bioinformatics - simbac · multiple copies of genome are broken up into fragments of 2-10k bases...
TRANSCRIPT

Bioinformatics

What can sequences tell us?
annotated sequence of human X chromosome
By themselves? Not a heck of a lot...*
AGACCTGAGATAACCGATAC
However, through comparison and analysis, combined with molecular and structural biology, they can reveal “vast amounts of evolutionary information hidden away within them” (Francis Crick, the less vocal eugenics advocate of the pair)
*Indeed, one of the key results learned from the Human Genome Project is that disease is much more complicated than a simple appeal to genome-based therapeutics as was originally promised
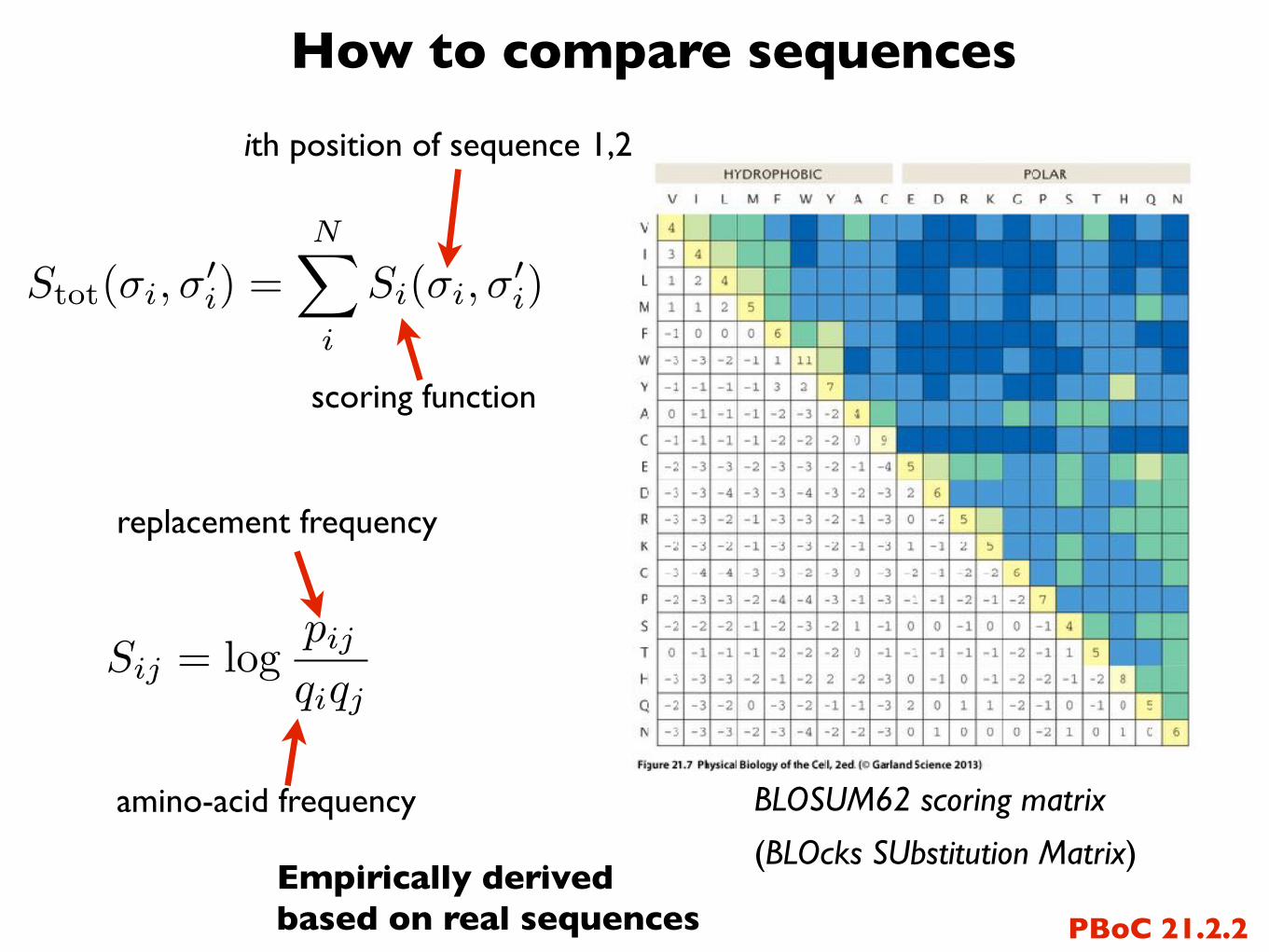
How to compare sequences
BLOSUM62 scoring matrix
(BLOcks SUbstitution Matrix)
Stot(σi, σ′
i) =N!
i
Si(σi, σ′
i)
ith position of sequence 1,2
scoring function
Sij = logpij
qiqj
replacement frequency
amino-acid frequency
PBoC 21.2.2Empirically derived based on real sequences

