.2 Yet, another ubiquitous source of oxygen, such as water (cf. water-gas shift reaction/WGS: H2 2 +CO2 or methanol reforming H2O+CH3 2 +CO2) has not been studied so far. Since these reactions result in the generation of molecular hydrogen their application in the field of mobile energy conversion (fuel cells) became a focus in latest research. Classical catalysts for the WGS are designed for large scale industrial applications, skeletal gold based catalysts in the form of coatings and structured catalysts are an attractive alternative. However, the reactivity of pure Au for the chemisorption and dissociation of H2O is very limited.3 One way to overcome this hurdle is to combine gold with a suitable oxide generating a bifunctional catalyst.
Most recently, our group reported that ceria deposits inside a nanoporous goldcatalyst resulted in an active and highly stable catalyst for the WGS under ambient pressure conditions at temperatures up to 500 °C. In the continuation of our research we pursued the question how to further improve the activity of this type of catalyst, for example, by deposits of mixed metal oxides of Ti and Ce. Incorporation of Ti atoms into the ceria lattice was demonstrated to result in a higher activity for the dissociation of water.4 We accordingly synthesized titania–ceria mixed oxides supported on nanoporous gold based on sol-gel chemistry. The resulting catalysts were investigated in a plug-flow laboratory scaled reactor for the water-gas shift reaction as well as the methanol reforming reaction. The observed activity of the catalysts was strongly dependent on the ration of Ti and Ce. Interestingly, while pure
421
Catalysis P11-06

TiO2 is less active than CeO2 it can increase the activity of the catalyst by nearly 100 % if added to the latter one in the right amount (Figure). In-situ infrared investigation (DRIFTS) and Raman spectroscopic characterization of the catalysts indicate a high density of defects inside the metal oxides which are active sites for the chemisorption of water.
Figure: On the left, scanning electron micrograph (x-SEM) of a cross section of a disk of nanoporous gold containing a mixture of Ti and Ce oxide (quantified by EDX). On the right: Production of CO2 from the oxidation of CO by water. The activity of the catalyst strongly depends on the ratio of Ti to Ce (5.8 vol% CO, 22.0 vol% H2O, 300 °C). As inset: A higher magnification x-SEM showing the gold ligaments and pores of the catalysts.
References
1. V. Zielasek, B. Jürgens, C. Schulz, J. Biener, M. M. Biener, A. V. Hamza and M. Bäumer, Angew. Chem.-Int. Edit., 2006, 45, 8241-8244.
2. A. Wittstock and M. Baumer, Accounts Chem. Res., 2014, 47, 731-739.3. J. A. Rodriguez, S. D. Senanayake, D. Stacchiola, P. Liu and J. Hrbek, Accounts Chem. Res.,
2014, 47, 773-782.4. J. Graciani and J. F. Sanz, Catal. Today, 2015, 240, Part B, 214-219.
422
CatalysisP11-06

Electroplating of polyurethane foams
S. Keck, A. Jung, R. Hempelmann, H. NatterSaarland University, Saarbrücken/D
Dwindling resources and increasing material costs are growing problems which influence future products and their production processes. So the future goal should be the decrease of sheer waste of resources by enhancing the material properties.Foams are a very interesting class of bio inspired light-weight materials [1].
In the present contribution we show a procedure for metallization of polymer foams by direct current plating. The essential electronically conductivity of the polymer foam was achieved in a priming process with conductive lacquer. The surface of the foam was coated with a 10 μm thick copper layer which allows electroplating ofnanocrystalline nickel or different alloys. By this procedure a highly strengthened material can be produced with the open foam structure of the used template.Compared to the bulk material the metallized foam shows a drastic weight decrease. These properties promise future applications for light-weight constructions in the automotive or aerospace industries.
The electrodeposition was performed in a specially designed plating cell which allows the electroplating of foams with a size of 240x240x40mm³. Some nickel alloys are tested on a smaller scale. The hydrodynamic conditions are one of the most important process parameter. For this reason a segmented flow cell was developed which allows a precise control of the electrolyte flow through the foam sample. The modular construction of the cell enables an easy upscaling.
[1] A. Jung, E. Lach, S. Diebels, New hybrid foam materials for impact protection,International Journal of Impact Engineering, 64 (2014), pp. 30–38
423
Electrochemistry and Energy P12-01

New catalyst supports for energy conversion prepared by surface modification of Graphene- and CNT-structures
Eun-Jin Oh; R. Hempelmann; H. Natter, Saarland University, Saarbrücken, Germany
A new, facile and green method was developed to prepare nitrogen-coated graphene- and carbon nanotubes (CNT)-based materials with a meso- and micro porous surface structure. New catalyst supports with different morphologies were synthesized by surface modification with nitrogen containing ionic liquids (IL). The surface modification was done by impregnating carbon structures in nitrogen containing ILs followed by a thermal treatment in air. The influence of the main experimental parameter such as IL amount, temperature and substrate morphology were investigated and the structure and the chemical composition of the resulting coating were analyzed by electron microscopic techniques (SEM, TEM), energy disperse X-ray analysis (EDX), X-ray photoelectron spectroscopy (XPS) and hot extraction analysis. It was found that the coating has a thickness of about 5 nm and a very high homogeneity. The modified surface has a nitrogen content of ~35 wt% which decreases strongly for temperatures above 600°C. Electrochemical characterization of polymer electrolyte membrane fuel cells (PEMFC) demonstrate that modified Graphene and CNT supported Pt catalysts (Pt loading: 0.29 mg cm-2)lead to a high performance (0.9 A cm-2 at 0.65 V) and low polarization loss compared to unmodified Graphene and CNT supported Pt catalysts.
TEM images of commercial (a) AO-2 graphene, (b) C150P nanotubes and nitrogen-coated (c) N-G, (d) N-CNT.
424
Electrochemistry and EnergyP12-02

Characterization and improvement of the active materials in thevanadium redox flow battery
K. Weißhaar a, H. Natter a, R. Hempelmann aa Institute of Physical Chemistry, Saarland University, Campus B2 2,
66123 Saarbrücken, Germany
The vanadium redox flow battery (VRFB) is an electrochemical energy storage device where the energy is stored in two separated liquid electrolytes. The electrolyte contains vanadium in four oxidation states, (II)/(III) on the negative side and (IV)/(V) on the positive side. The species undergo redox reactions in the half cells which provide the electrical energy [1]. Commonly graphite felts are used as electrodes separated by an ion-exchange membrane [2]. In order to increase the efficiency of the VRFB several graphite felts were tested without pre-treatment, with chemical and thermal oxidation and after modification with carbon materials like carbon nanotubes, graphene and carbon black. The electrochemical behaviour and the performance were investigated by cyclic voltammetry, polarization curve analysis and single cell charge/discharge tests. First investigations showed a significant increase of the energy efficiency from untreated felts (average value: 58%) to chemical and thermal treated ones (average value: 77%). The efficiency of the system also depends on the right choice of the membrane. Thus several membranes were characterized concerning their area resistance, proton conductivity, diffusion of the V-species through the membrane and performance at the charge/discharge tests.
[1] Z. González, C. Botas, C. Blanco, R. Santamaría, M. Granda, P. Álvarez, R. Menédez, J. Power Sources 241 (2013) 349-354.
[2] X. L. Zhou, T. S. Zhao, L. An, C. Zhang, Electrochim. Acta 153 (2015) 492-498.
425
Electrochemistry and Energy P12-03

Development of an improved state – of – charge indicator for the all - vanadium redox flow battery
J. Geiser, H. Natter, R. HempelmannSaarland University, Saarbrücken/D
One major challenge utilizing renewable energies for electricity generation is tobalance the discrepancy between power generation on the one hand and real demand of electric energy on the other hand by temporary storage. A potential technology is represented by the all-vanadium redox flow battery. For chemically energy storage this type of battery uses vanadium redox couples both at anode andcathode. However the determination of the battery charge condition is still not sufficiently solved yet. Therefore an improved state-of-charge indicator (SOC) is being developed. Severaltechniques have been applied to determine the state of charge of vanadium electrolytes. Based on a modified procedure of Corcuera et al. [1] electrochemical measuring procedures (potential-, conductivity measurements) were performed andSOC values were calculated from the resulting potential data. Furthermore relative contents of vanadium species obtained by optical methods (raman- and uv-visspectroscopy) were evaluated with a fitting routine. Potentiometric titration was used for absolute determination of the vanadium species. Therefore the relative methods can be calibrated using the potentiometric titration results in order to get absolute SOC- values. Both types of measurements were carried out in a new developed flow cell which will be tested in a 100 kWh pilot plant (electric charging station for electric cars combined with solar modules).
[1] S. Corcuera, M. Skyllas-Kazacos, Eur. Chem. Bull. 2012, 1(12), 511 - 519
426
Electrochemistry and EnergyP12-04

