apoptosis in mesenchymal stromal cells induces in vivo ... · apoptosis...
TRANSCRIPT

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
TRANSPLANTAT ION
1Regenerative Medicine, Division of Cancer Studies and Cancer Research UK King’sHealth Partners, King’s College London, London SE5 9NU, UK. 2Institute of Pharmaceu-tical Science, King’s College London, London SE1 9NH, UK. 3Institute of Immunityand Transplantation, University College London, London NW3 2QG, UK. 4CancerInstitute, University College London, London WC1E 6DD, UK. 5University Hospital CarlGustav Carus, 01307 Dresden, Germany. 6Medical Research Council Centre for Trans-plantation, King’s College London, London SE1 9RT, UK. 7Centre for Stem Cells andRegenerative Medicine, King’s College London, London SE1 9RT, UK. 8University Hos-pital Southampton NHS Foundation Trust, Southampton SO16 6YD, UK. 9Bristol Hae-matology and Oncology Centre, Bristol BS2 8ED, UK. 10Centre for Haematology,Imperial College London, London W12 0NN, UK.*These authors contributed equally to this work.†Corresponding author. Email: [email protected]
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
Copyright © 2017
The Authors, some
rights reserved;
exclusive licensee
American Association
for the Advancement
of Science. No claim
to original U.S.
Government Works
http://D
ownloaded from
Apoptosis in mesenchymal stromal cells induces in vivorecipient-mediated immunomodulationAntonio Galleu,1 Yanira Riffo-Vasquez,2 Cristina Trento,1 Cara Lomas,3,4 Luigi Dolcetti,1
Tik Shing Cheung,1 Malte von Bonin,5 Laura Barbieri,1 Krishma Halai,1 Sophie Ward,3,4
Ling Weng,1 Ronjon Chakraverty,3,4 Giovanna Lombardi,6 Fiona M. Watt,7 Kim Orchard,8
David I. Marks,9 Jane Apperley,10 Martin Bornhauser,1,5 Henning Walczak,4*Clare Bennett,3,4* Francesco Dazzi1,10†
The immunosuppressive activity of mesenchymal stromal cells (MSCs) is well documented. However, the thera-peutic benefit is completely unpredictable, thus raising concerns about MSC efficacy. One of the affecting factorsis the unresolved conundrum that, despite being immunosuppressive, MSCs are undetectable after administra-tion. Therefore, understanding the fate of infused MSCs could help predict clinical responses. Using a murinemodel of graft-versus-host disease (GvHD), we demonstrate that MSCs are actively induced to undergo perforin-dependent apoptosis by recipient cytotoxic cells and that this process is essential to initiate MSC-induced im-munosuppression. When examining patients with GvHD who received MSCs, we found a striking parallel, wherebyonly those with high cytotoxic activity against MSCs responded to MSC infusion, whereas those with low activitydid not. The need for recipient cytotoxic cell activity could be replaced by the infusion of apoptotic MSCs gener-ated ex vivo. After infusion, recipient phagocytes engulf apoptotic MSCs and produce indoleamine 2,3-dioxygenase,which is ultimately necessary for effecting immunosuppression. Therefore, we propose the innovative conceptthat patients should be stratified for MSC treatment according to their ability to kill MSCs or that all patients couldbe treated with ex vivo apoptotic MSCs.
stm
by guest on March 20, 2020.sciencem
ag.org/
INTRODUCTIONMesenchymal stromal cells (MSCs) have received center-stage attentionbecause they exhibit potent immunosuppressive and anti-inflammatoryactivities (1) that have been extensively tested in several medical con-ditions, ranging from autoimmune diseases to the immunological com-plications of clinical transplantation (2–5). The extensive clinical use hasbeen undeterred by the fact that themechanisms underlyingMSC ther-apeutic activity remain largely unresolved. MSC-mediated immuno-suppression is major histocompatibility complex (MHC)–independent,is non–antigen-specific (1), and targets virtually all immune cells byarresting cell cycle progression (6–8). Interference with amino acidmetabolism in the inflammatorymicroenvironment has been suggestedas a crucial mechanism, with indoleamine 2,3-dioxygenase (IDO) beingone of the major candidates (9, 10), along with transforming growthfactor–b1, hepatocyte growth factor (11), prostaglandin E2 (12), andsoluble human leukocyte antigen G (13, 14). Tumor necrosis factor–a(TNF-a)–stimulated gene 6 protein (TSG-6) has been identified as arecent addition to the anti-inflammatory armamentarium of humanMSCs (15). However, there is consensus that the immunosuppressiveactivity does not solely rely on MSCs but may involve the active en-gagement of other immunomodulatory cells, such as regulatory T cells(16, 17) and immunosuppressive macrophages (18, 19).
The imperfect knowledge ofMSC immunobiologymay well explainwhy the results of the clinical trials have often been controversial andwhy conclusive proof of efficacy has not yet been provided. Two majorunresolved challenges undermine progress in the field. The first is thatonly a proportion of patients, although affected by the same disease, re-sponds to MSC infusions, and this response cannot be predicted. Thesecond is that, to be efficacious, MSCs are not required to engraft. Thevast majority of infusedMSCs resides transiently in the lungs before be-coming undetectablewithin a fewhours (20). Because our current knowl-edge cannot provide an explanation to this paradox (6, 9, 10, 15), a betterunderstanding of the mechanisms underlyingMSC therapeutic activitywould be highly desirable to improve clinical efficacy and make thera-peutic achievements more reproducible. We selected to address thesechallenges in graft-versus-host disease (GvHD) because there is proof ofprinciple that MSCs are clinically efficacious (4, 21). By using a mousemodel and clinical samples, we have tested the hypothesis that MSCsundergo in vivo apoptosis after exposure to the GvHD environment.
RESULTSMSCs undergo apoptosis in recipient GvHD animalsWeused amousemodel of GvHD inwhich lethally irradiated C57BL/6male mice were transplanted on day 0 with bone marrow (BM), poly-clonal purifiedCD4+T cells from female syngeneic donors, and purifiedCD8+ T cells transgenic for a T cell receptor specific for themalemouseHYantigenUty [Matahari (Mh)] asGvHDeffectors (fig. S1A) (22). Theaddition of CD4+ T cells is necessary to facilitate the expansion of theCD8+ T cells. In thismodel, the expansion of the T cells effectingGvHD(CD8+Vb8.3+) can be precisely enumerated and correlateswith the clin-ical severity of the disease. HumanMSCswere injected 3 days after thetransplant. To explain themechanismbywhichMSCs are rapidly clearedafter injection (20, 23), we tested the hypothesis that MSCs undergoapoptosis.
1 of 11
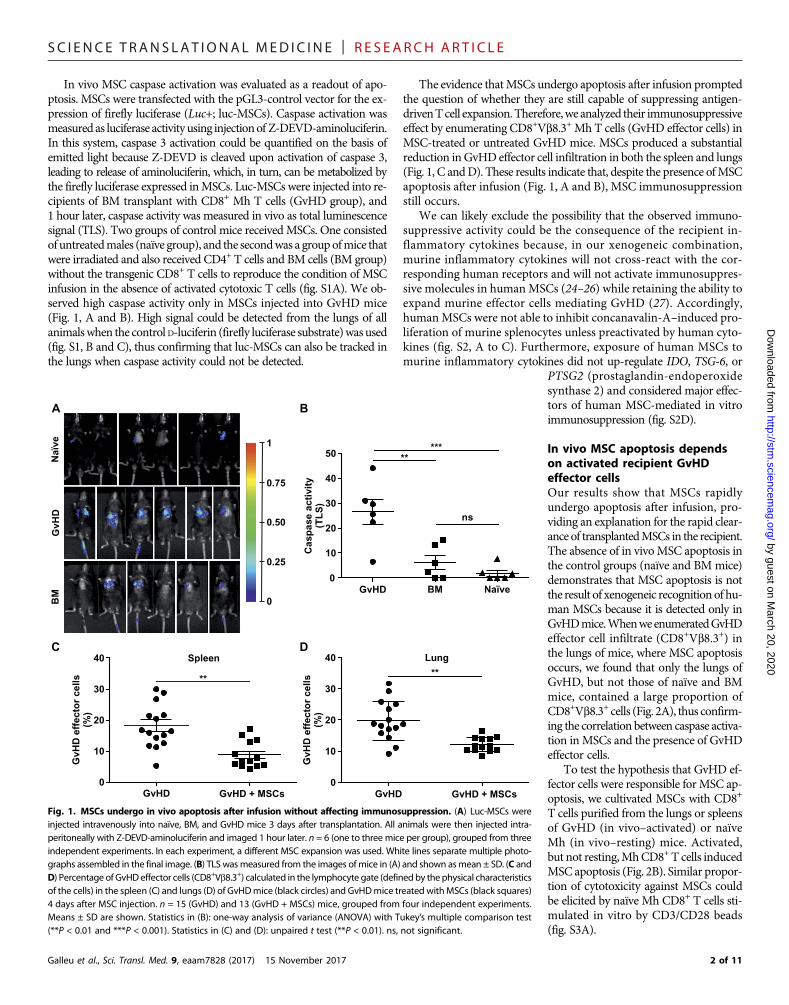
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
Dow
nload
In vivo MSC caspase activation was evaluated as a readout of apo-ptosis. MSCs were transfected with the pGL3-control vector for the ex-pression of firefly luciferase (Luc+; luc-MSCs). Caspase activation wasmeasured as luciferase activity using injectionof Z-DEVD-aminoluciferin.In this system, caspase 3 activation could be quantified on the basis ofemitted light because Z-DEVD is cleaved upon activation of caspase 3,leading to release of aminoluciferin, which, in turn, can be metabolized bythe firefly luciferase expressed inMSCs. Luc-MSCs were injected into re-cipients of BM transplant with CD8+ Mh T cells (GvHD group), and1 hour later, caspase activity was measured in vivo as total luminescencesignal (TLS). Two groups of control mice receivedMSCs. One consistedof untreatedmales (naïve group), and the secondwas a groupofmice thatwere irradiated and also received CD4+ T cells and BM cells (BM group)without the transgenic CD8+ T cells to reproduce the condition of MSCinfusion in the absence of activated cytotoxic T cells (fig. S1A). We ob-served high caspase activity only in MSCs injected into GvHD mice(Fig. 1, A and B). High signal could be detected from the lungs of allanimalswhen the control D-luciferin (firefly luciferase substrate)was used(fig. S1, B and C), thus confirming that luc-MSCs can also be tracked inthe lungs when caspase activity could not be detected.
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
The evidence thatMSCs undergo apoptosis after infusion promptedthe question of whether they are still capable of suppressing antigen-drivenTcell expansion.Therefore,we analyzed their immunosuppressiveeffect by enumerating CD8+Vb8.3+ Mh T cells (GvHD effector cells) inMSC-treated or untreated GvHD mice. MSCs produced a substantialreduction in GvHD effector cell infiltration in both the spleen and lungs(Fig. 1, C andD). These results indicate that, despite the presence ofMSCapoptosis after infusion (Fig. 1, A and B), MSC immunosuppressionstill occurs.
We can likely exclude the possibility that the observed immuno-suppressive activity could be the consequence of the recipient in-flammatory cytokines because, in our xenogeneic combination,murine inflammatory cytokines will not cross-react with the cor-responding human receptors and will not activate immunosuppres-sive molecules in humanMSCs (24–26) while retaining the ability toexpand murine effector cells mediating GvHD (27). Accordingly,humanMSCs were not able to inhibit concanavalin-A–induced pro-liferation of murine splenocytes unless preactivated by human cyto-kines (fig. S2, A to C). Furthermore, exposure of human MSCs tomurine inflammatory cytokines did not up-regulate IDO, TSG-6, or
by guest on March 20, 2020
http://stm.sciencem
ag.org/ed from
PTSG2 (prostaglandin-endoperoxidesynthase 2) and considered major effec-tors of human MSC-mediated in vitroimmunosuppression (fig. S2D).
