approximate lineage for probabilistic databases
DESCRIPTION
Approximate Lineage for Probabilistic Databases. Christopher Ré and Dan Suciu University of Washington. Approximate Lineage in One Slide. Lineage (Provenance) In QP used to track correlations Explain query/view results VLDBs have lots of lineage Chokes QP Hard for users to understand - PowerPoint PPT PresentationTRANSCRIPT

Approximate Lineage for Probabilistic Databases
Christopher Ré and Dan SuciuUniversity of Washington

Approximate Lineage in One Slide
• Lineage (Provenance) – In QP used to track correlations– Explain query/view results
• VLDBs have lots of lineage– Chokes QP– Hard for users to understand
• Obs: lineage contains a lot of redundancy!This work: Approximate the lineage, by keeping only the most important correlations
In a view, lineage is all derivations of a tuple
Especially with complex queries/views
probabilistic databases

Overview
• Motivation & Preliminaries
• An apx lineage approach: Sufficient Lineage
• Experiments
• Conclusions

id Description PX1 “Dr. Z told me” 0.9X2 “PubMed:123” 0.8X3 “Lab Experiment” 0.3
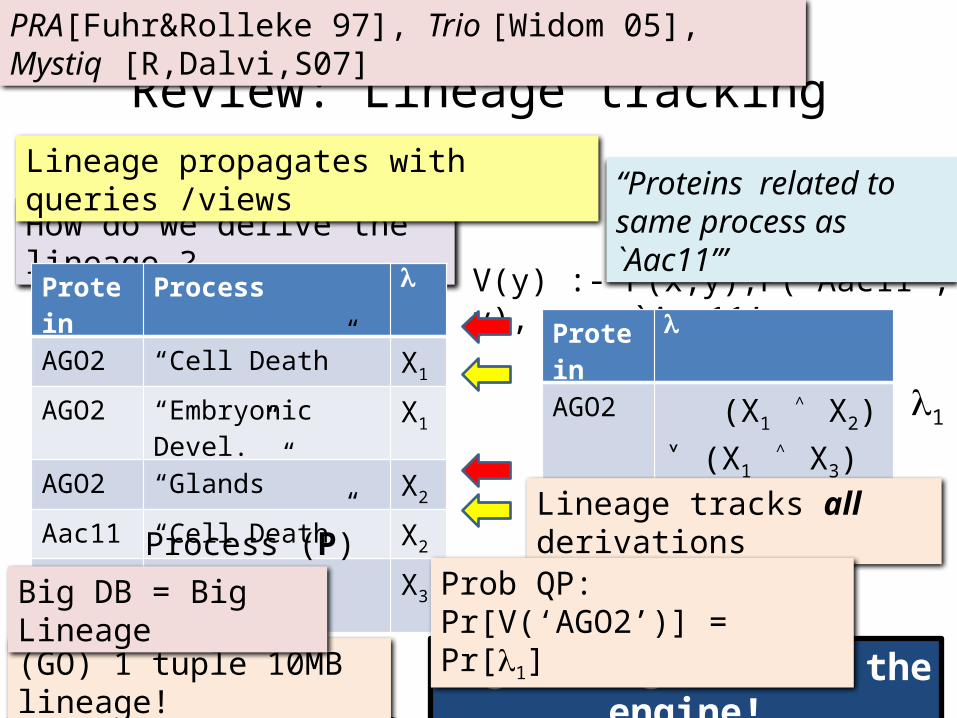
A Protein DatabaseProtein ProcessAGO2 “Cell Death”AGO2 “Embryonic Devel.”AGO2 “Glands”Aac11 “Cell Death”Aac11 “Embryonic Devel.”
Data are from somewhere
Lineage (l) is important
id DescriptionX1 “Dr. Z told me”
X2 “PubMed:123”
X3 “Lab Experiment”
Protein Process l
AGO2 “Cell Death” X1
AGO2 “Embryonic Devel.” X1
AGO2 “Glands” X2
Aac11 “Cell Death” X2
Aac11 “Embryonic Devel.” X3
AtomsManually Created
Machine inferredLineage from a wide variety of sources – not all trusted the same
Some with confidence, too!
Inspired by the Geneontology (GO) Database
Process (P)
Standard pDB, e.g. Mystiq, Trio
Protein lAGO2Protein lAGO2 (X1 ˄ X2)
Review: Lineage tracking
How do we derive the lineage ?
Protein Process l
AGO2 “Cell Death” X1
AGO2 “Embryonic Devel.” X1
AGO2 “Glands” X2
Aac11 “Cell Death” X2
Aac11 “Embryonic Devel.” X3
V(y) :- P(x,y),P(`Aac11’, y), x `Aac11’
Lineage propagates with queries /views
Protein l
AGO2 (X1 ˄ X2)˅ (X1 ˄ X3)
PRA[Fuhr&Rolleke 97], Trio [Widom 05], Mystiq [R,Dalvi,S07]
Lineage tracks all derivations
(GO) 1 tuple 10MB lineage!
Big DB = Big Lineage
Big Lineage chokes the engine!
Process (P)
“Proteins related to same process as `Aac11’”
Prob QP: Pr[V(‘AGO2’)] = Pr[l1]
l1

