aromatic...
TRANSCRIPT

Chapter 4
Part I
Aromatic Hydrocarbons Nomenclature, Structure,
Properties, and an Introduction to
Synthesis

Chapter 5 2
The discovery of benzene
In 1825, Michael Faraday isolated a pure compound of boiling point 80 °C from
the oily mixture that condensed from illuminating gas, the fuel burned in gaslights.
Elemental analysis showed an unusually small hydrogen-to-carbon ratio of 1:1,
corresponding to an empirical formula of CH. Faraday named the new compound
“bicarburet of hydrogen.”
Eilhard Mitscherlich synthesized the same compound in 1834 by heating
benzoic acid, isolated from gum benzoin, in the presence of lime. Like Faraday,
Mitscherlich found that the empirical formula was C6H6. He also used a vapor-
density measurement to determine a molecular weight of about 78, for a molecular
formula of C6H6. Since the new compound was derived from gum benzoin, he
named it benzin, now called benzene.
Many other compounds discovered in the 19th century seemed to be related to
benzene. These compounds also had low hydrogen-to-carbon ratios as well as
pleasant aromas, and they could be converted to benzene or related compounds.
This group of compounds was called aromatic because of their pleasant odors.
Other organic compounds without these properties were called aliphatic, meaning
“fatlike.” As the unusual stability of aromatic compounds was investigated, the
term aromatic came to be applied to compounds with this stability, regardless of
their odors.
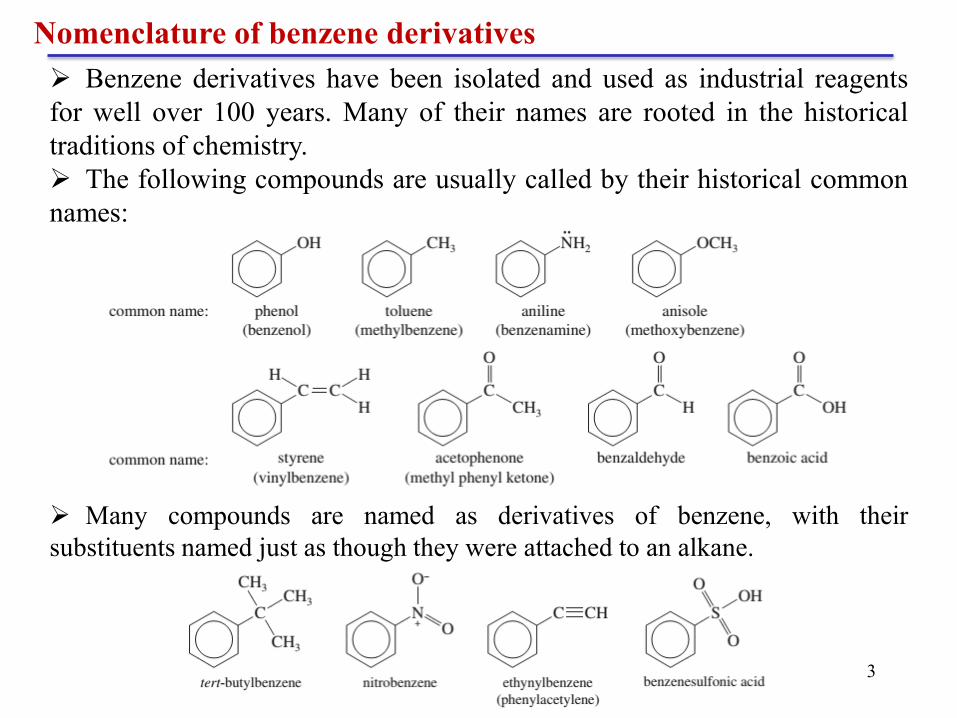
Many compounds are named as derivatives of benzene, with their
substituents named just as though they were attached to an alkane.
3
Nomenclature of benzene derivatives
Benzene derivatives have been isolated and used as industrial reagents
for well over 100 years. Many of their names are rooted in the historical
traditions of chemistry.
The following compounds are usually called by their historical common
names:

4
Nomenclature of benzene derivatives
Disubstituted benzenes are named using the prefixes ortho-, meta-, and
para- to specify the substitution patterns. These terms are abbreviated o-,
m-, and p-. Numbers can also be used to specify the substitution in
disubstituted benzenes.
5
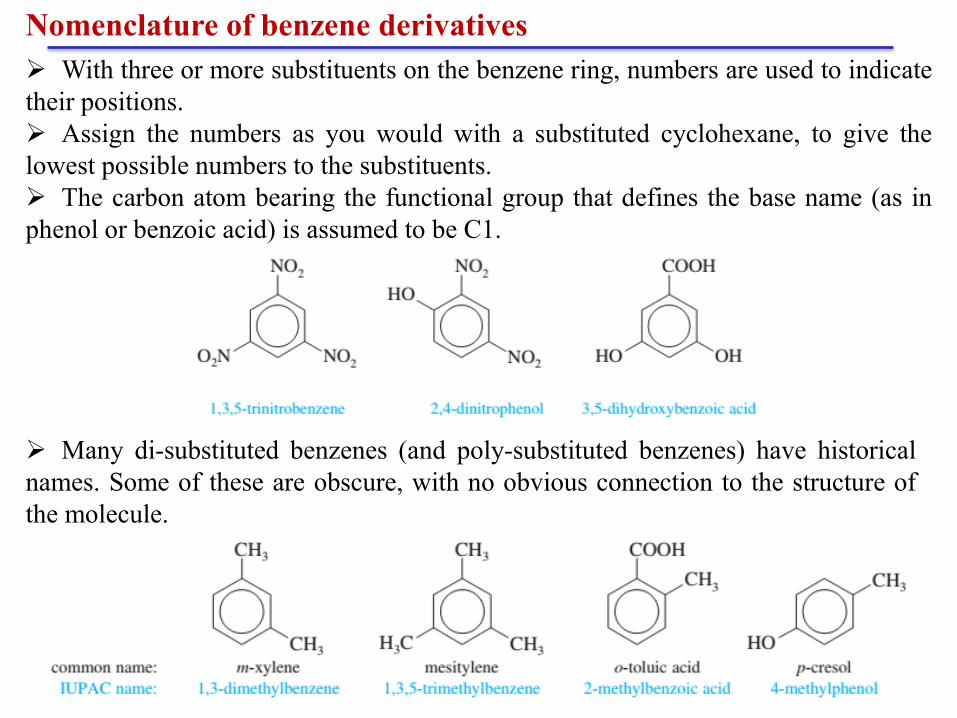
With three or more substituents on the benzene ring, numbers are used to indicate
their positions.
Assign the numbers as you would with a substituted cyclohexane, to give the
lowest possible numbers to the substituents.
The carbon atom bearing the functional group that defines the base name (as in
phenol or benzoic acid) is assumed to be C1.
Many di-substituted benzenes (and poly-substituted benzenes) have historical
names. Some of these are obscure, with no obvious connection to the structure of
the molecule.
Nomenclature of benzene derivatives

6
Physical properties
The seven-carbon unit consisting of a benzene ring and a
methylene group is often named as a benzyl group. + Be careful not to confuse the benzyl group (seven carbons) with the phenyl
group (six carbons).
Aromatic hydrocarbons are sometimes called arenes. An aryl group,
abbreviated Ar, is the aromatic group that remains after the removal of a
hydrogen atom from an aromatic ring.
The phenyl group, Ph, is the simplest aryl group.

Chapter 5 7
Problems

Chapter 5 8
Physical properties of benzene and its derivatives
Benzene derivatives tend to be more symmetrical than similar aliphatic
compounds, so they pack better into crystals and have higher melting points.
+ For example, benzene melts at 6 °C, while hexane melts at -95 oC.
Similarly, para-disubstituted benzenes are more symmetrical than the ortho
and meta isomers, and they pack better into crystals and have higher melting
points.
The relative boiling points of many benzene derivatives are related to their
dipole moments. For example:
+ Symmetrical p-dichlorobenzene has zero dipole moment and the lowest boiling
point.
+ m-Dichlorobenzene has a small dipole moment and a slightly higher boiling
point.
+ o-Dichlorobenzene has the largest dipole moment and the highest boiling point.
+ Even though p-dichlorobenzene has the lowest boiling point, it has the highest
melting point of the dichlorobenzenes because it packs best into a crystal.

Chapter 5 9
Physical properties of benzene and its derivatives
Benzene and other aromatic hydrocarbons are slightly denser than
the nonaromatic analogues, but they are still less dense than water.
The halogenated benzenes are denser than water.
Aromatic hydrocarbons and halogenated aromatics are generally
insoluble in water, although some derivatives with strongly polar
functional groups (phenol, benzoic acid, etc.) are moderately soluble in
water.

