archives of biochemistry and biophysics and pharmacological studies of tmps are ham-pered by their...
TRANSCRIPT

Archives of Biochemistry and Biophysics 564 (2014) 327–343
Contents lists available at ScienceDirect
Archives of Biochemistry and Biophysics
journal homepage: www.elsevier .com/ locate/yabbi
Review
Folding and stability of integral membrane proteins in amphipols
http://dx.doi.org/10.1016/j.abb.2014.10.0130003-9861/� 2014 Elsevier Inc. All rights reserved.
E-mail addresses: [email protected] (J.H. Kleinschmidt), [email protected] (J.-L. Popot)1 Abbreviations used: A8-35, a poly(sodium acrylate)-based amphipol comprising �35% free carboxylates, �25% octyl grafts, �40% isopropyl groups, whose number
molar mass is �4.3 kDa; APol, amphipol; BLT1 and BLT2, two human G protein-coupled receptors of leukotriene LTB4; BO, BR, respectively bacterio-opsin and bacteriorfrom Halobacterium salinarum; CAC, critical association concentration; CB1, human cannabinoid receptor 1; CD, circular dichroism; CHAPS, 3-[(3-cholamidopropyl)dammonio]-1-propanesulfonate; cmc, critical micellar concentration; 2D, 3D, two- and three-dimensional; DAGK, diacylglycerol kinase; dBO, delipidated BO, obtaisolubilizing purple membranes in organic solvents; DDM, n-dodecyl-b-D-maltoside; DHPC, dihexanoylphosphatidylcholine; DS, dodecylsulfate; EM, electron microscIMS-MS, electrospray ionization mass spectrometry coupled with ion mobility spectrometry; FomA, major outer membrane protein A from Fusobacterium nucleatum; Ghuman ghrelin receptor 1a; GPCR, G protein-coupled receptor; His-tag, polyhistidine tag; 5-HT4(a), human serotonin receptor; LDAO, N-lauryl-N,N-dimethylamine-N-oxidlight-harvesting complex II; LPS, lipopolysaccharide; MD, molecular dynamics; MOMP, major outer membrane protein from Chlamydia trachomatis; OG, n-octyl-b-D-gOmpA and OmpT, outer membrane proteins A and T from Escherichia coli; OTG, octylthioglucoside; PAGE, polyacrylamide gel electrophoresis; PagP, lipid A palmytoyltrafrom E. coli; PDS and SDS, potassium and sodium dodecylsulfate, respectively; PM, purple membrane; SEC, size-exclusion chromatography; SUV, small unilamellar vesictransmembrane protein; tOmpA, transmembrane domain of OmpA; UV, ultraviolet; Z3-14, 3-[dimethyl(tetradecyl)azaniumyl]propane-1-sulfonate (also called sulfobetaZwittergent 3-14).
Jörg H. Kleinschmidt a, Jean-Luc Popot b
a Abteilung Biophysik, Institut für Biologie, FB 10, Universität Kassel and Center for Interdisciplinary Nanostructure Science and Technology (CINSaT), Heinrich-Plett-Str. 40,D-34132 Kassel, Germanyb Laboratoire de Physico-Chimie Moléculaire des Protéines Membranaires, UMR 7099, Centre National de la Recherche Scientifique/Université Paris-7, Institut de BiologiePhysico-Chimique (FRC 550), 13, rue Pierre-et-Marie-Curie, F-75005 Paris, France
a r t i c l e i n f o a b s t r a c t
Article history:Received 30 July 2014and in revised form 11 October 2014Available online 30 October 2014
Keywords:Membrane proteinFoldingAmphipolsKineticsThermodynamic stabilityOuter membrane proteinsBacteriorhodopsinG protein-coupled receptorsA8-35
Amphipols (APols) are a family of amphipathic polymers designed to keep transmembrane proteins(TMPs) soluble in aqueous solutions in the absence of detergent. APols have proven remarkably efficientat (i) stabilizing TMPs, as compared to detergent solutions, and (ii) folding them from a denatured state toa native, functional one. The underlying physical–chemical mechanisms are discussed.
� 2014 Elsevier Inc. All rights reserved.
Introduction
Folding integral membrane proteins to their functionally active form:still a challenge
Integral membrane proteins – hereafter transmembrane pro-teins (TMPs)1 – are key biological players because of their roles inbioenergetics, transmembrane transport, signaling, cell and tissueorganization, etc. They are therefore primary targets for pharmaceu-tical drugs. Structural and pharmacological studies of TMPs are ham-pered by their low abundance and by difficulties in overexpressingthem in a functional form. TMPs can be overexpressed either homol-ogously or heterologously, for example in Escherichia coli, yeast, etc.
Plasmid-based overexpression can be designed to target TMPs eitherto a membrane, which often results in low yields, or to cytoplasmicinclusion bodies, which yields larger amounts of protein, but in amisfolded and aggregated form that has to be folded to a functionalstate – a difficult achievement. Folding outside the cell environmentis also necessary when TMPs are expressed in vitro in a cell-free sys-tem. Successful folding in detergent or mixed detergent/lipidmicelles has been demonstrated for both known structural classesof TMPs, the a-helix bundle and the b-barrel. Over 50 a-helical TMPshave been folded or refolded in vitro to date [1]. Among the first onesto be studied were bacteriorhodopsin (BR) [2–4], the light harvestingcomplex LHCII [5,6], several G protein-coupled receptors (GPCRs)such as olfactory receptors [7,8], the BLT1 receptor of leukotriene
-averagehodopsinimethyl-
ned afteropy; ESI-
HS-R1a,e; LHCII,
lucoside;nsferase
les; TMP,ine 3-14,

328 J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343
LTB4 [9], or the serotonin 5-HT4(a) receptor [10], channel proteinslike the homotetramer KcsA [11], the small multidrug transporterEmrE [12], and the enzyme diacylglycerol kinase (DAGK) [13,14](for reviews, see e.g. Refs. [1,15,16]). About 40 b-barrel TMPs havebeen (re)folded in vitro to date [1,17–20], ranging from simple 8-stranded b-barrels like OmpA [21,22], OmpX [23] or PagP [24–26]to large 22-stranded active transporters like FepA [27,28]. The listalso includes eukaryotic b-barrels such as the 19-stranded VDAChuman isoforms 1 [29] and 2 [30,31]. It is quite possible that all b-barrel TMPs can be folded in vitro without any cellular foldingmachinery. An unsuccessful attempt to fold a b-barrel in solutionhas been reported for the b-barrel domain of AIDA [32]. Howevereven for AIDA, folding was successful when the protein was boundto a nickel-bearing column [33].
a-Helical TMPs like BR or KcsA have been folded from dena-tured forms in organic solvent like trifluoroacetic acid or mixturesof formic acid and ethanol [2,11]. Folding of BR was initiated bytransferring denatured bacterio-opsin (BO, the apoprotein) firstinto micelles of the denaturing detergent sodium dodecylsulfate(SDS) and then, usually in the presence of retinal, BR’s cofactor, into‘‘mild’’ detergents, mixtures of detergent and lipids, or pure lipids[2–4]. Rather than diluting it into a large excess of the folding med-ium [2,3], dodecylsulfate (DS) can be precipitated as its potassiumsalt, PDS [4]. b-Barrel TMPs such as outer membrane protein A(OmpA, previously called Omp II) from E. coli were first refoldedfrom their SDS-denatured forms by replacing the SDS either withlipopolysaccharide [21] or octylglucoside [22]. Subsequently, most(re)folding protocols for b-barrel TMPs have been based on theirsolubilization and denaturation in concentrated solutions of urea(8–10 M) [34] or other chaotropic agents (reviewed in Ref. [1]).Denatured b-barrel TMPs are generally folded by transferring themto either detergent micelles or lipid membranes under concurrentstrong dilution of the denaturant.
The mechanisms of folding of TMPs of both structural classeshave been the subject of extensive research. For a-helical TMPs,BR (see e.g. Refs. [35,36]) or DAGK [37,38] have often served asmodels. Folding of a-helical membrane proteins is a sequential 3-stage process, in which helices insert independently in a first stageand associate laterally in a second stage, which is followed, in athird stage, by the formation of additional tertiary structure, suchas re-entrant loops, and/or by the binding of prosthetic groups(for discussions, see Refs. [35,39]). For b-barrel TMPs, a concertedmechanism has been reported for OmpA, in which folding andinsertion are coupled [40,41] (for reviews, see Refs. [20,42–44].
Using detergent- or lipid-based protocols, folding yields tend tobe lower than desired for many pharmacologically and physiolog-ically interesting TMPs, e.g. GPCRs. For example, the leukotriene B4
BLT1 receptor has been refolded to a moderate yield of �30% inmixed micelles of detergent and lipid [9]. To date, this is still oneof the highest yields reported using classic methods. A homologof BLT1, the BLT2 receptor, is an example of a GPCR that cannotbe folded with a decent yield (more than a few percents) in anydetergent or detergent/lipid mixture that has been tried to date[45]. The development of successful refolding strategies for novelTMPs of interest using classic approaches is extremely time-con-suming. It is therefore of great interest to examine new methods.
Amphipols as new tools for the refolding of integral membraneproteins
Over the last eight years, a new class of non-detergent surfac-tants, namely synthetic amphipathic polymers called amphipols(APols) [46], has emerged as very promising tools for folding dena-tured TMPs to their native state. APols are short polymers com-prised of both hydrophilic groups and hydrophobic chains. Theycan substitute to detergents, providing a milder environment to
TMPs while keeping them water-soluble (for reviews, see Refs.[47–51]). All of the three dozen TMPs tested to date form solublecomplexes with APols in the absence of detergent, whatever theirsize, origin, function and secondary, tertiary and quaternary struc-ture (reviewed in Ref. [51]). As compared to preparations in deter-gent solutions, most TMPs remain active for a much longer timewhen solubilized in the form of APol/TMP complexes (see e.g. Refs.[45,46,52–55]). APols bind to TMPs by adsorbing specifically ontotheir hydrophobic transmembrane surface, as demonstrated byNMR spectroscopy [56–60], electron microscopy (EM) [61–69],and molecular dynamics (MD) simulations [70]. Unless exchangedfor other surfactants, APols do not dissociate from the surface ofTMPs, even at very high dilution, keeping the protein water-soluble[47,71,72] (reviewed in Ref. [51]).
Several types of APols have been synthesized and validated(Fig. 1) (reviewed in Refs. [51,73]). Among these, A8-35 has beenmost extensively studied. A8-35 (Fig. 1A) is a poly(acrylic acid)partially amidated with octylamine and isopropylamine, leaving�35% of the carboxylic groups free [46]. In aqueous solution atpH > 7, A8-35 assembles into small micelle-like particles with amass of �40 kDa [74–76]. Other APols developed more recentlyinclude glycosylated nonionic APols (NAPols) [77,78] (Fig. 1B andC), sulfonated APols (SAPols) [79] (Fig. 1D) or phosphorylcholine-based APols (PC-APols) [80,81] (Fig. 1E) (reviewed in Refs.[50,51,73]). SAPols [79] are derived from a precursor of A8-35 lack-ing the amidation with isopropylamine. Instead, about �40% of thecarboxyl groups are amidated by taurine (2-aminoethanesulfonicacid) (Fig. 1D). The sulfonate groups of taurine do not protonateeven at pH 0, which keeps SAPols water-soluble at pH < 7, whereasunder such conditions A8-35 aggregates [74,75]. HomopolymericNAPols (Fig. 1B) are synthesized by homotelomerization of amonomer carrying two glycosyl residues [77]. HeteropolymericNAPols (Fig. 1C) have been obtained either by cotelomerizationof hydrophobic and hydrophilic monomers [82] or by randomlygrafting hydrophobic chains onto a glycosylated homotelomer[83]. PC-APols are zwitterionic at neutral and basic pH, cationicat acidic pH. NAPols and PC-APols, as SAPols, remain water-solubleunder acidic conditions.
APols, originally designed to keep TMPs water-soluble whilepreserving their activity [46], have been used for a wide range ofapplications (reviewed in Ref. [51]). Their properties and those ofTMP/APol complexes have been reviewed in Refs. [49–51] anddetailed protocols for their implementation provided in Ref. [84].This review focuses on their use for folding TMPs and for stabiliz-ing them against denaturation by heat or chaotropic agents. Sincea-helix bundle TMPs and b-barrel TMPs fold according to very dif-ferent principles (see e.g. Refs. [35,39,41,85]), our discussion is sub-divided into separate sections for these two categories of TMPs.Historically, APol-assisted folding of TMPs was first demonstratedfor three model TMPs, BR, an a-helical TMP, and two b-barrel TMPs,OmpA and FomA [86]. It was then extended to two more b-barrelTMPs and six GPCRs (Table 1).
Amphipol-assisted folding of a-helix bundle membraneproteins
Folding of BR in A8-35
BR, a light-driven proton pump [87], has served as a popularmodel protein in studies on TMP folding since the seminal workof Khorana and coworkers [2] (for reviews, see e.g. Refs.[1,35,88,89]). When overproduced, BR accumulates in the plasmamembrane of H. salinarum in the form of 2D protein/lipid crystals,the so-called purple membrane (PM). Its covalently but looselybound chromophore, retinal, confers it a characteristic purple