Perovskites containing protons, oxygen vacancies and holes: experiments and simulations for proton uptake and stoichiometry
relaxation kineticsR. Zohourian, R. Merkle, J. Maier, MPI for Solid State Research, Stuttgart, Germany
Fuel cells based on proton conducting ceramic electrolytes such as BaZrO3 are of increasing interest because such electrolytes offer a higher ionic conductivity at intermediate T (300-600°C) compared to oxide ion conductors. In this context we deal with oxides comprising three mobile carriers: protons, oxygen vacancies and holes. The presence of three carriers leads to interesting phenomena: (i) proton uptake can occur by water incorporation or by hydrogen incorporation [1], (ii) the stoichiometry relaxation after an increase in pH2O can develop a peculiar non-monotonic behavior because hydrogen and oxygen uptake are not necessarily directly coupled to each other [2]. This is analyzed in detail with numerical simulations [2,3].Experimental results (thermogravimetry) are presented for proton uptake into
perovskites such as Ba0.5Sr0.2Fe0.8Zn0.2O3- and BaCo0.4Fe0.4Zr0.2O3- which are considerd as potential cathode materials for proton conducting ceramic fuel cells.Regimes of proton uptake by hydration (low pO2) and hydrogen uptake (high pO2)are identified. The maximum proton concentrations in these perovskites are significantly lower than in BaZrO3-based electrolyte materials at same T and pH2O.The variation of proton concentration with cation composition in (Ba,Sr,La)(Fe,Co,Zn,Zr)O3- perovskites is discussed.
[1] D. Poetzsch, R. Merkle, J. Maier, Faraday Discussions 182 (2015) 129[2] D. Poetzsch, R. Merkle, J. Maier, Adv. Funct. Mater. 25 (2015) 1542[3] R. Merkle, R. Zohourian, J. Maier, Solid State Ionics (2015) accepted
427
Electrochemistry and Energy P12-05

Investigation of co-adsorption and side processes during underpotential deposition of Cu and Ag on Au(111) byelectrochemical microcalorimetry
S. Frittmann, V. Halka, R. Schuster, Institute of Physical Chemistry,
Karlsruhe Institute of Technology, Kaiserstraße 12, 76131 Karlsruhe, Germany
Heat effects in electrochemical reactions at the electrode/electrolyte interface provide direct information about the overall entropy change.[1] In our contribution, we presentmicrocalorimetric (galvanostatic) experiments which consecutively measure the involved entropy changes whilst stepping through the different stages of underpotential metal deposition (UPD).For Au(111) in CuSO4/H2SO4 the measured entropy changes showed co-adsorption of cations in addition to anion adsorption. Additionally, our measurements suggeststrong anion desorption just before Cu bulk deposition.For Ag bulk deposition on Au(111) we measured negative entropy changes in different aqueous solutions containing sulphate or perchlorate as expected for the immobilization of ions at the electrode. For the most pronounced Ag UPD waves the reaction entropy was essentially the same as for Ag bulk deposition, indicating that the dominating process is Faradaic Ag deposition. In particular, co-adsorption processes would lead to additional negative entropy contributions, which was not observed.Furthermore, during intermediate stages of Ag UPD at which only small amounts ofAg are deposited, a more positive reaction entropy was measured. From our data we suggest that an anion/hydroxide exchange with positive reaction entropy takes place. In consequence, we show that microcalorimetry in combination with classical electrochemical techniques provides complementary information on underpotential metal deposition elucidating side reactions of charge neutral species or co-adsorption.
[1] J.N. Agar, Thermogalvanic cells, in: P. Delahay (Ed.), Advances in Electro-chemistry and Electrochemical Engineering, vol. 3, Interscience, London, 1963.
428
Electrochemistry and EnergyP12-06

Designed architectures of electrode materials for advanced Li (Na)-ion batteries
Yan Yu,*,† Chao Wu ‡ Jun Liu,‡ Changbao Zhu, ‡ Joachim Maier†Department of Materials Science and Engineering, University of Science and
Technology of China, 230026, Hefei, Anhui, P. R. China.‡ Max Planck Institute for Solid State Research, Heisenbergstr. 1, Stuttgart, 70569,
Germany
Due to growing demand for hybird electric vehicles (HEVs) and smart grids, intensive efforts have been developed to design better electrode materials of lithium ion batteries (LIBs) and Sodium-ion batteries (SIBs) with high rate capability, long cycle life and high energy density. The key to the high performance batteries lies in the batteryʼs electrode materials. The search for better electrode materials has been the most dynamic motivation for the R&D of LIBs (SIBs). To obtain a noticeable improvement in the rate performance of LIBs (SIBs), it is essential to develop electrode materials with short pathway for both electrons and ions. To meet thekinetic and thermodynamic requirements for advanced batteries, the most popular approach is to develop better electrode materials with nanostructured that possesses kinetic advantages due to the shortened ions diffusion length. Another effective approach to improve the electrochemical performance of batteries is to design materials with conductive carbon network, providing an efficient electron pathway to offer better electrical contacts. In this talk, I will summarize recent progress of our group in the structural design, chemical synthesis, and electrochemical properties of electrode materials (i.e. MoS2, FeS2, Li4Ti5O12, Ge etc) with nanocarbon networks for LIBs.[1-4] By rational structure and carbon coating design, the lithium storage performance of these materials were significantly improved. Additionally, I will report our latest development in the double carbon coating design for Na3V2(PO4)3 cathode with excellent sodium storage performance and rate capability.[5]
Selected publications: 1. Zhu C.,Mu X. K.,van Aken P. A.,Yu Y.,* and Maier J. Angew. Chem. Int. Ed.
53(8), 2152–2156 (2014).
429
Electrochemistry and Energy P12-07

2. Liu J., Song K. P., Zhu C. , Chen C.-C., P. A. van Aken, Maier J. , Yu Y.,* ACS Nano 8(7), 7051–7059 (2014).
3. Liu J. , Wen Y.-R., Wang Y. , Aken P. A. van, Maier J. , Yu Y.,*, Adv. Mater., 26(34), 6025–6030 (2014).4. Liu J. , Song K.-P., P. A. van Aken, Maier J. , Yu Y.,* Nano Letters 14(5), 2597–2603 (2014).5. Zhu C. , Song K.-P., Aken P. A. van, Maier J. , Yu Y.,*, Nano Letters 14(4), 2175–2180 (2014).
430
Electrochemistry and EnergyP12-07

On the calculation of excess functions of electrolytesAlexander Maurer, Hartmut Krienke, Dominik Horinek, Georg Schmeer, University of
Regensburg, Regensburg, [email protected]
Thermodynamic and transport properties of aqueous and non-aqueous electrolyte solutions are subject of both experimental and theoretical considerations. In detail, the electrical conductivity is an important property of any electrolyte in many fields of research and development. Especially scientists developing new electrolyte solutions for primary and secondary electrochemical cells, fuel cells and so called supercapacitors are interested in electrolyte conductivity. Beside the great progress of experimental work in the field of electrochemistry [1], also theoretical investigationsexperienced a similar evolution of both theoretical description and numerical compliance with experimental measurements. From this point of view, a theory would be desirable, which gives practical scientists the ability to determine thermodynamic and transport properties of electrolytes simultaneously. We developed a theory of both thermodynamic [2] and transport properties [3,4] of aqueous and non-aqueous electrolyte solutions concerning ionic solvation and association. It is a combination of molecular dynamics simulations on Born-Oppenheimer level to get the potentials of mean forces (pmf) between ions, and integral equation techniques on McMillan-Mayer level, to calculate the excess functions with these pmfʼs. In nonequilibrium it is based on Onsagers continuity equation and an Ornstein-Zernike equation approach, which uses the concept of the direct correlation force.___________________________[1] C. Daniel, J. O. Besenhard, Handbook of Battery Materials, Wiley-VCH, 2011[2] G. Schmeer, A. Maurer, Phys. Chem. Chem. Phys., 2010, 12, 2407–2417[3] H. Krienke, G. Ahn-Ercan, A. Maurer, On the Influence of Molecular Structure on the Conductivity of Electrolyte Solutions – Sodium Chloride in Dioxane–Water Mixtures, Zeitschrift für Physikalische Chemie, 2013, 227, 285-302 [4] H. Krienke, Condensed Matter Physics, 16 (2013), 43006: 1 - 12
431
Electrochemistry and Energy P12-08

Manganese oxides for the oxygen evolution reactionJ. Doerfer, J. Schuch, B.Kaiser, W. Jaegermann
TU Darmstadt, Institut für Materialwissenschaften, Jovanka-Bontschits-Str. 2,64287 Darmstadt
[email protected]@surface.tu-darmstadt.de
The electrolysis of water into hydrogen and oxygen provides a promising solution to generate and store hydrogen from renewable energy sources. A major obstacle in this process is the slow reaction rate of the oxygen evolution reaction, which involves four electron transfer steps, each possesses a large activation energy, leading to ahigh overpotential. The benchmark catalysts for this reaction are still the rare and expensive IrOx and RuOx compounds.Our work focuses on the development of cheap and abundant manganese oxide as thin film catalysts via different deposition techniques such as chemical vapor deposition and physical vapor deposition. The electronic structure of the resulting films is characterized by X-ray photoelectron spectroscopy. The catalytic performance is obtained via a standardized electrochemical protocol using a three-electrode setup. The electrocatalytic activity is assessed from cyclovoltammograms,while the electrocatalytic stability is investigated by chronoamperometrymeasurements.
432
Electrochemistry and EnergyP12-09