Chapter 5 10
Physical properties of benzene and its derivatives

Chapter 5 11
The Kekulé Structure In 1866, Friedrich Kekulé proposed a cyclic
structure for benzene with three double bonds. Considering that multiple
bonds had been proposed only recently (1859), the cyclic structure with
alternating single and double bonds was considered somewhat bizarre.
The Kekulé structure
The Kekulé structure has its shortcomings, however. For example, it
predicts two different 1,2-dichlorobenzenes, but only one is known to
exist. Kekulé suggested (incorrectly) that a fast equilibrium interconverts
the two isomers of 1,2-dichlorobenzene.
Chapter 5
12
The structure and properties of benzene
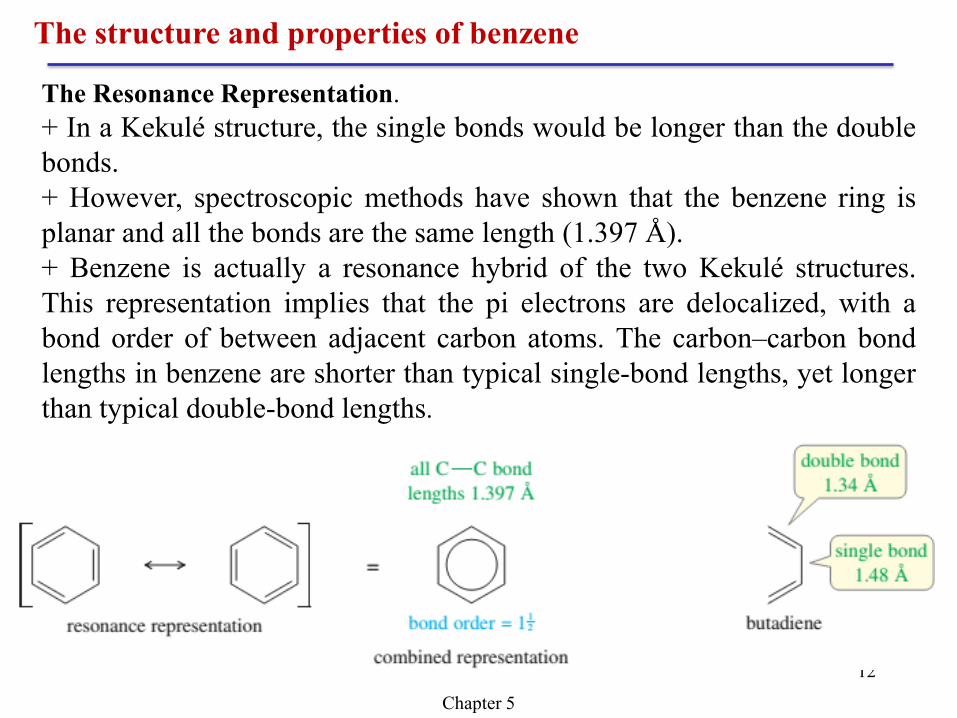
The Resonance Representation.
+ In a Kekulé structure, the single bonds would be longer than the double
bonds.
+ However, spectroscopic methods have shown that the benzene ring is
planar and all the bonds are the same length (1.397 Å).
+ Benzene is actually a resonance hybrid of the two Kekulé structures.
This representation implies that the pi electrons are delocalized, with a
bond order of between adjacent carbon atoms. The carbon–carbon bond
lengths in benzene are shorter than typical single-bond lengths, yet longer
than typical double-bond lengths.

Chapter 5 13
The structure and properties of benzene
Using the resonance picture, we can draw a more realistic
representation of benzene:
+ Benzene is a ring of six carbon atoms, each bonded to one hydrogen
atom.
+ All the carbon–carbon bonds are the same length, and all the bond angles
are exactly 120°.
+ Each carbon atom has an un-hybridized p orbital perpendicular to the
plane of the ring, and six electrons occupy this circle of p orbitals.
At this point, we can define an aromatic compound to be a cyclic
compound containing some number of conjugated double bonds and
having an unusually large resonance energy.

14
Structure of benzene
The Unusual Stability of Benzene
+ Benzene’s reluctance to undergo typical alkene reactions suggests that it must
be unusually stable. By comparing molar heats of hydrogenation, we can get a
quantitative idea of its stability:
1. Hydrogenation of cyclohexene is exothermic by 120 kJ mol (28.6 kcal
mol).
2. Hydrogenation of cyclohexa-1,4-diene is exothermic by 240 kJ mol
(57.4 kcal mol), about twice the heat of hydrogenation of cyclohexene.
The resonance energy of the isolated double bonds in cyclohexa-1,4-
diene is about zero.
3. Hydrogenation of cyclohexa-1,3-diene is exothermic by 232 kJ mol
(55.4 kcal mol), about 8 kJ (1.8 kcal) less than twice the value for
cyclohexene. A resonance energy of 8 kJ (1.8 kcal) is typical for a
conjugated diene.
4. Hydrogenation of benzene requires higher pressures of hydrogen and
a more active catalyst. This hydrogenation is exothermic by 208 kJ mol
(49.8 kcal mol), about 151 kJ (36.0 kcal) less than 3 times the value for
cyclohexene.

15
Chemical properties
The huge 151 kJ mol (36 kcal mol) resonance energy of
benzene cannot be explained by conjugation effects alone.

16
Hückel’s rule
Erich Hückel developed a shortcut for predicting which of the
annulenes and related compounds are aromatic and which are anti-
aromatic.

Chapter 5
17
Chemical properties
The Unusual Reactions of Benzene
Both the Kekulé structure and the resonance-delocalized picture show that
benzene is a cyclic conjugated triene. We might expect benzene to undergo
the typical reactions of polyenes. In fact, its reactions are quite unusual.
+ Benzene is actually much more stable than we would expect from the
simple resonance-delocalized picture.
+ For example, an alkene decolorizes potassium permanganate by reacting
to form a glycol. The purple permanganate color disappears, and a
precipitate of manganese dioxide forms.
+ When permanganate is added to benzene, however, no reaction occurs.
18
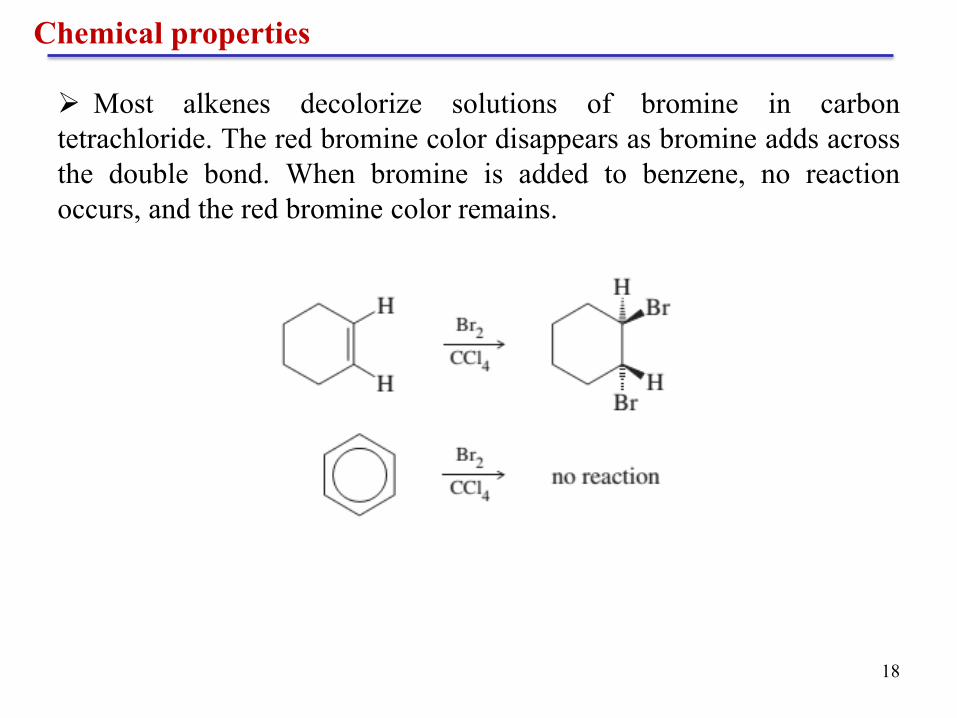
Most alkenes decolorize solutions of bromine in carbon
tetrachloride. The red bromine color disappears as bromine adds across
the double bond. When bromine is added to benzene, no reaction
occurs, and the red bromine color remains.
Chemical properties
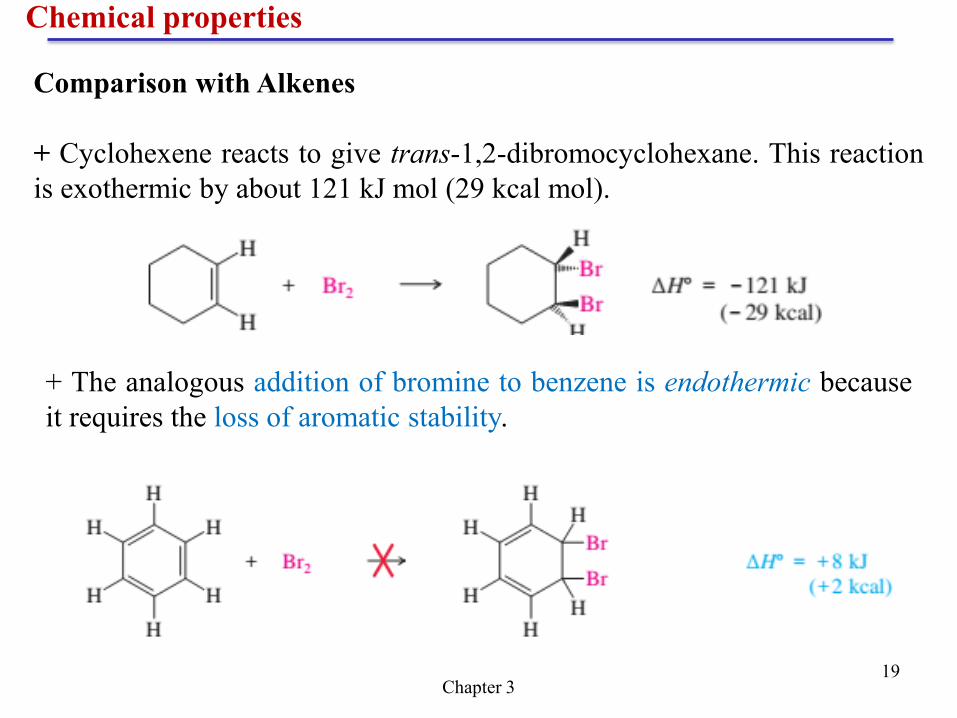
Chapter 3 19
Chemical properties
Comparison with Alkenes
+ Cyclohexene reacts to give trans-1,2-dibromocyclohexane. This reaction
is exothermic by about 121 kJ mol (29 kcal mol).
+ The analogous addition of bromine to benzene is endothermic because
it requires the loss of aromatic stability.

