atf3repressionofbcl-x determinesapoptotic...
TRANSCRIPT

Biology of Human Tumors
ATF3RepressionofBCL-XLDeterminesApoptoticSensitivity toHDAC Inhibitors acrossTumorTypesAnderly C. Ch€ueh1, Janson W.T. Tse1,2,3, Michael Dickinson4, Paul Ioannidis2,5,Laura Jenkins2,5, Lars Togel1,2,5, BeeShin Tan1, Ian Luk1,2,3,Mercedes Davalos-Salas1,2,5, Rebecca Nightingale1,2,5, Matthew R. Thompson6,Bryan R.G.Williams6, Guillaume Lessene7, Erinna F. Lee2,5,8,Walter D. Fairlie2,5,8,Amardeep S. Dhillon2,5, and John M. Mariadason1,2,5
Abstract
Purpose: Histone deacetylase inhibitors (HDACi) are epi-genome-targeting small molecules approved for the treatmentof cutaneous T-cell lymphoma and multiple myeloma. Theyhave also demonstrated clinical activity in acute myelogenousleukemia, non–small cell lung cancer, and estrogen receptor–positive breast cancer, and trials are underway assessing theiractivity in combination regimens including immunotherapy.However, there is currently no clear strategy to reliably predictHDACi sensitivity. In colon cancer cells, apoptotic sensitivityto HDACi is associated with transcriptional induction ofmultiple immediate-early (IE) genes. Here, we examinedwhether this transcriptional response predicts HDACi sensi-tivity across tumor type and investigated the mechanism bywhich it triggers apoptosis.
ExperimentalDesign: Fifty cancer cell lines fromdiverse tumortypeswere screened to establish the correlation between apoptotic
sensitivity, induction of IE genes, and components of the intrinsicapoptotic pathway.
Results:We show that sensitivity to HDACi across tumor typesis predicted by induction of the IE genes FOS, JUN, and ATF3, butthat only ATF3 is required for HDACi-induced apoptosis. Wefurther demonstrate that the proapoptotic function of ATF3 ismediated through direct transcriptional repression of the prosur-vival factor BCL-XL (BCL2L1). These findings provided the ratio-nale for dual inhibition of HDAC and BCL-XL, which we showstrongly cooperate to overcome inherent resistance to HDACiacross diverse tumor cell types.
Conclusions: These findings explain the heterogeneousresponses of tumor cells to HDACi-induced apoptosisand suggest a framework for predicting response and expand-ing their therapeutic use in multiple cancer types. Clin CancerRes; 23(18); 5573–84. �2017 AACR.
IntroductionHistone deacetylase inhibitors (HDACi) are epigenome-target-
ing anticancer therapeutics with established clinical activity inseveral hematological malignancies (1). A number of distinctchemical classes of HDACi have been identified or developedincluding short-chain fatty acids (butyrate, valproic acid),
hydroxamic acids (trichostatin A, vorinostat, belinostat, panobi-nostat, and pracinostat), tetrapeptides (romidepsin), and benza-midines (entinostat; ref. 2). Vorinostat and romidepsin areapproved for the treatment of cutaneous T-cell lymphoma (CTCL;ref. 3), and belinostat is approved for the treatment of peripheralT-cell lymphoma (PTCL). In addition, combinatorial use ofpanobinostat with the proteasome inhibitor bortezomib isapproved for refractory multiple myeloma (4), and pracinostatwas recently granted breakthrough therapy designation withazacytidine in acute myelogenous leukemia (AML; ref. 5).Although responses to single-agent HDACi are limited in solidtumors (6), studies in non–small cell lung cancer and estrogenreceptor–positive advanced breast cancer suggest they may haveefficacy in combination therapy regimens (7, 8).
HDACis inhibit class I (HDACs 1, 2, 3, and 8) and class IIHDACs (HDACs 4, 5, 6, 7, 9, and 10), which deacetylate lysineresidues on target proteins (2). HDACis activate gene expressionby inducing hyperacetylation of DNA-bound core histones, there-by increasing accessibility of the core transcriptional apparatus toDNA (1), or by hyperacetylating transcription factors, which caneither increase or decrease their transcriptional activity (1). Inaddition, HDACi can elicit cellular effects independent of tran-scription by acetylating cytoplasmic proteins such as Hsp90 andtubulin (9, 10).
Although HDACis induce multiple effects on tumor cells,including inhibiting proliferation and inducing differentiation
1Ludwig Institute for Cancer Research, Melbourne, Australia. 2Olivia Newton-John Cancer Research Institute, Heidelberg, Victoria, Australia. 3Department ofMedicine, University of Melbourne, Parkville, Victoria, Australia. 4Peter MacCal-lum Cancer Centre, Parkville, Victoria, Australia. 5School of Cancer Medicine, LaTrobe University, Bundoora, Victoria, Australia. 6Hudson Institute of MedicalResearch and Department of Molecular and Translational Sciences, MonashUniversity, Clayton, Victoria. 7The Walter and Eliza Hall Institute, Parkville,Victoria, Australia. 8Department of Biochemistry and Genetics, La TrobeUniversity, Bundoora, Victoria, Australia.
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
A.C. Ch€ueh and J.W.T. Tse contributed equally to this article.
Corresponding Author: John M. Mariadason, Olivia Newton-John CancerResearch Institute, Level 5, ONJ Centre, Austin Health, 145 Studley Road,Heidelberg, Victoria 3084, Australia. Phone: 613-9496-3068; Fax: 613-9496-5334; E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-17-0466
�2017 American Association for Cancer Research.
ClinicalCancerResearch
www.aacrjournals.org 5573
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466

(1), their primary mechanism of antitumor activity is through theinduction of apoptosis (2). In this regard, HDACis induce apo-ptosis primarily through the intrinsic/mitochondrial pathway(11), although in some tumor cell lines, the extrinsic/deathreceptor pathway is also activated (12, 13). HDACi-inducedapoptosis has been linkedwith altered expressionof key apoptoticregulators including upregulation of the proapoptotic moleculesBAX (14), BAK (15), APAF1 (16), BMF (17), BIM (18), and DR5(19), and downregulation of the antiapoptotic proteins SURVI-VIN (20), BCL-XL (21), and c-FLIP (22). However, HDACi regu-lation of these factors varies between cell type, and has not beensystematically linked to apoptotic response (23). Furthermore,themechanisms bywhichHDACis regulate the expression of pro-and antiapoptotic genes are only partially understood.
We previously identified a robust transcriptional responsespecifically associated with HDACi-induced apoptosis in colorec-tal cancer cell lines. This response involved the coordinate induc-tion of multiple immediate-early (IE) response genes (FOS, JUN,EGR1, EGR3, ATF3, ARC, and NR4A1) and stress response genes(NDRG4, MT1E, MT1F, and GADD45B; ref. 24). The goals of thisstudy were to determine whether this represents a generic tran-scriptional response that defines HDACi-induced apoptosisacross tumor types, including CTCL andmultiplemyelomawherethese agents currently have the greatest clinical activity. Second,we sought to determine whether this transcriptional responseunderpins HDACi-induced apoptosis by regulating expression ofkey apoptotic regulators.
Herein, we demonstrate that HDACis robustly induce expres-sion of the IE genes FOS, JUN, and ATF3 in multiple tumor celltypes, which correlated significantly with the magnitude ofHDACi-induced apoptosis. We also demonstrate induction ofthese genes in 2 patients with CTCL treated with panobinostat.Functional studies revealed that ATF3 but not FOS or JUN wasrequired for HDACi-induced apoptosis across tumor cell lines,and that the effects of ATF3 were mediated through repression ofthe prosurvival gene BCL-XL (BCL2L1). These data provided arationale for combining HDAC and BCL-XL inhibitors, whichsuccessfully overcame inherent resistance to HDACi in a range oftumor types. Our findings establish the induction of ATF3 andsubsequent repression of BCL-XL as a consistent and key deter-minant of HDACi-induced apoptosis independent of tumor type.They also define the molecular basis for differential sensitivity to
HDACi and identify avenues for predicting response and over-coming inherent resistance to HDACi through rational combina-tion therapy.
Materials and MethodsCell culture
All cell lines used for this study were obtained from theAmerican Type Tissue Culture Collection or as gifts from colla-borators listed in the Acknowledgments section. A total of 50human cancer cell lines derived from multiple tumor types wereused: Solid tumor cell lines used were PC-3, DU-145, LNCAP(prostate); HT-1197, HT-1376, 5637 (bladder); SK-MEL-3, SK-MEL-5, SK-MEL-28 (melanoma); MDA-MB-231, MDA-MB-468,MCF-7 (breast); A549, NCI-H292, NCI-H460, NCI-H358, NCI-H1650, NCI-H1975 (lung); RKO, LIM1215, Colo320, SW48,HCT116, SW948 (colon), IGROV1, SK-OV-3, JAM, OVCAR-8,OVCAR-5 (ovarian), OU-87 (glioblastoma); PANC-1 (pancreat-ic); ACHN (renal); 293T (embryonic kidney); A431 (epidermis);AGS (gastric), and Hep3B (hepatoma). Hematologic cancer celllines used were HH, HuT-78, HuT-102, MJ (cutaneous T-celllymphoma); Jurkat, Raji, U937 (lymphoma); LP-1, OPM-2,RPMI-8226, U266 (multiple myeloma); and K-562, KG-1, andKG-1A (leukemia). Cells weremaintained at 37�C and 5%CO2 inbase medium DMEM for solid tumor cell lines or RPMI forhematogic cancer cell lines. Base medium was supplementedwith 10% FCS, 2 mmol/L L-glutamine, 100 U/mL penicillin, and100 mg/mL streptomycin. Wild-type and Atf3�/� mouse embry-onic fibroblasts were maintained in low-glucose DMEM supple-mented with 10% FCS, 2 mmol/L L-glutamine, 100 U/mL pen-icillin, and 100 mg/mL streptomycin at 37�C in 10% CO2. Meth-ods for cellmaintenance have been previously described (25).WTand FLAG-tagged hBCL-XL–transduced mouse embryonic fibro-blasts were maintained in DMEM, high-glucose media supple-mented with 10% (v/v) FBS, 250 mmol/L L-asparagine, 50 mmol/L2-mercaptoethanol, and1mmol/LHEPES.Cell lineswere assessedfor mycoplasma status using the MycoAlert assay (Lonza) andmycoplasma-negative frozen stocks used for a maximum of2 months. Authenticity of frozen stocks of the A549, AGS,HCT116, PC3, U87, RPMI-8226, SKMEL28, MCF7, PANC1, HH,RKO, LIM1215, Colo320, SW48, and SW948 cell lines wasdetermined by short-tandem repeat profiling using the GenePrint10 system (Promega), and all found to be exact matches withpublished profiles.