In vivo MSC apoptosis dependson activated recipient GvHDeffector cellsOur results show that MSCs rapidlyundergo apoptosis after infusion, pro-viding an explanation for the rapid clear-ance of transplantedMSCs in the recipient.The absence of in vivoMSC apoptosis inthe control groups (naïve and BM mice)demonstrates that MSC apoptosis is notthe result of xenogeneic recognitionof hu-man MSCs because it is detected only inGvHDmice.WhenweenumeratedGvHDeffector cell infiltrate (CD8+Vb8.3+) inthe lungs of mice, where MSC apoptosisoccurs, we found that only the lungs ofGvHD, but not those of naïve and BMmice, contained a large proportion ofCD8+Vb8.3+ cells (Fig. 2A), thus confirm-ing the correlation between caspase activa-tion in MSCs and the presence of GvHDeffector cells.
To test the hypothesis that GvHD ef-fector cells were responsible forMSC ap-optosis, we cultivated MSCs with CD8+
T cells purified from the lungs or spleensof GvHD (in vivo–activated) or naïveMh (in vivo–resting) mice. Activated,but not resting,MhCD8+T cells inducedMSC apoptosis (Fig. 2B). Similar propor-tion of cytotoxicity against MSCs couldbe elicited by naïve Mh CD8+ T cells sti-mulated in vitro by CD3/CD28 beads(fig. S3A).
Naïv
eG
vH
DB
M
A
GvHD BM Naïve
0
10
20
30
40
50
Casp
ase a
cti
vit
y(T
LS
)
*****
ns
B
GvHD GvHD + MSCs
0
10
20
30
40
**
GvH
D e
ffecto
r cells
(%)
0
10
20
30
40
GvH
D e
ffecto
r cells
(%)
GvHD GvHD + MSCs
**
C D
0
0.25
0.50
0.75
1
Spleen Lung
Fig. 1. MSCs undergo in vivo apoptosis after infusion without affecting immunosuppression. (A) Luc-MSCs wereinjected intravenously into naïve, BM, and GvHD mice 3 days after transplantation. All animals were then injected intra-peritoneally with Z-DEVD-aminoluciferin and imaged 1 hour later. n = 6 (one to three mice per group), grouped from threeindependent experiments. In each experiment, a different MSC expansion was used. White lines separate multiple photo-graphs assembled in the final image. (B) TLS wasmeasured from the images of mice in (A) and shown asmean ± SD. (C andD) Percentage of GvHDeffector cells (CD8+Vb8.3+) calculated in the lymphocyte gate (definedby thephysical characteristicsof the cells) in the spleen (C) and lungs (D) of GvHDmice (black circles) and GvHDmice treated withMSCs (black squares)4 days after MSC injection. n = 15 (GvHD) and 13 (GvHD + MSCs) mice, grouped from four independent experiments.Means ± SD are shown. Statistics in (B): one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test(**P < 0.01 and ***P < 0.001). Statistics in (C) and (D): unpaired t test (**P < 0.01). ns, not significant.
2 of 11
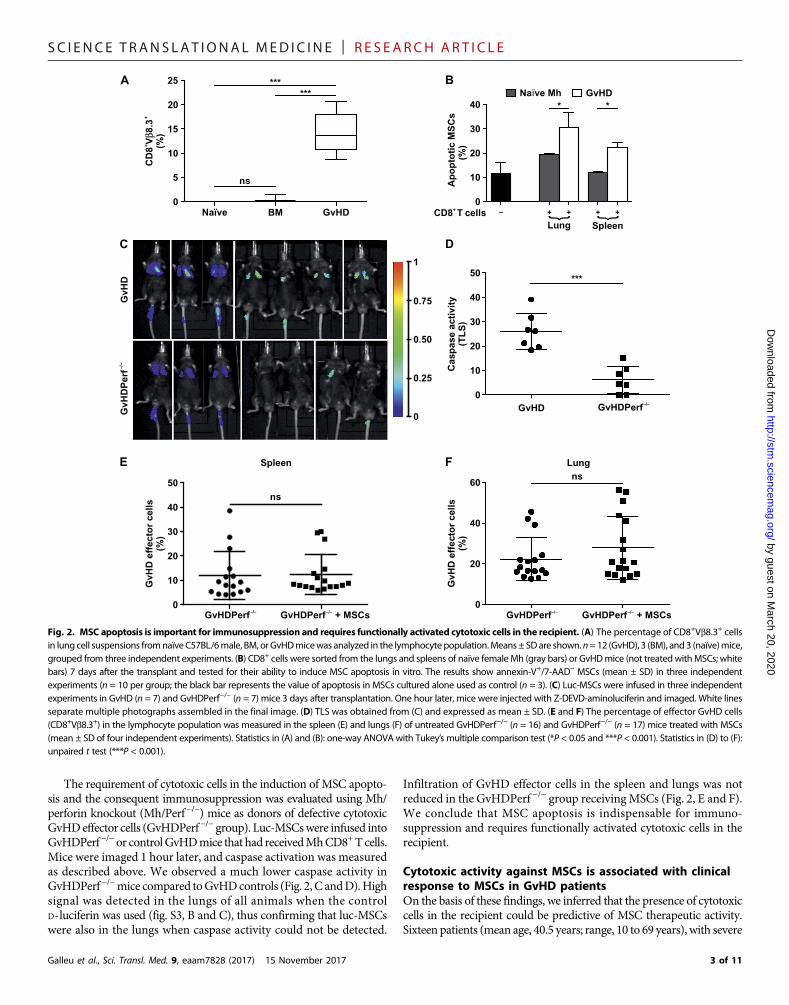
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on March 20, 2020
http://stm.sciencem
ag.org/D
ownloaded from
The requirement of cytotoxic cells in the induction of MSC apopto-sis and the consequent immunosuppression was evaluated using Mh/perforin knockout (Mh/Perf−/−) mice as donors of defective cytotoxicGvHDeffector cells (GvHDPerf−/− group). Luc-MSCswere infused intoGvHDPerf−/−or controlGvHDmice that had receivedMhCD8+Tcells.Mice were imaged 1 hour later, and caspase activation was measuredas described above. We observed a much lower caspase activity inGvHDPerf−/−mice compared toGvHDcontrols (Fig. 2, C andD).Highsignal was detected in the lungs of all animals when the controlD-luciferin was used (fig. S3, B and C), thus confirming that luc-MSCswere also in the lungs when caspase activity could not be detected.
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
Infiltration of GvHD effector cells in the spleen and lungs was notreduced in the GvHDPerf −/− group receivingMSCs (Fig. 2, E and F).We conclude that MSC apoptosis is indispensable for immuno-suppression and requires functionally activated cytotoxic cells in therecipient.
Cytotoxic activity against MSCs is associated with clinicalresponse to MSCs in GvHD patientsOn the basis of these findings, we inferred that the presence of cytotoxiccells in the recipient could be predictive of MSC therapeutic activity.Sixteen patients (mean age, 40.5 years; range, 10 to 69 years), with severe
GvH
D–/–
GvH
DP
erf
0
0.25
0.50
0.75
1
C
GvHD–/–
GvHDPerf
0
10
20
30
40
50***
Casp
ase a
cti
vit
y(T
LS
)
D
–/–GvHDPerf
–/–GvHDPerf + MSCs
0
10
20
30
40
50
ns
GvH
D e
ffecto
r cells
(%)
–/–GvHDPerf
–/–GvHDPerf + MSCs
0
20
40
60
GvH
D e
ffecto
r cells
(%)
ns
E F
***
***
Naïve BM GvHD0
5
10
15
20
25
++
CD
8V
β8.3
(%)
ns
A
+ CD8 T cells – a+
0
10
20
30
40
a+
* *
Ap
op
toti
c M
SC
s(%
)
BNaïve Mh GvHD
+ +
SpleenLung
{ {
Spleen Lung
Fig. 2. MSC apoptosis is important for immunosuppression and requires functionally activated cytotoxic cells in the recipient. (A) The percentage of CD8+Vb8.3+ cellsin lung cell suspensions fromnaïve C57BL/6male, BM, orGvHDmicewas analyzed in the lymphocyte population.Means± SDare shown.n=12 (GvHD), 3 (BM), and3 (naïve)mice,grouped from three independent experiments. (B) CD8+ cells were sorted from the lungs and spleens of naïve female Mh (gray bars) or GvHDmice (not treatedwithMSCs; whitebars) 7 days after the transplant and tested for their ability to induce MSC apoptosis in vitro. The results show annexin-V+/7-AAD− MSCs (mean ± SD) in three independentexperiments (n = 10 per group; the black bar represents the value of apoptosis in MSCs cultured alone used as control (n = 3). (C) Luc-MSCs were infused in three independentexperiments in GvHD (n = 7) and GvHDPerf−/− (n = 7) mice 3 days after transplantation. One hour later, mice were injected with Z-DEVD-aminoluciferin and imaged. White linesseparate multiple photographs assembled in the final image. (D) TLS was obtained from (C) and expressed as mean ± SD. (E and F) The percentage of effector GvHD cells(CD8+Vb8.3+) in the lymphocyte population was measured in the spleen (E) and lungs (F) of untreated GvHDPerf−/− (n = 16) and GvHDPerf−/− (n = 17) mice treated with MSCs(mean ± SD of four independent experiments). Statistics in (A) and (B): one-way ANOVA with Tukey’s multiple comparison test (*P < 0.05 and ***P < 0.001). Statistics in (D) to (F):unpaired t test (***P < 0.001).
3 of 11
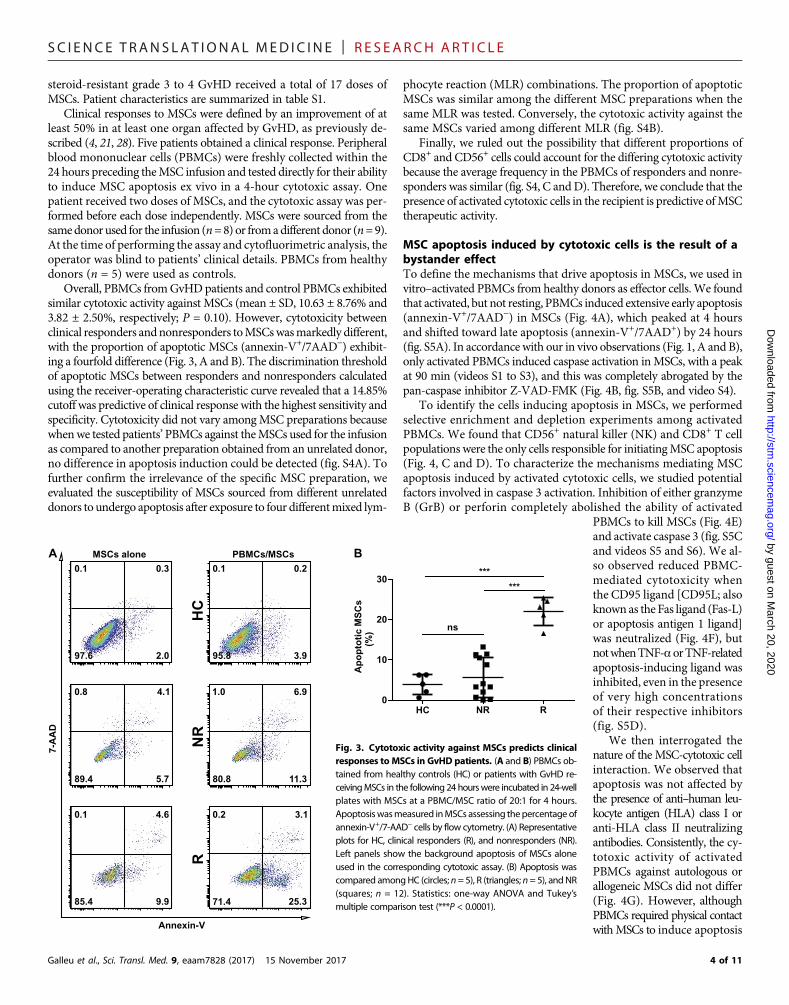
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
http://stm.sciencem
agD
ownloaded from
steroid-resistant grade 3 to 4 GvHD received a total of 17 doses ofMSCs. Patient characteristics are summarized in table S1.
