betsy baron, ba irb administrator office of research compliance stony brook university introduction...
TRANSCRIPT

Betsy Baron, BAIRB AdministratorOffice of Research ComplianceStony Brook University
• Introduction to the IRB Application Process at Stony
Brook University
• Overview of IRBNet Submission Process
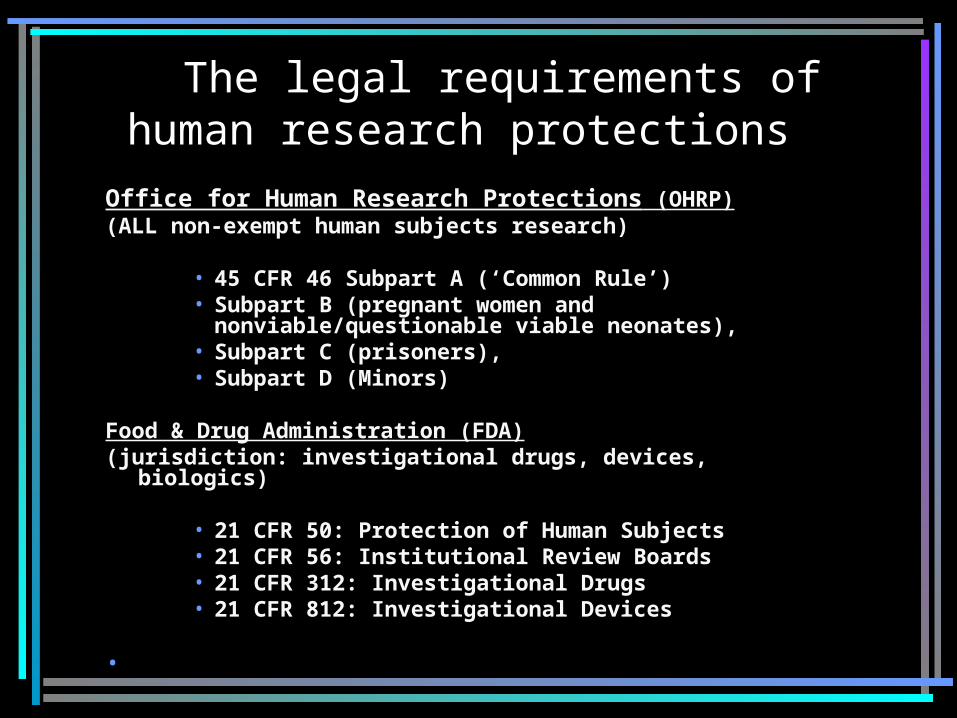
The legal requirements of human research protections
Office for Human Research Protections (OHRP)(ALL non-exempt human subjects research)
• 45 CFR 46 Subpart A (‘Common Rule’) • Subpart B (pregnant women and
nonviable/questionable viable neonates), • Subpart C (prisoners), • Subpart D (Minors)
Food & Drug Administration (FDA)(jurisdiction: investigational drugs, devices,
biologics)
• 21 CFR 50: Protection of Human Subjects • 21 CFR 56: Institutional Review Boards• 21 CFR 312: Investigational Drugs • 21 CFR 812: Investigational Devices
•

Institutional Review Boards
• Federally mandated• Locally, called Committees on
Research Involving Human Subjects (CORIHS)
• Must – facilitate safe and ethical research – ensure that human subjects’ rights and
welfare are protected– ensure compliance with federal regulations
governing the protection of human subjects including 45 CFR 46 and 21 CFR 50, 56 etc.

5 Possible Routes for IRB Review at SBU
SBU’s Committees on Research Involving Human Subjects (CORIHSa, CORIHSb)– Identical Charge (not ‘just SBS’ or ‘just ‘Biomed’)– Each has one deadline and meeting date per month
The Commercial (“Central”) IRB Option: – Chesapeake Research Review, Inc. (CRRI)
(Option is available only for Industry sponsored studies)
The National Cancer Institute Central IRB Option:– NCI’s Pediatric and Adult Central IRB’s– (available for certain oncology group trials: ECOG, GOG, COG,
NSABP, etc)

(OHRP)
IRBs review research…
– A systematic investigation including research development, testing and evaluation designed to contribute to generalizable knowledge
that involves human subjects…
– The actual human being, and/or– Identifiable human tissue, and/or– Identifiable human data

Clinical ResearchPatient Subject
What’s the difference between these two humans?

