biopharma lifecycle validation: beyond the · pdf filebiopharma lifecycle validation: beyond...
TRANSCRIPT

CONFIDENTIAL ©2014 PAREXEL INTERNATIONAL CORP. ALL RIGHTS RESERVED.
BioPharmaLifecycle
Validation: Beyond the Basics
Carmen Medina, MPH, Ph.D. ( c )
Vice President, Technical Services
Former FDA Investigator

Roadmap
• Life Cycle Phases for Process Validation
– Elements of Early Stage Process Design and Development
– Process Performance Qualification Strategy
• Risk Management: From Cell Bank to Finished Product Manufacturing
– Raw Material Characterization
– Univariate / Multivariate Experimentation (correlate multi-variables)
– Design Space / Control Space Relationship (DoE)
– Bioburden Mapping (EM Control Strategy)
– Viral Clearance Studies
– Resin Lifetime
• Monitor, Control and Improve
– Continuous Process Verification coupled with continuous improvement

Roadmap
Life Cycle Phases for Process Validation
3

Backdrop
• Biologics and Biosimilars are burgeoning markets
• Strong manufacturing capability is the flywheel of success
• Merck, Amgen , Pfizer, and many others are in both pioneer and biosimilar markets
• Manufacturing a biologic is a complex process
• PV rules apply, with an added twist

Current PV Standards
• FDA’s January 2011, Guidance for the Industry, Process Validation: General Principles and Practices– Aligns with product lifecycle concepts included in International
Conference on Harmonization –ICH Guidelines • Q8(R2) Pharmaceutical Development
• Q9 Quality Risk Management
• Q10 Pharmaceutical Quality System
– Promotes a 3-Stage Approach• Stage 1 – Process Design
• Stage 2 – Process Qualification
• Stage 3 – Continued Process Verification
Understand sources of variation// Detect degree of variation//Understand impact of variation//Control variation commensurate with risk it represents

Current PV Standards
• European Medicines Agency (EMA), March 2012 Guideline on Process Validation– Aligns with product lifecycle concepts included in International Conference on
Harmonization –ICH Guidelines
• Q8(R2) Pharmaceutical Development
• Q9 Quality Risk Management
• Q10 Pharmaceutical Quality System
– Promotes Continuous Process Verification • Risk-based, real-time verification
• Identification of CPPs, CQAs, etc.
• Deepen knowledge of process and heighten control of variability
Remember these stages, as they will become important when identifying potential landmines.

Current PV Standards
• cGMPs– 21 CFR 600, applies to biologics
– 21 CFR 211
• 100: Product and Process Controls
• 110: In-process Sampling and Testing
• 113: Validation of Sterilization Processes
• 160: Scientifically Sound Specs + Sampling
• 180 (e): Annual Product Reviews
• 198: Complaint Files
– Revised Compliance Policy Guide 7132c.08 / Sec. 490.100
– Promotes Continuous Process Verification and Control• Risk-based, real-time verification
• Identification of critical parameters in the process
• Monitor changes
• Evaluate status of control scheme at least annually
• Establish 360°feedback loop

Process Validation: A Product Life-cycle Management Program
Key phases: Should be delineated as such in the PV report.
Process Design:– Lab, pilot, small scale and commercial scale studies to establish
process
Process Qualification:– Facility, utilities and equipment
– Confirm commercial process design
– Includes technology transfer
Commercialization:– Monitor, collect information, assess
– Maintenance, continuous verification, process improvement.
8
Current PV Standards

Process Validation: A Product Life-cycle Management Program
Process Design:– RAW MATERIALS: Starting materials, such as cell banks, require extensive
characterization and risk assessment to establish the appropriate controlstrategy, which will ensure product consistency. Use-tests and risk mitigationplans for every lot received of RMs and starting materials must be in placeprior to filing and PAI.
Process Qualification:– Phase / Stage 2 entails two parts: Fitness of facilities and equipment; and
Process consistency and reproducibility. Both must be in place to legitimizePPQ batches.
Commercialization:– In order to mitigate the need for additional lots at the time of the PAI,
outputs of DoEs, MVAs, scale-down models, risk assessments, and processcapability studies during process characterization should be included in PartOne of PV report. Moreover, process risk assessments developed duringprocess characterization must be combined with risk assessments forprocess performance qualification.
9
Current PV Standards

