biochemical and reperfusion targeting strategies to improve brain
TRANSCRIPT

BIOCHEMICAL AND REPERFUSION TARGETING STRATEGIES TO IMPROVE BRAIN PROTECTION DURING PROLONGED HYPOTHERMIC CIRCULATORY ARREST
JUSSIRIMPILÄINEN
Department of Surgery,University of Oulu
OULU 2001
D 622

ACTA UNIVERS ITAT IS OULUENS I SD M e d i c a 6 2 2
JUSSI RIMPILÄINEN
BIOCHEMICAL AND REPERFUSION TARGETING STRATEGIES TO IMPROVE BRAIN PROTECTION DURING PROLONGED HYPOTHERMIC CIRCULATORY ARREST
Academic Dissertation to be presented with the assent ofthe Faculty of Medicine, University of Oulu, for publicdiscussion in the Auditorium 2 of the University Hospitalof Oulu, on February 23rd, 2001, at 12 noon.
OULUN YLIOPISTO, OULU 2001

Copyright © 2001Acta Univ. Oul. D 622, 2001
Manuscript received: 15 January 2001Manuscript accepted: 23 January 2001
Communicated byDocent Kari KuttilaProfessor Pertti Aarnio
ISBN 951-42-5885-1ISSN 0355-3221 (URL: http://herkules.oulu.fi/issn03553221/)
ALSO AVAILABLE IN ELECTRONIC FORMATISBN 951-42-5886-X (URL: http://herkules.oulu.fi/isbn951425886X/)
OULU UNIVERSITY PRESSOULU 2001

Rimpiläinen, Jussi, Biochemical and reperfusion targeting strategies to improve brainprotection during prolonged hypothermic circulatory arrest Department of Surgery, University of Oulu, P.O.Box 5000, FIN-90014 University of Oulu, Finland Acta Univ. Oul. D 622, 2001Oulu, Finland(Manuscript received: 15 January 2001)
Abstract
Ischaemic cerebral injury follows a well attested sequence of events including three phases, i.e. de-polarization, biochemical cascade and reperfusion injury. Glutamate excitotoxicity plays an impor-tant role in the development of ischaemic brain injury following prolonged hypothermic circulatoryarrest (HCA), and leukocyte infiltration and a cytokine-mediated inflammatory reaction are knownto play a pivotal role in the reperfusion phase. The aim of this series of experimental studies was todevelop biochemical and reperfusion-related strategies to improve brain protection. We tested the hy-potheses that the Na+ channel blocker lamotrigine (I) or the N-Methyl-D-Aspartate-receptor antago-nist memantine (III) could improve the cerebral outcome after HCA and studied whether a leukocyte-depletion filter (L-DF; LeukoGuard LG6®, Pall Biomedical, Portsmouth, U.K) could mitigate braininjury (II). The aim of the fourth study was to find out whether lamotrigine combined with the leuko-cyte-depleting filter can potentiate cerebral protection (IV).
A chronic porcine model was used, in which haemodynamic, electrophysiological, metabolic andtemperature monitoring were performed for four hours after the instigation of rewarming and S-100�measured up to 20 hours. Cytokines were measured, microdialysis was performed, and daily behav-ioural assessments were made until death or elective sacrifice on the seventh postoperative day, uponwhich a histopathological analysis of the brain was carried out.
The rate of EEG burst recovery was higher in the lamotrigine-treated animals, the median being40% of the baseline compared with 17% in the placebo group at 4 hours after the start of rewarming(p = 0.02) and 80% compared with 20% at 4 hours (p = 0.01). Complete behavioural recovery wasseen in 5/8 of cases (63%) after lamotrigine administration, compared with 1/8 (13%) in the placebogroup (p = 0.02). The median behavioural score among the animals that survived for 7 days was high-er in the lamotrigine group (8) than in the controls (7) (p = 0.02).
Mortality was 2/10 in the L-DF group and 5/10 in the controls, the median behavioural score onday 7 being higher in the L-DF group (8.5 vs. 3.5 p=0.04). The median of the total histopathologicalscore was 6.5 in the L-DF group and 15.5 in the control group (p=0.005).
In the memantine group 5/10 animals survived seven days, as compared with 9/10 in the placebogroup, and the median behavioural score on day 7 was 3.5 compared with 7.5 in the placebo group(p=0.39). The median of the total histopathological score was 16 in the memantine group and 14 inthe placebo group (p=0.25).
In the LD-F + lamotrigine group 7/8 animals survived for seven days, as compared with 4/8 in thelamotrigine only group and 3/8 among the controls. EEG burst recovery 7 hours after the start of re-warming was highest in the LDF + lamotrigine group, the median being 94% (p=0.024 vs. controls),compared with 81% in the lamotrigine group and 64% in the control group. The median behaviouralscore on day 7 was 9 in the LD-F + lamotrigine group (p=0.004 vs. controls), 4 in the lamotriginegroup and 0 in the control group, while the median of total histopathological score was 14 (p=0.046vs controls), 14.5 (p=.062 vs. controls) and 21, respectively. The control group had the highest intrac-erebral lactate, glutamate and glycerol levels after HCA.
In conclusion, the results indicate that the NA+ channel blocker lamotrigine improves the neuro-logical outcome after a prolonged period of HCA but that the NMDA receptor antagonist memantinedoes not have this property in the present setting. The leukocyte-depleting filter mitigates brain injuryafter a prolonged period of HCA, and lamotrigine can potentiate this effect.
Keywords: hypothermic circulatory arrest, lamotrigine, brain protection, leukocyte-deple-tion, memantine, reperfusion injury.

To Minna, Tuomas, Anniina and Salla


Acknowledgements
This work was carried out at the Cardiothoracic Research Laboratory of the Departmentof Surgery, Oulu University Hospital, during the years 1998 � 2000.
First of all, my greatest gratitude goes to my supervisor, Professor Tatu Juvonen, M.D.,Ph.D., Head of the Department of Surgery, for giving me the idea and possibility to studythis challenging aspect of surgery. It was his support and guidance that made this workpossible.
A thesis is not the result of the work of an individual, and in this sense I owe mywarmest thanks to my co-workers in the laboratory and co-authors Matti Pokela, M.S.,Vesa Anttila, M.D., Ph D., Pekka Romsi M.D., Docent Kai Kiviluoma, M.D., Ph.D.,Vilho Vainionpää, M.D., Ph.D., Kauko Korpi R.N., Seija Seljänperä R.N., VeikkoLähteenmäki, Janne Heikkinen M.S., Timo Kaakinen M.S., Erkka Rönkä M.S, ProfessorJorma Hirvonen, M.D., Ph.D., Docent Ville Jäntti, M.D., Ph.D., Minna Mäkiranta, M.Sc.,Hanna Heinonen, M.D., Elina Remes, M.S., Pasi Lepola, M.Sc, Ari Mennander, M.D.,Ph.D., and Pasi Ohtonen, M.Sc. We did this work together.
I am grateful to Professor Pertti Aarnio, M.D, Ph.D., and Docent Kari Kuttila, M.D.,Ph.D., for reviewing the present manuscript and for their constructive criticism.
I would like to thank Ari Ahola, M.D., and Simon Lister, Ph.D., of Glaxo-Wellcomefor providing the lamotrigine, Professor Osmo Hormi, Ph.D., and Anu Moilanen, Ph.D.,of the Department of Chemistry, Oulu University, for preparing the isethionate salt oflamotrigine, Sirpa Ämmälä, M.Sc. (Pharm.), and Outi Ryymin, M.Sc. (Pharm.), forpreparing and randomizing the ampoules, and Professor Wojchiech Danysz, for kindlyproviding us with memantine and the pharmacokinetic analyses. I would also express mygratitude to Heljä-Marja Surcel, M.D, PhD., for the cytokine analyses and to MalcolmHicks, M.A., for revising the English language of this thesis.
I would like to thank the previous head of Cardiothoracic and Vascular Surgery,Professor Pentti Kärkölä M.D., Ph.D., and the present head of Cardiothoracic Surgery,Docent Martti Lepojärvi M.D., for permitting me to concentrate on this research, andlatter together with Esa Salmela M.D. for helping my family with his surgical skills. I alsothank all my colleagues in the Division of Cardiothoracic and Vascular Surgery for theircooperation: Docent Risto Pokela, M.D., Ph.D., Kari Ylönen, M.D., Docent Pekka

Rainio, M.D., Ph.D., Docent Jari Satta, M.D., Ph.D., Jarmo Lahtinen, M.D., MarttiMosorin, M.D., Jouni Heikkinen, M.D. and Arjaleena Ilo, M.D.
My thanks also go to my friends Eino Ignatius, Antti Isokangas, Juho Kariniemi,Tommi Niku, Juha Oksanen, Mika Paavola, Seppo Pesola, Anssi Puutio and IsmoSaarenpää. It is a great privilege to have friends like you. I would also thank Keijo Nissiläand my many other friends along the Oulu, Tornio and Teno Rivers for helping me toforget scientific work temporarily.
I wish to thank my parents, Anneli and Olavi Rimpiläinen, and my sister JohannaRimpiläinen, for their never-failing love and support, and similarly my parents-in-law,Irma and Pekka Heikkinen, for their encouragement and help in domestic matters duringthis work.
Finally, I wish to express my loving thanks to my wife Minna and my childrenTuomas, Anniina and Salla. You are the most valuable things in my life and without yourlove this work would not have been successful.
This work was supported financially by Oulu University Hospital, the FinnishFoundation for Cardiovascular Research, the Sigrid Juselius Foundation and the Inkeriand Mauri Vänskä Foundation.
Oulu, December, 2000 Jussi Rimpiläinen

Abbreviations
AA Arachidonic acidAMPA µ-amino-3-hydroxy-5-methyl-4-isoxazoleproprionateANOVA Analysis of varianceATP Adenosine triphosphateAUC Area under the curveCABG Aortocoronary bypass graftingCMRO2 Cerebral metabolic rate of oxygenCPB Cardiopulmonary bypassDNA Deoxyribonucleic acidEAA Excitatory amino acidEEG ElectroencephalographyGluR Glutamate receptorHCA Hypothermic circulatory arrestIQR Interquartile rangeIL InterleukinLD-F Leukocyte-depleting filterNMDA N-Methyl-D-AspartateNMDAR N-Methyl-D-Aspartate receptorNO Nitric oxideNOS Nitric oxide synthaseONOO� Peroxynitrate PLA2 Phospholipase A2PMN Polymorphonuclear leukocytesRNA Ribonucleic AcidROS Reactive oxygen speciesRCP Retrograde cerebral perfusionSCP Selective cerebral perfusionSD Standard deviationTM Transmembrane domainTNF Tumour necrosing factor


List of original publications
This thesis is based on the following articles, which are referred to in the text by theirRoman numerals:
I Anttila V, Rimpiläinen J, Pokela M, Kiviluoma K, Mäkiranta M, Jäntti V, Vainion-pää V, Hirvonen J & Juvonen T (2000) Lamotrigine improves cerebral outcome afterhypothermic circulatory arrest: A study in a chronic porcine model. The Journal ofThoracic and Cardiovascular Surgery 120: 247�55.
II Rimpiläinen J, Pokela M, Kiviluoma K, Anttila V, Vainionpää V, Hirvonen J, Ohto-nen P, Mennander A, Remes E & Juvonen T (2000) Leukocyte filtration improvesbrain protection after prolonged period of hypothermic circulatory arrest: A study ina chronic porcine model. The Journal of Thoracic and Cardiovascular Surgery 120:1131�40.
III Rimpiläinen J, Pokela M, Kiviluoma K, Vainionpää V, Hirvonen J, Ohtonen P, JänttiV, Anttila V, Heinonen H & Juvonen T (2000) NMDA antagonist Memantine has noneuroprotective effect during hypothermic circulatory arrest: A study using achronic porcine model. The Journal of Thoracic and Cardiovascular Surgery, inpress.
IV Rimpiläinen J, Romsi P, Pokela M, Hirvonen J, Kiviluoma K, Vainionpää V, Ohto-nen P, Jäntti V, Anttila V & Juvonen T (2000) Lamotrigine with leukocyte filtrationprovides effective neuroprotection following hypotermic circulatory arrest in pig,submitted for publication.


Contents
Abstract Acknowledgements Abbreviations List of original publications 1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152 Review of the literature . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2.1 Pathogenesis of ischaemic brain injury . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.1.1 Depolarization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.1.2 Biochemical cascade . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2.1.2.1. Glutamate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182.1.2.2. Glutamatergic receptors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182.1.2.3. Consequences of calcium influx . . . . . . . . . . . . . . . . . . . . . . . 192.1.2.4. Nitric oxide (NO) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192.1.2.5. Calpains . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192.1.2.6. Gelsolin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 202.1.2.7. Apoptosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
2.1.3 Reperfusion injury . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 202.2 Strategies for mitigating ischaemic brain injury . . . . . . . . . . . . . . . . . . . . . . . . 21
2.2.1 Hypothermia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 212.2.2 Biochemical strategies for mitigating brain injury . . . . . . . . . . . . . . . . 24
2.2.2.1. Inhibitors of presynaptic glutamate release . . . . . . . . . . . . . . . 242.2.2.2. Inhibitors of postsynaptic glutamate release . . . . . . . . . . . . . . 252.2.2.3. Monosialogangliosides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 272.2.2.4. Ca2+ channel blockers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 272.2.2.5. Nitric oxide synthase inhibitors . . . . . . . . . . . . . . . . . . . . . . . . 282.2.2.6. Calpain inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 282.2.2.7. Gelsolin-like chemical agents . . . . . . . . . . . . . . . . . . . . . . . . . 292.2.2.8. Caspase inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.2.3 Strategies for mitigating reperfusion-related brain injury . . . . . . . . . . . 292.2.3.1. Pharmacological strategies . . . . . . . . . . . . . . . . . . . . . . . . . . . . 292.2.3.2. Modification of techniques or mechanical devices . . . . . . . . . 30
3 Aims of the present research . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 324 Materials and methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33

4.1 The chronic porcine model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 334.2 Preoperative Management . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 334.3 Pharmacokinetic pilot study (III) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 334.4 Drug Administration (I, III and IV) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 344.5 Anaesthesia and haemodynamic monitoring . . . . . . . . . . . . . . . . . . . . . . . . . . 344.6 Electroencephalography (EEG) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 354.7 Cardiopulmonary bypass . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 364.8 Experimental protocol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 364.9 Postoperative evaluation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 374.10 Histopathological analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 374.11 Serum S-100b . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 384.12 Cytokines (II) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 384.13 Microdialysis (III and IV) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 384.14 Other measurements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 394.15 Statistical analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
5 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 405.1 Lamotrigine improves cerebral outcome following hypothermic
circulatory arrest (I) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 405.2 Leukocyte filtration improves cerebral outcome following hypothermic
circulatory arrest (II) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 405.3 The NMDA antagonist memantine has no neuroprotective effect during
hypothermic circulatory arrest (III) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 415.4 Lamotrigine with leukocyte filtration provides effective neuroprotection
following HCA in pig (IV) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 426 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
6.1 General discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 456.2 Haemodynamic and metabolic data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 466.3 Leukocyte counts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 466.4 S-100b . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 466.5 Cytokines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 476.6 Behavioural outcome . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 476.7 Electroencephalography . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 476.8 Microdialysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 496.9 Mortality . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 496.10 Histopathological analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
7 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 538 References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54Original publications

1 Introduction
During operations on the aortic arch the normal antegrade perfusion must be interruptedto make it feasible to perform the technically complex procedures involved. There arethree methods currently used to protect the brain during circulatory arrest.
Hypothermic circulatory arrest (HCA) was introduced for this purpose (Niazi andLewis 1957) soon after the first clinical use of cardiopulmonary bypass (CPB)(Gibbon1954) and was later adopted for aortic arch surgery (Griepp et al. 1975, Gschnitzer 1973,Pierangeli et al. 1975), achieving acceptable mortality and morbidity rates (Griepp et al.1991, Kouchoukos et al. 1995). The major limitation of this method is the time constraint(Bellinger et al. 1995, Oates et al. 1995). A significant rate of cerebral oxygenmetabolism persists even in deep hypothermia (systemic temperature 10�15�C), and thesafe duration of HCA is limited to approximately 40 minutes (McCullough et al. 1999).
Selective cerebral perfusion (SCP) is a method of brain protection that has been usedin aortic arch surgery with good neurological results (Veeragandham et al. 1998). It istechnically more complicated than HCA, and its use predisposes the brain to embolicstroke in patients with supra-aortic arterial disease, so that it cannot be employed in casesof aortic dissection involving the arch vessels (Frist et al. 1986).
Retrograde cerebral perfusion (RCP) was introduced subsequently to prolong thepermissible interruption relative to HCA and to reduce the rate of embolic injury (Coselliand LeMaire 1997, Usui et al. 1997). In spite of the fact that neurological morbidity hasbeen shown to be acceptably low (Deeb et al. 1999), RCP itself has proved to involve thesame disadvantages as the other methods, e.g. cerebral oedema (Juvonen et al. 1998).Since RCP has failed to prolong the permissible interruption significantly, a renewedinterest is currently being shown in studying other strategies.
The aetiology of brain injury during aortic surgery is multifactorial, includingmicroemboli, macroemboli, haemorrhages and global ischaemia. These disparateaetiologies inevitably result in cellular ischaemia, and the underlying mechanism bywhich hypoxia and ischaemia lead to neuronal cell death is the same (Baumgartner et al.1997). In order to refine our strategies for improving cerebral protection during theseoperations, a better understanding of the pathogenesis of ischaemic brain injury isessential.