Fig. 1. Chemical structures of various amphipols. (A) A heteropolymeric, carboxylated and amidated anionic amphipathic polymer, amphipol A8-35. The indices x, y, z denotethe proportion of the various units: x � 35%, y � 25%, z � 40%. From Ref. [46]. (B) Homopolymeric, glycosylated nonionic amphipol (NAPol). From Ref. [77]. (C)Heteropolymeric, glycosylated nonionic amphipol (NAPol). From Refs. [82,83]. (D) Sulfonated anionic amphipol (SAPol) with x � 35%, y � 25%, z � 40%. From Ref. [79]. (E)Phosphorylcholine-based APol (PC-APol). From Ref. [81].
J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343 329
color. When PM is solubilized in SDS, BR denatures to BO, releasingits chromophore, which causes an absorption peak shift from�555 nm (dark-adapted BR) to �382 nm (free retinal). Theremoval of SDS from BO/retinal in the presence of APol A8-35 byprecipitating DS as PDS results in the regeneration of the character-istic purple color of BR within minutes, indicating folding of BO andrebinding of the retinal [86]. After a dialysis to remove residual DS,almost quantitative refolding of BR (P 90%) is observed for a massratio A8-35/BR P 5:1 [86,90]. When refolded at such mass ratios,BR migrates as an A8-35-trapped monomer, as determined bysize-exclusion chromatography (SEC). Control experiments carriedout in the same way using detergents instead of A8-35 show lowerlevels of BR refolding. Yields are in fact negligible with octylgluco-side (OG), octylthioglucoside (OTG) or C8E4, but reach �66% indodecylmaltoside (DDM).
More stringent experiments were performed in which BO wastotally unfolded in formic acid and transferred to SDS after lipidsand retinal had been removed by hydrophobic SEC. They estab-lished that completely delipidated BO (dBO) refolds in pureA8-35 to 60–80% [86,90]. BR refolded in A8-35 displayed a fully
functional photocycle. The light-adapted protein was excited at640 nm with a 5-ns laser flash. Transient absorption changes, mon-itored from 10 ns to 100 ms in a spectral range from 370 to500 nm, were found to be very similar to those observed withlipid-associated native BR trapped in A8-35 [86,90].
A comparison of the folding of BR into A8-35 from BO/retinal ordBO/retinal in SDS confirmed that slightly higher refolding yieldsare achieved when lipids were present [90]. However, in the pres-ence of lipids, larger amounts of A8-35 were necessary for optimalfolding yields. This suggested that lipids may favor BR aggregationat low A8-35 concentrations, in line with observations by SEC thatBR aggregates when refolded at 1:2 BO/A8-35 [86]. Interestingly,an attempt at direct refolding of dBO by transfer from trifluoroeth-anol (TFE) to A8-35, in aqueous buffer in the absence of SDS,resulted in a folding yield of �40% [90]. This approach, however,has not reached the stage of a protocol yielding reproducibleresults.
Folding yields were somewhat lower when folding of BR fromBO in SDS was initiated by dialysis of SDS instead of precipitatingDS as its potassium salt [90,91]. Upon SDS removal by dialysis,

Table 1Folding of membrane proteins in amphipols.
TMP Structure Denaturant Amphipol Lipids, cofactors Method Yield References
BRa 7-a b SDS 5% A8-35 (2–25 g/g)
PM lipids, retinal DS precipitation with KCl at pH 7,dialysis
Up to 87–92%
[78,86,128]
BR 7-a SDS 5% A8-35 (5 or10 g/g)
Retinal, no lipids DS precipitation with KCl at pH 7,dialysis
70–76% [128]
BR 7-a SDS 5% A8-35 (5 or10 g/g)
PM lipids, retinal Dialysis at pH 7, no NaCl 52–72% [128]
BR 7-a SDS 2.5% A8-35 (5 or10 g/g)
PM lipids, retinal Dialysis at pH 7, no NaCl 62–80% [128]
BR 7-a SDS 5% A8-35 (5 or10 g/g)
PM lipids, retinal Dialysis at pH 7, 100 mM NaCl �76% [128]
BR 7-a SDS 2.5% A8-35 (5 or10 g/g)
PM lipids, retinal Dialysis at pH 7, 100 mM NaCl 74–80% [128]
BR 7-a SDS 0.25% A8-35 (10 g/g) PM lipids, retinal 5� dilution at pH 7, no NaCl 40–50% [128]BR 7-a SDS 0.25% A8-35 (10 g/g) PM lipids, retinal 5� dilution at pH 7, 100 mM NaCl 70–80% [128]BR 7-a TFEc A8-35 (5 or
10 g/g)Retinal, no lipids Dialysis at pH 7 640% [128]
BLT1d 7-a SDS 0.8% A8-35 (1–20 g/g)
None DS precipitation with KCl at pH 8,dialysis
Up to �50% [45]
BLT1 7-a SDS 0.8% A8-35 (5 g/g) Asolectin/A8-35 1:5 w/w DS precipitation with KCl at pH 8,dialysis
65–70% [45]
BLT2e 7-a SDS 0.8% A8-35 (5 g/g) None DS precipitation with KCl at pH 8,dialysis
�50% [45]
BLT2 7-a SDS 0.8% A8-35 (5 g/g) Asolectin/A8-35 1:5 w/w DS precipitation with KCl at pH 8,dialysis
�70% [45,170]
CB1e 7-a SDS 0.8% A8-35 (5 g/g) None DS precipitation with KCl at pH 8,dialysis
�30% [45]
CB1 7-a SDS 0.8% A8-35 (5 g/g) Asolectin/A8-35 1:5 w/w DS precipitation with KCl at pH 8,dialysis
�40% [45]
5-HT4(a)e 7-a SDS 0.8% A8-35 (5 g/g) None DS precipitation with KCl at pH 8,
dialysis�30% [45]
5-HT4(a) 7-a SDS 0.8% A8-35 (5 g/g) Asolectin/A8-35 1:5 w/w DS precipitation with KCl at pH 8,dialysis
�60% [45]
GHSR-1ad 7-a SDS 0.8% NAPol (10 g/g) Asolectin/NAPol 1:5 w/w,0.2% (w/v) cholesterylhemisuccinate
DS precipitation with KCl at pH 8,dialysis
�40% [78]
FomAf 14-b g 10 M urea A8-35 (8.5 g/g) None 10� urea dilution at pH 10 �90% [86]OmpAh 8-b 8 M urea A8-35 (8 g/g) None 20� urea dilution at pH 10 �100% [86]tOmpAh,i 8-b 8 M urea SAPol (4 g/g) None 9� urea dilution at pH 7 �100% [79]OmpTh 10-b 8 M urea A8-35 (5 g/g) None Dilution, then dialysis, pH 8, 4 �C �100% [119]PagPh 8-b 8 M urea A8-35 (5 g/g) None Dilution, then dialysis, pH 8, 4 �C �60% [119]
a From Halobacterium salinarum.b 7-a: bundle of 7 transmembrane a-helices.c Experiment could not be reproduced.d From Homo sapiens.e From Mus musculus.f From Fusobacterium nucleatum.g n-b: barrel of n observed or predicted transmembrane b-strands.h From Escherichia coli.i Transmembrane domain of OmpA.
330 J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343
folding yields of BR increased with the A8-35/BO ratio, suggestingthat aggregation is a limiting factor [90].
BR was also successfully refolded by diluting a BO/SDS/A8-35mixture with SDS-free buffer, in which case larger mass ratios ofA8-35/BO were required than observed using either PDS precipita-tion or SDS dialysis: when folding was initiated by dilution, yieldswere highest at a mass ratio of 25 [90]. Two mechanisms may con-tribute to this effect. First, adding more A8-35 reduces the propor-tion of SDS in the environment of the refolding proteins. Second,the probability that two of them will come into contact and estab-lish intermolecular associations is reduced by diluting them withmore APol, making aggregation less likely (cf. Ref. [71]). Foldingkinetics were faster at higher concentrations of retinal, consistentwith the view that refolded BO can pick up retinal very rapidly ifit is present in the BO-associated APol belt, whereas the processis much slower if retinal uptake depends on the BO/APol complexcolliding with a retinal-containing free particle of APol (cf. Ref.[71]).
Folding of dBO in A8-35 by PDS precipitation was also success-ful in the absence of retinal [90]. However, refolded BO is less sta-
ble than refolded BR. The addition of retinal within an hour afteraddition of KCl results in similar yields as observed when foldingBO in the presence of retinal, but yields dropped to �30% when ret-inal was added after dialyzing refolded BO in A8-35 for three days,indicating that �2=3 of it had denatured again over this period. Thisobservation is consistent with the view that folding yields of fragileTMPs can be improved in the presence of their ligands.
Transfer from SDS to A8-35 can also be achieved by adsorbingSDS onto Bio-Beads, provided aggregation is limited by immobiliz-ing polyhistidine-tagged BR onto a nickel-bearing column [91].
A8-35-assisted folding of GPCRs
Successful folding of both a-helical and b-barrel model TMPs inA8-35 [86] suggested that the approach could be general and stim-ulated attempts at folding GPCRs, for which folding using classicaldetergent/lipid systems, when successful, is typically limited toyields of 30% or less (reviewed in Refs. [15,92]). The first study boreon BLT1, a GPCR involved in the control of inflammatory processes,using a similar strategy as that established for BR [86]: A8-35 was

J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343 331
added to the denatured GPCR in SDS solution, most of DS precipi-tated as PDS, and residual DS removed by dialysis. Ligand bindingassays indicated that the receptor folded in A8-35 was functional,with a dissociation constant KD � 9 nM, similar to that of nativeBLT1 expressed in membrane fractions [93]. Based on the numberof binding sites, the yield of folding was �50% in the absence of lip-ids and 65–70% in the presence of soybean asolectin (a mixture ofabout equal amounts of phosphatidylcholine, phosphatidyletha-nolamine and phosphatidylinositol, with smaller amounts of polarlipids like phosphatidic acid, phosphatidylglycerol, phosphatidyl-serine and sterols) (Table 1). The BLT1/A8-35/lipid mass ratiowas 1:5:1. Following folding, BLT1 showed a similar pharmacolog-ical profile as the membrane-bound receptor [45], indicating thatA8-35 is an efficient medium in which to fold BLT1 obtained frominclusion bodies.
The study was then extended to three other GPCRs: another leu-kotriene receptor, BLT2, the serotonin receptor 5-HT4(a), and thecannabinoid receptor CB1 [45]. In the presence of asolectin, A8-35 improved the folding yields of 5-HT4(a) previously observed bymore than a factor of two, namely from 20% to 25% in detergent/asolectin micelles [10] to �30% in pure A8-35 and �60% in A8-35 + asolectin [45]. For BLT2, which shares �45% sequence identitywith BLT1, the yield improved from 3–4% in detergent/asolectinmicelles to �50% in pure A8-35 and �70% in A8-35 + asolectin[45]. As regards CB1, which had not been folded to any significantextent in detergent/lipid mixtures at the time of these experiments(but has been folded to �30% since [94]), folding yields of �30%were achieved in pure A8-35 and of �40% in A8-35/asolectin mix-tures. Altogether, these data, obtained without any extensivesearch for optimal folding conditions, suggest an interesting poten-tial of A8-35 as a generally useful new tool for the folding of GPCRs.Indeed, more recently, two other GPCRs have been successfullyrefolded in APols, namely the arginine-vasopressin type 2 andghrelin GHSR-1a receptors (Table 1) [78,92].
Whereas in no case was a 100% yield reached, separation ofactive from inactive 5-HT4(a) could be achieved using aGR113808 affinity column [10], yielding �96% active receptor[45]. In this context, it is worth noting that, whereas 3D crystalli-zation of APol-trapped TMPs remains a difficult challenge (seeRef. [54]), A8-35-trapped BR has yielded highly organized crystals(diffracting to <2-Å resolution) following direct transfer to lipidicmesophases [95]. Combining APol-assisted folding of GPCRs withcrystallization in mesophases [96–98] might therefore open veryinteresting perspectives.
Folding of a-helical TMPs in NAPols
NAPols (Fig. 1B and C) have been used successfully to fold theghrelin GHSR-1a GPCR and BR to their native state [78]. Becauseof their nonionic character, NAPols may provide an even milderenvironment than polyanionic A8-35 [78], and they present theadvantage of being soluble over a broader pH-range [77], coveringthe mildly acidic regime favorable for NMR work. Folding of BR inhomopolymeric NAPols (Fig. 1B) was achieved using the samestrategy as previously used for folding in A8-35 [86], i.e. by supple-menting SDS-solubilized PM with NAPols, followed by PDS precip-itation and removal of residual DS by extensive dialysis.Quantitative analysis by ultraviolet (UV)/visible absorption spec-troscopy indicated a yield P90%. NAPol-refolded BR was homoge-neous, as shown by SEC [78].
Based on folding experiments with BR as a model, NAPols weresuccessfully used for folding the GHSR-1a receptor. Ghrelin, the pos-itively charged ligand of this receptor, binds non-specifically to A8-35 via charge interactions. This causes difficulties in assessing byligand binding the extent of folding of GHSR-1a in A8-35. Morefavorable conditions are achieved when the ghrelin receptor is
folded in NAPols, where the background is lower. Ligand-bindingmeasurements indicated a folding yield of �40%, and a receptor�97% active was obtained after affinity chromatography [78]. Thebinding properties of the folded GHSR-1a were then further exam-ined to assess the quality of folding. Fluorescence energy transferfrom GHSR-1a, labeled with Alexa Fluor 350, to a ghrelin peptidelabeled with fluorescein isothiocyanate was recorded in competi-tion experiments with synthetic antagonists. The competition pro-files obtained by this method are within the same range aspreviously inferred from radioactive and TagLite-based measure-ments of HEK cells transiently expressing GHSR-1a [99]. In addition,GHSR-1a folded in NAPols (i) is able to activate G proteins, (ii)recruits arrestin in an agonist-dependent manner, and (iii) adoptsa very similar equilibrium between active and inactive conforma-tions as in the membrane, confirming that it is fully functional [78].
The polyanionic APols A8-35 and SAPols have been found toblock in vitro synthesis of TMPs [100], but NAPols do not, as exem-plified by successful cell-free expression and folding of BR [78].This may open an interesting new route to producing hard-to-express TMPs.
Amphipol-assisted folding of b-barrel membrane proteins
The list of b-barrel TMPs that have been successfully folded bytransfer of their denatured forms in chaotropic denaturants todetergent micelles ranges from small 8-stranded b-barrels likeOmpA [21,22,34] to large 22-stranded transmembrane transport-ers like FepA [28], which contain an additional domain in the barrellumen. Indeed, most of the known b-barrel TMPs have been suc-cessfully (re)folded, the even-stranded TM b-barrels from bacteriaas well as the 19-stranded voltage-dependent anion-selectivechannel VDAC from the mitochondria of eukaryotic cells[29,30,101] (for reviews, see Refs. [1,19]). The mechanism of fold-ing of b-barrel membrane proteins into lipid bilayers has beenstudied for bacterial outer membrane proteins from E. coli, likeOmpA [40,85,102], OmpF [103], OmpG [104], OmpX [105], PagP[25,106] and others [26], for FomA from Fusobacterium nucleatum[107], and for VDAC isoform 1 from human mitochondria [29](for detailed reviews, see e.g. Refs. [20,42,44]). Whereas a foldingmachinery is required for the folding of b-barrel TMPs in cells,membranes or membrane biomimetics are not an absoluterequirement for folding in vitro. OmpA (Omp II [108]) is the firstb-barrel TMP for which successful refolding was demonstrated.More than 35 years ago, Schweizer et al. [21] showed that >90%of denatured OmpA regained its native structure in the presenceof lipopolysaccharide (LPS) and Triton-X-100 after dilution of thedenaturants SDS or urea. Dornmair et al. [22] established that afterheat-denaturation in SDS, OmpA, which comprises an 8-strandedtransmembrane b-barrel and a periplasmic domain, can refold inmicelles of OG in the absence of LPS. Further experiments demon-strated that OmpA can be (re)folded both in lipids and in a widerange of detergents, provided the detergent concentration is abovethe cmc [109]. OmpA can even fold from fragments in micelles ofOG [110], an observation first made in vivo for separatelyexpressed fragments [111].
Whereas the small 8-stranded b-barrel TMPs can usually befolded to nearly 100% using standard methods, this does not neces-sarily apply to larger barrels. To date, the yield of folding of the 14-stranded fatty acid transporter FADL does not seem to exceed 50%,irrespective of conditions [26]. Folding yields of VDAC (human iso-form 1, non-His-tagged) in detergent micelles depend strongly onthe pH. They are optimal at pH 3 [29]. The mere introduction ofa polyhistidine tag completely alters this pH-dependence of VDACfolding [29,30] (Shanmugavadivu & Kleinschmidt, unpublishedobservations).

