bispecific antibody therapy of acute...
TRANSCRIPT

Bispecific Antibody Therapy of Acute LeukemiaRoland B. Walter, MD, PhD, MSAssociate Member, Clinical Research DivisionFred Hutchinson Cancer Research CenterAssociate Professor of Medicine, Division of HematologyUniversity of Washington School of MedicineSeattle, WA

DisclosuresI disclose the following relationships
§ Grants/clinical trials support § Amgen, Inc.§ Amphivena Therapeutics, Inc.§ Covagen, Inc.§ Seattle Genetics, Inc.
§ Consultant fees § Amgen, Inc. § Amphivena Therapeutics, Inc. § Covagen, AG§ Emergent Biosolutions, Inc. § Janssen Research & Development, LLC § Pfizer, Inc. § Seattle Genetics, Inc.

Goals§ The purpose of this presentation is to
§ Provide an overview of acute leukemia treatment with bispecific antibodies
§ After this session, learners should be better able to§ Describe the mechanisms of action and resistance
pertinent for bispecific antibodies§ Appraise the benefits and toxicity profile of the BiTE
antibody, blinatumomab§ Describe the potential role of bispecific antibodies in future
treatment algorithms for acute leukemia

Bispecific antibody
=Molecule that contains two different antigen binding sites

Historical perspective
§ 1960: bispecific antibodies envisioned§ Low yield of early bispecific F(ab’)2, quadroma molecules
§ 1985: first bispecific antibody that recruits T-cells for cell-directed cytotoxicity§ Poor clinical results, concerning toxicities in early trials
§ Last 20 years: renewed enthusiasm with better protein engineering tools
Riethmüller. Cancer Immun. 2012;12:12