16
Ischaemic cerebral injury follows a well documented sequence of events, includingthree phases: depolarization, biochemical cascade and reperfusion injury. The importanceof the failure of neurotransmitter transport as a common pathway in the pathogenesis ofischaemic cerebral injury has been attested. This substantial step has increased ourunderstanding of the pathogenesis of ischaemic neuronal injury and at the same time hasopened up new therapeutic prospects for improving brain protection. One such strategy isto use appropriate pharmacological agents (Baumgartner et al. 1997, Lipton andRosenberg 1994). Previous studies also suggest that the reperfusion phase is of paramountimportance with regard to the pathogenesis of ischaemic brain injury. Leukocyteinfiltration can be detected following ischaemic insult to the brain, so that the bloodvessels will be filled with leukocytes (primarily neutrophils) and oedema will develop.The adhesion of leukocytes to the wall of blood vessels and their infiltration into theischaemic brain tissue can activate an inflammatory reaction driven by cytokines, whichexacerbates the degree of tissue injury on account of interference with normalmicrovascular perfusion and the release of cytotoxic enzymes (Feuerstein et al. 1998,Feuerstein et al. 1998).
In the present series of experimental studies we developed and tested biochemical andreperfusion-related strategies for improving brain protection during HCA (I, II and III).As emphasized above, the mechanism of ischaemic brain injury comprises these distinctphases, and targeting a single event may be insufficient. Therefore, the final series ofexperiments represented a combination of our most effective strategies in the hope thatthis approach could potentiate brain protection ( IV).

2 Review of the literature
2.1 Pathogenesis of ischaemic brain injury
As seen in Fig. 1, ischaemic brain injury involves three sequential phases: depolarization,biochemical cascade and reperfusion injury.
2.1.1 Depolarization
Although the mass of the brain is only 2% of the total body mass, its energy requirementis more than seven times higher than that of the other organs. The main source ofneuronal energy is the generation of ATP by the aerobic metabolism of glucose. The brainrequires a steady blood supply to provide both substrate and oxygen at a constant rate.Complete arrest of the cerebral circulation leads to cessation of neuronal electricalactivity and loss of the energy state and ion homeostasis within a few minutes. Depletionof high-energy phosphates, membrane ion pump failure, efflux of cellular potassium,influx of sodium, chloride and water, and membrane depolarization occur swiftly. If theinterruption in blood flow persists for longer than 5�10 min at normothermia, irreversiblecell damage is likely (Astrup et al. 1981). Depolarization leads to the failure ofneurotransmitter transport, which initiates a catastrophic biochemical cascade.
2.1.2 Biochemical cascade
Depolarization results in a failure to maintain ionic gradients and the normalhyperpolarized membrane potential. The cells of the presynaptic membrane depolarizeand release a flood of neurotransmitters, i.e. glutamate. This depolarization is mediatedby voltage-sensitive Na+ channels that carry electrical messages to the synapse. As aresult of depolarization, there is an influx of Ca2+ via the voltage-sensitive channels and aconcomitant secretion of glutamate ions. Although the principal targets of theseneurotransmitters are situated on the postsynaptic membrane, a number of presynaptic

18
excitatory amino acid (EAA) receptors and glutamate transporters receive the chemicalmessage and respond in various ways in order to regulate the actions of these messengers(Dunlap et al. 1995).
2.1.2.1 Glutamate
The accumulation of glutamate and the excessive activation of glutamate receptorsmediate neuronal injury and cell death. The intracellular glutamate concentration in braintissue is approximately 10 mmol per litre, and the extracellular concentration is estimatedto be 0.6 �mol per litre. Substantial excitotoxic damage to the cortical or hippocampalneurons in intact tissue is expected to occur when the extracellular glutamateconcentration reaches 2 to 5 �mol per litre (Lipton and Rosenberg 1994).
As glutamate is highly neurotoxic in the extracellular space, it is continually beingcleared in a normal environment, i.e. it is recycled by neuronal uptake through re-storagein the vesicles and re-release. Glial cells contain a similar, high-affinity, active transportthat ensures the efficient removal of glutamate from the synapse. Ischaemia impairs theability of cells to maintain such a glutamate uptake system (Rhoades and Tanner 1995).
2.1.2.2 Glutamatergic receptors
Most of the excitotoxicity is related to the destructive actions of high intracellular calci-um mediated by stimulation of the various subtypes of glutamatergic receptors. Glutama-te receptors (Glur) are either ionotropic or metabotropic. The ionotropic receptors are lig-and-gated ion channels which are further divided into three groups on the basis of whichligand preferentially activates the receptors: those that respond to N-methyl-D-aspartate(NMDA), µ-amino-3-hydroxy-5-methyl-4-isoxazoleproprionate (AMPA), and kainate(Small and Buchan 1996). The metabotropic receptors activate intracellular second mes-sengers via G-proteins (Pin and Duvoisin 1995).
NMDA receptors (NMDAR) are made up of various combinations of the subunitsNMDAR1A-G and NMDAR2A-D and exhibit a high Ca2+/Na+ permeability ratiocompared with other glutamate receptors which play a crucial role in Ca2+-drivenexcitotoxicity. The high Ca2+ permeability is believed to be caused in part by anasparagine residue at the �N site� in the second transmembrane domain (TM2) of theNMDAR1 and NMDAR2 subunits. It is this N site, which is analogous to the �Q/R site�of AMPA receptors, that is involved in the voltage-dependent blocking of the channel byMg2+ (the block that is relieved by depolarization). Pharmacologically, NMDA receptorshave a high affinity for glutamate and require glycine as a coagonist. They also have slowactivation and deactivation times, which are in strong contrast with the rapid kinetics ofthe AMPA receptors (Seeburg et al. 1995).
In many areas of the brain Na+ ions pass through AMPA receptors much more easilythan Ca2+, a property that is regulated at the nuclear level. Four subunits of AMPAreceptors exist, of which glutamate receptor 2 (GluR2) is the only one whose Q/R site inTM2 is edited by RNA. The prerequisite for high Na+/Ca2+ permeability of a pentameric

19
AMPA receptor, as seen under normal circumstances, is the existence of at least oneGluR2 subunit (Hollmann et al. 1991). After an ischaemic insult, however, there is adecrease in the ratio of GluR2 to other AMPA receptor subunits, leading to altered AMPAchannel kinetics and subsequently increased Ca2+ permeability. This is probably thesource of �unregulated� Ca2+-driven excitotoxicity (Gasic and Hollmann 1992).
2.1.2.3 Consequences of calcium influx
Massive release of glutamate and activation of glutamatergic receptors leads to Ca2+
influx, which starts numerous secondary processes that accelerate ischaemic neuronaldamage. These mechanisms include activation of proteases, phospholipase A2 (PLA2),nitric oxide synthases (NOS), reactive oxygen species (ROS) and calpains, which arecapable, either directly or indirectly, of destroying cellular structures (Chabrier et al.1999, Zipfel et al. 1999).
PLA2 catalyzes the breakdown of membrane lipids to form fatty acids and arachidonicacid (AA), the latter being capable of undergoing further metabolism by cyclo-oxygenase,thus producing oxygen radicals. There is a correlation between the release of AA andneuronal damage (Dumuis et al. 1990).
2.1.2.4 Nitric oxide (NO)
NO acts as a neuromodulator in the central nervous system and participates in manyphysiological processes such as brain development, pain perception, neuronal plasticity,memory function and behaviour. There are three isoforms of NO present in the brain:neuronal, endothelial and inducible. When overproduced, however,NO reverts from aneuromodulator to a neurotoxic mediator. Such an overproduction of NO may be aconsequence of persistent stimulation of EAA receptors, but it can also be driven bycytokines. NO reacts with superoxide anions to generate the toxic substance peroxynitrite(ONOO�), which in turn oxidates proteins, lipids and DNA. This reaction can produceeven more potent neurotoxins, such as hydroxyl radicals. In addition, ONOO� can disturbthe glutamate transport system and possibly impair the mitochondrial respiratory chain(Chabrier et al. 1999, Garthwaite et al. 1989). NO is also a potent dilator of the cerebralblood vessels (Faraci and Brian 1994).
2.1.2.5 Calpains
Calpains are cytosolic proteinases whose proteolytic activity is directed mainly againstthe cytoskeleton and regulatory proteins. Calpains are activated by an increase inintracellular Ca2+, and their proteolysis produces irreversible degradation of a number ofstructural and regulatory proteins (Bartus et al. 1998). Increased intracellular levels of

20
calpains have been demonstrated in areas of cerebral ischaemia in focal ischaemia models(Liebetrau et al. 1999).
2.1.2.6 Gelsolin
High levels of intracellular Ca2+ activate gelsolin, a ubiquitous enzyme that severs actinmicrofilaments, thereby inhibiting any further influx of Ca2+ mediated by NMDAreceptor-gated and voltage-gated Ca2+channels and acting as a neuroprotective factor(Endres et al. 1999). On the other hand, recent evidence suggests that gelsolin maypromote apoptosis by reducing Ca2+ influx in instances of Ca2+ starvation. Gelsolin alsoseems to serve as a substrate for the critical apoptosis enzyme caspase (Zipfel et al.1999).
2.1.2.7 Apoptosis
There is an increasing amount of evidence that severe ischaemic injury may leadimmediately to high glutamate levels, acute Ca2+ overload and cell death, whereasneurons with milder injury undergo apoptosis (Zipfel et al. 1999). Caspases have beenshown to mediate apoptotic neuronal cell death (Gottron et al. 1997). Apoptosis is anactive process requiring metabolic energy and protein and ribonucleic acid synthesis. Theapoptotic cell is characterized by shrinkage, nuclear collapse and the cleaving ofchromatin into nucleosomal fragments, while the organelles retain their integrity andthere is no associated inflammation (Arends and Wyllie 1991). Apoptotic pathways arecontrolled by several specific genes. This cascade leads to the engulfment of dying cellsby their neighbours and the degradation of deoxyribonucleic acid (DNA) in the dyingcells (Williams and Smith 1993). Early gene expression, heat shock protein expression,suppression of protein synthesis and probably other factors play a significant role inapoptosis, and the mitochondria are involved in several ways, including the release ofcaspase activators, changes in electron transport and the loss of mitochondrial membranepotential. The major difference between the two types of cell death is the existence ofgeneralized involvement of neighbouring cells within necrotic tissue, whereas inapoptosis cell death is detected without inflammatory cells. To prevent leakage ofexcitatory amino acids, proteolytic enzymes, DNA or oxidized lipids with a pro-inflammatory response, apoptotic cells condense their intracellular milieu by cross-linking membrane proteins. The cellular contents are sealed within the dying cells untilphagocytosis intervenes (Green 1998).
2.1.3 Reperfusion injury
The reperfusion injury that follows the biochemical cascade is driven by cytokines andleukocytes. The hallmark of an inflammatory reaction in the ischaemic brain is leukocyte

21
infiltration, which is dominated by polymorphonuclear leukocytes (PMN), primarilyneutrophils (Feuerstein et al. 1994). Ischaemic vessels in the brain are filled withleukocytes and many have oedema around them. The adhesion of leukocytes to the wallsof cerebral blood vessels and their infiltration into ischaemic brain tissue activates aninflammatory reaction, which results in the release of large amounts of oxygen-freeradicals, including superoxide anions, hydrogen peroxide, hydroxyl radicals and singletoxygen. This reaction exacerbates the degree of tissue injury. Leukocytes also interferewith normal microvascular perfusion (Feuerstein et al. 1998).
Cytokines are a large and rapidly expanding group of polypeptides produced by manycell types. They are mediators of immuno-endocrine interactions and are necessary for theoptimal functioning of leukocytes. The cytokines are key mediators of acute phaseresponses to tissue injury (Tonnesen et al. 1996), and the pro-inflammatory cytokinesinduce increased neutrophil and endothelial surface adhesive molecule expression,promoting neutrophil-endothelial adherence. This adherence and subsequent neutrophilorgan binding is thought to be a �common pathway� for organ injury (Hill et al. 1997).An important source of cytokines in the central nervous system (CNS) is the peripheralblood cells (e.g leukocytes) which may enter the brain after injury and ischaemia.Cytokines are also produced by resident brain cells, including glia, neurones andendothelial cells (Rothwell and Strijbos 1995). Interleukin 1 (IL-1) has been shown tomediate neurodegeneration in vivo (Rothwell and Strijbos 1995), and tumour necrosisfactor alpha (TNF-�) and IL-1� seem to play a significant role in brain immune andinflammatory activities and ischaemic brain injury (Feuerstein et al. 1998). Activatedneutrophils may induce multi-organ oedema, including cerebral oedema, via TNF-� andother regional cytokines (Dewanjee et al. 1998).
The presence of recruited leukocytes at the site of inflammation is critically dependenton co-ordinated expression of the adhesion molecules. The efficient �docking � ofactivated leukocytes to their respective receptors on inflammatory cells and the activatedcapillary is the primary step in the process of transendothelial migration. The candidatemolecules include ICAM-1, ELAM-1 and P-selectin on the endothelial side and CD11/CD18, MAC-1 and LFA-1 on the leukocyte side (Feuerstein et al. 1998).
2.2 Strategies for mitigating ischaemic brain injury
2.2.1 Hypothermia
The mechanism by which hypothermia postpones the depolarization of the presynapticmembrane and thereby prevents ischaemic brain injury are summarized in Table 1. Theintact mammalian brain covers its energy needs almost exclusively by the oxidation ofglucose, and a prerequesite for this is a steady blood flow. The brain will tolerate an acutereduction in blood flow below 20 ml/100g/min or even 10 ml/100g/min (i.e.,< 20% ofnormal values) at least temporarily in normothermia (Astrup et al. 1977), whereupon theneurons are depolarized and the biochemical cascade starts. Hypothermia reduces theoxygen demand and produces a state of decreased metabolic activity, thereby restrictingthe ischaemic and tissue damage by extending the period during which the circulation

22
may be "safely" interrupted. It also increases high-energy phosphates (Kramer et al.1968) and the intracellular pH in the brain tissue, which delays the onset of acidosis.Hypothermia inhibits the release of neurotransmitters and delays the onset of the fatalbiochemical cascade (Rokkas et al. 1995). These events suggest that the cerebralmetabolic rate of oxygen (CMRO2) is highly dependent on temperature (Govier et al.1984) and that cerebral oxygen consumption decreases progressively as the temperatureis reduced, being 39% of the baseline value at 18�C, 20% at 13�C and 5% at 8�C(Mezrow et al. 1994). The optimal temperature for HCA remains controversial, however.From an experimental point of view it seems that �the colder the better� in terms of brainprotection, and profound hypothermia (5� to 7�C) gives the best protection (Gillinov etal. 1993). In a clinical setting the situation is not so simple, because deeper hypothermiarequires longer CPB times and prolonged extracorporeal circulation entails aconsumption of clotting factors and interference with the coagulation pathway and mayresult in coagulation disturbances (Taylor et al. 1978). The platelet activation enzymesand the enzymatic activities of clotting factors decrease during and after hypothermia(Wilde 1997). In addition, the extended CPB itself may be a cause of neuronal damage(Taylor 1998).
A �safe� duration of HCA has not yet been clearly defined, and it is known to behighly dependent on temperature. HCA times longer than 40 minutes have led to anincreased incidence of stroke, and HCA longer than 65 minutes has been shown toincrease the mortality rate (Svensson et al. 1993). Acceptable clinical morbidity rateshave been reported provided that HCA is kept shorter than 60 minutes (Ergin et al. 1994),but it has also been claimed that HCA does not fully protect the brain and that there is alinear relationship between the amount of brain injury and duration of HCA (Oates et al.1995). The problem with clinical studies is the measurement of brain recovery, whichexplain the differences between reports, but the time constraint on HCA and itsdependende on temperature are well demonstrated in an experimental setting. aHistological evidence of brain injury was found in a canine model after 2 hours of HCA,even in profound hypothermia (Arroyo et al. 1993, Gillinov et al. 1993), and in anotherexperiment with puppies a better neurological outcome was seen following 90 minutes ofHCA at 13�C than at 18�C, but both groups had evidence of neurological injury (Mezrowet al. 1995).
McCullough and co-authors at the Mount Sinai Medical Center in New York measuredCMRO2 in 37 adult patients with the aid of an ultrasonic carotid flow probe and derivedthe best available estimate for the period of safe arrest from the calculated temperaturecoefficient (Q10), the results indicating that 25�40% of the baseline metabolism is stillpresent at 20�25 �C and the safe duration of HCA is approximately 15 minutes, while alittle over 40 minutes is safe at 10�C. These findings suggest that shorter intervals andlower temperatures that those currently used may be necessary to ensure adequatecerebral protection during HCA (McCullough et al. 1999).
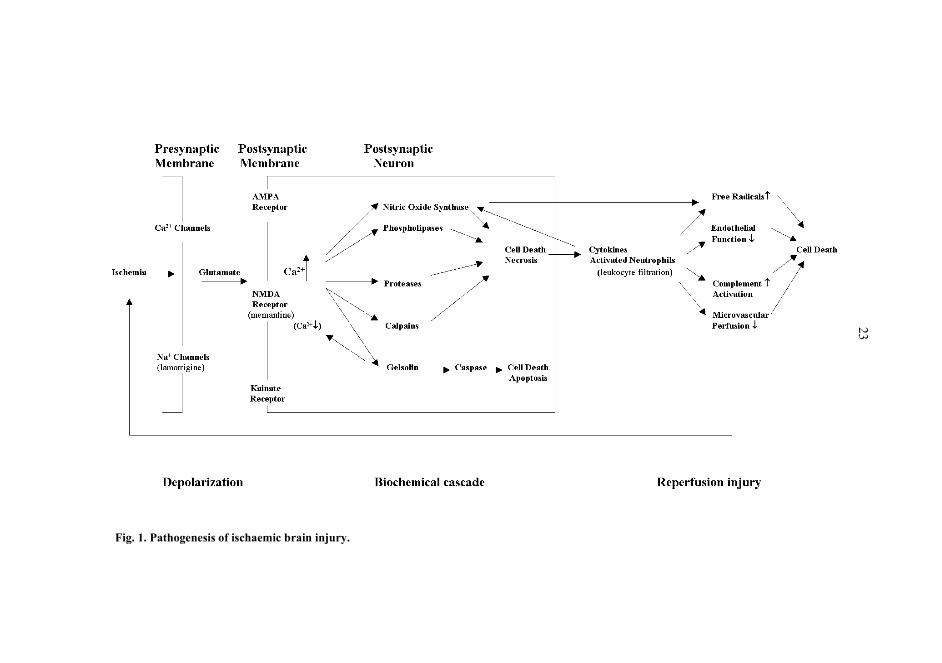
23
Fig. 1. Pathogenesis of ischaemic brain injury.