332 J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343
In spite of all progress, finding the right conditions for foldingunfolded b-barrel TMPs obtained from inclusion bodies hasremained a time-consuming and, more often than not, frustratingtask, which often requires very long incubation times. It is there-fore of great interest to develop new methodologies that can bemore widely applied, provide higher yields, may be more rapid,and/or facilitate a larger scale production of functionally active b-barrel TMPs.
OmpA [112,113] and FomA [107,114,115] were the first two b-barrel TMPs to be folded using an APol, A8-35, starting from theirunfolded forms in 8 M (OmpA) and 10 M urea (FomA) [86]. Foldingwas very efficient, with yields of �100% for OmpA and �90% forFomA (Table 1), and was achieved within 7 h for OmpA [116]and within 24 h for FomA. Several criteria were used to assess thatthese 8- and (predicted) 14-stranded b-barrels, respectively, hadachieved their native state:
(i) SDS–PAGE, taking advantage of the different electrophoreticmobilities of the folded and unfolded forms.
(ii) Protection of the folded b-barrels against proteolysis.(iii) Far-UV circular dichroism (CD) spectroscopy.(iv) Functional studies, that is single-channel conductance
recordings after transfer of A8-35-refolded OmpA and FomAto black lipid films (Fig. 2).
In these experiments the first three methods did not reveal anydifferences between the detergent- and A8-35-refolded forms.However, in single-channel recordings of the conductance of theb-barrels in black lipid bilayers, both OmpA and FomA initially dis-played smaller conductance when inserted from complexes with
Fig. 2. Single-channel recordings of refolded OmpA and FomA integrated into black lipid35-refolded OmpA was added to the cis side of a diphytanoyl phosphatidylcholine film. ThNo event was observed in the presence of A8-35 alone, whether added in trans (not show(5). (b) and (c) Distribution of small conductance states observed after addition of A8-35trans side. (B) FomA. (a) Channels formed by A8-35-refolded FomA added to the cis sexperiments with LDAO-refolded FomA (3), with A8-35 only (on the cis-side) (4) and wiafter addition of A8-35/FomA complexes to the cis side without (b) and with (c) A8-35
A8-35 rather than from detergent-refolded forms. This differencewas traced to an asymmetrical distribution of A8-35 in the blacklipid films. When A8-35 was present at equal concentrations onboth sides of the film, the expected transmembrane conductanceswere obtained: 75 pS and 290 pS for the small and of large conduc-tance channels of OmpA, respectively, 1.1 nS for FomA channels[86]. The spontaneous transfer of refolded OmpA and FomA fromA8-35 to black lipid bilayers is consistent with the transfer,observed earlier, of the a-helical TMP DAGK from another APol,OAPA-20, into lipid bilayers of 1-palmitoyl-2-oleoyl-phosphatidyl-choline (multilamellar vesicles) in a functionally active form [117].
Folding of a b-barrel TMP has also been demonstrated using sul-fonated APols (SAPols) [79]. After 2 days of incubation, the geneti-cally engineered transmembrane domain of OmpA (tOmpA),isolated after expression into inclusion bodies, folded to yieldsapproaching �100% (Table 1), as determined by shifts in the elec-trophoretic mobility from �16 kDa for the unfolded to �19 kDa forthe folded form [23].
More recently, two other b-barrel TMPs, namely OmpT [118], anintegral outer membrane protease with 10 transmembrane b-strands, and PagP [24], an 8-stranded b-barrel TMP that catalyzesthe transfer of a palmitate chain from a phospholipid to lipid A,were folded in A8-35 from their unfolded forms in 8 M urea. Usingelectrospray ionization mass spectrometry coupled with ionmobility spectrometry (ESI-IMS-MS), yields were shown to reach�100% for OmpT and �60% for PagP (Table 1) [119]. Folding wasconfirmed by electrophoretic mobility measurements, by far-UVCD spectroscopy and by functional studies. SEC showed a singlenarrow peak for OmpT/A8-35 complexes, indicating the presenceof a homogeneous single species of OmpT. A broader peak was
films. (A) OmpA. (a) Large (1) and small (2) conductance states observed when A8-e conductance of the small state increased when A8-35 was added the trans side (3).n), in cis (4), or on both sides of the film (not shown), nor in that of unfolded OmpA/OmpA complexes to the cis side in the absence (b) or presence (c) of A8-35 on theide with A8-35 present (1) or absent (2) in the trans compartment, and controlth denatured FomA (5). (b) and (c) Distribution of the conductance states observedadded to the trans side. Reprinted with permission from Ref. [86].

J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343 333
observed in SEC after folding of PagP in A8-35, indicating the pres-ence of a mixture of folded and unfolded PagP species, consistentwith the �60% folding yield observed by ESI-IMS-MS. RefoldedOmpT trapped in A8-35 was active, as evidenced by the decreaseof fluorescence self-quenching of a fluorogenic peptide upon pep-tide hydrolysis catalyzed by OmpT in the presence of LPS. Interest-ingly, a small level of activity of A8-35-refolded OmpT wasobserved even in the absence of LPS [119], which is not observedwhen OmpT is folded using classical methods [120]. Refolded PagPwas active when trapped in A8-35, hydrolyzing p-nitrophenol-palmitate to p-nitrophenol with an activity comparable to that ofPagP in detergent solution [25].
Recently, a detailed study [116] examined the kinetics of foldingof wild-type OmpA into APol A8-35, initiated by an 18-fold dilutionof urea and carried out at various mass ratios, ranging from 0.5 to16 g of A8-35 per g of OmpA, and at various temperatures [116].Analyses of the time course of electrophoretic mobility shifts fromthe unfolded form (Mr � 35 kDa) to the folded one (Mr � 30 kDa)indicated that folding takes �6–8 h at pH 10. The minimum massratio of A8-35/OmpA required for complete folding was 2 g/g. Alltime courses followed double exponential kinetics [116], likelycaused by the presence of two or more differently protonated statesof OmpA. These have been distinguished before by fluorescencespectroscopy [121]. At pH 10, these states of OmpA (pI � 5.5) coexistand are differently charged, as this pH corresponds to the pKa valuesof many lysine and tyrosine side-chains, which are present in pro-tonated and deprotonated states. The coexisting forms of OmpA foldvia parallel folding pathways both in negatively charged A8-35 par-ticles [116] and in negatively charged lipid bilayers [122]. It appearspossible that the parallel pathways are a consequence of charge–charge repulsion, which could be different for the coexisting formsof OmpA. However, parallel pathways have also been observed forthe folding of FomA into phosphatidylcholine bilayers, which donot carry a net charge [107]. When OmpA was folded into A8-35,the faster process contributed �65% to OmpA folding, with a half-time of �5 min, whereas the slower process had a half-time of�70 min [116]. Kinetics of folding of OmpA were obtained by fluo-rescence spectroscopy and by monitoring the formation of tertiarystructure by electrophoresis [43]. Independent of the method used,parallel pathways of OmpA folding were observed. SDS–PAGE didnot reveal any folding intermediates of OmpA during folding inA8-35 [116]. Such intermediates likely exist, but they do not surviveexposure to SDS. Interestingly, the rate constants of the two foldingprocesses did not depend on the concentration of A8-35, indicatingthat intermolecular interactions between proteins are not involved.
As compared to folding in detergents, an interesting feature offolding in A8-35 is that, because of the APol’s very low critical asso-ciation concentration (CAC) [123], it can be carried out under verydilute conditions.
The activation energy of the folding of OmpA into A8-35 wasdetermined from the temperature dependence of the folding kinet-ics. The SDS–PAGE assay yielded an activation energy of�5.9 ± 4.1 kJ�mol-1 (�1.4 kcal�mol-1) for the fast process,�36.5 ± 9.6 kJ�mol-1 (�8.7 kcal�mol-1) for the slow one. When mon-itored by fluorescence spectroscopy, these activation energies were�8.8 ± 2.3 kJ�mol-1 for the fast process and 28.9 ± 8.1 kJ�mol-1 forthe slower process, respectively. Within error margins, these val-ues are comparable. They are smaller than the activation energyof 46 kJ�mol-1 reported for the folding of OmpA into bilayers (SUVs)of dioleoylphosphatidylcholine [124].
2 Note added in proofs: a study has recently been published of the stability of thedimer of the transmembrane a-helix of glycophorin A in A8-35 and A8-35/SDSmixtures [171].
Stability of membrane proteins in amphipols
A large number of studies have shown that most TMPs of eitherthe a-helical or the b-barrel types inactivate more slowly or at
higher temperature when trapped in APols as compared to a deter-gent environment (see e.g. Refs. [45,46,52–55,78,125,126]; forrecent reviews, see Refs. [49–51]). Hereafter, we focus on the sta-bility of TMPs in APols against unfolding induced by either heator chemical denaturants2.
Stability of a-helical TMPs in APols
Heat-denaturation of BR has been studied in various environ-ments by monitoring its UV–visible spectrum as a function of tem-perature [127,128]. In PM, BR does not denature until �100 �C[129]. BR solubilized in OTG starts to unfold at 40 �C [128], as indi-cated by a decrease of the visible absorption peak of the holopro-tein and by an increase of the light scattering caused by proteinaggregation. Massive aggregation and a shift of the absorptionmaximum to �380 nm indicate complete denaturation at highertemperature. In contrast, both A8-35-trapped native BR and BRrefolded in A8-35 in the presence of PM lipids do not unfold untila temperature of �50 �C is reached, indicating a stabilization in A8-35 compared to OTG. A8-35-trapped BR denatures only to �70%after 20 min at 70 �C [128]. The thermostability of BR refolded inA8-35 in the absence of lipids was also relatively high, but aslightly larger amount of retinal was released at each temperature,indicating that lipids indeed bind to BR in the presence of A8-35and contribute to the observed stabilization [128].
A8-35 has also been shown to improve the thermostability ofBLT1 in comparison to fos-choline-16/asolectin mixed micelles[45]. Dahmane et al. compared the thermostability of BLT1 in prep-arations that had been folded either in lipid-detergent mixedmicelles, in pure A8-35, or in A8-35 supplemented with asolectin.The samples were heated for 30 min at various temperatures, afterwhich the folding state of BLT1 was examined by performing aligand binding assay [130]. A loss of ligand binding activity wasdetected above 27 �C for BLT1 in fos-choline-16/asolectin mixedmicelles. In contrast, BLT1 folded in A8-35 was still stable at�35 �C. An additional improvement of the thermostability up to�39 �C was observed for BLT1 folded in A8-35 in the presence oflipids.
In summary, all observations available to date indicate thatAPols increase the thermal stability of BLT1 and BR as comparedto that in a detergent or detergent/lipid environment. The higherthermal stability of a-helical TMPs in APols translates into animproved stability upon long-term storage, as observed for BR[46,78,126,128], the sarcoplasmic calcium pump SERCA1a[52,125], BLT1 [45], or cytochrome bc1 [54].
Less direct observations also suggest stabilization of a-helicalTMPs by APols as compared to detergents. A case in point is theion channel TRPV1, which has been studied by EM after trappingin A8-35. Galleries of images of negatively-stained particles indi-cate that their overall shape is much more reproducible in A8-35than it is in DDM, suggesting stabilization [62,64]. Similarly, theresolution of cryo-EM images of the human c-secretase complexis higher for A8-35-trapped than for digitonin-solubilized prepara-tions, suggesting improved conformational homogeneity [69].
Stability of b-barrel TMPs in APols
The heat stability of the major outer membrane protein fromthe pathogenic bacterium Chlamydia trachomatis (MOMP) in APolA8-35 vs. 0.05% of the detergent n-tetradecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate (zwittergent Z3-14) has been com-pared using CD spectroscopy [53,55]. MOMP represents �60% of

