building a platinum human genome assembly from single haplotype human genomes generated from long...
TRANSCRIPT

Building a platinum human genome assembly from single haplotype human genomes generated from long molecule sequencing
Karyn Meltz Steinberg ASHG 2015
@KMS_Meltzy
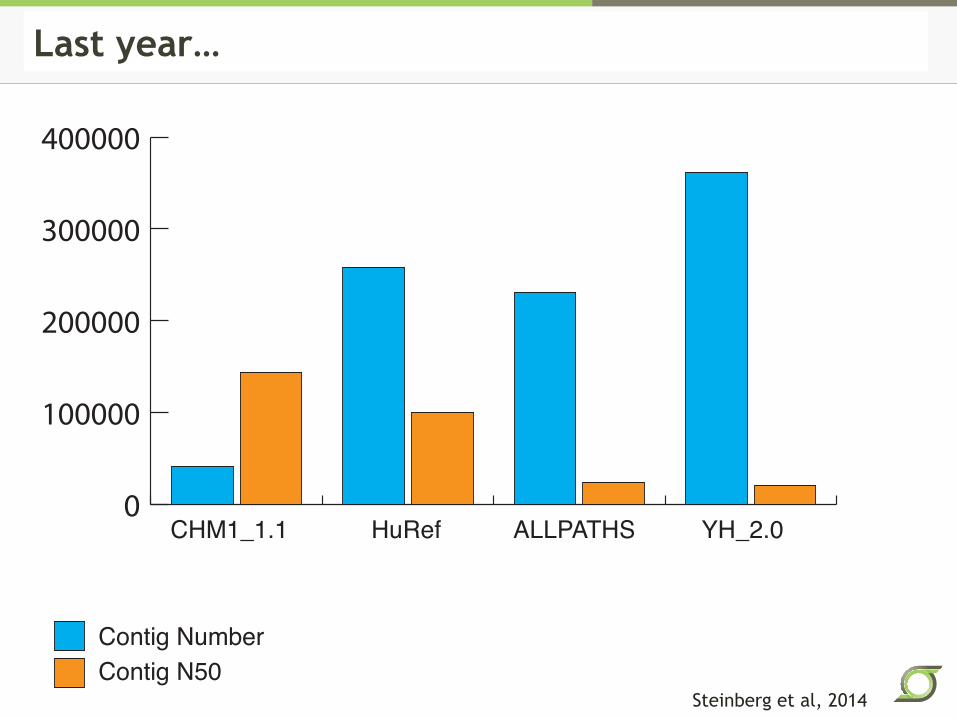
0
100000
200000
300000
400000
CHM1_1.1 HuRef ALLPATHS YH_2.0
Contig Number
Contig N50
Figure 1Last year…
Steinberg et al, 2014
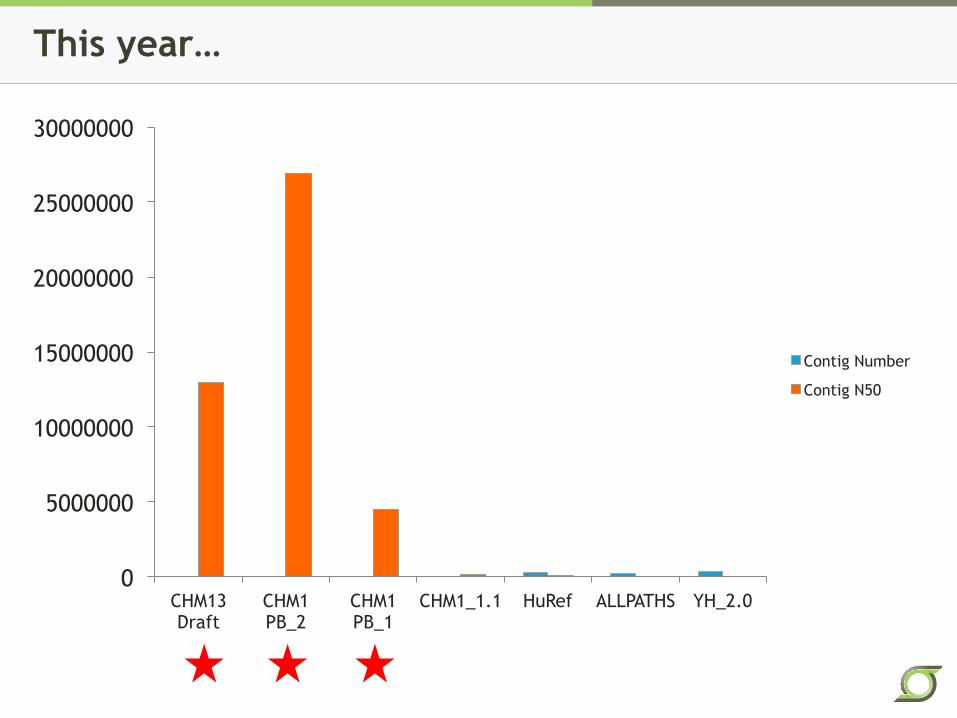
This year…
0
5000000
10000000
15000000
20000000
25000000
30000000
CHM13 Draft
CHM1 PB_2
CHM1 PB_1
CHM1_1.1 HuRef ALLPATHS YH_2.0
Contig Number
Contig N50