24
2.2.2 Biochemical strategies for mitigating brain injury
Most biochemical strategies for protecting the brain during HCA are based on theexperimental studies of normothermic ischaemia following a stroke or trauma. Althoughexperimental data of this kind have substantially increased our understanding of HCA-related injury and have opened up new therapeutic avenues for mitigating brain injuryfollowing hypothermic ischaemia, it must be borne in mind that not all pharmacologicalagents that are effective in normothermia necessarily protect the brain during HCA. Theliterature will be evaluated below in the same order as for the biochemical cascade in theprevious section.
2.2.2.1 Inhibitors of presynaptic glutamate release
SNX-111 is a presynaptic Ca2+ channel blocker that can reduce the influx of Ca2+ and thesubsequent release of neurotransmitters and has been shown in a rabbit model to beneuroprotective (Perez-Pinzon et al. 1997). On the other hand, it was recently found onlyto postpone neuronal injury in an ischaemic rat model and did not convey permanentprotection (Colbourne et al. 1999).
Lamotrigine (3,5-diamino-6-(dichlorophenol)-1,2,4-triazine) is a phenyltriazine deriv-ative that blocks voltage-gated Na+ channels and inhibits the ischaemia-induced releaseof glutamate. It is a commercially available, orally administered anticonvulsant with fewside effects, although rashes and cases of Stevens-Johnson syndrome have been reported(Bourgeois 1998, Dunn et al. 1999). Glutamate levels did not increase in the lamotriginegroup in an experimental rabbit model involving mild hypothermia (Bacher A 1997) andwere shown to decrease significantly in normothermic cerebral ischaemia (Conroy et al.1999). When lamotrigine and an NMDA receptor antagonist, MK-801, were compared inrats, both were found to effectively attenuate brain injury induced by 3-nitropropionicacid, a lamotrigine dose of 20 mg/kg providing a better neuroprotective effect than MK-801. In view of its better therapeutic effect and fewer side effects, the authors concludedthat lamotrigine is a more promising agent for potential clinical applications (Lee et al.2000).
Riluzole (2-amino-6-trigluoromethoxy benzothiazole) is a voltage-gated Na+ channelblocker that is also used clinically in cases of neurodegenerative diseases. It preventedischaemic spinal cord injury in a rabbit model (Lang-Lazdunski et al. 1999), in anotherexperiment animals treated with it had a better histopathological outcome after cardiacarrest (Kanthasamy et al. 1999).
Lubeluzole, a Na+ channel blocker that can inhibit the release of glutamate fromischaemic neurons, reducing postsynaptic excitotoxicity, may also inhibit postsynapticnitric oxide synthetase activity. It has been shown in a rat model to improve the structuraloutcome for brain cells after global cerebral ischaemia (Haseldonckx et al. 1997).Lubezole has also been studied in clinical setting for the treatment of acute stroke. In atrial employing 721 patients, its administration was associated with a trend towards areduction in mortality and statistically significant improvements in neurological recovery,

25
functional status and global disability measured three months after the stroke (Grotta1997).
Lifarizine is a similar substance that was found to be a promising neuroprotectant in anexperimental setting (Brown et al. 1995), but clinical trials were discontinued on accountof cardiac side effects such as hypotension and arrhythmia (Squire et al. 1995).
The voltage-dependent Na+ channel antagonist BW619C87 [4-amino-2-(4-methyl-1-piperazinyl)-5-(2,3,5-trichlorophenyl) pyrimidine] has proved to a potent neuroprotectant,reducing the volume of hemispheric ischaemic damage by 51% at a dose of 50 mg/kg in arat model (Kawaguchi et al. 1999). Another experimental Na+ channel antagonistBW1003C87 [5-(2,3,5-trichlorophenyl)-2,4-diamino-pyrimidine) mitigated brain damagein rats after carotid artery ligation (Gilland et al. 1994).
2.2.2.2 Inhibitors of postsynaptic glutamate release
These antagonists may be divided into non-competitive and competitive types, and alsointo compounds which modulate glycine sites and those which modulate polyamine sites.
Non-competitive NMDA receptor antagonists. In order to reach its binding site, a non-competitive NMDA receptor antagonist requires an ion channel to be opened up and thevoltage-dependent Mg2+ block to be released by postsynaptic membrane depolarization.As such drugs can only bind in an open ion channel, they accumulate in regions where theconcentration of glutamate is highest. Non-competitive NMDA receptor antagonists arehighly lipophilic and are rapidly distributed throughout the body, achieving a maximalconcentration in the ischaemic tissue with minimal delay (Muir and Lees 1995).
In the light of a neurological evaluation and cerebral histology, the prophylactic use ofa selective NMDA receptor antagonist, dizoclipine (MK-801), was found to have a clearneuroprotective effect on dogs subjected to 2 hours of HCA at 18�C (Redmond et al.1994). MK-801 crosses the blood-brain barrier easily and its effective dose can bedetermined from the disappearance of the EEG signal after administration.
NPS-1506 is a non-competitive NMDA receptor antagonist with a moderate affinity toits receptor. It was found to be neuroprotective in a rodent model for ischaemic stroke,haemorrhagic stroke and head trauma, being less neurotoxic than MK-801 (Mueller et al.1999).
Dextromethorphan and its analogue AHN649 are relatively selective, low-affinityNMDA antagonists, and have been shown to have neuroprotective efficacy in a rat modelutilizing temporal intraluminal filament occlusion of the middle cerebral artery (Britton etal. 1997, Tortella et al. 1999).
Aptiganel (CNS 1102) is a selective, non-competitive antagonist that acts on the ionchannel associated with the NMDA receptor and has been shown to be neuroprotective inneonatal lambs after HCA (Bokesch et al. 1997). The dose-limiting effects of aptiganelobserved in human volunteers were blood pressure increases and central nervous system(CNS) excitation or depression (Dyker et al. 1999).
Memantine is an non-competitive NMDA receptor antagonist that is widely used as avery well tolerated drug for the treatment of chronic neurodegenerative diseases such asParkinson�s disease, Alzheimer�s disease and AIDS, and also as a symptomatic treatment

26
for epilepsy and depression (Lipton 1994, Lipton 1996, Parsons et al. 1999). It has beenshown to ameliorate NMDA receptor-mediated neurotoxicity in vitro and in vivo innormothermia (Chen et al. 1992, Stieg et al. 1999), and has also proved to be a promisingdrug for the treatment of retinal ischaemia (Legreze et al. 1998, Osborne 1999). Resultsregarding the experimental use of memantine to prevent the injury induced by spinal cordischaemia have been controversial (Ehrlich et al. 1999, Euler et al. 1997, Miyamoto andMiyamoto 1999).
The anaesthetic ketamine bears surprising similarities to the other NMDA receptorinhibitors, but there is some controversy regarding its benefits for neurologicallycompromised patients (Cheng et al. 1997).
Remacemide hydrochloride and its principal active desglycinyl metabolite are low-affinity non-competitive NMDA receptor antagonists. Remacemide hydrochloride wasshown to be neuroprotective in an animal model of hypoxia and ischaemic stroke, andoverall postoperative neurological recovery in a clinical prospective study of 171 patientsundergoing aortocoronary bypass grafting (CABG) with CPB was more favourable in theremacemide group than in the controls (Arrowsmith et al. 1998).
Competitive NMDA receptor antagonists. In contrast to their non-competitivecounterparts, the penetration of competitive NMDA receptor antagonists through theblood-brain barrier is limited due to their low lipophility and delayed distribution to thebrain. Agents with higher lipophilic properties have been developed, the prototypecompound being CPP, of which d-CPPene and CGS 19755 (Selfotel) are derivatives.These drugs have been shown to limit neuronal damage in animal stroke models, butunfortunately they cause neuropsychological side-effects (Muir and Lees 1995). Aclinical trial of the drug Selfotel was terminated due to an increase in neurologicalmortality (Davis et al. 1997).
Glycine site antagonists. Glycine, a major inhibitory neurotransmitter in the spinalcord and brainstem of vertebrates, is collected in the synaptic vesicles via a proton-coupled transport system and is released into the synaptic cleft after depolarization of thepresynaptic terminal. It can participate in excitatory neurotransmission by modulating theactivity of the NMDA subtype of the glutamate receptor. The glycine site on the NMDAreceptor complex provides a therapeutic target for acute focal and global ischaemia,potentially avoiding most of the side-effects associated with competitive and non-competitive NMDA antagonists (Muir & Lees 1995).
A glysine receptor antagonist, ZD9379, administered before or after middle cerebralartery occlusion significantly reduced the infarct volume in permanent focal ischaemia inrats (Tatlisumak et al. 1998), and as the side-effects of ZD9379 have been found to beminimal, it undoubtedly warrants consideration for further development (Sun and Cheng1999).
7-Chlorokynuretic acid and its derivatives ACEA-1021 (Licostinel), ACEA-1031 andACEA-1416 have also been found to reduce infarct volume in focal ischaemia models(Warner et al. 1995). Licostinel may be a safer and better tolerated neuroprotective agentthan many of the previously evaluated NMDA antagonists (Albers et al. 1999).
Felbamate, a novel anticonvulsant that binds to the glycine site of the NMDA receptor,has been shown to have neuroprotective properties in vitro and in vivo. In a gerbil modelof global ischaemia, felbamate given after carotid occlusion prevented delayed apoptosis,

27
but the doses required to reach this effect were higher than those used for anticonvulsanttreatment (Wasterlain et al. 1996).
Non-NMDA receptor antagonists. In a study employing a rat model, the competitiveAMPA antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline (NBQX) pro-vided improved neurological recovery following middle cerebral artery occlusion (Lo etal. 1997). It had a disadvantageous effect on brain metabolism in a piglet model of HCAat 15�C (Aoki et al. 1994), but dogs treated with it showed better neurological recoveryafter 2 hours of HCA at 18�C than did their controls (Redmond et al. 1995).
The non-NMDA receptor antagonist 1-(4-aminophenyl)-4-methyl-7,8-methylenedi-oxy-5H-2,3-benzodiazepine hydrochloride (GYKI 52466) reduces ischaemia-inducedchanges in tissue concentrations of glutamate, aspartate and gamma-aminobutyric acid(GABA) and has been shown to be a neuroprotectant in rat models of acute normothermicischaemia (Arias et al. 1999, Arvin et al. 1994). Another AMPA antagonist, YM 90 K,proved to be neuroprotective in the same experimental model (Kawasaki-Yatsugi et al.1998, Umemura et al. 1997).
2.2.2.3 Monosialogangliosides
Monosialogangliosides play important roles in the physiological pathways of the nervoussystem, particularly in the brain. Changes in ganglioside composition occur in themammalian brain not only during development, but also upon ageing and in severalneuropathological situations. Monosialogangliosides may modulate the ability of thebrain to modify its response to signals from the surrounding environment, and additionalin vitro studies have shown that compounds such as GM1 provide protection againstexcitatory amino acid-related neurotoxicity by limiting the downstream consequences ofreceptor overstimulation. Monogangliosides do not interfere with the EAA receptorrecognition sites (Milani et al. 1991). The monoganglioside GM1 has also been shown toreduce cerebral injury following 2 hours of HCA at 18�C. In an experiment with adultdogs, a better behavioural outcome and less neuronal injury was seen in dogs pretreatedwith GM1 than in the controls (Redmond et al. 1993).
2.2.2.4 Ca2+ channel blockers
Calcium channel blockers may reduce Ca2+ influx into cells after the activation of EAAreceptors, and they also inhibit vasoconstriction after ischaemic insult (Lipton andRosenberg 1994). Calcium antagonists have been shown to improve the neurologicaloutcome after aneurysmal subarchnoidal haemorrhage. These results are mainly based ontrials with oral nimodipine, whereas the evidence for the Ca2+ antagonists nicardipine andAT877 is controversial (Feigin et al. 1998). In a rat model with transient focal cerebralischaemia via intraluminal thread occlusion of the middle cerebral artery, nimodipinereduced ischaemic damage by a maximum of 33% at a dose of 50 µg/kg, but BW619C89(a NA+ channel blocker) was found to be more effective in the same context (Kawaguchi

28
et al. 1999). Nimedipine has been shown to have a beneficial effect on the neurologicaloutcome after 3 hours of HCA at 10�C (Mazzoni M 1993).
AT877 pretreatment was effective in preventing brain injury during transient focalcerebral ischaemia in a rat model, improving the neurological status. This beneficialeffect is likely to be partly mediated by its primary action of increasing cerebral bloodflow (Ohtaki and Tranmer 1994). In a report on a prematurely terminated trial ofnimodipine as a neuroprotectant in cardiac valve replacement surgery, major adverseevents including deaths and strokes were documented shortly after surgery, the mainproblem being intracerebral haemorrhage. The data strongly suggest that the use of Ca2+
channel blockers during CPB should be avoided (Legault et al. 1996).
2.2.2.5 Nitric oxide synthase inhibitors
NO has been shown to mediate glutamate excitotoxicity, and the therapeutic potential ofNOS inhibitors have been emphasized. NOS inhibitors are divided into non-selective andselective ones. N-nitro �L-argine was the first non-selective NOS inhibitor to be studiedand has proved to be a potent neuroprotectant in an experimental setting (Barth et al.1997).
Conflicting results have also been reported concerning the potential benefits of non-selective NOS inhibitors in cases of ischaemia. Possible worsening of the condition canbe attributed to the inhibition of endothelial nitric oxide synthase, the activity of whichseems to be neuroprotective (O'Mahony and Kendall 1999). This has raised a need forselective NOS inhibitors (Chabrier et al. 1999). 7-Nitroindazole (7-NI), the first selectiveNOS inhibitor, reduced NO production and improved the neurological outcome afterHCA in dogs (Baumgartner et al. 1999). Aminoguanidine, a relatively selective inhibitorof inducible NO synthase, ameliorated neonatal hypoxic-ischaemic brain damage in rats(Tsuji et al. 2000), and 1-(2-trifluoromethylphenyl)imidazole reduced the infarct volumeafter transient middle cerebral artery occlusion in rats more effectively than did 7-Nitroindazole (Escott et al. 1998). ARL17477, a selective NOS inhibitor, also providedsome neuroprotection in a gerbil model, but the combination of MK-801 with ARL17477provided 21% or 44% higher protection than either alone, indicating that severalpathways contribute to ischaemic cell death and several agents targeting different pointsin the biochemical cascade may be of benefit (Hicks et al. 1999). N-(3-(aminomethyl)benzyl)acetamidine reduced the ischaemic lesion volume in rats by 31%and attenuated weight loss and neurological dysfunction after brain ischaemia(Parmentier et al. 1999). BN 80933 is a brand new selective NOS inhibitor which also hasantioxidant properties. In an experimental study with rats it reduced the infarct volume by60% (Chabrier et al. 1999).
2.2.2.6 Calpain inhibitors
One possibility for pharmacologically targeting a "downstream" event in the cascade is toinhibit activated calpain. The CNS-penetrating calpain inhibitor MDL 28,170 can reduce

29
infarct volume in rodents (Markgraf et al. 1998), as also can another inhibitor Cbz-Val-Phe-H (Hong et al. 1994).
2.2.2.7 Gelsolin-like chemical agents
Gelsolin expression at normal levels protects the brain from acute ischaemic injury withits ability to stabilize CA2+ influx. Cytochalacin D is a partial gelsolin analogue and canreduce ischaemic injury, although this can lead to apoptosis (Endres et al. 1999).
2.2.2.8 Caspase inhibitors
Caspase activation is required in many cases of apoptotic cell death, and follows afterhypoxic-ischaemia insult. The pan-caspase inhibitor boc-aspartyl(OMe)-fluoromethylke-tone significantly improved neuroprotection when injected into brain ventriceles 3 h aftercerebral hypoxic-ischaemia, and systemic injections of this molecule given in a delayedfashion resulted in significant neuroprotection. These findings suggest that caspase inhib-itors may provide benefit over a prolonged therapeutic window after ischaemia (Cheng etal. 1998). Treatment with the caspase inhibitors z-VAD.FMK and z-DEVD.FMK reducedDNA laddering after transient middle cerebral artery occlusion in a murine model(Endres et al. 1998).
2.2.3 Strategies for mitigating reperfusion-related brain injury
2.2.3.1 Pharmacological strategies
There are numerous potential sites to be targeted with drug action against the biochemicalmediators of reperfusion injury and the following section covers these.
Corticosteroids. Glucocorticoids have been shown to reduce the levels of circulatinginflammatory mediators (Hill et al. 1997). The release of interleukin-10, a potent anti-inflammatory cytokine, may play an important role in the anti-inflammatory effects ofcorticosteroids (Tabardel et al. 1996), but despite their anti-inflammatory potential, theyhave not turned out to be major neuroprotectants.
Free radical inhibition. Although the reason for neutrophil-related damage to theendothelial layer is the release of free oxygen radicals and proteases, studies focused onfree radical scavengers have yielded conflicting results. Critical evaluation of the data hasdemonstrated that free radical scavangers act more effectively at the level of the vascularendothelium than in the neuronal tissue per se (Hall et al. 1994). The use of alpha-phenyl-tert -butyl nitrone before HCA attenuated the normal response to ischaemia and improvedrecovery by affording protection from free radical-mediated damage (Langley et al.2000), while in another study of normothermic ischaemia U-101033E had better