In situ method for efficient catalyst layer production with very low catalyst loading
Roxanne Engstler, Dan Durneata, Harald Natter, Rolf Hempelmann, Saarland University, Saarbrücken/Deutschland
The catalyst layer (CL) on proton exchange membrane fuel cell (PEM-FC) contains electro catalysts, attached on different carbon structures (carbon black particles,carbon nanotubes, graphene multilayers etc.) and an ionomer. In order to assure the reactants distribution on entire structure it should be high porous and have a good electrical conductivity. Additionally, for a good electrochemical activity the catalyst should stay at the three phase boundary. In present work we use a new method for CL production by spraying a platinum precursor onto a commercial gas diffusion electrode (GDL) with a subsequent electrochemical reduction using a potentiostatic method. Parameters as spraying pressure, temperature and volume are varied. It was found that the best parameter combination is: 0.2 bar spraying pressure, 50°Csubstrate temperature and 2 mL platinum precursor solution (0.06 mol/L). Further, the CL structure was improved by intercalating the precursor in the ionomer layers. All prepared samples were characterized by optical (REM, TEM) and electrochemical methods (CV, EIS, single cell fuel cell test). New structured active layers show powerdensities over 0.9 A/cm2 (at 0.5V, 80°C, 95% RH, H2/O2).
433
Electrochemistry and Energy P12-10

BaTiO3 Modifications for Photochemical Hydrogen Evolution
Yan Xiong, Frank MarlowMax-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, Germany.
The perovskite-type material BaTiO3 is a wide-bandgap semiconductor and a prospective material for the photochemical hydrogen evolution via water splitting. In our work, we studied reduction-treatments of BaTiO3 and the effect of Pt as a co-catalyst on BaTiO3 for the hydrogen evolution reaction. We found that the catalytic activity of reduced-BaTiO3 is enhanced by a factor of 1.5 compared to the original material. The rates of hydrogen generation of reduced and bare BaTiO3 are 159 and 100 mol/h, respectively. The effect of Pt as a co-catalyst on BaTiO3 is non-linear. The photocatalytic activity increases up to 527 mol/h with the increasing Pt amount up to 1 wt.% and decreases above this amount. In our study reduced BaTiO3 with 1 wt.% Pt has shown the best performance.
434
Electrochemistry and EnergyP12-11

Long-term Stability of Silicon-Poisoned La0.6Sr0.4Co0.2Fe0.8O3-
SOFC-CathodesMartin Perza, Edith Buchera, Andreas Eggera, Christian Gspanb, Ferdinand Hoferb,
Werner Sittea
a Montanuniversitaet Leoben, Chair of Physical Chemistry, Austriab Institute for Electron Microscopy and Nanoanalysis (FELMI), Graz University of
Technology & Graz Centre for Electron Microscopy (ZFE), Austria
The long-term stability of the cathode is an important requirement for a wider commercial utilization of the solid oxide fuel cell (SOFC) technology. Among other impurities, silicon has a negative influence on the stability of a number of cathodes.In this study the influence of Si on porous La0.6Sr0.4Co0.2Fe0.8O3- (LSCF) cathodes was investigated in a long-term experiment. A symmetrical cell was fabricated by screen-printing porous LSCF layers on both sides of a solid electrolyte substrate of Ce0.9Gd0.1O2- . The evolution of the electrochemical behavior of the electrodes during 2000 hours at 700°C was characterized by impedance spectroscopy. In the first 1000 hours the as-produced cathodes were characterized. Afterwards an artificial Si-poisoning of the cathodes was induced by sputtering Si-layers of approx. 10 nm on both sides of the sample. Impedance measurements on the contaminated cell for additional 1000 hours revealed a dramatical decrease of the electrochemical performance, causing an increase of the area specific polarization resistance (ASR) from 1.4 to STEM analyses of the degraded sample a 50 nm thick secondary silicate phase layer was found in a surface-near region of the cathode. It can be concluded that contaminations with even small amounts of silicon drastically reduce the oxygen exchange reaction kinetics.
435
Electrochemistry and Energy P12-12

Development of a photocapacitor based on printed solar cells and supercapacitors
Katrin Anneser1, Stephan Braxmeier1, Andreas Baumann1, Gudrun Reichenauer1,Vladimir Dyakonov1,2
1Bavarian Center for Applied Energy Research, Würzburg/Germany2Julius Maximilian University, Würzburg/Germany
The main drawback of renewable energy sources, such as wind and solar energy, are unpredictable power fluctuations presenting a major challenge to storage devices with limited cycleability in terms of charging and discharging. For reducing the number of cycles initiated by high frequency fluctuations, such as caused by clouds in case of solar power, it is necessary to smooth the power before feeding it into a battery. This smoothing can be achieved without significant loss of energy by integration of a fast storage device with high cycle stability, e.g. an electrochemical double layer capacitor, also known as supercapacitor.
Rather than developing a modular system consisting of solar cell and supercapacitor our objective is an integrated layered system provided by printing the components from solution processed precursors. This hybrid system will provide a more constant power output compared to stand-alone photovoltaic systems and thus allow feeding into storage units with far slower kinetics or into the grid.
We show experimental data from solar cells measured for the first time at a high frequency (every second) and use these data to derive basic requirements in terms of area specific power and energy density required for the fast energy storage unit of the integrated system; in addition, we discuss different concepts for the implementation of the hybrid system, in particular printable solid electrolytes for the supercapacitor component.
436
Electrochemistry and EnergyP12-13

Electron bombardment induced cation transport in an ion conducting glass
Anneli Hein, Jan Wiemer and Karl-Michael Weitzel, Philipps Universität, Marburg
The technique of Bombardment Induced Ion Transport (BIIT) has recently been developed for measuring ionic conductivities [1, 2] and electro-diffusion profiles [3] of solid electrolytes. The original version is based on attaching a cation beam to the sample (cation-BIIT) inducing the transport of such cations towards a grounded backside electrode. In the current work the BIIT technique is extended to electron bombardment (e- - BIIT).Here, a low energy electron beam has been attached to a mixed sodium and potassium conducting borosilicate glass mounted on a single grounded backside electrode. Attachment of the electrons to the surface induces cation transport towards this surface. In the first part of the experiment current-voltage curves have been measured. The specific conductivities as well as the activation energy for ion hopping derived agree with cation-BIIT data. In the second part of the experiment we have performed a long term electron bombardment of the glass sample at -25 eV. Ex situ – after the bombardment - the sample has been analyzed by time-of-flight secondary ion mass spectrometry (ToF-SIMS). The depth profiles obtained from the ToF-SIMS analysis clearly demonstrate that sodium has been neutralized at the front side of the glass sample due to the recombination of electrons with sodium ions. Reduction of the less noble potassium ions appears to be suppressed. The electrochemical implications of this observation will be discussed.
[1] M. Schäfer, K.-M. Weitzel, Phys. Chem. Chem. Phys., 13, 20112-20122, (2011).[2] P. V. Menezes, J. Martin, M. Schäfer, H. Staesche, B. Roling and K.-M. Weitzel. Phys. Chem.
Chem. Phys., 13, 20123–20128, (2011).[3] L. Rossrucker, P.V. Menezes, J. Zakel, M. Schäfer, B. Roling, and K.-M. Weitzel. Z. Phys.
Chem. 226, 341–353, (2012).
437
Electrochemistry and Energy P12-14

Sulfide electrolytes based on Glyme-Lithium salt mixtures for new battery systems
Anne-Marie Bonsa, Andreas Appelhagen, Jochen K. Lehmann, and Ralf LudwigInstitute of Chemistry, Physical Chemistry, University of Rostock,
Dr.-Lorenz-Weg 1, D-18051 Rostock, Germany
Electrical energy storage by new battery systems has become a focus of interest due to the energy system transformation. Therefore, suitable and well performing electrolytes have to be designed to make new electrical cells applicable.Mixtures of the oligoethers monoglyme (G1), triglyme (G3) or tetraglyme (G4) with the salt lithium bis(trifluoromethylsulfonyl)amide (Li[NTf2]) show low volatility, high thermal stability, high ionic conductivity and a wide potential window. In particular thecomplex [Li(G3)1][NTf2] is presumed to be resistive against oxidation and hence the equimolar triglyme salt mixture is assumed to be a promising electrolyte for lithium batteries [1]. The addition of Li[NTf2] also increases the solubility of alkali sulfides in triglyme, as shown by Park et al., making such systems candidates for electrolytes in sulfide batteries [2].In this work we studied the transport properties of the (Gn)-lithium salt mixtures with a combination of experiments and molecular dynamics simulations. Particularly, viscosities and conductivities were measured as a function of temperature and salt concentration. One important goal is to understand the properties of all mixtures at molecular level, especially with respect to the influence of ion pair formation. The addition of small amounts of Li2S to the triglyme salt mixtures significantly increases the ionic conductivity whereas the viscosities do not change at all. These solutions are therefore promising and suitable to be used in sulfide battery applications [3].
[1] K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko, M. Watanabe, J. Phys. Chem. C, 2011, 115, 18384-18394.
[2] J. Park, K. Yamauchi, E. Takashima, N. Tachikawa, K. Ueno, K. Dokko, M. Watanabe, J. Phys. Chem. C 2013, 117, 4431-4440.
[3] P. Thiele, J. Neumann, A. Westphal, R. Ludwig, A.-M. Bonsa, A. Appelhagen, P. Malcher, M. Köckerling, to be submitted
438
Electrochemistry and EnergyP12-15