Drug sourceSodium-butyrate (NaBu) and valproate were obtained from
Sigma. Vorinostat, belinostat, depsipeptide, entinostat, ABT-737,andABT-199were obtained fromSelleckChemicals. ABT-263wasobtained from ApexBio. Synthesis of A-1331852was as describedpreviously (26).
Measurement of apoptosisApoptosis assays were performed as previously described by
propidium iodide (PI) staining and FACSanalysis (25). Cellswereseeded in triplicate in 24-well plates. Seeding densities variedbetween 30,000 and 90,000 cells per well and were calculatedsuch that control cell density approximated 80%confluence at thecompletion of the experimental period. Drug treatment wasperformed for 24 to 72 hours. Both attached and floating cellswere harvested by scraping, washed in cold PBS, and resuspendedin 50mg/mLPI, 0.1%sodium citrate, and 0.1%TritonX-100. Cells
Translational Relevance
The study identifies a novel mechanism by which HDACinhibitors induce apoptosis in tumor cells through inductionof the ATF3 transcription factor and subsequent repression ofBCL-XL. This mechanism transcends tumor type, is measur-able in patient samples in vivo, and defines the basis forsensitivity or resistance to HDAC inhibitors. These findingsestablish a strategy for overcoming inherent resistance toHDACi by rational combination with BCL-XL inhibitors, anddefine a framework for the identification of biomarkers pre-dictive of HDACi response, including rapid assessment ofATF3 induction. These findings have the potential to directlyaffect the clinical use ofHDACi for the approved indications ofcutaneous T-cell lymphoma and multiple myeloma and fortheir ongoing clinical development in multiple malignancies.
Ch€ueh et al.
Clin Cancer Res; 23(18) September 15, 2017 Clinical Cancer Research5574
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466

were stained overnight at 4�C, and 10,000 cells were analyzed forDNA content using a BD FACS Canto II (BD Biosciences). Thepercentage of cells with a subdiploid DNA content was quantifiedusing ModFit LT (Verity Software House).
Clinical trial samplesWhole blood was collected in sodium-heparin tubes from
2 patients diagnosed with cutaneous T-cell lymphoma whoparticipated in a single-arm, open-label, institutional phase IIpanobinostat trial (Clinicaltrials.gov identifier: NCT01658241).Patients received 30mgpanobinostat orally, 3 timesweekly for upto 4 weeks. Both patients had >70% tumor involvement inperipheral blood mononuclear cells (PBMC). PBMCs were iso-lated by density centrifugation (Lymphoprep), according to themanufacturer's instructions. RNA from PBMC was purified andsubjected to gene expression analysis using qRT-PCR. The clinicalprotocol, informed consent form, and other relevant study doc-umentation were approved by the Institutional Review Board ofthe Peter MacCallum Cancer Centre. All patients gave writteninformed consent prior to study entry.
Quantitative RT-PCRTotal RNA was extracted using the RNeasy Mini Kit (Qiagen)
and reverse-transcribed using random hexamers and the Tran-scriptor High Fidelity cDNA Synthesis Kit (Roche), according tothe manufacturer's instructions. Quantitative RT-PCR was per-formed using Power SYBR Green PCR Master Mix (AppliedBiosystems) on a 7500 Fast Real-Time PCR System (AppliedBiosystems) according to the manufacturer's instructions. cDNA(10 ng) was amplified with 75 nmol/L forward and reverseprimers in a 15 mL reaction. Primers used are listed in Supple-mentary Table S1.
Western blotWestern blot analysis was performed as previously described
(27). The source and dilutions of antibodies used are as follows:Rabbit anti-ATF3 (sc-188, Santa Cruz Biotechnology, 1:1,000),rabbit anti-FOS (cst-4384, Cell Signaling Technology, 1:1,000),mouse anti–c-JUN (cst-2315, Cell Signaling Technology,1:1,000), rabbit anti–Ac Histone H3 (06-599, Merck Millipore,1:10,000), goat anti–Histone H3 (sc8654, Santa Cruz Biotech-nology, 1:5,000), rabbit anti–beta Tubulin (ab6046, Abcam,1:20,000), mouse anti-actin (A5316, SIGMA, 1:10,000), andrabbit–anti-BCL-XL (54H6, Cell Signaling Technology, 1:1,000rat anti-FLAG antibody [WEHI, clone 9H1, 1:2500]).
Plasmids and luciferase reporter assaysThe ATF3 overexpression vector was provided by Dr. Dakang
Xu at Monash University (28). The AP-1 reporter construct wasobtained from Clontech, Sp1/Sp3 reporter constructs wereprovided by Dr. Yoshihiro Sowa (Kyoto Prefectural Universityof Medicine), and pGL3-BCL-XL reporter constructs were kind-ly provided by Dr. Ni Chen, Sichuan University, Chengdu,China (29).
Cell lines were transiently transfected with reporter constructsusing the Lipofectamine 2000 transfection reagent (Invitrogen).Transfected cells were treated with HDACi for 24 to 48 hours andluciferase reporter activity determined using the dual-luciferasereporter assay Kit from Promega. Due to the strong effects ofHDCAi treatment on TK-Renilla luciferase activity, reporter activ-ity was normalized to total protein.
RNAi-mediated knockdownsiRNAs targeting FOS, JUN, ATF3, and BCL-XL were obtained
from Dharmacon. siRNA transfection was performed using Lipo-fectamineRNAiMAX (Invitrogen) according to themanufacturer'sinstruction. Cells were harvested 24, 48, or 72 hours after trans-fection for subsequent analysis.
Xenograft studiesAnimal studies were performed with the approval of the
Austin Health Animal Ethics Committee. Eight-week-old femaleBALB/c nu/nu mice weighing approximately 16 g were obtainedfrom the Australian Resources Centre (ARC). U87 cells (3 � 106
cells) were injected subcutaneously into the right and left flank ofeach animal in a 150 mL suspension consisting of a 1:1 mixture ofDMEM (Invitrogen) and BD Matrigel Basement Matrix (BD Bios-ciences). Once palpable tumors developed, mice were random-ized into four groups to receive either vehicle [DMSO by intra-peritoneal injection, and Phosal50 (60% Phosal PG, 30%PEG400, and 10% EtOH) by oral gavage], 50 mg/kg vorinostatvia intraperitoneal injection, 25mg/kgABT-263 viaoral gavage, orthe combination. Mice were treated daily for 19 days. Tumorgrowth wasmonitored every second day by calliper measurementuntil the end of the experimental period or when tumors reached1 cm3 in size. At this point, animals were euthanized, and tumorswere excised and weighed.
Statistical analysisIn all cases, groups were compared using the Student's t test,
with P < 0.05 considered to be statistically significant. Correlationanalyses were performed using Pearson's correlationwith P < 0.05considered statistically significant.
ResultsHDACi sensitivity spectrum of human cancer cells
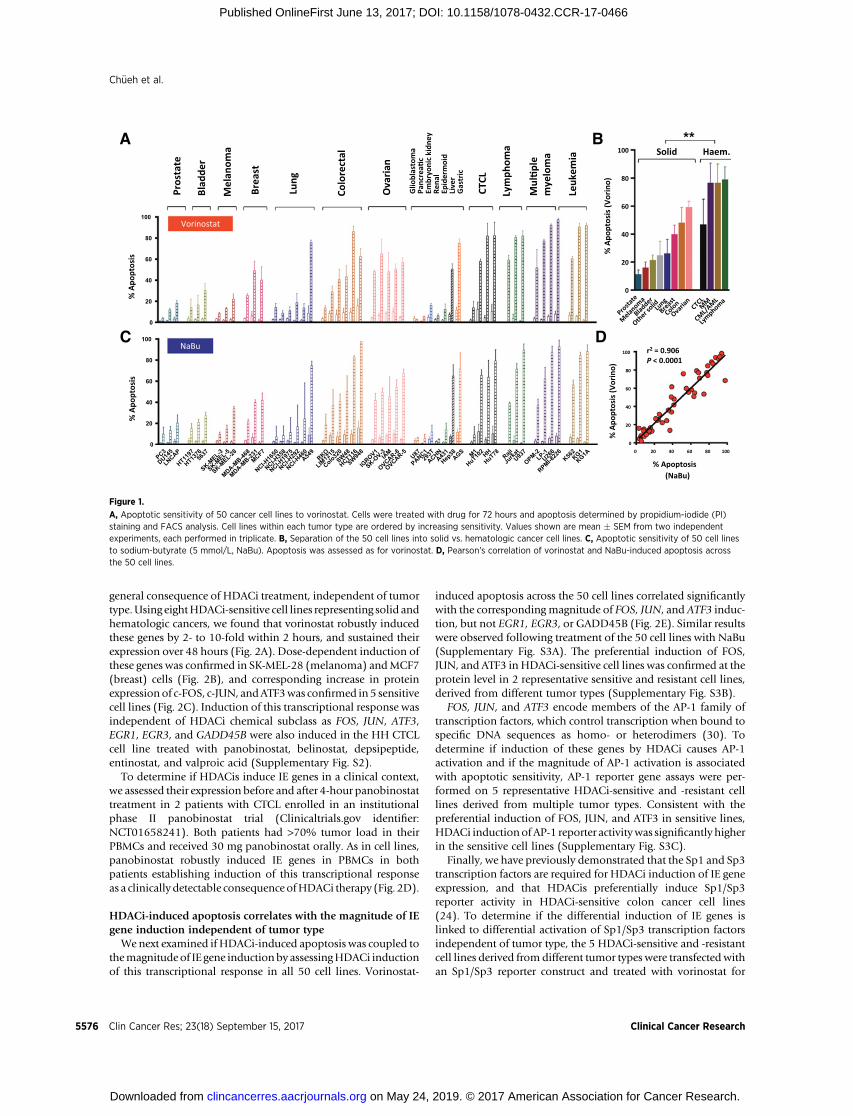
To identify the molecular mechanisms underlying HDACi-induced apoptosis, we first stratified vorinostat-induced apopto-tic responses in 50 human cancer cell lines representing commontumor types, including those displaying significant clinicalresponse to HDACi (CTCL, multiple myeloma, leukemia, breast,and lung cancers; refs. 3, 7, 8). Sensitivity of the cell lines tovorinostat was highly variable (ranging from 2.5% apoptosis inU87 cells to 97.4% in RPMI-8226 cells), enabling separation intostrong or weak responders (Fig. 1A). As observed clinically (3),vorinostat more potently induced apoptosis in hematologiccell lines (Fig. 1B). Among the solid tumor models, ovariancancer lines weremost sensitive, whereas prostate lines weremostresistant (Fig. 1B). This spectrum of antitumor responses wasreplicated using NaBu, a member of the short-chain fatty acidsubclass of HDACi (Fig. 1C and D). Differential sensitivity toHDACi was not due to differences in the extent of HDAC inhi-bition, as histone H3 acetylation was similarly increased byvorinostat in representative sensitive and resistant lines (Supple-mentary Fig. S1).