Bispecific antibodies as cancer therapeutics
§ Various bispecific antibody formats are currently explored clinically
conversion into a single-chain version (scDb) [26] and dimeric
tetravalent derivatives thereof, so-called ‘tandAb’ molecules with
two binding sites for each antigen [27], as well as disulfide-stabi-
lized variants, such as the dual-affinity retargeting molecules
(DART) [28]. The small size of these molecules, as well as the lack
of the Fc region, leads to rather rapid renal elimination in vivo.
Although the small size might be advantageous regarding tissue
penetration (e.g., in tumor therapy), the short plasma half-lives
affects dosing (i.e., frequent injections or infusions are required).
The implementation of half-life extension moieties, including
conjugation of polyethylene glycol (PEG), fusion of PEG-mimetic
polypeptides, or albumin-binding moieties, might be useful [29].
BsAbs can also be generated by fusing different antigen-binding
moieties (e.g., scFv or Fab) to other protein domains, which
enables further functionalities to be included. For example, two
scFv fragments have been fused to albumin, which endows the
antibody fragments with the long circulation time of serum albu-
min [30,31]. Another example is the ‘dock-and-lock’ approach
based on heterodimerization of cAMP-dependent protein kinase
A and A kinase-anchoring protein [32]. These domains can be
linked to Fab fragments and entire antibodies to form multivalent
bsAb [33].
Figure 1 shows schematically an overview of the different
strategies used to obtain bsAb formats by different groups in
academia, and the biotech and pharma industries. Most of these
formats are still in preclinical evaluation and many might remain
at this stage. However, some formats have made it already into
clinical development and are detailed in Table 1 and discussed
below. The composition and features of bsAb formats that are in
clinical development are depicted in Fig. 2.
Recruitment and activation of immune cellsQuadroma bsAbs for T cell recruitment: Trion PharmaThe anti-epithelial cell adhesion molecule (EpCAM)/anti-CD3
bsAb catumaxomab (Removab1) was the first bsAb to receive
market approval [34]. Trion Pharma developed catumaxomab as
REVIEWS Drug Discovery Today ! Volume 20, Number 7 ! July 2015
Triomab
DVD-Ig 2 in 1-IgG IgG-scFv scFv2-Fc
DNL-Fab3scFv-HSA-scFvDART-FcDARTtandAbsBiTEbi-Nanobody
kih IgGcommon LC CrossMab
ortho-FabIgG
Drug Discovery Today
FIG. 2
Various bispecific antibodies (bsAbs) are currently in clinical development or are already approved for cancer therapy. The upper two lines depict immunoglobulin(Ig)-like bsAbs comprising an IgG Fc region, either as bivalent or tetravalent molecules. Furthermore, several small bsAb and bsAb fusion proteins have enteredclinical trials. Abbreviations: BiTE, bispecific T cell engager; DART, Dual affinity retargeting; DNL, dock-and-lock; DVD-Ig, dual variable domain immunoglobulins;HSA, human serum albumin; kih, knobs into holes.
840 www.drugdiscoverytoday.com
Review
s!K
EYNOTEREVIEW
Kontermann, Brinkmann. Drug Discov Today. 2013;20(7):838-847
Adapted from: Blood Rev 2014;28:143-153
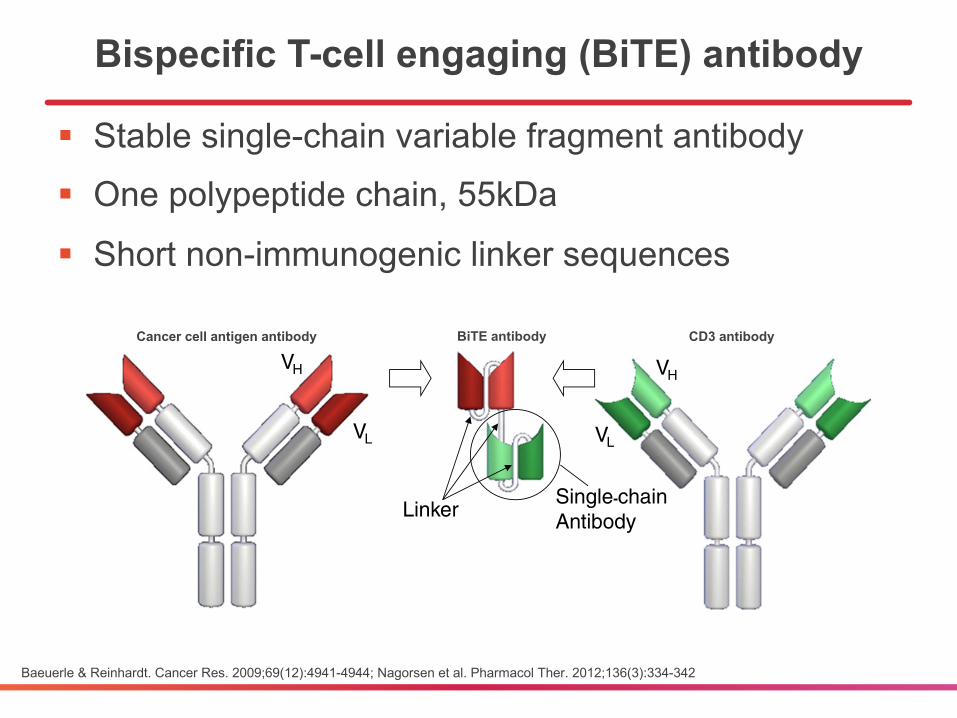
Bispecific T-cell engaging (BiTE) antibody
§ Stable single-chain variable fragment antibody§ One polypeptide chain, 55kDa
§ Short non-immunogenic linker sequences
Baeuerle & Reinhardt. Cancer Res. 2009;69(12):4941-4944; Nagorsen et al. Pharmacol Ther. 2012;136(3):334-342
CD19
MembraneBlebbing
Perforins
Granzymes
for a bispecific T cell-engaging antibody at the time (Loffler et al.,2000; Dreier et al., 2002; Hoffmann et al., 2005; Molhoj et al.,2007). The half maximal concentration of blinatumomab forredirected lysis of CD19+ target cells by T cells was in the range of10–100 pg/ml (pico- to femtomolar) BiTE® antibody when T cellsfrom multiple healthy donors were tested (Dreier et al., 2002).Peripheral mononuclear cell preparations or isolated CD3+, CD8+ orCD4+ T cells showed similar potency in lysis assays and mostly dif-fered in their kinetics of cell killing. With the exception of naïve Tcells, all other CD8+ or CD4+ T cell subpopulations including effectormemory α β T cells, γ δ T cells, and central memory T cells showedhigh activity of redirected lysis with CD8+ and CD4+ effector memo-ry T cells being most effective (Kischel et al., 2008). Of note, in no caseT cells did require prior activation, expansion or addition ofcostimulatory agents, but blinatumomab could fully activate a largeproportion of previously resting peripheral T cells initially devoid ofactivation markers on their surface. T cell activation by blinatumomabinvolved new expression of CD69, CD25, and upregulation of celladhesion molecules such as CD2, the transient release of inflammato-ry cytokines (e.g., interferon-gamma, TNF-alpha, interleukins 2, 6 and10), and robust T cell proliferation (Brandl et al., 2007). This washowever only observed in the presence of human CD19+ B cells, ma-lignant B cell lines or rodent CHO cells engineered to express humanCD19. Target cell-dependent T cell activation is a hallmark of allBiTE® antibodies (Brischwein et al., 2007; Baeuerle et al., 2009) andmay depend on clustering and activation of T cell receptors by onlythe small fraction of BiTE® antibodies presented to T cells on the sur-face of target cells.
A remarkable feature of blinatumomab and other BiTE® anti-bodies is that they support serial lysis by engaged T cells (Hoffmannet al., 2005). This has been studied by videomicroscopy andco-culture experiments performed at very low T effector-to-targetcell ratios. In support of blinatumomab inducing very tight cytolyticsynapses between T cells and CD19+ target cells, those T cellsengaged in serial lysis were observed to pick up CD19 antigen fromtarget cells. However, CD19 on T cells did not reach levels sufficientto cause their mutual lysis.
Target cell lysis by blinatumomab is not observed by monovalentbinding to CD19 but requires the presence of T cells expressinggranzymes and perforin. Naïve T cells with very low levels of suchtoxins can only be engaged after a costimulatory signal is provided(Kufer et al., 2001). Mature CD8+ and CD4+ T cells do not requirecostimulation but, after BiTE® antibody addition, take a several-hourlag phase before they fully engage in lysis duringwhich they upregulateperforin and granzyme synthesis. In the course of BiTE® antibody treat-ment in co-cultures, BiTE® antibody concentrations for half-maximal
lysis typically drop and dose response curves tend to become verysteep, indicative of a significant increase in the cytolytic performanceof BiTE®-antibody-engaged polyclonal T cells over time. At the end ofa co-culture experiment after several days, all target cells typically areeliminated, and T cells have significantly expanded in number due toseveral rounds of cell cycle.
Cell lysis by blinatumomab and other BiTE® antibodies involvestoxic proteins that are normally stored inside secretory vesicles ofall cytotoxic T cells and are discharged when the BiTE® antibodyforces formation of a cytolytic synapse between T cells and targetcells (Haas et al., 2009). Essential components of these vesicles arethe pore-forming protein perforin and a cocktail of granzymes,which are proteases of distinct substrate specificity that find multipletargets in the cytoplasm of target cells (Rousalova & Krepela, 2010).Cell lysis by blinatumomab can be prevented by an extracellular cal-cium chelator. Calcium ions are needed for both T cell signaling andassembly of perforin subunits into functional pores after their secre-tion. Likewise, concanamycin A, an inhibitor of perforin, can inhibitblinatumomab-induced target cell lysis, suggesting that redirectedlysis predominantly relies on cytotoxic vesicle fusion by T cells ratherthan on a Fas ligand, TRAIL or TNF-α mediated mechanism (Gruen etal., 2004). Activation of caspases and induction of apoptosis in targetcells by blinatumomab supports that in parallel to the insertion ofperforin pores into the target cell membrane granzyme B has beendelivered, which is known to potently elicit programmed cell death.This highly effective, redundant mode of target cell lysis by T cellshas developed during evolution to eliminate virus-infected cells. Itshigh importance for survival of virus-infected vertebrate species obvi-ously rendered it very robust against interception by any kind of in-hibitory mechanism during evolution. The basic mode of action ofblinatumomab and all other BiTE® antibodies is summarized in Fig. 2.
Blinatumomab has not only been characterized in heterologoussettings where T cells typically come from healthy PBMC donorsand target cells are human NHL or ALL cell lines but was also investi-gated for its activity in PBMC samples from CLL patients (Loffler et al.,2003). In 22 out of 25 patient samples tested, blinatumomab activat-ed the autologous T cells and caused a reduction or depletion of en-dogenous leukemic and normal B cells. The BiTE® antibody wasactive even at very low effector-to-target cell ratios, and in samplesfrom patients heavily pretreated with different chemotherapyregimen.
Blinatumomab was shown to act in concert with the CD20-specificchimeric IgG1mAb rituximab (d'Argouges et al., 2009). This combina-tion allows simultaneous targeting of CD19 and CD20 expressing lym-phoma cells and to engage T cells as well as natural killer cells andcomplement for target cell lysis. Clinical studies are needed to
α-CD19 MAb α-CD3 MAb
VH
VL
VH
VL
Linker
Blinatumomab
Single-chainAntibody
Fig. 1. Generation and structure of blinatumomab. Variable domains (VH and VL) of a CD19-specific monoclonal antibody (mAb) and a CD3-specific mAb were converted intosingle-chain antibodies (circle) recombinantly joined by non-immunogenic linker sequences as shown. The resulting bispecific antibody construct has with 55 kDa only a thirdof the size of a regular mAb.
336 D. Nagorsen et al. / Pharmacology & Therapeutics 136 (2012) 334–342
BiTE antibodyCancer cell antigen antibody CD3 antibody