334 J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343
the protein content of the outer membrane of C. trachomatis. Nohigh-resolution structure has been published to date. However,MOMP seems to share many structural features with the 16-stranded b-barrel porins from other bacteria [131]. At pH 7.4, themidpoint of the transition of heat-induced unfolding of the MOMPtrimer in Z3-14 is �52 �C, whereas A8-35-trapped MOMP does notunfold at all up to 78 �C, indicating an exceptionally strong stabil-ization [53], which translates into a remarkable stability uponextended storage [55].
Unless aggregation is prevented by APols [132,133] or an excessof detergent [22], heat denaturation of soluble or membrane pro-teins is usually not reversible, because the denatured protein pre-cipitates. To determine free energies of unfolding/folding,conditions must be found where unfolding/folding is fully revers-ible and where unfolded and folded forms are at equilibrium. Theclassic approach is to use chaotropic reagents like urea or guanid-inium chloride to unfold and solubilize proteins. Both are strongdenaturants that also prevent protein aggregation. When thesedenaturants are diluted, soluble proteins, as well as most b-barrelTMPs, refold if provided with a suitable folding environment: forTMPs, detergent micelles [21,22,109], lipid bilayers [34,85], orAPols such as A8-35 [86] or SAPols [79]. Under equilibrium condi-tions, a free energy for folding can be calculated from titrationswith urea [116,134] or similar denaturants. Reaching equilibrium,however, can be extremely lengthy (weeks).
Fig. 3. Unfolding (solid symbols) and refolding (open symbols) titrations of OmpA in LDAby fluorescence spectroscopy. Samples of OmpA in LDAO were incubated for 3 (d, s), 7samples of OmpA in A8-35 for 3 (d, s), 10 (j, h), 24 (N,4), 38 (.,5), 52 (solid and opethe folding and unfolding titrations shown in panels (A) and (B). Equilibrium was reaequilibrium, free energies of unfolding were �60 kJ/mol for OmpA in LDAO solution and
Equilibrium folding/unfolding has been compared for OmpA inA8-35 vs. the detergent LDAO [116], allowing the calculation andcomparison of the free energies of unfolding (Fig. 3). Unfoldingand folding titration curves superimposed only after very longincubation times: 30–40 days for OmpA/A8-35 complexes, 18–25 days for OmpA/LDAO ones. As shown in Fig. 3, it is the unfoldingreaction that imposes these long equilibration times. The activa-tion free energy for unfolding OmpA in urea, pH 10, is much higherin A8-35 than in LDAO, as indicated by the slower kinetics.
Titrations for folding indicate that equilibrium is reached muchfaster when the reaction is started with unfolded OmpA. This indi-cates that the activation energy for folding is lower than that forunfolding. Reversible folding/unfolding conditions were onlyachieved at pH 10, in line with an earlier study of the stability ofOmpA in sonicated lipid vesicles [134]. At pH 10, OmpA (pI = 5.5)is strongly negatively charged, because of the deprotonation ofsome of the 17 lysine (pK � 9.5–10.5) and 17 tyrosine (pK � 9.5–10) residues. The resulting increased negative charge of OmpAleads to intermolecular repulsion and a stronger side-chain hydra-tion, which prevents aggregation and ensures a better solubility.However, the increased net negative charge of OmpA may also leadto less stable complexes with the negatively charged A8-35,because of charge–charge repulsion. This might be the reason forthe reduced thermodynamic stability of folded OmpA observed inA8-35 as compared to LDAO (Fig. 3). Neutral APols can be expected
O (A) and in A8-35 (B) in the presence of various concentrations of urea, determined(j, h), 10 (N, 4), 17 (.,5), and 25 (solid and open bowties) days (panel A) and
n bowties) days (panel B) at 40 �C. (C) and (D) Energies of unfolding calculated fromched after 25 days for LDAO (C) and after 52 days for A8-35 (D), respectively. At�8 kJ/mol for A8-35-trapped OmpA. Adapted from Ref. [116].

J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343 335
to provide greater thermodynamic stabilization under suchextreme conditions as pH 10. Despite the thermodynamic destabi-lization of OmpA, the activation energy of unfolding of OmpA fromcomplexes with A8-35 is much higher, so that it takes longer forOmpA to unfold in A8-35 than in LDAO [116]. This is consistentwith the view that part of the stabilizing properties of APols resultfrom damping the conformational excursions of OmpA, whichslows down denaturation (see below).
In lipid bilayers (large unilamellar vesicles), an equilibriumunfolding titration of OmpA could not be observed even after12 days of incubation [135]. The folding titration had a midpointaround 2 M urea, similar to that observed for folding of OmpA intoA8-35 [116]. This midpoint is much lower than the midpointobserved for unfolding in LDAO. Since the presence of a surfactantis necessary for equilibrium unfolding studies of membrane pro-teins, it cannot be entirely excluded that the higher thermody-namic stability of OmpA seen in LDAO (or in octylmaltoside[136]) vs. A8-35 be due to surfactant-urea interactions. The activa-tion energy for unfolding of OmpA from lipid bilayers is so highthat it prevented the estimation of the free energy from unfoldingtitrations [135].
As a conclusion, these observations indicate that OmpA, atpH 10, is greatly stabilized by A8-35, as compared to LDAO, againstdenaturation by urea, not as a result of an increased thermody-namic stability, but because of a large increase of the activationenergy of unfolding.
Fig. 4. The hydrophobic sink effect. (A) Sedimentation velocity analysis of the associatdifferent concentrations of dodecylmaltoside (DDM). Samples of purified Chlamydomona(top) or 3 mM DDM (bottom) were centrifuged on sucrose gradients containing the samegels stained with silver. The areas shown correspond to the Rieske protein ( ) and to cytas a superdimer that has retained the Rieske protein and is functionally active. In the pretrails throughout the gradient, and, as a consequence, it has lost its electron-transfconcentrations of detergent supplemented or not with lipids. Purified b6f complex was inof egg PC to micellar detergent of 1:5 (*) or 1:10 (j), incubated in the dark at 4 �C, and
What is going on?
From the data summarized in the previous sections, it seemsreasonable to conclude that (i) as a rule, TMPs, whether of the a-helical or the b-barrel type, tend to be more stable after trappingwith APols than they are in detergent solution, and (ii) APolsappear as an efficient medium in which to fold TMPs to their nativestate. In the present section, we will discuss first various mecha-nisms that seem to be involved in stabilization, and try to relatethem to the physical–chemical properties of APols. We will nextexamine the issue of folding or refolding in APols.
Stabilization of membrane proteins by amphipols
Three mechanisms have been invoked as possible sources ofTMP stabilization by APols: (i) reduction of the hydrophobic sink;(ii) weak competition with protein/protein and protein/lipid inter-actions; (iii) damping of TMP dynamics (reviewed in Refs. [49–51]).Let us examine them in turn, provide some evidence in their favor,and discuss which properties of APols underlie them.
The hydrophobic sink effect. It is well known to membranebiochemists that the stability of a given TMP in a given detergentvery often depends on the concentration of micelles it is exposedto: the closer one is to the cmc of the detergent, the more stable the
ion state and composition of the cytochrome b6f complex in the presence of twos reinhardtii b6f complex in Tricine–NaOH buffer, pH 8.0, containing either 0.2 mMconcentration of DDM. Fractions were collected and analyzed by SDS–PAGE and theochrome b6 (|; diffuse band). In the presence of 0.2 mM DDM, the complex migratessence of 3 mM DDM, most of it has monomerized and lost the Rieske protein, whicher activity. (B) Kinetics of inactivation of the b6f complex incubated with highcubated with 50 mM Hecameg containing either no added lipids (�) or a molar ratiothe electron transfer activity measured as a function of time. From Ref. [137].

336 J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343
protein. This is illustrated in Fig. 4A in the case of the photosyn-thetic complex cytochrome b6f, a superdimer whose stability isexquisitely dependent on the detergent used and its concentration.Upon migration in a sucrose gradient containing 0.2 mM DDM, thatis just above the cmc of �0.17 mM, the complex migrates as adimer, retains all of its subunits, and it is active in electron transfer.At 3 mM DDM, a large fraction of the preparation disaggregatesinto inactive monomers, with the loss of cofactors and subunits[137]. Because the chemical potential of DDM monomers remainsessentially constant under these two conditions, buffered as it is bythe micelles, there is no reason to think that the physical–chemicalproperties of the DDM belt surrounding the complex vary signif-icantly upon increasing the DDM concentration from 0.2 to 3 mM.What does vary –enormously– is the concentration of micelles.Taking 0.17 mM to be the approximate cmc of DDM under theseconditions, the 0.2-mM gradient contains �0.03 mM of micellarDDM, the 3-mM one �2.87 mM, that is �0.015 and �1.5 g L-1,respectively. This corresponds to a �100� increase in the volumeof micellar detergent. This can obviously favor monomerization,either directly, by displacing a dimer/monomer equilibrium, orindirectly, by diluting hydrophobic or amphipathic molecules –lipids, cofactors, subunits– that are associated to the complex andmay stabilize its dimeric state. In the case of cytochrome b6f,delipidation has been identified as a major factor of destabilization.This is illustrated in Fig. 4B, where the electron transfer activity ofthe complex has been followed as a function of time in thepresence of a high concentration of Hecameg (a detergent similarto OG), supplemented or not with egg lecithin in a molar ratio tomicellar detergent of 1:10 or 1:5 [137].
That the hydrophobic sink effect also exists in preparations ofTMPs trapped in APols is suggested by Fig. 5, in which the stabilityof BR, solubilized from purple membrane (PM) along with PMlipids, has been compared at two temperatures and in the presenceof a variable excess of A8-35 or OTG (data from Ref. [138], figurefrom Ref. [50]). BR is known to bind �2 g A8-35 per g protein [126].The excess of APol over that actually bound to the protein istherefore, respectively, �2.5�, �5�, �10� and �25� for overallmass ratios of 1:5, 1:10, 1:20 and 1:50. It is also known that, in thefirst of these conditions –which is the one in which BR is usuallyhandled in A8-35–, PM lipids remain quantitatively associated to it,and that BR is mostly monomeric [126]. Experiments in which BRhas been refolded in A8-35 either in the presence or absence oflipids have shown that lipids improve its resistance to thermal
Fig. 5. Time stability of bacteriorhodopsin (BR) in amphipol A8-35 versus octylthioglmembrane (PM), along with PM lipids, trapped in A8-35 at various BR/A8-35 mass ratiocontaining 100 mM NaCl and 20 mM sodium phosphate, pH 7.0 ([BR] = 0.22 g L�1). Its abwas followed as a function of time. Control samples were kept in 18- or 25-mM OTGcorrespond to roughly the same mass concentration of aggregated detergent as that of Aratios. The absence of data points past 2 h in OTG at 40 �C is due to the aggregation of the[138].
denaturation [128]. Finally, because the CAC of A8-35 is very low,�0.002 g L�1 [123], and the concentration of free APol in theexperiments of Fig. 5 is much higher (�0.7–10 g L�1), nearly allAPol that is not bound to BR is present as particles, into whichlipids can partition. At room temperature, there is no clear effect ofthe concentration of APol on the stability of BR, the proteinremaining perfectly stable over a period of a week (Fig. 5, left). At40 �C, however, there is a tendency for BR to inactivate morerapidly at the highest concentrations of APol (Fig. 5, right). Themost likely interpretation of this data is that there is someredistribution of PM lipids between the surface of the proteinand free APol particles and that, as a result, inactivation sets insomewhat more rapidly in the presence of a large excess of APol.
In solution, it is recommended to keep a small excess of freeAPol in equilibrium with TMP/APol complexes, so as to avoid theformation of small oligomers (see Refs. [54,71,126,139,140]). Thisexcess is, however, of the same order of magnitude as the amountof TMP-bound APol, in our example �0.44 g L�1. When TMP/APolcomplexes cannot aggregate, as occurs upon immobilization ontochips or beads, free APol can be dispensed with entirely, becauseAPols will not spontaneously desorb unless they are displaced byother surfactants [47,141,142]. When working with detergentsolutions, on the contrary, not only must one keep the concentra-tion of the detergent above its cmc, but it is advisable to keep amargin of safety above it, e.g. 0.2 � cmc if the cmc is relatively wellknown, because the cmc is not a universal constant and it can beaffected by such factors as the presence of salt, pH, or thetemperature. With a detergent like OTG, whose cmc is �10 mM,this means working with �2 mM of micellar detergent, i.e.�0.6 g L�1. At such a high concentration of protein as that usedin Fig. 5 (�8 lM BR), the difference between the concentration offree, self-associated APol or detergent is not large, meaning thatthe volume of the hydrophobic sink is comparable (note that BR isnevertheless more stable in A8-35 than in OTG, indicating that, asdiscussed below, other factors must come into play). In many otherexperiments, however, e.g. spectroscopic or functional measure-ments, where the protein concentration is often a couple of ordersof magnitude lower, the concentration of free APol particles can belowered by as much, because what matters is the TMP/APol massratio, not the absolute concentration of APol. This does not apply tofree detergent micelles, which must be kept at a safe level abovethe cmc whatever the concentration of protein. Everything elsebeing equal, the distribution of lipids will therefore be strongly
ucoside (OTG). BR was extracted with OTG from Halobacterium salinarum purples, and stored in the dark either at room temperature (A) or at 40 �C (B) in a buffersorbance at 554 nm, which is proportional to the concentration of the holoprotein,(total OTG concentration, including bound detergent). These two concentrations
8-35 particles in the samples trapped respectively at 1:10 and 1:20 BR/A8-35 massprotein, accompanied by complete bleaching. From Ref. [50], original data from Ref.