HDACis induce sustained IE gene expression inmultiple tumorcell types and in CTCL patients in vivo
Using our comprehensive profile ofHDACi-induced apoptosis,we next investigated the mechanisms likely to underpin HDACiresponse across multiple cancers. Based on our prior findings incolon cancer cells (24), we investigated whether the induction ofthe IE genes FOS, JUN, ATF3, EGR1, EGR3, and GADD45B is a
ATF3 Drives HDACi-Induced Apoptosis
www.aacrjournals.org Clin Cancer Res; 23(18) September 15, 2017 5575
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466
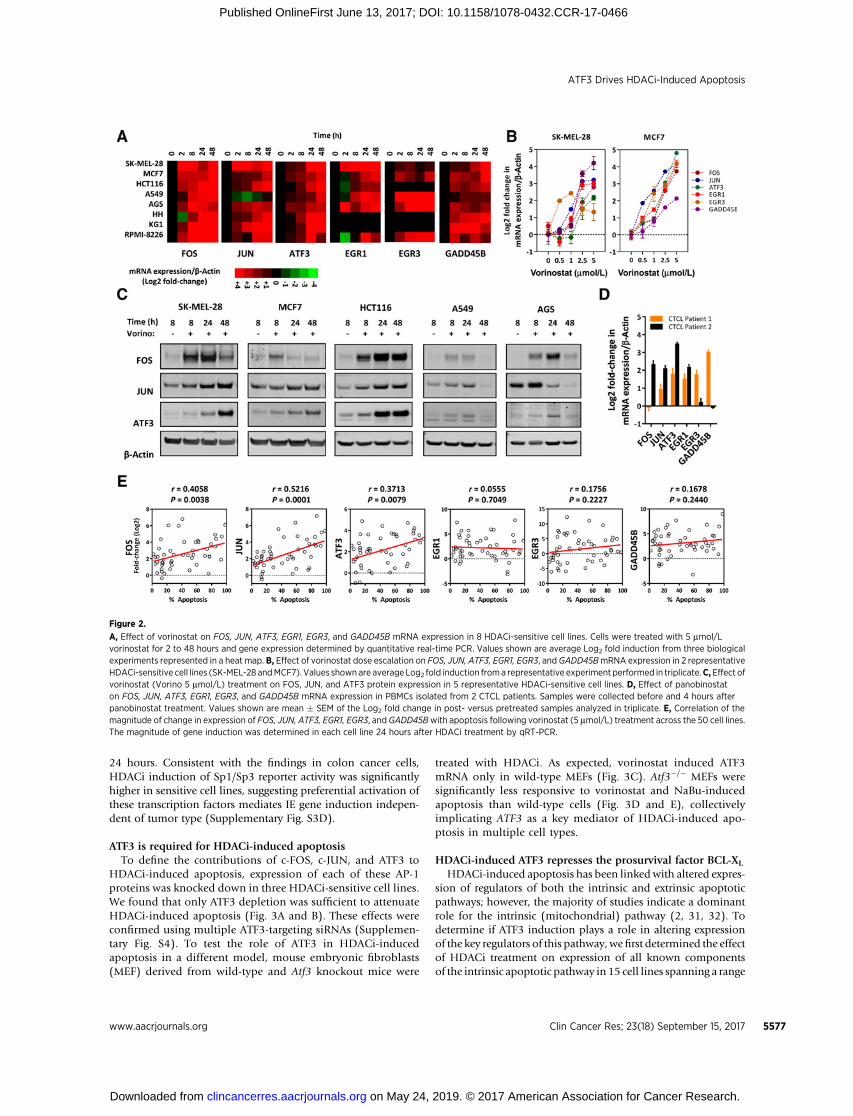
general consequence of HDACi treatment, independent of tumortype.Using eightHDACi-sensitive cell lines representing solid andhematologic cancers, we found that vorinostat robustly inducedthese genes by 2- to 10-fold within 2 hours, and sustained theirexpression over 48 hours (Fig. 2A). Dose-dependent induction ofthese genes was confirmed in SK-MEL-28 (melanoma) andMCF7(breast) cells (Fig. 2B), and corresponding increase in proteinexpression of c-FOS, c-JUN, andATF3was confirmed in 5 sensitivecell lines (Fig. 2C). Induction of this transcriptional response wasindependent of HDACi chemical subclass as FOS, JUN, ATF3,EGR1, EGR3, and GADD45B were also induced in the HH CTCLcell line treated with panobinostat, belinostat, depsipeptide,entinostat, and valproic acid (Supplementary Fig. S2).
To determine if HDACis induce IE genes in a clinical context,we assessed their expression before and after 4-hour panobinostattreatment in 2 patients with CTCL enrolled in an institutionalphase II panobinostat trial (Clinicaltrials.gov identifier:NCT01658241). Both patients had >70% tumor load in theirPBMCs and received 30 mg panobinostat orally. As in cell lines,panobinostat robustly induced IE genes in PBMCs in bothpatients establishing induction of this transcriptional responseas a clinically detectable consequence ofHDACi therapy (Fig. 2D).
HDACi-induced apoptosis correlates with the magnitude of IEgene induction independent of tumor type
Wenext examined if HDACi-induced apoptosis was coupled tothemagnitudeof IE gene inductionby assessingHDACi inductionof this transcriptional response in all 50 cell lines. Vorinostat-
induced apoptosis across the 50 cell lines correlated significantlywith the corresponding magnitude of FOS, JUN, and ATF3 induc-tion, but not EGR1, EGR3, or GADD45B (Fig. 2E). Similar resultswere observed following treatment of the 50 cell lines with NaBu(Supplementary Fig. S3A). The preferential induction of FOS,JUN, and ATF3 in HDACi-sensitive cell lines was confirmed at theprotein level in 2 representative sensitive and resistant cell lines,derived from different tumor types (Supplementary Fig. S3B).
FOS, JUN, and ATF3 encode members of the AP-1 family oftranscription factors, which control transcription when bound tospecific DNA sequences as homo- or heterodimers (30). Todetermine if induction of these genes by HDACi causes AP-1activation and if the magnitude of AP-1 activation is associatedwith apoptotic sensitivity, AP-1 reporter gene assays were per-formed on 5 representative HDACi-sensitive and -resistant celllines derived from multiple tumor types. Consistent with thepreferential induction of FOS, JUN, and ATF3 in sensitive lines,HDACi inductionof AP-1 reporter activitywas significantly higherin the sensitive cell lines (Supplementary Fig. S3C).
Finally, we have previously demonstrated that the Sp1 and Sp3transcription factors are required for HDACi induction of IE geneexpression, and that HDACis preferentially induce Sp1/Sp3reporter activity in HDACi-sensitive colon cancer cell lines(24). To determine if the differential induction of IE genes islinked to differential activation of Sp1/Sp3 transcription factorsindependent of tumor type, the 5 HDACi-sensitive and -resistantcell lines derived from different tumor types were transfected withan Sp1/Sp3 reporter construct and treated with vorinostat for
NaBu
Vorinostat
Lung
Mel
anom
a
Pros
tate
Glio
blas
tom
a
Panc
rea�
c Em
bryo
nic
kidn
ey
Rena
l Ep
ider
moi
d
Live
r G
astr
ic
CTCL
Brea
st
Mul
�ple
m
yelo
ma
Leuk
emia
Blad
der
Lym
phom
a
Ova
rian
Colo
rect
al
r2 = 0.906 P < 0.0001
D
A
0
20
40
60
80
100
% A
popt
osis
0
PC3
DU145
LNCAP
HT1197
HT137656
37
SK-MEL-3
SK-MEL-5
SK-MEL-28
MDA-MB-46
8
MDA-MB-23
1MCF7
NCI-H16
50
NCI-H35
8
NCI-H19
75
NCI-H29
2
NCI-H46
0A54
9RKO
LIM12
15
Colo320SW48
HCT116
SW948
IGROV1
SK-OV-3IA
M
OVCAR-8
OVCAR-5 U87
PANC129
3TACHN
A431
Hep38
AGS M1
HuT102 HH
HuT78 Raji
Jurka
tU93
7
OPM-2LP-1
U266
RPMI-822
6K56
2KG1
KG1A
20
40
60
80
100
% A
popt
osis
0 20 40 60 80 1000
20
40
60
80
100
% Apoptosis(NaBu)
% A
popt
osis
(Vor
ino)
C
Lymphoma
CML/AML
MMCTCL
Ovaria
n
ColonBreast
Lung
Other solid
Bladder
Melanoma
Prosta
te0
20
40
60
80
100
% A
popt
osis
(Vor
ino)
Solid Haem. ** B
Figure 1.
A, Apoptotic sensitivity of 50 cancer cell lines to vorinostat. Cells were treated with drug for 72 hours and apoptosis determined by propidium-iodide (PI)staining and FACS analysis. Cell lines within each tumor type are ordered by increasing sensitivity. Values shown are mean � SEM from two independentexperiments, each performed in triplicate. B, Separation of the 50 cell lines into solid vs. hematologic cancer cell lines. C, Apoptotic sensitivity of 50 cell linesto sodium-butyrate (5 mmol/L, NaBu). Apoptosis was assessed as for vorinostat. D, Pearson's correlation of vorinostat and NaBu-induced apoptosis acrossthe 50 cell lines.