Adapted from: Blood Rev 2014;28:143-153
BiTE antibody binding
§ Binds invariant epsilon subunit of CD3§ Recruits polyclonal CD3+ T-cells without
dependency on T-cell receptor specificity or MHC presentation
CD19
10/21/2016 www.discoverymedicine.com/Peter-Kufer/files/2009/08/kufer_23_fig4.jpg.jhtml?id=2
http://www.discoverymedicine.com/Peter-Kufer/files/2009/08/kufer_23_fig4.jpg.jhtml?id=2 1/1
Copyright 2016 Discovery Medicine. Print This Page
TCR/peptide-MHC interaction BiTE antibody binding
Kufer at al. Discov Med. 2004;4(23):325-332
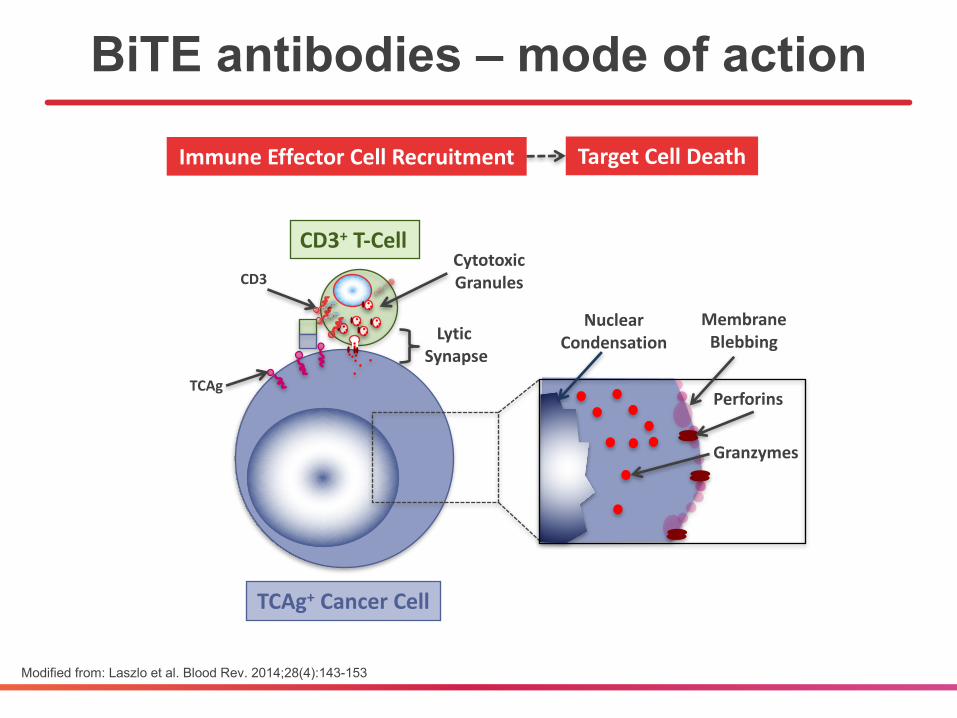
BiTE antibodies – mode of action
Modified from: Laszlo et al. Blood Rev. 2014;28(4):143-153
LyticSynapse
CD3
TCAg
CytotoxicGranules
ImmuneEffectorCellRecruitment
CD3+ T-Cell
MembraneBlebbing
Perforins
NuclearCondensation
TargetCellDeath
TCAg+ CancerCell
Granzymes

§ 21 adults aged 47 (20-77) years with B-ALL with molecular failure or molecular relapse (Ph- and Ph+)
§ Treatment: 15µg/m2/day over 4 weeks, q6 weeks, up to 4 cycles
§ MRD negativity in 16/20 (80%), all with 1 cycle of therapy
§ 61% relapse-free survival (at median follow-up of 33 months)
§ Long-term remission possible without further therapy (n=11)
Blinatumomab for MRD+ B-ALL
Topp et al. J Clin Oncol. 2011;29(18):2493-2498; Topp et al. Blood. 2012;120(26):5185-5187
10/24/2016 F1.large.jpg (1800×475)
http://www.bloodjournal.org/content/bloodjournal/120/26/5185/F1.large.jpg 1/1
10/24/2016 F2.large.jpg (1800×693)
http://www.bloodjournal.org/content/bloodjournal/120/26/5185/F2.large.jpg 1/1
All 11 patients 6 patients with MRD- response

§ 116 adults aged 45 (18-76) years with B-ALL with molecular failure or molecular relapse (Ph- and Ph+)
§ Treatment: 15µg/m2/day over 4 weeks, q6 weeks§ ≤4 cycles of treatment for responders or HCT after ≥1 cycle
§ MRD negativity in 90/113 (80%)§ 88/90 (98%) achieved MRD negativity with 1 cycle of therapy
BLAST: main findings
Gökbuget et al. Blood. 2014;124(21):379 [abstract]

Blinatumomab for R/R B-ALL§ 36 adults aged 32 (18-77) years with primary refractory
or relapsed B-ALL, >5% blasts (Ph- and Ph+)
§ Blinatumomab up to 30µg/m2/day (4 week cycles, q6 weeks, up to 5 cycles)
§ Best tolerability with lower dose during week 1
§ 25/36 (69%) achieved CR (42%) or CRh (28%), all within 2 treatment cycles§ 22/25 (88%) achieved MRD negativity
§ Median relapse-free survival: 7.9 months
§ Median overall survival: 9.8 months
Topp et al. J Clin Oncol. 2014;32(36):4134-4140