Chapter 5
20
Chemical properties
The substitution of bromine for a hydrogen atom gives an aromatic
product. The substitution is exothermic, but it requires a Lewis acid
catalyst to convert bromine to a stronger electrophile.
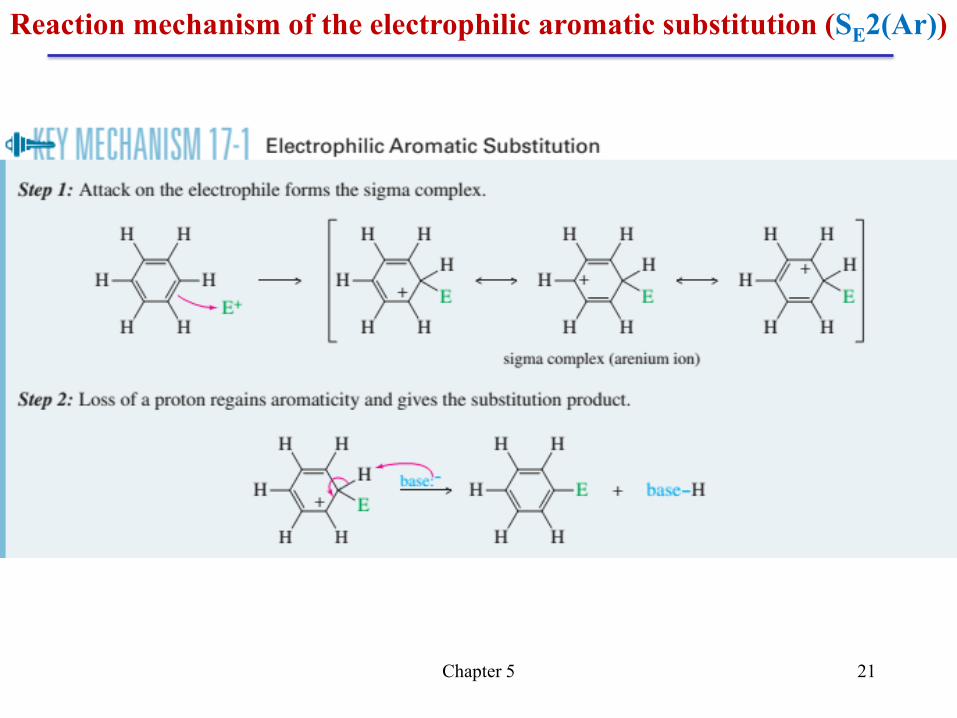
Chapter 5 21
Reaction mechanism of the electrophilic aromatic substitution (SE2(Ar))
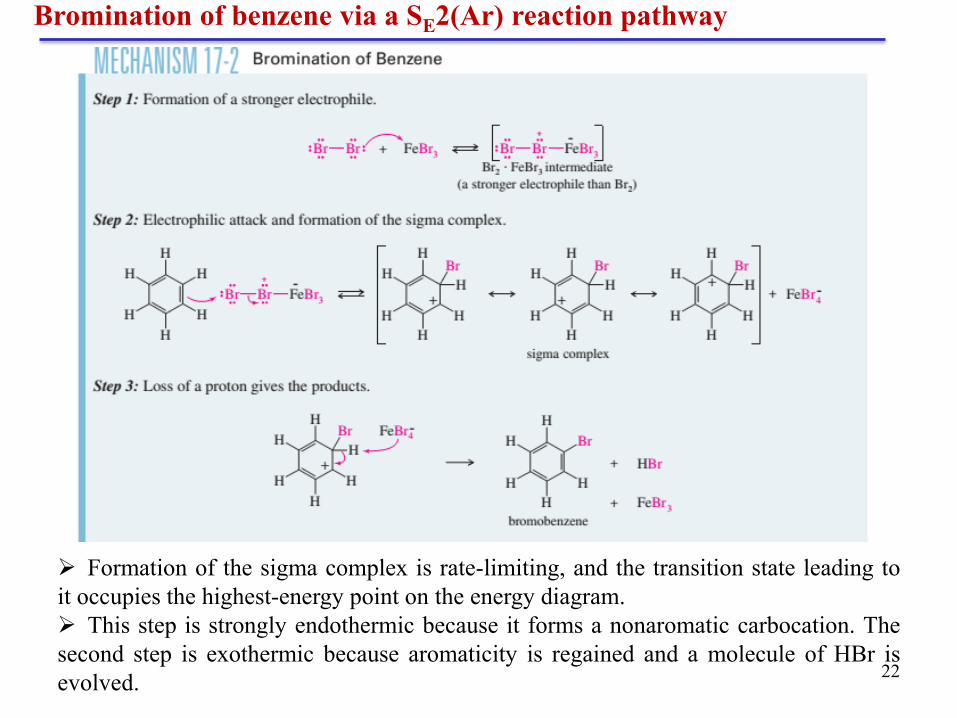
22
Bromination of benzene via a SE2(Ar) reaction pathway
Formation of the sigma complex is rate-limiting, and the transition state leading to
it occupies the highest-energy point on the energy diagram.
This step is strongly endothermic because it forms a nonaromatic carbocation. The
second step is exothermic because aromaticity is regained and a molecule of HBr is
evolved.

Chapter 5 23
Chlorination of benzene via the SE2(Ar) reaction pathway
Chlorination of benzene works much like bromination, except
that aluminum chloride is most often used as the Lewis acid
catalyst.

Chapter 5 24
Iodination of Benzene
Iodination of benzene requires an acidic oxidizing agent, such
as nitric acid.
Nitric acid is consumed in the reaction, so it is a reagent (an
oxidant) rather than a catalyst.
Iodination probably involves an electrophilic aromatic
substitution with the iodine cation acting as the electrophile. The
iodine cation results from oxidation of iodine by nitric acid.