Electrochemical study of Si-nanodroplets deposited on graphite using a LPCVD process
K. Nimmrich1, D. Klein1,T. Bartoldus2, S.Cap1 and R. Schlögl1,2
1Fritz-Haber-Institut der Max-Planck Gesellschaft, Berlin/Germany; 2Max-Planck-Institut für Chemische Energiekonversion, Mülheim an der Ruhr/Germany;
To treat the earthʼs natural resources with care, one approach is to increase the field of application of Lithium Ion Batteries. Therefore, efforts are underway to substitute the graphitic anodes used nowadays by silicon materials. This Li-alloying element exhibits an outstanding specific capacity and a well-developed infrastructure.[1]
Regrettably, classical Si-based anode materials feature a poor electrochemical performance, which can however be improved through nano-structuring[2].
We present a comparative study of composite anodes made of Si-nanoparticles (SiNPs) and artificial graphite (KS6) (Fig. 1a), and Si-based anodes obtained from Low Pressure Chemical Vapour Deposition (LPCVD) of SiH4 on KS6 (Fig. 1b). The silicon deposition is performed using a custom-made rotating reactor and is intended to favour a homogenous silicon distribution,decrease the mechanical stress within the material during (de-)lithiation and retain the contact of Si to the substrate during cycling.[3] The attention is drawn to the comparison of the electrochemical performance and stability of the LPCVD-obtained and composite anodes(Fig. 2). Moreover, further comprehension of the electrochemical processes is gleaned by intensive microscopic and spectroscopic characterization of the materials before and after electrochemical aging.
References:[1] M. T. McDowell et al., Adv. Mater. 2013, 25, 4966-4985.[2] X. H. Liu et al., ACS Nano. 2012, 6, 15522-1531.[3] M. Holzapfel et al., Electrochem. Solid-State Lett. 2005, 8,A516-A520.
Fig. 1: SEM image of a) KS6-SiNPs anode b) Si-nanodroplets on KS6 after SiH4 LPCVD.
Fig. 2: Specific capacity over 50 cycles of pure KS6, KS6-SiNPs composite and KS6 after SiH4 LPCVD.
1a)
1b)
Si
439
Electrochemistry and Energy P12-16

Development of Electrocatalysts for Alkaline Water ElectrolysisAnnette-E. Surkus, Mykola Polyakov, Nils Rockstroh, Henrik Junge,
Andreas Martin, Matthias Beller,Leibniz-Institute for Catalysis e.V. at the University of Rostock,
Albert-Einstein-Str. 29a, D-18059 Rostock, Germany
Utilization of renewable energy sources such as wind or solar energy is strongly related with the issue of storage and transport of this intermittently provided energy. Transformation of this energy to hydrogen or synthetic natural gas is considered to possess the highest potential regarding long-term storage capacity as well as transportation over long distances. Moreover, the energy stored in hydrogen can be converted back to electricity by fuel cells on demand.
Water electrolysis represents the first and most essential step in conversion of electricity to hydrogen or synthetic natural gas. Currently, this key technology is accomplished with either alkaline or PEM electrolysis. Thereby, the performance is strongly dependent on the type of electrocatalyst used. As basic requirements, a low overpotential as well as high current densities and long term stability are identified. However, noble metals as main components shall be replaced in order to conserve resources and to decrease the price.
Within the framework of the project “Noble metal free electrocatalysts and composite electrodes for water splitting – Sustainable Hydrogen (SusHy)” funded by the German Minsitry of Education and Research (BMBF), nanoparticulate electrocatalysts for the oxygen evolution reaction (OER) have been prepared and electrochemically tested. The OER catalysts were generated by wet-chemical and thermal treatment starting from transition metal precursors. Furthermore, noble metal free systems with varying nitrogen contents have been synthesized and investigated as potential cathode catalysts in the hydrogen evolution reaction (HER).
440
Electrochemistry and EnergyP12-17

Preparation of titanium dioxide thin films by atomic layer deposition applied on tandem solar cells for photoelectrochemical water
splittingThorsten Cottre, Jürgen Ziegler, Bernhard Kaiser, Wolfram Jaegermann, Technische
Universität Darmstadt, Institut für Materialwissenschaft, Jovanka-Bontschits-Str. 2,64287 Darmstadt
In this study catalytic effective photocathodes on the basis of thin-film silicon solar cells are presented for photoelectrochemical water splitting. To protect against corrosion, nanoscopic layers of titanium dioxide are applied as passivation. The titanium dioxide layers are generated by atomic layer deposition, in which titanium tetrachloride and distilled water are used as precursors. In order to ensure ahomogeneous coating, an ALD reactor was designed and the deposition parameters were systematically optimized. For the optimization of the chamber p-type silicon wafers were used, which were characterized by ellipsometry to determine the titanium dioxide layer thicknesses. After deposition the applied layers were investigated by X-ray photoelectron spectroscopy (XPS). To characterize the film morphology scanning electron microscopy (SEM) was used. After the deposition of the passivation layer platinum particles are applied electrochemically to reduce the overpotential for the hydrogen evolution reaction. The investigation of the photocathodes, with regard to their effectiveness and their stability, is performed by various electrochemical methods. The titanium dioxide passivation layer in combination with the catalytically active platinum particles has a high stability in 0.1 M KOH solution without decreasing the efficiency of the tandem structure significantly.
441
Electrochemistry and Energy P12-18

Metrological support for LNG custody transfer and transport fuel applications
Karine Arrhenius1, Michael Downey2, Peter Eilts3, Hugo Ent4,Bjoern Gieseking2, Oswin Kerkhof4, Jianrong Li4, P. Alberto Giuliano Albo5,
Arnas Lucassen6, Gerard Nieuwenkamp4, Jürgen Rauch6,Markus Richter7, Adriaan M.H. van der Veen4
1 – SP Sveriges Tekniska Forskningsinstitut AB (SP), Sweden; 2 – National Physical Laboratory (NPL), United Kingdom; 3 - Institut für Verbrennungskraftmaschinen, Technische Universität Braunschweig; 4 - VSL Dutch Metrology Institute, the Netherlands; 5 – Instituto Nazionale di Ricerca Metrologica (INRiM), Italy; 6 –Physikalisch-Technische Bundesanstalt (PTB); Braunschweig; 7 – Ruhr-UniversitätBochum
Liquefied Natural Gas (LNG) is an important energy source all over the world. Currently VSL is coordinating a European research project (EMRP) aiming at:
1) mid-scale calibration standard for LNG mass and volume flow measurement;2) reference standard for LNG sampling and composition measurement systems; 3) methane number; and4) developing and validating equation-of-state for LNG density prediction.
These calibration and reference standards, methods and models are indispensable for improving the custody transfer. The state-of-the art is primarily based on the tank level measurements for quantity and gas chromatographic composition measurements. The flow metering and use of improved methods for LNG sampling will lead to a reduction of the uncertainty in the energy traded.
The methane number (MN) qualifies LNG as transport fuel. The work undertaken in the project aims at improving the correlation between the LNG composition and the MN. For the use of LNG as transport fuel, not only the MN at the time of trading is relevant, but also its change due to ageing (decrease in the MN) of the fuel in storage tanks.
We present an overview over the issues concerned and the results obtained so far.
442
Industrial ApplicationsP13-01