Problems with Large Lineage in pDB
• Lineage is used to:– Process Queries
– Give explanations to users
– Find influential atoms
On VLDBs, helpful to shrink (approximate) the lineage
Large: Many redundant explanations
Large: chokes QP
Large: Needle in a haystack
This talk

Approximate Lineage Approach
Original VLDB
Level 1 Database(Big lineage)
Level 2 Database(Small lineage)
Protein lAGO2 (X1 ˄ X2)
˅ (X1 ˄ X3)
lProtein aAGO2 (X1 ˄ X2)
asmaller, approximate formula
All (most) querying on Level 2 database (using a instead of l)
Focus is on the Level 2 database
Protein aAGO2 0.5*x1 + 0.3
error, e

Overview
• Motivation & Preliminaries
• An apx lineage approach: Sufficient Lineage
• Experiments
• Conclusions

Sufficient lineage (SL)
Protein aAGO2 (X1 ˄ X2)
• Represent as?
• Use as to:– Answer queries?– Provide explanations?– Find influential tuples?
• Build good a, efficiently?
DNF formulae, that logically imply l
The remainder of this talk
Protein lAGO2 (X1 ˄ X2)
˅ (X1 ˄ X3)Reuse existing systems!
See paper
Nugget: An algorithm that always finds small, good SL
a is a lower bound l

Formalizing “good as”
id Description PX1 “Dr. Z told me” 0.9X2 “PubMed:123” 0.8X3 “Lab Experiment” 0.3
Atoms
Choosing an approximation a for a lineage function, l
Protein l
AGO2 (X1 ˄ X2)˅ (X1 ˄ X3)
Intuition: a should agree on most worlds
An atom is a Boolean proposition. A world is a set of the true atoms.
Formalizing this,
E[l – a] e Expectation of difference over all worlds, should be small
NB: really standard ℓ2 distance

Illustrating Good Lineage
id Description PX1 “Dr. Z told me” 0.9X2 “PubMed:123” 0.8X3 “Lab Experiment” 0.3
Protein lAGO2 (X1 ˄ X2)
˅ (X1 ˄ X3)
Protein aAGO2 (X1 ˄ X2)
0.9 *(1 - (1 - 0.8)(1-0.3)) 0.9 * 0.8 = 0.72= 0.9 *0.86 = .774
e = 0.054
E[l – a] = E[ ]l – E[a] e
Intuition: Pr[ ] a high means good lineage