30
neuroprotective properties than alpha-phenyl-tert -butyl nitrone (Schmid-Elsaesser et al.2000). Tirilazad mesylate, a non-glucocorticoid 21-aminosteroid lipid peroxidationinhibitor, has proved promising as a neuroprotectant in experimental models of focalcerebral ischaemia, but it failed to improve the functional outcome after cerebral stroke(Haley 1998).
Cytokine inhibition and anti-adhesion molecule antibodies. A challenging therapeuticprospect is to aim directly at cytokine suppressive agents. Soluble TNF receptor I (aphysiological inhibitor of TNF-�) has been shown to be neuroprotective, and TNF-� Mabtreatment has been found to effectively prevent the expansion of infarct size in the brain(Barone et al. 1997). Another possibility is to inhibit interactions between theendothelium and the leukocytes. The potential of the experimentally demonstrated anti-ischaemic effect of cytokine inhibition and anti-adhesion molecules for clinicallyeffective therapy is obscure, but these strategies evidently hold a promise for the future(Feuerstein et al. 1998).
2.2.3.2 Modification of techniques or mechanical devices
Improvements in biocompatibility. A systemic inflammatory response is increasinglybeing recognized as inevitable following the use of CPB, although this potentialmechanism for producing brain injury remains speculative (Taylor 1998). Surfacemodification of extracorporeal circuits with heparin has been shown to inhibit thealternative complement activation pathway and the activation of leukocytes (Allen 1997),but the connection between these strategies and possible brain protection is not clear.
Leukocyte depletion. A leukocyte-depleting filter is a commercially available devicethat has been used clinically. It is simple to add to a CPB circuit and it is potentially aneffective method, which requires no drug administration. It has been shown that leukocytedepletion can ameliorate myocardial reperfusion injury (Lazar et al. 1995, Schmidt et al.1996). The L-DF has also been shown to reduce ventricular dysfunction during prolongedpostischaemic reperfusion (Wilson et al. 1993) and to attenuate reperfusion injury inpatients with left ventricular hypertrophy (Sawa et al. 1996). When more than three bloodtransfusions are required after cardiac surgery, leukocyte depletion of the transfused bloodresults in a significant reduction in postoperative mortality (van de Watering et al. 1998).Leukocyte depletion during and after CPB has been shown to improve pulmonaryfunction (Gu et al. 1996) and to ameliorate free radical-mediated lung injury (Bando et al.1990, Bolling et al. 1997, Shiraishi et al. 1998). It has been demonstrated by means of apanel of leukocyte �associated monoclonal antibodies that the leukocyte-depleting filterselectively removes activated neutrophils but has little effect on the total leukocyte count(Thurlow et al. 1995). Since activated leukocytes, particularly neutrophils, play a pivotalrole in reperfusion injury, it can be presumed that white cell depletion is also potentiallyan effective method for mitigating reperfusion injury in the brain following HCA. Thisquestion has very recently been approached using an acute porcine model by Langley andassociates, who demonstrated that the results with respect to all the CPB and metabolismvariables measured were consistently better in the leukocyte filter group than in the

31
controls but that the difference was statistically of borderline significance (Langley et al.2000).
Table 1. Pharmacological strategies for mitigating cerebral injury.Stage Target Drug or Drug Class Compound CommentsPresynaptic Ca2+ channels
N-type �-conotoxins SNX-111 HypotensionNa+ channels Pyramidines: Lamotrigine Licensed for epilepsy
BW1003C87 In vitro and in vivo experimentsBW 619C87 In vitro and in vivo experiments
Lubeluzole; Studied clinically for strokeRiluzole In vitro and in vivo experimentsLifarizine Hypotension, cardiac side-effects
Postsynaptic NMDA receptorsNon-competitive Aptiganel CNS1102 (Cerestat) Hypotension, hallucinogenic
Dizoclipine MK801 ToxicityNPS 1506 Less toxic than MK801
Ketamine Anaesthetic, blood pressure effectsDextromethorphan AHN649 Blood pressure effectsMemantine Licensed for ParkinsonismRemacemide desglycine
FPL 12495 Clinical study with CABG
Competitive Selfotel CGS 19755 Toxicity / psychomimeticd-CPP In vitro and in vivo experiments studies
Glycine site Felbamate C-W 554 Lic.for epilepsy, protects in vitroZD9379 In vitro and in vivo experiments
Licostinel ACEA-1021 In vitro and in vivo experiments ACEA-1031 In vitro and in vivo experimentsACEA-1416 In vitro and in vivo experiments
AMPA receptors NBQX In vitro and in vivo experimentsGYKI 52466 In vitro and in vivo experiments YM 90 K In vitro and in vivo experiments
Monosialoganglio-sides
GM1 In vitro and in vivo experiments
Ca2+-channels Nimodipine Intracerebral haemorrhageAT 877 In vitro and in vivo experiments
Gelsolin Microfilaments Cytochalalacin D In vitro and in vivo experimentsCalpain Cbz-Val-Phe-H In vitro and in vivo experiments
MDL 28,170 In vitro and in vivo experiments NO NO synthase 7-nitroindazole In vitro and in vivo experiments
TRIM In vitro and in vivo experiments1400 W In vitro and in vivo experimentsBN 80933 In vitro and in vivo experiments
Reperfusion Cytokine inhibition Soluble TNF reseptor I
In vitro and in vivo experiments
TNF� Mab In vitro and in vivo experimentsEndothelium Anti-adhesion
moleculesIn vitro and in vivo experiments
Free radicals Lazeroids Tirilazad 74006F In clinical trialsU-101033E In vitro and in vivo experiments PBN In vitro and in vivo experiments studies
Apoptosis Caspases z-VAD-FMK In vitro and in vivo experiments z-DEVD.FMK In vitro and in vivo experiments

3 Aims of the present research
The aims of the present research were:1. to test the neuroprotective efficacy of lamotrigine during hypothermic circulatory
arrest (I)2. to find out whether depletion of activated leukocytes by filtration (Leukoguard
LG6�, Pall Biomedical, Portsmouth, U.K.) could mitigate ischaemic brain injuryfollowing hypothermic circulatory arrest (II)
3. to test the neuroprotective efficacy of memantine during hypothermic circulatoryarrest (III)
4. to find out whether lamotrigine and the leukocyte filter have additive brain protectiveproperties (IV)

4 Materials and methods
4.1 The chronic porcine model
The chronic porcine model was developed by Professor Randall Griepp`s group at theMount Sinai School of Medicine in New York and was adopted by Professor Juvonen andour group, who have been using it since 1997.
4.2 Preoperative Management
All the animals received care in accordance with the "Principles of Laboratory AnimalCare" formulated by the National Society for Medical Research and the "Guide for theCare and Use of Laboratory Animals" prepared by the Institute of Laboratory AnimalResources and published by the National Institutes of Health (NIH publication No. 85�23, revised 1985). The study was approved by the Research Animal Care and UseCommittee of the University of Oulu.
4.3 Pharmacokinetic pilot study (III)
The therapeutic range of plasma levels in patients treated with memantine (10�30 mg perday) is 0.2�1.0 µmol/L, and the free brain interstitial concentration is 20�30% lower thanin plasma (Parsons et al. 1999). It has been demonstrated previously that 1 µmol/Lplasma concentrations are achieved in laboratory animals after acute injections of 2.5�5.0mg/kg (Danysz t al. 1997). In order to determine the appropriate dose of memantine forpigs, a pharmacokinetic pilot study employing four pigs of weight 25 kg was performed.Doses of 3, 5, 7 and 10 mg/kg of memantine were given and blood samples wereobtained 30 minutes, 1 hour, 2 hours and 5 hours after the start of drug administration. Aplasma level of 1.49 µmol/L was recorded 30 minutes after a dose of 3 mg/kg. Animals inthis model are known to be exposed to fairly severe global cerebral ischaemia, but on theother hand, the plasma is diluted with CPB. Therefore a dose of 5 mg/kg, providing a

34
maximum blood level of 2.81 µmol/L 30 minutes after drug administration, was selectedfor this work.
4.4 Drug Administration (I, III and IV)
An isethionate (2-hydroxyethanesulphonate) salt of lamotrigine [3,5-diamino-6 (2,3-dichlorophenyl)-1,2,4-triazine] was diluted in saline to obtain a solution containinglamotrigine at 50 mg/ml, and this was packed in 10 ml ampoules in the PharmaceuticalLaboratory of our institution. Saline placebo ampoules were prepared analogously,diluting doses of 20 mg/kg in saline to eventual volumes of 50 ml. These volumes weregiven intravenously over a period of 20 minutes, starting two hours before HCA (I andIV). The concentrations of lamotrigine were the same in the brain and plasma (Bacher A1997). Intravenous administration was chosen because it is a more consistent method fordosing and timing medication in an experiment of this kind. A dose of 20 mg/kg wasselected because it should still be tolerated clinically even though it is higher than thedosage used for anti-epileptic medication (Ramsay et al. 1991).
Memantine was diluted to 10 mg/mL in 0.9% NaCl, and this was packed in 15 mLampoules in the Pharmaceutical Laboratory of our institution. Again the saline placeboampoules were prepared in an analogous manner by diluting doses of 5 mg/kg to 50 mLin saline solution. These volumes were given intravenously over a period of 20 minutes,starting 75 minutes before HCA. Randomization was carried out by the chemist in thePharmaceutical Laboratory. Pharmacokinetic analyses generously performed by Merz &Co. demonstrated that memantine concentrations were sustained at a therapeuticallyrelevant level throughout the critical period of the experiment.
4.5 Anaesthesia and haemodynamic monitoring
Anaesthesia was induced with ketamine hydrochloride (10 mg/kg intramuscularly) andmidatzolam (1 mg/kg intramuscularly) (I and II) or with medotomidine hydrochloride(0.4 mg/kg intramuscularly) (III and IV). Muscular paralysis was maintained withpancuronium bromide (0.1 mg/kg intravenously). Following endotracheal intubation, theanimals were maintained on positive pressure ventilation with 40% (II�IV) or100% (I)oxygen, and anaesthesia was maintained with Isoflurane (1.1�1.2 %). An arterial catheterwas positioned in the left femoral artery and a Swan-Ganz catheter was placed throughthe femoral vein to allow blood sampling and pressure monitoring in the pulmonaryartery and for the recording of cardiac output. Temperature probes were placed in theoesophagus, rectum and epidural space and a 10 F catheter inserted into the urinarybladder to monitor urine output.

35
4.6 Electroencephalography (EEG)
Cortical electrical activity was recorded via four stainless steel screw electrodes 5 mm indiameter implanted over the parietal and frontal areas of the cerebral cortex using adigital EEG recorder (Nervus®, Reykjavik, Iceland) and an amplifier (Magnus® EEG 32/8, Reykjavik, Iceland). The sampling frequency was 1024 Hz and the bandwidth 0.03 �256 Hz. All the EEG recordings are referenced to a frontal screw electrode which wasimplanted over the frontal sinuses together with a ground screw electrode. The isofluranelevel was adjusted so that the EEG showed a steady burst suppression pattern, after whichthe end tidal reading was kept at this steady level until the end of monitoring. EEG wasrecorded for 10 minutes to provide a baseline recording of steady burst suppressionactivity before the cooling period. After HCA, EEG recording was restarted andcontinued until the first postoperative day. EEG durations were measured from 5-minutesamples at fixed points in time, at half-hour intervals at first, and later at 1-hour intervals.Artefact periods were excluded from each 5-minute sample and the sum of the bursts wascounted from the rest as a percentage of the sum of all artefact-free bursts andsuppressions. This percentage was used as a measure of EEG activity in the analysis (Fig.2).
Fig. 2. An example of EEG burst recovery analysis in a pig after 75 minutes period of HCA.Characters a�f below depict EEG in different timepoints after HCA and above same charactersshow exact timepoints and percentage of EEG recovery.

36
4.7 Cardiopulmonary bypass
The heart and great vessels were exposed via a right thoracotomy in the fourth intercostalspace and the right mammarian artery was ligated. A membrane oxygenator (Midiflow D705, Dideco, Mirandola, Italy) was primed with 1-litre Ringer acetate and heparin (5000IU), and after heparinization (300 IU/kg), the ascending aorta was cannulated with a 16-French arterial cannula and the right atrial appendage with a single 24-French atrialcannula. Non-pulsatile CPB was initiated at a flow rate of 100 ml/kg per minute and theflow was subsequently adjusted to maintain a perfusion pressure of 50 mmHg. A 12-French intracardial sump cannula was positioned in the left ventricle for decompressionof the left heart during CPB. A heat exchanger was used for core cooling. The pH wasmaintained using alpha-stat principles at 7.40 � 0.05 with an arterial CO2 tension of 3.5to 4.0 kPa, uncorrected for temperature. A leukocyte-depleting filter (Leukoguard LG6�,Pall Biomedical, Portsmouth, U.K.) was used during CPB in randomly assigned groups(II and IV).
A cooling period of 60 minutes was allowed to attain both rectal and epiduraltemperatures of 20�C. Cardiac arrest was induced by injecting potassium chloride (1mEq/kg) into the aortic cannula, and topical cardiac cooling was then begun andmaintained throughout the aortic cross-clamp period. The ascending aorta was cross-clamped just proximally to the aortic cannula.
4.8 Experimental protocol
After cooling to 20�C and cross-clamping of the aorta, the animals underwent 75 minutesof HCA. When this was over, rewarming was initiated and the left ventricular vent wasremoved. Weaning from CPB occurred approximately 60 minutes after the start ofrewarming, with the administration of furosemide (40 mg), mannitol (15.0 g),methylprednisolone (80 mg) and lidocaine (40�150 mg) (Fig. 3). Cardiac support wasprovided with dopamine. The animals were kept under isoflurane anesthesia until the firstpostoperative day, extubated, and moved to a recovery room.
Haemodynamic and metabolic measurements were made at five points during theexperiments: 1. at baseline, after the Swan-Ganz catheter had been placed in position, 2.at the end of cooling (at 25�C), immediately prior to institution of the intervention, 3.during rewarming (at 30�C), and 4. 2 hours after the start of rewarming, 5. 4 hours afterthe start of rewarming.

37
Fig. 3. Experimental protocol.
4.9 Postoperative evaluation
All the animals were evaluated daily during the postoperative period using a species-specific quantitative behavioural score as reported earlier (Juvonen et al. 1998). Thisincluded quantifications of mental status (0=comatose, 1=stuporous, 2=depressed,3=normal); appetite (0=refuses liquids, 1=refuses solids, 2=decreased, 3=normal) andmotor function (0=unable to stand, 1=unable to walk, 2=unsteady gait, 3=normal) whichwere summed to provide a final score. The maximum of reflects apparently normalneurological function, while lower values indicate substantial neurological damage. Ascore of 8 means that the animals were able to stand unassisted and were likely to recoverfully.
Each surviving animal was sacrificed on day 7 after surgery. The entire brain wasimmediately harvested, weighed and stored for subsequent histological analysis.
4.10 Histopathological analysis
The brain was excised immediately upon death and the hemispheres were cut apart. Onehalf was immersed in 10% neutral formalin and fixed for one week en bloc. Thereafter 3-mm coronal slices were taken from the frontal lobe, thalamus (including the adjacentcortex) and hippocampus (including the adjacent brain stem and temporal cortex), andsagittal samples were taken from the posterior brain stem (medulla oblongata and pons)and cerebellum. The pieces were fixed in fresh formalin for another week. After fixation,the samples were rinsed in water for 20 minutes and immersed in 70% ethanol for 2hours, 94% ethanol for 4 hours and absolute ethanol for 9 hours. They were then kept inan absolute ethanol-xylene mixture for one hour and in xylene for 4 hours, followed byembedding in warm paraffin for 6 hours. The samples were sectioned at 6 �m and stainedwith haematoxylin eosin. The sections of the brain samples of each animal were screened

38
by a single experienced senior pathologist, blinded both to the experimental design and tothe identity and fate of the individual animals. Each section was carefully investigated forthe presence or absence of any infarctive or other damage.
Injuries were evaluated visually as follows: 0 = no morphological damage identified; 1= oedema and/or occasional dark neurons; 2 = numerous dark neurons (often also shrunk)and eosinophilic or dark/shrunk cerebellar Purkinje cells or haemorrhages, and 3 = clearlyinfarctive foci with neoformation of capillaries and the presence of macrophages and aglia reaction (I�III). 1 point: slight oedema, dark or eosinophilic neurons or cerebellarPurkinje cells, 2 points: moderate oedema, at least two haemorrhages in the section, 3points: extreme oedema, infarctive foci (local necrosis) (IV). To allow semiquantitativecomparisons between the animals, a total histological score was calculated by summingthe highest regional scores (I) or by summing all the regional scores (II, III and IV).
4.11 Serum S-100�
Concentrations of serum S-100� were determined in mixed venous blood samples using aluminescence immunoassay kit (Sangtec-100®, LIA-mat; Sangtec Medical AB, Bromma,Sweden). Serum S-100� protein levels were measured preoperatively and 2 hours, 4hours, 7 hours and 20 hours after the start of rewarming.
4.12 Cytokines (II)
Cytokine levels (IL-1�, IL-8 and TNF-�) were recorded up to the first postoperative day.Serum specimens were drawn, divided into aliquots and stored at -70�C until tested.Cytokine concentrations were determined by an ELISA method according to themanufacturer�s instructions. ELISA kits (Cytoscreen) for swine IL-1�, IL-8 and TNF-�were obtained from Biosource International (Camarillo, California, USA). The lowerdetection limits for the tests were 15 pg/ml for IL-1, 10pg/ml for IL-8 and 6 pg/ml forTNF-�.
4.13 Microdialysis (III and IV)
The microdialysis probe (CMA/20; CMA/Microdialysis, Stockholm, Sweden) wasintroduced into the brain cortex through a drill hole positioned 1 cm to the right of thesagittal joint above the parietal line. The microdialysis catheter was connected to a 2.5 mlsyringe placed in a microinfusion pump (CMA107; CMA Microdialysis, Stockholm,Sweden) and perfused with a Ringer solution (Perfusion Fluid; CMA Microdialysis,Stockholm, Sweden). Samples were collected every 30 minutes. Glucose, lactate,glutamate and glycerol concentrations were measured on a microdialysis analyzer (CMA600; CMA Microdialysis, Stockholm, Sweden) by ordinary enzymatic methods immedia.