443
LIST OF AUTHORS

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
444
Name Page
A
Abdi, F.F. P01-07, V14.01
Abel, B. V03.08
Abetz, V. P08-02
Adam, M. P07-02, P07-04, P07-21
Adel, P. P07-03
Ahlberg, S. P10-12
Ahlrichs, R. V04.04
Ahn-Ercan, G. P01-13
Ahrens, J. P01-20
Alberico, E. V01.13
Albert, S. P03-19
Albo, P.A.G. P13-01
Allehyani, B. P04-20
Al-Shamery, K. P07-19
Altnöder, J. V04.06
Altschäffel, J. P09-22
Alves da Rocha, M. P02-08, P02-09, P02-10
Amende, M. FL01.02, P09-04, P09-17
Andersen, J. P03-08
André, A. V07.05
Andrei, V. V01.21
Angelova, P. P08-02
Anneser, K. P12-13
Anta, J. V12.08
Antonsson, E. P03-09, P09-07, V09.07
Appelhagen, A. P12-15, V02.02
Arenas, B. P04-10
Arnas, L. P03-05, P03-21
Arrhenius, K. P13-01
Arrigo, R. V12.12
Assefa, T. P01-30
Auerbach, D. P03-17, P06-03, P09-24, P09-26,
V14.08
Awakowicz, P. P10-16
Awel, S. P05-12
Aziz, E. P01-06, P02-17, P03-15, P04-05
Aziz, S. P04-20
Name Page
B
Bächle, J. P01-02
Bachmann, J. P07-09, P09-12
Bachmann, P. P01-10, P09-12, V09.04
Bachmann, S. P10-12
Backus, E. FL09.01
Bahnemann, D.W. V01.23
Bajnoczi, E. P01-30
Bald, I. V10.04, V10.05
Baldauf, J. V07.06
Baldus, S. P10-16
Bande, A. P02-17, P03-11, P03-15, P04-05
Bäppler, F. P01-16
Barr, M. P07-09
Barsch, E. V13.06
Bartels, C. P09-20
Barthelmes, K. V01.16
Bartoldus, T. P12-16
Bauer, C. P07-21
Bauer, M. P05-01, V01.10, V06.04
Bauer, T. P09-21
Bauer, U. P01-05, P09-12, V09.04
Bauerecker, S. P02-20
Baumann, A. P12-13
Baumann, W. V01.13
Bäumer, A. P02-11
Bäumer, M. P11-01, P11-06
Baz, J. V08.03
Bebensee, F. P09-02, P09-27
Bechtel, M. P10-12
Becker, J.-P. P01-35
Behler, J. V09.11
Beitz, T. P05-08
Bekhtereva, E. P03-19
Beller, M. P01-29, P01-30, P01-31, P12-17, V01.10, V01.13
Benad, A. P07-21
Bendig, M. P10-19
Bentrup, U. V05.04
Name Page
Berger, R. V04.01
Berger, T. V09.08, V12.08
Bernardi, J. V07.04, V09.08
Bernhard, D. P04-06, V04.06
Bersenkowitsch, N.K. V03.05
Besold, D. P09-12
Bethke, K. V01.21
Beyer, A. P08-02
Beyer, M. V03.05
Bierbaum, S. V10.03
Bigall, N.-C. FL07.01, P01-17, P07-12, P07-17, P07-20, P07-25,
P07-26
Binder, R. P03-14
Bloh, J. P07-03
Bluhm, H. V14.01
Blume-Peytavi, U. P10-12
Bocklitz, T. P01-04
Böckmann, M. P01-27
Bohn, N. P10-19
Bokarev, S. P01-08, P01-30, P06-01
Bokareva, O. P01-08, V01.10
Boldt, K. P07-05, V07.06
Bolotova, I. P03-19, P04-15
Bonn, M. FL09.01, P02-01
Bonsa, A.-M. P02-19, P12-15
Borchert, H. V01.04
Börchert , J. P01-34
Börner, A. V11.01
Boubnov, A. P11-05, V11.04
Bourg, M. V08.08
Bourret, G. V12.08
Bräse, S. P01-16
Braun, S. P02-06
Bravic, I. P04-05
Braxmeier, S. P12-13
Bredenbeck, J. P03-07, P03-14, P04-19, P10-04, P10-15, P10-24,
V03.06
Bredol, M. V01.03
Bressel, L. P08-06
LIST OF AUTHORS

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
445
Name Page
Bressler, C. P01-30
Britz, A. P01-30
Broquier, M. P04-01
Brox, D. P10-14
Brückner, A. V05.04
Brückner, N. FL01.02
Brummel, O. P09-12, P09-21, V12.13
Bucher, E. P12-12
Buhro, W.E. V07.09
Bülter, H. V12.03
Bumüller, D. V04.04
Bünermann, O. P01-21, P09-24, V09.01
Burggraf, F. V13.04
Burghardt, I. P03-07, P03-14
Burke, U. P03-16
Burkhardt, L. P04-11
Buttersack, T. P02-20
Bykova, I. P10-12
C
Campen, R.K. P09-25
Cap, S. P05-03, P12-16
Casapu, M. P11-05, V11.04
Chanaewa, A. V12.09
Chandrasekhar, N. V12.02
Chen, C.-C. V12.04
Chen, L. P09-09
Chen, M. V09.06
Chen, R. V01.07
Chen, Y.-H. P09-16
Chen, Z. P04-15
Chiang, Y.-c. P03-15
Chiappisi, L. V08.01
Chinaryan, V. P08-02
Chmela, J. V01.17
Chmielewicz, W. V10.03, P10-23
Chuboda, R. P02-17, V14.05
Cinar, S. P10-09
Civale, K. P09-12
Clever, G.H. P01-20
Combe, S. P04-19
Name Page
Constantinescu, D. V14.07
Crumlin, E. V14.01
Czeslik, C. P10-09, P10-10
Czurlok, D. V02.08
D
Das, S. P01-28
DeBeer, S. PL04
Deibel, C. P01-24
Deichmöller, J. P05-04
Deiters, U.K. P06-07
Demir, H.V. P07-04
Deutschmann, O. V11.04
Dick, B. P03-23
Dierking, C. P02-16
Dietrich, F. P01-16, V04.06
Dietz, M. V05.02
Dietze, E. P06-02
Dietzek, B. P01-04, P01-28, P04-07, V01.16
Dilla, M. V01.11
Diller, R. P01-16
Dittmeyer, R. P05-10
Dittrich, R. V13.08
Diwald, O. V07.04, V09.08
Doerfer, J. P12-09
Doerfler, R. P02-22
Dohnal, V. V13.07
Dohnálek, Z. P09-02
Doltsinis, N. P01-27
Domratcheva, T. V05.02
Dong, R. V12.02
Döpper, T. P09-12, P09-17
Dorenkamp, Y. P01-21, P09-24, V09.01
Dorfs, D. P07-03, P07-12, P07-24, P07-25,
V07.01
Dörner, R. V04.01
Doronkin, D.E. P11-05
dos Santos Sardinha, E. V12.03
Doumy, G. P01-30
Downey, M. P13-01
Dräger, C. V01.19
Name Page
Drenckhan, W. V08.05
Drexler, H. V01.13
Dröge, F. P04-07
Düll, F. P09-28, V09.04
Duman, J.G. P02-11
Durneata, D. P12-10
Durrant, J. PL02
Dyakonov, V. P12-13
Dzubiella, J.
P02-17, V14.05
E
Ebbinghaus, S. V14.07
Eberl, K.B. P10-15, P10-24
Egger, A. P12-12, V13.02
Egorova, D. V03.04
Ehrenberg, H. V01.19
Ehrhard, F. P02-17
Eigler, S. V03.08
Eilts, P. P13-01
Einemann, M. V01.05
Eisbein, E. P02-14
Eisenhaber, S. P08-03, P08-04
El Khoury, Y. P10-04
Elsing, J. V08.05
Enders, F. P07-05
Endres, F. P09-03
Engstler, R. P12-10
Ent, H. P13-01
Erbe, A. P09-23
Erdem, T. P07-04
Erler, A. P05-08
Essig, M. P10-15
Euchner, H. P11-02
Eychmüller, A. P07-02, P07-04, P07-08, P07-21,
P09-29
F
Fabri, C. P04-15, V06.01
Fackler, O.T. V10.03
Faisal, F. V12.13
Fansa, E.K. P10-20
Fasshauer, E. P03-11
LIST OF AUTHORS

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
446
Name Page
Faßheber, N. P03-13
Fatima, M. P04-09
Favaro, M. V14.01
Fehre, K. P04-19
Feldhoff, A. V01.20
Feng, X. V12.02
Fernandes, R. P03-21, P06-06
Fiala, R. V12.13
Field, R. P03-06
Fingerle, M. V01.06
Fischer, H. P09-07
Fischer, S. V01.10
Flender, O. P01-03, V01.22, V03.03
Flesch, R. P10-12
Flyunt, R. V03.08
Forbert, H. V02.05
Forsting, T. P04-08
Frank, M. P01-20
Franke, R. V11.01
Freytag, A. P07-20
Friedrich, A. P01-31
Friedrich, K.A. V13.04
Friedrichs, G. P03-13, P04-14
Frittmann, S. P09-03, P12-06
Fu, L. V12.04
Fumino, K. P02-21
G
Gago, A. V13.04
Galchenko, M. P07-25
Galler, A. P01-30
Gaponik, N. P07-02, P07-04, P07-21, P09-29
Garidel, P. P10-06
Garling, T. P09-25
Gawelda, W. P01-30
Ge, Q. P09-02
Gebhardt, J. P10-07
Geiser, J. P12-04
Geist, N. P10-13, V10.07
Gellert, M. P10-13
Name Page
Gerhards, M. P01-16, P04-06, V04.06
Geweke, J. P09-20
Gheisi, A. V07.04
Gieseking, B. P13-01
Gil, M. P04-01, P04-02
Gilchrist, M. V08.05
Giuliani, A. V10.04
Gleichweit, C. FL01.02, V09.04
Gleim, J. V02.08
Gliemann, H. V14.06
Goerling, A. P09-17
Golka, L. P03-04
Golnak, R. P02-17
Golub, B. P02-03, P02-19
Gölzhäuser, A. P08-02
Goossen, J.-D. V01.03
Görling, A. P09-12
Görls, H. P04-07
Gottfried, J.M. V09.06
Gottschalk, H.C. P04-04
Gottwald, F. V06.02
Grabow, K. V05.04
Gradzielski, M. V08.01
Graf, I. P03-12, P03-16
Grätzel, M. OL01, V12.01
Gratzfeld, D. P03-02
Gregori, G. V12.01
Greisch, J.-F. V01.17, V05.01
Grell, G. P01-08, P06-01
Gremminger, A. V11.04
Groß, A. P11-04
Grunwaldt, J.-D. P05-10, P11-05, V11.04
Grüßer, M. P07-01
Grußmayer, K.S. P10-23, V10.03
Gspan, C. P12-12, V13.02
Guhrenz, C. P07-21
Günter, T. P11-05
Gusa, D. P05-12
Guthmuller, J. P01-04
Name Page
H
Haderspeck, A. P10-14
Hadler, T. P07-06
Hahn, H.W. P03-17, V14.08
Hajduk, A. P01-26
Halfpap, I. P03-09
Halka, V. P12-06
Haller, A. P03-15
Hänselmann, S. P10-23
Hansen, N. P10-07, V08.03
Hansen, N. V03.01
Harding, D. P03-17, V14.08
Harding, M.E. V01.17
Harms, C. V01.05
Hartmann, E. V05.02
Haubold, D. P07-08
Hausbrand, R. P09-05, V01.06
Hausen, F. P01-32
Hävecker, M. V12.12
Havenith, M. P01-22, P02-11, P02-13, P02-15, P05-04, P10-16, V02.06, V14.07
Haverkamp, J.-S. V12.14
Hebbeker, P. P08-01
Heck, C. V10.05
Heckel, A. P03-07, P03-14, V03.06
Heger, M. P03-08, V04.06
Hehn, A.-S. V04.04
Heib, F. V01.24
Hein, A. P12-14
Heister, P. V07.02
Heiz, U. P01-14, V01.02
Heiz, U. V07.02
Hellmann, R. V08.07
Hellström, M. V09.11
Hemberger, P. V14.02
Hemken, C. P03-16
Hempelmann, R. P12-01, P12-03, P12-04, P12-10,
V01.07
Hensen, E.J.M. V01.01
Herold, E. V08.07
LIST OF AUTHORS