When is the activity NOT research involving human subjects?
• Clinical Practice is not research
• Case Reports are not research
• Hospital-endorsed QA/QI activities are not research (as long as they do not meet the definition of research)

Which coat are you wearing?
• White coat on!
• Do no harm
• Treat the individual patient
• Convince the patient that the doctor knows ‘what’s best’
• White coat off!
• Do good (risk/benefit)
• Follow the study protocol and obtain data to answer the study question
• Convince the patient that you do not know ‘what’s best’: EQUIPOSE

The ‘application route’ you take depends on the level of risk and federally-determined
review category

• If no foreseeable risk and fits into an exemption review category, the ‘exempt’ application is completed. Review conducted by the ‘Institution’ (ORC staff)

If minimal risk, and fits into an expedited review category, the ‘full’ application is completed, but:
–Review conducted by 1 IRB member.
•
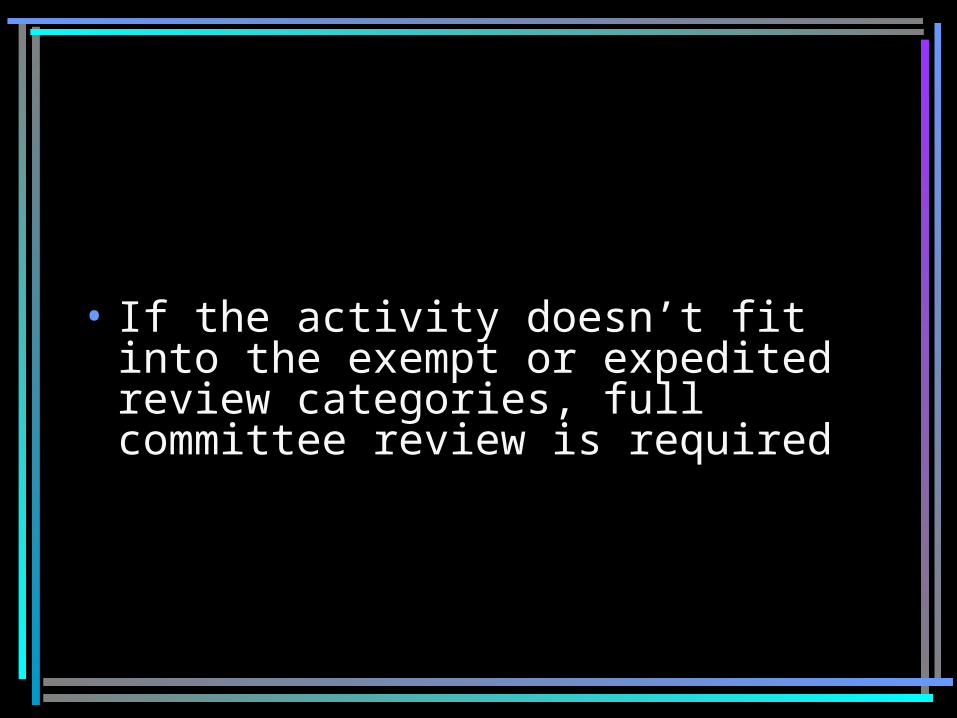
• If the activity doesn’t fit into the exempt or expedited review categories, full committee review is required

Once approval is granted:
• It is granted for no longer than one year• The study must be conducted exactly as approved
by the IRB. The only exception is where subject safety requires a change in protocol.
• Informed consent must be obtained (unless waived by the IRB) using ONLY the consent document(s) approved by CORIHS (i.e., displaying the CORIHS approval stamp)
• You must keep the IRB informed of:– Any proposed changes to any aspect of the protocol, and
you must receive approval before implementing them– Anything that happens relative to the study that is
unanticipated, and affects the risks to subjects or others

Procedures they think will answer the research hypothesis
Populations they think were correctly selected
Benefits they think subjects may experience
Risks they think subjects may experience
Researchers submit Proposals to IRBs…with best guesses as to…