1st step: Lineage DNFs to “graphs”
X1
X2
Xn
Y1
Ym
Y2
(X1 ˄ Y1) ˅(X2 ˄ Y1)
We can think of DNFs as graphs (k-DNF a k-hypergraph)
Goal: Given error e, find a subset of edges with error smaller than e and small size, i.e. a best lower bound;
Atoms = nodes
Monomials = edges
Trick: matching is an SL formula.
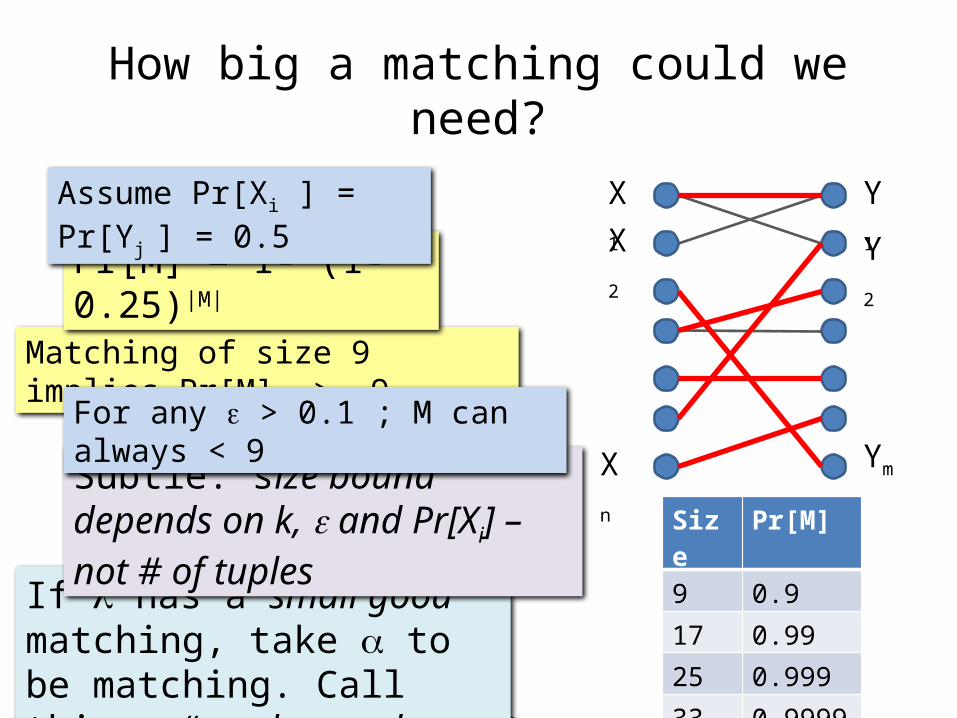
How big a matching could we need?
Matching of size 9 implies Pr[M] > .9
X1
X2
Xn
Y1
Ym
Y2Pr[M] = 1- (1-0.25)|M|
Assume Pr[Xi ] = Pr[Yj ] = 0.5
If l has a small good matching, take a to be matching. Call this a “good enough matching”
Subtle: size bound depends on k, e and Pr[Xi] – not # of tuples
For any e > 0.1 ; M can always < 9
Size Pr[M]
9 0.9
17 0.99
25 0.999
33 0.9999
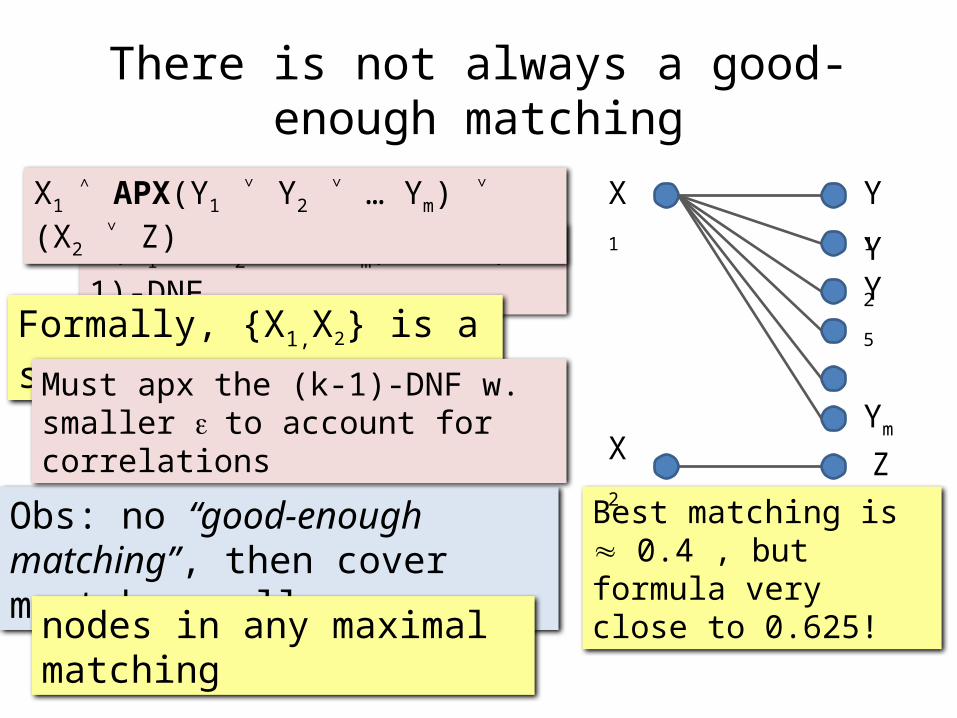
There is not always a good-enough matching
X1 Y1
Ym
Y2
Obs: no “good-enough matching”, then cover must be small
nodes in any maximal matching
(Y1 ˅ Y2 ˅ … Ym) – a (k-1)-DNF
Formally, {X1,X2} is a small cover
Best matching is 0.4 , but formula very close to 0.625!
X1 ˄ APX(Y1 ˅ Y2 ˅ … Ym) ˅ (X2 ˅ Z)
Must apx the (k-1)-DNF w. smaller e to account for correlations
ZX2
Y5