Ch€ueh et al.
Clin Cancer Res; 23(18) September 15, 2017 Clinical Cancer Research5576
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466
24 hours. Consistent with the findings in colon cancer cells,HDACi induction of Sp1/Sp3 reporter activity was significantlyhigher in sensitive cell lines, suggesting preferential activation ofthese transcription factors mediates IE gene induction indepen-dent of tumor type (Supplementary Fig. S3D).
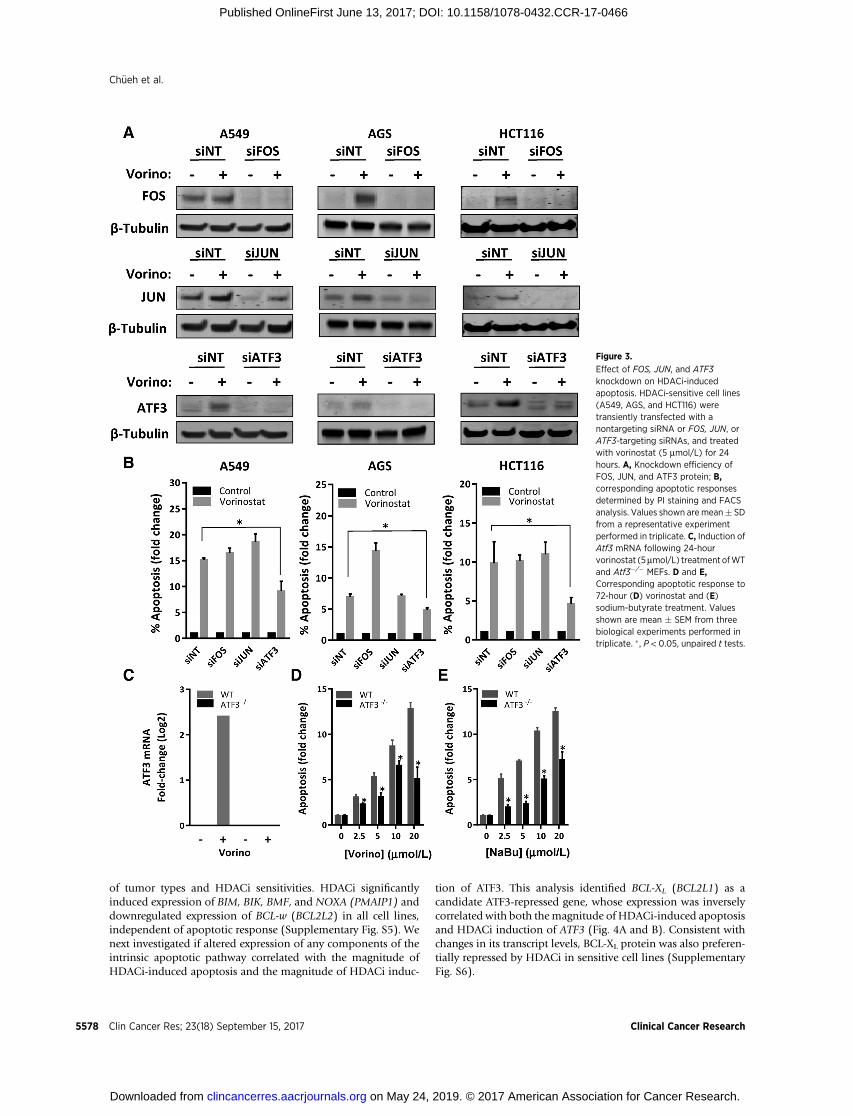
ATF3 is required for HDACi-induced apoptosisTo define the contributions of c-FOS, c-JUN, and ATF3 to
HDACi-induced apoptosis, expression of each of these AP-1proteins was knocked down in three HDACi-sensitive cell lines.We found that only ATF3 depletion was sufficient to attenuateHDACi-induced apoptosis (Fig. 3A and B). These effects wereconfirmed using multiple ATF3-targeting siRNAs (Supplemen-tary Fig. S4). To test the role of ATF3 in HDACi-inducedapoptosis in a different model, mouse embryonic fibroblasts(MEF) derived from wild-type and Atf3 knockout mice were
treated with HDACi. As expected, vorinostat induced ATF3mRNA only in wild-type MEFs (Fig. 3C). Atf3�/� MEFs weresignificantly less responsive to vorinostat and NaBu-inducedapoptosis than wild-type cells (Fig. 3D and E), collectivelyimplicating ATF3 as a key mediator of HDACi-induced apo-ptosis in multiple cell types.
HDACi-induced ATF3 represses the prosurvival factor BCL-XL
HDACi-induced apoptosis has been linkedwith altered expres-sion of regulators of both the intrinsic and extrinsic apoptoticpathways; however, the majority of studies indicate a dominantrole for the intrinsic (mitochondrial) pathway (2, 31, 32). Todetermine if ATF3 induction plays a role in altering expressionof the key regulators of this pathway, wefirst determined the effectof HDACi treatment on expression of all known componentsof the intrinsic apoptotic pathway in 15 cell lines spanning a range
Figure 2.
A, Effect of vorinostat on FOS, JUN, ATF3, EGR1, EGR3, and GADD45B mRNA expression in 8 HDACi-sensitive cell lines. Cells were treated with 5 mmol/Lvorinostat for 2 to 48 hours and gene expression determined by quantitative real-time PCR. Values shown are average Log2 fold induction from three biologicalexperiments represented in a heat map. B, Effect of vorinostat dose escalation on FOS, JUN, ATF3, EGR1, EGR3, andGADD45BmRNA expression in 2 representativeHDACi-sensitive cell lines (SK-MEL-28 andMCF7). Values shownare averageLog2 fold induction froma representative experiment performed in triplicate.C,Effect ofvorinostat (Vorino 5 mmol/L) treatment on FOS, JUN, and ATF3 protein expression in 5 representative HDACi-sensitive cell lines. D, Effect of panobinostaton FOS, JUN, ATF3, EGR1, EGR3, and GADD45B mRNA expression in PBMCs isolated from 2 CTCL patients. Samples were collected before and 4 hours afterpanobinostat treatment. Values shown are mean � SEM of the Log2 fold change in post- versus pretreated samples analyzed in triplicate. E, Correlation of themagnitude of change in expression of FOS, JUN, ATF3, EGR1, EGR3, andGADD45Bwith apoptosis following vorinostat (5 mmol/L) treatment across the 50 cell lines.The magnitude of gene induction was determined in each cell line 24 hours after HDACi treatment by qRT-PCR.
ATF3 Drives HDACi-Induced Apoptosis
www.aacrjournals.org Clin Cancer Res; 23(18) September 15, 2017 5577
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466
of tumor types and HDACi sensitivities. HDACi significantlyinduced expression of BIM, BIK, BMF, and NOXA (PMAIP1) anddownregulated expression of BCL-w (BCL2L2) in all cell lines,independent of apoptotic response (Supplementary Fig. S5). Wenext investigated if altered expression of any components of theintrinsic apoptotic pathway correlated with the magnitude ofHDACi-induced apoptosis and the magnitude of HDACi induc-
tion of ATF3. This analysis identified BCL-XL (BCL2L1) as acandidate ATF3-repressed gene, whose expression was inverselycorrelated with both themagnitude of HDACi-induced apoptosisand HDACi induction of ATF3 (Fig. 4A and B). Consistent withchanges in its transcript levels, BCL-XL protein was also preferen-tially repressed by HDACi in sensitive cell lines (SupplementaryFig. S6).
Figure 3.
Effect of FOS, JUN, and ATF3knockdown on HDACi-inducedapoptosis. HDACi-sensitive cell lines(A549, AGS, and HCT116) weretransiently transfected with anontargeting siRNA or FOS, JUN, orATF3-targeting siRNAs, and treatedwith vorinostat (5 mmol/L) for 24hours. A, Knockdown efficiency ofFOS, JUN, and ATF3 protein; B,corresponding apoptotic responsesdetermined by PI staining and FACSanalysis. Values shown are mean� SDfrom a representative experimentperformed in triplicate. C, Induction ofAtf3 mRNA following 24-hourvorinostat (5mmol/L) treatment ofWTand Atf3�/� MEFs. D and E,Corresponding apoptotic response to72-hour (D) vorinostat and (E)sodium-butyrate treatment. Valuesshown are mean � SEM from threebiological experiments performed intriplicate. �, P < 0.05, unpaired t tests.
Ch€ueh et al.