Clinical responses to MSCs were defined by an improvement of atleast 50% in at least one organ affected by GvHD, as previously de-scribed (4, 21, 28). Five patients obtained a clinical response. Peripheralblood mononuclear cells (PBMCs) were freshly collected within the24 hours preceding theMSC infusion and tested directly for their abilityto induce MSC apoptosis ex vivo in a 4-hour cytotoxic assay. Onepatient received two doses of MSCs, and the cytotoxic assay was per-formed before each dose independently. MSCs were sourced from thesamedonorused for the infusion (n=8) or fromadifferent donor (n=9).At the time of performing the assay and cytofluorimetric analysis, theoperator was blind to patients’ clinical details. PBMCs from healthydonors (n = 5) were used as controls.
Overall, PBMCs fromGvHDpatients and control PBMCs exhibitedsimilar cytotoxic activity against MSCs (mean ± SD, 10.63 ± 8.76% and3.82 ± 2.50%, respectively; P = 0.10). However, cytotoxicity betweenclinical responders andnonresponders toMSCswasmarkedly different,with the proportion of apoptotic MSCs (annexin-V+/7AAD−) exhibit-ing a fourfold difference (Fig. 3, A and B). The discrimination thresholdof apoptotic MSCs between responders and nonresponders calculatedusing the receiver-operating characteristic curve revealed that a 14.85%cutoff was predictive of clinical response with the highest sensitivity andspecificity. Cytotoxicity did not vary amongMSC preparations becausewhenwe tested patients’PBMCs against theMSCs used for the infusionas compared to another preparation obtained from an unrelated donor,no difference in apoptosis induction could be detected (fig. S4A). Tofurther confirm the irrelevance of the specific MSC preparation, weevaluated the susceptibility of MSCs sourced from different unrelateddonors to undergo apoptosis after exposure to four differentmixed lym-
0
10
20
30
Ap
op
toti
c M
SC
s(%
)
B
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
phocyte reaction (MLR) combinations. The proportion of apoptoticMSCs was similar among the different MSC preparations when thesame MLR was tested. Conversely, the cytotoxic activity against thesame MSCs varied among different MLR (fig. S4B).
Finally, we ruled out the possibility that different proportions ofCD8+ and CD56+ cells could account for the differing cytotoxic activitybecause the average frequency in the PBMCs of responders and nonre-sponders was similar (fig. S4, C andD). Therefore, we conclude that thepresence of activated cytotoxic cells in the recipient is predictive ofMSCtherapeutic activity.
MSC apoptosis induced by cytotoxic cells is the result of abystander effectTo define the mechanisms that drive apoptosis in MSCs, we used invitro–activated PBMCs from healthy donors as effector cells. We foundthat activated, but not resting, PBMCs induced extensive early apoptosis(annexin-V+/7AAD−) in MSCs (Fig. 4A), which peaked at 4 hoursand shifted toward late apoptosis (annexin-V+/7AAD+) by 24 hours(fig. S5A). In accordance with our in vivo observations (Fig. 1, A and B),only activated PBMCs induced caspase activation inMSCs, with a peakat 90 min (videos S1 to S3), and this was completely abrogated by thepan-caspase inhibitor Z-VAD-FMK (Fig. 4B, fig. S5B, and video S4).
To identify the cells inducing apoptosis in MSCs, we performedselective enrichment and depletion experiments among activatedPBMCs. We found that CD56+ natural killer (NK) and CD8+ T cellpopulations were the only cells responsible for initiatingMSC apoptosis(Fig. 4, C and D). To characterize the mechanisms mediating MSCapoptosis induced by activated cytotoxic cells, we studied potentialfactors involved in caspase 3 activation. Inhibition of either granzymeB (GrB) or perforin completely abolished the ability of activated
HC NR R
***
***
ns
by guest on March 20, 2020
.org/
PBMCs to kill MSCs (Fig. 4E)and activate caspase 3 (fig. S5Cand videos S5 and S6). We al-so observed reduced PBMC-mediated cytotoxicity whenthe CD95 ligand [CD95L; alsoknown as the Fas ligand (Fas-L)or apoptosis antigen 1 ligand]was neutralized (Fig. 4F), butnotwhenTNF-a orTNF-relatedapoptosis-inducing ligand wasinhibited, even in the presenceof very high concentrationsof their respective inhibitors(fig. S5D).
We then interrogated thenature of the MSC-cytotoxic cellinteraction. We observed thatapoptosis was not affected bythe presence of anti–human leu-kocyte antigen (HLA) class I oranti-HLA class II neutralizingantibodies. Consistently, the cy-totoxic activity of activatedPBMCs against autologous orallogeneic MSCs did not differ(Fig. 4G). However, althoughPBMCs required physical contactwithMSCs to induce apoptosis
7-A
AD
Annexin-V
25.3
3.10.2
71.4
3.9
0.20.1
95.82.0
0.30.1
97.6
MSCs alone PBMCs/MSCs
9.9
4.60.1
85.4
HC
R
5.7
4.10.8
89.4 11.3
6.91.0
80.8
NR
A
FretacpAapLuc(sm
ig. 3. Cytotoxic activity against MSCs predicts clinicalsponses to MSCs in GvHD patients. (A and B) PBMCs ob-ined from healthy controls (HC) or patients with GvHD re-eivingMSCs in the following 24 hourswere incubated in 24-welllates with MSCs at a PBMC/MSC ratio of 20:1 for 4 hours.poptosiswasmeasured inMSCs assessing thepercentage ofnnexin-V+/7-AAD− cells by flow cytometry. (A) Representativelots for HC, clinical responders (R), and nonresponders (NR).eft panels show the background apoptosis of MSCs alonesed in the corresponding cytotoxic assay. (B) Apoptosis wasompared amongHC (circles; n= 5), R (triangles; n= 5), andNRquares; n = 12). Statistics: one-way ANOVA and Tukey’sultiple comparison test (***P < 0.0001).
4 of 11
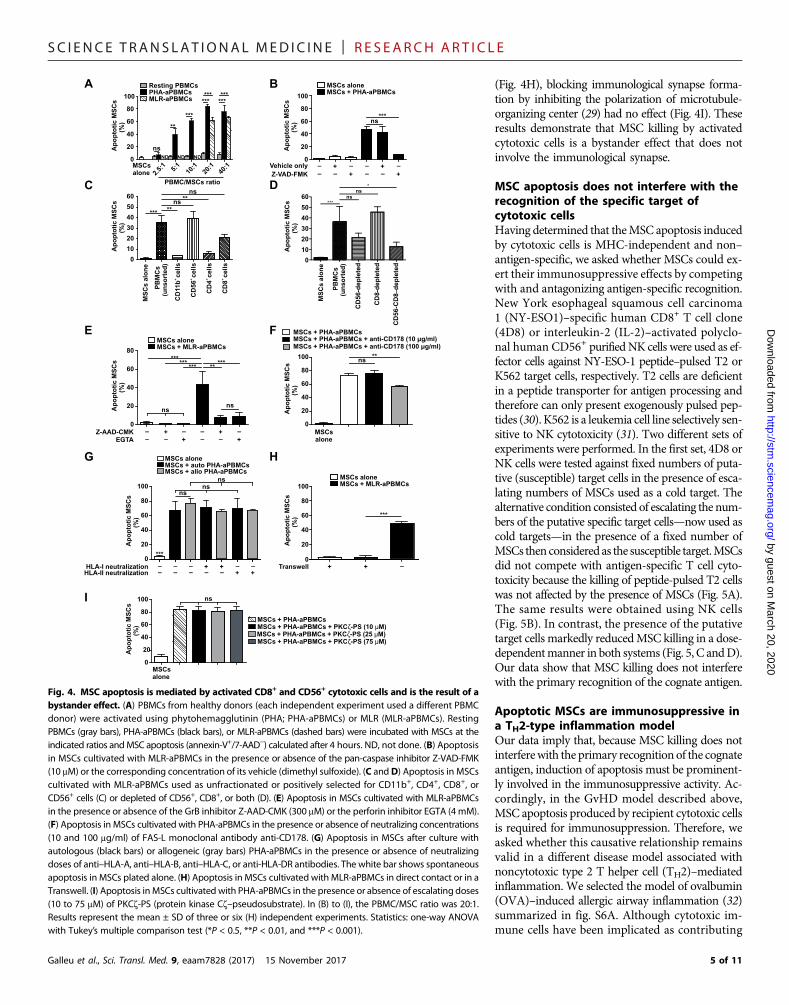
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
by guest on March 20, 2020
http://stm.sciencem
ag.org/D
ownloaded from
(Fig. 4H), blocking immunological synapse forma-tion by inhibiting the polarization of microtubule-organizing center (29) had no effect (Fig. 4I). Theseresults demonstrate that MSC killing by activatedcytotoxic cells is a bystander effect that does notinvolve the immunological synapse.
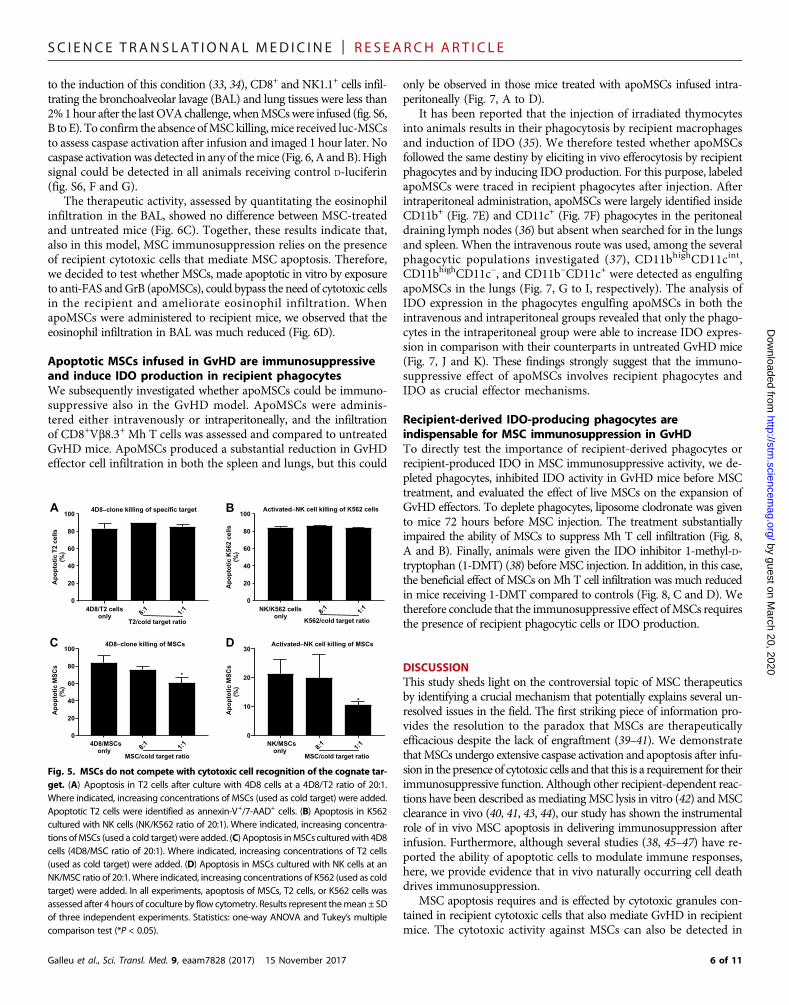
MSC apoptosis does not interfere with therecognition of the specific target ofcytotoxic cellsHaving determined that theMSC apoptosis inducedby cytotoxic cells is MHC-independent and non–antigen-specific, we asked whether MSCs could ex-ert their immunosuppressive effects by competingwith and antagonizing antigen-specific recognition.New York esophageal squamous cell carcinoma1 (NY-ESO1)–specific human CD8+ T cell clone(4D8) or interleukin-2 (IL-2)–activated polyclo-nal human CD56+ purified NK cells were used as ef-fector cells against NY-ESO-1 peptide–pulsed T2 orK562 target cells, respectively. T2 cells are deficientin a peptide transporter for antigen processing andtherefore can only present exogenously pulsed pep-tides (30). K562 is a leukemia cell line selectively sen-sitive to NK cytotoxicity (31). Two different sets ofexperiments were performed. In the first set, 4D8 orNK cells were tested against fixed numbers of puta-tive (susceptible) target cells in the presence of esca-lating numbers of MSCs used as a cold target. Thealternative condition consisted of escalating the num-bers of the putative specific target cells—now used ascold targets—in the presence of a fixed number ofMSCs then considered as the susceptible target.MSCsdid not compete with antigen-specific T cell cyto-toxicity because the killing of peptide-pulsed T2 cellswas not affected by the presence of MSCs (Fig. 5A).The same results were obtained using NK cells(Fig. 5B). In contrast, the presence of the putativetarget cells markedly reducedMSC killing in a dose-dependentmanner in both systems (Fig. 5, C andD).Our data show that MSC killing does not interferewith the primary recognition of the cognate antigen.