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
447
Name Page
Herritsch, J. V09.06
Herrmann, C. P06-02
Herrmann, C. V14.07
Herrmann, F. P01-28
Herrmann-Westendorf, F. P04-07
Herten, D.-P. P10-14, P10-23, V10.03
Herzog, B. P10-27
Hess, D. V11.01
Hesse, S. P03-13
Heufer, K.A. P03-16, P06-06
Heyda, J. V14.05
Heyden, M. V02.07
Hilal, R. P04-20
Hille, C. P10-03, P10-05, V10.01, V10.06
Hinrichs, D. P07-03, P07-25
Hippler, M. P10-26
Hirsch, A. P09-12
Hoberg, C. P02-13, P02-15
Hofer, F. P12-12, V13.02
Höfert, O. FL01.02
Hofmann, G. P05-10
Hofmann, J.P. V01.01
Hollenstein, H. P03-19
Hollmann, D. V05.04
Holzer, C. V04.06
Hölzer, J. V03.03
Horinek, D. P12-08
Horke, D. P05-12
Horny, L. P04-15, V06.01
Horstmann, B. V12.06
Hosseinpour, S. P01-11
Houtepen, A.j. V07.08
Huang, Z. P05-12
Hunger, J. P02-01
Hupfer, M. P01-28, P09-30
Hüter, O. P03-10, V03.04
I
Ignatiev, N. V13.07
Imoto, S. V02.05
Indris, S. V01.19
Name Page
Ismail, S.A. P10-20
Ivanov, S. V06.02
J
Jacob, C.R. P11-05
Jaegermann, W. P01-19, P01-23, P01-26, P01-35, P09-05, P12-09,
V11.03
Jahnke, T. V04.01
Jakob, M. V07.02
Jan, P. P01-17, P07-26
Janke, S. P06-03, P09-24
Jasper, A. V03.01
Jebreiil Khadem, S.M. V10.01
Jehser, M. P02-05, P08-03, P08-04
Jensen, L. P01-25
Jiang, H. P01-21, P09-24, V09.01
Jiang, J. P03-06
Jiao, H. V01.13
Jones, T.E. P03-01, V12.12
Joseph, Y. V13.08
Joswig, J.-O. P02-14, P07-15
Jung, A. P12-01
Junge, H. P01-29, P01-30, P01-31, P12-17,
V01.13
Jürß, A. P11-03
K
Kahnt, A. V03.08
Kaiser, B. P12-09
Kaiser, B. P01-19, P01-23, P01-26, P01-35,
V11.03
Kammler, M. P06-03
Kandratsenka, A. P06-03, P09-10, P09-22, P09-24,
V06.03
Kanis, M. V14.01
Kapoor, S. P10-20
Kappes, M. V01.17, V04.04, V05.01
Karnahl, M. P01-29
Name Page
Kartaschew, K. P05-04, P10-16
Kartouzian, A. V07.02
Kast, S. P06-04, P06-05
Kaufmann, S. P09-26
Kautek, W. P03-18, P05-09
Keck, S. P12-01
Kensy, U. P03-23
Kerkhof, O. P13-01
Kerlé, D. P02-09, P02-18, V02.02
Kern-Michler, D. P03-07, P03-14, P04-19, P10-24,
V03.06
Khabaz, F. P02-22
Khalakhan, I. V12.13
Khan, M. P01-06
Khare, R. P02-22
Khaybulkina, E. P05-09
Kiefer, J. P02-08, P02-10, P05-06, P05-07,
V02.04
Kijak, M. P04-01, P04-02
Kinge, S. V07.08
Kipp, T. P07-06, P07-07, V07.09, V09.09
Kirchner, B. P04-19
Kirkwood, N. V07.06
Kitsopoulos, T.N. V14.08, P03-17
Klein, D. P12-16
Klein, J.R. P01-03, V01.22
Klein, P. V10.03
Klepzig, L. P07-26
Klink, S. V05.04
Klinke, C. V07.11
Klinkhammer, C. P01-22
Klopper, W. V01.17, V04.06
Klossek, A. P10-12
Klüner, T. P11-01
Knake, L. V02.06
Knapp, M. V01.07
Knolle, W. V03.08
Knop-Gericke, A. P03-01, V12.12
Knorke, H. P04-13
Knorr, A. P02-21, V02.03
LIST OF AUTHORS

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
448
Name Page
Kobylinski, M. P07-14
Kodanek, T. P07-03, P07-25
Kogelheide, F. P10-16
Kohnke, H.-J. V13.01
Kohse-Höinghaus, K. P03-03, P03-12, P03-16
Kolditz, A. P07-18
Kollmannsberger, S. P01-14, V01.02
Kolny-Olesiak, J. P07-19
Kondati Natarajan, S. V09.11
König, M. V11.01
Kornyshev, A.A. FL01.01
Kottke, T. V05.02
Kovács, G. V12.13
Kozlov, S. V12.13
Kraska, T. P02-06
Krause, C. V01.04
Krein, V. V04.03
Krienke, H. P12-08
Kröger, E. P07-27
Kroon, M. P02-10
Kropp, T. V09.05
Krüger, B. P09-08, P09-10, P09-11, P09-15,
V06.03
Kruss, S. P10-01, V07.10
Kübel, J. V01.16
Kubis, C. V11.01
Kucera, J. P11-04
Kühn, O. P01-08, P01-30, P06-01, V01.10,
V06.02
Kulkarni, A. V07.08
Kulke, M. P10-08
Kumar, N. P10-25
Kundu, A. V01.25
Kunitski, M. V04.01
Kunz, S. V11.02
Kupfer, S. P01-04
Küpper, J. P05-12, V04.02
Kus, J.A. P03-10
Name Page
L
Lackmann, A. P11-06
Lackmann, J.-W. P10-16
Lambert, C. V01.14, V01.16
Langel, W. P10-08, P10-13, V10.07
Langer, B. V09.07
Latz, A. V12.05, V12.06
Lauth, J. V07.08
Lebedev, M. P01-26
Lehmann, J.K. P12-15
Lennox, A. V01.10, V01.13
Lenz, D. P07-15
Lenzer, T. P01-03, V01.22, V03.03
Lesnyak, V. P09-29
Lettenmeier, P. V13.04
Levin, A. P10-10
Li, J. P13-01
Li, X. P09-01, V09.02
Liang, H. V12.02
Libuda, J. FL01.02, P09-04, P09-12, P09-17, P09-21, V12.13
Lillig, C.H. P10-13
Lindner, J. P01-09, P09-03
Lindner, J. V02.08, V03.02
Litke, A. V01.01
Liu, S. V12.02
Livingstone, R. FL09.01
Lochbrunner, S. P01-29, P01-30, P01-31
Lode, A. P03-11
Loerting, T. P02-05
Löffler, J.G. P10-15
Lohmann, S.-H. P07-07
Löhmannsröben, H.-G. P05-08, V10.01
Lopez, J.-C. V04.07
Lorenz, M. FL01.02
Lorenz, T. P02-14, P07-15
Lox, J.F.L. P07-04, P07-21
Lubitz, W. PL03
Lübkemann, F. P07-12, P07-20
Name Page
Lucassen, A. P13-01
Lück, J. V12.05
Ludwig, R. P02-02, P02-03, P02-12, P02-18, P02-19, P02-21, P04-12, P12-15, V01.10, V02.03, V11.01, V13.06
Lunkenbein, T. P03-01
Lünskens, T. V07.02
Luo, S.-P. P01-31
Luong, T. V10.02
M
Ma, X. P07-01
Maawad, E. V01.07
Magerl, K. P10-18
Maginn, E. P02-22
Maier, J. P12-05, V12.01, V12.04
Mallouk, T.E. P01-25, V13.03
March, A. P01-30
Marchenko, D. V09.07
Markland, T. P02-07
Markthaler, D. P10-22
Marquardt, S. V04.01
Marquardt, S. V04.01
Márquez, A. V12.08
Marsalek, O. P02-07
Marschall, R. V01.23
Martin, A. P12-17
Martin, J. P01-18
Martinez Uriarte, L. P01-24
Marx, D. V02.05, V09.10, V14.03
Mata, R.A. P01-36
Matolín, V. V12.13
Maurer, A. P12-08
Medcraft, C. V04.06
Meerbach, C. P07-02
Mehl, S. P09-21
Meier, F. P01-24
Meling, A. P09-11, P09-13
Merkle, R. P12-05
Merschjann, C. V01.18
LIST OF AUTHORS