Chapter 5 25
Nitration of benzene via the SE2(Ar) reaction mechanism
Benzene reacts with hot, concentrated nitric acid to give
nitrobenzene. This sluggish reaction is hazardous because a hot
mixture of concentrated nitric acid with any oxidizable material
might explode.
A safer and more convenient procedure uses a mixture of
nitric acid and sulfuric acid. Sulfuric acid is a catalyst, allowing
nitration to take place more rapidly and at lower temperatures.
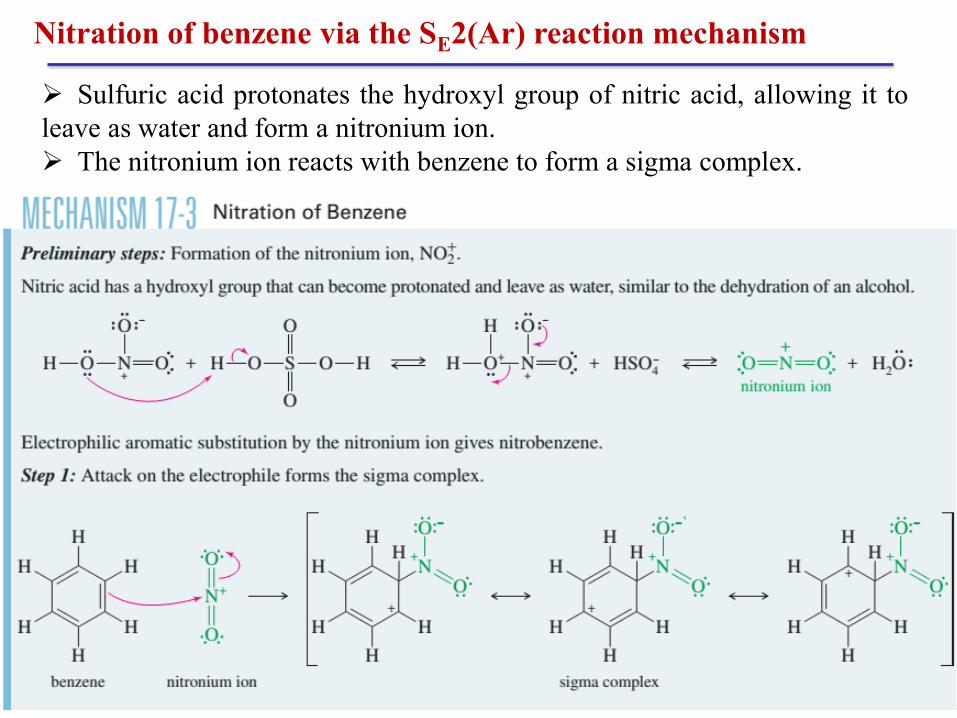
Chapter 3 26
Sulfuric acid protonates the hydroxyl group of nitric acid, allowing it to
leave as water and form a nitronium ion.
The nitronium ion reacts with benzene to form a sigma complex.
Nitration of benzene via the SE2(Ar) reaction mechanism
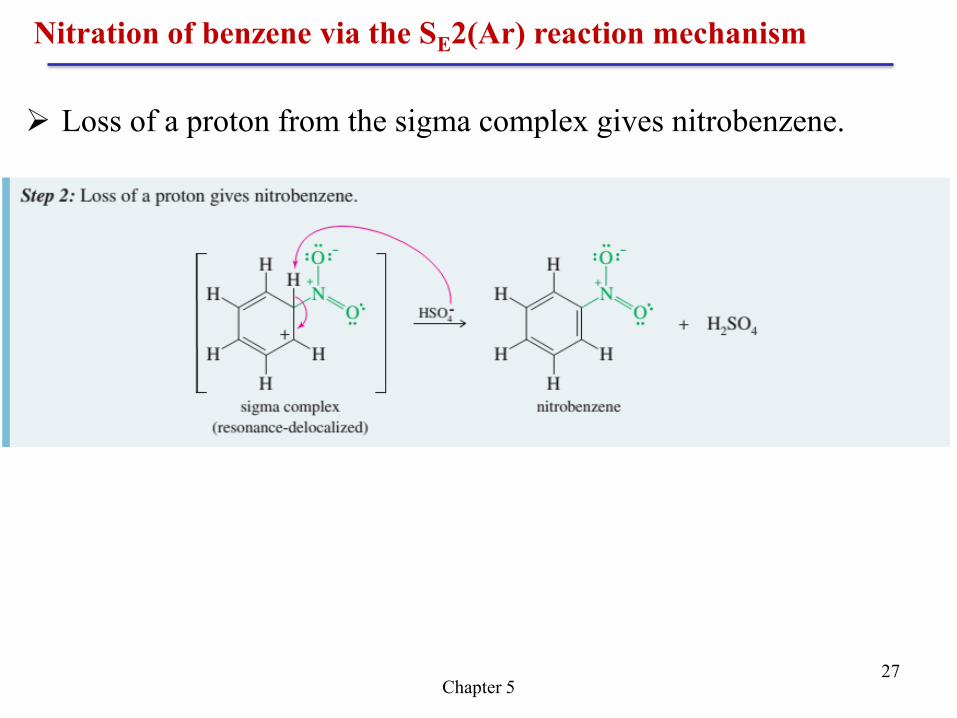
Chapter 5 27
Nitration of benzene via the SE2(Ar) reaction mechanism
Loss of a proton from the sigma complex gives nitrobenzene.

Chapter 5 28
Sulfonation of benzene via the SE2(Ar) pathway
p-Toluenesulfonic acid is an example of an arylsulfonic acid (general
formula ArSO3H), which are often used as strong acid catalysts that are
soluble in nonpolar organic solvents.
Arylsulfonic acids are easily synthesized by sulfonation of benzene
derivatives, an electrophilic aromatic substitution using sulfur trioxide
(SO3) as the electrophile.

Chapter 5 29
Sulfonation of benzene via the SE2(Ar) pathway
“Fuming sulfuric acid” is the common name for a solution of 7% SO3
in H2SO4 Sulfur trioxide is the anhydride of sulfuric acid, meaning that
the addition of water to gives
Although it is uncharged, sulfur trioxide is a strong electrophile, with
three sulfonyl bonds drawing electron density away from the sulfur
atom.
Benzene attacks sulfur trioxide, forming a sigma complex. Loss of a
proton on the tetrahedral carbon and reprotonation on oxygen gives
benzenesulfonic acid.

Chapter 3 30
Reaction mechanism of the sulfonation of benzene

Chapter 5 31
Sulfonation is economically important because alkylbenzene
sulfonates are widely used as detergents. Sulfonation of an
alkylbenzene gives an alkylbenzenesulfonic acid, which is
neutralized with base to give an alkylbenzene sulfonate detergent.
Sulfonation of benzene via the SE2(Ar) pathway

Chapter 5 32
Desulfonation – a reverse reaction of the sulfonation of benzene
Sulfonation is reversible, and a sulfonic acid group may be removed
from an aromatic ring by heating in dilute sulfuric acid. In practice,
steam is often used as a source of both water and heat for
desulfonation.
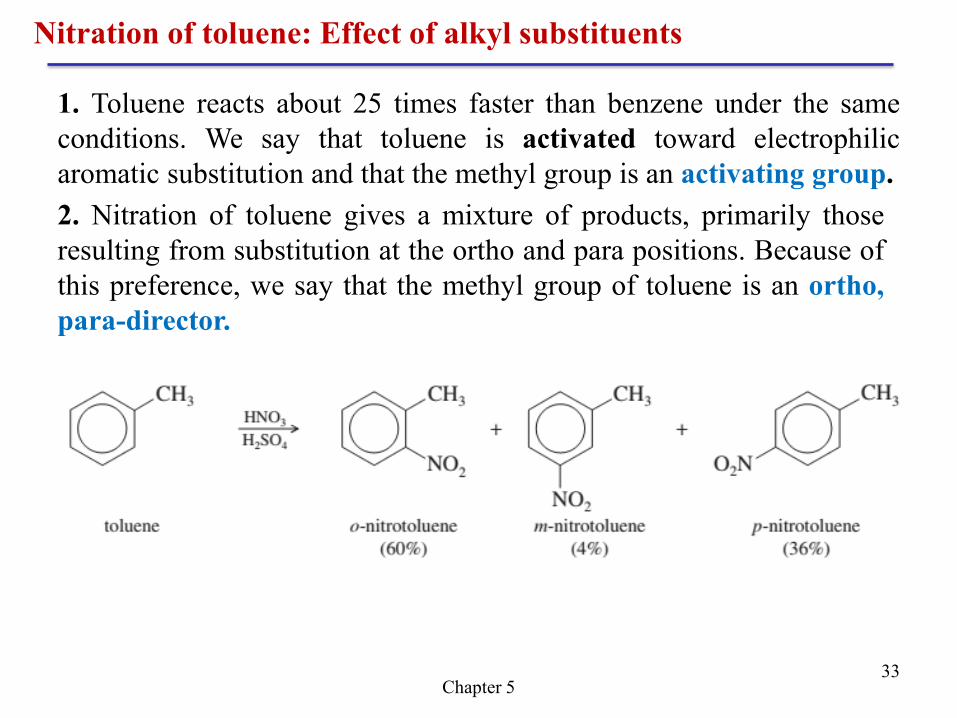
Chapter 5 33
Nitration of toluene: Effect of alkyl substituents
1. Toluene reacts about 25 times faster than benzene under the same
conditions. We say that toluene is activated toward electrophilic
aromatic substitution and that the methyl group is an activating group.
2. Nitration of toluene gives a mixture of products, primarily those
resulting from substitution at the ortho and para positions. Because of
this preference, we say that the methyl group of toluene is an ortho,
para-director.