Apoptotic MSCs are immunosuppressive ina TH2-type inflammation modelOur data imply that, because MSC killing does notinterferewith the primary recognition of the cognateantigen, induction of apoptosis must be prominent-ly involved in the immunosuppressive activity. Ac-cordingly, in the GvHD model described above,MSC apoptosis produced by recipient cytotoxic cellsis required for immunosuppression. Therefore, weasked whether this causative relationship remainsvalid in a different disease model associated withnoncytotoxic type 2 T helper cell (TH2)–mediatedinflammation. We selected the model of ovalbumin(OVA)–induced allergic airway inflammation (32)summarized in fig. S6A. Although cytotoxic im-mune cells have been implicated as contributing
a a
0
20
40
60
80
100
PBMC/MSCs ratio
2.5:
15:
140
:110
:120
:1
ND ND ND
a
***
******
******
**
nsAp
op
toti
c M
SC
s(%
)
A
Vehicle only
Z-VAD-FMK
– – – –+ +– – – –+ +
0
20
40
60
80
100
***ns
Ap
op
toti
c M
SC
s
(%)
B
+
CD
4cells
PB
MC
s(u
nso
rted
)
+
CD
11b
cells
+
CD
56
cells
+
CD
8cells
0
10
20
30
40
50
60ns
**
**
***
ns
Ap
op
toti
c M
SC
s
(%)
CD
56-C
D8
–d
ep
lete
d
CD
56-d
ep
lete
d
CD
8-d
ep
lete
d
0
10
20
30
40
50
60
Ap
op
toti
c M
SC
s(%
)
***ns
ns*C D
Z-AAD-CMK
EGTA
–
– –
– –
– –+
+
–+
0
20
40
60
80
Ap
op
toti
c M
SC
s
(%)
******
***
ns
*****
ns
E
0
20
40
60
80
100ns
**
Ap
op
toti
c M
SC
s(%
)
F
0
20
40
60
80
100ns
***
nsns
Ap
op
toti
c M
SC
s(%
)
HLA-I neutralizationHLA-II neutralization
––
–– –
– +–
+– +
–+–
G
Transwell + –+0
20
40
60
80
100
***
Ap
op
toti
c M
SC
s(%
)
H
0
20
40
60
80
100
Ap
op
toti
c M
SC
s(%
)
nsI
+
Resting PBMCs PHA-aPBMCs MLR-aPBMCs
MSCsalone
a
MSCs alone MSCs + PHA-aPBMCs
MS
Cs a
lon
e
MS
Cs a
lon
e
PB
MC
s(u
nso
rted
)a
MSCs alone MSCs + MLR-aPBMCs
MSCsalone
MSCs + PHA-aPBMCs MSCs + PHA-aPBMCs + anti-CD178 (10 µg/ml)
MSCs + PHA-aPBMCs + anti-CD178 (100 µg/ml)
a
MSCs alone MSCs + auto PHA-aPBMCs MSCs + allo PHA-aPBMCs
a
MSCs alone MSCs + MLR-aPBMCs
MSCsalone
a
MSCs + PHA-aPBMCs + PKCζ-PS (25 μM) MSCs + PHA-aPBMCs + PKCζ-PS (10 μM) MSCs + PHA-aPBMCs
MSCs + PHA-aPBMCs + PKCζ-PS (75 μM)
Fig. 4. MSC apoptosis is mediated by activated CD8+ and CD56+ cytotoxic cells and is the result of abystander effect. (A) PBMCs from healthy donors (each independent experiment used a different PBMCdonor) were activated using phytohemagglutinin (PHA; PHA-aPBMCs) or MLR (MLR-aPBMCs). RestingPBMCs (gray bars), PHA-aPBMCs (black bars), or MLR-aPBMCs (dashed bars) were incubated with MSCs at theindicated ratios andMSC apoptosis (annexin-V+/7-AAD−) calculated after 4 hours. ND, not done. (B) Apoptosisin MSCs cultivated with MLR-aPBMCs in the presence or absence of the pan-caspase inhibitor Z-VAD-FMK(10 mM) or the corresponding concentration of its vehicle (dimethyl sulfoxide). (C andD) Apoptosis in MSCscultivated with MLR-aPBMCs used as unfractionated or positively selected for CD11b+, CD4+, CD8+, orCD56+ cells (C) or depleted of CD56+, CD8+, or both (D). (E) Apoptosis in MSCs cultivated with MLR-aPBMCsin the presence or absence of the GrB inhibitor Z-AAD-CMK (300 mM) or the perforin inhibitor EGTA (4 mM).(F) Apoptosis in MSCs cultivated with PHA-aPBMCs in the presence or absence of neutralizing concentrations(10 and 100 mg/ml) of FAS-L monoclonal antibody anti-CD178. (G) Apoptosis in MSCs after culture withautologous (black bars) or allogeneic (gray bars) PHA-aPBMCs in the presence or absence of neutralizingdoses of anti–HLA-A, anti–HLA-B, anti–HLA-C, or anti-HLA-DR antibodies. The white bar shows spontaneousapoptosis in MSCs plated alone. (H) Apoptosis in MSCs cultivated with MLR-aPBMCs in direct contact or in aTranswell. (I) Apoptosis inMSCs cultivated with PHA-aPBMCs in the presence or absence of escalating doses(10 to 75 mM) of PKCz-PS (protein kinase Cz–pseudosubstrate). In (B) to (I), the PBMC/MSC ratio was 20:1.Results represent the mean ± SD of three or six (H) independent experiments. Statistics: one-way ANOVAwith Tukey’s multiple comparison test (*P < 0.5, **P < 0.01, and ***P < 0.001).
5 of 11
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
http://stm.sci
Dow
nloaded from
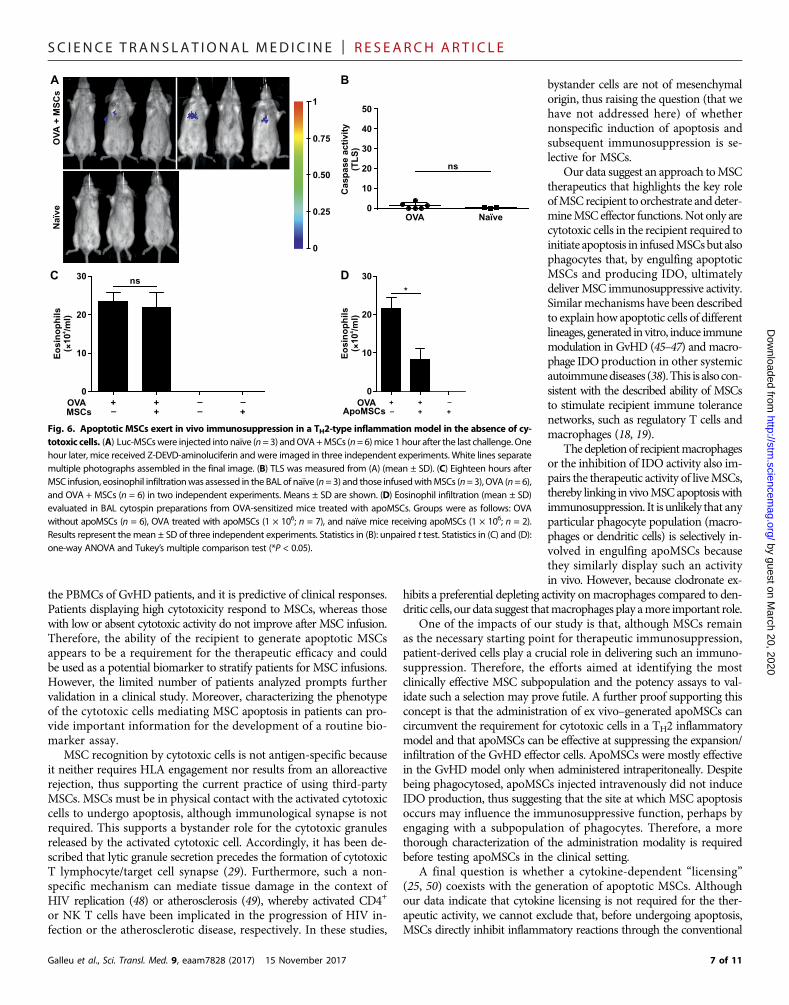
to the induction of this condition (33, 34), CD8+ and NK1.1+ cells infil-trating the bronchoalveolar lavage (BAL) and lung tissues were less than2%1hour after the lastOVAchallenge, whenMSCswere infused (fig. S6,B to E). To confirm the absence ofMSCkilling,mice received luc-MSCsto assess caspase activation after infusion and imaged 1 hour later. Nocaspase activationwas detected in any of themice (Fig. 6, A andB). Highsignal could be detected in all animals receiving control D-luciferin(fig. S6, F and G).
The therapeutic activity, assessed by quantitating the eosinophilinfiltration in the BAL, showed no difference between MSC-treatedand untreated mice (Fig. 6C). Together, these results indicate that,also in this model, MSC immunosuppression relies on the presenceof recipient cytotoxic cells that mediate MSC apoptosis. Therefore,we decided to test whether MSCs, made apoptotic in vitro by exposureto anti-FAS andGrB (apoMSCs), could bypass the need of cytotoxic cellsin the recipient and ameliorate eosinophil infiltration. WhenapoMSCs were administered to recipient mice, we observed that theeosinophil infiltration in BAL was much reduced (Fig. 6D).
Apoptotic MSCs infused in GvHD are immunosuppressiveand induce IDO production in recipient phagocytesWe subsequently investigated whether apoMSCs could be immuno-suppressive also in the GvHD model. ApoMSCs were adminis-tered either intravenously or intraperitoneally, and the infiltrationof CD8+Vb8.3+ Mh T cells was assessed and compared to untreatedGvHD mice. ApoMSCs produced a substantial reduction in GvHDeffector cell infiltration in both the spleen and lungs, but this could
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
by guest on March 2
encemag.org/
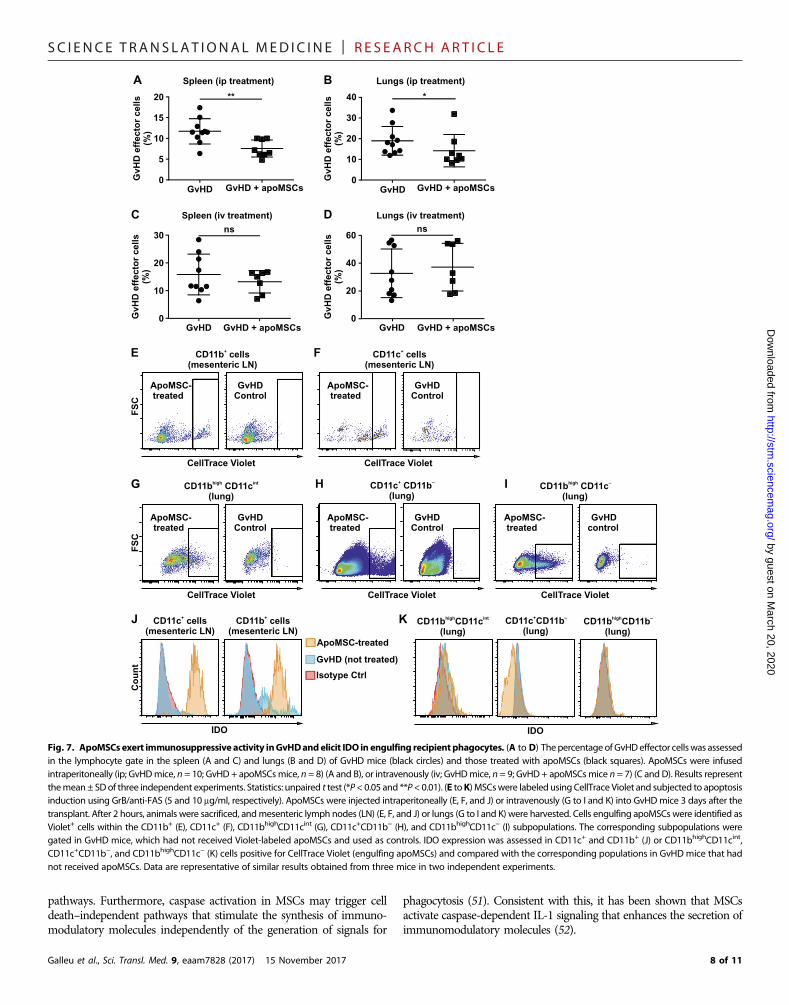
only be observed in those mice treated with apoMSCs infused intra-peritoneally (Fig. 7, A to D).