*Only cycle 1, days 1 to 7: 9 μg/day
Blinatumomab registration study§ Multicenter phase 2 study in 189 adults aged 39 (18-79)
years with Ph- B-ALL§ Primary refractory disease, untreated 1st relapse with CR <12 months, relapse
<12 months after allogeneic HCT, any disease >1st salvage§ ≥10% bone marrow blasts (i.e. measurable disease)§ No history of clinically relevant CNS pathology
Topp et al. Lancet Oncol. 2015;16(1):57-66
Scre
enin
g an
d En
rollm
ent
Follo
w-u
p(u
p to
24
mon
ths)
HSCT Offered to Patients in CR/CRh
Blinatumomab28 μg/day*
cIV infusion4 weeks on,2 weeks off
Up to 2 cycles
Induction
Blinatumomab28 μg/day
cIV infusion4 weeks on,2 weeks off
Up to 3 cycles
Consolidation
Study EndpointsPrimary• CR/CRh during the first two
cycles
Secondary• CR, CRh• Relapse-free survival• Overall survival• HSCT realization• Incidence of adverse events
Exploratory• Minimal residual disease
response by PCR during the first two cycles
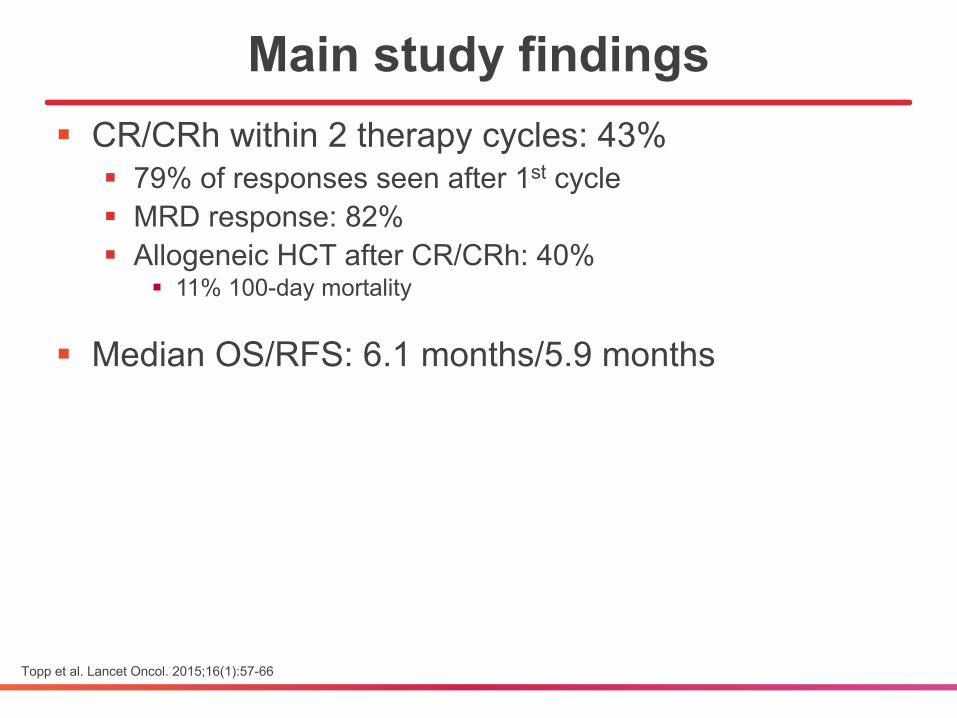
§ CR/CRh within 2 therapy cycles: 43%§ 79% of responses seen after 1st cycle§ MRD response: 82% § Allogeneic HCT after CR/CRh: 40%
§ 11% 100-day mortality
§ Median OS/RFS: 6.1 months/5.9 months
Main study findings
Topp et al. Lancet Oncol. 2015;16(1):57-66

§ Comparison of single-arm phase 2 study with historical cohort (for CR: n=694; for OS: n=1,112)
§ Higher CR rate with blinatumomab§ 33% (27-41%) vs. 24% (20-27%)
§ Longer OS with blinatumomab§ Median OS: 6.1 (4.2-7.5) vs. 3.3 (2.8-3.6) months§ HR: 0.54 (0.39-0.73)
Blinatumomab vs. standard therapy
Gökbuget et al. Blood Cancer J. 2016;6:e473
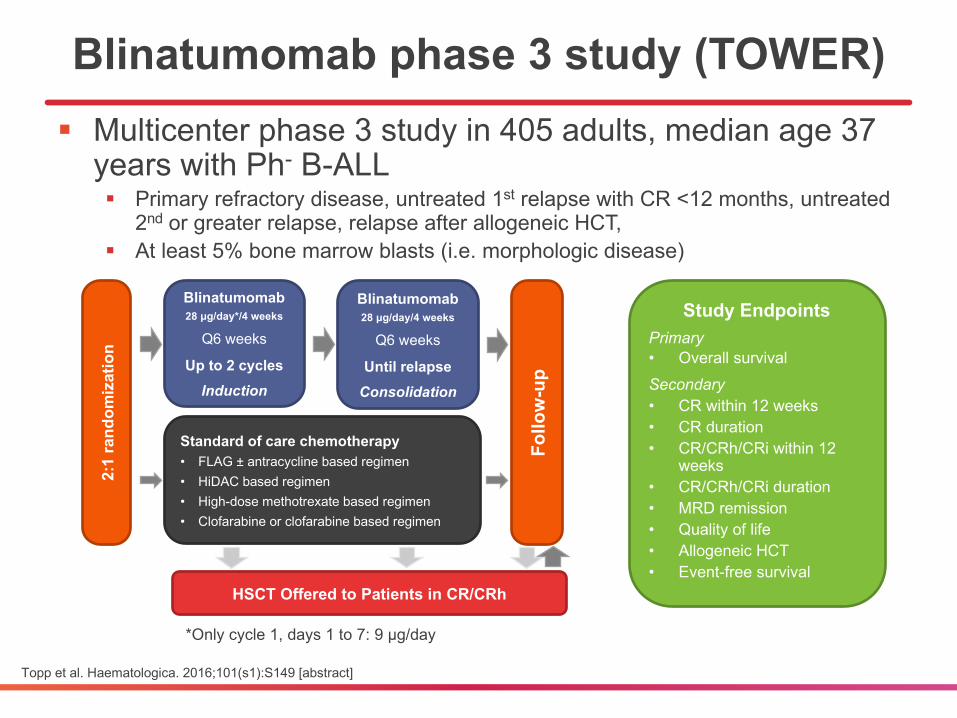
*Only cycle 1, days 1 to 7: 9 μg/day
Blinatumomab phase 3 study (TOWER)§ Multicenter phase 3 study in 405 adults, median age 37
years with Ph- B-ALL§ Primary refractory disease, untreated 1st relapse with CR <12 months, untreated
2nd or greater relapse, relapse after allogeneic HCT, § At least 5% bone marrow blasts (i.e. morphologic disease)
Topp et al. Haematologica. 2016;101(s1):S149 [abstract]
2:1
rand
omiz
atio
n
Follo
w-u
p
HSCT Offered to Patients in CR/CRh
Blinatumomab28 μg/day*/4 weeks
Q6 weeks
Up to 2 cyclesInduction
Blinatumomab28 μg/day/4 weeks
Q6 weeks
Until relapseConsolidation
Study EndpointsPrimary• Overall survivalSecondary• CR within 12 weeks• CR duration• CR/CRh/CRi within 12
weeks• CR/CRh/CRi duration• MRD remission• Quality of life• Allogeneic HCT• Event-free survival
Standard of care chemotherapy• FLAG ± antracycline based regimen• HiDAC based regimen• High-dose methotrexate based regimen• Clofarabine or clofarabine based regimen