Phylogenetic analysis
sequence similarity can be used to trace ancestral lineages
hemoglobin sequences
PBoC 21.4.1
phylogenetic tree

-based on 16S rRNA taxonomy
-demonstrates most diversity is in the microbes
-first proof of archaea as a separate evolutionary domain (only accepted a decade after first published!)
-determined by Carl Woese, who was dubbed “Microbiology's Scarred Revolutionary” Woese, C. R.; G. E. Fox (1977-11-01). "Phylogenetic structure of the
prokaryotic domain: The primary kingdoms".PNAS 74 (11): 5088–5090.
Tree of life

Neutral mutations and evolution
presence or absence of retroviral sequences inserted into DNA match phylogenetic tree (similar for transposons)
frequency of neutral amino acid (and codon redundant) mutations also agree with expectations
Human 2
Chimp 2q
Chimp 2p
all great apes have 24 pairs of chromosomes, while humans have 23 pairs
genetic analysis shows human chromosome 2 resulted from ancestral fusion of two chromosomes

From sequence to structure
PBoC 21.2
sequences of actin-like proteins in bacteria (MreB, ParM) and eukaryotes (actin) - almost ZERO similarity
...yet the structures look nearly identical!
sequence conservation
➔ structure conservation
(BUT NOT THE CONVERSE)

How to compare structuresSimple RMSD no longer works when sequence lengths differ
QH scores alignment based on residue-residue distances AND gaps (no sequence information!)
Patrick O'Donoghue, Zaida Luthey-Schulten, Evolutionary Profiles Derived from the QR Factorization of Multiple Structural Alignments Gives an Economy of Information, (2005) JMB, 346: 875-894,
Sequence-based and structure-based phylogenetic trees are in agreement ➔ structure encodes evolutionary information as well!!!

Molecular paleontologyancient protein sequences can be
reconstructed via phylogenetic analysis (NOT the same as Jurassic Park, but close!)
absorbance spectra of dinosaur rhodopsin demonstrates what it could see
PBoC 21.4.1
Beta-lactamase (modern, ancestor 1, ancestor 2)
Valeria A. Risso et al. 2013. Hyperstability and Substrate Promiscuity in Laboratory Resurrections of Precambrian β-Lactamases. J. Am. Chem. Soc., 135 (8), pp. 2899–2902
Eric Gaucher at GT structurally characterized 3-4 billion-year-old versions of an antibiotic-resistance protein (www.gauchergroup.biology.gatech.edu/)

Horizontal gene transfer
genes are shared horizontally between species instead of solely vertically
common (even dominant?) among bacteria; can lead to, e.g., extremely fast spread of antibiotic resistance genes
even eukaryotes may acquire some genes horizontally, including entire cells (mitochondria and chloroplasts)
complicates attempts to draw a universal tree of life with a unique common ancestor (but does not erase it completely!)

Human accelerated regions
Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, Kern AD, Dehay C, Igel H, Ares M Jr, Vanderhaeghen P, Haussler D (2006). "An RNA gene expressed during cortical development evolved rapidly in humans". Nature 443: 167–172.
If chimps and humans share > 98% of our DNA, where are the important differences? In the so-called “Human Accelerated Regions” (HARs)
~200 identified so far, mostly in non-coding regions, NOT genes for proteins
For example, HAR1, the most accelerated region, codes for a novel RNA gene expressed during neocortical development codes ➔ it’s all about regulation!