Clin Cancer Res; 23(18) September 15, 2017 Clinical Cancer Research5578
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466
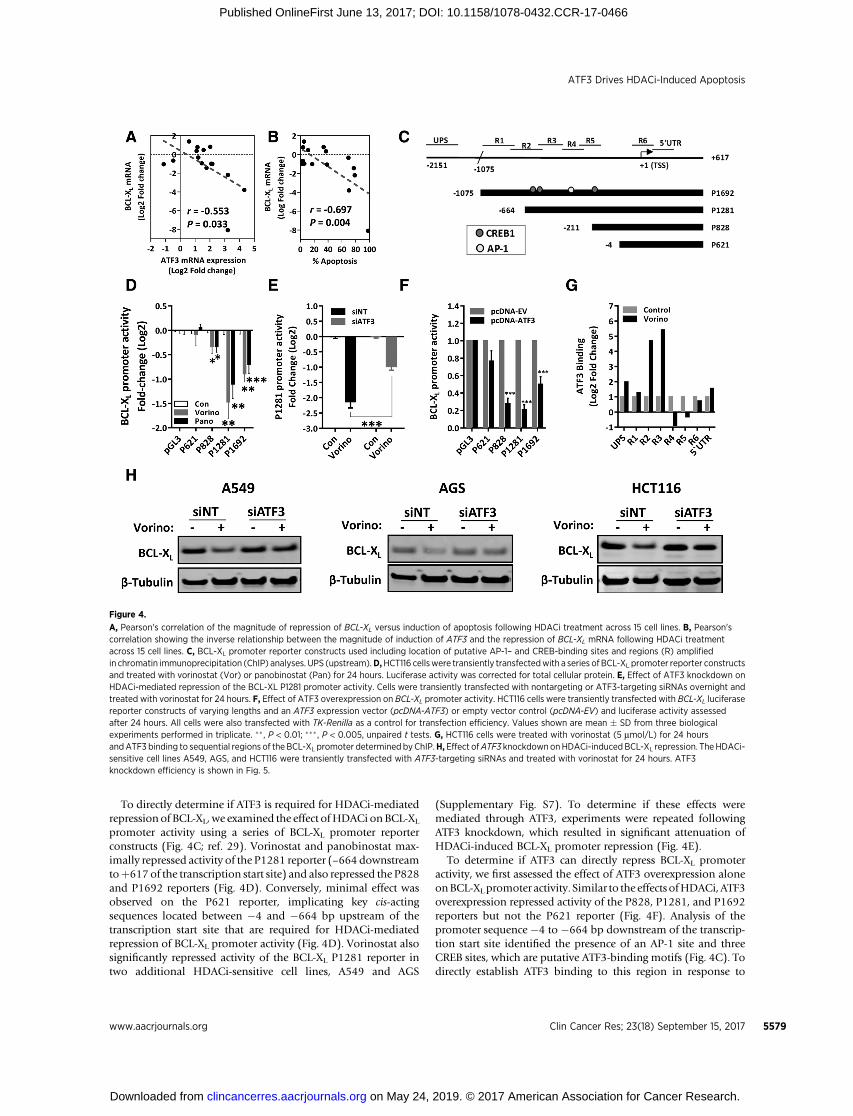
To directly determine if ATF3 is required for HDACi-mediatedrepression of BCL-XL, we examined the effect ofHDACi onBCL-XL
promoter activity using a series of BCL-XL promoter reporterconstructs (Fig. 4C; ref. 29). Vorinostat and panobinostat max-imally repressed activity of the P1281 reporter (–664 downstreamtoþ617 of the transcription start site) and also repressed the P828and P1692 reporters (Fig. 4D). Conversely, minimal effect wasobserved on the P621 reporter, implicating key cis-actingsequences located between �4 and �664 bp upstream of thetranscription start site that are required for HDACi-mediatedrepression of BCL-XL promoter activity (Fig. 4D). Vorinostat alsosignificantly repressed activity of the BCL-XL P1281 reporter intwo additional HDACi-sensitive cell lines, A549 and AGS
(Supplementary Fig. S7). To determine if these effects weremediated through ATF3, experiments were repeated followingATF3 knockdown, which resulted in significant attenuation ofHDACi-induced BCL-XL promoter repression (Fig. 4E).
To determine if ATF3 can directly repress BCL-XL promoteractivity, we first assessed the effect of ATF3 overexpression aloneonBCL-XLpromoter activity. Similar to the effects ofHDACi, ATF3overexpression repressed activity of the P828, P1281, and P1692reporters but not the P621 reporter (Fig. 4F). Analysis of thepromoter sequence �4 to �664 bp downstream of the transcrip-tion start site identified the presence of an AP-1 site and threeCREB sites, which are putative ATF3-binding motifs (Fig. 4C). Todirectly establish ATF3 binding to this region in response to
Figure 4.
A, Pearson's correlation of the magnitude of repression of BCL-XL versus induction of apoptosis following HDACi treatment across 15 cell lines. B, Pearson'scorrelation showing the inverse relationship between the magnitude of induction of ATF3 and the repression of BCL-XL mRNA following HDACi treatmentacross 15 cell lines. C, BCL-XL promoter reporter constructs used including location of putative AP-1– and CREB-binding sites and regions (R) amplifiedin chromatin immunoprecipitation (ChIP) analyses. UPS (upstream).D,HCT116 cellswere transiently transfectedwith a series of BCL-XL promoter reporter constructsand treated with vorinostat (Vor) or panobinostat (Pan) for 24 hours. Luciferase activity was corrected for total cellular protein. E, Effect of ATF3 knockdown onHDACi-mediated repression of the BCL-XL P1281 promoter activity. Cells were transiently transfected with nontargeting or ATF3-targeting siRNAs overnight andtreated with vorinostat for 24 hours. F, Effect of ATF3 overexpression on BCL-XL promoter activity. HCT116 cells were transiently transfected with BCL-XL luciferasereporter constructs of varying lengths and an ATF3 expression vector (pcDNA-ATF3) or empty vector control (pcDNA-EV) and luciferase activity assessedafter 24 hours. All cells were also transfected with TK-Renilla as a control for transfection efficiency. Values shown are mean � SD from three biologicalexperiments performed in triplicate. �� , P < 0.01; ��� , P < 0.005, unpaired t tests. G, HCT116 cells were treated with vorinostat (5 mmol/L) for 24 hoursandATF3 binding to sequential regions of the BCL-XL promoter determined byChIP.H,Effect ofATF3 knockdown onHDACi-inducedBCL-XL repression. TheHDACi-sensitive cell lines A549, AGS, and HCT116 were transiently transfected with ATF3-targeting siRNAs and treated with vorinostat for 24 hours. ATF3knockdown efficiency is shown in Fig. 5.
ATF3 Drives HDACi-Induced Apoptosis
www.aacrjournals.org Clin Cancer Res; 23(18) September 15, 2017 5579
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466
D
C
- + - + siNT siBCL-XL
PC3
- + - + siNT siBCL-XL
U87
BCL-XL
Vorino:
β-Tubulin
siNT siBCL-XL
- + - +
Vorino:siNT siBCL-XL
+ + --
PANC1A
***
*
**
*
**
siNT siBCL-XL
+ + --siNT siBCL-XL
+ + --
U87PC3 HCT116PANC1
F
E
B
Flag Tag
BCL-XL
β-Tubulin
% A
popt
osis
0 200
20
40
60 WTBCL-XL O/E
*
[Vorino] (µmol/L)
O/EEndog.
0
10
20
% A
po
pto
sis
0
Vorino: – –
– – +
+ +
+
– –
– – +
+ +
+
– –
– – +
+ +
+
– –
– – +
+ +
+
– –
– – +
+ +
+
– –
– – +
+ +
+
– –
– – +
+ +
+
– –
– – +
+ +
+
ABT-263:
Vorino:
A1331852:
20
40
60
80
100
% A
po
pto
sis
0
20
40
60
80
100
% A
po
pto
sis
0
20
40
60
80
100
% A
po
pto
sis
0
20
40
60
80
100
% A
po
pto
sis
0
20
40
60
80
100
% A
po
pto
sis
0
20
40
60
80
100
% A
po
pto
sis
0
20
40
60
80
100
% A
po
pto
sis
0
20
40
60
80
100
% A
po
pto
sis
30
40
0
10
20
% A
po
pto
sis 30
40
0
10
20
% A
po
pto
sis 30
40
******
***
***
***
******
*
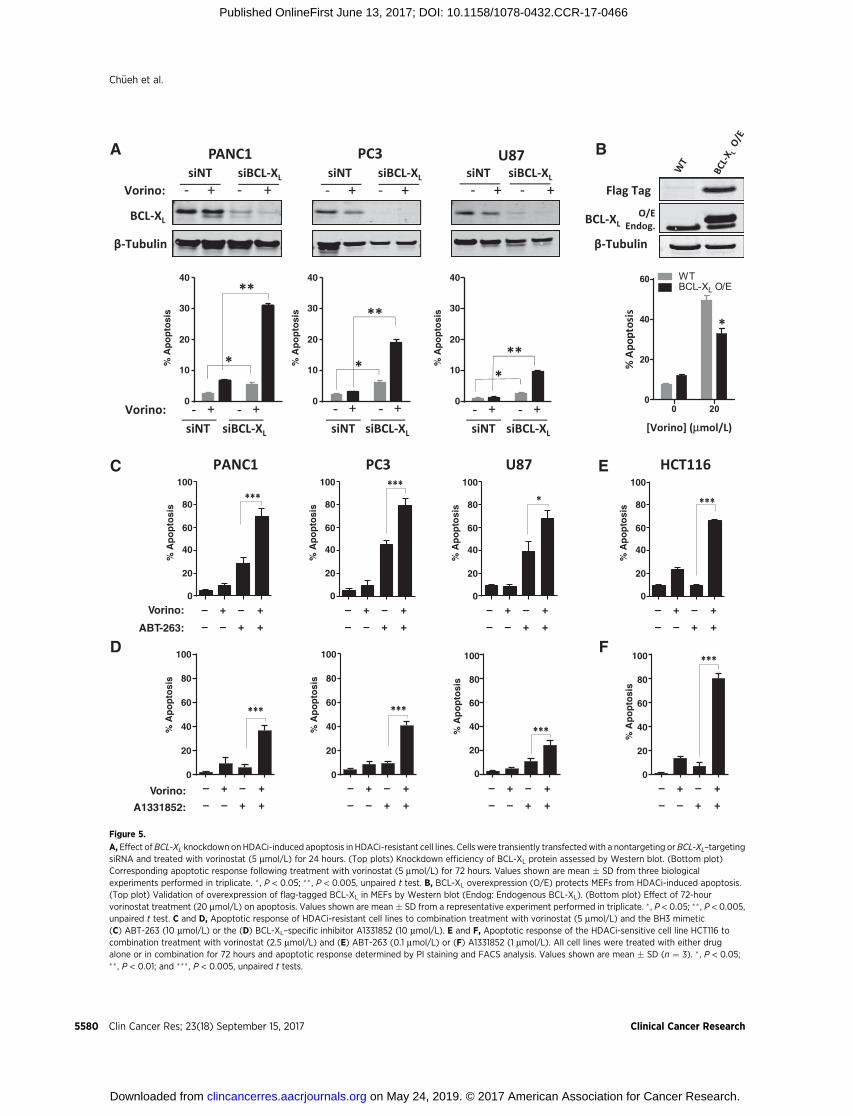
Figure 5.