Chapter 5 34
Effect of alkyl substituents
These product ratios show that the orientation of substitution is
not random.
If each position were equally reactive, there would be equal
amounts of ortho and meta substitution and half as much para
substitution: 40% ortho, 40% meta, and 20% para. This is the
statistical prediction based on the two ortho positions, two meta
positions, and just one para position available for substitution.

Chapter 3 35
The rate-limiting step for electrophilic aromatic substitution is the first
step, formation of the sigma complex. This step is where the electrophile
bonds to the ring, determining the substitution pattern.
When benzene reacts with the nitronium ion, the resulting sigma
complex has the positive charge distributed over three secondary (2°) carbon
atoms.
In ortho or para substitution of toluene, the positive charge is spread
over two secondary carbons and one tertiary (3°) carbon.
Effect of alkyl substituents

Chapter 5 36
Activating, ortho, para-directing substituents: Alkyl Groups
The results observed with toluene are general for any alkylbenzene
undergoing electrophilic aromatic substitution.
Substitution ortho or para to the alkyl group gives a
transition state and an intermediate with the positive charge shared by the
tertiary carbon atom.
As a result, alkylbenzenes undergo electrophilic aromatic substitution faster
than benzene, and the products are predominantly ortho- and para-substituted.
An alkyl group is therefore an activating substituent, and it is ortho, para-
directing.
Shown next is the reaction of ethylbenzene with bromine, catalyzed by ferric
bromide. As with toluene, the rates of formation of the ortho- and para-
substituted isomers are greatly enhanced with respect to the meta isomer.

Chapter 5 37
Activating, ortho, para-directing substituents: Alkyl Groups

Chapter 5
38
Activating, ortho, para-directing substituents:
Substituents with non-bonding electrons - Alkoxy Groups
The EPM of anisole shows the aromatic ring to be
electron-rich (red), consistent with the observation that
anisole is strongly activated toward reactions with
electrophiles.
Anisole (methoxybenzene) undergoes nitration about 10,000 times
faster than benzene and about 400 times faster than toluene. This result
seems curious because oxygen is a strongly electronegative group, yet it
donates electron density to stabilize the transition state and the sigma
complex.

Chapter 5 39
Activating, ortho, para-directing substituents: Alkoxy groups
The second resonance form puts the positive charge on the
electronegative oxygen atom, but it has more covalent bonds, and it
provides each atom with an octet in its valence shell. This type of
stabilization is called resonance stabilization, and the oxygen atom is
called resonance-donating or pi-donating because it donates electron
density through a pi bond in one of the resonance structures.
Like alkyl groups, the methoxy group of anisole preferentially
activates the ortho and para positions.
Recall that the nonbonding electrons of an oxygen atom adjacent
to a carbocation stabilize the positive charge through resonance.
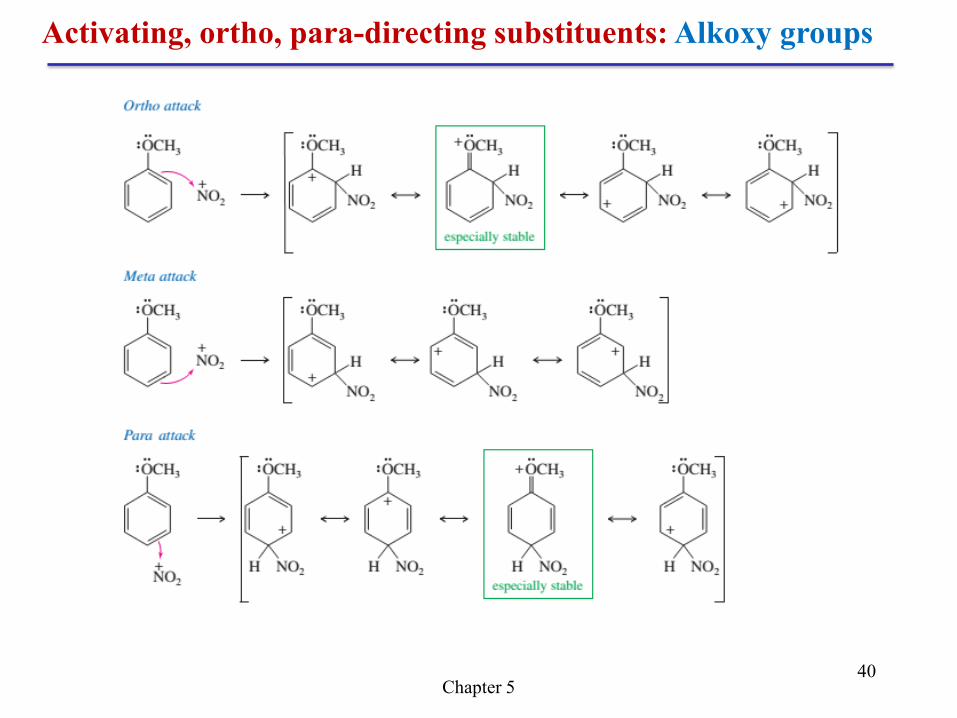
Chapter 5 40
Activating, ortho, para-directing substituents: Alkoxy groups

Chapter 5 41
Activating, ortho, para-directing substituents: Amine groups
Amine Groups Like an alkoxyl group, a nitrogen atom
with a nonbonding pair of electrons serves as a
powerful activating group. For example, aniline
undergoes a fast bromination (without a catalyst) in
bromine water to give the tribromide. Sodium
bicarbonate is added to neutralize the HBr formed and
to prevent protonation of the basic amino group.
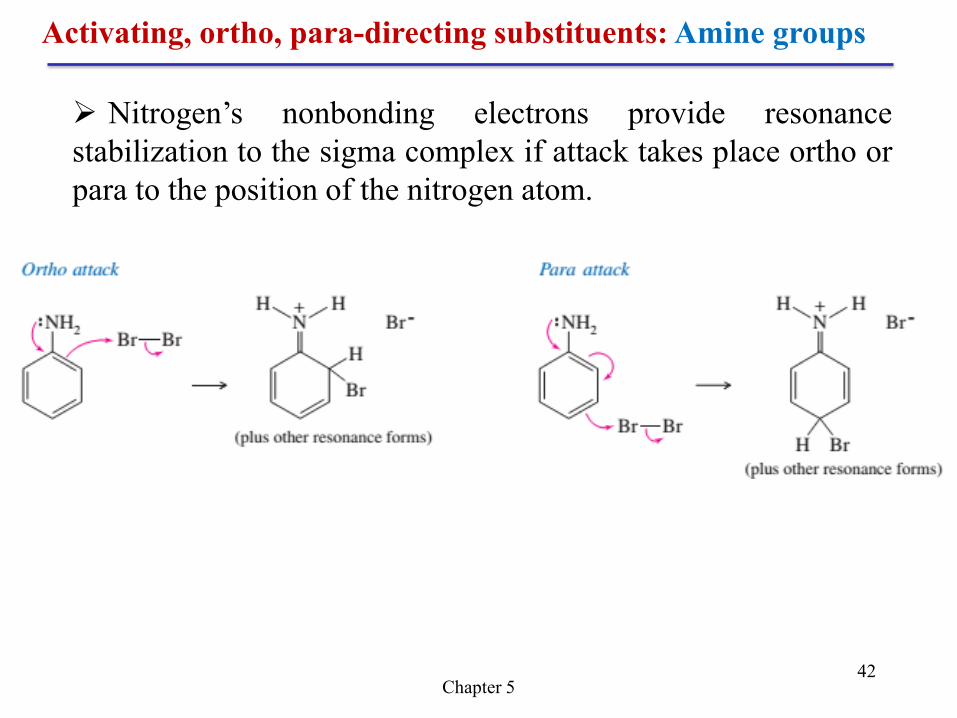
Chapter 5 42
Activating, ortho, para-directing substituents: Amine groups
Nitrogen’s nonbonding electrons provide resonance
stabilization to the sigma complex if attack takes place ortho or
para to the position of the nitrogen atom.
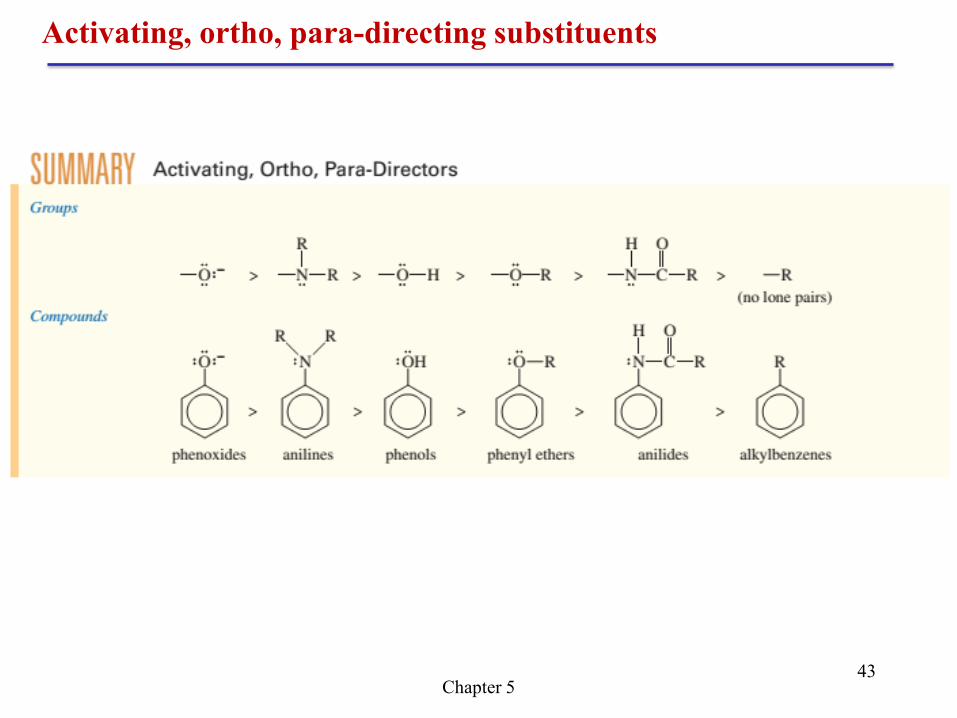
Chapter 5 43
Activating, ortho, para-directing substituents