39
4.14 Other measurements
Systemic arterial and venous blood samples were obtained to determine pH, oxygentension, carbon dioxide tension, oxygen saturation, oxygen content, haematocrit,haemoglobin and glucose (Ciba-Corning 288 Blood Gas System, Ciba-CorningDiagnostic Corp, Medfield, MA, USA). Lactate was analyzed with a YSI 1500 analyzer(Yellow Springs Instrument Co, Yellow Springs, OH, USA), and leukocyte counts weretaken with a Cell-Dyn analyzer (Abbot, Santa Clara, California, USA) (I and IV).Temperatures were recorded at intervals throughout the procedures. Haemodynamics,temperatures and respiratory gases were assessed using a Datex AS/3 anaesthesia monitor(Datex Inc., Espoo, Finland).
4.15 Statistical analysis
Summary statistics for continuous or ordinal variables are expressed as medians withinterquartile ranges (IQR, 25th and 75th percentiles) or means with standard deviation(SD). All values in the figures are shown as medians with IQR.
An analysis of variance was used for repeated measurements, and relevant time-pointsand baseline (reference) levels were compaired by means of the paired sample t-test or theWilcoxon matched pairs signed rank test. Differences between groups were determinedwith the t-test or Mann-Whitney U-test. The two-tailed Fisher exact test was used todetermine the significances of differences in mortality rates between the groups. The AreaUnder the Curve (AUC) was calculated for the microdialysis measurements, andKendall�s rank correlation was used to estimate correlation coefficients. Significancelevels for comparisons are reported with 2-tailed p < 0.05. Analyses were performedusing a standard commercially available statistical program (SPSS v. 9.0, SPSS Inc.,Chicago, Ill., USA).

5 Results
The major findings are summarized in Table 2 and Fig. 4. All the animals accepted forthese series of experiments were stable during the surgical procedures and survived untilat least the first postoperative day.
5.1 Lamotrigine improves cerebral outcome following hypothermic circulatory arrest (I)
No significant differences in haemodynamic measurements were found between thegroups, and complete behavioural recovery was seen in 5/8 (63%) following lamotrigineadministration, compared with 1/8 (13%) in the placebo group (p=0.02). Altogether 6/8animals in the lamotrigine group survived for seven days, compared with 5/8 in theplacebo group (p>0.2), the median behavioural score for the surviving animals washigher in the lamotrigine group (8 vs. 7, p=0.02).
The medians of the recovering EEG bursts 4½ hours after the start of rewarming werehigher in the lamotrigine group than in the placebo group (p=0.01), while the median S-100� level 20 hours after the start of rewarming was lower in the lamotrigine group (0.01�g/l vs. 0.1 �g/l in the placebo controls, p=0.01). The median of total histopathologicalscore was 5.5 in the lamotrigine group and 7.5 in the placebo group (p=0.06).
5.2 Leukocyte filtration improves cerebral outcome following hypothermic circulatory arrest (II)
Mean arterial pressure 2 hours after the start of rewarming was higher in the L-DF group(p<0.01), but cardiac output 4 hours after the start of rewarming was higher in thecontrols (p<0.01). The mortality rate was 2/10 in the L-DF group and 5/10 among thecontrols. Thus the risk of early death was 2.5 (95% CI 0.63�10.0) times higher in thecontrol animals than in the LD-F animals. The median behavioural score on day 7 was

41
higher in the L-DF group (8.5 vs. 3.5 p=0.04), but that for the animals surviving until thatday was 9 in the LD-F group and 8 in the control group (p=0.08).
No statistically significant differences in the rate of EEG burst recovery were detectedbetween the groups. TNF-� levels increased in both groups after the intervention, themedians being 18 pg/ml at 30�C during rewarming and 107pg/ml after 2 hours ofrewarming in the L-DF group and 3 pg/ml and 861 pg/ml respectively in the controlgroup. The changes relative to the baselines were found to be statistically significant (p<0.05), but there were no statistically significant differences between the groups. IL-1� andIL-8 did not differ between the groups at any time during the experiment. The median ofthe total histopathological score was 8.5 in the L-DF group and 15.5 in the control group(p=0.005).
5.3 The NMDA antagonist memantine has no neuroprotective effect during hypothermic circulatory arrest (III)
There were no differences in either the haemodynamic or metabolic data between thegroups, 5/10 animals in the memantine group surviving seven days compared with 9/10in the placebo group (p=0.14). The median behavioural score on day 7 was 3.5 in thememantine group and 7.5 in the placebo group (p=0.39), and those among the survivinganimals 9.0 and 8.0 respectively (p>0.2). The median numberss of recovering EEG burstswere equal in both groups.
The intracerebral glucose concentration decreased during cooling and during HCA butreturned to the baseline in both groups, with no differences between them. Lactateconcentration increased 30 minutes after HCA in both groups, the highest peak beingfound one hour after the start of rewarming. Values for the AUC (Area Under the Curve)were calculated from the start of HCA until 7 hours after the start of rewarming, but thesedid not differ between the groups (p=0.60). Median glutamate levels exhibited peakvalues 30 minutes after the start of rewarming and subsequently decreased virtually tozero. The median glutamate concentration 30 minutes after the start of rewarming washigher in the memantine group (49 �M/l) than in the placebo group (23 �M/l), but thedifference was not found to be statistically significant (p=0.40). The AUC for glutamatedid not differ between the groups (p=0.39). Glycerol also showed a peak after HCA, butthis occurred slightly later than for glutamate. The AUC for glycerol similarly did notdiffer between the groups (p=0.74).
Since there were no statistically significant differences between the groups in the AUCvalues derived from the microdialysis data, the correlation coefficients were calculatedusing all the animals. A positive correlation was found between the AUC values forglutamate and glycerol (�=0.474, p=0.004), but there was no correlation betweenglutamate and lactate. A one-hour lag was found in the correlation between the peaks forglutamate (30 minutes after the start of rewarming) and glycerol (from 1.5 hours after thestart of rewarming onwards), giving the highest correlation with a lag of 1.5 or 2 hours(�=0.537, p=0.001). There was an inverse correlation between the highest individualglutamate levels and EEG burst recovery 3 hours after the start of rewarming, the highestcorrelation coefficient being (�=-0.377, p=0.043, Fig. 7). In addition, there was a positive

42
correlation between the highest individual glutamate value and the histopathologic score(� =0.336, p=0.045)
The median of the total histopathological score was 16 in the memantine group and 14in the placebo group (p=0.25).
5.4 Lamotrigine with leukocyte filtration provides effective neuroprotection following HCA in pig (IV)
All the animals were stable prior to, during and after CPB. Cardiac output 2 hours afterthe start of rewarming was better in the LD-F + lamotrigine group than in the lamotrigineor control group (p=0.051), and PaCO2 at the same point in time was lower this groupthan in the controls (p=0.008). By 4 hours after the start of rewarming PaCO2 was lowerin both treatment groups than in the controls (p=0.011 and p=0.051).
EEG burst recovery was highest in the LDF + lamotrigine group, the median 7 hoursafter the start of rewarming being 94% compared with 81% in the lamotrigine group and64% in the control group (LD-F + lamotrigine vs. controls p=0.024). As many as 7/8(88%) of the LD-F + lamotrigine animals survived for seven days, compared with 4/8(50%) in the lamotrigine group and 3/8 (38%) in the controls. The median behaviouralscore on day 7 was 9 in the LD-F + lamotrigine group, 4 in the lamotrigine group and 0 inthe control group ( LD-F + lamotrigine vs. controls p=0.005).
The intracerebral glucose concentration decreased during cooling and during HCA andsubsequently returned to the baseline in all the groups, the differences between the groupspossibly being due to chance(??) (p>0.2). The lactate concentration increased 30 minutesafter HCA in all the groups, the highest peak being found one hour after the start ofrewarming, beyond which point lactate decreased to just above the baseline level in allthe groups, although later in the control group. Thus differences were seen 2½ hours and3 hours after the start of rewarming, the medians being 5.4 �mol/L and 4.3�mol/L in thecontrol group and 2.2�mol/L and 1.9�mol/L in the LD-F + lamotrigine group,respectively (p=0.014 and p=0.021). A difference was also detected between the controlsand the lamotrigine group 2½ hours after the start of rewarming, the median being 2.3 inthe lamotrigine group (difference vs. controls p=0.005). The minor differences betweenthe LD-F + lamotrigine and lamotrigine groups could be due to chance (p>0.2). As seenin Fig. 5, median glutamate levels exhibited peak values 40 minutes after the start ofrewarming and subsequently decreased virtually to zero, although the decrease occurredlater in the control group. The medians of the glutamate concentrations 1 hour 40 minutesafter the start of rewarming were 4.5 �mol/L in the LD-F + lamotrigine group, 2.0 �mol/L in the lamotrigine group and 15.0 �mol/L in the controls (lamotrigine vs. controlsp=0.038). The other differences between the groups could be due to chance (p>0.2).Glycerol also showed a peak after HCA, but it occurred slightly later than that forglutamate. As seen in Fig. 4, glycerol concentrations in the controls remained at a higherlevel and the medians at 5 and 6 hours after the start of rewarming were 49.2 �mol/L and34.1�mol/L in the LD-F + lamotrigine group, 34.4 �mol/L and 26.8 �mol/L in thelamotrigine group and 92.2�mol/L and 69.6�M/L in the control group (lamotrigine vs.control group p = 0.015 and 0.083). As depicted in Fig. 4, the lamotrigine and LD-F

43
groups behaved very similarly, but the other differences between the groups could be dueto chance (p>0.2).
The median of the total histopathological score was 14 in the LD-F + lamotriginegroup (p=0.046 vs. controls), 14.5 the lamotrigine group (p=0.062 vs. controls) and 20 inthe control group.
Table 2. Survival, median behavioural score on day seven, median EEG burst recoveryand median histopathological scores in 80 pigs undergoing 75 minutes of hypothermiccirculatory arrest at 20�C.
Group N SurvivalMedian behavioural
score on day 7Median EEG burst
recoveryMedian
histopathological score
Lamotrigine 8 6/8 (75%) 8 80% (at 4 ½ hours) 5.5
Controls I 8 5/8 (63%) 7 20% 7.5
p value ns ns 0.01 0.06
LD-F 10 8/10 (80%) 8.5 40% (at 7 hours) 8.5
Controls II 10 5/10 (50%) 3.5 40% (at 7 hours) 15.5
p value ns 0.04 ns 0.005
Memantine 10 5/10 (50%) 3.5 48% (at 7 hours) 16
Controls III 10 9/10 (90%) 7.5 53% (at 7 hours) 14
p value 0.14 ns ns ns
Lamotrigine + LD-F 8 7/8 (88%)* 9* 94% (at 7 hours)* 14*
Lamotrigine 8 4/8 (50%) 4 81% 14.5
Controls IVp value vs controls 8 3/8 (38%)
*0.120
*0.00464%*0.02
20*0.046
Abbreviations: LD-F = Leukocyte depleting filter, EEG = Electroencephalography, ns = not signinificant

44
Fig. 4. Median concentrations of intracerebral lactate, glycerol, glutamate and glucose in 44pigs undergoing hypothermic circulatory arrest (HCA) at 20�C ( III and IV).

6 Discussion
6.1 General discussion
Cerebral morbidity remains a significant problem in aortic surgery when conventionalCPB is not possible, and an understanding of the mechanism of ischaemic brain injuryfollowing HCA may provide information that is germane to all types of cardiac surgery.The key issue without any doubt is good surgical technique, in order to avoidneurological complications. Antegrade selective perfusion and RCP techniques combinedwith HCA have yielded good results (Murase et al. 1993, Veeragandham et al. 1998), butbrain protection is often compromised in complex operations or when dealing with riskpatients. In addition, neurological outcomes are often overestimated in clinical series, sothat whether or not the harmful consequences are detected depends on the evaluationcriteria and the methods and timing of the investigation. Even routine cardiac surgery canbe seen to cause neurological problems if studied sufficiently carefully (Sotaniemi 1995).Basic research into neuroscience has increased our understanding of the fundamentalmechanism of neuron cell death, and this has opened up new therapeutic avenues forrestricting and mitigating brain injury, but experimental and clinical studies are stillneeded to improve brain protection strategies during cardiac and aortic surgery.
Our chronic porcine model, adopted from Mt Sinai by Professor Juvonen, has beenused earlier to study RCP. In this setting a lack of competent jugular valves, which existin only 20% of pigs, may cause problems (Juvonen et al. 1998, Juvonen et al. 1998). Inthe present series of experiments, however, when we were studying only a prolongedperiod of HCA, this was not problem. A chronic model is more demanding than an acutemodel, but we think that it comes closer to the clinical setting. Other animal models suchas dog models have been used with good results, but certainly not better ones(Baumgartner et al. 1998), while smaller animals such as canines are not suitable forchronic models. Non-human primates would be ideal for studying brain protection, butthis would be difficult on both ethical and economic grounds. Some research of this kindhas been carried out, however (Boeckxstaens 1996).
The strength of our model lies in the number of end points. One problem withexperimental surgery is always the number of animals required to achieve statisticalpower. When we are able to examine neurological recovery, survival, EEG, biochemical

46
markers, microdialysis and histopathologic findings at the same time, we can see the datain line and in that way gather more information. In view of the differences between theanimals in different series, it is very important to use prospective randomized protocols inconnection with animal models, as the results are not comparable with each other in termsof absolute numbers. Conclusions can only be reached on the basis of different studies bycomparing summaries of the outcome data.
6.2 Haemodynamic and metabolic data
Summarizing the whole body of haemodynamic and metabolic data, there were fewdifferences between the groups in these series of experiments, indicating that thisdemanding animal model was technically successful and that the biochemical agents werewell tolerated.
6.3 Leukocyte counts
The fact that the leukocyte counts used when testing the LD-F method were very similarin all the groups is not surprising, since Thurlow and his colleagues demonstrated wheninvestigating the expression of antigens on neutrophils that an LG-6 filter selectivelyremoves activated neutrophils but has no significant effect in reducing the total leukocytecount (Thurlow et al. 1995).
6.4 S-100�
S-100� levels were measured for Papers I and II. The S-100 protein is a small (21 kDa)dimetric cytosolic calcium-binding protein that exists in various forms depending on itschain structure (� or �). The �� form occurs predominantly in astroglial and Schwanncells, and the �� form in astroglial cells, the concentrations in other cells beingnegligible, and the protein is involved in promoting axonal growth, glial proliferation,neuronal differentiation and calcium homeostasis (Johnsson 1996) It has been suggestedthat serum S-100� protein may be a neurobiochemical marker of brain injury aftercardiac and aortic arch surgery (Westaby et al. 1996), as its release has been associatedwith the duration of HCA (Bhattacharya et al. 1999). Tt has been demonstrated recentlythat there is correlation between early changes in neuropsychological performance and S-100b formation after HCA (Svensson et al. 2000), with levels increasing significantly inresponse to ischaemic brain insult shortly after the start of rewarming in both groups. Thefinding that S-100� concentrations decreased and were sustained at a lower level on dayone after surgery in lamotrigine-treated animals (I) is in line with other outcome data andsupports the findings of better brain protection in these animals than in controls. In PaperII, where the LD-F was the only neuroprotection strategy, there were no differences in S-100� release between the groups. The reason for this remains open, but one explanation

47
could be that L-DF exercises its effect on the reperfusion injury and not on thebiochemical cascade.
6.5 Cytokines
Cytokines TNF-�, IL-1 and IL-8 were measured in Paper II. TNF-� increased after HCAin both groups, but tended to be lower in the L-DF group than in the controls. There wereno differences in IL-1� and IL-8 between the groups, and the changes relative to thebaseline were much smaller than in TNF-�. CPB is known to release proinflammatorycytokines, including TNF and IL-1, IL-6, IL-8 (Hattler et al. 1995, Hill et al. 1997) andTNF-� and IL-1� have been shown to play an important role in brain immune andinflammatory activities and ischaemic brain injury. CPB per se releases cytokines, andthe cytokines measured in the present series were probably released from many organs onaccount of HCA. It is interesting, however, that TNF-� tended to be lower in the L-DFgroup than in the controls, since this cytokine in particular is known to play an importantrole in ischaemic brain injury(Feuerstein et al. 1998, Feuerstein et al. 1994, Feuerstein etal. 1998).
6.6 Behavioural outcome
Among the animals that survived for 7 days, the behavioural score was better in thelamotrigine group (I), the median score of 8 for which indicates full recovery or only amild disturbance. The control animals showed more severe initial neurologicalimpairment, and recovery to a score of 8 was seen in only one animal in this group. Thebehavioural outcome was also better in the L-DF group (II). The memantine trial failed topoint to any differences in behavioural outcome between the surviving animals (III). Inthe final trial with three groups, the LD-F + lamotrigine group had best neurologicalrecovery and the controls the worse, with the lamotrigine group in an intermediateposition. The difference between the LD-F + lamotrigine group and the controls reachedstatistical significance (Table II)(IV). The behavioural outcome data are in line with theother findings and confirm that LD-F and lamotrigine are both neuroprotective whereasmemantine does not have this property in the present setting. The data also support thehypothesis that combining two effective strategies provides the best outcome.
6.7 Electroencephalography
Electrical activity in the brain is readily accessible for evaluation in the form of an EEGrecording in experimental animals as it is in humans. Continuous recordings can beobtained with relatively low-cost equipment with a time resolution of a fraction of amilliseconds for many hours and days (Prior and Maynard 1986). Burst suppression is apattern of high amplitude EEG activity interrupted by relatively low amplitude activity,

