basil rigas and khosrow kashfi division of cancer...
TRANSCRIPT

JPET #80564
1
Cancer prevention: A new era beyond COX-2
Basil Rigas and Khosrow Kashfi
Division of Cancer Prevention, Department of Medicine, SUNY at Stony Brook, Stony Brook, NY
11794-5200 (BR); Department of Physiology and Pharmacology, City University of New York
Medical School, New York, NY 10031 (KK)
JPET Fast Forward. Published on April 1, 2005 as DOI:10.1124/jpet.104.080564
Copyright 2005 by the American Society for Pharmacology and Experimental Therapeutics.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
2
Running Title: Cancer prevention beyond COX-2
Corresponding authors:
Basil Rigas, MD
Division of Cancer Prevention, Department of Medicine
SUNY at Stony Brook
Stony Brook, NY 11794-8160
Tel: (631) 632-9035; Fax: (631) 632-1992; e-mail: [email protected]
Number of:
Text pages: 32
Tables: 1
Figures: 2
References: 65
Words in the Abstract: 238
Words in Text: 5,685
Abbreviations: NSAIDs, nonsteroidal anti-inflammatory drugs; COX, cyclooxygenase; LOX,
lipoxygenase; PG, prostaglandin; FAP, familial adenomatous polyposis
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
3
ABSTRACT
The seminal epidemiological observation that nonsteroidal anti-inflammatory drugs (NSAIDs)
prevent colon and possibly other cancers has spurred novel approaches to cancer prevention.
The known inhibitory effect of NSAIDs on the eicosanoid pathway prompted studies focusing on
cyclooxygenase (COX) and its products. The increased PGE2 levels and the overexpression of
COX-2 in colon and many other cancers provided the rationale for clinical trials with COX-2
inhibitors for cancer prevention or treatment. Their efficacy in the prevention of sporadic colon
and other cancers remains unknown; one COX-2 inhibitor has been withdrawn due to side
effects and there are concerns about whether this effect is class-specific. There is evidence to
suggest that COX-2 may not be the only or the ideal eicosanoid pathway target for cancer
prevention. Six sets of observations support this notion: the relatively late induction of COX-2
during carcinogenesis; the finding that NSAIDs may not require inhibition of COX-2 for their
effect; the modest effect of coxibs in cancer prevention; that currently available coxibs have
multiple non-COX-2 effects which may account for at least some of their efficacy; the possibility
that concurrent inhibition of COX-2 in non-neoplastic cells may be harmful; and the possibility
that COX-2 inhibition may modulate alternative eicosanoid pathways in a way promoting
carcinogenesis. Given the limitations of COX-2 specific inhibitors and the biological evidence
mentioned above, we suggest that targets other than COX-2 should be pursued as alternative
or complementary approaches to cancer prevention.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
4
Cancer, a major medical challenge during the last 100 years, has touched the life of
almost every American family and absorbs an inordinate amount of health care resources. In
the US cancer has moved to the first place among the causes of death, surpassing
cardiovascular disease. That cancer prevention is better than treatment is almost axiomatic in
medicine. In contrast to cardiovascular and infectious diseases, whose prevention has had a
substantial impact on their associated morbidity and mortality, gains in cancer prevention have
been limited. The main reasons for this can be found in two areas: identification of informative
biomarkers and development of safe and effective chemopreventive agents. In this
commentary, we present a brief overview of current efforts to develop effective
chemopreventive drugs focused on cyclooxygenase (COX) inhibition and, taking a rather
iconoclastic approach, we suggest that a search for alternative targets for drug development
may be in order.
The clear message of NSAIDs for cancer prevention and its limitations
The year 1988 marked a watershed point for cancer prevention; it was then that the first
epidemiological demonstration that nonsteroidal anti-inflammatory drugs (NSAIDs) prevent
human colon cancer established the feasibility of human cancer chemoprevention (Kune et al.,
1988). That long-term use of NSAIDs prevents an array of cancers, is now firmly established.
This conclusion is based on two sets of data: a) epidemiological studies, which document an
association between NSAID use and cancer risk; and b) interventional clinical trials
demonstrating that the administration of NSAIDs can actually prevent cancer. The
epidemiological studies reported to date (Thun et al., 2002), describing collectively results on >1
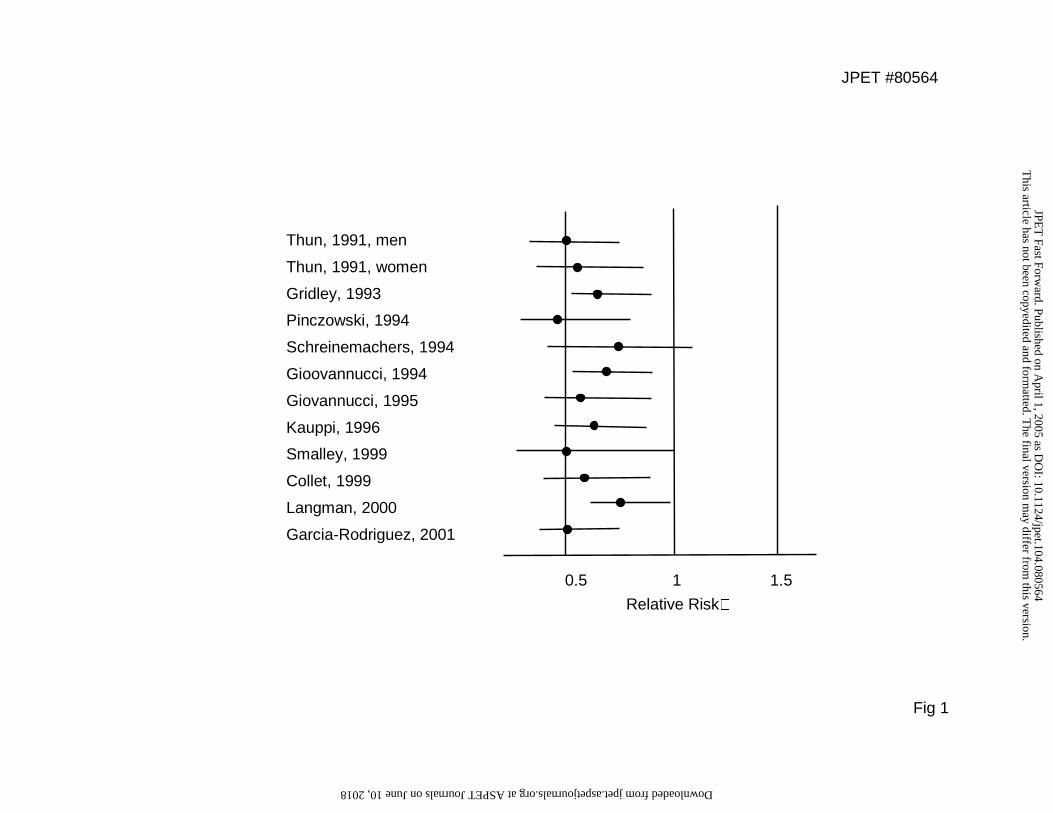
million subjects, have pointed out the protective effect of NSAIDs against colon (Fig. 1),
esophageal, gastric, breast (estrogen receptor positive) and perhaps pancreatic and ovarian
cancers. For colon cancer, two randomized interventional studies using polyp recurrence as a
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
5
general end point demonstrated the preventive effect of aspirin (Baron et al., 2003; Sandler et
al., 2003). The relative risks following administration of aspirin ranged between 0.59 and 0.96,
depending on the specific end point and aspirin dose. Several aspects of this effect appear
unclear at this point, but the overall significance of these landmark studies proving the general
concept of chemoprevention by NSAIDs cannot be overstated.