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
449
Name Page
Mette, K. P03-01
Mews, A. P07-06, P07-07, P07-13, P07-14, P07-16, P10-19,
V07.09, V09.09
Meyer, S. P09-08, P09-10, P09-15
Michalik, D. P02-19
Miethe, J. P01-17, P07-17
Mikorski, M. P10-24
Milosavljevic, A. V10.04
Minnich, C. P04-05
Miranti, R. V01.04
Mischo, M. P10-16
Möbitz, S. P10-20
Moehle, T. P06-01
Mohamed, T. P07-24
Mohr, S. P09-04, P09-12, P09-17
Mohrhusen, L. P07-19
Möller, D. P10-13
Moors, M. P09-27
Moshammer, K. V03.01
Moskaleva, L. P11-01
Movsesyan, L. P01-35
Mrugalla, F. P06-04
Müh, F. V01.15
Müller, C. P04-16, P04-18, V10.08
Müller, P. V06.04
Müller, P. V13.06
Müller-Werkmeister, H.M. P10-15
Mulvaney, P. V07.06
Muñoz -Santiburcio, D. V09.10
Murphy, S. V07.06
N
Nack, A. V07.07, V08.06
Nagata, Y. FL09.01
Nagy, T.O. P03-18, P05-09
Namayandeh Jorabchi, M. P02-18
Naskar, S. P07-17, P07-25
Natter, H. P12-01, P12-03, P12-04, P12-10
Nattland, D. P05-02
Name Page
Naumov, S. V03.08
Nefedov, A. P09-02, P09-27, V09.03
Neff, A. P01-24, V01.09
Nemeth, Z. P01-30
Neubauer, A. P01-29
Neugebohren, J. P03-17, V14.08
Neumann, C. P03-07, P03-14, V03.06
Neumann, H. V03.04
Neumann, J. P02-02
Neumann, L. V01.13
Neyman, K. V12.13
Neymeyr, K. P11-03, V11.01
Niedermaier, M. V07.04
Niefind, F. V09.06
Nielsen, A. V07.09
Nielson, M. V01.13
Niemann, V. V01.03
Nieuwenkamp, G. P13-01
Niktin, K. V01.23
Nimmrich, K. P12-16
Noack, J. P03-01
Noack, K. V02.04
Novák, P. P01-09
O
Oezaslan, M. V12.14
Oh, E.-J. P12-02
Olejko, L. V10.05
Olzmann, M. P03-02, P03-04
Ondo, D. V13.07
Oskam, G. V12.08
Osmic, M. P01-34, P07-19
Ossenbrüggen, T. P05-12
Ostapko, J. P04-01
Oswald, S. P04-03
Otto, K.E. P04-08
Oum, K. P01-03, V01.22, V03.03
Overbeck, V. P02-19
Name Page
P
Pacher, U. P03-18, P05-09
Pacholski, C. V08.02
Paier, J. P09-01, P09-18, V09.02, V09.05
Papp, C. FL01.02, P09-12, V09.04
Parab, P. P06-06
Parisi, J. V01.04
Park, B. P03-06, P09-15
Paschek, D. P02-02, P02-03, P02-18, P02-19
Passow, C. V07.07, V08.06
Patoka, P. P10-12
Pellet, N. V12.01
Penschke, C. P09-18
Pereira, R. P01-14
Perez, C. V04.07
Pérez de Tudela, R. V14.03
Perz, M. P12-12
Peter, E. V10.07
Petrenko, A. P01-01
Petri, A. V13.06
Petrosyan, A. P02-12
Peukert, S. P04-01, P04-02
Pfeifer, V. P03-01, V12.12
Pfuhl, S. P08-06
Pieper, J. P03-12
Pitzer, M. V04.01
Plamper, F. P08-01, V08.04
Plumeré, N. P01-22
Poblotzki, A. V04.05, V04.06
Pohako-Esko, K. P09-21
Polet, W.S.K. P04-12
Polo, E. P07-23
Polyakov, M. P12-17, V05.04
Post, A. P10-05
Pougin, A. V01.11
Pramann, A. V05.03
Presselt, M. P01-28, P04-07
Prinz, J. V10.05
Prisner, L. P10-19
Puskar, L. P04-05
LIST OF AUTHORS

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
450
Name Page
Q
Quack, M. P03-19, P04-15, V06.01
Quaranta, V. V09.11
Quitevis, E. P02-22
Quoika, P. P10-15
R
Rabe, M. P09-23
Rabeah, J. V05.04
Rackwitz, J. V10.04
Rademann, K. V01.21
Rahinov, I. P09-11, P09-20
Rancan, F. P10-12
Raschpichler, C. P03-09, V09.07
Rathke, B. P02-08, P02-09, P02-10, P05-06, P05-07, V02.01
Rau, S. P01-04
Rauch, J. P13-01
Ravi, F. P03-05
Redder, T. P07-13
Reich, O. P08-06
Reichardt, C. P04-07
Reichenauer, G. P12-13
Reichhelm, A. P07-08
Reinfelds, M. P03-07, P03-14, V03.06
Reiter, E. P02-04
Rengers, C. P07-02, P09-29
Reusch, N. V04.03
Richter, M. P13-01
Riebe, D. P05-08
Rienitz, O. V05.03
Rijs, A. V04.07
Rinke, G. P05-10
Risse, T. P05-03
Ritter, E. P04-05
Rockstroh, N. P01-29, P01-30, P12-17, V01.10
Roder, P. V10.06
Rodrigues Correia, A. V03.06
Rodríguez-Gattorno, G. V12.08
Name Page
Rodríguez-Pérez, M.J. V12.08
Rohnke, M. P10-21
Roling, B. P09-06
Rösel, A. V01.10
Rostas, A. P05-04
Roth, N. P05-12
Roy, S. P02-01
Ruben, M. V01.17, V05.01
Rudolph, J. P11-05
Rühl, E. P03-09, P09-07, P10-12, V09.07
Ruhmlieb, C. P07-16
Russina, O. V02.01
Ruwe, L. P03-03, P03-16
S
Sadiek, I. P04-14
Safo, I.A. V12.14
Sakade, N. P06-06
Sakai, Y. P06-06
Sala, M. V03.04
Saladrigas, C. P03-06
Samadi Khoshkhoo, M. V07.05
Sánchez-Paradinas, S. P07-17
Santinacci, L. P07-09
Sarfraz, A. P09-23
Sauer, J. P09-18, V01.25, V09.05
Savolainen, J. P02-13, P02-15
Sawall, M. P11-03, V11.01
Schade, U. P04-05
Schäfer, B. V01.17, V05.01
Schäfer, M. P01-18, P07-11
Schäfer, S. P02-13, P02-15
Schäfer, T. P09-08, P09-10, P09-11, P09-13, P09-15, V06.03
Schäfer-Korting, M. P10-12
Schaumberg, C. V07.03
Schedel-Niedrig, T. V12.10
Scheele, M. V07.05
Schepper, R. P05-01
Schernich, S. FL01.02
Name Page
Scheunemann, D. V01.04
Schindler, J. P01-04, P04-07
Schins, J.M. V07.08
Schlag, K. P05-02
Schleicher, E. P05-04
Schlichting, I. V05.02
Schlögl, R. P03-01, P05-03, P12-16, V12.12
Schmeer, G. P12-08
Schmid, M. V09.06
Schmid , M. P01-09
Schmidt-Böcking, H. V04.01
Schmidt-Engler, J. P10-04
Schmiedekind, S. P01-34
Schmiedel, A. V01.16
Schmitt, M. P07-10, V01.24
Schmode, S. P02-12
Schneider, J. V01.23
Schneider, J. V09.08
Schneider, J. P07-09
Schneider, M. P03-23
Schneider, S. P08-01
Schnell, M. V04.06, V04.07
Schöffler, M. V04.01
Scholz, M. P01-03, V01.22, V03.03
Schön, D. P02-17
Schooss, D. V01.17, V04.04, V05.01
Schöttner, L. P09-27
Schrade, P. P10-12
Schran, C. P02-07
Schreiner, P.R. P04-19
Schröder, H. P11-03
Schrödl, N. V13.02
Schröer, W. P02-09, P02-23, P05-07, V02.01
Schubert, U.S. V01.16
Schuch, J. P12-09
Schuhmann, W. V05.04
Schuld, S. P07-11
Schulz, B. V14.05
Schulz, M. P04-07
LIST OF AUTHORS