48
typically under 20 µV peak-to-peak. The periods of high activity, the bursts, and those oflow activity, the suppressions, are usually of a few seconds� duration and occur in anunpredictable, irregular fashion. The bursts are also characterized by negative DC shift,and suppression correspondingly by a positive DC level. Burst suppression can beproduced by deep anaesthesia with several intravenous or volatile anaesthetics, and alsoby brain damage. It can also be seen in cases of local brain damage if the cortex isdissociated from the underlying brain structures. Hypothermia causes a decrease in EEGamplitude and an increase in the duration of the suppressions. The effects of anaesthetics,brain damage and hypothermia are additive, as each increases the duration of thesuppressions and reduces that of the bursts (Wennberg et al. 1997). The EEG in humanscan be inactive after ischaemic brain damage due to cardiac arrest, i.e. it can besuppressed for up to 3 hours, and the patient may still make a full recovery. EEG recoveryin these patients starts with occasional slow waves, which develop into burst suppression,then into a continuous EEG. In the burst suppression phase stimulation will producebursts, and a painful stimulus may turn burst suppression into a continuous EEG pattern(Jorgensen and Malchow-Moller 1981).
For the purpose of monitoring EEG recovery, we anaesthetized the pigs to the level ofburst suppression with isoflurane, whereupon hypothermia caused the EEG to becontinuously suppressed. During rewarming the EEG recovered first with short, lowamplitude bursts, which then bursts became increasingly higher in amplitude and durationuntil finally a continuous EEG was observed in those pigs which recovered. This enabledus to use the percentage duration of bursts as a measure of recovery, a measure which iseasier to calculate accurately than amplitude or frequency, for instance, when the EEG isof low amplitude and there are artefacts present. EEG recovery and the correspondingburst suppression ratio are illustrated in Fig. 2.
The additive effect of ischaemic damage plus isoflurane-induced burst suppressionproved to be a sensitive measure of recovery in the present experiments. The bursts arereadily detectable and their duration can even be measured when EEG recordings are verylow in amplitude as a result of hypothermia, i.e. close to the noise level of the recordingsystem, when other quantification of EEG recordings is questionable. Our previousstudies using EEG after prolonged periods have also suggested that a change in electricalactivity in the brain approximately 3 hours after HCA correctly distinguishes betweenpigs who will suffer severe damage and those who will fare better (Anttila et al. 1999,Anttila et al. 2000). The fact that the burst suppression pattern became more severe uponischaemic damage, and that EEG recovery was in line with other measures of braindamage suggests that it correctly reflects the neuroprotective effect of lamotrigine (I andIV). When we studied LD-F as a single strategy there were no differences between thegroups (II), but when we combined lamotrigine with LD-F we were also able to achievebetter EEG recovery (IV). This improvement in the EEG data supports our hypothesisthat combining two effective strategies results in better neurological recovery. Theabsence of any differences in EEG recovery in the memantine trial (III) is in line with theother findings (III). Lamotrigine and other neuroprotective drugs have an effect on thebiochemical cascade, while L-DF exercises an effect on reperfusion injury, and this mayexplain why the latter alone could not provide any improvement in EEG recovery. AnEEG may also be able to pinpoint accurately the time when lamotrigine-protected brainsare recovering better than those in the placebo group. EEG recordings made for 16 hours

49
in the LD-F trial (II) detected one control animal in which there were no EEG bursts afterHCA and two animals (one in each group) whose EEG bursts decreased after an initialrecovery. All three animals died soon after extubation. This is in line with other data andsupports the hypothesis that early mortality was due to neurological damage. EEG wasthe only method which could point to the probable time of cerebral cortex death.
6.8 Microdialysis
Changes in the chemical microenvironment are important for obtaining an understandingof the mechanisms of cerebral ischaemia. Microdialysis enables changes in theextracellular biochemical microenvironment in body tissues to be monitored in vivo(Benveniste 1989, Benveniste et al. 1984, Tossman and Ungerstedt 1986). The pioneeringwork on microdialysis was performed on animals, and the first clinical studies werepublished in 1990 (Di Chiara 1990, Meyerson et al. 1990, Persson and Hillered 1992).Since then there has been extensive interest in brain chemistry, especially in neurosurgerypatients. Microdialysis in vivo has been used with patients suffering from head injury(Bullock et al. 1998, Menzel et al. 1999), subarchnoidal haemorrhage (Persson andHillered 1992, Saveland et al. 1996) and epilepsy (Fried et al. 1999), and also with HCApatients (Mendelowitsch et al. 1998), and we found it very promising for the continuousmonitoring of brain biochemistry in our model (III and IV). A summary of themicrodialysis data is provided in Fig. 3.
The lack of any differences between the groups in the memantine trial (III) confirmedthe fact that memantine was unable to protect the brain, but microdialysis did enable us todemonstrate a close correlation between glutamate levels and glycerol levels, with thepeak in the latter following a little later. This is in line with previous findings that thebiochemical cascade starts with the release of glutamate and that cell damage followslater (Lipton and Rosenberg 1994). There was also a correlation between the highestindividual glutamate levels and the histopathological scores, and a negative correlationbetween glutamate levels and EEG burst recovery. These findings support the observationthat glutamate accumulation in the intercellular space is a neurotoxic feature(Baumgartner et al. 1997).
In the last of these series of experiments (IV) lamotrigine and lamotrigine combinedwith LD-F were both found to reduce glutamate, glycerol and lactate levels, the curvesbehaving in very similar fashions (Fig. 4). This is in line with other data obtained in thattrial and supports the hypothesis that the neuroprotective strategies were effective. Thecombination of lamotrigine and LD-F was not superior to lamotrigine alone in terms ofmicrodialysis, however, possibly on account of the fact that reperfusion injury occursafter the biochemical cascade (Fauerstein, et al 1998).
6.9 Mortality
The mortality rate in the first trial with lamotrigine was 2/8 in the lamotrigine group and3/8 in the control group. Thus the animals treated with lamotrigine did a little better, but

50
the difference between the groups could be due to chance (I). In the second trial, withLD-F, mortality was higher in the control group than in the lamotrigine group (5/10 vs. 2/10), which is in line with other data (Table II), but the difference did not reach statisticalsignificance. In the memantine trial the mortality rate tended to be higher in memantinegroup (5/10 vs. 1/10, p=0.14), but the explanation for this remains obscure (III). Webelieve that the trial is of particular importance in this respect, as it has been suggestedthat memantine can improve the neurological outcome during operations on the thoracicaorta (Ehrlich et al. 1999). The last trial pointed to a high mortality rate among thecontrols (5/8), with a lower rate in the lamotrigine group (4/8) and the lowest mortalityassociated with lamotrigine combined with LD-F (1/8). This outcome confirms ourhypothesis that it is possible to improve brain protection and survival by combiningstrategies targeted at different phases of ischaemic brain injury. The other reason for thegood survival rate in the LDF + lamotrigine group may have been the protective effectagainst reperfusion injury that the LD-F has been shown to have in other organs and inthe recent literature (Bando et al. 1990, Bolling et al. 1997, Gu et al. 1996, Lazar et al.1995), although the early deaths were very probably due to brain damage as there wereno differences in the haemodynamic data or any specific co-morbidity findings at theautopsies. The mortality rates cannot be compared directly between the series reportedhere, of course, because the experiments were carried out at different times. In ourexperience, it seems that experimental animals differ in quality from time to time, eventhere may be no signs of illness or other co-morbidity findings preoperatively or atautopsy. The animals in some series simply withstand prolonged HCA better than others,and as seen in Fig. 3, the control animals in the memantine trial (III) had lower glycerolconcentrations than those in the last series (IV) even though the HCA protocol was thesame. It is therefore of particular importance to use randomized prospective protocols.
6.10 Histopathological analysis
The total histopathological scores are not directly comparable between the series because,as seen in the methods section, the analytical methods changed in the meantime. Thereason for this is that we have been constantly trying to improve our model, and we thinkthat our present scoring is more sensitive and better able to depict the differences in brainpathology between the animals. The made histopathological analyses performed in ablinded fashion by a senior pathologist constitute the most important outcome of theseseries of experiments, and their results are summarized below.
The histopathological scores recorded in the memantine trial were almost equalbetween the groups, convincing us that memantine does not have any neuroprotectiveeffect in this animal model (Table II). The median behavioural score and survival werepoorer than in the controls, and the microdialysis data and EEG recovery level weresimilar (Table II and Fig. 4). All the other data are in line with these histopthologicalfindings, and it may be said that in the light of these results memantine cannot beconsidered a candidate for clinical use in connection with hypothermic ischaemia. This issomewhat surprising, since it is claimed in the recent literature that memantineameliorates NMDA receptor-mediated neurotoxicity in vitro and in vivo in normothermia

51
(Chen et al. 1992, Stieg et al. 1999), and it has also proved to be a promising drug for thetreatment of retinal ischaemia (Legreze et al. 1998, Osborne 1999). On the other hand, theresults of experiments with its use for the prevention of spinal cord ischaemia-inducedinjury have been controversial (Ehrlich et al. 1999, Euler et al. 1997, Miyamoto andMiyamoto 1999). The reason for the failuire of memantine as a neuroprotectant may alsobe related to the hypothermia, as our study was the first to test its efficacy in hypothermicbrain ischaemia. The finding is nevertheless surprising, and it can be speculated that thedose used here may have been too low. It must be borne in mind, however, that side-effects like ataxia, myorelaxation and amnesia can be encountered following higher acutedoses of memantine, i.e. doses in the region of 20 mg/kg (Parsons et al. 1999), and alsothat, as depicted under Methods, the memantine concentrations were at an effective levelthroughout.
As memantine had failed as a neuroprotectant in our chronic porcine model, we choselamotrigine for use in the combined strategy in our last trial. Papers I, II and IV considerlamotrigine and LD-F, and their results can be summarized together. In the first trial andin one group of the last trial lamotrigine was used as a single neuroprotective strategyduring hypothermic ischaemia. In both groups the median of the total histopathologicalscore was lower than in the control group, although the differences were on the borderlineof significance. Neurological recovery, survival and EEG burst recovery were in line withthe histopathological findings in both trials, although not all the differences reachedstatistical significance (Table II). In addition, the S-100� data on the first series and themicrodialysis data on the latter (Fig. 4) confirm the other findings that lamotrigine isneuroprotective in our long-term pig model for HCA. This finding verifies the results ofthe other authors studying brain ischaemia using different animal models (Bacher andZornow 1997, Conroy et al. 1999, Lee et al. 2000). The good results achieved here raisesthe question of the clinical use of lamotrigine in aortic surgery when HCA is needed. Thepossibility of skin reactions (Bourgeois 1998, Dunn et al. 1999) and the fact that nointravenous form of lamotrigine is available neverthgeless lead us to the conclusion thatits use in aortic surgery still requires some further research.
LD-F was considered alone as a preventive method in the second paper, where a cleardifference emerged between the groups in terms of their histopathology (Table II).Neurological recovery and mortality were in line with these data, indicating thatreperfusion injury plays a significant role in the pathogenesis of ischaemic brain injuryand that it is possible to mitigate it with an LD-F (Table II). Langley and associates reporta borderline improvment in recovery in terms of cerebral blood flow in an acute HCAmodel with neonatal pigs (Langley et al. 2000) and mention the need for furtherreesearch. We believe that our trial with a chronic model employing more end pointsanswers that question, confirming the beneficial effect of an LD-F in providingneuroprotection after HCA. A LD-F is much easier to use clinically than lamotrigine orany other chemical agent, as it is a commercially available device and has already been inclinical use. It is simple to add to a CPB circuit and it is potentially an effective methodwhich requires no drug administration. It has no published adverse effects; on thecontrary, as mentioned in the review of the literature, it mitigates reperfusion injury inmany organs, e.g. the myocardium (Lazar et al. 1995, Sawa et al. 1996, Schmidt et al.1996, Wilson et al. 1993). It has also been shown that when more than three bloodtransfusions are required after cardiac surgery, leukocyte filtration of the transfused blood

52
results in a significant reduction in postoperative mortality (van de Watering et al. 1998).Leukocyte depletion during and after CPB has been shown to improve pulmonaryfunction (Gu et al. 1996) and to ameliorate free radical-mediated lung injury (Bando et al.1990, Bolling et al. 1997). To summarize the recent literature and our findings, LD-F canbe recommended for clinical use, and we have already used it with patients who needHCA.
Both neuroprotection protocol groups in the last of these series of experiments had abetter outcome in terms of histopathological data than the controls. It was the LDF +lamotrigine group that achieved the best outcome of all, although the lamotrigine grouphad almost equal histopathological results and we could not demonstrate any differencebetween the neuroprotective protocols histopathologically (IV). The reason for thisremains open, but it does seem that when we combine two effective neuroprotectivestrategies and compare this combination with either of these strategies alone, it may bedifficult to show any difference between the groups. This finding also encourages us toseek more sensitive ways of measuring brain damage in terms of histopathology, e.g. interms of apoptosis. The combination of strategies led to the best outcome when all the endpoints were summarized, however, and this finding inspires us to conduct furtherinvestigations into the pathogenesis of brain ischaemia and to develop step by step a brainprotection protocol that is capable of targeting different phases of the ischaemic cascade.

7 Conclusions
Recalling the purpose of the present investigation, the results can be summed up asfollows:1. The Na+ channel blocker lamotrigine improves the neurological outcome following a
prolonged period of hypothermic circulatory arrest in this chronic porcine model.2. Leukocyte filtration improves brain protection following a prolonged period of
hypothermic circulatory arrest in this chronic porcine model.3. The NMDA antagonist memantine has no neuroprotective effect during a prolonged
period of hypothermic circulatory arrest in this chronic porcine model.4. The Na+ channel blocker lamotrigine mitigates brain injury following hypothermic
ischaemia and its combination with leukocyte-depleting filter makes it possible toimprove the cerebral outcome and survival rate even further.

8 References
Albers GW, Clark WM, Atkinson RP, Madden K, Data JL & Whitehouse MJ (1999) Dose escalationstudy of the NMDA glycine-site antagonist licostinel in acute ischemic stroke. Stroke 30 (3): 508�513.
Allen S (1997) The Role of Leukocytes in the Systemic Inflammatory Response and the Potential Im-pact of Leukocyte Depletion. Cardiovascular Engineering 12: 34�54.
Anttila V, Kiviluoma K, Pokela M, Rimpilainen J, Mäkiranta M, Jantti V, Hirvonen J & Juvonen T(1999) Cold retrograde cerebral perfusion improves cerebral protection during moderate hypo-thermic circulatory arrest: A long-term study in a porcine model. Journal of Thoracic and Cardi-ovascular Surgery 118 (5): 938�945.
Anttila V, Pokela M, Kiviluoma K, Mäkiranta M, Hirvonen J & Juvonen T (2000) Is the maintainedcranial hypothermia the only factor leading to improved outcome following retrograde cerebralperfusion? An experimental study using a chronic porcine model. Journal of Thoracic & Cardio-vasc Surg 119 (5): 1021�1029
Aoki M, Nomura F, Stromski ME, Tsuji MK, Fackler JC, Hickey PR, Holtzman D & Jonas RA(1994) Effects of MK-801 and NBQX on acute recovery of piglet cerebral metabolism after hy-pothermic circulatory arrest. Journal of Cerebral Blood Flow & Metabolism 14 (1): 156�165.
Arends MJ & Wyllie AH (1991) Apoptosis: mechanisms and roles in pathology. International Re-view of Experimental Pathology 32: 223�254.
Arias RL, Tasse JR & Bowlby MR (1999) Neuroprotective interaction effects of NMDA and AMPAreceptor antagonists in an in vitro model of cerebral ischemia. Brain Research 816 (2): 299�308.
Arrowsmith JE, Harrison MJ, Newman SP, Stygall J, Timberlake N & Pugsley WB (1998) Neuro-protection of the brain during cardiopulmonary bypass: a randomized trial of remacemide duringcoronary artery bypass in 171 patients. Stroke 29 (11): 2357�2362.
Arroyo S, Lesser RP, Gillinov AM, Redmond M, Zehr KJ, Troncoso JC, Jackson D, BaumgartnerWA & Cameron DE (1993) EEG and prognosis of neurologic recovery of dogs under profoundhypothermic circulatory arrest. Electroencephalography & Clinical Neurophysiology 87 (4): 242�249.
Arvin B, Lekieffre D, Graham JL, Moncada C, Chapman AG & Meldrum BS (1994) Effect of thenon-NMDA receptor antagonist GYKI 52466 on the microdialysate and tissue concentrations ofamino acids following transient forebrain ischaemia. Journal of Neurochemistry 62 (4): 1458�1467.
Astrup J, Siesjo BK & Symon L (1981) Thresholds in cerebral ischemia � the ischemic penumbra.Stroke 12 (6): 723�725.

55
Astrup J, Symon L, Branston NM & Lassen NA (1977) Cortical evoked potential and extracellularK+ and H+ at critical levels of brain ischemia. Stroke 8 (1): 51�57.
Bacher A & Zornow MH (1997) Lamotrigine inhibits extracellular glutamate accumulation duringtransient global cerebral ischemia in rabbits. Anesthesiology 86 (2): 459�463.
Bando K, Pillai R, Cameron DE, Brawn JD, Winkelstein JA, Hutchins GM, Reitz BA & BaumgartnerWA (1990) Leukocyte depletion ameliorates free radical-mediated lung injury after cardiopulmo-nary bypass. Journal of Thoracic & Cardiovascular Surgery 99 (5): 873�877.
Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG & Feuerstein GZ (1997)Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 28 (6): 1233�1244.
Barth A, Newell DW, Nguyen LB, Winn HR, Wender R, Meno JR & Janigro D (1997) Neurotoxicityin organotypic hippocampal slices mediated by adenosine analogues and nitric oxide. Brain Re-search 762 (1�2): 79�88.
Bartus RT, Dean RL, Mennerick S, Eveleth D & Lynch G (1998) Temporal ordering of pathogenicevents following transient global ischemia. Brain Research 790 (1�2): 1�13.
Baumgartner WA, Redmond JM, Zehr KJ, Brock MV, Tseng EE, Blue ME, Troncoso JC & JohnstonMV (1998) The role of the monosialoganglioside, GM1 as a neuroprotectant in an experimentalmodel of cardiopulmonary bypass and hypothermic circulatory arrest. Annals of the New YorkAcademy of Sciences 845: 382�390.
Baumgartner WA, Redmond M, Brock M, Tseng E, Blue ME, Troncoso JC & Johnston MV (1997)Pathophysiology of cerebral injury and future management. Journal of Cardiac Surgery 12 (2 Sup-pl): 300�310.
Baumgartner WA, Walinsky PL, Salazar JD, Tseng EE, Brock MV, Doty JR, Redmond JM, BlueME, Goldsborough MA, Troncoso JC & Johnston MV (1999) Assessing the impact of cerebralinjury after cardiac surgery: will determining the mechanism reduce this injury? Annals of Tho-racic Surgery 67 (6): 1871�1873.
Bellinger DC, Jonas RA, Rappaport LA, Wypij D, Wernovsky G, Kuban KC, Barnes PD, HolmesGL, Hickey PR, Strand RD & et al. (1995) Developmental and neurologic status of children afterheart surgery with hypothermic circulatory arrest or low-flow cardiopulmonary bypass. New Eng-land Journal of Medicine 332 (9): 549�555.
Benveniste H (1989) Brain microdialysis. Journal of Neurochemistry 52 (6): 1667�1679.Benveniste H, Drejer J, Schousboe A & Diemer NH (1984) Elevation of the extracellular concentra-
tions of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitoredby intracerebral microdialysis. Journal of Neurochemistry 43 (5): 1369�1374.
Bhattacharya K, Westaby S, Pillai R, Standing SJ, Johnsson P & Taggart DP (1999) Serum S100Band hypothermic circulatory arrest in adults. Annals of Thoracic Surgery 68 (4): 1225�1229.
Boeckxstaens CJ (1996) Retrograde cerebral perfusion: An alternative for cerebral protection duringaortic arch surgery? A study on non-human primates. Thesis, Univ Antwerpen, DepartementGeneeskunde.
Bokesch PM, Halpin DP, Ranger WR, Drummond-Webb JJ, Marchand JE, Bronson RT, Warner KG& Kream RM (1997) Immediate-early gene expression in ovine brain after hypothermic circula-tory arrest: effects of aptiganel. Annals of Thoracic Surgery 64 (4): 1082�1087.
Bolling KS, Halldorsson A, Allen BS, Rahman S, Wang T, Kronon M & Feinberg H (1997) Preven-tion of the hypoxic reoxygenation injury with the use of a leukocyte-depleting filter. Journal ofThoracic & Cardiovascular Surgery 113 (6): 1081�1089.
Bourgeois BF (1998) New antiepileptic drugs. Archives of Neurology 55 (9): 1181�1183.Britton P, Lu XC, Laskosky MS & Tortella FC (1997) Dextromethorphan protects against cerebral
injury following transient, but not permanent, focal ischemia in rats. Life Sciences 60 (20): 1729�1740.