What these studies have failed to provide is the details of the big picture: which one of
the nearly 30 NSAIDs is the most effective, what is the optimal dose and what is the ideal
schedule of administration? Even if such information existed, however, it would probably be of
limited or no practical value. The reason is that, although we have unassailable proof of
principle for chemoprevention, current NSAIDs cannot overcome two prohibitive limitations
concerning their safety and efficacy. For example, for colon cancer, the one most thoroughly
studied, NSAIDs can prevent at best 50% of the cases (Thun et al., 2002). Although no precise
numbers are available for the incidence of the side effects of NSAIDs, it seems that among
patients using NSAIDs, up to 4% per year suffer serious gastrointestinal complications
(Bjorkman, 1999). The issue of safety is clearly underscored by the report that in 1998 in the US
the number of deaths from NASAID-induced gastrointestinal complications was virtually equal to
the number of deaths from AIDS (Singh and Triadafilopoulos, 1999).
These considerations should be viewed against the fundamental distinction between
chemotherapy and chemoprevention. In chemotherapy both patient and physician are prepared
(and justified) to accept substantial treatment-related toxicity to save the patient’s life from a fully
developed cancer. In contrast, chemoprevention agents will be often prescribed to a healthy
subject for a cancer he/she may never develop. Thus, safety and efficacy assume a different
value and criteria for them ought to be more demanding than those applied to chemotherapy.
The ideal chemopreventive agent ought to have an efficacy approaching 100% and a safety
profile that is nearly perfect. Such perfection is, however, probably unattainable and the
challenge may be simply to define what would constitute an acceptable deviation from the ideal
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
6
compound. Agents with imperfect performance will be judged on the basis of their individual
characteristics and against competing agents. Aspirin, for example, is widely employed for the
prevention of cardiovascular thrombotic events, although its efficacy is far from perfect (e.g. it is
only 28% for primary prevention) and the side effects even of the low dose of 81 mg daily are
not insignificant [reviewed in (Kimmey, 2004)].
Efficacy, safety, cost and ease of administration will likely be the main criteria for
candidate chemoprevention agents. A fifth criterion that may be employed in the future is the
degree of risk of developing a given cancer. It is quite possible that genetic testing will be able
to quantify risk as well as the likelihood for a given subject to respond to an agent, thus making
possible individualized cancer prevention. Whether articulated or not, considerations of safety
end efficacy have spurred the search for a “better NSAID”, with coxibs, selective inhibitors of
COX-2, being the most notable outcome.
COX-2 as a target for cancer prevention: Pros and cons
The development of coxibs has been based on the notion that inhibition of COX-2, the
induced isoform of COX, will diminish the proinflammatory activities of COX, whereas sparing
COX-1, the constitutive isoform of COX, will diminish the gastrointestinal and perhaps other side
effects of nonsteroidal anti-inflammatory drugs (Simmons et al., 2004). The observation that
COX-2 is overexpressed in cancer led to the idea that coxibs could serve as excellent
chemoprevention agents. The pros and cons for this concept are discussed below.
The pros
The best known biochemical effect of NSAIDs is inhibition (suicide or competitive) of
COX, an important enzyme in the eicosanoid cascade that ultimately leads to prostaglandins
(PGs) and related compounds (Simmons et al., 2004). This provided an immediate and
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
7
plausible explanation of the epidemiological data on NSAIDs and cancer prevention. To
substantiate this conjecture, we determined the levels of various eicosanoids in human colon
cancers showing that the most striking abnormality was elevated PGE2 levels compared to
uninvolved mucosa, confirming earlier findings by Bennett et al, who had used somewhat crude
analytical methodologies (Bennett et al., 1987; Rigas et al., 1993). Subsequently, DuBois and
colleagues demonstrated overexpression of COX-2 in 45% of colon adenomas and 85% of
colon carcinomas (Eberhart et al., 1994). Overexpression of COX-2 has been demonstrated for
several human cancers, besides colorectal adenocarcinomas , including gastric, breast, lung,
esophageal and hepatocellular carcinomas (Turini and DuBois, 2002). Given the potentially
enormous significance of this finding, a recent study assessed the possibility that single
nucleotide COX-2 polymorphisms may be associated with disease or with individual responses
to drug therapies. Lin et al have reported reduced an association between the COX-2 V511A
polymorphism and reduced susceptibility to colon cancer in African Americans (Lin et al., 2002).
Sequencing of the COX-2 gene of 72 individuals identified rare polymorphisms, including the
V511A polymorphism which, however, was not functionally important (Fritsche et al., 2001).
Neither hypothesis, that polymorphism in the COX-2 gene could contribute to individual
susceptibility to cancer nor that COX-2 polymorphisms may have an impact on individual
response to NSAIDs, appears to be supported. Polymorphisms in other genes of the eicosanoid
pathway may account for inter-individual differences in cancer susceptibility or response to
NSAIDs.
In terms of mechanism, we showed that PGE2 increased colon cancer cell proliferation
(Qiao et al., 1995), while DuBois’ group showed that it suppressed apoptosis (Sheng et al.,
1998). The role of eicosanoids in carcinogenesis has been further expanded by studies
demonstrating that in certain cases, lipoxygenase (LOX) products may also play a role in
carcinogenesis (Shureiqi and Lippman, 2001) (Fig 2). It was, therefore, reasonable to conclude
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
8
that inhibition of COX-2 would arrest carcinogenesis and thus would a) prevent cancer
development, and b) regress cancer once developed.
A series of detailed cell culture, animal studies and published clinical trials on the use of
coxibs in patients with familial adenomatous polyposis (FAP) and in patients with cancer,
culminated in one COX-2 inhibitor, celecoxib, receiving FDA approval for cancer prevention in
patients with FAP.
The supportive animal studies Two sets of animal studies support the role
of COX-2 in carcinogenesis a) studies using genetically modified animals, and b) studies using
pharmacological inhibitors of COX-2. They are summarized below.
Although COX enzymes may not be acting simply as oncogenes in tumor development
[reviewed in (Simmons et al., 2004)], several studies have indicated that they may be required
for tumorigenesis. Oshima et al showed that deletion of COX-2 decreased significantly the
number of intestinal tumors in Apc 716 mice (Oshima et al., 1996). Deletion of COX-1, however,
also attenuated tumor formation in the same mice (Chulada et al., 2000). A stronger case was
made by the studies of Hla’s group, who showed that multiparous but not virgin female
transgenic mice that overexpress the human COX-2 gene in the mammary glands exhibited a
greatly exaggerated incidence of focal mammary gland hyperplasia, dysplasia, and
transformation into metastatic tumors (Liu et al., 2001). The clear implication from these data is
that enhanced COX-2 expression is sufficient to induce mammary gland tumorigenesis. Studies
have also addressed whether COX-2 is critical for skin carcinogenesis. Overexpression of COX-
2 in basal epidermal cells of transgenic mice (using the keratin 5 promoter), which leads to high
levels of epidermal PGs, is insufficient for tumor induction but sensitizes the tissue to genotoxic
carcinogens (Muller-Decker et al., 2002). In contrast, transgenic mice that overexpress COX-2
(under control of the keratin 14 promoter), in the epidermis and some other epithelia, developed
tumors at a much lower frequency than did their littermate controls; tumors were induced by an
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
9
initiation/promotion protocol (Bol et al., 2002). The latter results raised questions regarding the
role of COX-2 in skin tumor development.