A, Effect of BCL-XL knockdown onHDACi-induced apoptosis in HDACi-resistant cell lines. Cells were transiently transfectedwith a nontargeting or BCL-XL–targetingsiRNA and treated with vorinostat (5 mmol/L) for 24 hours. (Top plots) Knockdown efficiency of BCL-XL protein assessed by Western blot. (Bottom plot)Corresponding apoptotic response following treatment with vorinostat (5 mmol/L) for 72 hours. Values shown are mean � SD from three biologicalexperiments performed in triplicate. � , P < 0.05; �� , P < 0.005, unpaired t test. B, BCL-XL overexpression (O/E) protects MEFs from HDACi-induced apoptosis.(Top plot) Validation of overexpression of flag-tagged BCL-XL in MEFs by Western blot (Endog: Endogenous BCL-XL). (Bottom plot) Effect of 72-hourvorinostat treatment (20 mmol/L) on apoptosis. Values shown are mean � SD from a representative experiment performed in triplicate. � , P < 0.05; �� , P < 0.005,unpaired t test. C and D, Apoptotic response of HDACi-resistant cell lines to combination treatment with vorinostat (5 mmol/L) and the BH3 mimetic(C) ABT-263 (10 mmol/L) or the (D) BCL-XL–specific inhibitor A1331852 (10 mmol/L). E and F, Apoptotic response of the HDACi-sensitive cell line HCT116 tocombination treatment with vorinostat (2.5 mmol/L) and (E) ABT-263 (0.1 mmol/L) or (F) A1331852 (1 mmol/L). All cell lines were treated with either drugalone or in combination for 72 hours and apoptotic response determined by PI staining and FACS analysis. Values shown are mean � SD (n ¼ 3). � , P < 0.05;�� , P < 0.01; and ��� , P < 0.005, unpaired t tests.
Ch€ueh et al.
Clin Cancer Res; 23(18) September 15, 2017 Clinical Cancer Research5580
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466

HDACi treatment, we performed ATF3 chromatin immunopre-cipitation (ChIP) experiments which sequentially interrogatedATF3 binding along the BCL-XL promoter. The most robustenrichment of ATF3 binding following vorinostat treatment wasobserved at regions R2 and R3 (Fig. 4G), overlapping the keyregulatory region (�4 to�664) identified in the promoter report-er assays. Notably, HDACi and ATF3 overexpressions were able torepress the P828 promoter despite the lack of ATF3 binding to thisregion, suggesting that ATF3 may also indirectly repress BCL-XL
promoter activity.Finally, to establish the requirement of ATF3 induction for
HDACi-mediated repression of BCL-XL at the endogenous level,ATF3 knockdown was performed in three sensitive cell lines priorto HDACi treatment. In each case, ATF3 knockdown markedlyattenuated BCL-XL repression in response to HDACi treatment(Fig. 4H), establishingATF3 induction as a critical requirement forHDACi-mediated BCL-XL repression.
BCL-XL inhibition overcomes inherent resistance to HDACi-induced apoptosis
We next examined the importance of BCL-XL repression inHDACi-induced apoptosis. Knockdown of BCL-XL in HDACi-refractory PANC1, U87, and PC3 cells significantly enhancedHDACi-induced apoptosis, implicating BCL-XL repression as akey determinant of HDACi response (Fig. 5A). Conversely, BCL-XL overexpression in FLAG-tagged hBCL-XL MEFs conferred resis-tance to vorinostat-induced apoptosis compared with WT MEFs(Fig. 5B), collectively establishing BCL-XL repression as a keydeterminant of HDACi-induced apoptosis.
These findings suggested that therapeutic targeting of BCL-XL
may have similar effects. To test this, the HDACi-resistant celllines PANC1, PC3, and U87 were treated with vorinostat aloneand in combination with the BH3 mimetics ABT-263 (navito-clax), which inhibits BCL-2, BCL-XL, and BCL-w. Combinationtreatment significantly enhanced apoptosis compared witheither agent alone in each cell line (Fig. 5C). Similar effectswere obtained using its precursor compound, ABT-737 (Sup-plementary Fig. S8A). To directly determine the role of BCL-XL,we next examined the effects of combining HDACi with thenovel BCL-XL–specific inhibitor, A-1331852 (33). Combina-tion treatment significantly enhanced apoptosis in all 3 celllines compared with either agent alone (Fig. 5D). In contrast,combination treatment with the BCL-2–specific inhibitor ABT-199 (venetoclax) resulted in modest to no enhancement ofHDACi-induced apoptosis (Supplementary Fig. S8B). We nextdetermined whether this combination could also be utilized inHDACi-sensitive cell lines, by enabling each drug to be used atsignificantly lower concentrations. Treatment of the HDACi-sensitive cell line HCT116 with a 2-fold lower concentration ofvorinostat (2.5 mmol/L) and a 100-fold lower concentration ofABT-263 (0.1 mmol/L), or a 10-fold lower concentration of A-1331852 (1 mmol/L) to that used in resistant cells was stillsufficient to induce >60% apoptosis (Fig. 5E and F).
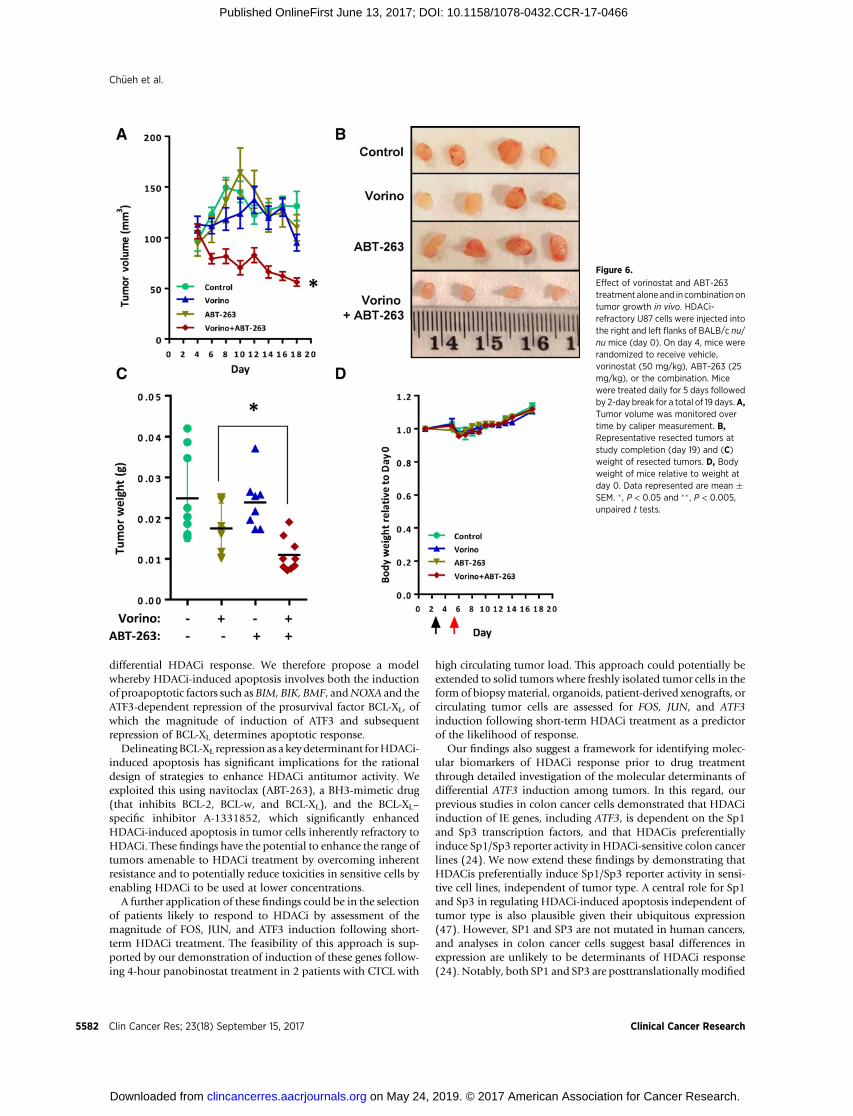
As A-1331852 is not suitable for use in vivo, we next tested theeffect of combination treatment with vorinostat and ABT-263 ongrowth of HDACi-refractory U87 xenografts in vivo. Daily treat-ment with the combination significantly inhibited tumor growthcompared with control or either agent alone (Fig. 6A–C). Impor-tantly, no differences in body weight were observed in either thesingle agent or combination treatment arms compared withcontrol (Fig. 6D).
DiscussionHDACis are an established treatment for hematologic malig-
nancies (CTCL, multiple myeloma) and continue to be tested,mostly in combination, for activity in other tumor types (1).Comparatively, the activity of these agents in solid tumors ismorelimited. The goal of this study was to define the mechanisms ofHDACi action in tumor cells in order to provide a framework forthe rational design of drug combinations involving their use, andthe identification of molecular determinants of sensitivity.
We previously demonstrated that HDACi-induced apoptosis incolon cancer cells is associated with a specific transcriptionalresponse involving the induction of multiple IE response genes,including 3members of the AP-1 transcription factor family, FOS,JUN and ATF3. We now demonstrate that this transcriptionalresponse provides a robust and early readout of HDACi-inducedapoptosis which transcends tumor cell type.
Although HDACi treatment preferentially induces expressionof three AP-1 familymembers in sensitive cell lines, we found thatonly ATF3 is required for HDACi-driven apoptosis. The proapop-totic role for ATF3 identified herein is consistent with ATF3overexpression alone being sufficient to induce apoptosis inprostate (34) and ovarian cancer cells (35), and the resistance ofAtf3 knockout MEFs to UV-induced apoptosis (36). Furthermore,ATF3 is required for apoptosis induced by ER stress (37), anoxia(38), and the chemotherapeutic agents 5FU, etoposide, andcisplatin (39–41). Finally, ATF3 is required for apoptotic sensi-tization to HDACi combination therapy with cisplatin and ago-nistic anti-DR5 antibodies (41, 42) and for HDACi-inducedapoptosis in bladder cancer cells (43). However, the subsequentmechanisms of apoptosis induction have not been investigated.
Prior studies have linked HDACi-induced apoptosis withaltered expression of a number of pro- and antiapoptotic genes,particularly components of the intrinsic apoptotic pathway (43).However, these effects have not been investigated in the context ofsensitivity across tumor cell type, and the mechanisms whichunderpin altered expression of these genes have not been system-atically addressed. The current study identifies a uniform mech-anism that determines HDACi-induced apoptosis, involvingATF3-mediated repression of BCL-XL, which transcends tumorcell type. The role of ATF3 as a transcriptional repressor isconsistent with prior reports (44), and our ChIP and reportergene analysis indicate that repression of BCL-XL inHDACi-treatedtumor cells involves direct binding of ATF3 to the BCL-XL pro-moter. Furthermore, we demonstrate that repression of BCL-XL iscentral in HDACi-induced apoptosis, as both molecular andpharmacologic inhibition of this prosurvival factor markedlyenhanced HDACi-induced apoptosis in vitro and in vivo, andBCL-XL overexpression protects cells from HDAC-induced apo-ptosis, consistent with previous studies (18, 45).