Chapter 5 44
Deactivating, meta-directing substituents: nitro group

Chapter 5 45
Activating, ortho, para-directing substituents: nitro group
This selective deactivation leaves the meta positions the most reactive, and
meta substitution is seen in the products. Meta-directors, often called
meta-allowing substituents, deactivate the meta position less than the ortho
and para positions, allowing meta substitution.
We can show why the nitro group is a strong deactivating group by
considering its resonance forms. No matter how we position the electrons
in a Lewis dot diagram, the nitrogen atom always has a formal positive
charge.

Chapter 5 46
Activating, ortho, para-directing substituents: nitro group
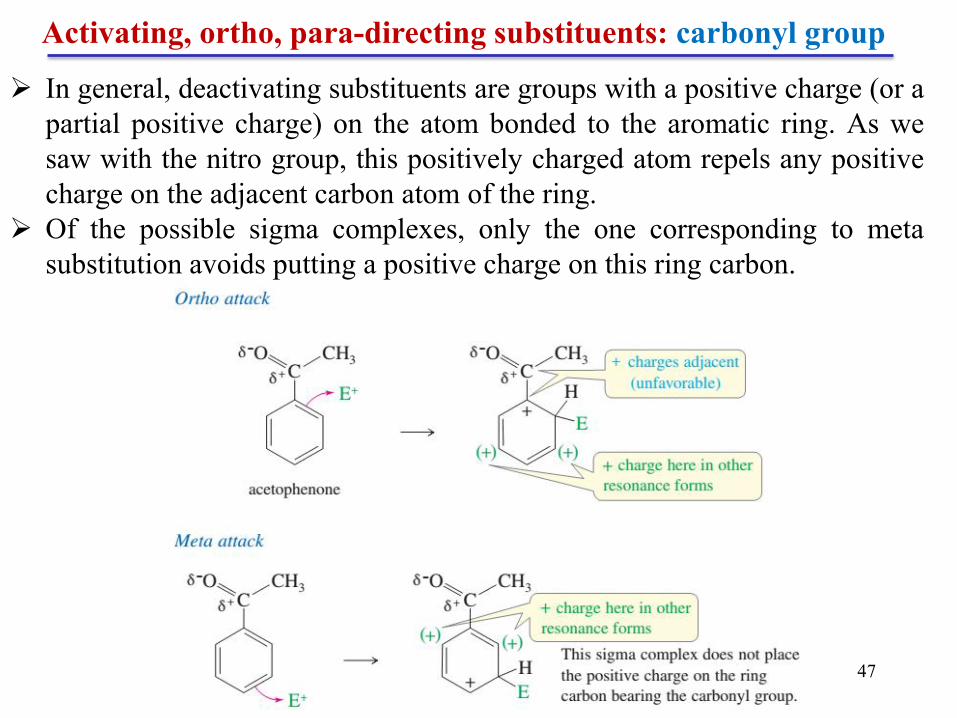
Chapter 5 47
Activating, ortho, para-directing substituents: carbonyl group
In general, deactivating substituents are groups with a positive charge (or a
partial positive charge) on the atom bonded to the aromatic ring. As we
saw with the nitro group, this positively charged atom repels any positive
charge on the adjacent carbon atom of the ring.
Of the possible sigma complexes, only the one corresponding to meta
substitution avoids putting a positive charge on this ring carbon.

Chapter 5 48
Activating, ortho, para-directing substituents


50
Chapter 5
Halogen substituents: deactivating, but orthor, para directing
The halobenzenes are exceptions to the general rules: Halogens are
deactivating groups, yet they are ortho, para-directors.
We can explain this unusual combination of properties by considering
that
+ The halogens are strongly electronegative, withdrawing electron density
from a carbon atom through the sigma bond (inductive withdrawal). The
carbon–halogen bond is strongly polarized, with the carbon atom at the
positive end of the dipole. This polarization draws electron density away
from the benzene ring, making it less reactive toward electrophilic
substitution.
+ The halogens have nonbonding electrons that can donate electron
density through pi bonding (resonance donation).
+ These inductive and resonance effects oppose each other.

Chapter 5
51
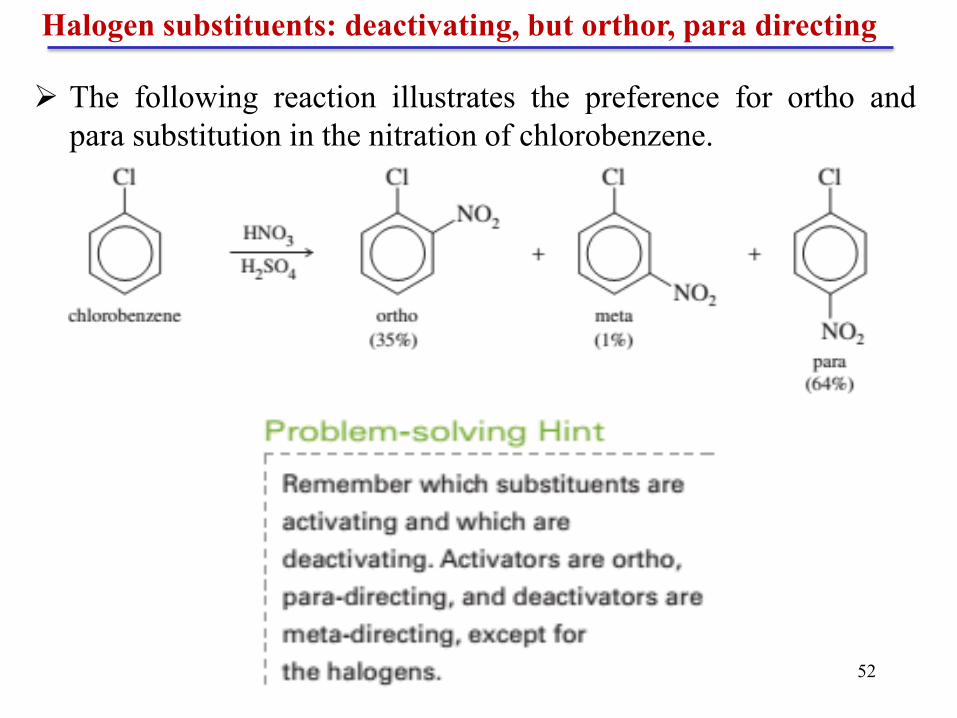
If an electrophile reacts at the ortho or para position, the
nonbonding electrons of the halogen can further delocalize the
charge onto the halogen, giving a halonium ion structure.
This resonance stabilization allows a halogen to be pi-donating,
even though it is sigma-withdrawing.
Reaction at the meta position gives a sigma complex whose
positive charge is not delocalized onto the halogen-bearing carbon
atom. Therefore, the meta intermediate is not stabilized by the
halonium ion structure .
Halogen substituents: deactivating, but orthor, para directing
Chapter 5 52
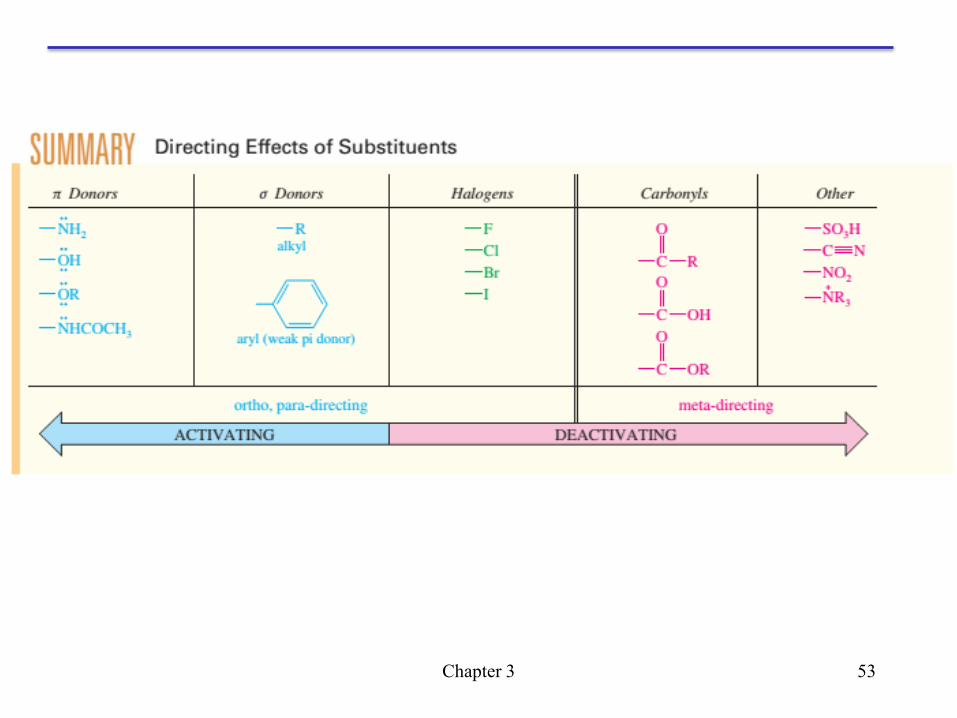
Halogen substituents: deactivating, but orthor, para directing
The following reaction illustrates the preference for ortho and
para substitution in the nitration of chlorobenzene.
Chapter 3 53