There have been numerous animal studies using specific COX-2 inhibitors providing
support for the concept that COX-2 inhibition both prevents and regresses tumors arising from a
variety of tissues, including colon, lung, breast, pancreas, skin and others [reviewed in
(Dannenberg and Subbaramaiah, 2003)]. Perhaps the most dramatic result was reported by
Reddy’s group who studied the effect of celecoxib on colon carcinogenesis in the azoxymethane
rat model. Remarkably, dietary administration of celecoxib inhibited both the incidence and
multiplicity of colon tumors by about 93% and 97%, respectively. It also suppressed the overall
colon tumor burden by more than 87%.
Taken together these studies (along with many more not reviewed here) provide a
spectrum of evidence for the role of COX-2 in carcinogenesis, ranging from the compelling
(breast carcinogenesis) to the rather weak and controversial (skin carcinogenesis). Overall, the
evidence from animal studies suggests strongly that COX enzymes, particularly COX-2,
participate in a significant way in carcinogenesis.
The cons
There are six sets of observations that challenge the intellectual beauty and utter
simplicity of the notion that COX-2 is central to the pathogenesis of several cancers, and that its
inhibition would prevent them and regress those already established. They include a) the
relatively late induction of COX-2 during carcinogenesis; b) the finding that NSAIDs, which
prevent cancer, may not require inhibition of COX-2 for their effect; c) the modest effect of
coxibs in cancer prevention; d) the fact that currently available coxibs have multiple non-COX-2
effects which may account for at least some of their efficacy; e) the possibility that concurrent
inhibition of COX-2 in non-neoplastic cells may be harmful; and f) the possibility that COX-2
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
10
inhibition may modulate alternative eicosanoid pathways in a manner that promotes
carcinogenesis. Below, we discuss briefly these points as well as the potential role of the large
family of eicosanoids in carcinogenesis.
Pattern of COX-2 expression The pattern of COX-2 expression during colon
carcinogenesis does not entirely fit the model that COX-2 is central to carcinogenesis. There is
no COX-2 expression in human aberrant crypt foci, the earliest recognizable premalignant
lesion in the colon (Nobuoka et al., 2004). For COX-2 to be the ideal chemoprevention target, its
expression in aberrant crypt foci would be highly advantageous, if not a requirement, as
chemoprevention would be easiest at this stage of lower complexity. Moreover, the expression
of COX-2 commences only at the adenoma stage and in less than half of them, increasing as
the transition to cancer progresses and becoming positive in 85% of them (Eberhart et al.,
1994).
This pattern of COX-2 overexpression is subject to an alternative interpretation, namely
that COX-2 expression is the result of and not a dominant contributor to carcinogenesis. The
strongest argument against this interpretation are the animal studies demonstrating that
disruption of either the COX-1 or COX-2 gene reduces the development of tumors in mice.
Interestingly, Chulada et al proposed that since COX-1 (as well as COX-2) plays a key role in
intestinal tumorigenesis it may also be a chemotherapeutic target for NSAIDs (Chulada et al.,
2000). A counterargument to this is the recently reported study in which targeted
overexpression of human microsomal PGE synthase-1 (mPGES) in the alveolar type II cells of
transgenic mice, accompanied by highly elevated PGE2 production (12.2 fold over control), was
not sufficient to induce lung tumors (Blaine et al., 2004).
An interesting idea has been advanced by Takeda et al who demonstrated that most
COX-2-expressing cells in the polyps of Apc∆716 mice are stromal fibroblasts in which COX-1,
COX-2 and mPGES colocalized. COX-2 was induced only in polyps >1 mm in diameter, but
COX-1 was found in polyps of any size. Based on these data, they proposed that COX-1
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
11
expression in the stromal cells secures the basal level of PGE2 that can support polyp growth to
~1 mm, and that simultaneous inductions of COX-2 and mPGES support the polyp expansion
beyond ~1 mm by boosting the stromal PGE2 production (Takeda et al., 2003).
COX-independent effects of NSAIDs At variance with the notion that COX-2 has
a central role in carcinogenesis is the concept, originally proposed by us, that NSAIDs do not
require the presence of COX-2 to prevent cancer (Hanif et al., 1996). This was based on the
finding that in vitro NSAIDs display effects compatible with cancer prevention such as inhibition
of cell proliferation, induction of apoptosis, inhibition of angiogenesis and many others, in the
absence of COX-1 or -2. This initial observation is now firmly established by many studies
pointing to an array of molecular targets affected by NSAIDs (Shiff and Rigas, 1999). It is
unclear which one(s), if any, of them is the pathway(s) mediating the chemopreventive effect of
NSAIDs. Quite astonishing in this regard has been our recent observation that a modified
aspirin inhibited cell proliferation, induced apoptosis, inhibited NF-κB activation and Wnt
signaling while inducing COX-2 expression without affecting COX-1 expression (Williams et al.,
2003). Of note, such COX-2 was catalytically competent as evidenced by the proportional
increase in PGE2 production.
A reservation concerning the non-COX effects of NSAIDs is that they occur only at
“industrial strength” concentrations of NSAIDs (Marx, 2001). This is not, however, entirely
accurate or necessarily relevant. For example, some NSAIDs, have IC50s for the inhibition of
colon cancer cell growth in the µM range, with probably sulindac sulfide, the active metabolite of
sulindac, having the lowest (about 175 µM) and ASA the highest (2.5 mM); the IC50 for sulindac
approaches 1 mM (Shiff et al., 1995). The corresponding IC50 for some COX-2 inhibitors
appears to be around 30 µM (Grosch et al., 2001). Thus there is a clear discrepancy between
cell culture results and pharmacological efficacy. These observations prompt a reminder of the
limitations of the cell culture system, whose findings should be viewed for what they are: a mere
indication of what may happen in the complexity of the human organism. For example, as
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
12
discussed below, sulindac is more effective in the prevention of colon cancer in patients with
FAP than coxibs, a complete reversal of the conclusions that might have been reached from the
cell culture data. It should also be kept in mind that cell culture systems are by their very nature
time sensitive. Most studies using cell culture systems have to be terminated by 72 to 96 hours
and thus high concentrations of an agent are at times necessary to detect an effect. In contrast
in in vivo chemoprevention studies comparatively lower doses of an agent are administered for
much longer periods of time and the effect of the agent may thus become apparent. The “low
dose - long duration” or “high dose - short duration” balance needs to be considered so that
potentially useful agents are not abandoned unjustifiably.