How to sequence DNA?“shotgun sequencing”
multiple copies of genome are broken up into fragments of 2-10k bases
PCR-like method can read 0.5-1k bases of each fragment (from both directions)
random short sequences examined for overlaps and computationally reassembled into one long sequence
Human Genome Project (public) used “hierarchical” shotgun, where libraries of 100-300k bases were first created and then shotgun-sequenced
Celera Genomics project (private) used “whole-genome” shotgun sequencing

Next (and next)-gen sequencing methods
Human Genome Project cost $2.7 billion and took 10 years
goal for personalized medicine is (was) $1000
challenge now met, new goal is < $100
One promising technique: nanopore sequencing, e.g., using alpha-hemolysin (left) or MsbA (above)
computational modeling/simulation necessary to interpret experiments (group of Alek Aksimentiev, UIUC)

Epigenetics - beyond sequence aloneModifications to DNA other than sequence changes can also influence expression
in some cases are even heritable (some definitions include heritability as a requirement)

Epigenetics
same genes, different tail kink
hypermethylation involved in some cancers
DNA methylation a key example of epigenetic control
methylation can both increase and decrease stability of DNA strands depending on spacing, frequency
Recognition of methylated DNA through methyl-CpG binding domain proteins Xueqing Zou, Wen Ma, Ilia Solov'yov, Christophe Chipot and Klaus Schulten Nucleic Acids Research, 40:2747-2758,2012

And now for something completely different…

Radiating genomes of cichlid fish
-over 2000 species in just three lakes
-500 species in Lake Victoria arose in only 100k years
-exhibit a diversity of morphological and ecological traits, e.g., what they eat
five species have been sequenced to help answer it
How did the cichlid fish evolve so quickly?
Brawand et al. The genomic substrate for adaptive radiation in African cichlid fish. (2014) Nature. 513: 375-381.

How many biologists does it take to sequence a fish?

Molecular mechanisms of evolution at work-burst of gene duplication (20% of
new genes expressed in a completely new, tissue-specific domain)
-accelerated evolution in protein-coding genes (the “boring” answer), e.g., opsin in the eye and a signaling protein involved in jaw development
# of duplicationsspecies divergence
Brawand et al. The genomic substrate for adaptive radiation in African cichlid fish. (2014) Nature. 513: 375-381.

Molecular mechanisms of evolution at work
-high rate of change in gene regulatory elements, which changes how and where certain genes are expressed
CNE: conserved non-coding element
-high turnover of microRNAs (including 40 new genes) for suppressing gene expression to stabilize/refine new expression patterns
microRNAs: about 22 nucleotides long, interfere with mRNA AFTER transcription but BEFORE translation
Brawand et al. The genomic substrate for adaptive radiation in African cichlid fish. (2014) Nature. 513: 375-381.

“Three waves of TE insertions were detected in each of the cichlid genomes”TE: transposable
elementsBrawand et al. The genomic substrate for adaptive radiation in African cichlid fish. (2014) Nature. 513: 375-381.
Transposable elements (jumping genes)

Neutral drift or positive selection?The authors attribute the great diversity of changes seen across these genomes to a period of relaxed selection that occurred early in the radiation. During this time, the selective pressures that maintained the stability of the genome were reduced, thereby allowing genetic variation to accumulate and produce subsequent diversification into the lineages we observe today. However, accelerated evolution can result either from neutral evolution due to relaxed selection, or from positive natural selection acting through new selective pressures. Most of the genomic signatures in the paper do not strongly distinguish between these two possibilities. Indeed, it seems most likely that the retention of gene duplicates and rapid genetic divergence were primarily driven by positive natural selection, as species adapted to the great diversity of ecological niches available in the lakes. Subsequent extinction of early lineages could have led to an apparent burst of rapid change on the branch leading to the extant species. There may be no need to invoke a genetic revolution when plain old natural selection can explain the observed patterns.
C.D. Jiggins. Evolutionary biology: Radiating genomes. (2014) Nature. 513: 318-319.