54
Effects of multiple substituents on electrophilic aromatic substitution
Two or more substituents exert a combined effect on the reactivity of an
aromatic ring. If the groups reinforce each other, the result is easy to predict.
+ For example, we can predict that all the xylenes (dimethylbenzenes) are
activated toward electrophilic substitution because the two methyl groups are both
activating.
+ In the case of a nitrobenzoic acid, both substituents are deactivating, so we
predict that a nitrobenzoic acid is deactivated toward attack by an electrophile.

Chapter 5 55
The orientation of addition is easily predicted in many cases.
+ For example, in m-xylene there are two positions ortho to one of the methyl
groups and para to the other. Electrophilic substitution occurs primarily at
these two equivalent positions. There may be some substitution at the position
between the two methyl groups (ortho to both), but this position is sterically
hindered, and it is less reactive than the other two activated positions.
+ In p-nitrotoluene, the methyl group directs an electrophile toward its ortho
positions. The nitro group directs toward the same locations because they are
its meta positions.
Effects of multiple substituents on electrophilic aromatic substitution

Chapter 5 56
Effects of multiple substituents on electrophilic aromatic substitution

Chapter 5 57
Effects of multiple substituents on electrophilic aromatic substitution
When the directing effects of two or more substituents conflict, it is more
difficult to predict where an electrophile will react. In many cases, mixtures
result. For example, o-xylene is activated at all the positions, so it gives
mixtures of substitution products.
When there is a conflict between an activating group and a deactivating
group, the activating group usually directs the substitution. We can make
an important generalization:

Chapter 5 58
Effects of multiple substituents on electrophilic aromatic substitution
In fact, it is helpful to separate substituents into three classes, from
strongest to weakest.
1. Powerful ortho, para-directors that stabilize the sigma complexes
through resonance. Examples are –OH, -OR, and –NR2 groups.
2. Moderate ortho, para-directors, such as alkyl groups and halogens.
3. All meta-directors.
If two substituents direct an incoming electrophile toward different
reaction sites, the substituent in the stronger class predominates.
If both are in the same class, mixtures are likely.

Chapter 5
59
Effects of multiple substituents on electrophilic aromatic substitution
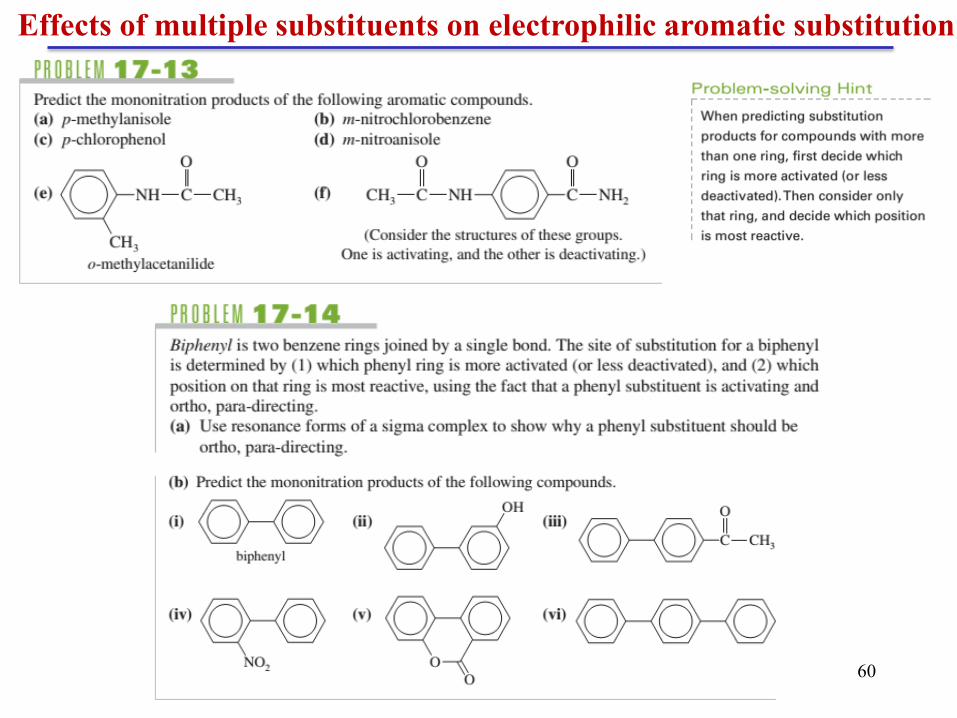
Chapter 5 60
Effects of multiple substituents on electrophilic aromatic substitution
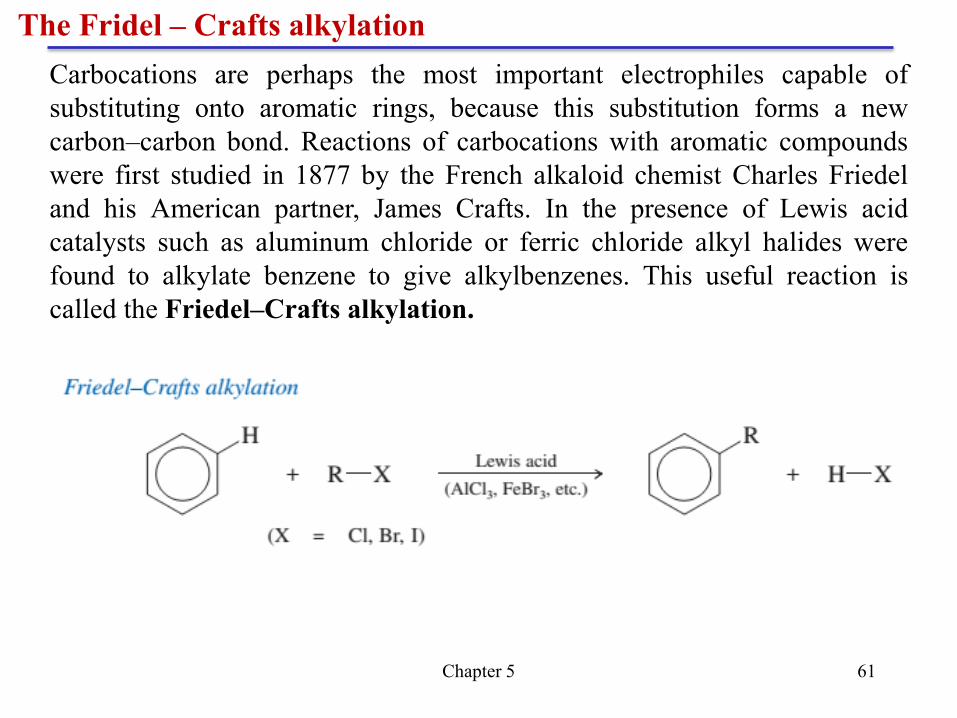
Chapter 5 61
The Fridel – Crafts alkylation
Carbocations are perhaps the most important electrophiles capable of
substituting onto aromatic rings, because this substitution forms a new
carbon–carbon bond. Reactions of carbocations with aromatic compounds
were first studied in 1877 by the French alkaloid chemist Charles Friedel
and his American partner, James Crafts. In the presence of Lewis acid
catalysts such as aluminum chloride or ferric chloride alkyl halides were
found to alkylate benzene to give alkylbenzenes. This useful reaction is
called the Friedel–Crafts alkylation.