The limited clinical efficacy of coxibs When the COX-2 inhibitor celecoxib
was used in FAP, suppression of the neoplastic process was modest. After six months, FAP
patients receiving 400 mg of celecoxib twice a day had a 28.0% reduction in the mean number
of colorectal polyps (P=0.003 for the comparison with placebo) and a 30.7% reduction in the
polyp burden (the sum of polyp diameters) (P=0.001), as compared with reductions of 4.5% and
4.9%, respectively, in the placebo group (Steinbach et al., 2000). Rofecoxib had a statistically
significant but marginal effect on the number of polyps in FAP patients (6.8% reduction from
baseline values) (Higuchi et al., 2003). Another study assessed the effect of celecoxib on
duodenal polyps in FAP patients (Phillips et al., 2002). There was a trend to improvement in
duodenal disease when absolute adenoma numbers were assessed but this was not statistically
significant. However, when taking a global overview of the state of the duodenum, overall,
patients taking celecoxib 400 mg twice daily for 6 months had a 14.5% reduction in involved
areas compared with a 1.4% for placebo (p=0.436). However, patients with clinically significant
disease at baseline (greater than 5% covered by polyps) showed a 31% reduction in involved
areas with celecoxib 400 mg twice daily compared with 8% on placebo (p=0.049)
These results could be interpreted in one of two ways: either COX-2 is not central to the
neoplastic process or celecoxib is not a sufficiently strong COX-2 inhibitor. However, all
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
13
available clinical and preclinical evidence, suggests that it is a strong COX-2 inhibitor [reviewed
in (Dannenberg and Subbaramaiah, 2003)]. Of interest, the dose required to inhibit COX-2
activity by 80% also causes a 60% inhibition in COX-1 activity (Warner et al., 1999).
Sulindac, an NSAID, had a pronounced effect on colorectal polyps in FAP patients. A
statistically significant decrease in the mean number of polyps and their mean diameter
occurred in patients treated with sulindac (150 mg orally twice a day), as compared with those
given placebo. When treatment was stopped at nine months, compared to baseline values, the
number of polyps had decreased by 56% and the diameter of the polyps by 65% (Giardiello et
al., 1993). Furthermore, Winde et al performed a pilot study using rectally applied sulindac in
colectomized FAP patients (Winde et al., 1995). All patients responded to therapy within 6 to 24
weeks. Sixty and 87% of patients achieved complete adenoma reversion after 12 months at 53
and 67 mg of sulindac per day per patient on average, respectively. Reversion was evident
compared with the control group. Tissue PGE2 levels were greatly reduced. It has also been
shown that long-term use of sulindac is effective in reducing polyp number and preventing
recurrence of higher-grade adenomas in the retained rectal segment of most FAP patients
(Winde et al., 1997; Cruz-Correa et al., 2002). Although the coxib and sulindac studies are not
directly comparable, given the different duration of drug administration, it appears that sulindac
may have a stronger effect than coxibs, at least compared to rofecoxib. These findings suggest
that inhibition of targets other than or in addition to COX-2, may contribute to sulindac’s stronger
chemopreventive effect in the colon.
Another disappointing result, although not directly related to chemoprevention, was the
recently reported failure of celecoxib combined with trastuzumab to have an effect on patients
with HER2/neu-overexpressing, trastuzumab-refractory metastatic breast cancer (Dang et al.,
2004). Preclinical studies had demonstrated a link between overexpression of HER-2/neu and
COX-2 activity. Similarly, in a phase II study of metastatic colon cancer, rofecoxib in
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
14
combination with chemotherapy showed increased toxicity and no efficacy (Becerra et al.,
2003).
COX-2-independent effects of coxibs There have been extensive data
establishing that coxibs have several COX-2 independent activities which may account, at least
for part of their cancer preventive properties (Table 2). Relevant to this argument are studies
showing that, for example, celecoxib inhibits the growth of various cancer cell lines (Maier et al.,
2004) including hematopoietic cell lines (Waskewich et al., 2002) that are COX-2 deficient.
Moreover, celecoxib inhibited the growth of COX-2 deficient colon cancer xenografts in nude
mice, providing strong evidence for its COX-2 independent antitumor effect (Grosch et al.,
2001). Quite recent data suggest a disturbing COX-2 independent effect of selective COX-2
inhibitors. While in COX-2 positive pancreatic cancer, a selective COX-2 inhibitor reduced tumor
growth and angiogenesis, the opposite effect was observed in COX-2 negative pancreatic
cancer. That is to say, the selective COX-2 inhibitor increased angiogenesis and tumor growth
(Eibl et al., 2005).
These findings allow an alternative interpretation of the existing data, one that is at
variance with the idea that COX-2 is a preeminent, if not altogether the target for cancer
prevention. Thus the chemopreventive effect of COX-2 specific inhibitors may be due to their
effect on these targets and not on COX-2. If this reasoning is correct (and it may not be), it may
reflect the classic limitation encountered when evaluating biology using pharmacological
inhibitors: additional effects by the inhibitor can never be ruled out, making conclusions always
subject to re-interpretation.
A few years ago we proposed a model that attempted to resolve the apparent
contradiction between data suggesting that both COX-2 and non-COX-2 inhibition were
preventing cancer (Rigas and Shiff, 2000). Analysis of the then-available data led us to suggest
that inhibition of either the COX-2 pathway alone or of a non-COX-2 pathway(s) alone could
prevent cancer. We are still missing some of the information needed to validate or reject this
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
15
model, mainly results of trials evaluating the efficacy of COX-2 inhibitors and their effects on
relevant molecular targets. The limited efficacy of COX-2 inhibitors in FAP and the in vitro
evidence that COX-2 inhibitors act also beyond COX-2 inhibition suggest that the first part of our
conclusion, i.e., that COX-2 inhibition alone is sufficient to arrest carcinogenesis, may be wrong.
In fact, it would not be altogether surprising if it is COX-2 that is the “collateral target” in cancer
prevention.
Non-neoplastic COX-2 expression The expression of COX-2 is not restricted to
tumor cells. While COX-2 is undetectable in most tissues in the absence of stimulation, it is
induced in a limited repertoire of cells, notably in monocytes, macrophages, neutrophils, and
endothelial cells (Simmons et al., 2004). Among the stimulants of COX-2 induction are bacterial
lipopolysaccharides (LPS), growth factors, cytokines, and phorbol esters. Relevant to the
cardiovascular side effects of coxibs may be the observations that COX-2, although detectable
at only low levels in vascular endothelium, may be expressed in response to normal blood flow
(Topper et al., 1996). A study in healthy humans after administration of celecoxib or ibuprofen
suggests that COX-2 is a major source of systemic prostacyclin biosynthesis (McAdam et al.,
1999). High levels of COX-2 are detected in activated and proliferating vascular tissues, such as
angiogenic microvessels and atherosclerotic tissues. Atheromatous lesions contain both COX-1
and COX-2, colocalizing mainly with macrophages of the shoulder region and lipid core
periphery, whereas smooth muscle cells show lower levels (Schonbeck et al., 1999).
There is an interesting corollary to these observations, which may impact the design of
pharmacological strategies aimed at this extensive cascade of enzyme-catalyzed reactions.
Eicosanoids often form “opposing pairs” whose balance determines the final result. A classic
pair consists of thromboxane A2 and prostacyclin, which have opposite effects on platelets and
vascular tone (Simmons et al., 2004). Shifting the balance of such a pair could have either
beneficial (e.g., prevention of cardiovascular events by low dose aspirin) or catastrophic effects.
The latter possibility, discussed below, may account for the cardiovascular side effects of
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
16
coxibs. This may be particularly relevant to chemoprevention, in which a chemopreventive agent
against cancer will be administered on a long-term basis to older subjects, i.e., those likely to
have atheromatous lesions.