It has been reported that the injection of irradiated thymocytesinto animals results in their phagocytosis by recipient macrophagesand induction of IDO (35). We therefore tested whether apoMSCsfollowed the same destiny by eliciting in vivo efferocytosis by recipientphagocytes and by inducing IDO production. For this purpose, labeledapoMSCs were traced in recipient phagocytes after injection. Afterintraperitoneal administration, apoMSCs were largely identified insideCD11b+ (Fig. 7E) and CD11c+ (Fig. 7F) phagocytes in the peritonealdraining lymph nodes (36) but absent when searched for in the lungsand spleen. When the intravenous route was used, among the severalphagocytic populations investigated (37), CD11bhighCD11cint,CD11bhighCD11c−, and CD11b−CD11c+ were detected as engulfingapoMSCs in the lungs (Fig. 7, G to I, respectively). The analysis ofIDO expression in the phagocytes engulfing apoMSCs in both theintravenous and intraperitoneal groups revealed that only the phago-cytes in the intraperitoneal group were able to increase IDO expres-sion in comparison with their counterparts in untreated GvHD mice(Fig. 7, J and K). These findings strongly suggest that the immuno-suppressive effect of apoMSCs involves recipient phagocytes andIDO as crucial effector mechanisms.
Recipient-derived IDO-producing phagocytes areindispensable for MSC immunosuppression in GvHDTo directly test the importance of recipient-derived phagocytes orrecipient-produced IDO in MSC immunosuppressive activity, we de-pleted phagocytes, inhibited IDO activity in GvHD mice before MSCtreatment, and evaluated the effect of live MSCs on the expansion ofGvHD effectors. To deplete phagocytes, liposome clodronate was givento mice 72 hours before MSC injection. The treatment substantiallyimpaired the ability of MSCs to suppress Mh T cell infiltration (Fig. 8,A and B). Finally, animals were given the IDO inhibitor 1-methyl-D-tryptophan (1-DMT) (38) before MSC injection. In addition, in this case,the beneficial effect of MSCs on Mh T cell infiltration was much reducedin mice receiving 1-DMT compared to controls (Fig. 8, C and D). Wetherefore conclude that the immunosuppressive effect ofMSCs requiresthe presence of recipient phagocytic cells or IDO production.
0, 2020
DISCUSSIONThis study sheds light on the controversial topic of MSC therapeuticsby identifying a crucial mechanism that potentially explains several un-resolved issues in the field. The first striking piece of information pro-vides the resolution to the paradox that MSCs are therapeuticallyefficacious despite the lack of engraftment (39–41). We demonstratethatMSCs undergo extensive caspase activation and apoptosis after infu-sion in the presence of cytotoxic cells and that this is a requirement for theirimmunosuppressive function. Although other recipient-dependent reac-tions have been described as mediatingMSC lysis in vitro (42) andMSCclearance in vivo (40, 41, 43, 44), our study has shown the instrumentalrole of in vivo MSC apoptosis in delivering immunosuppression afterinfusion. Furthermore, although several studies (38, 45–47) have re-ported the ability of apoptotic cells to modulate immune responses,here, we provide evidence that in vivo naturally occurring cell deathdrives immunosuppression.MSC apoptosis requires and is effected by cytotoxic granules con-tained in recipient cytotoxic cells that also mediate GvHD in recipientmice. The cytotoxic activity against MSCs can also be detected in
8/1
A
Ap
op
toti
c K
562 c
ells
(%)
B
C
8:1
1:1
Ap
op
toti
c M
SC
s(%
)
D
0
10
20
30
*
0
20
40
60
80
100
8:1
1:1
MSC/cold target ratio
4D8/MSCs only
NK/MSCs only
MSC/cold target ratio
Ap
op
toti
c M
SC
s(%
)
*
4D8–clone killing of MSCs Activated–NK cell killing of MSCs
0
20
40
60
80
100
8:1
1:1
T2/cold target ratio
4D8/T2 cells only
4D8–clone killing of specific target
0
20
40
60
80
100
Ap
op
toti
c T
2 c
ells
(%)
NK/K562 cells only
8:1
1:1
K562/cold target ratio
Activated–NK cell killing of K562 cells
Fig. 5. MSCs do not compete with cytotoxic cell recognition of the cognate tar-get. (A) Apoptosis in T2 cells after culture with 4D8 cells at a 4D8/T2 ratio of 20:1.Where indicated, increasing concentrations of MSCs (used as cold target) were added.Apoptotic T2 cells were identified as annexin-V+/7-AAD+ cells. (B) Apoptosis in K562cultured with NK cells (NK/K562 ratio of 20:1). Where indicated, increasing concentra-tions ofMSCs (used a cold target)were added. (C) Apoptosis inMSCs culturedwith 4D8cells (4D8/MSC ratio of 20:1). Where indicated, increasing concentrations of T2 cells(used as cold target) were added. (D) Apoptosis in MSCs cultured with NK cells at anNK/MSC ratio of 20:1. Where indicated, increasing concentrations of K562 (used as coldtarget) were added. In all experiments, apoptosis of MSCs, T2 cells, or K562 cells wasassessed after 4 hours of coculture by flow cytometry. Results represent themean ± SDof three independent experiments. Statistics: one-way ANOVA and Tukey’s multiplecomparison test (*P < 0.05).
6 of 11
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on March 20, 2020
http://stm.sciencem
ag.org/D
ownloaded from
the PBMCs of GvHD patients, and it is predictive of clinical responses.Patients displaying high cytotoxicity respond to MSCs, whereas thosewith low or absent cytotoxic activity do not improve after MSC infusion.Therefore, the ability of the recipient to generate apoptotic MSCsappears to be a requirement for the therapeutic efficacy and couldbe used as a potential biomarker to stratify patients for MSC infusions.However, the limited number of patients analyzed prompts furthervalidation in a clinical study. Moreover, characterizing the phenotypeof the cytotoxic cells mediating MSC apoptosis in patients can pro-vide important information for the development of a routine bio-marker assay.
MSC recognition by cytotoxic cells is not antigen-specific becauseit neither requires HLA engagement nor results from an alloreactiverejection, thus supporting the current practice of using third-partyMSCs. MSCs must be in physical contact with the activated cytotoxiccells to undergo apoptosis, although immunological synapse is notrequired. This supports a bystander role for the cytotoxic granulesreleased by the activated cytotoxic cell. Accordingly, it has been de-scribed that lytic granule secretion precedes the formation of cytotoxicT lymphocyte/target cell synapse (29). Furthermore, such a non-specific mechanism can mediate tissue damage in the context ofHIV replication (48) or atherosclerosis (49), whereby activated CD4+
or NK T cells have been implicated in the progression of HIV in-fection or the atherosclerotic disease, respectively. In these studies,
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
bystander cells are not of mesenchymalorigin, thus raising the question (that wehave not addressed here) of whethernonspecific induction of apoptosis andsubsequent immunosuppression is se-lective for MSCs.
Our data suggest an approach toMSCtherapeutics that highlights the key roleofMSC recipient to orchestrate anddeter-mineMSC effector functions.Not only arecytotoxic cells in the recipient required toinitiate apoptosis in infusedMSCsbut alsophagocytes that, by engulfing apoptoticMSCs and producing IDO, ultimatelydeliverMSC immunosuppressive activity.Similarmechanisms have been describedto explain how apoptotic cells of differentlineages, generated invitro, induce immunemodulation in GvHD (45–47) and macro-phage IDOproduction in other systemicautoimmunediseases (38).This is alsocon-sistent with the described ability of MSCsto stimulate recipient immune tolerancenetworks, such as regulatory T cells andmacrophages (18, 19).
Thedepletionof recipientmacrophagesor the inhibition of IDO activity also im-pairs the therapeutic activity of liveMSCs,thereby linking in vivoMSCapoptosis withimmunosuppression. It is unlikely that anyparticular phagocyte population (macro-phages or dendritic cells) is selectively in-volved in engulfing apoMSCs becausethey similarly display such an activityin vivo. However, because clodronate ex-
hibits a preferential depleting activity onmacrophages compared to den-dritic cells, our data suggest thatmacrophages play amore important role.
One of the impacts of our study is that, although MSCs remainas the necessary starting point for therapeutic immunosuppression,patient-derived cells play a crucial role in delivering such an immuno-suppression. Therefore, the efforts aimed at identifying the mostclinically effective MSC subpopulation and the potency assays to val-idate such a selection may prove futile. A further proof supporting thisconcept is that the administration of ex vivo–generated apoMSCs cancircumvent the requirement for cytotoxic cells in a TH2 inflammatorymodel and that apoMSCs can be effective at suppressing the expansion/infiltration of the GvHD effector cells. ApoMSCs were mostly effectivein the GvHD model only when administered intraperitoneally. Despitebeing phagocytosed, apoMSCs injected intravenously did not induceIDO production, thus suggesting that the site at which MSC apoptosisoccurs may influence the immunosuppressive function, perhaps byengaging with a subpopulation of phagocytes. Therefore, a morethorough characterization of the administration modality is requiredbefore testing apoMSCs in the clinical setting.
A final question is whether a cytokine-dependent “licensing”(25, 50) coexists with the generation of apoptotic MSCs. Althoughour data indicate that cytokine licensing is not required for the ther-apeutic activity, we cannot exclude that, before undergoing apoptosis,MSCs directly inhibit inflammatory reactions through the conventional
0
0.25
0.50
0.75
1
OV
A +
MS
Cs
Naïv
e
A
OVA Naïve0
10
20
30
40
50
Casp
ase a
cti
vit
y(T
LS
)
ns
B
OVAMSCs
+ ++–
0
10
20
30
Eo
sin
op
hil
s4
( ×1
0/m
l)
–
+
––
C
0
10
20
30
OVAApoMSCs
+
–
+
+ +
–
*
Eo
sin
op
hil
s4
( ×1
0/m
l)
Dns
Fig. 6. Apoptotic MSCs exert in vivo immunosuppression in a TH2-type inflammation model in the absence of cy-totoxic cells. (A) Luc-MSCswere injected into naïve (n=3) andOVA+MSCs (n=6)mice 1 hour after the last challenge. Onehour later, mice received Z-DEVD-aminoluciferin and were imaged in three independent experiments. White lines separatemultiple photographs assembled in the final image. (B) TLS was measured from (A) (mean ± SD). (C) Eighteen hours afterMSC infusion, eosinophil infiltrationwas assessed in the BAL of naïve (n=3) and those infusedwithMSCs (n=3), OVA (n=6),and OVA + MSCs (n = 6) in two independent experiments. Means ± SD are shown. (D) Eosinophil infiltration (mean ± SD)evaluated in BAL cytospin preparations from OVA-sensitized mice treated with apoMSCs. Groups were as follows: OVAwithout apoMSCs (n = 6), OVA treated with apoMSCs (1 × 106; n = 7), and naïve mice receiving apoMSCs (1 × 106; n = 2).Results represent the mean ± SD of three independent experiments. Statistics in (B): unpaired t test. Statistics in (C) and (D):one-way ANOVA and Tukey’s multiple comparison test (*P < 0.05).
7 of 11
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on March 20, 2020
http://stm.sciencem
ag.org/D
ownloaded from
pathways. Furthermore, caspase activation in MSCs may trigger celldeath–independent pathways that stimulate the synthesis of immuno-modulatory molecules independently of the generation of signals for
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
phagocytosis (51). Consistent with this, it has been shown that MSCsactivate caspase-dependent IL-1 signaling that enhances the secretion ofimmunomodulatory molecules (52).