J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343 337
displaced towards binding to the protein in the APol-trapped vs.the detergent-solubilized preparation, which is a stabilizing factor.Needless to say, when APol-trapped TMPs are immobilized ontosolid supports and flushed with surfactant-free solutions, thehydrophobic sink effect disappears altogether.
The poorly dissociating character of APols. In the experiments ofFig. 5, the volume of aggregated surfactant is roughly the same inthe presence of 18 mM OTG (resp. 25 mM) and at 1:10 (resp. 1:20)BR/A8-35 mass ratios. Yet, BR inactivates much faster in OTG thanin A8-35. Under such conditions, the hydrophobic sink effectcannot account for the stabilization of BR by A8-35 as compared toOTG. Several lines of observations suggest that, as compared todetergents, APols are poor competitors for protein/protein andprotein/lipid interactions. This is reflected in their inability tosolubilize biological membranes [47], and even, in most cases, purelipid vesicles [143]: according to this criterion, APols are not, oronly extremely weak detergents. The issue is somewhat complex,in the sense that dispersion of lipids can occasionally be observed,given the right combination of APol chemical structure, lipidcomposition, ionic strength, temperature and, typically, longperiod of incubation (days) (see Ref. [143], and Refs. therein).Nevertheless, APols can be applied to lipid vesicles [117] or blacklipid membranes [86] without breaking them, and to live cellswithout lysing them [47,50].
Another line of evidence is provided by studies of the functionalcycles of BR [128] and, possibly, the nicotinic acetylcholinereceptor [144]. Upon excitation of its chromophore, retinal, bygreen light, BR undergoes a complex series of transconformations,the photocycle, which results in building up a transmembraneproton gradient [145,146]. Solubilization by detergents affects thekinetics of the photocycle: the early steps are accelerated, an effectthat largely persists after transfer to A8-35 and may reflect the lossof the 2D lattice, whereas late phases are slowed down (see Ref.[128], and therein). When detergent-solubilized BR is supple-mented with A8-35 and the detergent removed, the late kineticsbecomes similar to that in PM. If PM is solubilized in SDS, themixture supplemented with A8-35, and dodecylsulfate precipi-tated as PDS, renatured BR exhibits a photocycle identical to that ofBR trapped with A8-35 without having been denatured. If, how-ever, BR is renatured in A8-35 in the absence of lipids, itsphotocycle resembles that in OTG. The most likely interpretationof these observations is that interactions with lipids control thekinetics of the late steps of BR photocycle, that these interactionsare absent or perturbed in OTG solution, and that they reformwhen OTG is replaced with A8-35. The implication is that OTGefficiently competes with lipids for binding to the surface of BR,whereas A8-35 does not [128].
The evidence regarding the nicotinic acetylcholine receptor(nAChR) is less detailed, but points in the same direction. ThenAChR is a chemically controlled cation-specific channel that liesin the postsynaptic membrane of the neuromuscular junction andother synapses. Upon binding the neuromediator acetylcholine, itsconformation shifts from a non-conducting, low-affinity state toone in which the channel is open, and then to non-conducting,high-affinity, desensitized states [147]. In native receptor-richmembrane fragments from the electric organ of the electric rayTorpedo marmorata, the nAChR, in the absence of ligands, pre-exists in an allosteric equilibrium where a majority of receptors isin the low-affinity resting state, ready to be activated, and aminority of them pre-exists in one of the high-affinity desensitizedstates. Upon solubilization with CHAPS, the equilibrium shifts infavor of the desensitized states. Upon addition of A8-35 anddilution under the cmc of CHAPS, the equilibrium comes back to
that in the original membranes [144] (Fig. 6). The most straight-forward interpretation of this observation is that CHAPS displaceslipids from the surface of the nAChR. Upon dilution in the presenceof A8-35, it desorbs. Recovery of the original allosteric equilibriumcould be due either to the desorption of CHAPS and its replacementwith A8-35 or, as in the case of BR, to the rebinding of lipids.
Altogether, and even though more systematic studies would bedesirable, it seems therefore likely that part of the stabilizingcharacter of APols could be due to their inability to efficientlycompete with the protein/protein and protein/lipid interactionsthat keep biological membranes together and stabilize the nativestructure of TMPs. This may be due to the fact that the relativelyshort hydrophobic chains of APols are bound to a polymericbackbone, making it more difficult for them to intrude into protein/lipid assemblies and disrupt their structure than it is for detergentmolecules. The underlying mechanism may be enthalpic or,perhaps more likely, entropic.
Amphipols damp the dynamics of membrane proteins: the‘‘Gulliver effect’’. APols as a rule do not perturb the function ofTMPs (reviewed in Refs. [49–51]). There is however one remark-able exception, the sarcoplasmic calcium pump from twitchmuscle (SERCA1a) [148], whose enzymatic cycle is reversiblyinhibited by APols with respect to what it is both in permeabilizedsarcoplasmic vesicles and in detergent solution [52]. The inhibitionis not due to a trivial mechanism such as interference with calciumor ATP binding. It is accompanied by a strong stabilization againstthe irreversible denaturation that is observed upon calciumremoval in detergent solution. Later studies revealed anotherinteresting feature: when different APols are compared, or whenmixtures of A8-35 and detergent are used, a correlation appearsbetween the level of inhibition and the degree of stabilization: thegreater the stabilization, the stronger the inhibition [125]. Thisgave a first hint that the same mechanism might underlie botheffects. Indeed, i) the enzymatic cycle of SERCA1a involves large(nanometric) rearrangements of its transmembrane helices [149],which is not the case for the other TMPs whose function has beenstudied in the APol-trapped state to date; and ii) calcium is knownto bridge SERCA1a transmembrane helices, which leads to thesuggestion that the rapid inactivation of the enzyme upon calciumremoval in the presence of detergent [150] may result from theopening of the transmembrane helix bundle, a first step on the wayto irreversible denaturation.
These observations led to a hypothesis, dubbed the ‘‘Gullivereffect’’, according to which APols, because they attach to thetransmembrane surface of TMPs at multiple points, may damplarge-scale (nanometric) rearrangements of the surface of trans-membrane protein domains [47,50,51,125]. A possible mechanismwould be that conformational transitions of the protein that entaila rearrangement of the backbone of the polymer are affected by afree energy penalty that makes them less probable than they are indetergent solution. This could account both for the slowing downof the enzymatic cycle of SERCA1a and for lowering the frequencyand amplitude of transient openings of the transmembrane helixbundle, purported to be the first step towards denaturation,potentially explaining the observed correlation between the twoeffects.
Long highly speculative, this hypothesis has been progressivelybuttressed by a number of observations:
(i) MD [76,151] and inelastic neutron scattering [151] dataindicate that A8-35 particles have a hydrophobic core whoseviscosity is comparable to that of the hydrophobic region ofbiological membranes, whereas their interface with the

Fig. 6. Allosteric transitions of the nicotinic acetylcholine receptor (nAChR) in three different environments. Kinetics of binding of a fluorescent ligand to nAChR (A and B) innative membrane fragments from Torpedo marmorata electric organ; (C and D) after solubilization in detergent solution (CHAPS); (E and F) after addition of A8-35 anddilution below the cmc of CHAPS. In its native membrane environment, the nAChR pre-exists to the addition of ligands in an equilibrium between a low-affinity resting stateand high-affinity, inactive states, in a proportion of about 9:1. Upon addition of a low concentration of fluorescent agonist, only the receptors in the high-affinity states bindthe ligand (panel A), relaxation of those in the resting state to high-affinity ones, followed by ligand binding, occurring more slowly (panel B). After solubilization, the ratiobetween high- and low-affinity states becomes about 1:1, explaining the higher level of fast binding seen in panel C. When most of CHAPS is replaced by A8-35 in theenvironment of the receptor, the allosteric equilibrium comes back to a situation similar to that in the membrane (E and F). From Ref. [144].
338 J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343
aqueous solution, where most of the APol backbone lies(Fig. 7, top), is more viscous than that of detergent micelles,and even that of lipid bilayers.
(ii) According to MD data, conformational excursions of A8-35-trapped OmpX (Fig. 7, middle) are damped as compared tothose in a detergent or a lipid environment [70] (Fig. 7,bottom).
(iii) As described above, the stabilization of OmpA by A8-35against urea-induced denaturation at pH 10 is not due to athermodynamic stabilization, but to a higher energy barrieron the way to unfolding [116].
Taken together, these observations suggest that there is prob-ably some truth in the view that one of the mechanisms of TMPstabilization by APols is a relative freezing of their movements ascompared to those in detergent solution.
Stabilization of membrane proteins by amphipols has not a uniqueorigin
It will be apparent from the above that stabilization of TMPs byAPols has a multifactorial origin. Depending on the protein consid-ered, on the APol, and on experimental conditions, one mechanismmay be more important than another. For instance, it is generally
observed that trapping TMPs with APols in the presence of lipidsadds to their stability as compared to APol alone (see e.g. Refs.[45,128]). It is likely that, in the presence of lipids, stabilizationdue to the reduction of the hydrophobic sink will lose some of itsimportance, as illustrated in Fig. 4B for the b6f complex in deter-gent solution.
There are suggestions that, as is generally the case for deter-gents, the more highly charged an APol is, the less stabilizing. Thus,SERCA1a is more stable in A8-35 (which carries �35% of chargedunits) than in SAPols (�75% of charged units) [125], and cyto-chrome b6f is stabilized by NAPols (no charges), but not by A8-35[46,78]. This can be understood if electrostatic repulsion favorsextended or dissociated states of TMP/APol complexes. Yet, theion channel TRPA1 is reported to be stabilized by SAPols as com-pared to A8-35 [67]. Clearly, more systematic studies are neededbefore a general view can emerge.
Amphipol-assisted folding of membrane proteins
As described above, folding TMPs in APols has been, to date,remarkably successful. This statement must be qualified byremarking that only a limited range of structural types has been
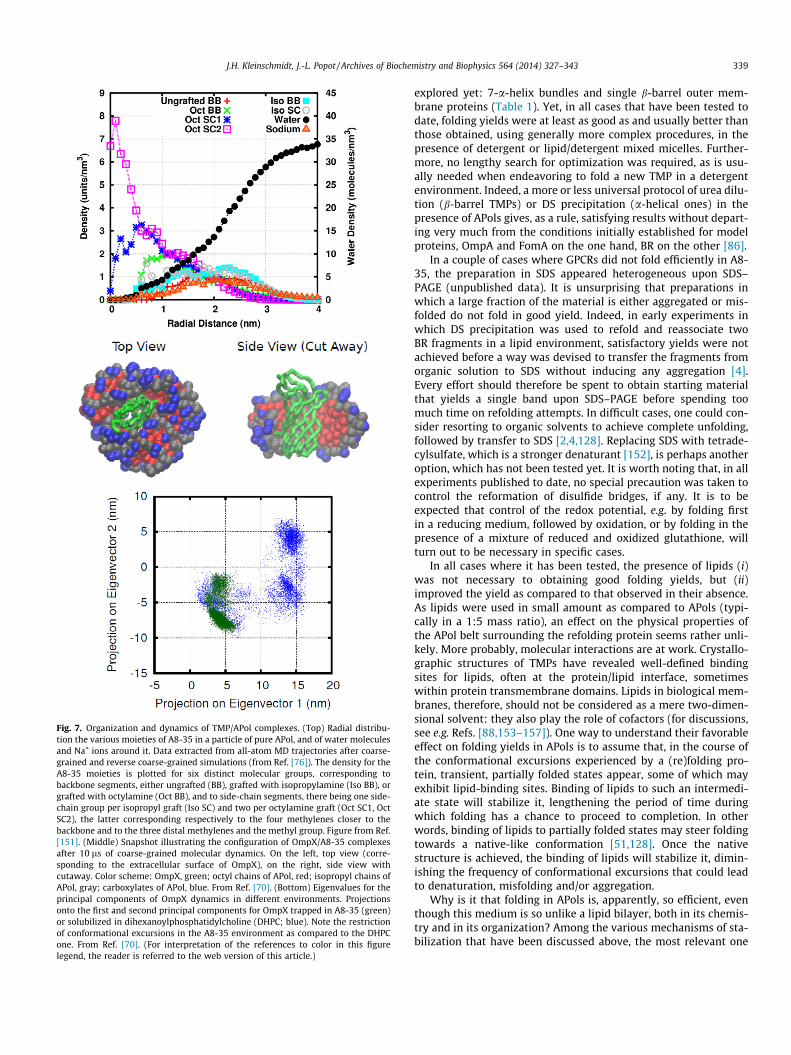
Fig. 7. Organization and dynamics of TMP/APol complexes. (Top) Radial distribu-tion the various moieties of A8-35 in a particle of pure APol, and of water moleculesand Na+ ions around it. Data extracted from all-atom MD trajectories after coarse-grained and reverse coarse-grained simulations (from Ref. [76]). The density for theA8-35 moieties is plotted for six distinct molecular groups, corresponding tobackbone segments, either ungrafted (BB), grafted with isopropylamine (Iso BB), orgrafted with octylamine (Oct BB), and to side-chain segments, there being one side-chain group per isopropyl graft (Iso SC) and two per octylamine graft (Oct SC1, OctSC2), the latter corresponding respectively to the four methylenes closer to thebackbone and to the three distal methylenes and the methyl group. Figure from Ref.[151]. (Middle) Snapshot illustrating the configuration of OmpX/A8-35 complexesafter 10 ls of coarse-grained molecular dynamics. On the left, top view (corre-sponding to the extracellular surface of OmpX), on the right, side view withcutaway. Color scheme: OmpX, green; octyl chains of APol, red; isopropyl chains ofAPol, gray; carboxylates of APol, blue. From Ref. [70]. (Bottom) Eigenvalues for theprincipal components of OmpX dynamics in different environments. Projectionsonto the first and second principal components for OmpX trapped in A8-35 (green)or solubilized in dihexanoylphosphatidylcholine (DHPC; blue). Note the restrictionof conformational excursions in the A8-35 environment as compared to the DHPCone. From Ref. [70]. (For interpretation of the references to color in this figurelegend, the reader is referred to the web version of this article.)
J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343 339
explored yet: 7-a-helix bundles and single b-barrel outer mem-brane proteins (Table 1). Yet, in all cases that have been tested todate, folding yields were at least as good as and usually better thanthose obtained, using generally more complex procedures, in thepresence of detergent or lipid/detergent mixed micelles. Further-more, no lengthy search for optimization was required, as is usu-ally needed when endeavoring to fold a new TMP in a detergentenvironment. Indeed, a more or less universal protocol of urea dilu-tion (b-barrel TMPs) or DS precipitation (a-helical ones) in thepresence of APols gives, as a rule, satisfying results without depart-ing very much from the conditions initially established for modelproteins, OmpA and FomA on the one hand, BR on the other [86].
In a couple of cases where GPCRs did not fold efficiently in A8-35, the preparation in SDS appeared heterogeneous upon SDS–PAGE (unpublished data). It is unsurprising that preparations inwhich a large fraction of the material is either aggregated or mis-folded do not fold in good yield. Indeed, in early experiments inwhich DS precipitation was used to refold and reassociate twoBR fragments in a lipid environment, satisfactory yields were notachieved before a way was devised to transfer the fragments fromorganic solution to SDS without inducing any aggregation [4].Every effort should therefore be spent to obtain starting materialthat yields a single band upon SDS–PAGE before spending toomuch time on refolding attempts. In difficult cases, one could con-sider resorting to organic solvents to achieve complete unfolding,followed by transfer to SDS [2,4,128]. Replacing SDS with tetrade-cylsulfate, which is a stronger denaturant [152], is perhaps anotheroption, which has not been tested yet. It is worth noting that, in allexperiments published to date, no special precaution was taken tocontrol the reformation of disulfide bridges, if any. It is to beexpected that control of the redox potential, e.g. by folding firstin a reducing medium, followed by oxidation, or by folding in thepresence of a mixture of reduced and oxidized glutathione, willturn out to be necessary in specific cases.
In all cases where it has been tested, the presence of lipids (i)was not necessary to obtaining good folding yields, but (ii)improved the yield as compared to that observed in their absence.As lipids were used in small amount as compared to APols (typi-cally in a 1:5 mass ratio), an effect on the physical properties ofthe APol belt surrounding the refolding protein seems rather unli-kely. More probably, molecular interactions are at work. Crystallo-graphic structures of TMPs have revealed well-defined bindingsites for lipids, often at the protein/lipid interface, sometimeswithin protein transmembrane domains. Lipids in biological mem-branes, therefore, should not be considered as a mere two-dimen-sional solvent: they also play the role of cofactors (for discussions,see e.g. Refs. [88,153–157]). One way to understand their favorableeffect on folding yields in APols is to assume that, in the course ofthe conformational excursions experienced by a (re)folding pro-tein, transient, partially folded states appear, some of which mayexhibit lipid-binding sites. Binding of lipids to such an intermedi-ate state will stabilize it, lengthening the period of time duringwhich folding has a chance to proceed to completion. In otherwords, binding of lipids to partially folded states may steer foldingtowards a native-like conformation [51,128]. Once the nativestructure is achieved, the binding of lipids will stabilize it, dimin-ishing the frequency of conformational excursions that could leadto denaturation, misfolding and/or aggregation.
Why is it that folding in APols is, apparently, so efficient, eventhough this medium is so unlike a lipid bilayer, both in its chemis-try and in its organization? Among the various mechanisms of sta-bilization that have been discussed above, the most relevant one