COX-2 inhibition may modulate alternative eicosanoid pathways The concept
of the role of COX-2 in cancer should be viewed against the accumulating appreciation of the
role of the eicosanoid pathways in carcinogenesis. An intricate system of talented enzymes,
including phospholipases, cytochrome P450, COX, lipoxygenases (LOX) and the so-called
“terminal enzymes”, i.e., those converting endoperoxides to end products, generates an array of
biologically active eicosanoids from polyunsaturated fatty acids such as arachidonic and linoleic
acids (Fig. 2). At times these eicosanoids have antithetic functions. Besides products of COX
isoforms, LOX products may be important in carcinogenesis. Some LOX products have pro-
tumorigenic activities while others are anti-tumorigenic (Shureiqi and Lippman, 2001). Some of
the “terminal enzymes” may have their own distinctive role in cancer. For example, increased
pulmonary production of PGI2 (prostacyclin) by lung-specific overexpression of prostacyclin
synthase, which operates downstream of COX, decreases lung tumor incidence and multiplicity
in both chemically induced murine lung cancer models and in a tobacco smoke exposure model
(Keith et al., 2002; Keith et al., 2004). Such findings suggest the possibility that COX inhibition
may not be always desirable for cancer control.
Inhibition of COX may shift its substrate fatty acid to a non-COX pathway and generate a
pro-carcinogenic end product. There is little evidence for substrate channeling from the COX
active site to the peroxidase active site of the COX monomer. Functional coupling between
COXs and phospholipase A2s has, however, been described (Murakami et al., 2002) as has
been colocalization of COX isoforms with other enzymes of the eicosanoid cascade [e.g., (Liou
et al., 2001)]. Thus a case can be considered where inhibition of COX-2 could shift arachidonic
acid to the LOX pathway thereby suppressing apoptosis -- not a desirable effect in cancer
prevention. A recent human study suggested that oral celecoxib increased LTB4 production in
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
17
the lung microenvironment, under physiologic conditions, although the functional significance of
this effect was uncertain (Mao et al., 2004).
These seemingly subtle issues emphasize the complexity of the system and suggest the
need for caution in devising chemopreventive strategies. Viewed more creatively, this situation
may also represent a pharmacological opportunity; indeed, multi-pathway inhibition may be the
next wave in chemoprevention. Such considerations have led to the development of dual
inhibitors of both COX and 5-LOX. Licofelone, a compound blocking both a LOX and COX is
already in clinical trials.
It is apparent from these considerations that the discovery of COX-2 overexpression in
various cancers has provided a strong stimulus in the last decade to unravel many pathways
likely related to cancer pathogenesis and sharpen the focus on cancer prevention as a realistic
option. However, the central concept of a dominant role of COX-2 in cancer prevention may
have significant limitations that necessitate its reappraisal. In terms of practical applications, its
Achilles’ heel may be the fact that COX-2 expression is not cancer-restricted, and thus long-
term inhibition of COX-2 for cancer control may be associated with limiting or even
unacceptable side effects. It is conceivable that cancer prevention achieved by current coxibs is
to some extent due to their non-COX-2 effects, while their cardiovascular toxicity is due to their
COX-2 effects. This notion could provide the basis for an alternative look at the wealth of
available data.
The withdrawal of rofecoxib and its implications for cancer prevention
In some patients, NSAIDs inhibit growth of colorectal adenomas. Hence, the APPROVe
(Adenomatous Polyp Prevention on Vioxx) study was launched to evaluate the efficacy of
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
18
rofecoxib in this group of patients. Two thousand six hundred subjects with previously removed
colorectal polyps were randomly assigned to receive rofecoxib or placebo. The data showed
that 3.5% of rofecoxib recipients and 1.9% of placebo recipients suffered myocardial infarctions
or strokes during the trial. This prompted the termination of this and all related trials and the
permanent withdrawal of rofecoxib. This has been a setback to the use of COX-2 inhibitors to
prevent colon and perhaps other cancers. At this time, it is still unclear whether or not the COX-
2 inhibitor celecoxib shares this side effect and is still marketed. Although the CLASS
(Celecoxib Long Term Arthritis Safety Study) trial showed this agent to have fewer
gastrointestinal side effects and no increase in cardiovascular risk at 6 months, the 12 month
data suggest that celecoxib did not differ from the traditional NSAIDs in its GI effects and,
importantly, a retrospective analysis of the data suggests signs of increased cardiovascular risk
(Silverstein et al., 2000). This has prompted concerns for a “class (side) effect” (Fitzgerald,
2004). The mechanism of the increased cardiovascular risk is not yet clear. It has been
suggested that since COX-2 is the principal enzyme involved in the production of PGI2
(prostacyclin), its inhibition by COX-2 inhibitors could increase cardiovascular risk by tipping the
balance toward platelet aggregation and vasoconstriction (Fitzgerald, 2004). Of note, an elegant
recent study showed that COX-2 derived prostacyclin confers atheroprotection on female mice,
suggesting that chronic treatment of patients with selective inhibitors of COX-2 could undermine
protection from cardiovascular disease in premenopausal females (Egan et al., 2004).
Two important considerations merit mention here. First, strictly speaking, the COX-2
specific inhibitors may be only COX-2 preferential inhibitors (Vane and Warner, 2000). Second,
individual variations between COX-2 inhibitors may be due to their varied effect on molecular
targets outside COX-2. Parenthetically, this may account for the presumed differential behavior
of celecoxib, compared to, for example, rofecoxib. Indeed, as mentioned earlier, these agents
do have COX-2 independent activities that may account for their cancer preventive properties.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
19
Targets beyond COX-2: The rationale and the promise
That NSAIDs and COX-2 specific inhibitors alike modulate targets other than COX-2 is
now beyond doubt and should be factored in when analyzing their effects on cancer prevention.
The data discussed above make it unlikely that inhibition of COX-2 alone could prevent cancer;
even COX-2 specific inhibitors may require the contribution of their non-COX-2 effects for their
(probably suboptimal) effect on cancer prevention. Combined with the recent withdrawal of one
COX-2 inhibitor, these data make a strong case to evaluate for drug development targets
beyond COX-2. Of note, Niitsu and his group in Sapporo have actively pursued the idea that
modulating targets other than COX-2 can prevent cancer. In a series of elegant studies, they
have shown that the phase II enzyme glutathione-S-transferase P1-1 may be an appropriate
target for chemoprevention (Niitsu et al., 2004; Nobuoka et al., 2004).
Whenever compounds, such as the various NSAIDs, modulate multiple pathways, it is
crucial to assess the mechanistic relevance of each pathway. This is more so in the case of
cancer that represents a process rather than an abrupt transition from normalcy to malignancy.