SL is always small
Two Cases:1. Small-good matching 2. Small-cover of important nodes
We’re done!Recurse on k-1 DNF
THM (SL is always small) Size of SL is constant in data.
Problem: Maximum matching in general hypergraphs is NP-hard
Apx NP-hard!need a maximal matching – pick greedily!
Requires “non-vanishing” probs In datasets, usually, Pr > 10-3
Exponential in query Similar to data-complexity

Summary of Constructing SL
• For SL, good lineage = big lineage– Not true in general.
• Gave an algorithm that always finds small SL– Constant in the data– Exponential in almost everything else
• Main trick: Don’t try to find optimal solutions, when sloppy is good enough!

Other fun results in the paper
• Sufficient Lineage (SL)– Error bounds for QP– Finding influential tuples
• Polynomial Lineage (PL): DNF to polynomial– Use Taylor/Fourier approximation of poly– Algos for QP, explanations and influential tuples
Leverage extensive prior art!
PL smaller than SL, but not usable in pDBs (Mystiq, Trio).

Overview
• Motivation & Preliminaries
• An apx lineage approach: Sufficient Lineage
• Experiments
• Conclusions

Experiments
• Geneontology Database– Publically available– Predefined views– Atoms = “evidence codes”
• Discuss a single view– 6 tables– 2 sources of evidence– 1119 tuples – 141MB
Similar results on IMDB data not presented
“All proteins associated with a single protein”