GvHD GvHD + apoMSCs0
5
10
15
20 **
B
Gv
HD
eff
ec
tor
ce
lls
(%)
0
10
20
30
40 *
A
GvHD GvHD + apoMSCs
20
30
DC
0
10
ns
GvHD GvHD + apoMSCs0
20
40
60ns
GvHD GvHD + apoMSCs
high intCD11b CD11c
(lung)
+ –CD11c CD11b
(lung)
high –CD11b CD11b
(lung)
K
CellTrace Violet
E F
G H I
J
Gv
HD
eff
ec
tor
ce
lls
(%)
Spleen (ip treatment) Lungs (ip treatment)
Gv
HD
eff
ec
tor
ce
lls
(%)
Gv
HD
eff
ec
tor
ce
lls
(%)
Spleen (iv treatment) Lungs (iv treatment)
FS
C
CellTrace Violet
+CD11b cells
(mesenteric LN)
ApoMSC- treated
GvHD Control
ApoMSC- treated
GvHD Control
+CD11c cells
(mesenteric LN)
CellTrace VioletCellTrace Violet
FS
C
ApoMSC-treated
GvHD Control
ApoMSC- treated
GvHD Control
CellTrace Violet
high intCD11b CD11c
(lung)
+ –CD11c CD11b
(lung)
high –CD11b CD11c
(lung)
ApoMSC- treated
GvHD control
IDO
+CD11c cells
(mesenteric LN)
+CD11b cells
(mesenteric LN)
IDO
Co
un
t
ApoMSC-treated
GvHD (not treated)
Isotype Ctrl
Fig. 7. ApoMSCsexert immunosuppressive activity inGvHDandelicit IDO in engulfing recipientphagocytes. (A to D) Thepercentage ofGvHDeffector cellswas assessedin the lymphocyte gate in the spleen (A and C) and lungs (B and D) of GvHD mice (black circles) and those treated with apoMSCs (black squares). ApoMSCs were infusedintraperitoneally (ip; GvHDmice, n = 10; GvHD + apoMSCsmice, n = 8) (A and B), or intravenously (iv; GvHDmice, n = 9; GvHD + apoMSCsmice n = 7) (C and D). Results representthemean± SDof three independent experiments. Statistics: unpaired t test (*P< 0.05 and **P<0.01). (E toK) MSCswere labeled using CellTrace Violet and subjected to apoptosisinduction using GrB/anti-FAS (5 and 10 mg/ml, respectively). ApoMSCs were injected intraperitoneally (E, F, and J) or intravenously (G to I and K) into GvHDmice 3 days after thetransplant. After 2 hours, animals were sacrificed, andmesenteric lymph nodes (LN) (E, F, and J) or lungs (G to I and K) were harvested. Cells engulfing apoMSCs were identified asViolet+ cells within the CD11b+ (E), CD11c+ (F), CD11bhighCD11cint (G), CD11c+CD11b− (H), and CD11bhighCD11c− (I) subpopulations. The corresponding subpopulations weregated in GvHD mice, which had not received Violet-labeled apoMSCs and used as controls. IDO expression was assessed in CD11c+ and CD11b+ (J) or CD11bhighCD11cint,CD11c+CD11b−, and CD11bhighCD11c− (K) cells positive for CellTrace Violet (engulfing apoMSCs) and compared with the corresponding populations in GvHD mice that hadnot received apoMSCs. Data are representative of similar results obtained from three mice in two independent experiments.
8 of 11

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on March 20, 2020
http://stm.sciencem
ag.org/D
ownloaded from
Our study constitutes a paradigm shift in MSC therapeutics,whereby their apoptotic demise is a key step in the effector mechanismof immunosuppression exerted by MSCs. A further impact of our dis-covery is that the principle underpinning this mechanism could beused as a biomarker to predict clinical responses to MSCs and there-fore stratify GvHD patients for MSC treatment. We therefore believethat the next generation of clinical trials should shift the focus fromchoosing the best MSC population to choosing the patients most likelyto respond. Furthermore, the intriguing possibility that apoMSCs maybe effective in patients refractory to MSCs paves the way to newavenues in the clinical manufacturing of MSCs.
MATERIALS AND METHODSStudy designThis study aimed to verify whether MSCs undergo apoptosis afterinfusion and to test the role played by MSC apoptosis in the initia-tion of recipient-derived tolerogenic immune response. A mousemodel of GvHD, in which the disease is mediated by the expansionand activation of Mh CD8+ T cells in the recipient, was chosen forthree important reasons: It recapitulates a minor mismatch betweendonor and recipient, T cells effecting GvHD can be precisely enu-merated, and there is proof of principle that MSCs are effective intreating GvHD. Furthermore, human MSCs were used to avoid theconfounding effects of recipient cytokines on MSC immunomo-dulating function (10, 50). In this system, murine inflammatory cy-tokines will not cross-react with the corresponding human receptorsand will not activate immunosuppressive molecules in human MSCs
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
(24–26) while retaining the ability to expandmurine effector cells mediat-ing GvHD. Depletion of phagocytes and inhibition of IDO productionwere conceived as loss-of-function experiments to assess the requirementof these factors in the delivery of MSC apoptosis–dependent immuno-suppression. A mouse model of OVA-induced allergic airway inflam-mation was used to assess whether the causative relationship betweencytotoxic cells and MSC apoptosis in the delivery of MSC immuno-suppression is valid in a disease associatedwith TH2-type inflammation.All animal procedures were carried out in compliance with the UKHome Office Animals (Scientific Procedures) Act of 1986.
In all experiments, animals were randomly allocated to controlor experimental groups. No blinding approach was adopted. Nostatistical method was used to predetermine sample size, which wasestimated only on previous experience with assay sensitivity and thedifferent animal models. Unless otherwise specified, three independentexperimental replicates were performed.
To demonstrate that the presence of cytotoxic cells against MSCsin GvHD patients could be predictive of MSC therapeutic activity,samples from GvHD patients were collected and tested for their abilityto induce MSC apoptosis in a cytotoxic assay within 24 hours beforeMSC infusion. At the time of performing the assay and cytofluori-metric analysis, the operator was blind to patients’ clinical details.All patients were affected by steroid-resistant GvHD and received MSCsfor compassionate use. PBMCs from healthy donors were used ascontrols. All samples were collected after informed consent was ob-tained in accordance with the local ethics committee requirements.Primary data are located in table S2.
Mice and disease modelsAcute GvHD was induced as previously described (22). Briefly, afterlethal irradiation (11 gray), recipient C57BL/6 male mice were trans-planted intravenously with 1 × 106 purified CD8+ T cells from fe-male Mh mice, together with 5 × 106 unfractionated BM and 2 ×106 purified CD4+ T cells from female C57BL/6 wild-type donors.The control group received BM and purified CD4+ T cells only.C57BL/6-Prf1tm1Sdz/J (Perforin−/−) mice were purchased from theJackson Laboratory and bred with Mh Rag2−/− mice, and the resultingoffspring was intercrossed for two generations to obtain Mh Rag2−/−
Perf knockout F3 mice. For the depletion of all phagocytes, mice re-ceived 1 mg of liposome clodronate (https://clodronateliposomes.com/)intravenously 72 hours before MSC infusion (35). Recipient IDO ac-tivity was inhibited by using 1-DMT treatment (2 mg/ml; Sigma-Aldrich)in the drinking water starting from 6 days before MSC injection untilanimals were sacrificed (38).
OVA-induced airway inflammation was induced as previously de-scribed (32). Briefly, female Balb/C mice (Harlan Laboratories) wereinjected intraperitoneally with 30 mg of chicken egg albumin (OVA typeV; Sigma-Aldrich) on days 0 and 7. Controls received vehicle (alumi-num hydroxide) only. On days 14, 15, and 16, animals were challengedwith an aerosolized solution of OVA (3%) administered with a DeVilbissUltraNeb 90 for 25min.MSCs or apoMSCswere injected 1 hour after thelast challenge. After additional 18 hours, mice were terminally anesthe-tized, a cannula was inserted into the exposed trachea, and three aliquotsof sterile saline were injected into the lungs to obtain BAL fluid. The totalnumber of cells in the lavage fluid was counted.
MSC preparationsClinical-grade BM-derived human MSCs were generated from BMaspirates collected from the iliac crest of healthy donors. The cells
0
10
20
30
0
10
20
30
40
50
Gv
HD
eff
ec
tor
ce
lls
(%
)
A
ns
ns
B
GvHD GvHD+MSCs GvHD GvHD+MSCs
Gv
HD
eff
ec
tor
ce
lls
(%
)
0
10
20
30
40
50
GvHD GvHD+MSCs
ns
0
5
10
15
20
25
GvHD GvHD+MSCs
ns
C D
Spleen Lung
Spleen Lung
Fig. 8. Recipient phagocytes and IDOproduction are required forMSC immuno-suppressive activity in GvHD. (A and B) GvHD mice were treated with liposomalclodronate 10 min after the transplant. Where indicated, MSCs were infused 3 dayslater. The percentage of GvHD effector cells (CD8+Vb8.3+) was calculated in the lym-phocyte gate in the spleen (A) or lungs (B) after 4 additional days. Mean ± SD wasobtained by grouping three independent experiments with n = 12 (GvHD) and 10(GvHD + MSCs) mice per group. (C and D) GvHD effector cell infiltration was studiedin the spleen (C) and lungs (D) of GvHD mice treated with the IDO inhibitor 1-DMT. Inthe treated mice, MSCs were infused 3 days after the transplant (n = 11). Controls con-sisted of GvHDmice that did not receive MSCs (n = 9). Percentage of CD8+Vb8.3+ cellsrefers to the lymphocyte population. Results refer to the mean ± SD of threeindependent experiments. Statistics: unpaired t test (*P < 0.05 and **P < 0.01).
9 of 11

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on March 20, 2020
http://stm.sciencem
ag.org/D
ownloaded from
were plated at a density of 10 to 25 million/636 cm2. After 3 days at37°C and 5% CO2, nonadherent cells were discarded. When cellconfluence of 90 to 100% was achieved, cells were detached withtrypsin-EDTA and reseeded at a density of 5000 cells/cm2. MSCs fromdifferent donors were used at passage 2 for all in vivo experiments,whereas they were used at passage 8 for the in vitro experiments. Inthe latter case, we did not observe any difference in terms of apoptosissusceptibility between different passages. In each experiment, MSCswere derived from a single expansion and not pooled.
PatientsBetween November 2012 and July 2016, 16 patients affected by steroid-resistant GvHD were treated with MSCs according to Regulation(European Commission) no. 1394/2007. All patients received GvHDprophylaxis. Of the 16 patients included in the study, 13 developedGvHDafter hematopoietic stem cell transplantation, and the remaining3 developed GvHD after donor lymphocyte infusion. Twelve patientswere affected by acuteGvHD, 3were affected by late onset acuteGvHD,and 1was affected by chronicGvHD.The diagnosis ofGvHDwasmadeon histological criteria and GvHD staged according to standard criteria(53, 54). Patient characteristics are summarized in table S1. Sampleswere collected within 24 hours before MSC injection.
StatisticsResults were expressed as mean ± SD. The unpaired Student’s t testwas performed to compare two mean values. One-way ANOVA andTukey’s multiple comparison tests were used to compare three ormore mean values. A probability of null hypothesis less than 5%(P < 0.05; two-sided) was considered statistically significant. Seethe Supplementary Materials for the full experimental procedures.