This year…
Log scale
1
10
100
1000
10000
100000
1000000
10000000
100000000
CHM13 Draft
CHM1 PB_2
CHM1 PB_1
CHM1_1.1 HuRef ALLPATHS YH_2.0
Contig Number
Contig N50

We combine PacBio with other technologies to construct the assembly
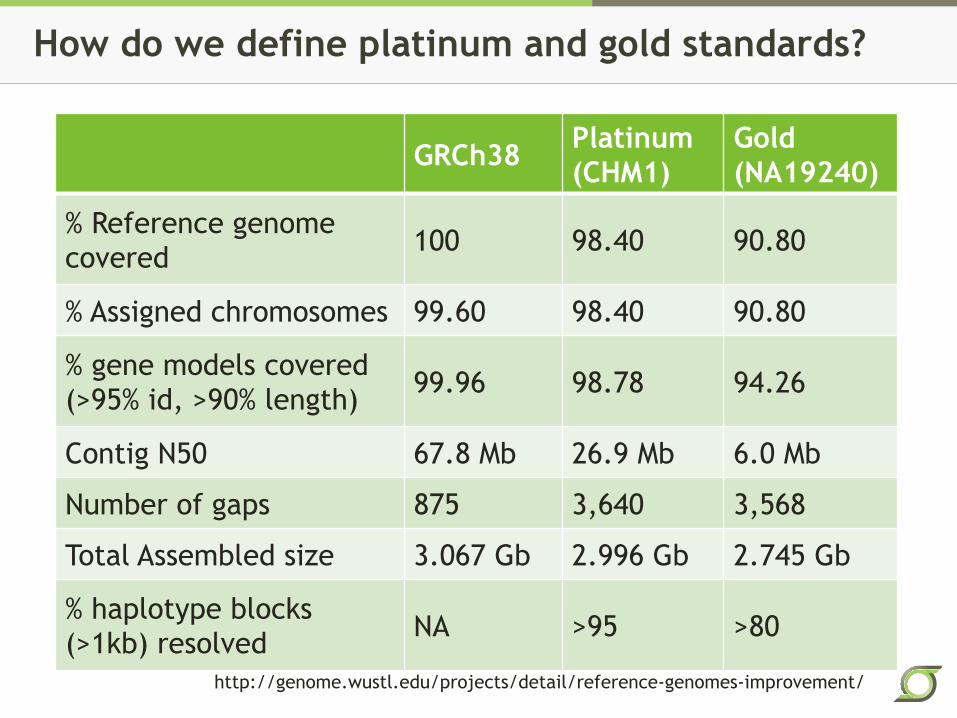
How do we define platinum and gold standards?
GRCh38 Platinum (CHM1)
Gold (NA19240)
% Reference genome covered 100 98.40 90.80
% Assigned chromosomes 99.60 98.40 90.80
% gene models covered (>95% id, >90% length) 99.96 98.78 94.26
Contig N50 67.8 Mb 26.9 Mb 6.0 Mb
Number of gaps 875 3,640 3,568
Total Assembled size 3.067 Gb 2.996 Gb 2.745 Gb
% haplotype blocks (>1kb) resolved NA >95 >80
http://genome.wustl.edu/projects/detail/reference-genomes-improvement/