56
Brown CM, Calder C, Linton C, Small C, Kenny BA, Spedding M & Patmore L (1995) Neuroprotec-tive properties of lifarizine compared with those of other agents in a mouse model of focal cerebralischaemia. British Journal of Pharmacology 115 (8): 1425�1432.
Bullock R, Zauner A, Woodward JJ, Myseros J, Choi SC, Ward JD, Marmarou A & Young HF (1998)Factors affecting excitatory amino acid release following severe human head injury. Journal ofNeurosurgery 89 (4): 507�518.
Chabrier PE, Auguet M, Spinnewyn B, Auvin S, Cornet S, Demerle-Pallardy C, Guilmard-Favre C,Marin JG, Pignol B, Gillard-Roubert V, Roussillot-Charnet C, Schulz J, Viossat I, Bigg D & Mon-cada S (1999) BN 80933, a dual inhibitor of neuronal nitric oxide synthase and lipid peroxidation:a promising neuroprotective strategy. Proc Natl Acad Sci U S A 96 (19):10824�10829.
Chabrier PE, Demerle-Pallardy C & Auguet M (1999) Nitric oxide synthases: targets for therapeuticstrategies in neurological diseases. Cellular and Molecular Life Sciences 55 (8�9):1029�1035.
Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE & Lipton SA (1992) Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantageagainst NMDA receptor-mediated neurotoxicity. Journal of Neuroscience 12 (11): 4427�4436.
Cheng MA, Theard MA & Tempelhoff R (1997) Intravenous agents and intraoperative neuroprotec-tion. Beyond barbiturates. Critical Care Clinics 13 (1):185�199.
Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, JohnsonEM & Holtzman DM (1998) Caspase inhibitor affords neuroprotection with delayed administra-tion in a rat model of neonatal hypoxic-ischemic brain injury. Journal of Clinical Investigation 101(9):1992�1999.
Colbourne F, Li H, Buchan AM & Clemens JA (1999) Continuing postischemic neuronal death inCA1: influence of ischemia duration and cytoprotective doses of NBQX and SNX-111 in rats.Stroke 30 (3): 662�668.
Conroy BP, Black D, Lin CY, Jenkins LW, Crumrine RC, DeWitt DS & Johnston WE (1999) Lamo-trigine attenuates cortical glutamate release during global cerebral ischemia in pigs on cardiopul-monary bypass. Anesthesiology 90 (3): 844�54.
Coselli JS & LeMaire SA (1997) Experience with retrograde cerebral perfusion during proximal aor-tic surgery in 290 patients. Journal of Cardiac Surgery 12 (2 Suppl): 322�325.
Danysz W, Parsons CG, Kornhuber J, Schmidt WJ & Quack G (1997) Aminoadamantanes as NMDAreceptor antagonists and antiparkinsonian agents � preclinical studies. Neuroscience & Biobehav-ioral Reviews 21 (4): 455�468.
Davis SM, Albers GW, Diener HC, Lees KR & Norris J (1997) Termination of Acute Stroke StudiesInvolving Selfotel Treatment. ASSIST Steering Committed. Lancet 349 (9044): 32.
Deeb GM, Williams DM, Quint LE, Monaghan HM & Shea MJ (1999) Risk analysis for aortic sur-gery using hypothermic circulatory arrest with retrograde cerebral perfusion. Annals of ThoracicSurgery 67 (6):1883�1886.
Dewanjee MK, Wu SM, Burke GW, 3rd & Hsu LC (1998) Tumor necrosis factor-alpha in plasmaduring cardiopulmonary bypass in a pig model: correlation with marginated neutrophils and cer-ebral edema by magnetic resonance imaging. ASAIO Journal 44 (3): 212�218.
Di Chiara G (1990) In-vivo brain dialysis of neurotransmitters. Trends in Pharmacological Sciences11 (3): 116�121.
Dumuis A, Pin JP, Oomagari K, Sebben M & Bockaert J (1990) Arachidonic acid released from stri-atal neurons by joint stimulation of ionotropic and metabotropic quisqualate receptors. Nature 347(6289):182�184.
Dunlap K, Luebke JI & Turner TJ (1995) Exocytotic Ca2+ channels in mammalian central neurons.Trends in Neurosciences 18 (2): 89�98.
Dunn N, Wilton L & Shakir S (1999) Stevens-Johnson syndrome and antiepileptics. Lancet 354(9183): 1033�1034.

57
Dyker AG, Edwards KR, Fayad PB, Hormes JT & Lees KR (1999) Safety and tolerability study ofaptiganel hydrochloride in patients with an acute ischemic stroke. Stroke 30 (10): 2038�2042.
Ehrlich M, Knolle E, Ciovica R, Bock P, Turkof E, Grabenwoger M, Cartes-Zumelzu F, Kocher A,Pockberger H, Fang WC, Wolner E & Havel M (1999) Memantine for prevention of spinal cordinjury in a rabbit model. Journal of Thoracic and Cardiovascular Surgery 117 (2): 285�291.
Endres M, Fink K, Zhu J, Stagliano NE, Bondada V, Geddes JW, Azuma T, Mattson MP,Kwiatkowski DJ & Moskowitz MA (1999) Neuroprotective effects of gelsolin during murinestroke. Journal of Clinical Investigation 103 (3): 347�354.
Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT &Moskowitz MA (1998) Attenuation of delayed neuronal death after mild focal ischemia in miceby inhibition of the caspase family. Journal of Cerebral Blood Flow & Metabolism 18 (3): 238�247.
Ergin MA, Galla JD, Lansman s L, Quintana C, Bodian C & Griepp RB (1994) Hypothermic circu-latory arrest in operations on the thoracic aorta. Determinants of operative mortality and neuro-logic outcome. Journal of Thoracic & Cardiovascular Surgery 107 (3): 788�797.
Escott KJ, Beech JS, Haga KK, Williams SC, Meldrum BS & Bath PM (1998) Cerebroprotective ef-fect of the nitric oxide synthase inhibitors, 1-(2-trifluoromethylphenyl) imidazole and 7-nitro in-dazole, after transient focal cerebral ischemia in the rat. Journal of Cerebral Blood Flow &Metabolism 18 (3): 281�287.
Euler Mv, Mo L-L, Whittemore S, Seiger Å & Sundström E (1997) No protective effect of the NMDAantagonist memantine in experimental spinal cord injuries. Journal of Neurotrauma 14: 53�61.
Faraci FM & Brian JE, Jr. (1994) Nitric oxide and the cerebral circulation. Stroke 25 (3): 692�703.Feigin VL, Rinkel GJ, Algra A, Vermeulen M & van Gijn J (1998) Calcium antagonists in patients
with aneurysmal subarachnoid hemorrhage: a systematic review. Neurology 50 (4): 876�83.Feuerstein G, Wang X & Barone FC (1998) Cytokines in brain ischemia � the role of TNF alpha. Cel-
lular & Molecular Neurobiology 18 (6): 695�701.Feuerstein GZ, Liu T & Barone FC (1994) Cytokines, inflammation, and brain injury: role of tumor
necrosis factor-alpha. Cerebrovascular & Brain Metabolism Reviews 6 (4): 341�360.Feuerstein GZ, Wang X & Barone FC (1998) The role of cytokines in the neuropathology of stroke
and neurotrauma. Neuroimmunomodulation 5 (3�4): 143�159.Fried I, Wilson CL, Maidment NT, Engel J, Jr., Behnke E, Fields TA, MacDonald KA, Morrow JW
& Ackerson L (1999) Cerebral microdialysis combined with single-neuron and electroencephalo-graphic recording in neurosurgical patients. Technical note. Journal of Neurosurgery 91 (4): 697�705.
Frist WH, Baldwin JC, Starnes VA, Stinson EB, Oyer PE, Miller DC, Jamieson SW, Mitchell RS &Shumway NE (1986) A reconsideration of cerebral perfusion in aortic arch replacement. Annalsof Thoracic Surgery 42 (3): 273�281.
Garthwaite J, Garthwaite G, Palmer RM & Moncada S (1989) NMDA receptor activation induces ni-tric oxide synthesis from arginine in rat brain slices. European Journal of Pharmacology 172 (4�5): 413�416.
Gasic GP & Hollmann M (1992) Molecular neurobiology of glutamate receptors. Annual Review ofPhysiology 54: 507�536.
Gibbon JH (1954) Application of a mechanical heart and lung apparatus to cardiac surgery. Minne-sota MED 34: 171�185.
Gilland E, Puka-Sundvall M, Andine P, Bona E & Hagberg H (1994) Hypoxic-ischemic injury in theneonatal rat brain: effects of pre- and post-treatment with the glutamate release inhibitorBW1003C87. Brain Research. Developmental Brain Research 83 (1): 79�84.
Gillinov AM, Redmond JM, Zehr KJ, Troncoso JC, Arroyo S, Lesser RP, Lee AW, Stuart RS, ReitzBA, Baumgartner WA & et al. (1993) Superior cerebral protection with profound hypothermiaduring circulatory arrest. Annals of Thoracic Surgery 55 (6): 1432�1439.

58
Gottron FJ, Ying HS & Choi DW (1997) Caspase inhibition selectively reduces the apoptotic com-ponent of oxygen-glucose deprivation-induced cortical neuronal cell death. Molecular & CellularNeurosciences 9 (3): 159�169.
Govier AV, Reves JG, McKay RD, Karp RB, Zorn GL, Morawetz RB, Smith LR, Adams M & Free-man AM (1984) Factors and their influence on regional cerebral blood flow during nonpulsatilecardiopulmonary bypass. Annals of Thoracic Surgery 38 (6): 592�600.
Green DR (1998) Apoptosis. Death deceiver. Nature 396 (6712): 629�630.Griepp RB, Ergin MA, Lansman SL, Galla JD & Pogo G (1991) The physiology of hypothermic cir-
culatory arrest. Seminars in Thoracic & Cardiovascular Surgery 3 (3):188�393.Griepp RB, Stinson EB, Hollingsworth JF & Buehler D (1975) Prosthetic replacement of the aortic
arch. Journal of Thoracic & Cardiovascular Surgery 70 (6): 1051�1063.Grotta J (1997) Lubeluzole treatment of acute ischemic stroke. The US and Canadian Lubeluzole
Ischemic Stroke Study Group. Stroke 28 (12):2338�2346.Gschnitzer F (1973) [Resection of a syphilitic aneurysm of the aortic arch using left heart bypass, pro-
found hypothermia, and circulatory arrest]. Thoraxchirurgie Vaskulare Chirurgie 21 (2): 87�90.Gu YJ, de Vries AJ, Boonstra PW & van Oeveren W (1996) Leukocyte depletion results in improved
lung function and reduced inflammatory response after cardiac surgery. Journal of Thoracic &Cardiovascular Surgery 112 (2): 494�500.
Haley EC, Jr. (1998) High-dose tirilazad for acute stroke (RANTTAS II). RANTTAS II Investigators.Stroke 29 (6): 1256�1257.
Hall ED, McCall JM & Means ED (1994) Therapeutic potential of the lazaroids (21-aminosteroids)in acute central nervous system trauma, ischemia and subarachnoid hemorrhage. Advances inPharmacology (New York) 28: 221�268.
Haseldonckx M, Van Reempts J, Van de Ven M, Wouters L & Borgers M (1997) Protection withlubeluzole against delayed ischemic brain damage in rats. A quantitative histopathologic study.Stroke 28 (2): 428�432.
Hattler BG, Zeevi A, Oddis CV & Finkel MS (1995) Cytokine induction during cardiac surgery: anal-ysis of TNF-alpha expression pre- and postcardiopulmonary bypass. Journal of Cardiac Surgery10 (4 Suppl): 418�422.
Hicks CA, Ward MA, Swettenham JB & O'Neill MJ (1999) Synergistic neuroprotective effects bycombining an NMDA or AMPA receptor antagonist with nitric oxide synthase inhibitors in globalcerebral ischaemia. European Journal of Pharmacology 381 (2�3): 113�119.
Hill GE, Whitten CW & Landers DF (1997) The influence of cardiopulmonary bypass on cytokinesand cell-cell communication. Journal of Cardiothoracic & Vascular Anesthesia 11 (3): 367�375.
Hollmann M, Hartley M & Heinemann S (1991) Ca2+ permeability of KA-AMPA--gated glutamatereceptor channels depends on subunit composition. Science 252 (5007): 851�853.
Hong SC, Goto Y, Lanzino G, Soleau S, Kassell NF & Lee KS (1994) Neuroprotection with a calpaininhibitor in a model of focal cerebral ischemia. Stroke 25 (3): 663�669.
Johnsson P (1996) Markers of cerebral ischemia after cardiac surgery. Journal of Cardiothoracic andVascular Anesthesia 10 (1): 120�126.
Jorgensen O & Malchow-Moller A (1981) Natural history of global and critical brain ischemia. Re-suscitation 9: 133�188.
Juvonen T, Weisz DJ, Wolfe D, Zhang N, Bodian CA, McCullough JN, Mezrow CK & Griepp RB(1998) Can Retrograde Perfusion Mitigate Cerebral Injury Following Particulate Embolization?Journal of Thoracic and Cardiovascular Surgery 115: 1142�1159.
Juvonen T, Zhang N, Wolfe D, Weisz DJ, Bodian CA, Shiang HH, McCullough JN & Griepp RB(1998) Retrograde cerebral perfusion enhances cerebral protection during prolonged hypothermiccirculatory arrest: a study in a chronic porcine model. Annals of Thoracic Surgery 66 (1): 38�50.

59
Kanthasamy AG, Yun RJ, Nguyen B & Truong DD (1999) Effect of riluzole on the neurological andneuropathological changes in an animal model of cardiac arrest-induced movement disorder. Jour-nal of Pharmacology & Experimental Therapeutics 288 (3):1340�1348.
Kawaguchi K, Henshall DC & Simon RP (1999) Parallel dose-response studies of the voltage-de-pendent Na+ channel antagonist BW619C89, and the voltage-dependent Ca2+ channel antagonistnimodipine, in rat transient focal cerebral ischaemia. European Journal of Pharmacology 364 (2�3): 99�105.
Kawasaki-Yatsugi S, Shimizu-Sasamata M, Yatsugi S & Yamaguchi T (1998) Delayed treatmentwith YM90K, an AMPA receptor antagonist, protects against ischaemic damage after middle cer-ebral artery occlusion in rats. Journal of Pharmacy & Pharmacology 50 (8): 891�898.
Kouchoukos NT, Daily BB, Rokkas CK, Murphy SF, Bauer S & Abboud N (1995) Hypothermic by-pass and circulatory arrest for operations on the descending thoracic and thoracoabdominal aorta.Annals of Thoracic Surgery 60 (1): 67�76.
Kramer RS, Sanders AP, Lesage AM, Woodhall B & Sealy WC (1968) The effect profound hypo-thermia on preservation of cerebral ATP content during circulatory arrest. Journal of Thoracic &Cardiovascular Surgery 56 (5): 699�709.
Lang-Lazdunski L, Heurteaux C, Vaillant N, Widmann C & Lazdunski M (1999) Riluzole preventsischemic spinal cord injury caused by aortic crossclamping. Journal of Thoracic & CardiovascularSurgery 117 (5): 881�889.
Langley S, Chai P, Chui S, Jaggers J & Ungerleider R (2000) The effects of a leukocyte-depletingfilter on cerebral and renal recovery after deep hypothermic circulatory arrest. The Journal of Tho-racic and Cardiovascular Surgery 119: 1262�1269.
Langley SM, Chai PJ, Jaggers JJ & Ungerleider RM (2000) The free radical spin trap alpha-phenyl-tert-butyl nitrone attenuates the cerebral response to deep hypothermic ischemia. Journal of Tho-racic & Cardiovascular Surgery 119 (2): 305�313.
Lazar HL, Zhang X, Hamasaki T, Treanor P, Rivers S, Bernard S & Shemin RJ (1995) Role of leu-kocyte depletion during cardiopulmonary bypass and cardioplegic arrest. Annals of Thoracic Sur-gery 60 (6): 1745�1748.
Lee WT, Shen YZ & Chang C (2000) Neuroprotective effect of lamotrigine and MK-801 on rat brainlesions induced by 3-nitropropionic acid: evaluation by magnetic resonance imaging and in vivoproton magnetic resonance spectroscopy. Neuroscience 95 (1): 89�95.
Legault C, Furberg CD, Wagenknecht LE, Rogers AT, Stump DA, Coker L, Troost BT & HammonJW (1996) Nimodipine neuroprotection in cardiac valve replacement: report of an early terminat-ed trial. Stroke 27 (4): 593�598.
Legreze W, Knorle R, Bach M & Feurstein T (1998) Memantine is neuroprotective in a rat model ofpressure-induced retinal ischemia. Invest Ophtal Vis Sci 39: 1063�1066.
Liebetrau M, Staufer B, Auerswald EA, Gabrijelcic-Geiger D, Fritz H, Zimmermann C, PfefferkornT & Hamann GF (1999) Increased intracellular calpain detection in experimental focal cerebralischemia. Neuroreport 10 (3): 529�534.
Lipton SA (1994) Neuronal injury associated with HIV-1 and potential treatment with calcium-chan-nel and NMDA antagonists. Developmental Neuroscience 16 (3�4): 145�151.
Lipton SA (1996) Similarity of neuronal cell injury and death in AIDS dementia and focal cerebralischemia: potential treatment with NMDA open-channel blockers and nitric oxide-related species.Brain Pathology 6 (4): 507�517.
Lipton SA & Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologicdisorders. New England Journal of Medicine 330 (9): 613�622.
Lo EH, Pierce AR, Mandeville JB & Rosen BR (1997) Neuroprotection with NBQX in rat focal cer-ebral ischemia. Effects on ADC probability distribution functions and diffusion-perfusion rela-tionships. Stroke 28 (2): 439�446.