Compression Ratio v. Error
0.5 0.1
0.05000000000000010.01
0.0010
100200300400
e, error level (smaller is more conservative)
Compress Ratio
Good compression ratio even for
stringent error
30x compression
141MB to 4MB
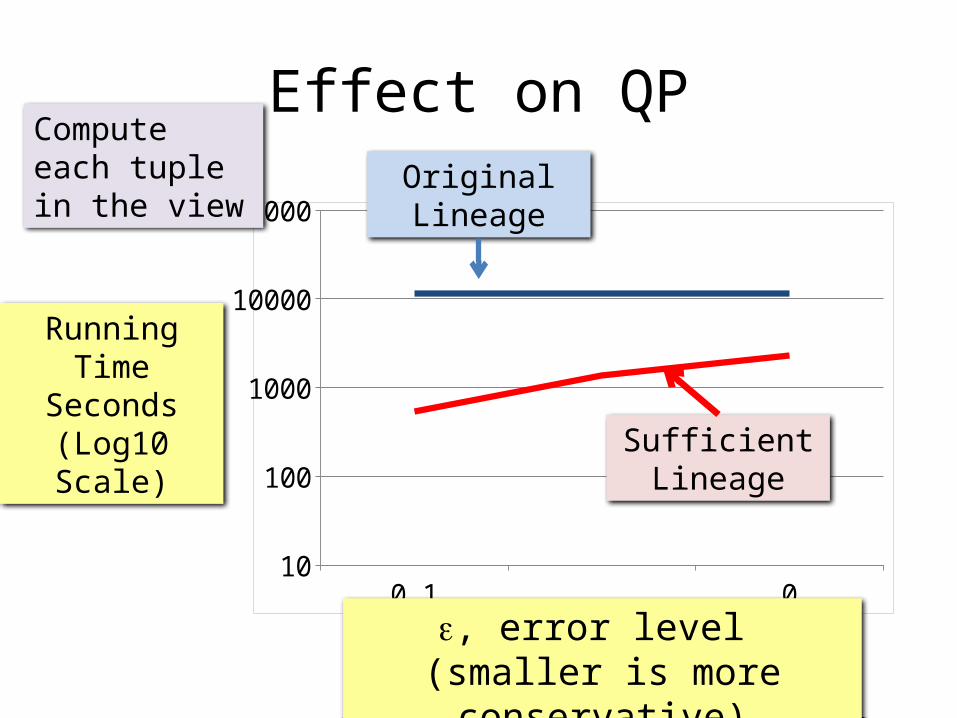
Effect on QP
0.1 0.01 0.00110
100
1000
10000
100000
Running TimeSeconds
(Log10 Scale)
e, error level (smaller is more conservative)
Sufficient Lineage
Original Lineage
Compute each tuple in the view
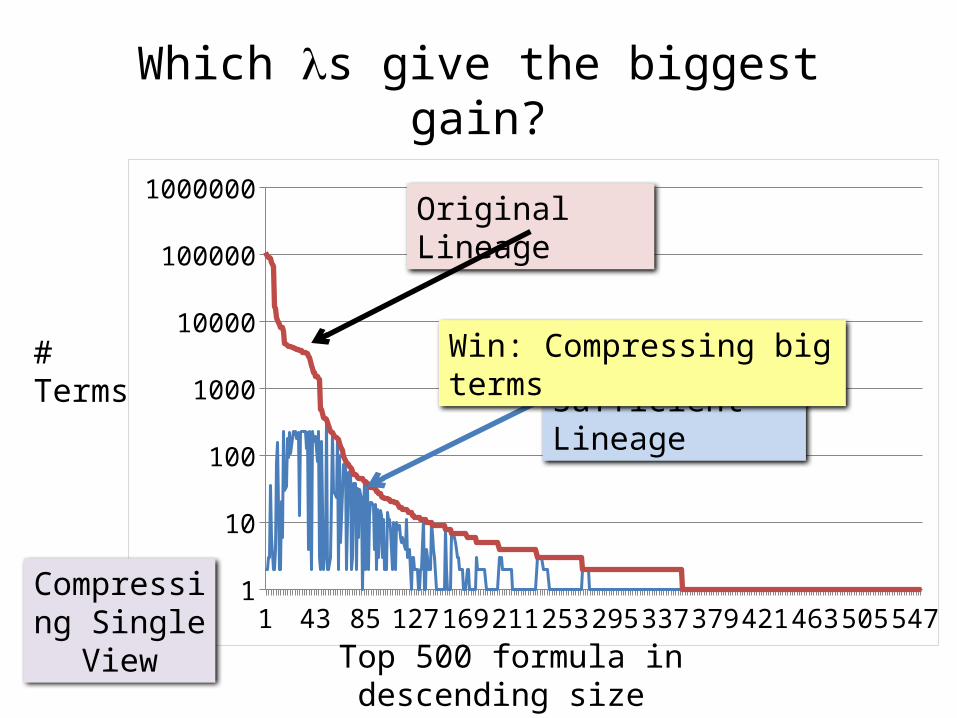
Which ls give the biggest gain?
1 37 73 1091451812172532893253613974334695055411
10
100
1000
10000
100000
1000000
Top 500 formula in descending size (# is rank)
# Terms
Original Lineage
Sufficient Lineage
Win: Compressing big terms
Compressing Single View

Conclusion
• Discussed approximate lineage approach– Goal: Fast QP, Explanations
• Sufficient Lineage– Can be used by standard QPs– Improves QP dramatically
• Apx lineage is more general, e.g. Polynomial