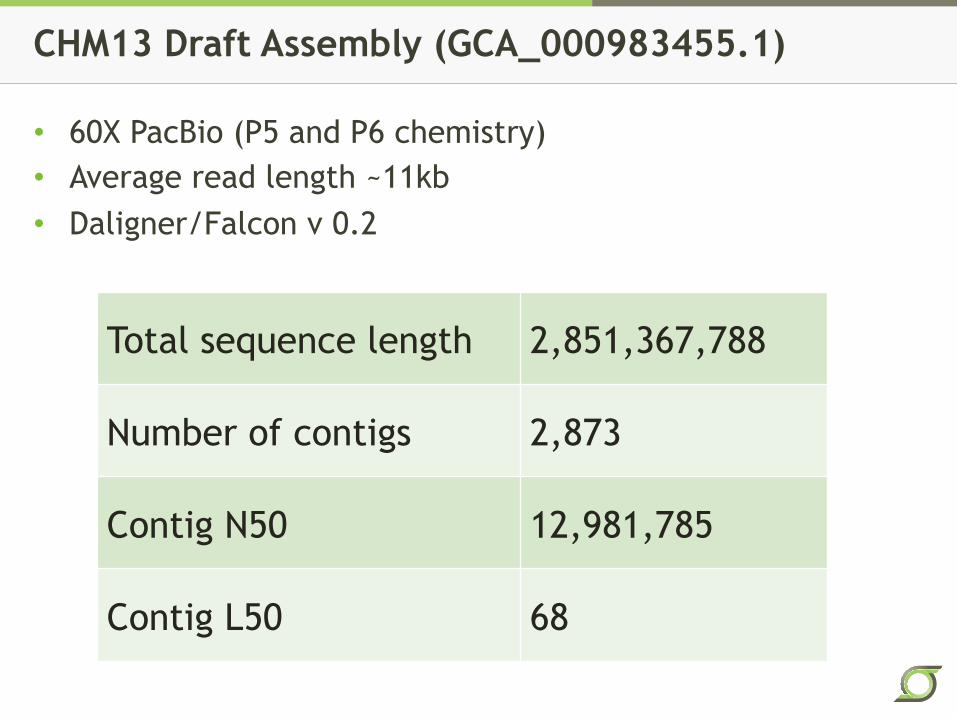
CHM13 Draft Assembly (GCA_000983455.1)
• 60X PacBio (P5 and P6 chemistry) • Average read length ~11kb • Daligner/Falcon v 0.2
Total sequence length 2,851,367,788
Number of contigs 2,873
Contig N50 12,981,785
Contig L50 68

Gene Model (RefSeq) Analysis
GRCh38 CHM1_1.1 CHM1_PB1 CHM1_PB2 CHM13
Number of sequences not aligning
21 88 67 67 125
Split Transcripts 8 35 1,245 1,131 285
CDS coverage <95% 17 266 1,339 1,212 265
Total Sequences Retrieved from Entrez 49,680

Short read sequence analysis
• 100X Illumina sequence • Align with BWA-MEM to ordered and
oriented assembly • Variant calling via SpeedSeq (Chiang et al,
2015) • SNVs, indels: FreeBayes • SVs: LUMPY, SVTyper • CNV: CNVnator