In this context, and examined from the viewpoint of the drug, it is important to resolve the issues
of dominance versus cooperation versus redundancy of pathways. It is conceivable that only
one of the pathways affected by the drug may actually mediate the effect under study
(dominance), the remaining being either minor accessories or simple downstream effects. In
case of cooperation, concerted modulation of more than one pathway is needed to achieve the
result, while in case of redundancy each of several pathways could autonomously arrest the
process. Redundancy is of particular interest when dealing with cancer, where several sub-
clones, each with different biochemical profiles, can develop “under pressure”, mainly from the
host or from treatment. An agent displaying mechanistic redundancy could affect multiple sub-
clones and thus is better equipped to arrest or abort the carcinogenic process.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
20
This line of reasoning underscores the need to explore targets beyond COX-2,
especially in view of emerging limitations of the COX-2 approach. Developing a rational
approach to cancer prevention would likely require a strategy capitalizing on two facts: a)
NSAIDs do prevent cancer in humans, and b) NSAIDs act on multiple molecular targets, of
which COX-2 is but one. With this as a point of departure, it is clear that the study of the non-
COX-2 targets is likely to point to opportunities for cancer prevention hitherto unappreciated.
Inherent in this approach is the need to resolve issues of mechanistic dominance, cooperation
and redundancy and then make informed choices for drug development. This represents the
promise of “molecular targets beyond COX-2”.
Concluding remarks
The preceding analysis makes three conclusions clear. First, conventional NSAIDs
prevent colon and other cancers most likely by modulating several molecular targets; their
safety and efficacy limitations, however, preclude their application to humans for this purpose.
Second, focusing exclusively on COX-2 inhibition may not be an effective strategy for cancer
prevention. Third, exploring targets beyond COX-2 is likely to yield a productive approach to
cancer prevention.
The most pressing need in cancer chemoprevention is to identify agents or combinations
of agents that combine high efficacy with minimal toxicity. Although this may represent a difficult
goal, the enormity of the “cancer burden” to our society should compel us to strive for it. The
development of successful chemoprevention agents seems to require a strategy that not only
generates mechanistic insights, but also advances them rapidly to their clinical evaluation. The
work on COX-2 to date constitutes an excellent example of an effort based on this premise. As
the results of trials using coxibs for sporadic colon cancer prevention are not available, the
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
21
ultimate jury on COX-2 inhibitors is still out. The balance of their risk/benefit relationship will
need to be assessed. In case these studies do not provide compelling results or COX-2
inhibitors are limited by side effects, we believe that expanding our focus to targets beyond
COX-2 will provide alternative or complementary approaches, the latter representing
mechanism-driven drug combinations.
In addition to its apparent rationality, this approach rests on the solid fact that traditional
NSAIDs prevent several cancers in humans. Thus it is a reasonable inference that if the
molecular targets mediating their effect are identified, it will be feasible to enhance this effect
with new agents that will meet the general criteria of a successful chemopreventive agent. A
new era may have already begun.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
22
REFERENCES
Badawi AF, Eldeen MB, Liu Y, Ross EA and Badr MZ (2004) Inhibition of rat mammary gland
carcinogenesis by simultaneous targeting of cyclooxygenase-2 and peroxisome
proliferator-activated receptor gamma. Cancer Res 64:1181-1189.
Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R, McKeown-Eyssen G,
Summers RW, Rothstein R, Burke CA, Snover DC, Church TR, Allen JI, Beach M, Beck
GJ, Bond JH, Byers T, Greenberg ER, Mandel JS, Marcon N, Mott LA, Pearson L, Saibil
F and van Stolk RU (2003) A randomized trial of aspirin to prevent colorectal adenomas.
N Engl J Med 348:891-899.
Becerra CR, Frenkel EP, Ashfaq R and Gaynor RB (2003) Increased toxicity and lack of efficacy
of Rofecoxib in combination with chemotherapy for treatment of metastatic colorectal
cancer: A phase II study. Int J Cancer 105:868-872.
Bennett A, Civier A, Hensby CN, Melhuish PB and Stamford IF (1987) Measurement of
arachidonate and its metabolites extracted from human normal and malignant
gastrointestinal tissues. Gut 28:315-318.
Bjorkman DJ (1999) Current status of nonsteroidal anti-inflammatory drug (NSAID) use in the
United States: risk factors and frequency of complications. Am J Med 107:3S-8S;
discussion 8S-10S.
Blaine SA, Meyer AM, Hurteau G, Wick M, Hankin JA, Murphy RC, Subbaramaiah K,
Dannenberg AJ, Geraci MW and Nemenoff RA (2004) Targeted overexpression of
mPGES-1 and elevated PGE2 production is not sufficient for lung tumorigenesis in mice.
Carcinogenesis.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
23
Bol DK, Rowley RB, Ho CP, Pilz B, Dell J, Swerdel M, Kiguchi K, Muga S, Klein R and Fischer
SM (2002) Cyclooxygenase-2 overexpression in the skin of transgenic mice results in
suppression of tumor development. Cancer Res 62:2516-2521.
Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG,
Smithies O and Langenbach R (2000) Genetic disruption of Ptgs-1, as well as Ptgs-2,
reduces intestinal tumorigenesis in Min mice. Cancer Res 60:4705-4708.
Cruz-Correa M, Hylind LM, Romans KE, Booker SV and Giardiello FM (2002) Long-term
treatment with sulindac in familial adenomatous polyposis: a prospective cohort study.
Gastroenterology 122:641-645.
Dang CT, Dannenberg AJ, Subbaramaiah K, Dickler MN, Moasser MM, Seidman AD, D'Andrea
GM, Theodoulou M, Panageas KS, Norton L and Hudis CA (2004) Phase II study of
celecoxib and trastuzumab in metastatic breast cancer patients who have progressed
after prior trastuzumab-based treatments. Clin Cancer Res 10:4062-4067.
Dannenberg AJ and Subbaramaiah K (2003) Targeting cyclooxygenase-2 in human neoplasia:
rationale and promise. Cancer Cell 4:431-436.
Ding H, Han C, Zhu J, Chen CS and D'Ambrosio SM (2005) Celecoxib derivatives induce
apoptosis via the disruption of mitochondrial membrane potential and activation of
caspase 9. Int J Cancer 113:803-810.
Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S and Dubois RN (1994) Up-
regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and
carcinomas. Gastroenterology 107:1183-1188.
Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM and Fitzgerald GA (2004) COX-2
Derived Prostacyclin Confers Atheroprotection on Female Mice. Science.
Eibl G, Takata Y, Boros LG, Liu J, Okada Y, Reber HA and Hines OJ (2005) Growth stimulation
of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res
65:982-990.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
24
Fitzgerald GA (2004) Coxibs and cardiovascular disease. N Engl J Med 351:1709-1711.
Fritsche E, Baek SJ, King LM, Zeldin DC, Eling TE and Bell DA (2001) Functional
characterization of cyclooxygenase-2 polymorphisms. J Pharmacol Exp Ther 299:468-
476.
Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind LM, Celano P, Booker SV,
Robinson CR and Offerhaus GJ (1993) Treatment of colonic and rectal adenomas with
sulindac in familial adenomatous polyposis. The New England Journal of Medicine
328:1313-1316.
Grosch S, Tegeder I, Niederberger E, Brautigam L and Geisslinger G (2001) COX-2
independent induction of cell cycle arrest and apoptosis in colon cancer cells by the
selective COX-2 inhibitor celecoxib. Faseb J 15:2742-2744.
Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI and Rigas B (1996)
Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of
apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem
Pharmacol 52:237-245.
Higuchi T, Iwama T, Yoshinaga K, Toyooka M, Taketo MM and Sugihara K (2003) A
randomized, double-blind, placebo-controlled trial of the effects of rofecoxib, a selective
cyclooxygenase-2 inhibitor, on rectal polyps in familial adenomatous polyposis patients.