§ Pre-specified interim analysis after 248 deaths on study (75%) § 405 patients randomized to blinatumomab (n=271) or SOC (n=134)
§ Balanced baseline characteristics
§ Longer median OS with blinatumomab§ 7.8 (5.7-10) months vs. 4.0 (2.9-5.4) months; HR=0.71, p=0.011§ OS improvement consistent across subgroups (age, prior salvage
chemotherapy, prior allo-HCT)
§ Higher CR rate with blinatumomab: 39% vs. 19%, p=0.001
§ Similar safety outcomes
§ Study stopped for efficacy before planned final analysis
TOWER: main findings
Topp et al. Haematologica. 2016;101(s1):S149 [abstract]

Adapted from: Blood Rev 2014;28:143-153
Toxicities with blinatumomab
§ Grade 3 and 4 toxicities in 38% and 29% of patients§ Febrile neutropenia, cytopenias, hyperglycemia, fever
§ Grade 5 infection in 11% of patients§ Safety outcomes comparable to SOC chemotherapy§ Unique toxicities usually reversible but can be
dose/treatment-limiting§ Cytokine release syndrome (2% grade 3)§ Neurologic events (11% grade 3, 2% grade 4)
§ Disorientation, seizure, apraxia, memory impairment, encephalopathy
§ Cost
Topp et al. Lancet Oncol. 2015;16(1):57-66; Topp et al. Haematologica. 2016;101(s1):S149 [abstract]

Blinatumomab: approval status§ United States
§ December 2014: Adults with Ph- relapsed/refractory B-ALL (accelerated approval)
§ September 2016: Pediatric patients with Ph-
relapsed/refractory B-ALL (accelerated approval)
§ European Union§ November 2015: Adults with Ph- relapsed/refractory B-ALL
(conditional approval)

Other bispecific antibodies for ALL
§ MGD011 (JNJ-64052781, duvortuxizumab): humanized CD19/CD3 Dual-Affinity Re-Targeting [DART] antibody1,2
§ AFM11: CD19/CD3 tandem diabody (TandAb)3,4
§ REGN1979: human CD20/CD3 bispecific antibody5
§ Phase 1 trials ongoing
1Liu et al. Blood. 2014;124(21):1775 [abstract]; 2Liu et al. Clin Cancer Res. 2016;in press; 3Reusch et al. Mabs. 2015;7(3):584-604; 4www.affimed.com; 5Smith et al. Sci Rep. 2015;5:17943
CD19VL CD3VH E Fc KnobChain 1
Fc Hole
Chain 2
Chain 3
CD19VHCD3VL K
S
S
Figure 1. Design, production, and purification of recombinant AFM11-His. (A) Scheme of AFM11-His gene design. The gene construct is shown from 50 to30-end encoding the monomeric TandAb subunit with N-terminal signal sequence, VH and VL domains separated by identical (Gly2Ser)2 linkers (L1, L2,L3), and the C-terminal hexa-histidine-tag (His6-tag). Upon expression, 2 polypeptide gene products dimerize in a head-to-tail fashion. (B) AFM11-Hiswas expressed in 10-day fed-batch cultures of stably-transfected CHO cells which grow to high cell densities, maintain high viability, and secrete theAFM11-His product into the cell culture supernatant (CSS). (C) AFM11-His was purified by Ni-NTA Superflow chromatography followed by preparativesize exclusion chromatography (SEC) on Superdex 200. High purity of the AFM11-His TandAb homodimer was demonstrated by analytical SEC. PurifiedAFM11-His runs in SEC as a single peak with a retention time that is consistent with its expected molecular weight of 106 kDa. (D) Integrity of the purifiedAFM11-His protein was analyzed by SDS-PAGE. Under denaturing, reducing and non-reducing conditions, AFM11-His monomers run at the expected sizeof »50 kDa. (E) Molecular forms of AFM11-His were analyzed by analytical SEC after incubation at 4!C, 21!C, or 37!C for the indicated numbers of days.High Molecular weight Forms (HMF) appear as a mixture of aggregates and species with an apparent molecular weight of »200 kDa, consistent with tet-rameric forms; AFM11-His homodimer represents the main chromatographic peak under all conditions assayed; Low Molecular weight Forms (LMF) rep-resent a minor fraction under all conditions and are consistent with the monomeric form of the AFM11 with molecular weights <60 kDa.
www.tandfonline.com 587mAbs
10/28/2016 Affimed
http://www.affimed.com/technologie-developement.php 1/2
Technology Development
Affimed is continuing to push the boundaries of cancer immunotherapies.
Technology Development and Other Targets
Affimed continues to invest in the further development of its proprietary
technologies and seeks to expand its intellectual property. For example,
preclinical proof of concept has been demonstrated for Trispecific Abs
recognizing two distinct tumor targets. Such molecules offer the
opportunity to target tumors expressing structures which are also
exposed on healthy cells as long as healthy cells do not coexpress both
cancerassociated antigens. Therefore, this technology has the potential
to widen the therapeutic window compared to classic single targeting.
One such Trispecific Abs is currently being developed for a hematologic
malignancy. Affimed has also compiled a short list of attractive targets for
the development of future TandAbs or Trispecific Abs. Furthermore,
Affimed is now wellpositioned to increase the leverage of its cutting edge
technologies in the context of research collaborations with pharmaceutical
industry partners.
Likewise, AbCheck, Affimed’s wholly owned subsidiary devoted to the
generation and optimization of antibodies using phage display, yeast
display and exclusive bioinformatics software is continuously upgrading its
technologies in order to provide stateoftheart services for industrial
partners, in addition to providing candidates for Affimed projects.
Construction of a TandAb
Antibodies for construction of TandAbs
are either derived from IgGs (e.g.
coming from a yeast display screen) or
from single chain Fv’s (e.g. coming from
a phage display screen). Because
Affimed has developed TandAbs for a
large number of targets, the company
has both the expertise and experience
for designing and constructing these
molecules.
Fully human antibodies are generated
by Affimed’s strategic partner AbCheck,
which uses a broad spectrum of state
oftheart antibody generation
technologies, including affinity
maturation and in silico optimization
tools.
Impressum Disclaimer
Technologies NKcell TandAbs Tcell TandAbs Trispecific Abs Technology Development
About Affimed Technologies Products Collaborations Investors & Media Contact

§ Clinically much less advanced than for ALL
§ Several compounds in phase 1 testing§ AMG 330§ MGD006§ MCLA-117
§ Many more in preclinical testing§ Expanding range of target antigens§ Variety of antibody formats
Bispecific antibodies for AML

AMG 330§ CD3-directed humanized BiTE antibody
§ Targets CD33§ Expressed on AML blasts in almost all patients§ Possibly expressed on leukemic stem cells in some
patients
§ Potent preclinical anti-AML activity§ Active across entire cytogenetic/molecular disease
spectrum§ Preserved activity in presence of ABC transporter
proteins
§ Phase 1 study ongoingFriedrich et al. Mol Cancer Ther. 2014;13(6):1549-1557; Laszlo et al. Blood. 2014;123(4):554-561; Harrington et al. PLoS One. 2015;10:e0135945