Fig. 8. Hypothetical scheme of events leading to either (re)folding to the native state r or to the formation of inactive forms due to either incomplete (re)folding s,misfolding t, or aggregation u, upon removal of the denaturant from a solution of denatured membrane protein in the presence of (A) detergent or mixed lipid/detergentmicelles or (B) amphipols or amphipol/lipid mixtures. As compared to detergents, APols are postulated to favor pathway r over, in particular, pathway s, because of theirlesser detergency. See text. Adapted from Ref. [86].
340 J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343
may be their poorly dissociating character. When folding is carriedout in detergent, or in lipid/detergent mixtures, the detergent –amolecule that was initially selected for its dissociating proper-ties–, competes with reforming protein/protein and protein/lipidinteractions. In the scheme of Fig. 8A, this means that folding tothe native structure r has to compete with partial folding s, inwhich protein/surfactant interactions replace some protein/pro-tein ones, misfolding t, in which non-native-like interactions form
intramolecularly, and aggregation u, induced by intermolecularprotein/protein interactions. APols, because of their low deter-gency, can be expected to favor the productive path r, which,being more accessible, should more efficiently compete with theunproductive paths s, t and u (Fig. 8B). Furthermore, most TMPsare not particularly stable in detergent solutions, so that a proteinthat has managed to reach a native-like state stands a chance todenature again at a later stage. In SDS or urea, it would go back

J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343 341
to the ‘‘denatured’’ state, from which it can fold again. In a ‘‘non-denaturing’’ detergent, which is not so dissociating, it is at risk ofreaching a misfolded t and/or aggregated u state, from which itmay not recover (Fig. 8A). In an APol environment, chances thata protein that has reached a partially folded state s will move tothe native state r can be expected to be higher, because of thelesser competition of protein/surfactant interactions with native-like protein/protein and protein/lipid ones. This, in turn, shoulddiminish the risks of moving to the irreversible states t and u
(Fig. 8B).According to this view, APols could provide a good folding med-
ium because, on the one hand, they adsorb onto hydrophobic sur-faces, keeping unfolded TMPs from aggregating (or slowing downtheir aggregation), while, on the other hand, they do not competeefficiently with the protein/protein and protein/lipid interactionsthat define the native structure. Thus, they would substitute forSDS or urea at the surface of the unfolded protein, keeping it solu-ble, but be progressively displaced from those protein surfaces thatcan form stronger interactions either with other proteic elementsor with lipids. Although the term of ‘‘molecular chaperones’’ hasbeen overused and misused, it may be to some extent appropriatehere, in the sense that APols may slow down the formation of non-specific, unproductive interactions between hydrophobic seg-ments, which would lead to misfolding and/or aggregation, whilemoving out of the way when specific interactions establishthemselves.
Three types of APols have led to successful (re)folding of TMPsto date: A8-35, SAPols and NAPols (Table 1). Except for BR, forwhich comparable folding yields are achieved in A8-35 and in NAP-ols [78,107,128], no comparative studies of the folding yieldsachieved for one given TMP using one or the other APol have beencarried out yet. On the basis of our early experiments, we had pro-posed that A8-35 formed around refolding TMPs a sort of protec-tive ‘‘bubble’’ that would allow folding to proceed while slowingdown the formation of non-productive intermolecular interactions[86] (Fig. 8). One possible mechanism providing a relative isolationof refolding proteins one from another could be the electrostaticrepulsion between complexes incorporating either A8-35 or SAP-ols: both of them are polyanions, whose interactions indeedstrongly depend on the ionic strength [71]. The fact that the totallyuncharged NAPols also allow folding in good yield [78] seems toindicate that such an electrostatic mechanism, if present, is notessential.
Detailed protocols for folding TMPs in APols are given in Ref.[84].
Perspectives
Once trapped or folded in APols, TMPs can be purified, handledand studied using most of the approaches classically used in deter-gent solutions, plus some extra ones made possible, in particular,by the use of labeled or functionalized APols (reviewed in Refs.[49–51]). The stability conferred to them by the APol environmentfacilitates experiments that are lengthy and/or must be carried outunder destabilizing conditions, such as high concentrations ofdetergent and/or high temperature (for a general discussion, seeRef. [51]). A case in point is solution NMR, whose conditions tendto be harsh (reviewed in Refs. [60,91,158,159]). In EM single-parti-cle image reconstruction, the use of APols facilitates the study offragile TMPs and TMP complexes (see e.g. Refs. [61,62,64,67–69,160], and Refs. therein). As noted above, an interesting observa-tion is that, in the case of the ion channel TRPV1, more homoge-neous images are obtained in A8-35 than in detergent [64],which may be related to stabilization. In vaccination, better protec-tion against a bacterial disease is obtained following immunization
against an APol-trapped than a detergent-solubilized outer TMPused as immunogen, which may be related either to its stabiliza-tion and/or to a better presentation to the immune system[53,55,161]. Also worth noting is the developing use of taggedAPols for immobilizing TMPs onto solid surfaces [141,142,162–164]. In such experiments, several advantages of APols areexploited simultaneously: they stabilize the TMP during extendedscreening sessions, they mediate its immobilization, and, becauseof the stability of TMP/APol interactions, they make it possible towork in surfactant-free media (reviewed in Ref. [165]).
As noted above, trapping TMPs in APols in the presence of lipidsgenerally increases their stability. This can of course be combinedwith other stabilizing strategies, such as adding ligands, a case inpoint being BR [128]. It is interesting that, in this particular case,the ligand, retinal, is extremely hydrophobic. When added to therefolded apoprotein in the presence of APol, it must distributebetween protein-bound APol belts and free APol particles. It islikely delivered to retinal-free BO/APol complexes when they col-lide with retinal-containing particles (for a discussion, see Refs.[71,128]).
An interesting question is whether TMPs that have been madedetergent-resistant by accumulating stabilizing point mutations(see e.g. Refs. [166–169], and therein) will show improved stabilityin APols as compared to wild-type proteins. This may not be thecase if the stabilization mechanism involves the selection of deter-gent-binding sites at the surface of the protein. Note also thatdetergent-resistant GPCRs have been engineered primarily withthe view of forming 3D crystals in detergent solution, whereascrystallizing in aqueous solution is not the easiest application ofAPols (see Refs. [51,54,95], and therein).
One of the many open questions is which types of TMPs will beamenable to APol-assisted folding. A particularly difficult case isthat of oligomeric TMPs. There are however reasons to be cau-tiously optimistic:
(i) Many (currently close to 20) oligomeric TMPs have beenfolded or expressed in vitro using classic surfactants(reviewed in Ref. [1]).
(ii) As illustrated by the delivery of retinal after BO has folded,being trapped with APols does not prevent a TMP to interactwith a molecule carried by another APol particle [128] (seealso Ref. [171]).
(iii) By increasing the TMP/APol ratio, APol-trapped TMPs can beinduced to interact one with another [71,126,139].
(iv) There are indirect indications that GPCRs folded in A8-35can dimerize. Whereas no strong, direct demonstration hasbeen provided yet, this is suggested by the fact that thereis an optimum protein/A8-35 ratio for the BLT1 leukotrienereceptor to fold [45]. In the absence of lipids or any othercofactor that could become diluted, the most straightfor-ward interpretation of this observation is that newly foldedreceptors become stabilized by dimerization, which occursless efficiently in the presence of too large an excess of APol.
One of the main challenges in folding oligomeric TMPs is that, inmost cases, unassembled monomers can be expected to be onlymarginally stable. Conditions must be found that favor their fold-ing and assembly while discouraging the formation of improperintermolecular interactions. Given that it can be done in detergentor mixed lipid/detergent micelles (see Ref. [1]), there is every rea-son to hope that APols will make it easier. It is likely that, in mostcases, simultaneous (re)folding of all subunits will turn out to bethe best strategy. In cases where unassembled monomers havevery different stabilities, however, alternative routes could beexperimented with, such as folding or expressing the most instablesubunit in the presence of its already folded partner(s).