CHM13 Illumina data aligned to CHM13 assembly
202,016 SNVs/indels on unplaced scaffolds
SV_TYPES >10kb 5-10kb 1-5kb <1kb DELETIONS 174 131 430 2582 INVERSIONS 5 0 2 7
DUPLICATIONS 151 112 309 113 TOTAL 330 243 741 2702

BioNano SV calls can be used to identify misassembly
Colla
pse
Expa
nsio
n in
Ass
embl
y
Gap in Sequence PacBio Assembly
BioNano Map
SV_TYPES DELETIONS 41 INVERSIONS 10 INSERTIONS 15
TOTAL 66
BioNano alignment to CHM13

BioNano reveals collapse in PacBio assembly
PacBio Assembly
BioNano Map

Illumina data aligned to PacBio assembly also shows collapse

BioNano reveals collapse in PacBio assembly due to highly homologous segmental duplications
SD = 96%
CHR1 46746040 46857004 40 W LBHZ01000938.1 110965
CHR1 46857005 47034202 41 N 177198 gap
CHR1 47034203 52157695 42 W LBHZ01000245.1 5123493
PacBio Assembly
BioNano Map
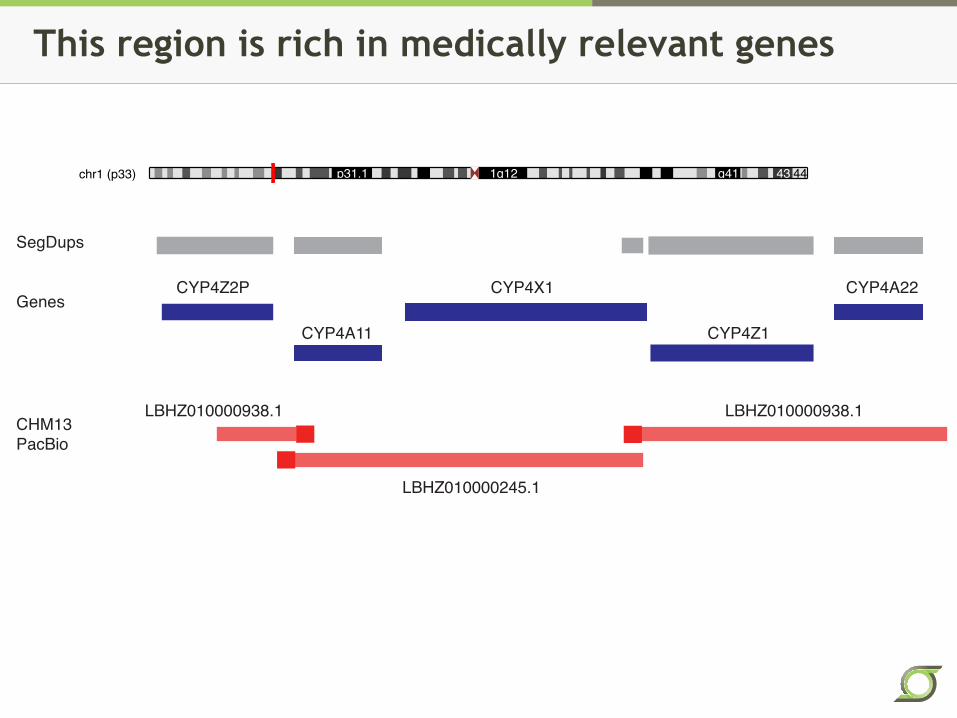
This region is rich in medically relevant genes
chr1 (p33) p31.1 1q12 q41 43 44
CYP4Z2P
CYP4A11
CYP4X1
CYP4Z1
CYP4A22
SegDups
Genes
CHM13
PacBio
LBHZ010000938.1 LBHZ010000938.1
LBHZ010000245.1