MGD006 (S80880)§ CD3-directed humanized DART
antibody
§ Targets CD1231
§ Found on AML blasts in almost all patients§ Overexpressed on leukemic stem cells relative to normal
hematopoietic progenitor/stem cells
§ Potent preclinical anti-AML activity2,3
§ No effect on colony formation of CD34+ cord blood cells§ Little in vivo effect on monkey marrow progenitor cells
§ Phase 1 study ongoing
demonstrated similar binding affinitiesand kinetics to human and cynomolgusmonkey CD3 and CD123 antigens (Fig. 1Band fig. S1E). Furthermore,MGD006simul-taneously bound both antigens in a bispecif-ic enzyme-linked immunosorbent assay(ELISA) format that used human or mon-key CD123 for capture and CD3 for detec-tion (Fig. 1C) and exhibited similar cellsurface binding to human and monkey Tlymphocytes (Fig. 1D).
MGD006 activity in primary AMLpatient samplesLeukemic blasts from anAMLpatient sam-ple were identified as CD45med+/CD33+
cells in the peripheral blood mononu-clear cells (PBMCs) and demonstratedrobust CD123 expression (Fig. 2A andfig. S2). Treatment with MGD006 over a6-day period resulted in a dose-dependentdepletion of leukemic blasts accompa-nied by a concomitant expansion of au-tologous T cells, up-regulation of theproliferation marker Ki-67, and a pro-portionally greater expansion of CD8+
cells. Greater activation (CD25 expres-sion) of CD4+ than CD8+ cells was ob-served, with IFN-g (interferon-g) and IL-6being the predominant cytokines produced.Expression of granzyme B and perforin,however, was elevated in CD8+ cells withonly a modest elevation in CD4+ cells (Fig.2A). Similar datawere obtainedwith a bonemarrow sample from a separate AML pa-tient whose blasts expressed moderatelevels of CD123 (fig. S3). These data showthat MGD006 is capable of expandingand redirecting autologous T cells fromAML patients toward leukemic blast cellkilling.
Antitumor activity in humanPBMC–reconstituted tumor-bearingmice continuously exposedto MGD006We previously demonstrated that redi-rected T cell tumor killing can be recapitu-lated by systemic DART protein injectionsin xenograft mouse models in which hu-man PBMCs are provided as a sourceof effector cells (14). On the basis of itsdesign, the short circulating half-life ofMGD006 allows close regulation of theexposure inpatientsbut requires continuousinfusion to ensure prolonged and adequateexposure. To ascertain MGD006 antitu-mor activityunder conditions recapitulating
Fig. 1. MGD006 binding to human and cynomolgusmonkey CD3 andCD123. (A) Schematic represen-tation of MGD006. (B) Equilibrium dissociation constants (KD) for the binding of MGD006 to human andcynomolgus (Cyno) monkey CD3 and CD123 determined by surface plasmon resonance analysis. (C) Bi-functional ELISA demonstrates simultaneous engagement of both target antigens byMGD006. ELISA plateswere coated with human CD123 (left) or cynomolgus monkey CD123 (right). DART at a range of concentra-tions was added, and binding was detected with human CD3–biotin. OD, optical density. (D) Cell surfacebinding of MGD006 to CD123+ Molm-13 target cells (left), human T cells (middle), and cynomolgus T cells(right) was detected by fluorescence-activated cell sorting (FACS) with a mAb specific to E-coil and K-coilregions of the DART (a-EK).
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 27 May 2015 Vol 7 Issue 289 289ra82 2
on February 27, 2016http://stm
.sciencemag.org/
Dow
nloaded from
1Testa et al. Biomark Res. 2014;2(1):4; 2Chichili et al. Sci Transl Med. 2015;7(289):289ra82; 3Al Hussaini et al. Blood. 2016;127(1):122-131
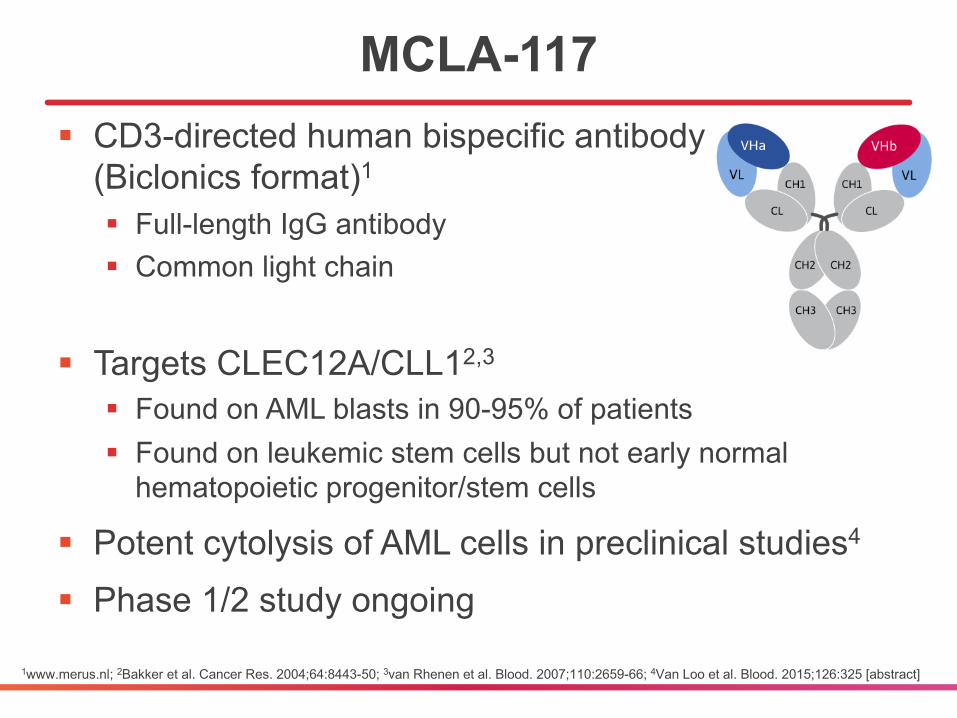
MCLA-117§ CD3-directed human bispecific antibody
(Biclonics format)1
§ Full-length IgG antibody§ Common light chain
§ Targets CLEC12A/CLL12,3
§ Found on AML blasts in 90-95% of patients§ Found on leukemic stem cells but not early normal
hematopoietic progenitor/stem cells
§ Potent cytolysis of AML cells in preclinical studies4
§ Phase 1/2 study ongoing
1www.merus.nl; 2Bakker et al. Cancer Res. 2004;64:8443-50; 3van Rhenen et al. Blood. 2007;110:2659-66; 4Van Loo et al. Blood. 2015;126:325 [abstract]
10/25/2016 Technology - MERUS
http://www.merus.nl/technology/ 2/4