342 J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343
Acknowledgments
Particular thanks are due to Jean-Louis Banères, Laurent Catoire,Melanie Cocco, Tassadite Dahmane, Yann Gohon, Jason Perlmutter,Cosmin Pocanschi, and Manuela Zoonens for discussions and/orcomments on the manuscript. Work performed in JHK’s laboratorywas funded by the Deutsche Forschungsgemeinschaft, the Univer-sity of Konstanz and the University of Kassel. Work performed inJLP’s laboratory was funded principally by the French CentreNational de la Recherche Scientifique, University Paris-7 Denis Did-erot, the Human Frontier Scientific Program Organization, theEuropean Community, the Agence Nationale pour la Rechercheand the US National Institutes of Health.
References
[1] J.-L. Popot, Arch. Biophys. Biochem. 564 (2015) 314–326.[2] K.-S. Huang, H. Bayley, M.-J. Liao, E. London, H.G. Khorana, J. Biol. Chem. 256
(1981) 3802–3809.[3] E. London, H.G. Khorana, J. Biol. Chem. 257 (1982) 7003–7011.[4] J.-L. Popot, S.E. Gerchman, D.M. Engelman, J. Mol. Biol. 198 (1987) 655–676.[5] H. Paulsen, U. Rümler, W. Rüdiger, Planta 181 (1990) 204–211.[6] H. Rogl, K. Kosemund, W. Kühlbrandt, I. Collinson, FEBS Lett. 432 (1998) 21–
26.[7] H. Kiefer, J. Krieger, J.D. Olszewski, G. Von Heijne, G.D. Prestwich, H. Breer,
Biochemistry 35 (1996) 16077–16084.[8] H. Kiefer, K. Maier, R. Vogel, Biochem. Soc. Trans. 27 (1999) 908–912.[9] J.-L. Banères, A. Martin, P. Hullot, J.P. Girard, J.C. Rossi, J. Parello, J. Mol. Biol.
329 (2003) 801–814.[10] J.-L. Banères, D. Mesnier, A. Martin, L. Joubert, A. Dumuis, J. Bockaert, J. Biol.
Chem. 280 (2005) 20253–20260.[11] F.I. Valiyaveetil, R. MacKinnon, T.W. Muir, J. Am. Chem. Soc. 124 (2002) 9113–
9120.[12] Y.J. Chen, O. Pornillos, S. Lieu, C. Ma, A.P. Chen, G. Chang, Proc. Natl. Acad. Sci.
U.S.A. 104 (2007) 18999–19004.[13] F.W. Lau, J.U. Bowie, Biochemistry 36 (1997) 5884–5892.[14] B.M. Gorzelle, J.K. Nagy, K. Oxenoid, W.L. Lonzer, D.S. Cafiso, C.R. Sanders,
Biochemistry 38 (1999) 16373–16382.[15] H. Kiefer, Biochim. Biophys. Acta 1610 (2003) 57–62.[16] N.J. Harris, P.J. Booth, Biochim. Biophys. Acta 1818 (2012) 1055–1066.[17] S.K. Buchanan, Curr. Opin. Struct. Biol. 9 (1999) 455–461.[18] A.M. Stanley, K.G. Fleming, Arch. Biochem. Biophys. 469 (2008) 46–66.[19] S.K. Buchanan, S. Yamashita, K.G. Fleming, in: L.K. Tamm (Ed.),
Comprehensive Biophysics, Academic Press, Oxford, 2012, pp. 139–163.[20] D.E. Otzen, K.K. Andersen, Arch. Biochem. Biophys. 531 (2013) 34–43.[21] M. Schweizer, I. Hindennach, W. Garten, U. Henning, Eur. J. Biochem. 82
(1978) 211–217.[22] K. Dornmair, H. Kiefer, F. Jähnig, J. Biol. Chem. 265 (1990) 18907–18911.[23] A. Pautsch, J. Vogt, K. Model, C. Siebold, G.E. Schulz, Proteins 34 (1999) 167–
172.[24] P.M. Hwang, W.Y. Choy, E.I. Lo, L. Chen, J.D. Forman-Kay, C.R. Raetz, G.G. Privé,
R.E. Bishop, L.E. Kay, Proc. Natl. Acad. Sci. U.S.A. 99 (2002) 13560–13565.[25] G.H. Huysmans, S.E. Radford, D.J. Brockwell, S.A. Baldwin, J. Mol. Biol. 373
(2007) 529–540.[26] N.K. Burgess, T.P. Dao, A.M. Stanley, K.G. Fleming, J. Biol. Chem. 283 (2008)
26748–26758.[27] C.S. Klug, W. Su, J. Liu, P.E. Klebba, J.B. Feix, Biochemistry 34 (1995) 14230–
14236.[28] S.K. Buchanan, Biochem. Soc. Trans. 27 (1999) 903–908.[29] B. Shanmugavadivu, H.J. Apell, T. Meins, K. Zeth, J.H. Kleinschmidt, J. Mol. Biol.
368 (2007) 66–78.[30] H. Engelhardt, T. Meins, M. Poynor, V. Adams, S. Nussberger, W. Welte, K.
Zeth, J. Membr. Biol. 216 (2007) 93–105.[31] S.R. Maurya, R. Mahalakshmi, PLoS One 9 (2014) e87701.[32] J.E. Mogensen, J.H. Kleinschmidt, M.A. Schmidt, D.E. Otzen, Protein Sci. 14
(2005) 2814–2827.[33] J.E. Mogensen, D. Tapadar, M.A. Schmidt, D.E. Otzen, Biochemistry 44 (2005)
4533–4545.[34] T. Surrey, F. Jähnig, Proc. Natl. Acad. Sci. U.S.A. 89 (1992) 7457–7461.[35] D.M. Engelman, Y. Chen, C.N. Chin, A.R. Curran, A.M. Dixon, A.D. Dupuy, A.S.
Lee, U. Lehnert, E.E. Matthews, Y.K. Reshetnyak, A. Senes, J.-L. Popot, FEBSLett. 555 (2003) 122–125.
[36] P.J. Booth, Curr. Opin. Struct. Biol. 22 (2012) 469–475.[37] J.K. Nagy, W.L. Lonzer, C.R. Sanders, Biochemistry 40 (2001) 8971–8980.[38] M. Lorch, P.J. Booth, J. Mol. Biol. 344 (2004) 1109–1121.[39] J.-L. Popot, D.M. Engelman, Biochemistry 29 (1990) 4031–4037.[40] J.H. Kleinschmidt, P.V. Bulieris, J. Qu, M. Dogterom, T. den Blaauwen, J. Mol.
Biol. 407 (2011) 316–332.
[41] J.H. Kleinschmidt, T. den Blaauwen, A. Driessen, L.K. Tamm, Biochemistry 38(1999) 5006–5016.
[42] J.H. Kleinschmidt, Chem. Phys. Lipids 141 (2006) 30–47.[43] J.H. Kleinschmidt, Cell. Mol. Life Sci. 60 (2003) 1547–1558.[44] L.M. McMorran, D.J. Brockwell, S.E. Radford, Arch. Biochem. Biophys. (2014),
http://dx.doi.org/10.1016/j.abb.2014.1002.1011.[45] T. Dahmane, M. Damian, S. Mary, J.-L. Popot, J.-L. Banères, Biochemistry 48
(2009) 6516–6521.[46] C. Tribet, R. Audebert, J.-L. Popot, Proc. Natl. Acad. Sci. U.S.A. 93 (1996)
15047–15050.[47] J.-L. Popot, E.A. Berry, D. Charvolin, C. Creuzenet, C. Ebel, D.M. Engelman, M.
Flötenmeyer, F. Giusti, Y. Gohon, P. Hervé, Q. Hong, J.H. Lakey, K. Leonard, H.A.Shuman, P. Timmins, D.E. Warschawski, F. Zito, M. Zoonens, B. Pucci, C. Tribet,Cell. Mol. Life Sci. 60 (2003) 1559–1574.
[48] C.R. Sanders, A. Kuhn Hoffmann, D.N. Gray, M.H. Keyes, C.D. Ellis,ChemBioChem 5 (2004) 423–426.
[49] J.-L. Popot, Annu. Rev. Biochem. 79 (2010) 737–775.[50] J.-L. Popot, T. Althoff, D. Bagnard, J.-L. Banères, P. Bazzacco, E. Billon-Denis, L.J.
Catoire, P. Champeil, D. Charvolin, M.J. Cocco, G. Crémel, T. Dahmane, L.M. dela Maza, C. Ebel, F. Gabel, F. Giusti, Y. Gohon, E. Goormaghtigh, E. Guittet, J.H.Kleinschmidt, W. Kühlbrandt, C. Le Bon, K.L. Martinez, M. Picard, B. Pucci, J.N.Sachs, C. Tribet, C. van Heijenoort, F. Wien, F. Zito, M. Zoonens, Annu. Rev.Biophys. 40 (2011) 379–408.
[51] M. Zoonens, J.-L. Popot, J. Membr. Biol. 247 (2014) 759–796, http://dx.doi.org/10.1007/s00232-00014-09666-00238.
[52] P. Champeil, T. Menguy, C. Tribet, J.-L. Popot, M. le Maire, J. Biol. Chem. 275(2000) 18623–18637.
[53] D.F. Tifrea, G. Sun, S. Pal, G. Zardeneta, M.J. Cocco, J.-L. Popot, L.M. de la Maza,Vaccine 29 (2011) 4623–4631.
[54] D. Charvolin, M. Picard, L.-S. Huang, E.A. Berry, J.-L. Popot, J. Membr. Biol. 247(2014) 981–996, http://dx.doi.org/10.1007/s00232-00014-09694-00234.
[55] H.E. Feinstein, D. Tifrea, G. Sun, J.-L. Popot, L.M. de la Maza, M.J. Cocco, J.Membr. Biol. 247 (2014) 1053–1065, http://dx.doi.org/10.1007/s00232-00014-09693-00235.
[56] M. Zoonens, L.J. Catoire, F. Giusti, J.-L. Popot, Proc. Natl. Acad. Sci. U.S.A. 102(2005) 8893–8898.
[57] L.J. Catoire, M. Zoonens, C. van Heijenoort, F. Giusti, E. Guittet, J.-L. Popot, Eur.Biophys. J. 39 (2010) 623–630.
[58] L.J. Catoire, M. Zoonens, C. van Heijenoort, F. Giusti, J.-L. Popot, E. Guittet, J.Magn. Reson. 197 (2009) 91–95.
[59] M. Etzkorn, M. Zoonens, L.J. Catoire, J.-L. Popot, S. Hiller, J. Membr. Biol. 247(2014) 965–970, http://dx.doi.org/10.1007/s00232-00014-09657-00239.
[60] N. Planchard, E. Point, T. Dahmane, F. Giusti, M. Renault, C. Le Bon, G. Durand,A. Milon, E. Guittet, M. Zoonens, J.-L. Popot, L.J. Catoire, J. Membr. Biol. 247(2014) 827–842, http://dx.doi.org/10.1007/s00232-00014-09654-z.
[61] T. Althoff, D.J. Mills, J.-L. Popot, W. Kühlbrandt, EMBO J. 30 (2011) 4652–4664.[62] E. Cao, M. Liao, Y. Cheng, D. Julius, Nature 504 (2013) 113–118.[63] B.M. Kevany, Y. Tsybovsky, I.D. Campuzano, P.D. Schnier, A. Engel, K.
Palczewski, J. Biol. Chem. 288 (2013) 36272–36284.[64] M. Liao, E. Cao, D. Julius, Y. Cheng, Nature 504 (2013) 107–112.[65] Y. Tsybovsky, T. Orban, R.S. Molday, D. Taylor, K. Palczewski, Structure 21
(2013) 854–860.[66] A. Vahedi-Faridi, B. Jastrzebska, K. Palczewski, A. Engel, Microscopy (Oxf.) 62
(2013) 95–107.[67] K.W. Huynh, M.R. Cohen, V.Y. Moiseenkova-Bell, J. Membr. Biol. 247 (2014)
843–851, http://dx.doi.org/10.1007/s00232-00014-09684-00236.[68] M. Liao, E. Cao, D. Julius, Y. Cheng, Curr. Opin. Struct. Biol. 27 (2014) 1–7.[69] P. Lu, X.C. Bai, D. Ma, T. Xie, C. Yan, L. Sun, G. Yang, Y. Zhao, R. Zhou, S.H.
Scheres, Y. Shi, Nature (2014).[70] J.D. Perlmutter, J.-L. Popot, J.N. Sachs, J. Membr. Biol. 247 (2014) 883–895,
http://dx.doi.org/10.1007/s00232-00014-09690-00238.[71] M. Zoonens, F. Giusti, F. Zito, J.-L. Popot, Biochemistry 46 (2007) 10392–
10404.[72] C. Tribet, C. Diab, T. Dahmane, M. Zoonens, J.-L. Popot, F.M. Winnik, Langmuir
25 (2009) 12623–12634.[73] C. Le Bon, J.-L. Popot, F. Giusti, J. Membr. Biol. 247 (2014) 797–814, http://
dx.doi.org/10.1007/s00232-00014-09655-y.[74] Y. Gohon, G. Pavlov, P. Timmins, C. Tribet, J.-L. Popot, C. Ebel, Anal. Biochem.
334 (2004) 318–334.[75] Y. Gohon, F. Giusti, C. Prata, D. Charvolin, P. Timmins, C. Ebel, C. Tribet, J.-L.
Popot, Langmuir 22 (2006) 1281–1290.[76] J.D. Perlmutter, W.J. Drasler 2nd, W. Xie, J. Gao, J.-L. Popot, J.N. Sachs,
Langmuir 27 (2011) 10523–10537.[77] K.S. Sharma, G. Durand, F. Gabel, P. Bazzacco, C. Le Bon, E. Billon-Denis, L.J.
Catoire, J.-L. Popot, C. Ebel, B. Pucci, Langmuir 28 (2012) 4625–4639.[78] P. Bazzacco, E. Billon-Denis, K.S. Sharma, L.J. Catoire, S. Mary, C. Le Bon, E.
Point, J.-L. Banères, G. Durand, F. Zito, B. Pucci, J.-L. Popot, Biochemistry 51(2012) 1416–1430.
[79] T. Dahmane, F. Giusti, L.J. Catoire, J.-L. Popot, Biopolymers 95 (2011) 811–823.[80] C. Diab, F.M. Winnik, C. Tribet, Langmuir 23 (2007) 3025–3035.[81] C. Diab, C. Tribet, Y. Gohon, J.-L. Popot, F.M. Winnik, Biochim. Biophys. Acta
1768 (2007) 2737–2747.[82] K.S. Sharma, G. Durand, F. Giusti, B. Olivier, A.S. Fabiano, P. Bazzacco, T.
Dahmane, C. Ebel, J.-L. Popot, B. Pucci, Langmuir 24 (2008) 13581–13590.