CHM13 Hybrid Scaffold Hybrid Scaffold
PacBio Contigs
BioNano Contigs

CHM13 Hybrid Scaffolds
BioNano Map PacBio Assmbly Hybrid Scaffold
# of Contigs 3593 1590 * 254
Min Contig Length 0.08 Mb 0 0.27 Mb
Median Contig Length 0.61 Mb 0.06 Mb 4.35 Mb
Mean Contig Length 0.78 Mb 1.78 Mb 9.68 Mb
Contig N50 1.02 Mb 13.46 Mb 20.79 Mb
Max Contig Length 5.27 Mb 63.15 Mb 82.83 Mb
Total Contig Length 2.812 Gb 2.824 Gb 2.458 Gb
*Number of contigs used in hybrid scaffolding
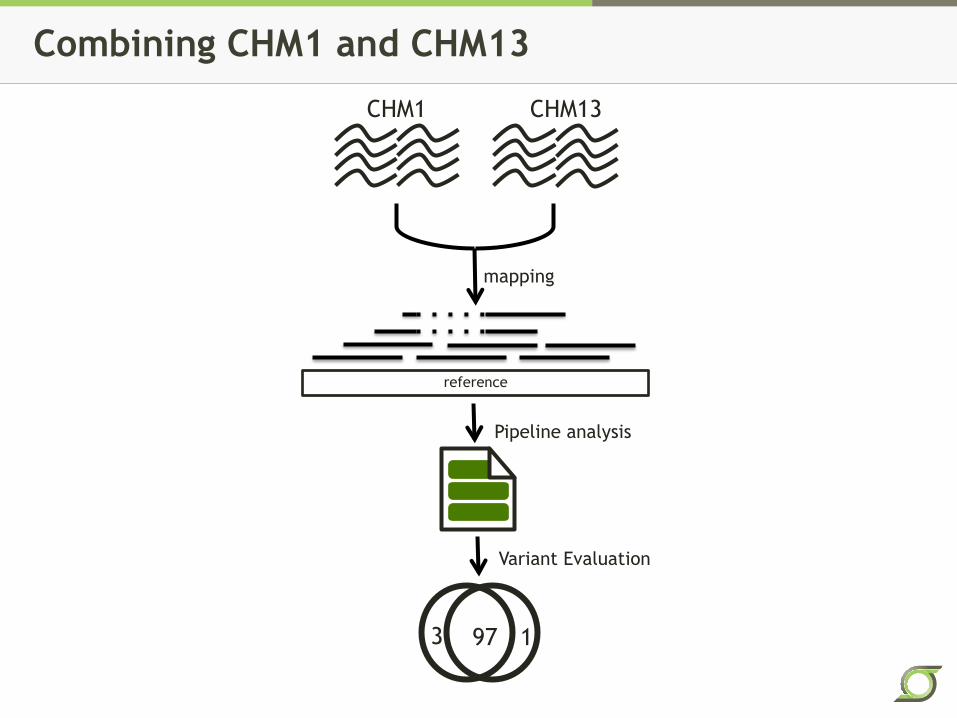
Combining CHM1 and CHM13
reference
mapping
CHM1 CHM13
Pipeline analysis
Variant Evaluation
97 1 3

Acknowledgements
The McDonnell Genome Institute at Washington University in St. Louis
Rick Wilson Bob Fulton Wes Warren Tina Graves-Lindsay Vince Magrini Sean McGrath Derek Albracht Milinn Kremitzki Susan Rock Debbie Scheer Aye Wollam
The Finishing and Bioinformatics Teams at The Genome Institute
University of Washington Evan Eichler John Huddleston Archana Raja
NCBI Valerie Schneider
University of Pittsburgh School of Medicine (CHM13 cell line)
Urvashi Surti
Personalis Deanna Church
BioNano Genomics Palak Sheth
Pacific Biosciences Jason Chin Nick Sisneros