Process Validation: A Product Life-cycle Management Program
• Make clear which CPP and CQA process requirements must be supported byfacility components, such as water system, equipment, automation platforms,coupled with the appropriate engineering-based control strategy to ensure risksto process requirements are properly controlled and mitigated. These processrequisites should be referred to as “critical engineering aspects” and included inthe CPV or some other Process Control Strategy (PCS). (Risk ranking strategiesmay be used, uni and multivariate analysis (MVA), FMEA may be used.)
• Ensure the CQAs that contribute the most process variability receive enhancedsampling and monitoring. Critical equipment interfaces and process steps, (viarisk assessments) should also receive enhanced sampling and monitoring.
This is all part of Continuous Verification requirements!
10
Current PV Standards

Process Validation: A Product Life-cycle Management Program
• Part Two of PV report should correspond to FDA’s and EU’s Phase 2, related toprocess performance qualification and should target process confidence andcapability, by linking objective measures of variability (process performance andprocess confidence) scientifically justifying the manufacture of three PPQbatches. (ISPE offers several published methods with which to accomplish this.)
• Part Three of the PV report should also correspond to FDA’s and EU’s Phase 3:Continuous Process Verification (CPV), which is intended to ensure and proveprocess remains in a state of control throughout it lifecycle. In order to show thatQuality attributes, especially CQAs, are being appropriately controlled andmonitored throughout the process, the CPV must take into account allinformation related to raw material characterization.
• This means the CPV plan will evolve as information becomes available. Moreover,over time, raw material quality attributes will vary; making it necessary to onceagain enhance the CPV, to ensure impact to product quality is adequatelymonitored.
11
Current PV Standards

Roadmap
Risk Management:
From Cell Bank to Finished Product Manufacturing
12
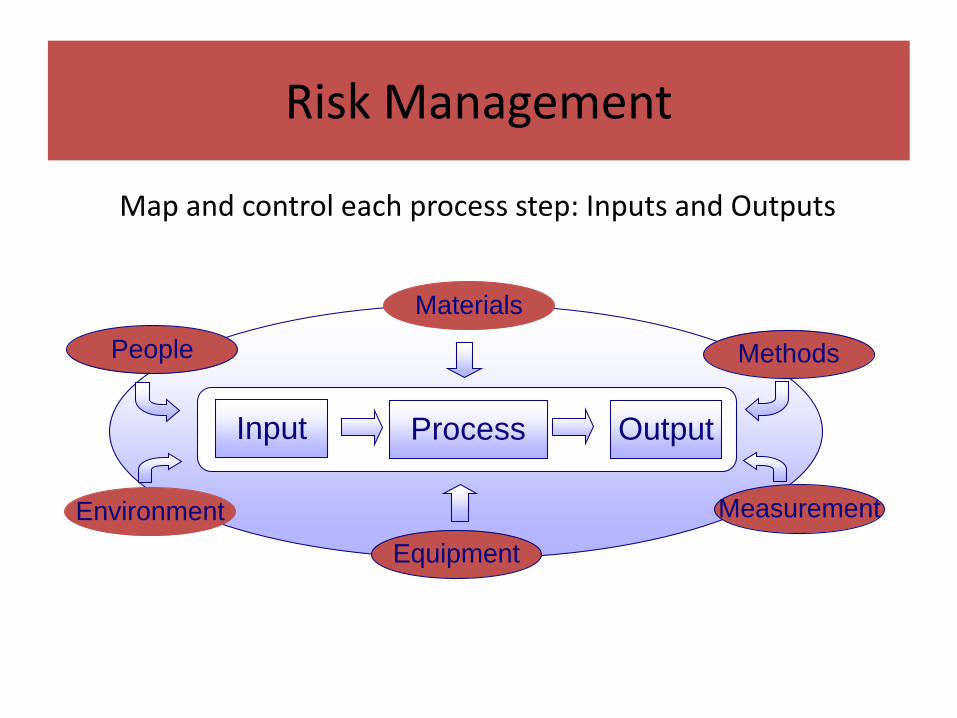
Process Characterization
Map and control each process step: Inputs and Outputs
Materials
MethodsPeople
Environment
Equipment
Measurement
Input Process Output
Risk Management