However, we note that the molecular or pharmacologic inhi-bition of BCL-XL alone did not induce apoptosis to the sameextent as when BCL-XL was inhibited in the presence of HDACi,suggesting the requirement for additionalHDACi-inducedmolec-ular changes to drive apoptosis. In this regard, we did identifyconsistent induction of the proapoptotic BH3-only genes BIM,BIK, BMF, andNOXA in response to HDACi treatment, several ofwhich has been shown to be required for HDACi-induced apo-ptosis (32, 46). Notably however, induction of these genesoccurred uniformly across the cell lines, independent of apoptoticsensitivity, implying their altered expression is not the basis for
ATF3 Drives HDACi-Induced Apoptosis
www.aacrjournals.org Clin Cancer Res; 23(18) September 15, 2017 5581
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466
differential HDACi response. We therefore propose a modelwhereby HDACi-induced apoptosis involves both the inductionof proapoptotic factors such asBIM, BIK, BMF, andNOXA and theATF3-dependent repression of the prosurvival factor BCL-XL, ofwhich the magnitude of induction of ATF3 and subsequentrepression of BCL-XL determines apoptotic response.
Delineating BCL-XL repression as a keydeterminant forHDACi-induced apoptosis has significant implications for the rationaldesign of strategies to enhance HDACi antitumor activity. Weexploited this using navitoclax (ABT-263), a BH3-mimetic drug(that inhibits BCL-2, BCL-w, and BCL-XL), and the BCL-XL–
specific inhibitor A-1331852, which significantly enhancedHDACi-induced apoptosis in tumor cells inherently refractory toHDACi. These findings have the potential to enhance the range oftumors amenable to HDACi treatment by overcoming inherentresistance and to potentially reduce toxicities in sensitive cells byenabling HDACi to be used at lower concentrations.
A further application of these findings could be in the selectionof patients likely to respond to HDACi by assessment of themagnitude of FOS, JUN, and ATF3 induction following short-term HDACi treatment. The feasibility of this approach is sup-ported by our demonstration of induction of these genes follow-ing 4-hour panobinostat treatment in 2 patients with CTCL with
high circulating tumor load. This approach could potentially beextended to solid tumors where freshly isolated tumor cells in theform of biopsymaterial, organoids, patient-derived xenografts, orcirculating tumor cells are assessed for FOS, JUN, and ATF3induction following short-term HDACi treatment as a predictorof the likelihood of response.
Our findings also suggest a framework for identifying molec-ular biomarkers of HDACi response prior to drug treatmentthrough detailed investigation of the molecular determinants ofdifferential ATF3 induction among tumors. In this regard, ourprevious studies in colon cancer cells demonstrated that HDACiinduction of IE genes, including ATF3, is dependent on the Sp1and Sp3 transcription factors, and that HDACis preferentiallyinduce Sp1/Sp3 reporter activity in HDACi-sensitive colon cancerlines (24). We now extend these findings by demonstrating thatHDACis preferentially induce Sp1/Sp3 reporter activity in sensi-tive cell lines, independent of tumor type. A central role for Sp1and Sp3 in regulating HDACi-induced apoptosis independent oftumor type is also plausible given their ubiquitous expression(47). However, SP1 and SP3 are not mutated in human cancers,and analyses in colon cancer cells suggest basal differences inexpression are unlikely to be determinants of HDACi response(24). Notably, both SP1 and SP3 are posttranslationallymodified
Figure 6.
Effect of vorinostat and ABT-263treatment aloneand in combination ontumor growth in vivo. HDACi-refractory U87 cells were injected intothe right and left flanks of BALB/c nu/nu mice (day 0). On day 4, mice wererandomized to receive vehicle,vorinostat (50 mg/kg), ABT-263 (25mg/kg), or the combination. Micewere treated daily for 5 days followedby 2-day break for a total of 19 days.A,Tumor volume was monitored overtime by caliper measurement. B,Representative resected tumors atstudy completion (day 19) and (C)weight of resected tumors. D, Bodyweight of mice relative to weight atday 0. Data represented are mean �SEM. � , P < 0.05 and �� , P < 0.005,unpaired t tests.
Ch€ueh et al.
Clin Cancer Res; 23(18) September 15, 2017 Clinical Cancer Research5582
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466

by a number of mechanisms including acetylation, ubiquitina-tion, and phosphorylation which can alter their activity (48).Exploration of whether such posttranslational modificationsoccur in response to HDACi treatment and identification of thefactors regulating these changes, which may vary between sensi-tive and resistant cells,mayprovide novel insight into the basis fordifferential HDACi response. Notably, several other mechanismsof ATF3 induction have also been described, including inductionby p53 (49), activation of JNK, ERK, and p38 signaling (50), andby ATF4 subsequent to activation of the ER stress/unfoldedprotein response pathway (37). In addition to modulating SP1and SP3, HDACi can also affect these pathways which maycontribute to the differential induction of ATF3 among tumors.
In summary, we have identified a specific transcriptionalresponse associated with HDACi-induced apoptosis that trans-cends tumor type, involving the coordinate induction of FOS,JUN, andATF3.We identify the inductionofATF3 and subsequentrepression of BCL-XL as a central mechanism of HDACi-inducedapoptosis and applied these findings to develop rational drugcombinations which overcome inherent resistance and enhancethe activity of HDACi in a range of tumor types.
Disclosure of Potential Conflicts of InterestM. Dickinson reports receiving other commercial research support from,
reports receiving speakers bureau honoraria from, and is a consultant/advisoryboard member for Novartis. No potential conflicts of interest were disclosed bythe other authors.
Authors' ContributionsConception and design: A.C. Ch€ueh, J.W.T. Tse, P. Ioannidis, M.R. Thompson,W.D. Fairlie, A.S. Dhillon, J.M. MariadasonDevelopment of methodology: A.C. Ch€ueh, J.W.T. Tse, P. Ioannidis, L. Togel,E. Lee, J.M. Mariadason
Acquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): A.C. Ch€ueh, J.W.T. Tse, M. Dickinson, P. Ioannidis,L. Jenkins, B. Tan, I. Luk, R. Nightingale, J.M. MariadasonAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): A.C. Ch€ueh, J.W.T. Tse, M. Dickinson, P. Ioannidis,A.S. Dhillon, J.M. MariadasonWriting, review, and/or revision of the manuscript: A.C. Ch€ueh, J.W.T. Tse,M. Dickinson, L. Togel, B.R.G. Williams, G. Lessene, E.F. Lee, W.D. Fairlie,A.S. Dhillon, J.M. MariadasonAdministrative, technical, or material support (i.e., reporting ororganizing data, constructing databases): A.C. Ch€ueh, J.W.T. Tse, L. Togel,M. Davalos-Salas, J.M. MariadasonStudy supervision: A.C. Ch€ueh, A.S. Dhillon, J.M. Mariadason
AcknowledgmentsWe thank Paul G. Ekert (Murdoch Children's Research Institute), Kaye
Wycherley (Walter Eliza Hall Institute for Medical Research), Andrew Wei(Alfred Hospital), and Michael H. Kershaw (Peter Mac Cancer Centre) forproviding us with the leukemia and multiple myeloma cell lines used in thisstudy.
Grant SupportThis studywas funded by theNational Health andMedical Research Council
(NHMRC) of Australia (1008833 and 1066665), The National Institutes ofHealth (NIH1RO1 CA123316), an Australian Research Council Future Fellow-ship (FT0992234), an NHMRC Senior Research Fellowship (1046092) to J.M.Mariadason, Ludwig Cancer Research, and the Operational Infrastructure Sup-port Program, Victorian Government, Australia. J.W.T. Tse was supported by anAustralian Postgraduate Award.
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received February 17, 2017; revised April 27, 2017; accepted June 9, 2017;published OnlineFirst June 13, 2017.
References1. Ch€ueh AC, Tse JW, Togel L, Mariadason JM. Mechanisms of histone
deacetylase inhibitor-regulated gene expression in cancer cells. Antioxi-dants Redox Signal 2015;23:66–84.
2. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone dea-cetylase inhibitors. Nat Rev Drug Discov 2006;5:769–84.
3. Prince HM, BishtonMJ, Harrison SJ. Clinical studies of histone deacetylaseinhibitors. Clin Cancer Res 2009;15:3958–69.
4. FenichelMP. FDA approves new agent formultiplemyeloma. JNatl CancerInst 2015;107:djv165.
5. Garcia-ManeroG,AtallahE, Khaled SK, ArellanoM,PatnaikMM,Butler TA,et al. Final results from a phase 2 study of pracinostat in combination withazacitidine in elderly patients with acute myeloid leukemia (AML). Blood2015;126:A453.
6. Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM, et al.Vorinostat in solid and hematologic malignancies. J Hematol Oncol2009;2:31.
7. Juergens RA,Wrangle J, Vendetti FP, Murphy SC, ZhaoM, Coleman B, et al.Combination epigenetic therapy has efficacy in patients with refractoryadvanced non-small cell lung cancer. Cancer Discov 2011;1:598–607.
8. Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, KleinPM, et al. Randomized phase II, double-blind, placebo-controlled study ofexemestane with or without entinostat in postmenopausal women withlocally recurrent or metastatic estrogen receptor-positive breast cancerprogressing on treatment with a nonsteroidal aromatase inhibitor. J ClinOncol 2013;31:2128–35.
9. Kovacs JJ,Murphy PJ, Gaillard S, Zhao X,Wu JT, Nicchitta CV, et al. HDAC6regulates Hsp90 acetylation and chaperone-dependent activation of glu-cocorticoid receptor. Mol Cell 2005;18:601–7.
10. Hubbert C,Guardiola A, ShaoR, Kawaguchi Y, ItoA,NixonA, et al.HDAC6is a microtubule-associated deacetylase. Nature 2002;417:455–8.