Clin Cancer Res 9:4756-4760.
Keith RL, Miller YE, Hoshikawa Y, Moore MD, Gesell TL, Gao B, Malkinson AM, Golpon HA,
Nemenoff RA and Geraci MW (2002) Manipulation of pulmonary prostacyclin synthase
expression prevents murine lung cancer. Cancer Res 62:734-740.
Keith RL, Miller YE, Hudish TM, Girod CE, Sotto-Santiago S, Franklin WA, Nemenoff RA, March
TH, Nana-Sinkam SP and Geraci MW (2004) Pulmonary prostacyclin synthase
overexpression chemoprevents tobacco smoke lung carcinogenesis in mice. Cancer
Res 64:5897-5904.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
25
Kimmey MB (2004) Cardioprotective effects and gastrointestinal risks of aspirin: maintaining the
delicate balance. Am J Med 117 Suppl 5A:72S-78S.
Kune G, Kune S and Watson L (1988) Colorectal cancer risk, chronic illnesses, operations, and
medications: case-control results from the Melbourne Colorectal Cancer Study. Cancer
Research 48:4399-4404.
Lin HJ, Lakkides KM, Keku TO, Reddy ST, Louie AD, Kau IH, Zhou H, Gim JS, Ma HL, Matthies
CF, Dai A, Huang HF, Materi AM, Lin JH, Frankl HD, Lee ER, Hardy SI, Herschman HR,
Henderson BE, Kolonel LN, Le Marchand L, Garavito RM, Sandler RS, Haile RW and
Smith WL (2002) Prostaglandin H synthase 2 variant (Val511Ala) in African Americans
may reduce the risk for colorectal neoplasia. Cancer Epidemiol Biomarkers Prev
11:1305-1315.
Liou JY, Deng WG, Gilroy DW, Shyue SK and Wu KK (2001) Colocalization and interaction of
cyclooxygenase-2 with caveolin-1 in human fibroblasts. J Biol Chem 276:34975-34982.
Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, Haudenschild C, Lane TF and Hla T
(2001) Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in
transgenic mice. J Biol Chem 276:18563-18569.
Maier TJ, Schilling K, Schmidt R, Geisslinger G and Grosch S (2004) Cyclooxygenase-2 (COX-
2)-dependent and -independent anticarcinogenic effects of celecoxib in human colon
carcinoma cells. Biochem Pharmacol 67:1469-1478.
Mao JT, Tsu IH, Dubinett SM, Adams B, Sarafian T, Baratelli F, Roth MD and Serio KJ (2004)
Modulation of pulmonary leukotriene B4 production by cyclooxygenase-2 inhibitors and
lipopolysaccharide. Clin Cancer Res 10:6872-6878.
Marx J (2001) Cancer research. Anti-inflammatories inhibit cancer growth--but how? Science
291:581-582.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
26
McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA and FitzGerald GA (1999)
Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human
pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A 96:272-277.
Muller-Decker K, Neufang G, Berger I, Neumann M, Marks F and Furstenberger G (2002)
Transgenic cyclooxygenase-2 overexpression sensitizes mouse skin for carcinogenesis.
Proc Natl Acad Sci U S A 99:12483-12488.
Murakami M, Kambe-Ohkura T and Kudo I (2002) Functional coupling between phospholipase
A2S and cyclooxygenases in immediate and delayed prostanoid biosynthetic pathways.
Adv Exp Med Biol 507:15-19.
Niitsu Y, Takayama T, Miyanishi K, Nobuoka A, Hayashi T, Kukitsu T, Takanashi K, Ishiwatari
H, Abe T, Kogawa T, Takahashi M, Matsunaga T and Kato J (2004) Chemoprevention of
colorectal cancer. Cancer Chemother Pharmacol.
Nobuoka A, Takayama T, Miyanishi K, Sato T, Takanashi K, Hayashi T, Kukitsu T, Sato Y,
Takahashi M, Okamoto T, Matsunaga T, Kato J, Oda M, Azuma T and Niitsu Y (2004)
Glutathione-S-transferase P1-1 protects aberrant crypt foci from apoptosis induced by
deoxycholic acid. Gastroenterology 127:428-443.
Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF
and Taketo MM (1996) Suppression of intestinal polyposis in ApcÐ716 knockout mice by
inhibition of cyclooxygenase 2 (COX-2). Cell 87:803-809.
Phillips RK, Wallace MH, Lynch PM, Hawk E, Gordon GB, Saunders BP, Wakabayashi N, Shen
Y, Zimmerman S, Godio L, Rodrigues-Bigas M, Su LK, Sherman J, Kelloff G, Levin B
and Steinbach G (2002) A randomised, double blind, placebo controlled study of
celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial
adenomatous polyposis. Gut 50:857-860.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
27
Qiao L, Kozoni V, Tsioulias GJ, Koutsos MI, Hanif R, Shiff SJ and Rigas B (1995) Selected
eicosanoids increase the proliferation rate of human colon carcinoma cell lines and
mouse colonocytes in vivo. Biochim Biophys Acta 1258:215-223.
Rigas B, Goldman IS and Levine L (1993) Altered eicosanoid levels in human colon cancer.
Journal of Laboratory and Clinical Medicine 122:518-523.
Rigas B and Shiff SJ (2000) Is inhibition of cyclooxygenase required for the chemopreventive
effect of NSAIDs in colon cancer? A model reconciling the current contradiction. Med
Hypotheses 54:210-215.
Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, Petrelli N, Pipas JM, Karp
DD, Loprinzi CL, Steinbach G and Schilsky R (2003) A randomized trial of aspirin to
prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med
348:883-890.
Schonbeck U, Sukhova GK, Graber P, Coulter S and Libby P (1999) Augmented expression of
cyclooxygenase-2 in human atherosclerotic lesions. Am J Pathol 155:1281-1291.
Sheng HM, Shao JY, Morrow JD, Beauchamp RD and Dubois RN (1998) Modulation of
apoptosis and bcl-2 expression by prostaglandin E-2 in human colon cancer cells.
Cancer Research 58:362-366.
Shiff SJ, Qiao L, Tsai LL and Rigas B (1995) Sulindac sulfide, an aspirin-like compound, inhibits
proliferation, causes cell cycle quiescence, and induces apoptosis in HT-29 colon
adenocarcinoma cells. J Clin Invest 96:491-503.
Shiff SJ and Rigas B (1999) The role of cyclooxygenase inhibition in the antineoplastic effects of
nonsteroidal antiinflammatory drugs (NSAIDs) [comment]. J Exp Med 190:445-450.
Shureiqi I and Lippman SM (2001) Lipoxygenase modulation to reverse carcinogenesis. Cancer
Res 61:6307-6312.
Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, Makuch R, Eisen G,
Agrawal NM, Stenson WF, Burr AM, Zhao WW, Kent JD, Lefkowith JB, Verburg KM and
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
28
Geis GS (2000) Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory
drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized
controlled trial. Celecoxib Long-term Arthritis Safety Study. Jama 284:1247-1255.
Simmons DL, Botting RM and Hla T (2004) Cyclooxygenase isozymes: the biology of
prostaglandin synthesis and inhibition. Pharmacol Rev 56:387-437.
Singh G and Triadafilopoulos G (1999) Epidemiology of NSAID induced gastrointestinal
complications. J Rheumatol 26:18-24.
Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N,
Saunders B, Shen Y, Fujimura T, Su LK and Levin B (2000) The effect of celecoxib, a
cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 342:1946-
1952.
Sun Y, Tang XM, Half E, Kuo MT and Sinicrope FA (2002) Cyclooxygenase-2 overexpression
reduces apoptotic susceptibility by inhibiting the cytochrome c-dependent apoptotic
pathway in human colon cancer cells. Cancer Res 62:6323-6328.
Takeda H, Sonoshita M, Oshima H, Sugihara K, Chulada PC, Langenbach R, Oshima M and
Taketo MM (2003) Cooperation of cyclooxygenase 1 and cyclooxygenase 2 in intestinal
polyposis. Cancer Res 63:4872-4877.
Teh SH, Hill AK, Foley DA, McDermott EW, O'Higgins NJ and Young LS (2004) COX inhibitors
modulate bFGF-induced cell survival in MCF-7 breast cancer cells. J Cell Biochem
91:796-807.
Thun MJ, Henley SJ and Patrono C (2002) Nonsteroidal anti-inflammatory drugs as anticancer
agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst 94:252-266.
Topper JN, Cai J, Falb D and Gimbrone MA, Jr. (1996) Identification of vascular endothelial
genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2,
manganese superoxide dismutase, and endothelial cell nitric oxide synthase are
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
29
selectively up-regulated by steady laminar shear stress. Proc Natl Acad Sci U S A
93:10417-10422.
Turini ME and DuBois RN (2002) Cyclooxygenase-2: a therapeutic target. Annu Rev Med
53:35-57.
Vane JR and Warner TD (2000) Nomenclature for COX-2 inhibitors. Lancet 356:1373-1374.
Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA and Vane JR (1999) Nonsteroid drug
selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with
human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A 96:7563-
7568.
Waskewich C, Blumenthal RD, Li H, Stein R, Goldenberg DM and Burton J (2002) Celecoxib
exhibits the greatest potency amongst cyclooxygenase (COX) inhibitors for growth
inhibition of COX-2-negative hematopoietic and epithelial cell lines. Cancer Res
62:2029-2033.
Weber A, Casini A, Heine A, Kuhn D, Supuran CT, Scozzafava A and Klebe G (2004)
Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib:
new pharmacological opportunities due to related binding site recognition. J Med Chem
47:550-557.
Williams JL, Nath N, Chen J, Hundley TR, Gao J, Kopelovich L, Kashfi K and Rigas B (2003)
Growth inhibition of human colon cancer cells by nitric oxide (NO)-donating aspirin is
associated with cyclooxygenase-2 induction and beta-catenin/T-cell factor signaling,
nuclear factor-kappaB, and NO synthase 2 inhibition: implications for chemoprevention.
Cancer Res 63:7613-7618.
Winde G, Schmid KW, Brandt B, Muller O and Osswald H (1997) Clinical and genomic influence
of sulindac on rectal mucosa in familial adenomatous polyposis. Diseases of the Colon &
Rectum 40:1156-1168.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
30
Winde G, Schmid KW, Schlegel W, Fischer R, Osswald H and Bunte H (1995) Complete
reversion and prevention of rectal adenomas in colectomized patients with familial
adenomatous polyposis by rectal low-dose sulindac maintenance treatment. Advantages
of a low-dose nonsteroidal anti-inflammatory drug regimen in reversing adenomas
exceeding 33 months. Dis Colon Rectum 38:813-830.
Yamaguchi K, Lee SH, Eling TE and Baek SJ (2004) Identification of nonsteroidal anti-
inflammatory drug-activated gene (NAG-1) as a novel downstream target of
phosphatidylinositol 3-kinase/AKT/GSK-3beta pathway. J Biol Chem 279:49617-49623.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
31
Grant Support: This work was supported by the NIH Grants CA92423 and CA34527 and PSC-
CUNY Grant 65201-00 34
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

JPET #80564
32
FIGURE LEGENDS
Fig. 1. Epidemiological cohort studies of the association between NSAID use and colorectal
cancer or adenomatous polyps. Circles: relative risk estimates; lines: 95% confidence
intervals. Adapted from (Thun et al., 2002).
Fig 2. Overview of the eicosanoid pathway. Arachidonic acid, the substrate of three major
biosynthetic pathways, is derived from diet, is released from membrane phospholipids
through a series of reactions requiring phospholipases, or is synthesized from linolenic
acid. The COX pathway produces various eicosanoids and thromboxane; the LOX
pathways produce leukotrienes and hydroxyeicosatetraenoic acids; and the cytochrome
P450 pathways produce EET and dihydroxyacids.
Abbreviations: Phospholipases A2, C, and D, PLA2; PLC; PLD; Prostaglandins, PGE2,
PGF2α, PGD2; Prostacyclin, PGI2; Thromboxane A2, TxA2; Leukotrienes, LTA4, LTB4,
LTC4, LTD4, LTE4; 13-S-hyroxyoctadecadienoic acid, 13-S-HODE; Epoxyeicosatrienoic
acid, EET.
T-shaped arrows denote inhibition. Broken arrow: putative pathway.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from

Table 1
Selected COX-2 independent effects of coxibs relevant to cancer
Effect Reference
Inhibition of cell proliferation (Badawi et al., 2004)
Induction of apoptosis (Ding et al., 2005)
Induction of cell cycle block (Maier et al., 2004)
Survivin expression (Teh et al., 2004)
Cytochrome c release (Sun et al., 2002)
NAG-1 induction (Yamaguchi et al., 2004)
Carbonic anhydrase inhibition (Weber et al., 2004)
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on April 1, 2005 as DOI: 10.1124/jpet.104.080564
at ASPE
T Journals on June 10, 2018
jpet.aspetjournals.orgD
ownloaded from
0.5 1 1.5
Relative Risk�
Thun, 1991, men
Thun, 1991, women
Gridley, 1993
Pinczowski, 1994
Schreinemachers, 1994
Gioovannucci, 1994
Giovannucci, 1995
Kauppi, 1996
Smalley, 1999
Collet, 1999
Langman, 2000
Garcia-Rodriguez, 2001
Fig 1
JPET #80564
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
JPET
Fast Forward. Published on A
pril 1, 2005 as DO
I: 10.1124/jpet.104.080564 at ASPET Journals on June 10, 2018 jpet.aspetjournals.org Downloaded from

Arachidonic Acid
PLA2PLCPLD
Activation
Stimuli
EET Dihydroxy
acids
Cyt P450epoxygenase
COX-1Constitutive
COX-2Inducible
PGE2, PGF2α, PGI2, TxA2
Homeostasis
PGE2, PGI2Inflammation
NSAIDs Coxibs
5-LOX
LTA4
LTB4
Adhesion
LTC4, D4 ,E4
Vasoconstriction
15-LOX-1
Linoleic Acid
13-S-HODE
Proliferation
COX-3Brain
PGD2
Fever, Pain
Paracetamol
?
ZileutonCaffeic acid
Membrane Phospholipids
Licofelone
Steroids
JPET #80564
Fig 2
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
JPET
Fast Forward. Published on A
pril 1, 2005 as DO
I: 10.1124/jpet.104.080564 at ASPET Journals on June 10, 2018 jpet.aspetjournals.org Downloaded from