SUPPLEMENTARY MATERIALSwww.sciencetranslationalmedicine.org/cgi/content/full/9/416/eaam7828/DC1Materials and MethodsFig. S1. MSCs can be traced in the lungs of mice after infusion.Fig. S2. Human MSC immunosuppression is not licensed by murine cytokines.Fig. S3. MSC apoptosis is induced by cytotoxic cells.Fig. S4. Cytotoxicity against MSCs varies among PBMC donors but is independent on thepercentage of CD8+ or CD56+ in GvHD patients.Fig. S5. MSC killing is mediated by caspase 3 and effected by GrB and perforin.Fig. S6. Infused MSCs can be imaged in the lungs of mice with TH2-type lung inflammation.Table S1. Clinical features of GvHD patients.Table S2. Primary data.Video S1. Living-cell imaging of fluorescence resonance energy transfer (FRET)–MSCs plated alone.Video S2. Living-cell imaging of FRET-MSCs plated with PHA-aPBMCs.Video S3. Living-cell imaging of FRET-MSCs plated with resting PBMCs.Video S4. Living-cell imaging of FRET-MSCs plated with PHA-aPBMCs in the presence of thepan-caspase inhibitor Z-VAD-FMK.Video S5. Living-cell imaging of FRET-MSCs plated with PHA-aPBMCs in the presence of theGrB inhibitor Z-AAD-CMK.Video S6. Living-cell imaging of FRET-MSCs plated with PHA-aPBMCs in the presence of theperforin inhibitor EGTA.References (55–57)
REFERENCES AND NOTES1. M. Krampera, S. Glennie, J. Dyson, D. Scott, R. Laylor, E. Simpson, F. Dazzi, Bone marrow
mesenchymal stem cells inhibit the response of naive and memory antigen-specificT cells to their cognate peptide. Blood 101, 3722–3729 (2003).
2. R. Ciccocioppo, M. E. Bernardo, A. Sgarella, R. Maccario, M. A. Avanzini, C. Ubezio,A. Minelli, C. Alvisi, A. Vanoli, F. Calliada, P. Dionigi, C. Perotti, F. Locatelli, G. R. Corazza,Autologous bone marrow-derived mesenchymal stromal cells in the treatment offistulising Crohn’s disease. Gut 60, 788–798 (2011).
3. F. Dazzi, M. Krampera, Mesenchymal stem cells and autoimmune diseases. Best Pract. Res.Clin. Haematol. 24, 49–57 (2011).
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
4. F. von Dalowski, M. Kramer, M. Wermke, R. Wehner, C. Röllig, N. Alakel, F. Stölzel,S. Parmentier, K. Sockel, M. Krech, M. Schmitz, U. Platzbecker, J. Schetelig, M. Bornhäuser,M. von Bonin, Mesenchymal stromal cells for treatment of acute steroid-refractorygraft versus host disease: Clinical responses and long-term outcome. Stem Cells34, 357–366 (2016).
5. J. Tan, W. Wu, X. Xu, L. Liao, F. Zheng, S. Messinger, X. Sun, J. Chen, S. Yang, J. Cai, X. Gao,A. Pileggi, C. Ricordi, Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: A randomized controlled trial. JAMA 307, 1169–1177 (2012).
6. S. Glennie, I. Soeiro, P. J. Dyson, E. W.-F. Lam, F. Dazzi, Bone marrow mesenchymal stem cellsinduce division arrest anergy of activated T cells. Blood 105, 2821–2827 (2005).
7. R. Ramasamy, H. Fazekasova, E. W.-F. Lam, I. Soeiro, G. Lombardi, F. Dazzi, Mesenchymalstem cells inhibit dendritic cell differentiation and function by preventing entryinto the cell cycle. Transplantation 83, 71–76 (2007).
8. R. Ramasamy, C. K. Tong, H. F. Seow, S. Vidyadaran, F. Dazzi, The immunosuppressiveeffects of human bone marrow-derived mesenchymal stem cells target T cellproliferation but not its effector function. Cell. Immunol. 251, 131–136 (2008).
9. R. Meisel, A. Zibert, M. Laryea, U. Göbel, W. Däubener, D. Dilloo, Human bone marrowstromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase–mediated tryptophan degradation. Blood 103, 4619–4621 (2004).
10. J. Su, X. Chen, Y. Huang, W. Li, J. Li, K. Cao, G. Cao, L. Zhang, F. Li, A. I. Roberts,H. Kang, P. Yu, G. Ren, W. Ji, Y. Wang, Y. Shi, Phylogenetic distinction of iNOS and IDOfunction in mesenchymal stem cell-mediated immunosuppression in mammalianspecies. Cell Death Differ. 21, 388–396 (2014).
11. M. Di Nicola, C. Carlo-Stella, M. Magni, M. Milanesi, P. D. Longoni, P. Matteucci, S. Grisanti,A. M. Gianni, Human bone marrow stromal cells suppress T-lymphocyte proliferationinduced by cellular or nonspecific mitogenic stimuli. Blood 99, 3838–3843 (2002).
12. S. Aggarwal, M. F. Pittenger, Human mesenchymal stem cells modulate allogeneicimmune cell responses. Blood 105, 1815–1822 (2005).
13. F. Morandi, L. Raffaghello, G. Bianchi, F. Meloni, A. Salis, E. Millo, S. Ferrone, V. Barnaba,V. Pistoia, Immunogenicity of human mesenchymal stem cells in HLA-class I-restrictedT-cell responses against viral or tumor-associated antigens. Stem Cells 26, 1275–1287 (2008).
14. Z. Selmani, A. Naji, I. Zidi, B. Favier, E. Gaiffe, L. Obert, C. Borg, P. Saas, P. Tiberghien,N. Rouas-Freiss, E. D. Carosella, F. Deschaseaux, Human leukocyte antigen-G5 secretion byhuman mesenchymal stem cells is required to suppress T lymphocyte and natural killerfunction and to induce CD4+CD25highFOXP3+ regulatory T cells. StemCells 26, 212–222 (2008).
15. H. Choi, R. H. Lee, N. Bazhanov, J. Y. Oh, D. J. Prockop, Anti-inflammatory proteinTSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis bydecreasing TLR2/NF-kB signaling in resident macrophages. Blood 118, 330–338 (2011).
16. C. Prevosto, M. Zancolli, P. Canevali, M. R. Zocchi, A. Poggi, Generation of CD4+ or CD8+
regulatory T cells upon mesenchymal stem cell-lymphocyte interaction. Haematologica92, 881–888 (2007).
17. K. English, J. M. Ryan, L. Tobin, M. J. Murphy, F. P. Barry, B. P. Mahon, Cell contact,prostaglandin E2 and transforming growth factor beta 1 play non-redundant roles inhuman mesenchymal stem cell induction of CD4+CD25High forkhead box P3+ regulatoryT cells. Clin. Exp. Immunol. 156, 149–160 (2009).
18. S. M. Melief, E. Schrama, M. H. Brugman, M. M. Tiemessen, M. J. Hoogduijn, W. E. Fibbe,H. Roelofs, Multipotent stromal cells induce human regulatory T cells through a novelpathway involving skewing of monocytes toward anti-inflammatory macrophages.Stem Cells 31, 1980–1991 (2013).
19. K. Németh, A. Leelahavanichkul, P. S. T. Yuen, B. Mayer, A. Parmelee, K. Doi, P. G. Robey,K. Leelahavanichkul, B. H. Koller, J. M. Brown, X. Hu, I. Jelinek, R. A. Star, É. Mezey, Bone marrowstromal cells attenuate sepsis via prostaglandin E2–dependent reprogramming of hostmacrophages to increase their interleukin-10 production. Nat. Med. 15, 42–49 (2009).
20. R. H. Lee, A. A. Pulin, M. J. Seo, D. J. Kota, J. Ylostalo, B. L. Larson, L. Semprun-Prieto,P. Delafontaine, D. J. Prockop, Intravenous hMSCs improve myocardial infarction in micebecause cells embolized in lung are activated to secrete the anti-inflammatory proteinTSG-6. Cell Stem Cell 5, 54–63 (2009).
21. I. B. Resnick, C. Barkats, M. Y. Shapira, P. Stepensky, A. I. Bloom, A. Shimoni, D. Mankuta,N. Varda-Bloom, L. Rheingold, M. Yeshurun, B. Bielorai, A. Toren, T. Zuckerman,A. Nagler, R. Or, Treatment of severe steroid resistant acute GVHD with mesenchymalstromal cells (MSC). Am. J. Blood Res. 3, 225–238 (2013).
22. T. Toubai, I. Tawara, Y. Sun, C. Liu, E. Nieves, R. Evers, T. Friedman, R. Korngold,P. Reddy, Induction of acute GVHD by sex-mismatched H-Y antigens in the absenceof functional radiosensitive host hematopoietic-derived antigen-presenting cells. Blood119, 3844–3853 (2012).
23. J. A. Ankrum, J. F. Ong, J. M. Karp, Mesenchymal stem cells: Immune evasive, not immuneprivileged. Nat. Biotechnol. 32, 252–260 (2014).
24. M. Krampera, L. Cosmi, R. Angeli, A. Pasini, F. Liotta, A. Andreini, V. Santarlasci,B. Mazzinghi, G. Pizzolo, F. Vinante, P. Romagnani, E. Maggi, S. Romagnani, F. Annunziato,Role for interferon-g in the immunomodulatory activity of human bone marrowmesenchymal stem cells. Stem Cells 24, 386–398 (2006).
10 of 11

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on March 20, 2020
http://stm.sciencem
ag.org/D
ownloaded from
25. G. Ren, L. Zhang, X. Zhao, G. Xu, Y. Zhang, A. I. Roberts, R. C. Zhao, Y. Shi, Mesenchymalstem cell-mediated immunosuppression occurs via concerted action of chemokinesand nitric oxide. Cell Stem Cell 2, 141–150 (2008).
26. R. S. Waterman, S. L. Tomchuck, S. L. Henkle, A. M. Betancourt, A new mesenchymalstem cell (MSC) paradigm: Polarization into a pro-inflammatory MSC1 or anImmunosuppressive MSC2 phenotype. PLOS ONE 5, e10088 (2010).
27. J. Fu, D. Wang, Y. Yu, J. Heinrichs, Y. Wu, S. Schutt, K. Kaosaard, C. Liu, K. Haarberg,D. Bastian, D. G. McDonald, C. Anasetti, X.-Z. Yu, T-bet is critical for the development ofacute graft-versus-host disease through controlling T cell differentiation and function.J. Immunol. 194, 388–397 (2015).
28. M. von Bonin, A. Kiani, U. Platzbecker, J. Schetelig, K. Hölig, U. Oelschlägel, C. Thiede,G. Ehninger, M. Bornhäuser, Third-party mesenchymal stem cells as part of themanagement of graft-failure after haploidentical stem cell transplantation. Leuk. Res. 33,e215–e217 (2009).
29. F. Bertrand, S. Muller, K. H. Roh, C. Laurent, L. Dupre, S. Valitutti, An initial and rapidstep of lytic granule secretion precedes microtubule organizing center polarizationat the cytotoxic T lymphocyte/target cell synapse. Proc. Natl. Acad. Sci. U.S.A. 110,6073–6078 (2013).
30. N. A. Hosken, M. J. Bevan, Defective presentation of endogenous antigen by a cell lineexpressing class I molecules. Science 248, 367–370 (1990).
31. B. B. Lozzio, C. B. Lozzio, Properties and usefulness of the original K-562 humanmyelogenous leukemia cell line. Leuk. Res. 3, 363–370 (1979).
32. Y. Riffo-Vasquez, A. R. M. Coates, C. P. Page, D. Spina, Mycobacterium tuberculosischaperonin 60.1 inhibits leukocyte diapedesis in a murine model of allergic lunginflammation. Am. J. Respir. Cell Mol. Biol. 47, 245–252 (2012).
33. E. Hamelmann, A. Oshiba, J. Paluh, K. Bradley, J. Loader, T. A. Potter, G. L. Larsen,E. W. Gelfand, Requirement for CD8+ T cells in the development of airwayhyperresponsiveness in a marine model of airway sensitization. J. Exp. Med. 183,1719–1729 (1996).
34. M. Korsgren, C. G. A. Persson, F. Sundler, T. Bjerke, T. Hansson, B. J. Chambers, S. Hong,L. Van Kaer, H.-G. Ljunggren, O. Korsgren, Natural killer cells determine developmentof allergen-induced eosinophilic airway inflammation in mice. J. Exp. Med. 189,553–562 (1999).
35. T. L. McGaha, Y. Chen, B. Ravishankar, N. van Rooijen, M. C. I. Karlsson, Marginal zonemacrophages suppress innate and adaptive immunity to apoptotic cells in the spleen.Blood 117, 5403–5412 (2011).