11. Heerdt BG, Houston MA, Augenlicht LH. Short-chain fatty acid-initiatedcell cycle arrest and apoptosis of colonic epithelial cells is linked tomitochondrial function. Cell Growth Differ 1997;8:523–32.
12. Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, et al.Inhibitors of histone deacetylases induce tumor-selective apoptosisthrough activation of the death receptor pathway. Nat Med 2005;11:71–6.
13. Rosato RR, Almenara JA, Dai Y, Grant S. Simultaneous activation of theintrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitorsand tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) syn-ergistically induces mitochondrial damage and apoptosis in human leu-kemia cells. Mol Cancer Ther 2003;2:1273–84.
14. Chirakkal H, Leech SH, Brookes KE, Prais AL, Waby JS, Corfe BM. Upre-gulation of BAK by butyrate in the colon is associated with increased Sp3binding. Oncogene 2006;25:7192–200.
15. Ruemmele FM, Dionne S, Qureshi I, Sarma DS, Levy E, Seidman EG.Butyratemediates Caco-2 cell apoptosis via up-regulation of pro-apoptoticBAK and inducing caspase-3 mediated cleavage of poly-(ADP-ribose)polymerase (PARP). Cell Death Differ 1999;6:729–35.
16. Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, et al. Thehistone deacetylase inhibitors LAQ824 and LBH589 do not require deathreceptor signaling or a functional apoptosome tomediate tumor cell deathor therapeutic efficacy. Blood 2009;114:380–93.
17. Zhang Y, Adachi M, Kawamura R, Imai K. Bmf is a possible mediator inhistone deacetylase inhibitors FK228 and CBHA-induced apoptosis. CellDeath Differ 2006;13:129–40.
18. Chen S, Dai Y, Pei XY, Grant S. Bim upregulation by histone deacetylaseinhibitors mediates interactions with the Bcl-2 antagonist ABT-737: evi-dence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol Cell Biol2009;29:6149–69.
www.aacrjournals.org Clin Cancer Res; 23(18) September 15, 2017 5583
ATF3 Drives HDACi-Induced Apoptosis
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466

19. Kim YH, Park JW, Lee JY, Kwon TK. Sodium butyrate sensitizes TRAIL-mediated apoptosis by induction of transcription from the DR5 genepromoter through Sp1 sites in colon cancer cells. Carcinogenesis2004;25:1813–20.
20. Facchetti F, Previdi S, Ballarini M, Minucci S, Perego P, La Porta CA.Modulation of pro- and anti-apoptotic factors in human melanoma cellsexposed to histone deacetylase inhibitors. Apoptosis 2004;9:573–82.
21. Ruemmele FM, Schwartz S, Seidman EG, Dionne S, Levy E, Lentze MJ.Butyrate induced Caco-2 cell apoptosis is mediated via the mitochondrialpathway. Gut 2003;52:94–100.
22. Hernandez A, Thomas R, Smith F, Sandberg J, Kim S, Chung DH, et al.Butyrate sensitizes human colon cancer cells to TRAIL-mediated apoptosis.Surgery 2001;130:265–72.
23. Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics2008;3:28–37.
24. Wilson AJ, Ch€ueh AC, Togel L, Corner GA, Ahmed N, Goel S, et al.Apoptotic sensitivity of colon cancer cells to histone deacetylase inhibitorsis mediated by an Sp1/Sp3-activated transcriptional program involvingimmediate-early gene induction. Cancer Res 2010;70:609–20.
25. Mariadason JM, Arango D, Shi Q, Wilson AJ, Corner GA, Nicholas C, et al.Gene expressionprofiling-based prediction of responseof colon carcinomacells to 5-FU and camptothecin. Cancer Res 2003;63:8791–812.
26. Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, et al.Exploiting selective BCL-2 family inhibitors to dissect cell survival depen-dencies and define improved strategies for cancer therapy. Sci Transl Med2015;7:279ra40.
27. Wilson AJ, Byun DS, Popova N, Murray LB, L'Italien K, Sowa Y, et al.Histone deacetylase 3 (HDAC3) andother class IHDACs regulate colon cellmaturation and p21 expression and are deregulated in human coloncancer. J Biol Chem 2006;281:13548–58.
28. Yuan X, Yu L, Li J, Xie G, Rong T, Zhang L, et al. ATF3 suppresses metastasisof bladder cancer by regulating gelsolin-mediated remodeling of the actincytoskeleton. Cancer Res 2013;73:3625–37.
29. ChenN, Chen X,Huang R, ZengH, Gong J,MengW, et al. BCL-xL is a targetgene regulated by hypoxia-inducible factor-1{alpha}. J Biol Chem2009;284:10004–12.
30. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat RevCancer 2003;3:859–68.
31. Vrana JA, Decker RH, Johnson CR, Wang Z, Jarvis WD, Richon VM, et al.Induction of apoptosis in U937 human leukemia cells by suberoylanilidehydroxamic acid (SAHA) proceeds through pathways that are regulated byBcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene1999;18:7016–25.
32. Wiegmans AP, Alsop AE, Bots M, Cluse LA, Williams SP, Banks KM, et al.Deciphering the molecular events necessary for synergistic tumor cellapoptosis mediated by the histone deacetylase inhibitor vorinostat andthe BH3 mimetic ABT-737. Cancer Res 2011;71:3603–15.
33. Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, et al.Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol2013;9:390–7.
34. Huang X, Li X, Guo B. KLF6 induces apoptosis in prostate cancer cellsthrough up-regulation of ATF3. J Biol Chem 2008;283:29795–801.
35. Syed V, Mukherjee K, Lyons-Weiler J, Lau KM, Mashima T, Tsuruo T, et al.Identification of ATF-3, caveolin-1, DLC-1, and NM23-H2 as putative
antitumorigenic, progesterone-regulated genes for ovarian cancer cells bygene profiling. Oncogene 2005;24:1774–87.
36. Lu D, Wolfgang CD, Hai T. Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J Biol Chem2006;281:10473–81.
37. EdagawaM, Kawauchi J, HirataM, GoshimaH, InoueM, Okamoto T, et al.Role of activating transcription factor 3 (ATF3) in endoplasmic reticulum(ER) stress-induced sensitization of p53-deficient human colon cancer cellsto tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis through up-regulation of death receptor 5 (DR5) byzerumbone and celecoxib. J Biol Chem 2014;289:21544–61.
38. Ameri K, Hammond EM, Culmsee C, Raida M, Katschinski DM, WengerRH, et al. Induction of activating transcription factor 3 by anoxia isindependent of p53 and the hypoxic HIF signalling pathway. Oncogene2007;26:284–9.
39. Sato A, Nakama K, Watanabe H, Satake A, Yamamoto A, Omi T, et al. Roleof activating transcription factor 3 protein ATF3 in necrosis and apoptosisinduced by 5-fluoro-20-deoxyuridine. FEBS J 2014;281:1892–900.
40. Mashima T, Udagawa S, Tsuruo T. Involvement of transcriptional repressorATF3 in acceleration of caspase protease activation during DNA damagingagent-induced apoptosis. J Cell Physiol 2001;188:352–8.
41. StGermainC,O'BrienA,Dimitroulakos J. Activating Transcription Factor 3regulates in part the enhanced tumour cell cytotoxicity of the histonedeacetylase inhibitor M344 and cisplatin in combination. Cancer Cell Int2010;10:32.
42. Liu J, EdagawaM, GoshimaH, InoueM, Yagita H, Liu Z, et al. Role of ATF3in synergistic cancer cell killing by a combination of HDAC inhibitors andagonistic anti-DR5 antibody through ER stress in human colon cancer cells.Biochem Biophys Res Commun 2014;445:320–6.
43. Sooraj D, Xu D, Cain JE, Gold DP, Williams BR. Activating TranscriptionFactor 3 Expression as a Marker of Response to the Histone DeacetylaseInhibitor Pracinostat. Mol Cancer Ther 2016;15:1726–39.
44. Thompson MR, Xu D, Williams BR. ATF3 transcription factor and itsemerging roles in immunity and cancer. J Mol Med 2009;87:1053–60.
45. Zhang XD, Gillespie SK, Borrow JM, Hersey P. The histone deacetylaseinhibitor suberic bishydroxamate regulates the expression of multipleapoptotic mediators and induces mitochondria-dependent apoptosis ofmelanoma cells. Mol Cancer Ther 2004;3:425–35.
46. Xargay-Torrent S, Lopez-GuerraM, Saborit-Villarroya I, Rosich L, CampoE,Roue G, et al. Vorinostat-induced apoptosis in mantle cell lymphoma ismediated by acetylation of proapoptotic BH3-only gene promoters. ClinCancer Res 2011;17:3956–68.
47. Wierstra I. Sp1: emerging roles–beyond constitutive activation of TATA-lesshousekeeping genes. Biochem Biophys Res Commun 2008;372:1–13.
48. Waby JS, Bingle CD, Corfe BM. Post-translational control of sp-familytranscription factors. Curr Genomics 2008;9:301–11.
49. Zhang C, Gao C, Kawauchi J, Hashimoto Y, Tsuchida N, Kitajima S.Transcriptional activation of the human stress-inducible transcriptionalrepressor ATF3 gene promoter by p53. Biochem Biophys Res Commun2002;297:1302–10.
50. St Germain C, Niknejad N, Ma L, Garbuio K, Hai T, Dimitroulakos J.Cisplatin induces cytotoxicity through the mitogen-activated proteinkinase pathways and activating transcription factor 3. Neoplasia2010;12:527–38.
Clin Cancer Res; 23(18) September 15, 2017 Clinical Cancer Research5584
Ch€ueh et al.
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466

2017;23:5573-5584. Published OnlineFirst June 13, 2017.Clin Cancer Res Anderly C. Chüeh, Janson W.T. Tse, Michael Dickinson, et al. HDAC Inhibitors across Tumor Types
Determines Apoptotic Sensitivity toLATF3 Repression of BCL-X
Updated version
10.1158/1078-0432.CCR-17-0466doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2017/06/13/1078-0432.CCR-17-0466.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/23/18/5573.full#ref-list-1
This article cites 50 articles, 21 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/23/18/5573To request permission to re-use all or part of this article, use this link
on May 24, 2019. © 2017 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0466