Process Characterization
• Biologics manufacturing offers limited data for intended commercial scale due to duration of process, cost of goods, extensive raw material characterization
• Design of Experiments (DOE) methodologies are often used to limit the number of required experiments and to determine the effects of parameter interactions
• Process Characterization studies focus on parameters that were deemed critical through risk assessment
• Studies are performed using lab-scale models
• Pre-approved acceptance criteria are NOT required for process characterization studies
• Results should be summarized in process characterization reports.
Process Characterization

Process Characterization
• Recommend having clinical safety data profile on hand as it relates to raw materials. (Toxicity studies)
• Uncertainty around CPPs and CQA (which will be linked to raw material characterization) will result in a higher number of high-risk (via FMEA) parameters, which will require formal characterization via uni or multivariate experimentation.
• Raw material characterization tends to be very complex, especially for larger molecules and blends. Their impact to product quality variability must be established during Phase 1 (Process Characterization), product quality substantively verified in Phase 2 (Process Performance Qualification), and most importantly, monitored throughout the process’ lifecycle during Phase 3(Continuous Verification).
Focus on risks, statistics, and sampling plans.
Process Characterization

Important Definitions
ICH Q5E states,
Quality Attribute: A molecular or product characteristic that is selected for its
ability to help indicate the quality of the product. Collectively, the quality
attributes define identity, purity, potency, and stability of the product, and
safety with respect to adventitious agents. Specifications measure a selected
subset of the quality attributes.

Important Definitions
ICH Q8 (R2)states,
Critical Quality Attribute (CQA): A physical, chemical, biological or
microbiological property or characteristic that should be within an
appropriate limit, range, or distribution to ensure the desired product quality.

Important Definitions
Impact/No impact: Assessment of impact is focused to drug product safety,
efficacy, and quality based on scientific knowledge. The statement "No
impact to drug product" means that the failure mode is not impacting the
safety, efficacy, and quality on the final drug product.
Process Performance Attribute (PPA): An in-process measurement that is
used to evaluate the performance of the process. Process Performance
Attributes are not directly controlled and are typically considered process
outputs. An example would be a process step yield measurement. Two types
of Process Performance Attributes are defined; critical, and key.
Critical Performance Attribute (CPA): A direct measure of the functionality orobjective of a step especially as it relates to product quality.
Key Performance Attribute (KPA): An attribute that is used to assess processconsistency for a particular process step or stage.

Important Definitions
Process Parameter (Operational Parameter): A parameter that can be directly manipulated. Operational parameters are typically considered process inputs. Three types of Process Parameters are defined; critical, key, and non-key (Key and Non-Key are subsets of Non-Critical).
Critical Process Parameter (CPP): A process parameter whose variability has an impact on one or more critical quality attributes and therefore should be monitored or controlled to ensure the desired product quality.
Key Process Parameter (KPP): A process parameter that, when varied within the Characterization Range, has a significant impact on process consistency or on a key or critical performance attribute.
Non-key Process Parameter (Non-KPP): A process parameter that, when varied within the Characterization Range, will not have a significant impact on either process consistency or product quality.

Important Definitions
Set point: A target value of the process parameter during operation, around which the control system regulates the value. Not all process parameters have a set point. The maintenance of the set point by the equipment defines the control range of the process parameter.
Control Range: The ability of the control system to regulate a defined set point.
Operational Range: A range within the license that defines the optimal process control. In absence of a defined operational range the license range is used for process control.
License: The operating value or range for the process parameters that are defined in the TD/ regulatory submission. Within these ranges, the process produces the specified product.

Important Definitions
Evaluated Range: A given process parameter range that has been evaluated for parameter's impact on CQAs. The data may be from development studies, process validations, or commercial-scale studies or excursions.
Impacted CQA: Primary CQA impacted when CPP exceeds the evaluated range.
High-Risk: Quality impact is high if the failure has a potential impact on patient health or disrupts the supply of product to the patient.
Low Risk: Quality impact is low if the failure results in no negative impact to patient health and does not disrupt the supply of product to the patient.

Risk Management
Let’s look at potential landmines from Cell Culture, to Purification, to Final Product Manufacturing:
– Creating a cell line
– Qualification of cell culture
– Bioreactors / transfer lines / Temperature / Dissolved Oxygen
– Purified growth media
– Buffer prep
– Harvesting controls
– Filtration
– Filling process: vial bunching, glass fragments, etc.
– Glass variations in vials or syringes (protein aggregation)
– Reagents with trace amounts of copper, metals, etc.
– Column packing, pressure
– Column cleaning, storage
– Resin reuse
Manufacturing changes are not created equal.