Bispecific antibodies in AML – open questions
§ Ideal target antigen(s)?
§ Purpose of therapy?§ Gross reduction of leukemia cell burden?§ Elimination of minimal residual disease§ Conditioning before hematopoietic cell transplantation
§ Sequence in which therapeutics are used?
§ Optimization with conventional chemotherapeutics?
§ Supportive care needs?§ “On-target, off-AML” effects (cytopenias, immune effector cell
activation)
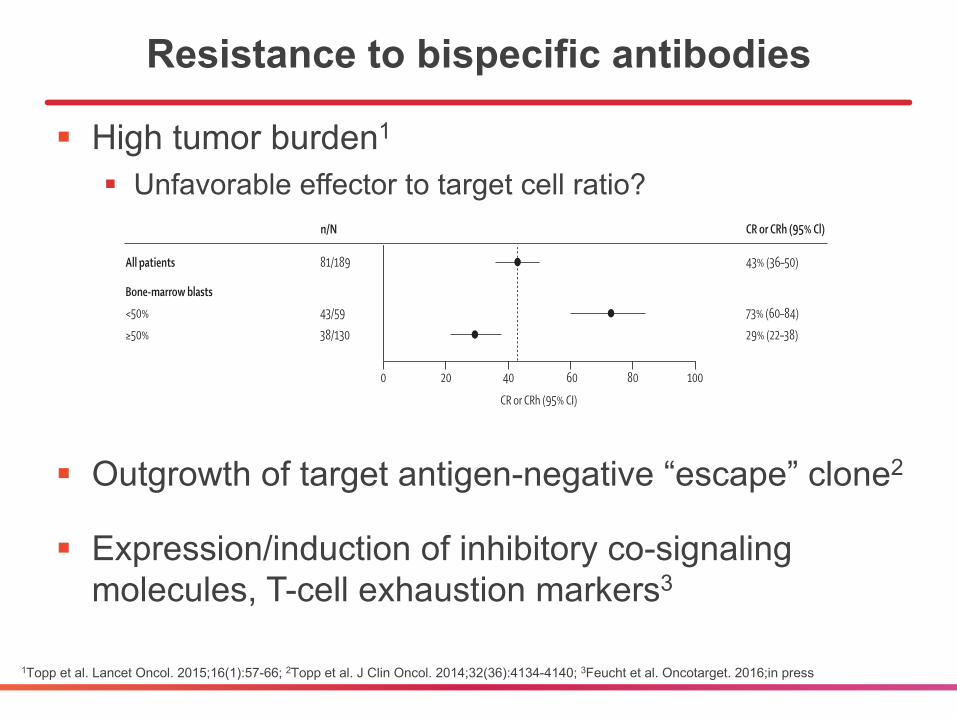
Resistance to bispecific antibodies
§ High tumor burden1
§ Unfavorable effector to target cell ratio?
§ Outgrowth of target antigen-negative “escape” clone2
§ Expression/induction of inhibitory co-signaling molecules, T-cell exhaustion markers3
1Topp et al. Lancet Oncol. 2015;16(1):57-66; 2Topp et al. J Clin Oncol. 2014;32(36):4134-4140; 3Feucht et al. Oncotarget. 2016;in press
Articles
www.thelancet.com/oncology Vol 16 January 2015 61
or CRh during the core study period were still alive and in remission. The remaining 45 patients had either relapsed (37 patients) or died without documented relapse (seven patients, six of whom died after allogeneic HSCT; one patient without allogeneic HSCT died of infection). One patient relapsed during cycle one of blinatumomab therapy, and fi ve patients relapsed during cycle two. Median relapse-free survival was 5∙9 months (95% CI 4∙8–8∙3) for the 82 patients in CR or CRh (fi gure 3A), 6∙9 months (95% CI 4∙2–10∙1) for patients in CR, and 5∙0 months (95% CI 1∙4–6∙2) for those in CRh, with a median follow-up of 8·9 months (IQR 4∙6–11∙1). Median overall survival was 6∙1 months (95% CI 4∙2–7∙5; fi gure 3B) for all 189 patients, with a median follow-up of 9∙8 months (IQR 6∙0–12∙9). In Mantel-Byar analysis, the relative odds ratio for survival benefi t of achieving remission was 0∙13 (95% CI 0∙08–0∙22; p<0∙0001).
CR or CRh occurred in similar proportions of patients with (29 [45%] of 64) and without (52 [42%] of 125) previous allogeneic HSCT. After blinatumomab treatment, 32 (40%) patients who achieved CR or CRh within the fi rst two treatment cycles proceeded to receive allogeneic HSCT while still in remission (table 2),
including fi ve (17%) of 29 patients who had received allogeneic HSCT before study entry and 27 (52%) of 52 patients who had not. The overall 100-day mortality after allogeneic HSCT (counting from the day of HSCT after blinatumomab-induced response) was 11% (95% CI 0–23). When censored for allogeneic HSCT, the Kaplan-Meier curves of overall survival largely overlapped and the medians were similar (appendix). Of the 49 responders (including 14 CRh responders) who did not proceed to allogeneic HSCT, 32 (65%) had either received previous allogeneic HSCT (including nine patients with CRh) or were 65 years of age or older (including one patient with CRh).
73 patients who achieved CR or CRh within the fi rst two cycles were evaluable for MRD. Of those, 60 (82%) patients achieved an MRD response (ie, MRD negativity; table 2), 59 in cycle one and one in cycle two. Among CR or CRh responders, median relapse-free survival for MRD responders was 6∙9 months (95% CI 5∙5–10∙1) versus 2∙3 months (95% CI 1∙2–not estimable) for MRD non-responders. Median overall survival for MRD responders versus MRD non-responders was 11∙5 months (95% CI 8∙5–not estimable) versus 6∙7 months (95% CI 2∙0–not estimable).
Figure 2: Overall responses (CR or CRh) after two treatment cycles among prespecifi ed patient subgroupsThe dashed line represents the point estimate for CR or CRh for the entire patient population. CR=complete remission. CRh=CR with partial recovery of peripheral blood counts. HSCT=haemopoietic stem-cell transplantation.
n/N
All patientsSexWomenMenGeographical regionEuropeUSAAge group (years)18 to <3535 to <5555 to <65≥65Previous salvage therapyNo previous salvage1 previous salvage2 previous salvage>2 previous salvageDisease statePrevious HSCTNo previous HSCTNo previous HSCT, no previous salvageNo previous HSCT, 1 previous salvageNo previous HSCT, ≥2 previous salvageBone-marrow blasts<50%≥50%
81/189
32/70 49/119
39/95 42/94
39/90 21/46 10/28 11/25
19/38 36/77 15/42 11/32
29/64 52/125 12/29 27/55 13/41
43/59 38/130
43% (36–50)
46% (34–58) 41% (32–51)
41% (31–52) 45% (34–55)
43% (33–54) 46% (31–61)36% (19–56) 44% (24–65)
50% (33–67)47% (35–58)36% (22–52)34% (19–53)
45% (33–58)42% (33–51)41% (24–61)49% (35–63)32% (18–48)
73% (60–84)29% (22–38)
0 20 8060
CR or CRh (95% Cl)
40 100
CR or CRh (95% CI)
Articles
www.thelancet.com/oncology Vol 16 January 2015 61
or CRh during the core study period were still alive and in remission. The remaining 45 patients had either relapsed (37 patients) or died without documented relapse (seven patients, six of whom died after allogeneic HSCT; one patient without allogeneic HSCT died of infection). One patient relapsed during cycle one of blinatumomab therapy, and fi ve patients relapsed during cycle two. Median relapse-free survival was 5∙9 months (95% CI 4∙8–8∙3) for the 82 patients in CR or CRh (fi gure 3A), 6∙9 months (95% CI 4∙2–10∙1) for patients in CR, and 5∙0 months (95% CI 1∙4–6∙2) for those in CRh, with a median follow-up of 8·9 months (IQR 4∙6–11∙1). Median overall survival was 6∙1 months (95% CI 4∙2–7∙5; fi gure 3B) for all 189 patients, with a median follow-up of 9∙8 months (IQR 6∙0–12∙9). In Mantel-Byar analysis, the relative odds ratio for survival benefi t of achieving remission was 0∙13 (95% CI 0∙08–0∙22; p<0∙0001).
CR or CRh occurred in similar proportions of patients with (29 [45%] of 64) and without (52 [42%] of 125) previous allogeneic HSCT. After blinatumomab treatment, 32 (40%) patients who achieved CR or CRh within the fi rst two treatment cycles proceeded to receive allogeneic HSCT while still in remission (table 2),
including fi ve (17%) of 29 patients who had received allogeneic HSCT before study entry and 27 (52%) of 52 patients who had not. The overall 100-day mortality after allogeneic HSCT (counting from the day of HSCT after blinatumomab-induced response) was 11% (95% CI 0–23). When censored for allogeneic HSCT, the Kaplan-Meier curves of overall survival largely overlapped and the medians were similar (appendix). Of the 49 responders (including 14 CRh responders) who did not proceed to allogeneic HSCT, 32 (65%) had either received previous allogeneic HSCT (including nine patients with CRh) or were 65 years of age or older (including one patient with CRh).
73 patients who achieved CR or CRh within the fi rst two cycles were evaluable for MRD. Of those, 60 (82%) patients achieved an MRD response (ie, MRD negativity; table 2), 59 in cycle one and one in cycle two. Among CR or CRh responders, median relapse-free survival for MRD responders was 6∙9 months (95% CI 5∙5–10∙1) versus 2∙3 months (95% CI 1∙2–not estimable) for MRD non-responders. Median overall survival for MRD responders versus MRD non-responders was 11∙5 months (95% CI 8∙5–not estimable) versus 6∙7 months (95% CI 2∙0–not estimable).
Figure 2: Overall responses (CR or CRh) after two treatment cycles among prespecifi ed patient subgroupsThe dashed line represents the point estimate for CR or CRh for the entire patient population. CR=complete remission. CRh=CR with partial recovery of peripheral blood counts. HSCT=haemopoietic stem-cell transplantation.
n/N
All patientsSexWomenMenGeographical regionEuropeUSAAge group (years)18 to <3535 to <5555 to <65≥65Previous salvage therapyNo previous salvage1 previous salvage2 previous salvage>2 previous salvageDisease statePrevious HSCTNo previous HSCTNo previous HSCT, no previous salvageNo previous HSCT, 1 previous salvageNo previous HSCT, ≥2 previous salvageBone-marrow blasts<50%≥50%
81/189
32/70 49/119
39/95 42/94
39/90 21/46 10/28 11/25
19/38 36/77 15/42 11/32
29/64 52/125 12/29 27/55 13/41
43/59 38/130
43% (36–50)
46% (34–58) 41% (32–51)
41% (31–52) 45% (34–55)
43% (33–54) 46% (31–61)36% (19–56) 44% (24–65)
50% (33–67)47% (35–58)36% (22–52)34% (19–53)
45% (33–58)42% (33–51)41% (24–61)49% (35–63)32% (18–48)
73% (60–84)29% (22–38)
0 20 8060
CR or CRh (95% Cl)
40 100
CR or CRh (95% CI)