Unanticipated Problems (UPs) involving risks to subjects or others must be reported to the IRB:
Refer to any problem, event, or new information that:
• Is unexpected (in terms of nature, severity, or frequency) given the research procedures that are described in the protocol-related documents, such as the CORIHS-approved research protocol and informed consent documents; and the characteristics of the subject population being studied; and
• Related or possibly related (reasonable possibility) to participation in the research, and
• Indicates that subjects or others are at a greater risk of harm (including physical, psychological, economic, or social harm) than was previously known or recognized.
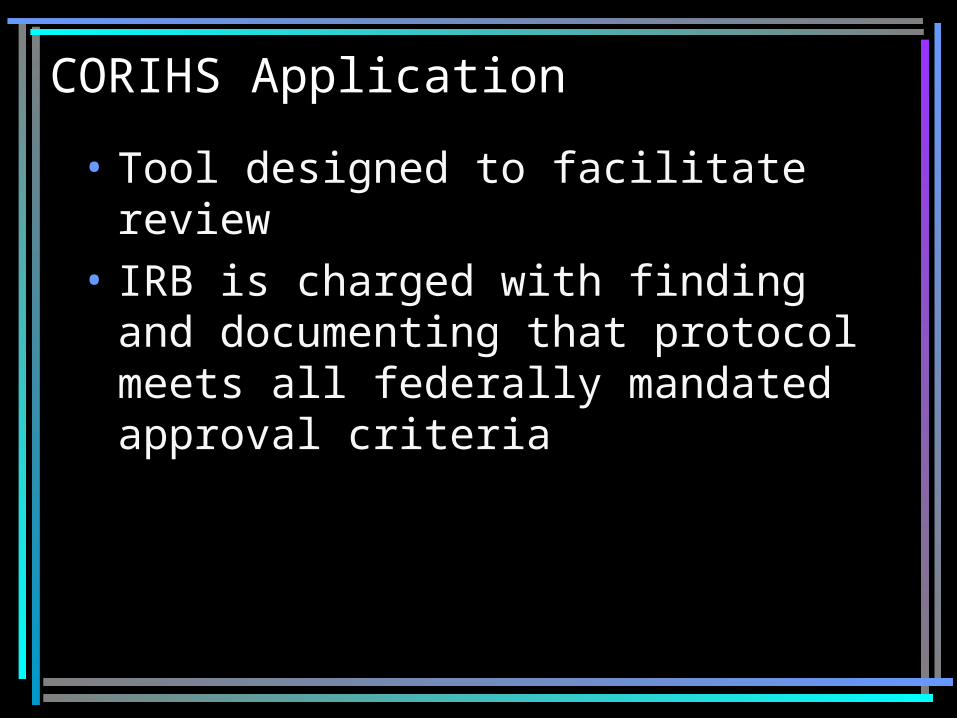
CORIHS Application
• Tool designed to facilitate review• IRB is charged with finding and
documenting that protocol meets all federally mandated approval criteria

Informed Consent: Respect for Persons

The first step in the consent process
Recruitment (Truth in Advertising)

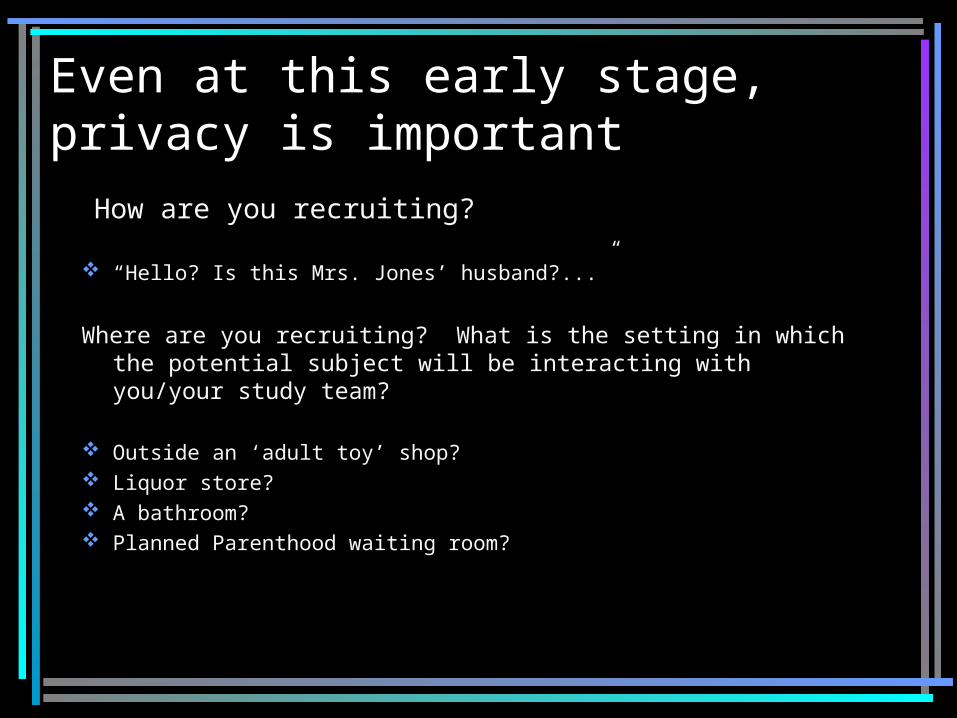
Even at this early stage, privacy is important
How are you recruiting? “Hello? Is this Mrs. Jones’ husband?...”
Where are you recruiting? What is the setting in which the potential subject will be interacting with you/your study team?
Outside an ‘adult toy’ shop? Liquor store? A bathroom? Planned Parenthood waiting room?