36. C. P. Parungo, D. I. Soybel, Y. L. Colson, S.-W. Kim, S. Ohnishi, A. M. De Grand,R. G. Laurence, E. G. Soltesz, F. Y. Chen, L. H. Cohn, M. G. Bawendi, J. V. Frangioni,Lymphatic drainage of the peritoneal space: A pattern dependent on bowel lymphatics.Ann. Surg. Oncol. 14, 286–298 (2007).
37. M. Guilliams, I. De Kleer, S. Henri, S. Post, L. Vanhoutte, S. De Prijck, K. Deswarte,B. Malissen, H. Hammad, B. N. Lambrecht, Alveolar macrophages develop from fetalmonocytes that differentiate into long-lived cells in the first week of life via GM-CSF.J. Exp. Med. 210, 1977–1992 (2013).
38. B. Ravishankar, H. Liu, R. Shinde, P. Chandler, B. Baban, M. Tanaka, D. H. Munn,A. L. Mellor, M. C. I. Karlsson, T. L. McGaha, Tolerance to apoptotic cells is regulated byindoleamine 2,3-dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 109, 3909–3914 (2012).
39. L. von Bahr, I. Batsis, G. Moll, M. Hägg, A. Szakos, B. Sundberg, M. Uzunel, O. Ringden,K. Le Blanc, Analysis of tissues following mesenchymal stromal cell therapy in humansindicates limited long-term engraftment and no ectopic tissue formation. Stem Cells30, 1575–1578 (2012).
40. N. Eliopoulos, J. Stagg, L. Lejeune, S. Pommey, J. Galipeau, Allogeneic marrow stromalcells are immune rejected by MHC class I- and class II-mismatched recipient mice.Blood 106, 4057–4065 (2005).
41. A. J. Nauta, G. Westerhuis, A. B. Kruisselbrink, E. G. A. Lurvink, R. Willemze, W. E. Fibbe,Donor-derived mesenchymal stem cells are immunogenic in an allogeneic hostand stimulate donor graft rejection in a nonmyeloablative setting. Blood 108,2114–2120 (2006).
42. G. M. Spaggiari, A. Capobianco, S. Becchetti, M. C. Mingari, L. Moretta, Mesenchymalstem cell-natural killer cell interactions: Evidence that activated NK cells are capable ofkilling MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood107, 1484–1490 (2006).
43. G. Moll, I. Rasmusson-Duprez, L. von Bahr, A.-M. Connolly-Andersen, G. Elgue, L. Funke,O. A. Hamad, H. Lönnies, P. U. Magnusson, J. Sanchez, Y. Teramura, K. Nilsson-Ekdahl,O. Ringden, O. Korsgren, B. Nilsson, K. Le Blanc, Are therapeutic human mesenchymalstromal cells compatible with human blood?. Stem Cells 30, 1565–1574 (2012).
44. Y. Li, F. Lin, Mesenchymal stem cells are injured by complement after their contact withserum. Blood 120, 3436–3443 (2012).
45. K. Mizrahi, I. Yaniv, S. Ash, J. Stein, N. Askenasy, Apoptotic signaling through Fas and TNFreceptors ameliorates GVHD in mobilized peripheral blood grafts. Bone MarrowTransplant. 49, 640–648 (2014).
Galleu et al., Sci. Transl. Med. 9, eaam7828 (2017) 15 November 2017
46. A. E. Morelli, A. T. Larregina, Concise review: Mechanisms behind apoptotic cell-basedtherapies against transplant rejection and graft versus host disease. Stem Cells 34,1142–1150 (2016).
47. M. Florek, E. I. Sega, D. B. Leveson-Gower, J. Baker, A. M. S. Müller, D. Schneidawind,E. Meyer, R. S. Negrin, Autologous apoptotic cells preceding transplantation enhancesurvival in lethal murine graft-versus-host models. Blood 124, 1832–1842 (2014).
48. J. Couturier, A. T. Hutchison, M. A. Medina, C. Gingaras, P. Urvil, X. Yu, C. Nguyen, P. Mahale,L. Lin, C. A. Kozinetz, J. E. Schmitz, J. T. Kimata, T. C. Savidge, D. E. Lewis, HIV replicationin conjunction with granzyme B production by CCR5+ memory CD4 T cells: Implications forbystander cell and tissue pathologies. Virology 462–463, 175–188 (2014).
49. Y. Li, P. Kanellakis, H. Hosseini, A. Cao, V. Deswaerte, P. Tipping, B.-H. Toh, A. Bobik,T. Kyaw, A CD1d-dependent lipid antagonist to NKT cells ameliorates atherosclerosis inApoE–/– mice by reducing lesion necrosis and inflammation. Cardiovasc. Res. 109,305–317 (2016).
50. M. Duijvestein, M. E. Wildenberg, M. M. Welling, S. Hennink, I. Molendijk, V. L. van Zuylen,T. Bosse, A. C. W. Vos, E. S. M. de Jonge-Muller, H. Roelofs, L. van der Weerd,H. W. Verspaget, W. E. Fibbe, A. A. te Velde, G. R. van den Brink, D. W. Hommes,Pretreatment with interferon-g enhances the therapeutic activity of mesenchymalstromal cells in animal models of colitis, Stem Cells 29, 1549–1558 (2011).
51. D. Wallach, T.-B. Kang, S.-H. Yang, A. Kovalenko, The in vivo significance ofnecroptosis: Lessons from exploration of caspase-8 function. Cytokine Growth Factor Rev.25, 157–165 (2014).
52. T. J. Bartosh, J. H. Ylostalo, N. Bazhanov, J. Kuhlman, D. J. Prockop, Dynamic compactionof human mesenchymal stem/precursor cells into spheres self-activates caspase-dependent IL1 signaling to enhance secretion of modulators of inflammation and immunity(PGE2, TSG6, and STC1). Stem Cells 31, 2443–2456 (2013).
53. D. Przepiorka, D. Weisdorf, P. Martin, H. G. Klingemann, P. Beatty, J. Hows, E. D. Thomas,1994 Consensus Conference on acute GVHD grading. Bone Marrow Transplant. 15,825–828 (1995).
54. A. H. Filipovich, D. Weisdorf, S. Pavletic, G. Socie, J. R. Wingard, S. J. Lee, P. Martin, J. Chien,D. Przepiorka, D. Couriel, E. W. Cowen, P. Dinndorf, A. Farrell, R. Hartzman,J. Henslee-Downey, D. Jacobsohn, G. McDonald, B. Mittleman, J. D. Rizzo, M. Robinson,M. Schubert, K. Schultz, H. Shulman, M. Turner, G. Vogelsang, M. E. D. Flowers, NationalInstitutes of Health consensus development project on criteria for clinical trials in chronicgraft-versus-host disease: I. Diagnosis and staging working group report. Biol. BloodMarrow Transplant. 11, 945–956 (2005).
55. A. Valujskikh, O. Lantz, S. Celli, P. Matzinger, P. S. Heeger, Cross-primed CD8+ T cellsmediate graft rejection via a distinct effector pathway. Nat. Immunol. 3, 844–851 (2002).
56. J.-L. Chen, A. J. Morgan, G. Stewart-Jones, D. Shepherd, G. Bossi, L. Wooldridge,S. L. Hutchinson, A. K. Sewell, G. M. Griffiths, P. A. van der Merwe, E. Y. Jones, A. Galione,V. Cerundolo, Ca2+ release from the endoplasmic reticulum of NY-ESO-1–specificT cells is modulated by the affinity of TCR and by the use of the CD8 coreceptor.J. Immunol. 184, 1829–1839 (2010).
57. L. He, X. Wu, F. Meylan, D. P. Olson, J. Simone, D. Hewgill, R. Siegel, P. E. Lipsky,Monitoring caspase activity in living cells using fluorescent proteins and flow cytometry.Am. J. Pathol. 164, 1901–1913 (2004).
Acknowledgments: We would like to thank D. Spina (Institute of Pharmaceutical Science,King’s College London) for the help and support for the statistical analysis. Funding:This work was supported by the Bloodwise Specialist Programme (14019 and 12006).A.G. is a recipient of a Bloodwise Clinical Research Training Fellowship (15029). Authorcontributions: A.G, H.W., and F.D. conceptualized the study. A.G., F.D, C.B., Y.R.-V.,and R.C. developed the methodology. A.G. and L.D. carried out the formal analysis. A.G.,Y.R.-V., C.T., C.L., L.B., S.W., K.H., T.S.C., and L.W. conducted the investigation. F.D., K.O.,D.I.M., J.A., M.v.B., and M.B. provided the resources. A.G. and L.D. carried out thevisualization. A.G. and F.D. wrote the original draft of the manuscript. A.G., C.B., G.L., F.M.W.,H.W., and F.D. reviewed and edited the manuscript. F.D. acquired the funding andsupervised the study. Competing interests: The authors declare that they have nocompeting interests. Data and materials availability: Reasonable requests for additionaldata or materials will be fulfilled under appropriate agreements.
Submitted 16 January 2017Resubmitted 23 June 2017Accepted 16 August 2017Published 15 November 201710.1126/scitranslmed.aam7828
Citation: A. Galleu, Y. Riffo-Vasquez, C. Trento, C. Lomas, L. Dolcetti, T. S. Cheung, M. von Bonin,L. Barbieri, K. Halai, S. Ward, L. Weng, R. Chakraverty, G. Lombardi, F. M. Watt, K. Orchard,D. I. Marks, J. Apperley, M. Bornhauser, H. Walczak, C. Bennett, F. Dazzi, Apoptosis inmesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl.Med. 9, eaam7828 (2017).
11 of 11

immunomodulationApoptosis in mesenchymal stromal cells induces in vivo recipient-mediated
Orchard, David I. Marks, Jane Apperley, Martin Bornhauser, Henning Walczak, Clare Bennett and Francesco DazziLaura Barbieri, Krishma Halai, Sophie Ward, Ling Weng, Ronjon Chakraverty, Giovanna Lombardi, Fiona M. Watt, Kim Antonio Galleu, Yanira Riffo-Vasquez, Cristina Trento, Cara Lomas, Luigi Dolcetti, Tik Shing Cheung, Malte von Bonin,
DOI: 10.1126/scitranslmed.aam7828, eaam7828.9Sci Transl Med
therapy before transfer.provide important mechanistic insight but also suggest that patients could be screened for responsiveness to MSCfrom patients that responded to MSC therapy had more cytotoxic activity against MSCs. These findings not only
cellsof graft-versus-host disease that cytotoxic cells rendered the MSCs apoptotic shortly after transfer. Moreover, mystery, but Galleu and colleagues now suggest that MSC apoptosis is crucial. They observed in a mouse modelundetectable shortly after transfer. The immunosuppressive mechanism of MSCs has been somewhat of a
Transfer of mesenchymal stromal cells (MSCs) induces immunosuppression, although the cells areMSC sacrifice for immunosuppression
ARTICLE TOOLS http://stm.sciencemag.org/content/9/416/eaam7828
MATERIALSSUPPLEMENTARY http://stm.sciencemag.org/content/suppl/2017/11/13/9.416.eaam7828.DC1
CONTENTRELATED
http://science.sciencemag.org/content/sci/363/6431/1050.fullhttp://stm.sciencemag.org/content/scitransmed/11/480/eaat2189.fullhttp://stm.sciencemag.org/content/scitransmed/10/432/eaai8524.fullhttp://stm.sciencemag.org/content/scitransmed/9/386/eaag2513.fullhttp://stm.sciencemag.org/content/scitransmed/9/372/eaai8269.fullhttp://stm.sciencemag.org/content/scitransmed/9/408/eaan3085.fullhttp://stm.sciencemag.org/content/scitransmed/8/339/339ra71.full
REFERENCES
http://stm.sciencemag.org/content/9/416/eaam7828#BIBLThis article cites 57 articles, 23 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
registered trademark of AAAS. is aScience Translational MedicineScience, 1200 New York Avenue NW, Washington, DC 20005. The title
(ISSN 1946-6242) is published by the American Association for the Advancement ofScience Translational Medicine
of Science. No claim to original U.S. Government WorksCopyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement
by guest on March 20, 2020
http://stm.sciencem
ag.org/D
ownloaded from