Risk Management
• Creating and characterizing cell line: This is your IP
• Cultures are then transferred to successively larger vessels
• Harvesting controls: Temperature, Dissolved Oxygen, etc.
• Filtration: clogging, manual variations, limit number of filtrations
• Filled into vials or syringes: vial bunching, glass fragments, etc.
• Reagents: purchased from a different supplier may present trace elements such as trace amounts of copper
• Pre-filled syringes: found to be cloudy; client learned that a part per million of tungsten was in the syringes, which caused the protein to aggregate.

Risk Management
• Purification and filtration process changes can lead to big difference in the FP
• Column packing, pressure
• Column cleaning, storage
• Resin reuse requires extensive study
• Hold times: Dirty Hold Time and Clean Hold Time included in the CV plan– Holding tanks, times, temperatures, and conditions of storage are described in Hold-
time studies.
• Procedures used to protect microbiological quality of the bulk drug during all holding periods are referenced.
• Verification of maintenance of the microbiological quality during holding periods is required.

Risk Management
VIRAL CLEARANCE: Ensure there is a clear link to the lab-scale viral clearance model and PPQ conditions.
BIOBURDEN: Ensure Bioburden control and bioburden mapping are demonstrated at PPQ scale and conditions.
RESIN Re-USE: Ensure studies are depicted in the PV report’s Section 2, in terms of linking the scaled-down model to PPQ scale. Resin re-use must be monitored in real time for its impact to quality attributes. PAREXEL recommends site commit to on-going verification of chromatography resin and membrane cleaning (lifetime).
MIXING STUDIES: At time of filing, full-scale required for all mixing studies.
PV Report should demonstrate clearance of all impurities at PPQ scale to belowLOQ (Supplemented w/ small scale “spiking data” of fold clearance at current andnext step.)

Risk Management
Ways to diffuse potential landmines:
• Process mapping
• Characterize and control raw materials
• Justify sampling and testing plans
• Gather data efficiently and continuously
• Analyze data using statistical tools
• Intercept and mitigate variability
• Continuous monitoring of CQAs and CPPs
• Leverage key quality systems and metrics
• Establish alert and action limits, where possible
• Prospective and retrospective control charts

Process Mapping
Process Legend for any step in a process
Process Parameters:
• CPP = Critical Process Parameter
• CQA = Critical Quality Attribute
• L = Licensed Process Parameter
Equipment or Instrument
Material (Tested by Inspection Plan)
Material (Untested)
◊ In-Process Quality Control Testing

Risk Management Systems
Organization: multidisciplinary lenses, evaluations and conclusions Design Space for new products Quality Mission / Policy Purchasing Controls / Supplier Management Management Review [211.180(f)] Deviations and Corrective and Preventive Actions Complaint Management Statistical Techniques
Interpretation and appropriate application of quality systems during the entire lifecycle of the product—concept to commerce.

In summary…
• Prioritize and control most costly and risky CQAs, CPPs, CPAs, KPAs, etc.
• Develop predictive models for each process
• Tie to development knowledge
• Quality Triad or grouping that provides the most insight into process fluctuations
• Make PV Lifecycle management the flywheel of QA activities
– Monitor process using pertinent quality metrics
– Establish alert & action limits around key metrics
– Establish a multidisciplinary feedback loop

In summary…
• Monitor, Correct, Improve• Deepen process understanding batch-to-batch
– Identify and measure variation
– Quarterly monitoring
– APRs
• Enhance sampling and testing scheme– May need to increase, or reduce, over time
• Develop risk-based, real-time profiles for each process
• Maintain validated state using continuous monitoring of both quality and process metrics– Change control trends
– Deviation Trends and CAPAs
– Complaint Trends
– Equipment Histories (OOTs)

In summary…
Process Design: Basis of process knowledge and understanding of quality risks, which lead to a comprehensive process control strategy. Key is to characterize variability using the appropriate tools: DoE, MVA, scale-down models, etc. Use risk assessments to assign risks, but realize that the risk rating may change over time: up or down.
Process Qualification: Part 1 (P=facility and equipment), plus Part 2 (P=process qualification)= Process Performance Qualification, PPQ.
Continued Process Verification: Statistically based, continual process monitoring of CPPs, CQAs, KPPs, PARs, etc., with close attention to raw material variability over the lifetime of the process.

Q & A
Thank you