Before you sit down to initiate the consent process with a prospective subject…
• Do you know the protocol COLD, including the inclusion/exclusion criteria and recruitment parameters?
Do you have the time to consent the prospective subject properly? (the more risky or complicated the protocol, the more time needs to be taken for the consent process)
Do you speak the language of the prospective subject?

and…
…How do you know if the person in front of you is even capable of making the decision to participate in your research?

Capacity to consent=a demonstrated appreciation:
1. that the activity is research, not standard treatment
2. of the risks and benefits of a study
3. of the alternatives that are available if s/he does not participate
4. that, if s/he chooses not to participate, this decision will be accepted without penalty, i.e., without jeopardizing e.g., status or clinical care

Capacity to consent…cont.
The potential subject needs to demonstrate an ability to use
this information in a rational manner, applying these factors appropriately.
‘Yeah, whatever’
vs.
‘YES! YES! A THOUSAND TIMES, YES!’

Ready?
• Adults consent for themselves
• Parents give permission for their children
• Children assent for themselves
And, where the IRB permits the inclusion of adult subjects who cannot consent for themselves,
• Surrogates give permission for the subject, and whenever possible, the subject themselves assent.

Consent Form vs. Consent Process
Is the signing of the consent form a monumental event?
*but if the IRB approved documented consent, you better have them for every subject.
A signed consent form is proof that…a consent form was
signed. Big whoop.*
The Informed Consent Process
Repeat after me: Your mother, your father, your
aunt, your uncle…
Assess the capacity to consent of ALL individuals
Give the subject TIME to decide
‘I won’t be your doctor if you don’t consent to this’
Me falta práctica en Español!!!

What information MUST be included to ensure informed consent?
• Statement: The study involves research, not standard of care.
• Purpose: Why is this study being done? • Duration: How long does the subject have to
be in this study? • Procedures: What will the subject have to do
that s/he wouldn’t otherwise have to do if s/he wasn’t a subject?

And…
• What are the reasonably foreseeable risks or discomforts the subject may experience?
• What are the benefits to the subject that s/he may reasonably expect from participation?

And…
• If the person says no to being in this study, what other procedures or treatments are available to him/her?
• Will the subject’s participation in this study be kept confidential? What steps will be taken to make sure his/her confidentiality will be preserved?

And…
• If the subject gets injured while on study…
• Who does s/he tell?• Where should s/he go?• Who will pay for any treatment or hospitalization?
• Who can the subject speak with if s/he has any questions about the study?
• Who can the subject speak with if s/he has a question about his/her rights as a research subject?

And finally, the best part…
The subject doesn’t have to be in this study if she doesn’t want to be.
• S/he can say no, and there will be no penalty whatsoever.
• S/he can say yes now, and then change his/her mind without penalty and without giving a reason.

What information MAY be included to ensure informed consent?
• risks to the subject which are currently unforeseeable (should be included when testing investigational drugs);
• Why a subject's participation may be terminated by the investigator;
• Costs to participation;• Consequences of withdrawal • the approximate number of subjects involved
in the study.