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
451
Name Page
Schulze, S. P05-06
Schulze, S. P10-03
Schumacher, S. P04-17
Schummel, P. P10-11
Schuster, R. FL12.01, P01-09, P05-02, P07-01, P09-03, P12-06
Schwarz, M. P09-04, P09-12, P09-17
Schwarzer, D. P01-20, P09-09, P09-26
Schweigert, C. P05-11
Seibert, N. P03-07, P03-14, V03.06
Seidel, S. P04-16, P04-18
Seidl, M. P02-05
Seifert, G. P02-14, V12.02
Seifert, J. V08.06
Seitz, M. P04-18
Selent, D. V11.01
Senocrate, A. V12.01
Senske, M. V14.07
Sepioł, J. P04-01, P04-02
Seyfang, G. P04-15, P05-05
Shaker, M. P01-06
Sheppard, T. P05-10
Shi, J. P11-06
Shirhatti, P. P09-20
Shishatskiy, S. P08-02
Shuai, Q. P09-26
Siebbeles, L.D.A. V07.08
Siebels, J. V09.09
Siefermann, K. P01-24, V01.09
Sieghard, A. P04-15
Sillar, K. V01.25
Simon, M. V08.01
Single, F. V12.06
Sitte, W. P12-12, V13.02
Smith, T.W. P10-26
Sokolov, I. V10.01
Sommer, C. V05.02
Sonntag, L. P07-22
Southworth, S. P01-30
Späth, F. V09.04
Name Page
Späth, T. P09-05
Spiccia, L. P01-06
Spoor, F.C.M. V07.08
Stambolic, I. P10-17
Stamm, A. P04-06, V04.06
Stange, P. P02-21, V02.03
Stapf, S. P01-22
Starr, D.E. V14.01
Steber, A. V04.07
Stein, M. P01-01
Steinrück, H.-P. FL01.02, P09-12, V09.04
Steinschulte, A. P08-01, V08.04
Steinsiek, C. P09-20
Stolper, T. P01-36
Strauch, M. P02-19
Strelow, C. P07-07
Strunk, J. V01.11
Stubenrauch, C. V08.05
Stuckenholz, M. P05-07
Studzinski, H. V03.04
Stutzmann, M. P01-14
Suhm, M.A. P03-08, P04-03, P04-04, P04-08, V04.05, V04.06
Sukrus, A.-E. P12-17, V05.04
T
Tabary, F. P01-23
Taffa, D.H. V12.11
Tao, S. P01-19
Temps, F. P03-10, V03.04
Tesch, M. P01-06, P02-17
Teschmit, N. P05-12
Thämer, M. V07.02
Tielker, N. P06-05
Timm, A.E. P05-02
Tofighi, G. P05-10
Toimil-Molares, M.E. P01-35
Tomazic, D. P06-05
Tong, Y. P09-25
Tonner, R. V09.06
Torres-Alacan, J. V03.07
Name Page
Tran, L.-S. P03-12
Tranca, D. V12.02
Tranca, I. V12.02
Triolo, A. V02.01
Trippel, S. P03-20, V04.02
Trunschke, A. P03-01
Tschierlei, S. P01-29
Tschurl, M. P01-14, V01.02
Turchanin, A. P09-19
U
Uhlemann, T. P04-16, P04-18
Ulenikov, O. P03-19
Ulrich, G. P10-12
Urbain, F. P01-35
Urbanski, M. V08.08
Usiskin, R. V12.04
Usvyat, D. V14.04
V
Vallabhuni, S. P03-05, P03-21
van de Krol, R. P01-07, V14.01
van der Linde, C. V03.05
van der Veen, A. P13-01
van Wilderen, L.J.G.W. P03-07, P03-14, P10-04, P10-24,
V03.06
Vankó, G. P01-30
Vargas-Barbosa, N. P01-25, P09-06, V13.03
Velasco-Vélez, J.J. V12.12
Vieker, H. P08-02
Vogel, S. V10.04
Vogt, A. P10-12
Vogt, J. P09-14
Vöhringer, P. V02.08, V03.02
Voitekhovich, S. P09-29
Volz, D. P01-16
von Cosel, J. P03-07, P03-14
von Randow, C. P03-09
Von Weber, A. V07.02
Vondracek, H. V02.06
Vorokhta, M. V12.13
Vu, H. V09.09
LIST OF AUTHORS

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
452
Name Page
W
Wächtler, M. V01.16
Wagner, J. P08-05, V07.07, V08.06, V08.07
Wagner, R. P09-13
Wagner, S. V11.03
Waidhas, F. V12.13
Waldt, E. V04.04, V05.01
Walenta, C. P01-14, V01.02
Wallesch, M. P01-16
Walsh, P.S. V10.08
Waluk, J. P04-01
Wang, Y. P09-02, P09-27, V09.03
Wark, M. V01.05, V12.11
Wasserscheid, P. FL01.02, P09-21
Weber, F. P03-11
Weber, I. P03-04
Weber, M. V07.05
Weber, S. P05-04
Weber, T. V01.01
Weidler, N. P01-15
Weigand, M. P10-12
Weiler, M. V08.02
Weimerskirch, M. P03-18
Weingärtner, H. V14.07
Weise, K. P10-20
Weiss, T. P11-01
Weißhaar, K. P12-03
Weiszer, S. P01-14
Weitzel, K.-M. P01-18, P07-11, P12-14, V04.03
Weller, H. P01-23
Werner, K. P09-04, P09-12, P09-17
Werner, P. P08-07
Wesp, V. P01-18
Wete, E.P. V13.08
Wezisla, B. V03.02
Widmann, D. V01.12
Wiebeler, C. P04-17
Wiemer, J. P12-14
Wiese, J. V04.02
Name Page
Wilken, S. V01.04
Wind, J. P08-02
Winnerl, A. P01-14
Winter, A. V01.16
Winter, R. P10-11, P10-20, V10.02
Wirtz, H. P02-13, P02-15
Wittern, L. P01-23
Witthöft, P. P07-13
Wittinghofer, A. P10-20
Wittstock, A. P11-06
Wittstock, G. V12.03
Wodtke, A. P01-21, P03-17, P06-03, P09-08, P09-09, P09-10, P09-11, P09-13, P09-15, P09-20, P09-22, P09-24, P09-26, V06.03, V09.01,
V14.08
Wolf, A. P09-29
Wolf, A. V07.01
Wolf, E.H. P05-03
Wöll, C. P09-02, P09-27, V09.03, V14.06
Wollscheid, N. V04.03
Wrabetz, S. P03-01
Wu, C. V12.07
Wu, Y. P07-09
Wugt Larsen, R. P03-08
X
Xiao, J. P01-06, P02-17
Xiong, Y. P12-11
Xu, J. P01-09
Xu, Q. PL01
Xu, T. P09-04, P09-12
Xue, L. P02-22
Y
Yamamoto, K. P10-12
Yang, C. P09-02, V09.03
Yang, F. P01-19, P01-35
Yang, T.-Y. V12.01
Yavuz, M. V01.07
Yazdi Nezhad, S. P06-07
Yserentant, K. P10-23
Name Page
Yu, X. P09-02
Yu, Y. P12-07
Z
Zachäus, C. P01-07
Zander, S. V01.07
Zedler, L. P01-04
Zehentbauer, F. P02-08, P02-10, V02.04
Zehnacker-Rentien, A. P04-01
Zeng, M. P03-12
Zeuch, T. P02-16
Zhang, J. V12.02
Zhang, S. V03.04
Zhang, X. P08-02
Zhang, Y. P01-04, P04-07
Zhang, Y. P02-22
Zhang, Z. P09-02
Zhao, W. FL01.02
Zheng, Z. V12.02
Ziegert, F. P08-05
Ziegler, C. P07-21
Ziegler, J. P01-23, V11.03
Zifferer, G. P02-05, P08-03, P08-04
Zimina, A. V05.05
Zimmer, M. P01-16
Zindel, D. P04-15
Zinn, S. V04.06
Zohourian Aboutorabi, R. P12-05
Zubeir, L. P02-10
Zugermeier, M. V09.06
Zwier, T.S. V10.08
LIST OF AUTHORS

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
453

B U N S E N T A G U N G 2 0 1 6 · R O S T O C K
454

B A S I C M E C H A N I S M S I N E N E R G Y C O N V E R S I O N
455
IMPRINT
Content: Dr. Susanne Kühner, Deutsche Bunsen-Gesellschaft für physikalische Chemie e.V., Frankfurt am Main/DEditorial Office: Matthias Neumann, DECHEMA Gesellschaft für Chemische Technik und Biotechnologie e.V., Frankfurt am Main/D Carmen Weidner-Friedrich, Deutsche Bunsen-Gesellschaft für physikalische Chemie e.V., Frankfurt am Main/D ISBN 978-3-9809691-8-5
Conception: Peter Mück, PM-GrafikDesign, Wächtersbach/D
Catering
Exhibition
Train Station “Parkstraße”
(S-Bahn, Tram)
Entrance Parking
Haus 1
Saal 224 · Saal 323 · Conference Office (SR 022) · Meetingrooms SR 020-SR 025
Tent
Welcome ReceptionPoster Sessions
VENUE OVERVIEW / IMPRINT