62
For example, aluminum chloride catalyzes the alkylation of benzene by
tert-butyl chloride. HCl gas is evolved.
This alkylation is a typical electrophilic aromatic substitution, with the tert-
butyl cation acting as the electrophile.
(i) The tert-butyl cation is formed by reaction of tert-butyl chloride with the
catalyst, aluminum chloride.
(ii) The tert-butyl cation reacts with benzene to form a sigma complex.
(iii) Loss of a proton gives the product, tert-butylbenzene. The aluminum
chloride catalyst is regenerated in the final step.
The Fridel – Crafts alkylation

Chapter 5 63
With primary alkyl halides, the free primary carbocation is too
unstable. The actual electrophile is likely a complex of
aluminum chloride with the alkyl halide.
In this complex, the carbon–halogen bond is weakened (as
indicated by dashed lines) and there is considerable positive
charge on the carbon atom.
The Fridel – Crafts alkylation

Chapter 5 64

Chapter 5 65
We have seen several ways of generating carbocations, and most of these
can be used for Friedel–Crafts alkylations. Two common methods are
protonation of alkenes and treatment of alcohols with BF3.
+ Alkenes are protonated by HF to give carbocations. Fluoride ion is a weak
nucleophile and does not immediately attack the carbocation.
+ If benzene (or an activated benzene derivative) is present, electrophilic
substitution occurs. The protonation step follows Markovnikov’s rule, forming
the more stable carbocation, which alkylates the aromatic ring.
Friedel–Crafts Alkylation Using Other Carbocation Sources
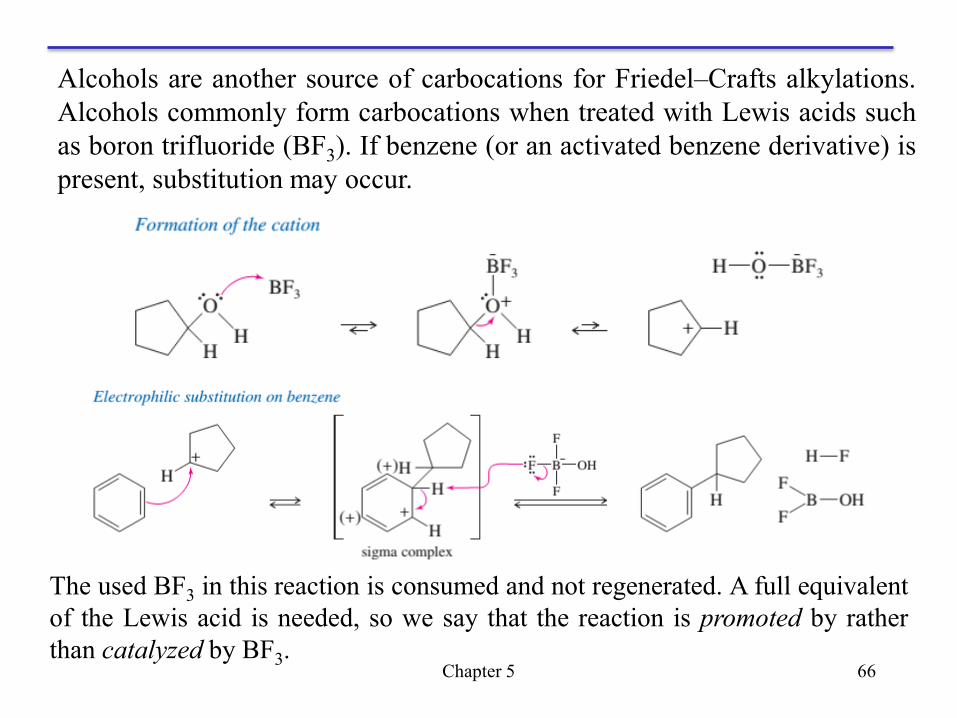
Chapter 5 66
Alcohols are another source of carbocations for Friedel–Crafts alkylations.
Alcohols commonly form carbocations when treated with Lewis acids such
as boron trifluoride (BF3). If benzene (or an activated benzene derivative) is
present, substitution may occur.
The used BF3 in this reaction is consumed and not regenerated. A full equivalent
of the Lewis acid is needed, so we say that the reaction is promoted by rather
than catalyzed by BF3.

Chapter 5 67
Although the Friedel–Crafts alkylation looks good in principle, it
has three major limitations that severely restrict its use.
Limitation 1
Friedel–Crafts reactions work only with benzene, activated benzene
derivatives, and halobenzenes.
They fail with strongly deactivated systems such as nitrobenzene,
benzenesulfonic acid, and phenyl ketones.
In some cases, we can get around this limitation by adding the
deactivating group or changing an activating group into a deactivating group
after the Friedel–Crafts step.
Limitations of the Friedel–Crafts alkylation

Chapter 5 68
Limitations of the Friedel–Crafts alkylation

Chapter 5 69
Limitation 2
Like other carbocation reactions, the Friedel–Crafts alkylation is
susceptible to carbocation rearrangements. As a result, only certain
alkylbenzenes can be made using the Friedel–Crafts alkylation.
+ tert-Butylbenzene, isopropylbenzene, and ethylbenzene can be
synthesized using the Friedel–Crafts alkylation because the corresponding
cations are not prone to rearrangement.
+ However, n-propylbenzene cann’t be prepared by the Friedel–Crafts
alkylation.
Limitations of the Friedel–Crafts alkylation

Chapter 5 70
Limitation 3
Because alkyl groups are activating substituents, the product of the
Friedel–Crafts alkylation is more reactive than the starting material. Multiple
alkylations are hard to avoid.
As some ethylbenzene is formed, however, it is activated, reacting even faster than
benzene itself. The product is a mixture of some (ortho and para) diethylbenzenes,
some triethylbenzenes, a small amount of ethylbenzene, and some leftover
benzene.
The problem of overalkylation can be minimized by using a large excess of benzene.
For example, if 1 mole of ethyl chloride is used with 50 moles of benzene, the
concentration of ethylbenzene is always low, and the electrophile is more likely to
react with benzene than with ethylbenzene.

Chapter 5 71
Problems

Chapter 5 72
The Friedel–Crafts Acylation
An acyl group is a carbonyl group with an alkyl group attached. Acyl
groups are named systematically by dropping the final -e from the
alkane name and adding the -oyl suffix.
An acyl chloride is an acyl group bonded to a chlorine atom. Acyl
chlorides are made by reaction of the corresponding carboxylic acids
with thionyl chloride. Therefore, acyl chlorides are also called acid
chlorides.

Chapter 5 73
The Friedel–Crafts Acylation
In the presence of aluminum chloride, an acyl chloride reacts with
benzene to give a phenyl ketone: an acylbenzene. The Friedel–Crafts acylation is analogous to the Friedel–Crafts alkylation.

Chapter 5 74
Mechanism of Acylation
The mechanism of Friedel–Crafts acylation resembles that for alkylation,
except that the electrophile is a resonance-stabilized acylium ion.
The acylium ion reacts with benzene or an activated benzene derivative
via an electrophilic aromatic substitution to form an acylbenzene.

Chapter 5 75
Mechanism of Acylation

Chapter 5 76
The Friedel–Crafts Acylation
The product of acylation (the acylbenzene) is a ketone.
The electrophile in the Friedel–Crafts acylation appears to be a large,
bulky complex, such as RC+=O-AlCl4. Para substitution usually
prevails when the aromatic substrate has an ortho, para-directing
group, possibly because the electrophile is too bulky for effective
attack at the ortho position.

Chapter 5 77
The acylbenzene has a carbonyl group (a deactivating group)
bonded to the aromatic ring.
Since Friedel–Crafts reactions do not occur on strongly deactivated
rings, the acylation stops after one substitution.
Thus, Friedel–Crafts acylation overcomes two of the three limitations of
the alkylation:
+ The acylium ion is resonance-stabilized, so that no rearrangements occur;
+ and the acylbenzene product is deactivated, so that no further reaction occurs.
The Friedel–Crafts Acylation

Chapter 5 78
Comparison of the the Friedel–Crafts alkylation and acylation

Chapter 5 79
How do we synthesize alkylbenzenes that cannot be made by Friedel–Crafts
alkylation?
+ We use the Friedel–Crafts acylation to make the acylbenzene,
+ then we reduce the acylbenzene to the alkylbenzene using the Clemmensen
reduction: treatment with aqueous HCl and amalgamated zinc (zinc treated with
mercury salts).
The Clemmensen Reduction: Synthesis of Alkylbenzenes