And finally, the subject must be assured that…
New Information that is discovered during the course of the research which may relate to the subject's willingness to continue to be in the study will be provided to the subject

IRB Mandate: Vulnerable Populations45 CFR 46.111 (b)
When some or all of the subjects are likely to be vulnerable to coercion or undue influence, such as children… additional safeguards have been included in the study to protect the rights and welfare of these subjects.
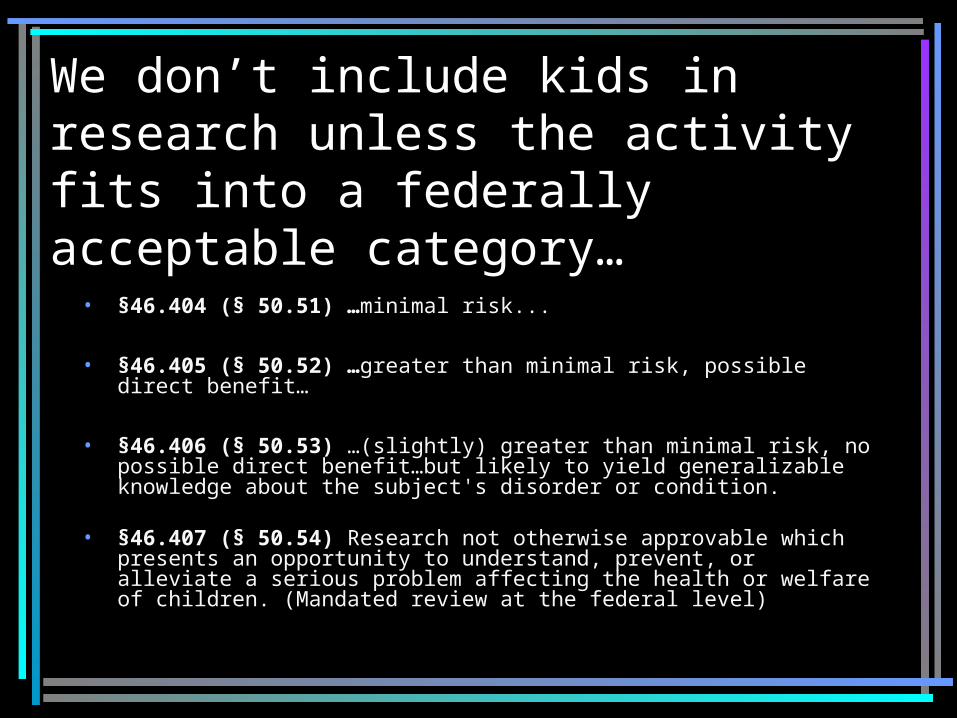
We don’t include kids in research unless the activity fits into a federally acceptable category…
• §46.404 (§ 50.51) …minimal risk...
• §46.405 (§ 50.52) …greater than minimal risk, possible direct benefit…
• §46.406 (§ 50.53) …(slightly) greater than minimal risk, no possible direct benefit…but likely to yield generalizable knowledge about the subject's disorder or condition.
• §46.407 (§ 50.54) Research not otherwise approvable which presents an opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of children. (Mandated review at the federal level)

We don’t include kids unless we get parental permission, except when…
…the IRB determines that parental or guardian permission is not a reasonable requirement to protect the subjects (for example, neglected or abused children).

We don’t include kids unless we get their assent, except when…
– the capability of some or all of the children is so limited that they cannot reasonably be consulted
Or – the intervention or procedure involved in the
research holds out a prospect of direct benefit that is important to the health or well-being of the children and is available only in the context of the research,

If the IRB requires assent, it means:
Obtaining Obtaining verbal assent documented
assent starting at age 8-9 starting at age
11-12
AND:If the child does not assent, the child is not enrolled in the study.

How do we (i.e., IRB and YOU) additionally protect adult subjects who cannot consent for themselves?
• Do such individuals have to be in the research study? JUSTICE!
•Does the research involve minimal risk, or more than minimal risk, with possibility of benefit?
•Are ALL or SOME unable to consent?

Other Vulnerable Populations
• Prisoners• Pregnant Women• Fetuses• Questionably Viable Neonates• Non-Viable Neonates• Elderly• Economically/Educationally
Disadvantaged• Students• Subordinates• Those who have just received a medical
diagnosis…

If PHI is being used or generated…
IPAA Impacts Research Activities

HIPAA AUTHORIZATION
• Authorization can be addressed in ONE of the following ways: – Signed consent document (must
contain required HIPAA authorization language)
– De-identified data– Limited Data Set (LDS)

IRBNet: www.irbnet.org
• Select a library – Human Subjects• Instructions• Submission requirements• Registration form• Application forms• Consent form templates• Protocol template

IRBNet (con’t)
• Share with study team members• Obtain required signatures• Ensure all required materials are
uploaded• Submit for review!
Review time
• Allow 2-3 weeks for initial review