60
Markgraf CG, Velayo NL, Johnson MP, McCarty DR, Medhi S, Koehl JR, Chmielewski PA & LinnikMD (1998) Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia inrats. Stroke 29 (1):152�158.
Mazzoni M MC, Shiang HH, Griepp RB (1993) Protective effects of nimodipine on neurological out-come after prolonged hypotermic circulatory arrest. Drugs in Development (2): 369�375.
McCullough JN, Zhang N, Reich D, Juvonen T, Klein J, Spielvoge D & Ergin MA (1999) Cerebralmetabolic suppression during hypothermic circulatory arrest in humans. Annals of Thoracic Sur-gery 67: 1895�1899.
Mendelowitsch A, Mergner GW, Shuaib A & Sekhar LN (1998) Cortical brain microdialysis andtemperature monitoring during hypothermic circulatory arrest in humans. Journal of Neurology,Neurosurgery & Psychiatry 64 (5): 611�618.
Menzel M, Doppenberg EM, Zauner A, Soukup J, Reinert MM & Bullock R (1999) Increased in-spired oxygen concentration as a factor in improved brain tissue oxygenation and tissue lactatelevels after severe human head injury. Journal of Neurosurgery 91 (1): 1�10.
Meyerson BA, Linderoth B, Karlsson H & Ungerstedt U (1990) Microdialysis in the human brain:extracellular measurements in the thalamus of parkinsonian patients. Life Sciences 46 (4): 301�308.
Mezrow CK, Midulla PS, Sadeghi AM, Gandsas A, Wang W, Bodian C, Shing HH, Zappulla R, Da-punt OE & Griepp RB (1995) Quantitative electroencephalography: a method to assess cerebralinjury after hypothermic circulatory arrest. Journal of Thoracic & Cardiovascular Surgery 109 (5):925�934.
Mezrow CK, Midulla PS, Sadeghi AM, Gandsas A, Wang W, Dapunt OE, Zappulla R & Griepp RB(1994) Evaluation of cerebral metabolism and quantitative electroencephalography after hypo-thermic circulatory arrest and low-flow cardiopulmonary bypass at different temperatures. Journalof Thoracic & Cardiovascular Surgery 107 (4): 1006�1019.
Milani D, Guidolin D, Facci L, Pozzan T, Buso M, Leon A & Skaper SD (1991) Excitatory aminoacid-induced alterations of cytoplasmic free Ca2+ in individual cerebellar granule neurons: rolein neurotoxicity. Journal of Neuroscience Research 28 (3): 434�441.
Miyamoto TA & Miyamoto KJ (1999) A word of caution in extrapolating the spinal cord protectiveeffects of memantine obtained in a rabbit model under ketamine anesthesia. Journal of Thoracic& Cardiovascular Surgery 118 (4): 770�771.
Mueller AL, Artman LD, Balandrin MF, Brady E, Chien Y, Delmar EG, George K, Kierstead A, Mar-riott TB, Moe ST, Newman MK, Raszkiewicz JL, Sanguinetti EL, van Wagenen BC & Wells D(1999) NPS 1506, a novel NMDA receptor antagonist and neuroprotectant. Review of preclinicaland clinical studies. Annals of the New York Academy of Sciences 890: 450�457.
Muir KW & Lees KR (1995) Clinical experience with excitatory amino acid antagonist drugs. Stroke26 (3): 503�513.
Murase M, Maeda M, Koyama T, Tomida Y, Murakami F, Teranishi K, Ogawa Y, Seki A, OkamotoH, Hoshino M & et al. (1993) Continuous retrograde cerebral perfusion for protection of the brainduring aortic arch surgery. European Journal of Cardio-Thoracic Surgery 7 (11): 597�600.
Niazi SA & Lewis FJ (1957) Profound hypothermia: Report of a case. Annals of Surgery 147: 264�266.
Oates RK, Simpson JM, Turnbull JA & Cartmill TB (1995) The relationship between intelligence andduration of circulatory arrest with deep hypothermia. Journal of Thoracic & Cardiovascular Sur-gery 110 (3): 786�792.
Ohtaki M & Tranmer B (1994) Pretreatment of transient focal cerebral ischemia in rats with the cal-cium antagonist AT877. Stroke 25 (6): 1234�1239.
O'Mahony D & Kendall MJ (1999) Nitric oxide in acute ischaemic stroke: a target for neuroprotec-tion. Journal of Neurology, Neurosurgery & Psychiatry 67 (1): 1�3.

61
Osborne N (1999) memantine reduces alternations to the mammalian retina, insitu, induced byischemia. Vis Neurosci 1: 45�52.
Parmentier S, Bohme GA, Lerouet D, Damour D, Stutzmann JM, Margaill I & Plotkine M (1999) Se-lective inhibition of inducible nitric oxide synthase prevents ischaemic brain injury. British Jour-nal of Pharmacology 127 (2): 546�552.
Parsons CG, Danysz W & Quack G (1999) Memantine is a clinically well tolerated N-methyl-D-as-partate (NMDA) receptor antagonist � a review of preclinical data. Neuropharmacology 38 (6):735�767.
Perez-Pinzon MA, Yenari MA, Sun GH, Kunis DM & Steinberg GK (1997) SNX-111, a novel, pre-synaptic N-type calcium channel antagonist, is neuroprotective against focal cerebral ischemia inrabbits. Journal of the Neurological Sciences 153 (1): 25�31.
Persson L & Hillered L (1992) Chemical monitoring of neurosurgical intensive care patients usingintracerebral microdialysis. Journal of Neurosurgery 76 (1): 72�80.
Pierangeli A, Coli G, Donati A, Galli R, Mikus P & Turinetto B (1975) Treatment of aortic arch an-eurysms with deep hypothermia and circulatory arrest. Journal of Cardiovascular Surgery 16 (4):409�414.
Pin JP & Duvoisin R (1995) The metabotropic glutamate receptors: structure and functions. Neurop-harmacology 34 (1): 1�26.
Prior PF & Maynard DE (1986): Monitoring cerebral function. Long term monitoring of EEG andevoked potentials. Elsevier, Amsterdam.
Ramsay RE, Pellock JM, Garnett WR, Sanchez RM, Valakas AM, Wargin WA, Lai AA, Hubbell J,Chern WH, Allsup T & et al. (1991) Pharmacokinetics and safety of lamotrigine (Lamictal) in pa-tients with epilepsy. Epilepsy Research 10 (2�3): 191�200.
Redmond JM, Gillinov AM, Blue ME, Zehr KJ, Troncoso JC, Cameron DE, Johnston MV & Baum-gartner WA (1993) The monosialoganglioside, GM1, reduces neurologic injury associated withhypothermic circulatory arrest. Surgery 114 (2): 324�332.
Redmond JM, Gillinov AM, Zehr KJ, Blue ME, Troncoso JC, Reitz BA, Cameron DE, Johnston MV& Baumgartner WA (1994) Glutamate excitotoxicity: a mechanism of neurologic injury associat-ed with hypothermic circulatory arrest. Journal of Thoracic & Cardiovascular Surgery 107 (3):776�786.
Redmond JM, Zehr KJ, Blue ME, Lange MS, Gillinov AM, Troncoso JC, Cameron DE, Johnston MV& Baumgartner WA (1995) AMPA glutamate receptor antagonism reduces neurologic injury afterhypothermic circulatory arrest. Annals of Thoracic Surgery 59 (3): 579�584.
Rhoades Ra & Tanner GA (1995) Medical Physiology. Little, Brown and Company, Boston, NewYork, Toronto, London, p 45�54.
Rokkas CK, Cronin CS, Nitta T, Helfrich LR, Jr., Lobner DC, Choi DW & Kouchoukos NT (1995)Profound systemic hypothermia inhibits the release of neurotransmitter amino acids in spinal cordischemia. Journal of Thoracic & Cardiovascular Surgery 110 (1): 27�35.
Rothwell NJ & Strijbos PJ (1995) Cytokines in neurodegeneration and repair. International Journalof Developmental Neuroscience 13 (3�4): 179�185.
Sawa Y, Taniguchi K, Kadoba K, Nishimura M, Ichikawa H, Amemiya A, Kuratani T & Matsuda H(1996) Leukocyte depletion attenuates reperfusion injury in patients with left ventricular hyper-trophy. Circulation 93 (9): 1640�1646.
Saveland H, Nilsson OG, Boris-Moller F, Wieloch T & Brandt L (1996) Intracerebral microdialysisof glutamate and aspartate in two vascular territories after aneurysmal subarachnoid hemorrhage.Neurosurgery 38 (1): 12�19.
Schmid-Elsaesser R, Hungerhuber E, Zausinger S, Baethmann A & Reulen HJ (2000) Neuroprotec-tive effects of the novel brain-penetrating antioxidant U-101033E and the spin-trapping agent al-pha-phenyl-N-tert-butyl nitrone (PBN). Experimental Brain Research 130 (1): 60�66.

62
Schmidt FE, Jr., MacDonald MJ, Murphy CO, Brown WM, 3rd, Gott JP & Guyton RA (1996) Leu-kocyte depletion of blood cardioplegia attenuates reperfusion injury. Annals of Thoracic Surgery62 (6):1691�1696.
Seeburg PH, Burnashev N, Kohr G, Kuner T, Sprengel R & Monyer H (1995) The NMDA receptorchannel: molecular design of a coincidence detector. Recent Progress in Hormone Research 50:19�34.
Shiraishi Y, Lee JR, Laks H, Waters PF, Meneshian A, Marelli D, Blitz A & Chang P (1998) Use ofleukocyte depletion to decrease injury after lung preservation and rewarming ischemia: an exper-imental model. Journal of Heart & Lung Transplantation 17 (3): 250�258.
Small DL & Buchan AM (1996) Mechanisms of cerebral ischemia: intracellular cascades and thera-peutic interventions. Journal of Cardiothoracic & Vascular Anesthesia 10 (1) 139�146.
Sotaniemi KA (1995) Long-term neurologic outcome after cardiac operation. Annals of ThoracicSurgery 59 (5): 1336�1339.
Squire IB, Lees KR, Pryse-Phillips W, Kertesz A & Bamford J (1995) Efficacy and tolerability oflifarizine in acute ischemic stroke. A pilot study. Lifarizine Study Group. Annals of the New YorkAcademy of Sciences 765: 317�318.
Stieg PE, Sathi S, Warach S, Le DA & Lipton SA (1999) Neuroprotection by the NMDA receptor-associated open-channel blocker memantine in a photothrombotic model of cerebral focalischemia in neonatal rat. European Journal of Pharmacology 375 (1�3): 115�120.
Sun A & Cheng J (1999) Novel targets for therapeutic intervention against ischemic brain injury.Clinical Neuropharmacology 22 (3): 164�171.
Svensson LG, Crawford ES, Hess KR, Coselli JS, Raskin S, Shenaq SA & Safi HJ (1993) Deep hy-pothermia with circulatory arrest. Determinants of stroke and early mortality in 656 patients. Jour-nal of Thoracic & Cardiovascular Surgery 106 (1):19�28.
Svensson LG, Husain A, Penney DL, Swanson RA, Margolis DS, Kimmel WA, Nadolny E & Sha-hian DM (2000) A prospective randomized study of neurocognitive function and s-100 protein af-ter antegrade or retrograde brain perfusion with hypothermic arrest for aortic surgery. Journal ofThoracic and Cardiovascular Surgery 119 (1): 163�167.
Tabardel Y, Duchateau J, Schmartz D, Marecaux G, Shahla M, Barvais L, Leclerc JL & Vincent JL(1996) Corticosteroids increase blood interleukin-10 levels during cardiopulmonary bypass inmen. Surgery 119 (1): 76�80.
Tatlisumak T, Takano K, Meiler MR & Fisher M (1998) A glycine site antagonist, ZD9379, reducesnumber of spreading depressions and infarct size in rats with permanent middle cerebral artery oc-clusion. Stroke 29 (1): 190�195.
Taylor KM (1998) Brain damage during cardiopulmonary bypass. Annals of Thoracic Surgery 65 (4Suppl): 20�26.
Taylor KM, Bain WH, Maxted KJ, Hutton MM, McNab WY & Caves PK (1978) Comparative stud-ies of pulsatile and nonpulsatile flow during cardiopulmonary bypass. I. Pulsatile system em-ployed and its hematologic effects. Journal of Thoracic & Cardiovascular Surgery 75 (4): 569�573.
Thurlow PJ, Doolan L, Sharp R, Sullivan M, Smith B & Andersen LW (1995) Studies of the effectof Pall leucocyte filters LG6 and AV6 in an in vitro simulated extracorporeal circulatory system.Perfusion 10 (5) 291�300.
Tonnesen E, Christensen VB & Toft P (1996) The role of cytokines in cardiac surgery. InternationalJournal of Cardiology 53 (Suppl): 1�10.
Tortella FC, Britton P, Williams A, Lu XC & Newman AH (1999) Neuroprotection (focal ischemia)and neurotoxicity (electroencephalographic) studies in rats with AHN649, a 3-amino analog ofdextromethorphan and low-affinity N-methyl-D-aspartate antagonist. Journal of Pharmacology &Experimental Therapeutics 291 (1): 399�408.

63
Tossman U & Ungerstedt U (1986) Microdialysis in the study of extracellular levels of amino acidsin the rat brain. Acta Physiol Scand 128 (1): 9�14.
Tsuji M, Higuchi Y, Shiraishi K, Kume T, Akaike A & Hattori H (2000) Protective effect of amino-guanidine on hypoxic-ischemic brain damage and temporal profile of brain nitric oxide in neonatalrat. Pediatric Research 47 (1): 79�83.
Umemura K, Shimakura A & Nakashima M (1997) Neuroprotective effect of a novel AMPA receptorantagonist, YM90K, in rat focal cerebral ischaemia. Brain Research 773 (1�2): 61�65.
Usui A, Abe T, Murase M, Tanaka M, Takeuchi E, Ishihara T, Hoshino M, Ogawa Y, Seki A, Okamo-to H & Moriya H (1997) Early experience of retrograde cerebral perfusion. Cardiovascular Sur-gery 5 (5): 510�515.
Van de Watering LM, Hermans J, Houbiers JG, van den Broek PJ, Bouter H, Boer F, Harvey MS,Huysmans HA & Brand A (1998) Beneficial effects of leukocyte depletion of transfused blood onpostoperative complications in patients undergoing cardiac surgery: a randomized clinical trial.Circulation 97 (6): 562�568.
Warner DS, Martin H, Ludwig P, McAllister A, Keana JF & Weber E (1995) In vivo models of cer-ebral ischemia: effects of parenterally administered NMDA receptor glycine site antagonists.Journal of Cerebral Blood Flow & Metabolism 15 (2): 188�96.
Wasterlain CG, Adams LM, Wichmann JK & Sofia RD (1996) Felbamate protects CA1 neurons fromapoptosis in a gerbil model of global ischemia. Stroke 27 (7): 1236�1240.
Veeragandham RS, Hamilton IN, Jr., O'Connor C, Rizzo V & Najafi H (1998) Experience with ante-grade bihemispheric cerebral perfusion in aortic arch operations. Annals of Thoracic Surgery 66(2): 493�499.
Wennberg R, Quesney F, Olivier A & Dubeau F (1997) Induction of burst-suppression and activationof epileptiform activity after methohexital and selective amygdalo-hippocampectomy. Electroen-cephalography & Clinical Neurophysiology 102 (5): 443�451.
Westaby S, Johnsson P, Parry AJ, Blomqvist S, Solem JO, Alling C, Pillai R, Taggart DP, GrebenikC & Stahl E (1996) Serum S100 protein: a potential marker for cerebral events during cardiopul-monary bypass. Annals of Thoracic Surgery 61 (1): 88�92.
Wilde JT (1997) Hematological consequences of profound hypothermic circulatory arrest and aorticdissection. Journal of Cardiac Surgery 12 (2 Suppl): 201�206.
Williams GT & Smith CA (1993) Molecular regulation of apoptosis: genetic controls on cell death.Cell 74 (5): 777�779.
Wilson IC, Gardner TJ, DiNatale JM, Gillinov AM, Curtis WE & Cameron DE (1993) Temporaryleukocyte depletion reduces ventricular dysfunction during prolonged postischemic reperfusion.Journal of Thoracic & Cardiovascular Surgery 106 (5): 805�810.
Zipfel GJ, Lee JM & Choi DW (1999) Reducing calcium overload in the ischemic brain. New Eng-land Journal of Medicine 341 (20): 1543�1544.