Checkpoint proteins modulate BiTE antibodies
§ PD-L1 increased in ALLs resistant to blinatumomab1
§ Expression of T-cell ligands modulates BiTE antibody activity in vitro2
§ Activation of T-cell co-signaling increases efficacy of BiTE antibodies2
1Feucht et al. Oncotarget. 2016;in press; 2Laszlo et al. Blood Cancer J. 2015; 5:e340
PD-L1 PD-L2 CD80 CD860
10000
20000
50000
100000
Exp
ress
ion
(MFI
)
n=3
TF-1 Parental TF-1 CellsEngineered Sublines
PD-L1 PD-L2 CD80 CD860
5000
10000
15000
20000
Exp
ress
ion
(MFI
)
n=3
ML-1 Parental ML-1 CellsEngineered Sublines
25 50 1000
20
40
60
80
100
AMG 330 (pg/mL)
%S
peci
fic C
ytot
oxic
ity E:T=1:1
10 25 500
20
40
60
80
100
AMG 330 (pg/mL)
%S
peci
fic C
ytot
oxic
ity E:T=1:2.5
25 50 1000
20
40
60
80
100
AMG 330 (pg/mL)
%S
peci
fic C
ytot
oxic
ity
Parental+PD-L1+PD-L2+CD80+CD86
E:T=2.5:1
10 25 500
20
40
60
80
100
AMG 330 (pg/mL)
%S
peci
fic C
ytot
oxic
ity
Parental+PD-L1+PD-L2+CD80+CD86
E:T=1:1
A
B
ML-1 TF-10
20
40
60
80
100
%S
peci
fic C
ytot
oxic
ity
Control+CD28 Antibody

Future directions – look for§ Further definition of role for blinatumomab in B-ALL
§ Blinatumomab combination therapy § Other chemotherapeutics§ Checkpoint inhibitors
§ Initial results with bispecific antibodies in AML
§ Widening range of leukemia cell targets, antibody formats
§ Better definition of relevant resistance mechanisms

Key points§ Bispecific antibodies offer new option for B-ALL
§ Blinatumomab improves outcome for R/R ALL§ Unique toxicities
§ Alternative antibodies in early clinical testing
§ Role in AML will be defined in next years
Ø My take: exciting field, but many important open questions