J.H. Kleinschmidt, J.-L. Popot / Archives of Biochemistry and Biophysics 564 (2014) 327–343 343
[83] P. Bazzacco, K.S. Sharma, G. Durand, F. Giusti, C. Ebel, J.-L. Popot, B. Pucci,Biomacromolecules 10 (2009) 3317–3326.
[84] M. Zoonens, F. Zito, K.L. Martinez, J.-L. Popot, in: I. Mus-Veteau (Ed.),Membrane Protein Production for Structural Analysis, Springer, New York,2014, pp. 173–203.
[85] J.H. Kleinschmidt, L.K. Tamm, J. Mol. Biol. 324 (2002) 319–330.[86] C.L. Pocanschi, T. Dahmane, Y. Gohon, F. Rappaport, H.-J. Apell, J.H.
Kleinschmidt, J.-L. Popot, Biochemistry 45 (2006) 13954–13961.[87] D. Oesterhelt, W. Stoeckenius, Nat. New Biol. 233 (1971) 149–152.[88] J.-L. Popot, D.M. Engelman, Annu. Rev. Biochem. 69 (2000) 881–922.[89] O. Tastan, A. Dutta, P. Booth, J. Klein-Seetharaman, Biochim. Biophys. Acta
1837 (2014) 656–663.[90] T. Dahmane, F. Rappaport, J.L. Popot, Eur. Biophys. J. 42 (2013) 85–101.[91] S. Elter, T. Raschle, S. Arens, A. Viegas, V. Gelev, M. Etzkorn, G. Wagner, J.
Membr. Biol. 247 (2014) 957–964, http://dx.doi.org/10.1007/s00232-00014-09669-00235.
[92] J.-L. Banères, J.-L. Popot, B. Mouillac, Trends Biotechnol. 29 (2011) 314–322.[93] M. Damian, A. Martin, D. Mesnier, J.P. Pin, J.-L. Banères, EMBO J. 25 (2006)
5693–5702.[94] K. Michalke, C. Huyghe, J. Lichière, M.E. Gravière, M. Siponen, G. Sciara, I.
Lepaul, R. Wagner, C. Magg, R. Rudolph, C. Cambillau, A. Desmyter, Anal.Biochem. 401 (2010) 74–80.
[95] V. Polovinkin, I. Gushchin, T. Balandin, P. Chervakov, E. Round, V.Schevchenko, A. Popov, V. Borshchevskiy, J.-L. Popot, V. Gordeliy, J. Membr.Biol. 247 (2014) 997–1004, http://dx.doi.org/10.1007/s00232-00014-09700-x.
[96] M. Caffrey, Biochem. Soc. Trans. 39 (2011) 725–732.[97] V. Cherezov, Curr. Opin. Struct. Biol. 21 (2011) 559–566.[98] E. Ishchenko, V. Abola, V. Cherezov, in: I. Mus-Veteau (Ed.), Membrane
Protein Production for Structural Analysis, Springer, New York, 2014, pp.289–314.
[99] J.P. Leyris, T. Roux, E. Trinquet, P. Verdie, J.A. Fehrentz, N. Oueslati, S. Douzon,E. Bourrier, L. Lamarque, D. Gagne, J.C. Galleyrand, C. M’Kadmi, J. Martinez, S.Mary, J.-L. Banères, J. Marie, Anal. Biochem. 408 (2011) 253–262.
[100] K.H. Park, E. Billon-Denis, T. Dahmane, F. Lebaupain, B. Pucci, C. Breyton, F.Zito, N. Biotechnol. 28 (2011) 255–261.
[101] D.C. Bay, J.D. O’Neil, D.A. Court, Biophys. J. 94 (2008) 457–468.[102] G.J. Patel, J.H. Kleinschmidt, Biochemistry 52 (2013) 3974–3986.[103] T. Surrey, A. Schmid, F. Jähnig, Biochemistry 35 (1996) 2283–2288.[104] M. Damaghi, S. Köster, C.A. Bippes, O. Yildiz, D.J. Müller, Angew. Chem. Int. Ed.
Engl. 50 (2011) 7422–7424.[105] S.R. Maurya, D. Chaturvedi, R. Mahalakshmi, Sci. Rep. 3 (2013) 1989.[106] G.H. Huysmans, S.E. Radford, S.A. Baldwin, D.J. Brockwell, J. Mol. Biol. 416
(2012) 453–464.[107] C.L. Pocanschi, H.-J. Apell, P. Puntervoll, B.T. Høgh, H.B. Jensen, W. Welte, J.H.
Kleinschmidt, J. Mol. Biol. 355 (2006) 548–561.[108] U. Henning, I. Sonntag, I. Hindennach, Eur. J. Biochem. 92 (1978) 491–498.[109] J.H. Kleinschmidt, M.C. Wiener, L.K. Tamm, Protein Sci. 8 (1999) 2065–2071.[110] D. Debnath, K.L. Nielsen, D.E. Otzen, Biophys. Chem. 148 (2010) 112–120.[111] R. Koebnik, EMBO J. 15 (1996) 3529–3537.[112] A. Pautsch, G.E. Schulz, Nat. Struct. Biol. 5 (1998) 1013–1017.[113] A. Arora, F. Abildgaard, J.H. Bushweller, L.K. Tamm, Nat. Struct. Biol. 8 (2001)
334–338.[114] P. Puntervoll, M. Ruud, L.J. Bruseth, H. Kleivdal, B.T. Høgh, R. Benz, H.B. Jensen,
Microbiology 148 (2002) 3395–3403.[115] V. Anbazhagan, N. Vijay, J.H. Kleinschmidt, D. Marsh, Biochemistry 47 (2008)
8414–8423.[116] C.L. Pocanschi, J.-L. Popot, J.H. Kleinschmidt, Eur. Biophys. J. 42 (2013) 103–
118.[117] J.K. Nagy, A. Kuhn Hoffmann, M.H. Keyes, D.N. Gray, K. Oxenoid, C.R. Sanders,
FEBS Lett. 501 (2001) 115–120.[118] L. Vandeputte-Rutten, R.A. Kramer, J. Kroon, N. Dekker, M.R. Egmond, P. Gros,
EMBO J. 20 (2001) 5033–5039.[119] A.C. Leney, L.M. McMorran, S.E. Radford, A.E. Ashcroft, Anal. Chem. (2012).[120] R.A. Kramer, D. Zandwijken, M.R. Egmond, N. Dekker, Eur. J. Biochem. 267
(2000) 885–893.[121] J. Qu, C. Mayer, S. Behrens, O. Holst, J.H. Kleinschmidt, J. Mol. Biol. 374 (2007)
91–105.[122] G.J. Patel, S. Behrens-Kneip, O. Holst, J.H. Kleinschmidt, Biochemistry 48
(2009) 10235–10245.[123] F. Giusti, J.-L. Popot, C. Tribet, Langmuir 28 (2012) 10372–10380.[124] J.H. Kleinschmidt, L.K. Tamm, Biochemistry 35 (1996) 12993–13000.[125] M. Picard, T. Dahmane, M. Garrigos, C. Gauron, F. Giusti, M. le Maire, J.-L.
Popot, P. Champeil, Biochemistry 45 (2006) 1861–1869.[126] Y. Gohon, T. Dahmane, R.W. Ruigrok, P. Schuck, D. Charvolin, F. Rappaport, P.
Timmins, D.M. Engelman, C. Tribet, J.-L. Popot, C. Ebel, Biophys. J. 94 (2008)3523–3537.
[127] C.G. Brouillette, R.B. McMichens, L.J. Stern, H.G. Khorana, Proteins 5 (1989)38–46.
[128] T. Dahmane, F. Rappaport, J.-L. Popot, Eur. Biophys. J. 42 (2013) 85–101.[129] M.B. Jackson, J.M. Sturtevant, Biochemistry 17 (1978) 911–915.
[130] M. Damian, S. Perino, A. Polidori, A. Martin, L. Serre, B. Pucci, J.-L. Banères,FEBS Lett. 581 (2007) 1944–1950.
[131] V.A. Feher, A. Randall, P. Baldi, R.M. Bush, L.M. de la Maza, R.E. Amaro, PLoSOne 8 (2013) e68934.
[132] N. Martin, D. Ma, A. Herbet, D. Boquet, F.M. Winnik, C. Tribet,Biomacromolecules 15 (2014) 2952–2962.
[133] D. Ma, N. Martin, A. Herbet, D. Boquet, C. Tribet, F.M. Winnik, Chem. Lett. 41(2012) 1380–1382.
[134] H. Hong, L.K. Tamm, Proc. Natl. Acad. Sci. U.S.A. 101 (2004) 4065–4070.[135] C.L. Pocanschi, G.J. Patel, D. Marsh, J.H. Kleinschmidt, Biophys. J. 91 (2006)
L75–L78.[136] K.K. Andersen, H. Wang, D.E. Otzen, Biochemistry 51 (2012) 8371–8383.[137] C. Breyton, C. Tribet, J. Olive, J.-P. Dubacq, J.-L. Popot, J. Biol. Chem. 272 (1997)
21892–21900.[138] T. Dahmane, Protéines membranaires et amphipols: stabilisation, fonction,
renaturation, et développement d’amphipols sulfonatés pour la RMN dessolutions, Université Paris-7, Paris, 2007, 229 p.
[139] W. Arunmanee, J.R. Harris, J.H. Lakey, J. Membr. Biol. 247 (2014) 949–956,http://dx.doi.org/10.1007/s00232-00014-09640-00235.
[140] A. Sverzhinsky, S. Qian, L. Yang, M. Allaire, I. Moraes, D. Ma, J.W. Chung, M.Zoonens, J.-L. Popot, J.W. Coulton, J. Membr. Biol. 247 (2014) 1005–1018,http://dx.doi.org/10.1007/s00232-00014-09678-00234.
[141] D. Charvolin, J.-B. Perez, F. Rouvière, F. Giusti, P. Bazzacco, A. Abdine, F.Rappaport, K.L. Martinez, J.-L. Popot, Proc. Natl. Acad. Sci. U.S.A. 106 (2009)405–410.
[142] E.A. Della Pia, J.V. Holm, N. Lloret, C. Le Bon, J.-L. Popot, M. Zoonens, J. Nygård,K.L. Martinez, ACS Nano 8 (2014) 1844–1853.
[143] E. Marie, S. Sagan, S. Cribier, C. Tribet, J. Membr. Biol. 247 (2014) 861–881,http://dx.doi.org/10.1007/s00232-00014-09679-00233.
[144] K.L. Martinez, Y. Gohon, P.-J. Corringer, C. Tribet, F. Mérola, J.-P. Changeux, J.-L. Popot, FEBS Lett. 528 (2002) 251–256.
[145] D. Oesterhelt, W. Stoeckenius, Proc. Natl. Acad. Sci. U.S.A. 70 (1973) 2853–2857.
[146] J.K. Lanyi, Biochim. Biophys. Acta 1757 (2006) 1012–1018.[147] J.-P. Changeux, Drug Discov. Today Technol. 10 (2013) e223–228.[148] J.V. Møller, C. Olesen, A.M. Winther, P. Nissen, Q. Rev. Biophys. 43 (2010)
501–566.[149] J.V. Møller, C. Olesen, A.M. Winther, P. Nissen, Methods Mol. Biol. 604 (2010)
119–140.[150] J.M. Merino, J.V. Møller, C. Gutiérrez-Merino, FEBS Lett. 343 (1994) 155–159.[151] M. Tehei, J.D. Perlmutter, F. Giusti, J. Sacher, G. Zaccai, J.-L. Popot, J. Membr.
Biol. 247 (2014) 897–908, http://dx.doi.org/10.1007/s00232-00014-09725-00231.
[152] A.A. Moosavi-Movahedi, J. Chamanil, Y. Goto, G.H. Hakimelahi, J. Biochem.133 (2003) 93–102.
[153] A.G. Lee, Biochim. Biophys. Acta 1612 (2003) 1–40.[154] A.G. Lee, in: P. Yeagle (Ed.), Structure of Biological Membranes, Taylor and
Francis, Boca Raton, Florida, USA, 2010, pp. 273–313.[155] L. Adamian, H. Naveed, J. Liang, Biochim. Biophys. Acta 1808 (2011) 1092–
1102.[156] C. Aponte-Santamaríaa, R. Brionesa, A.D. Schenk, T. Walz, B.L. de Groot, Proc.
Natl. Acad. Sci. U.S.A. 109 (2012) 9887–9892.[157] P.J. Stansfeld, E.E. Jeffreys, M.S.P. Sansom, Structure 21 (2013) 810–819.[158] D.E. Warschawski, A.A. Arnold, M. Beaugrand, A. Gravel, E. Chartrand, I.
Marcotte, Biochim. Biophys. Acta 1808 (2011) 1957–1974.[159] L.J. Catoire, X.L. Warnet, D.E. Warschawski, in: I. Mus-Veteau (Ed.),
Membrane Protein Production for Structural Analysis, Springer, New York,2014, pp. 315–346.
[160] T.L. Cvetkov, K.W. Huynh, M.R. Cohen, V.Y. Moiseenkova-Bell, J. Biol. Chem.286 (2011) 38168–38176.
[161] D.F. Tifrea, S. Pal, J.-L. Popot, M.J. Cocco, L.M. de la Maza, J. Immunol. 192(2014) 5201–5213.
[162] H. Basit, S.K. Sharma, A. Van der Heyden, C. Gondran, C. Breyton, P. Dumy,F.M. Winnik, P. Labbé, Chem. Commun. (2012).
[163] Y. Ferrandez, M. Dezi, M. Bosco, A. Urvoas, M. Valério, C. Le Bon, F. Giusti, I.Broutin, G. Durand, A. Polidori, J.-L. Popot, M. Picard, P. Minard, J. Membr.Biol. 247 (2014) 925–940, http://dx.doi.org/10.1007/s00232-00014-09707-00233.
[164] C. Le Bon, E.A. Della Pia, F. Giusti, N. Lloret, M. Zoonens, K.L. Martinez, J.-L.Popot, Nucleic Acids Res. (2014), http://dx.doi.org/10.1093/nar/gku1250.
[165] E.A. Della Pia, R. Westh Hansen, M. Zoonens, K.L. Martinez, J. Membr. Biol. 247(2014) 815–826, http://dx.doi.org/10.1007/s00232-00014-09663-y.
[166] Y. Zhou, J.U. Bowie, J. Biol. Chem. 275 (2000) 6975–6979.[167] M.J. Serrano-Vega, F. Magnani, Y. Shibata, C.G. Tate, Proc. Natl. Acad. Sci.
U.S.A. 105 (2008) 877–882.[168] Y. Shibata, J.F. White, M.J. Serrano-Vega, F. Magnani, A.L. Aloia, R.
Grisshammer, C.G. Tate, J. Mol. Biol. 390 (2009) 262–277.[169] J.L. Miller, C.G. Tate, J. Mol. Biol. 413 (2011) 628–638.[170] L.J. Catoire, M. Damian, F. Giusti, A. Martin, C. van Heijenoort, J.-L. Popot, E.
Guittet, J.-L. Banères, J. Am. Chem. Soc. 132 (2010) 9049–9057.[171] M. Stangl, S. Unger, S. Keller, D. Schneider, PLOS One 9 (2014) e110970.